
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A solution-processable dissymmetric porous organic cage†
A. G.
Slater
 *a,
M. A.
Little
a,
M. E.
Briggs
a,
K. E.
Jelfs
b and
A. I.
Cooper
a
*a,
M. A.
Little
a,
M. E.
Briggs
a,
K. E.
Jelfs
b and
A. I.
Cooper
a
aDepartment of Chemistry and Materials Innovation Factory, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: anna.slater@liverpool.ac.uk
bDepartment of Chemistry, Imperial College London, South Kensington, London SW7 2AZ, UK
First published on 22nd November 2017
Abstract
Two dissymmetric racemic analogues of the chiral porous organic cage, CC3, were isolated and unambiguously characterised as a racemate pair of the R,R,R,S,S,S and S,S,S,R,R,R-diastereomers (CC3-RS and CC3-SR). CC3-RS/CC3-SR equals the highest porosity measured for CC3 but is an order of magnitude more soluble, making it an excellent candidate for incorporation into a membrane for separation applications.
Design, System, ApplicationThe porous organic cage (POC), CC3, is a promising material for a variety of industrial separations, and has been shown to separate noble gases, hexane isomers, racemic alcohols, and SF6 from N2. CC3 is remarkably hydrolytically stable, but is formed from expensive chiral starting materials, and has limited solubility for further processing (up to 3 mg mL−1 in chloroform) – clear disadvantages when considering industrial applications. We sought to design a cheaper route to CC3 using racemic precursors (£1.36 g−1vs. £42.90 g−1 for racemic vs. chiral diamine), and in the process serendipitously discovered a new dissymmetric isomer of CC3 that is sixteen times more soluble (48 mg mL−1) and retains the desirable porosity characteristics of the original analogue in the solid state. We anticipate this will make the industrial use of porous organic cages more feasible, and open new routes to processing POCs into membranes for separation technologies. This discovery also has implications for high-throughput screening workflows for designing and discovering new POCs, underlining the importance of in situ reaction monitoring for even seemingly simple reactions. |
Introduction
The separation of chemicals and gases from crude mixtures accounts for 10–15% of the world's energy consumption, chiefly due to distillation processes.1 Membrane-based separations can use up to 90% less energy than distillation,2 and hence there is a demand for new materials that can be processed into separation membranes. Molecular cage compounds3,4 are attractive materials for membrane applications, in part due to their solution processability, which can be used to tune their porosity by crystal engineering,5 to deposit them onto a range of substrates, and to blend them with other materials, such as polymers, to form mixed matrix membranes.6 The porous organic cage (POC) CC3 (ref. 7) shows remarkable hydrolytic stability and can separate noble gases,4 SF6 from N2,8 hexane9 and xylene isomers,10 and racemic alcohols and amines.4,9 To date, CC3 is the subject of more than 20 publications.CC3 is a chiral imine cage and is synthesised from 1,3,5-triformylbenzene (TFB) and either R,R- or S,S-cyclohexanediamine (CHDA), resulting in CC3-R or -S, respectively. The sorption and separation capabilities of CC3 in a chiral crystalline form arises from the window-to-window packing of the cages in the structure, which affords a diamondoid pore network, CC3-α, with a narrow pore size distribution (static pore diameter = 3.6 Å).4 The pore topology found in chirally pure CC3 (CC3-R or CC3-S) is also found in its racemate, CC3-R/CC3-S.