
pH responsive glycopolymer nanoparticles for targeted delivery of anti-cancer drugs†
Gokhan
Yilmaz
abc,
Emine
Guler
d,
Caner
Geyik
 e,
Bilal
Demir
d,
Melek
Ozkan
f,
Dilek
Odaci Demirkol
d,
Serdar
Ozcelik
f,
Suna
Timur
*dg and
C. Remzi
Becer
*c
e,
Bilal
Demir
d,
Melek
Ozkan
f,
Dilek
Odaci Demirkol
d,
Serdar
Ozcelik
f,
Suna
Timur
*dg and
C. Remzi
Becer
*c
aDepartment of Chemistry, University of Warwick, CV4 7AL, Coventry, UK
bDepartment of Basic Sciences, Turkish Military Academy, Ankara, Turkey
cPolymer Chemistry Laboratory, School of Engineering and Materials Science, Queen Mary, University of London, E1 4NS, London, UK. E-mail: r.becer@qmul.ac.uk
dDepartment of Biochemistry, Faculty of Science, Ege University, 35100, Bornova, Izmir, Turkey. E-mail: suna.timur@ege.edu.tr
eInstitute of Drug Abuse, Toxicology & Pharmaceutical Sciences, Ege University, 35100, Bornova, Izmir, Turkey
fDepartment of Chemistry, Faculty of Science, Izmir Institute of Technology, 35430, Urla, Izmir, Turkey
gCentral Research Testing and Analysis Laboratory Research and Application Center, Ege University, 35100, Bornova, Izmir, Turkey
First published on 18th October 2017
Abstract
Over the past decade, there has been a great deal of interest in the integration of nanotechnology and carbohydrates. The advances in glyconanotechnology have allowed the creation of different bioactive glyconanostructures for different types of medical applications, especially for drug delivery and release systems. Therefore, the use of more efficient biocompatible nanocarriers with high loading capacity, low overall toxicity and receptor-mediated endocytosis specificity is still in focus for the enhancement of the therapeutic effect. Conjugation of sugar derivatives onto gold nanoparticles presents unique properties that include a wide array of assembling models and size-related electronic, magnetic and optical properties. Here, pH-responsive drug-conjugated glycopolymer-coated gold nanoparticles were prepared by functionalization of gold nanoparticles with thiol-terminated glycopolymers and then subsequent conjugation of doxorubicin (DOX). Among the four different glycopolymers, their drug release, physicochemical characterization (spectroscopy, particle size and surface charge) and in vitro bioapplications with four different cell lines were compared. As a result, pH-sensitive drug delivery via sugar-coated AuNPs was performed thanks to hydrazone linkages between glycopolymers and DOX. Comparative viability tests also demonstrated the efficiency of glycopolymer–DOX conjugates by fluorescence cell imaging. The obtained results reveal that AuNP homoglycopolymer DOX conjugates (P4D) have significant potential, especially in human neuroblastoma cells in comparison to cervical cancer cells and lung cancer cells.
Design, System, ApplicationGlycopolymer coated gold nanoparticles have been designed as pH responsive anticancer drug carriers through biological media and to release the drug under acidic conditions. Functional ‘Sweet or Sugar Coated Gold’ nanostructures were fabricated via functionalization of gold nanoparticles with thiol-terminated glycopolymers and then doxorubicin as an anticancer drug model was conjugated onto the surfaces via pH-sensitive hydrazone bonds. After characterization of the resulting nanoparticle drug conjugates, their cytotoxic properties and cell affinities towards various cancerous cell lines were evaluated. This design could be adapted to the controlled-release of various chemotherapeutics and these ‘Sweet nano structures’ could be applied for both imaging and therapeutic applications. |
Introduction
Advances in the nanotechnology field have been guided by an increasing demand for nanotools that can be applied by way of therapy and non-invasive diagnostics, especially in the field of biomedicine. In the past few years, many different materials have been proposed as biomedical architectures, which have been developed ambitiously for use in disease diagnosis and therapy. Along this purpose, organic-based and metallic-based materials and their composite versions have been studied by numerous researchers and novel techniques for creating these materials have been established to increase the efficacy, robustness, stability or other desirable physical and chemical properties. Among them, organic–inorganic hybrids,1,2 polymers,3,4 liposomes,5,6 hydrogels,7 emulsions8 and metallic-based functionalized materials9,10 have been presented with outstanding abilities.Metal nanoparticle-based materials are appealing alternatives to fluorescence- or radiolabelled protein substrates, owing to their photo-stability, facile synthesis and ability to conjugate with biological molecules.11,12 Among these metallic nanoparticles, gold nanoparticles have paved the way thanks to the advantages such as chemical inertness, facile surface functionalization, amenable surface plasmon resonance and suitable optical properties for imaging.13–17