‡ The latter racemate precipitates immediately when a solution of CC3-R is mixed with a solution of CC3-S (Fig. 1).11 Previous gas phase density functional theory (DFT) dimer calculations showed that heterochiral dimer pairs were more stable than homochiral dimer pairs (−169 kJ mol−1versus −150 kJ mol−1), explaining the rapid precipitation of a stable network on the mixing of enantiomers in solution.11 The marked solid state stabilization of the racemic crystalline CC3-R/CC3-S material with respect to homochiral CC3 was also demonstrated by crystal structure prediction.12 More generally, this chiral window pairing extends to a range of other [4 + 6] tetrahedral imine POCs,11,13,14 and also to non-tetrahedral POCs bearing the same chiral windows, such as linear, tubular POCs.15
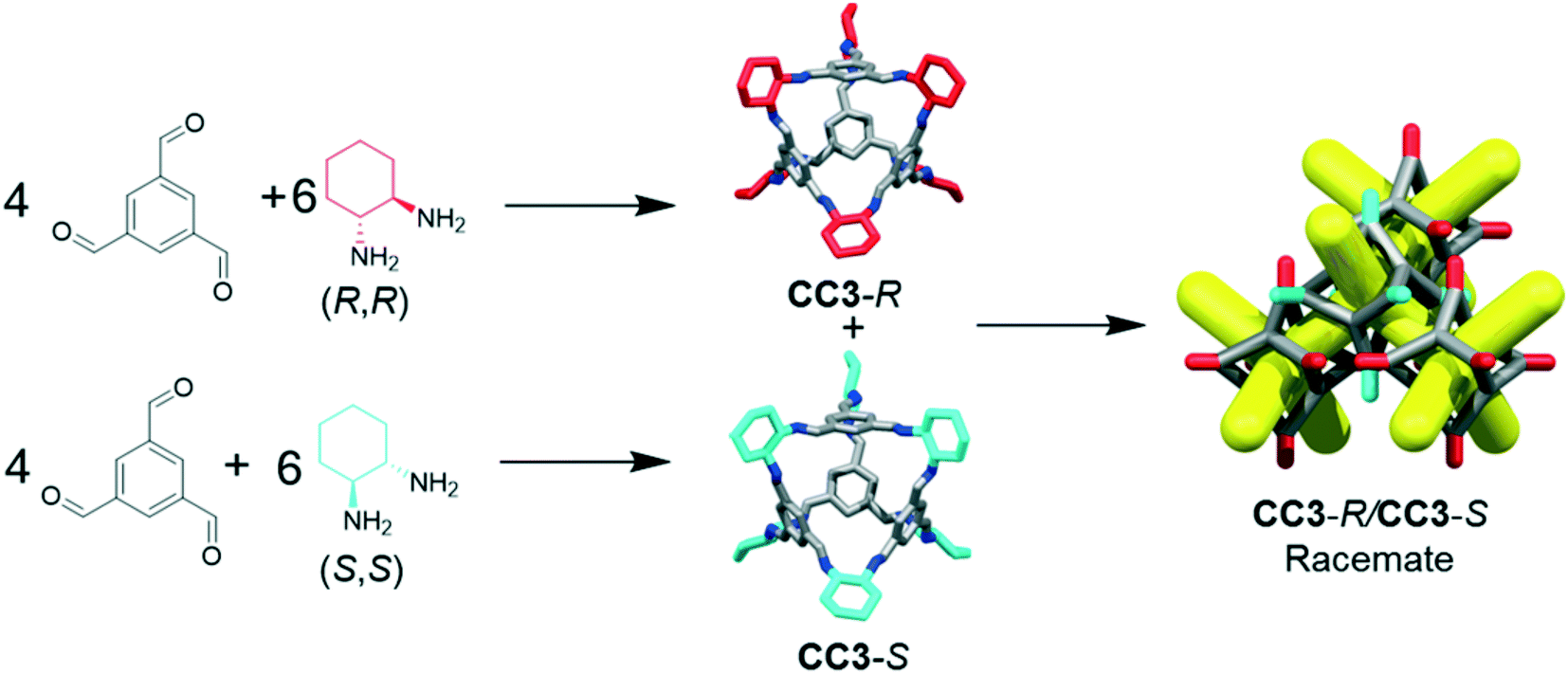 | ||
| Fig. 1 Reaction scheme for formation of CC3-R and CC3-S, which co-crystallises immediately upon mixing to form the racemate CC3-R/CC3-S. For CC3-R, the cyclohexane groups are shown in red; for CC3-S, in turquoise; other C, grey; N, blue; H omitted. The structure on the right is a schematic representation of the desolvated racemic CC3-R/CC3-S co-crystal; diamondoid pore network shown in yellow, simplified cage frame in grey, simplified cyclohexyl vertices in red (−R) and turquoise (−S). | ||
CC3 and its racemate have good hydrolytic stability16 and aminal derivatives of this POC are even stable to strong acids and bases.17 However, for CC3 or similar materials to find use in industrial applications, the synthesis must also be cost-efficient. Enantiomerically pure CHDA is much more expensive than racemic CHDA (£214.50 for 5 g and £12.90 for 10 mL respectively; prices obtained from Sigma-Aldrich®)18 and hence the use of the racemic diamine would provide a significant cost benefit. The racemic POC CC3-R/CC3-S can be accessed by directly reacting TFB with (±)-trans-1,2-cyclohexanediamine (rac-CHDA)19 and we thus sought to synthesise a porous solid in this manner (Fig. 1). The racemic co-crystal was found to precipitate rapidly from the reaction solution as a poorly-soluble crystalline solid (408 mg isolated, 47% yield, ESI† section 2.1; Fig. 4bii for PXRD) — a clear disadvantage for further processing, such as incorporation into membranes. Others have reported the isolation of new stereoisomers of CC3 by collecting the precipitate resulting from the reaction of racemic CHDA with TFB.12 In our experiments, we only observed CC3-R/CC3-S precipitating from solution (characterised by PXRD, Fig. 4bi and ii). However, analysis of the supernatant revealed the presence of two previously unreported diastereomers of CC3, CC3-RS and CC3-SR‡ (Fig. 2; 312 mg isolated, 36% yield, ESI† section 2.1): that is, an asymmetric racemic cage that, unlike CC3-R/CC3-S, retains its solution processability.
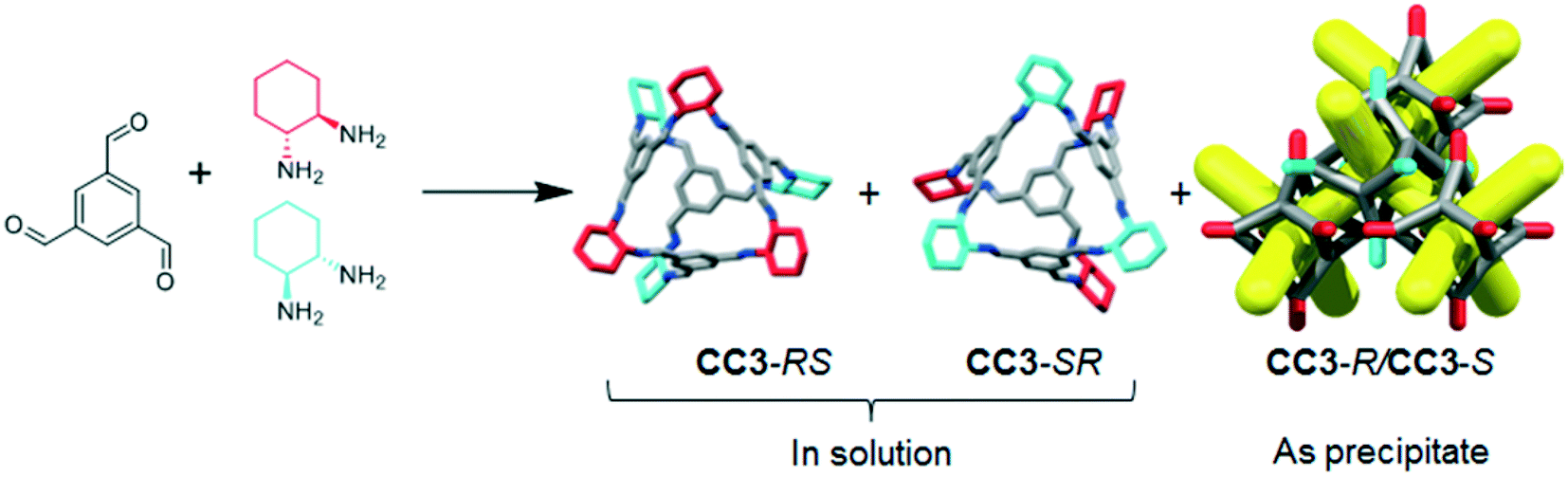 | ||
| Fig. 2 TFB and rac-CHDA react to form CC3-RS and CC3-SR, which are soluble and remain in solution, as well as CC3-R and CC3-S, which immediately co-crystallise to form the racemate, CC3-R/CC3-S, as a white precipitate. Colours as in Fig. 1. | ||
NMR analysis (Fig. 3, ESI† section 2.1.1 and 2.4) suggested that both enantiomers, CC3-RS and CC3-SR, contain three R,R-CHDA vertices and three S,S-CHDA vertices, and that these cages were the majority enantiomers present in solution. The NMR spectra of the asymmetric species, CC3-RS and CC3-SR, are markedly different to homochiral CC3, which enables identification of the two cage geometries. To probe this in more detail and to ascertain if any other species were formed, even transiently, the reaction between TFB and rac-CHDA was analysed by 1H NMR immediately after mixing, and then at 1, 2, 4, and 24 hours (Fig. 3). A solid precipitate formed over the course of the reaction, which was identified by PXRD analysis as the known racemate, CC3-R/CC3-S (Fig. 4bii, red line). NMR analysis of the solution after 1 hour indicated a complex mixture of products that could not be identified unambiguously (Fig. 3 and ESI† Fig. S3 and S4). After 24 hours, an imine-containing cage-like product was the major component remaining in solution.