Concomitantly, there are other significant issues for NPs consisting of their stability and improvement of enhanced permeability and retention effect (EPR) without facing renal elimination and recognition by phagocytes.18 Hence, functionalization of AuNPs by constructing a core–shell architecture with polymeric layers including other recognition elements to facilitate the uptake of NPs has gained attention. To enable these specialties for AuNPs, glycofunctionalization has been a breakthrough both in surface modification for cell adhesion and potential anticancer activities applied with radiation therapy.19,20 Furthermore, prior to their modification on the surface of NPs, glyco-based structures as supramolecules were synthesized and used in bioapplications, successfully.21 The specific recognition properties of carbohydrates and unique physiochemical properties of gold nanoparticles are a desirable combination for the creation of multivalent nanoparticles for the treatment of chemotherapy/radiotherapy and fluorescence imaging. In our previous study, we presented polymethacrylic acid (PMAA) coated AuNPs and their post-modification with doxorubicin (DOX) to create a drug delivery system based on a pH-responsive manner.22 Instead of direct coating via thiol linkages, an additional polymer layer covered the surface of AuNPs before conjugation of DOX molecules and post-modified AuNP–PMAA particles via pH-sensitive hydrazone bonds.
Herein, we successfully synthesized mannose (Man) tagged AuNPs by covering the metallic core with methacrylic acid derivatives as a toolbox for pH-sensitive drug delivery. After defining bioconjugates in terms of size, surface charge and spectrophotometric properties, they were applied to four different cell lines including three cancerous and one healthy ones and their cytotoxic properties and cellular uptake were evaluated by fluorescence cell imaging. The efficacy of polymer-coated AuNPs with Man was also tested in a comprehensive comparison by using only polymer coated AuNPs without Man and free DOX.
Results and discussion
Synthesis of D-mannose methacrylate glycomonomer (ManMac)
The reversible addition-fragmentation chain transfer (RAFT) polymerization is a commonly used technique to polymerize methacrylates. Hence, mannose methacrylate glycomonomers were polymerized via RAFT polymerization. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) was performed for the synthesis of mannose methacrylate monomers. Azide functionalized methacrylate was clicked to mannose alkyne, successfully. The CuAAC click reaction was carried out in the presence of CuSO4·5H2O and (+)-sodium L-ascorbate as a catalyst system in a MeOH/H2O mixture at room temperature for 24 h. The click reaction was monitored and confirmed by 1H- NMR, 13C-NMR and ESI-MS.As depicted in Fig. S1,† the new peak at 8.02 ppm appeared due to the formation of a triazole ring, which confirmed the click reaction between azide and alkyne groups. In addition, the existence of D-mannose C-1 peaks at 102.3 & 104.7 ppm revealed that the monomer is an anomeric mixture according to the 13C-NMR data. In the ESI-MS spectra, there is a clear peak at 387.4 m/z that corresponds to the molecular weight of the mannose glycomonomer with a theoretical mass of 387.2 m/z. The product was obtained as a white solid with a 44% yield. The main reason behind the low yield is the poor solubility of 3-azidopropyl methacrylate in the MeOH/H2O mixture which affects the efficiency of purification on the column.
Synthesis of homo/co-polymers via RAFT
RAFT polymerization of the homo/co-monomers was carried out in the presence of 2-cyano-2-propyl benzodithioate (CPBDT) as a RAFT agent and 4,4′-azobis-(2-methylpropio-nitrile) AIBN as an initiator in a methanol and water mixture (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 70 °C for 12 h. Briefly, a Schlenk tube was charged with targeted monomers (MAA, OEGMA and ManMac) (in total, 100 eq.), CPBDT (1 eq.), AIBN (0.1 eq.) and solvent (3.0 mL), which was degassed by gently bubbling argon gas for 30 min. The Schlenk tube was sealed and the mixed solution was allowed to polymerize. The RAFT homo/copolymerizations have been achieved with good control indicated by the narrow dispersity indices in accordance with the targeted molar mass. After purification, DMF SEC analysis based on PMMA standards revealed a single peak of each polymer without any considerable tailing or shoulder with an apparent Mn = 12.1–32.7 kDa and Mw/Mn = 1.18–1.21 (Table 1). However, some obtained polymers including carboxylic acid moieties showed a small amount of tailing in the low molar mass range in the SEC analysis. This is most likely due to the interaction of the carboxylic acid moieties of the polymers with the SEC column's packing material. In addition, the obtained polymer containing OEGMA presented a small shoulder due to possible coupling reactions. OEGMA was used to prepare a reference material (P3) due to its biocompatibility and hydrophilicity. Additionally, polymers containing OEGMA are suitable for biological applications since they do not trigger an immune response. In order to understand the difference in lectin binding and cell recognition, the composition of the glyco-copolymers was designed to have a minimum of 50% of the carbohydrate units of the overall repeating units.