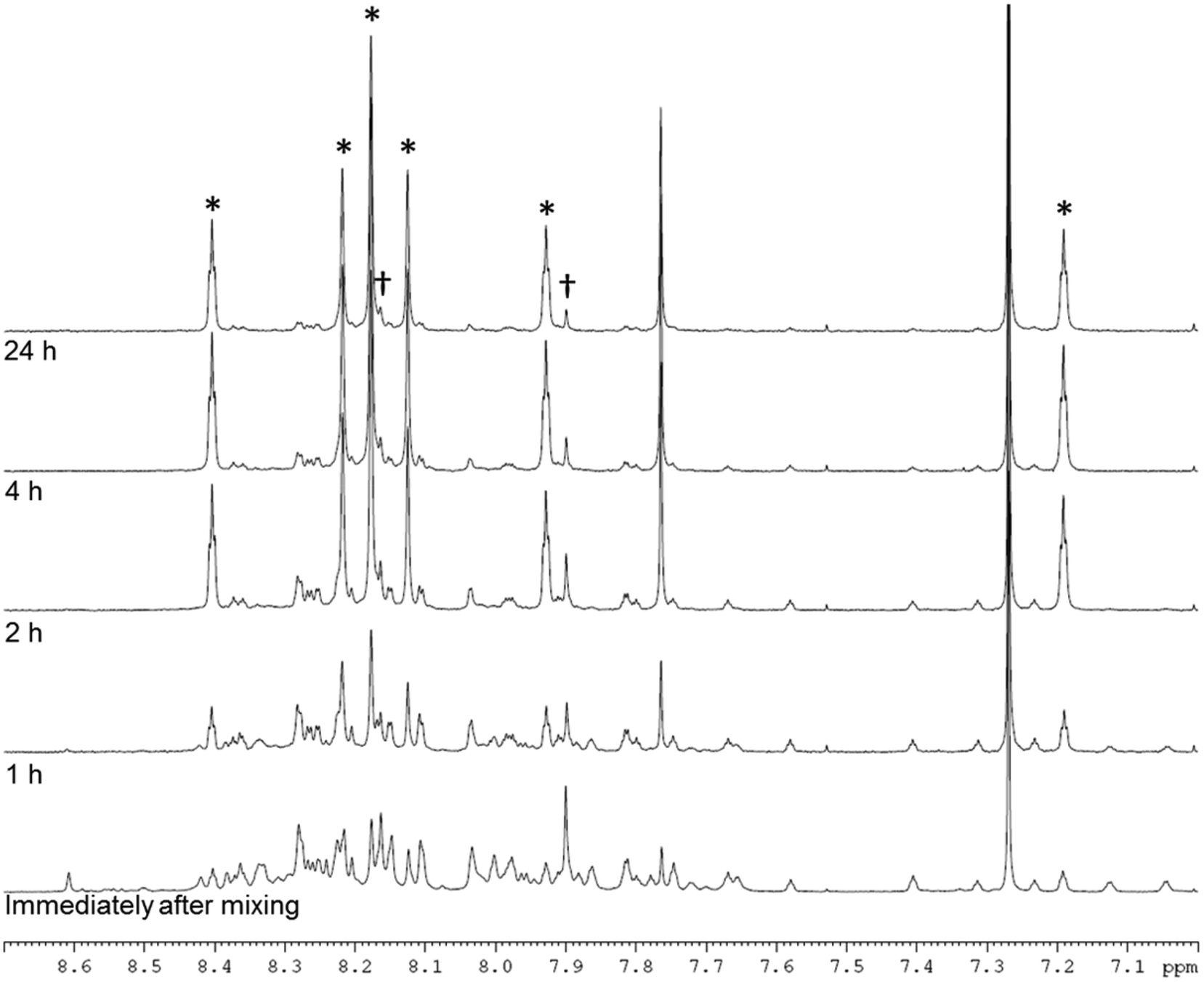 | ||
| Fig. 3 NMR spectrum (400 MHz, CDCl3) between 7.0 and 8.7 ppm of the reaction of TFB + rac-CHDA immediately after mixing, and after 1, 2, 4, and 24 hours. Asterisks indicate peaks arising from CC3-RS and CC3-SR, confirmed by 2D NMR, MS, and analysis of pure cage sample (ESI† section 2); daggers indicate peaks assigned to CC3-R- or -S. | ||
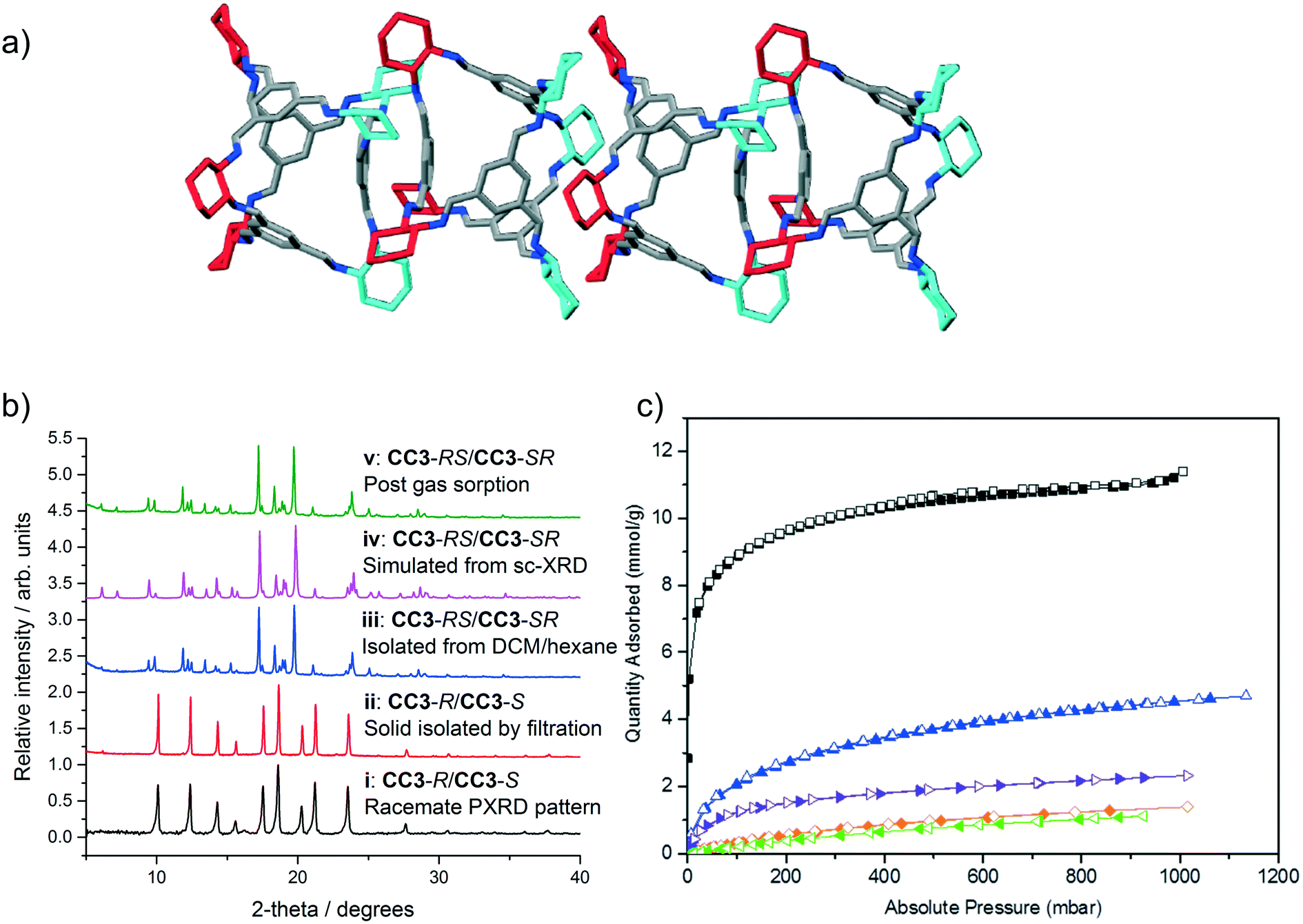 | ||
| Fig. 4 a) Four cages are shown with both arene-to-arene (leftmost cage pair) and window-to-window packing (central cage pair), taken from the single crystal structure of CC3-RS/CC3-SR (ESI† section 2.8). Hydrogen atoms removed for clarity; colouring as in Fig. 1. b) PXRD patterns of (i) known racemate CC3-R/CC3-S, (ii) CC3-R/CC3-S precipitate isolated via filtration; (iii) CC3-RS/CC3-SR isolated by precipitation with hexane, (iv) simulated PXRD from sc-XRD data for CC3-RS/CC3-SR and (v) CC3-RS/CC3-SR post gas sorption measurements. c) Gas sorption isotherms for CC3-RS/CC3-SR for N2 (77 K, black), H2 (77 K, blue), CO2 (295 K, orange), Xe (273 K, purple), and Kr (273 K, green). Filled and open symbols represent adsorption and desorption isotherms respectively. | ||
We have shown previously that the reaction of TFB with a mixture of diamines, such as a binary mixture of cyclohexanediamine and ethylenediamine, results in the formation of a statistical mixture of scrambled cage products.20 In that example, 12 mixed cage products were formed. By contrast, only four of the possible 12 products were isolated in these experiments: CC3-R, CC3-S, CC3-RS, and CC3-SR.
DFT calculations were carried out to assess the relative stabilities of the two isomers (ESI† section 3). CC3-RS was calculated to be 10 kJ mol−1 less stable than CC3-R. As previously reported,11 precipitation of the CC3-R/CC3-S co-crystal (Fig. 1) provides an additional solid state stabilization. That CC3-R/CC3-S is not the only product suggests that both CC3-RS and CC3-SR are kinetically trapped in this configuration, and that the process is insufficiently dynamic, potentially due to the evaporation of catalytic TFA.
To explore whether the presence of CC3-RS and CC3-SR over other possible enantiomers of CC3 was a result of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of R,R- and S,S-CHDA, reactions were performed with varying proportions of each enantiomer of CHDA (5:1, 4:2, 2:4, and 1:5 of R,R-CHDA:S,S-CHDA respectively; each set repeated twice; ESI† section 2.2 and Fig. S5 and S6). After 24 hours, all reactions resulted in either mixtures of homochiral CC3 and CC3-RS/CC3-SR, or, in one case, homochiral CC3, CC3-RS and -SR, and aldehyde starting materials. This strongly suggests that homochiral CC3 and CC3-RS/CC3-SR are more stable than other potential diastereomers such as, for example, a cage containing 1 S,S-CHDA and 5 R,R-CHDA vertices. Such chiral self-sorting has been reported recently in salicylimine cages;21 in that case, both products could be retained in solution in certain solvents, thus allowing a detailed analysis of their relative energies. Such analysis is not possible in our system due to the very low solubility (<1 mg mL−1) of the CC3-R/CC3-S racemate.
:1 ratio of R,R- and S,S-CHDA, reactions were performed with varying proportions of each enantiomer of CHDA (5:1, 4:2, 2:4, and 1:5 of R,R-CHDA:S,S-CHDA respectively; each set repeated twice; ESI† section 2.2 and Fig. S5 and S6). After 24 hours, all reactions resulted in either mixtures of homochiral CC3 and CC3-RS/CC3-SR, or, in one case, homochiral CC3, CC3-RS and -SR, and aldehyde starting materials. This strongly suggests that homochiral CC3 and CC3-RS/CC3-SR are more stable than other potential diastereomers such as, for example, a cage containing 1 S,S-CHDA and 5 R,R-CHDA vertices. Such chiral self-sorting has been reported recently in salicylimine cages;21 in that case, both products could be retained in solution in certain solvents, thus allowing a detailed analysis of their relative energies. Such analysis is not possible in our system due to the very low solubility (<1 mg mL−1) of the CC3-R/CC3-S racemate.
POCs with different geometries have divergent physical properties,7,15 which results from their different crystal packings and the impact of this on pore connectivity, internal cavity size, window configuration, surface area, and porosity. However, it is rare to be able to study two cages with identical chemical composition, but different geometries. Here, we can identify differences that arise purely from the shape of the cage molecules. We therefore investigated the solid-state crystal packing and physical properties of this new CC3-RS/CC3-SR racemate compare to the previously reported racemate, CC3-R/CC3-S.