:1) at 70 °C for 12 h. Briefly, a Schlenk tube was charged with targeted monomers (MAA, OEGMA and ManMac) (in total, 100 eq.), CPBDT (1 eq.), AIBN (0.1 eq.) and solvent (3.0 mL), which was degassed by gently bubbling argon gas for 30 min. The Schlenk tube was sealed and the mixed solution was allowed to polymerize. The RAFT homo/copolymerizations have been achieved with good control indicated by the narrow dispersity indices in accordance with the targeted molar mass. After purification, DMF SEC analysis based on PMMA standards revealed a single peak of each polymer without any considerable tailing or shoulder with an apparent Mn = 12.1–32.7 kDa and Mw/Mn = 1.18–1.21 (Table 1). However, some obtained polymers including carboxylic acid moieties showed a small amount of tailing in the low molar mass range in the SEC analysis. This is most likely due to the interaction of the carboxylic acid moieties of the polymers with the SEC column's packing material. In addition, the obtained polymer containing OEGMA presented a small shoulder due to possible coupling reactions. OEGMA was used to prepare a reference material (P3) due to its biocompatibility and hydrophilicity. Additionally, polymers containing OEGMA are suitable for biological applications since they do not trigger an immune response. In order to understand the difference in lectin binding and cell recognition, the composition of the glyco-copolymers was designed to have a minimum of 50% of the carbohydrate units of the overall repeating units.
| Code | Composition | M n,Theo (g mol−1) | M n,GPC (g mol−1) | Đ |
|---|---|---|---|---|
| P1 | P((MAA)96) | 8400 | 12100 |
1.18 |
| P2 | P((ManMac)46-r-(MAA)44) | 21800 |
24600 |
1.21 |
| P3 | P((ManMac)45-r-(OEGMA)43) | 30500 |
29400 |
1.19 |
| P4 | P((ManMac)94) | 37300 |
32700 |
1.21 |
The conversion values of monomers were calculated using the 1H NMR data by comparing the integrated signal intensity due to the aromatic protons of the RAFT agent at 7.8–7.9 ppm with the vinyl protons of monomers at 5.8–6.2 ppm. 1H NMR characterization revealed that quantitative conversions were achieved for each monomer. The Mn,SEC calculated by SEC measurement is slightly higher or lower than the theoretical molar mass mainly due to the different structures of the obtained polymers according to PMMA calibration standards. The theoretical number average molar mass of the synthesized polymers, Mn,Theo, was calculated by using the equation: Mn,Theo = ([M]0/[RAFT]0 × conversion × Mmon) + MRAFT, where [RAFT]0 is the initial RAFT agent concentration, [M]0 is the initial monomer concentration, Mmon is the monomer molecular weight, and MRAFT is the RAFT agent's molecular weight.
Reduction of the RAFT end group of homo/co-polymers
The RAFT agent terminal group of the obtained glycopolymers was reduced to a thiol-terminal in order to immobilize the synthesized polymers onto gold nanoparticles in the presence of aqueous NaBH4. 1H NMR and SEC were used to analyze the polymers before and after reduction. The disappearance of the aromatic protons of the RAFT agent at 7.8–7.9 ppm after treatment indicated that all end groups in the CPBDT units have been successfully reduced.As depicted in Fig. 1 and S2,† no obvious changes in the SEC traces were observed and the Mn,SEC was exactly similar to that before treatment according to RI detection of SEC. However, variable wavelength detection of SEC at 308 nm did not show any significant peak after treatment suggesting that the polymer end groups are successfully modified to –SH. Moreover, the color of polymers which is due to the RAFT agent changed from pink to white after the successful cleavage of the RAFT end group.