CC3-RS/CC3-SR was isolated from the reaction mixture in solution by filtration to remove the CC3-R/CC3-S precipitate. The filtrate was then reduced in volume and poured into hexane, resulting in a white precipitate that was isolable by filtration. Unlike CC3-R/CC3-S (solubility of <1 mg mL−1 in CHCl3), and to a lesser extent CC3-R (solubility of 3 mg mL−1),22 the solid product was highly soluble in chloroform (48 mg mL−1, ESI† section 1). The solid CC3-RS/CC3-SR precipitate was found to be crystalline and easily distinguished from CC3-R/CC3-S by PXRD (Fig. 4biii and ii, blue and red line respectively, ESI† section 2.9). Single crystals of the filtrate were grown from DCM/hexane, DCM/acetone, and DCM/MeOH by vial-in-vial crystallisation experiments. Single crystal X-ray diffraction (SCXRD) unambiguously identified the product as a racemate of CC3-RS and CC3-SR which has crystallised in the trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (Fig. 4a, ESI† section 2.8) and the simulated PXRD diffraction pattern of the crystal structure matches with the PXRD pattern of the hexane precipitated filtrate (Fig. 4biii and iv, pink and blue line respectively). CC3-RS and CC3-SR both have one triangular window with an equilateral shape, identical to the windows found in CC3-R (or -S). However, unlike CC3-R (or -S), in CC3-RS and CC3-SR there are three windows with an acute triangle shape. In addition, one aromatic ring is angled towards the centre of the cage cavity (ESI† section 2.8). During crystallisation, CC3-RS and CC3-SR self-sort into 1-D packed arrangements. In the structure, CC3-RS packs in an alternating window-to-window and arene-to-arene packing arrangement (Fig. 4a, central two cages). 1-D packed arrangements of CC3-SR are equivalent and related by inversion symmetry. By contrast, there are no arene-to-arene contacts in chiral CC3 or in the previously reported racemate, CC3-R/CC3-S.
(Fig. 4a, ESI† section 2.8) and the simulated PXRD diffraction pattern of the crystal structure matches with the PXRD pattern of the hexane precipitated filtrate (Fig. 4biii and iv, pink and blue line respectively). CC3-RS and CC3-SR both have one triangular window with an equilateral shape, identical to the windows found in CC3-R (or -S). However, unlike CC3-R (or -S), in CC3-RS and CC3-SR there are three windows with an acute triangle shape. In addition, one aromatic ring is angled towards the centre of the cage cavity (ESI† section 2.8). During crystallisation, CC3-RS and CC3-SR self-sort into 1-D packed arrangements. In the structure, CC3-RS packs in an alternating window-to-window and arene-to-arene packing arrangement (Fig. 4a, central two cages). 1-D packed arrangements of CC3-SR are equivalent and related by inversion symmetry. By contrast, there are no arene-to-arene contacts in chiral CC3 or in the previously reported racemate, CC3-R/CC3-S.
Gas sorption measurements (Fig. 4c) showed CC3-RS/CC3-SR has an apparent BET surface area of 800 m2 g−1 (ESI† section 2.10), as compared to values of 409–819 m2 g−1 and up to 696 m2 g−1 obtained for CC3-R and CC3-R/CC3-S, respectively (the higher values were obtained from rapidly precipitated samples with a high proportion of structural defects).11 As such, the dissymmetric crystalline racemate is more porous than either crystalline form of CC3 reported so far, and its surface area is similar to that reported previously for amorphous CC3.11,23 As the CC3-RS/CC3-SR used for gas sorption was isolated by rapid precipitation, there is likely to be a contribution to porosity from crystal defects in the sample, as seen for rapidly precipitated CC3.11 As much higher concentrations of the dissymmetric cage can be achieved in solution than for CC3, it is likely that crystallisation conditions favouring higher defect inclusion can be achieved via rapid precipitation, potentially achieving even higher gas sorption capacities. The isolated crystalline solid is stable to >300 °C, and it is also stable in neutral solutions in DCM for at least 6 days, thus it can be solution processed without conversion to alternative diastereomers of CC3.
Conclusions
A new diastereomer of the well-known homochiral POC, CC3, has been isolated and identified as CC3-RS. CC3-RS surpasses crystalline CC3-R and CC3-R/CC3-S in terms of both solubility and microporosity, and it does not require the use of expensive homochiral amines. An oft-cited advantage of imine condensation routes to POCs is that the reactions are one-pot in nature. This study shows, however, that the cage-forming mechanism can be more complex than implied by the majority product (Fig. 3). In turn, this suggests that it might be useful to pay more attention to in situ reaction monitoring for these systems in the future.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the EPSRC (EP/N004884/1) and European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013) through grant agreement n. 321156 (ERC-AG-PE5-ROBOT) for funding. KEJ thanks the Royal Society for a University Research Fellowship; AGS thanks the Royal Society and the EPSRC for a Royal Society-EPSRC Dorothy Hodgkin Fellowship. The authors thank Dr Tom Hasell for useful discussions. We acknowledge Stephen Moss and the MicroBioRefinery for assistance with QTOF-MS measurements.Notes and references
- D. S. Sholl and R. P. Lively, Nature, 2016, 533, 316 CrossRef PubMed.
- M. Mulder, Basic principles of membrane technology, Kluwer Academic, Dordrecht, Boston, 2nd edn, 1996 Search PubMed.
- M. Mastalerz, Angew. Chem., Int. Ed., 2010, 49, 5042–5053 CrossRef CAS PubMed.
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed.
- M. A. Little, M. E. Briggs, J. T. A. Jones, M. Schmidtmann, T. Hasell, S. Y. Chong, K. E. Jelfs, L. J. Chen and A. I. Cooper, Nat. Chem., 2015, 7, 153–159 CrossRef CAS PubMed.
- Q. Song, S. Jiang, T. Hasell, M. Liu, S. Sun, A. K. Cheetham, E. Sivaniah and A. I. Cooper, Adv. Mater., 2016, 28, 2629–2637 CrossRef CAS PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- T. Hasell, M. Miklitz, A. Stephenson, M. A. Little, S. Y. Chong, R. Clowes, L. Chen, D. Holden, G. A. Tribello, K. E. Jelfs and A. I. Cooper, J. Am. Chem. Soc., 2016, 138, 1653–1659 CrossRef CAS PubMed.
- A. Kewley, A. Stephenson, L. J. Chen, M. E. Briggs, T. Hasell and A. I. Cooper, Chem. Mater., 2015, 27, 3207–3210 CrossRef CAS.
- T. Mitra, K. E. Jelfs, M. Schmidtmann, A. Ahmed, S. Y. Chong, D. J. Adams and A. I. Cooper, Nat. Chem., 2013, 5, 276–281 CrossRef CAS PubMed.
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 588–598 CrossRef CAS PubMed.
- J. T. A. Jones, T. Hasell, X. F. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed.
- T. Hasell, M. A. Little, S. Y. Chong, M. Schmidtmann, M. E. Briggs, V. Santolini, K. E. Jelfs and A. I. Cooper, Nanoscale, 2017, 9, 6783–6790 RSC.
- T. Hasell, S. Y. Chong, M. Schmidtmann, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 7154–7157 CrossRef CAS PubMed.
- A. G. Slater, M. A. Little, A. Pulido, S. Y. Chong, D. Holden, L. Chen, C. Morgan, X. Wu, G. Cheng, R. Clowes, M. E. Briggs, T. Hasell, K. E. Jelfs, G. M. Day and A. I. Cooper, Nat. Chem., 2017, 9, 17–25 CAS.
- T. Hasell, M. Schmidtmann, C. A. Stone, M. W. Smith and A. I. Cooper, Chem. Commun., 2012, 48, 4689–4691 RSC.
- M. Liu, M. A. Little, K. E. Jelfs, J. T. A. Jones, M. Schmidtmann, S. Y. Chong, T. Hasell and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 7583–7586 CrossRef CAS PubMed.
- http://www.sigmaaldrich.com/catalog/search?interface=All&term=cyclohexanediamine&N=0&focus=product&lang=en®ion=GB, accessed on 7th August 2017.
- G. Zhu, C. D. Hoffman, Y. Liu, S. Bhattacharyya, U. Tumuluri, M. L. Jue, Z. Wu, D. S. Sholl, S. Nair, C. W. Jones and R. P. Lively, Chem. – Eur. J., 2016, 22, 10743–10747 CrossRef CAS PubMed.
- S. Jiang, J. T. Jones, T. Hasell, C. E. Blythe, D. J. Adams, A. Trewin and A. I. Cooper, Nat. Commun., 2011, 2, 207 CrossRef PubMed.
- D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2017, 56, 1244–1248 CrossRef CAS PubMed.
- R. L. Greenaway, D. Holden, E. G. B. Eden, A. Stephenson, C. W. Yong, M. J. Bennison, T. Hasell, M. E. Briggs, S. L. James and A. I. Cooper, Chem. Sci., 2017, 8, 2640–2651 RSC.
- S. Jiang, K. E. Jelfs, D. Holden, T. Hasell, S. Y. Chong, M. Haranczyk, A. Trewin and A. I. Cooper, J. Am. Chem. Soc., 2013, 135, 17818–17830 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1571867 and 1571866. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7me00090a |
| ‡ Notation used throughout the text for CC3 is as follows: CC3-R and CC3-S are enantiomerically pure cages; CC3-R/CC3-S is the racemic co-crystal of CC3-R and CC3-S; CC3-RS and CC3-SR, the new cages reported here, are a racemate pair of diastereomers of CC3; CC3-RS/CC3-SR refers to the co-crystal containing both CC3-RS and CC3-SR. |
| This journal is © The Royal Society of Chemistry 2018 |