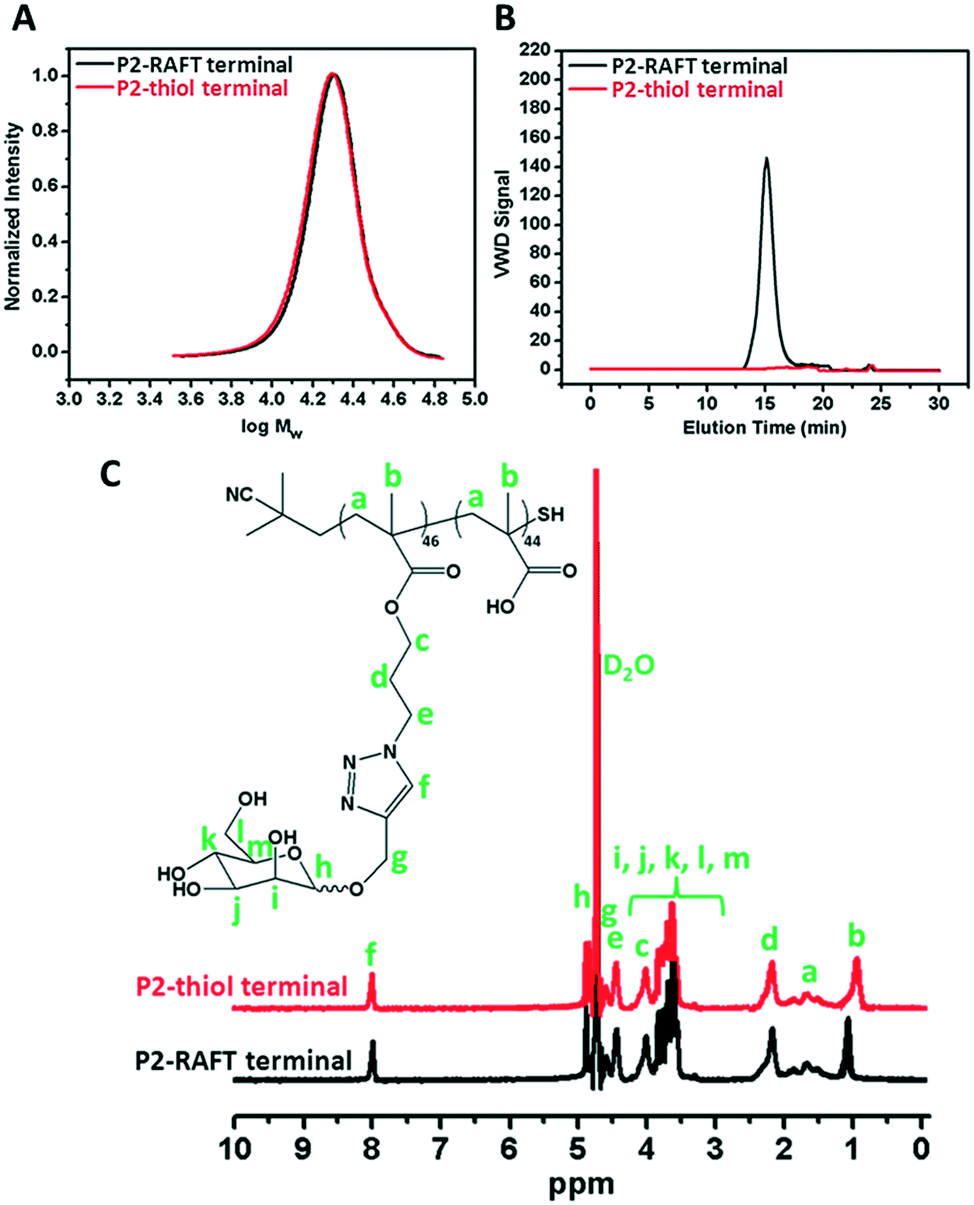 | ||
| Fig. 1 A) SEC analysis via RI detection; B) via VWD; C) 1H NMR characterization of the synthesized P2 homopolymer before and after reduction of the RAFT group. | ||
Preparation of glycopolymer-substituted AuNPs
Prior to polymer functionalization, AuNP solution was centrifuged. The supernatant was removed and replaced with the same volume of water. Then, the polymer coated gold nanoparticles were prepared by mixing the polymer and AuNPs in the dark overnight. Firstly, the obtained polymer-coated AuNPs were characterized via DLS and UV/vis spectroscopy in terms of investigating the size and surface plasmon resonance maximum band (SPRmax) value change. The SPRmax band of AuNPs is usually dependent on the nanoparticle size, shape, aggregation and also their dielectric environment. As seen in Fig. 2 and S4,† the position of the SPRmax band shifted to higher wavelengths due to adsorption of the polymer onto the surface. Moreover, the UV absorbance peaks confirmed that the AuNPs coated with thiol-terminated polymers were still spherical in shape and also did not show any aggregation due to the absence of any large broad shift in the UV absorbance. The general scheme of the reactions related to the synthesis of AuNPs coated with glycopolymers is provided in Scheme 1.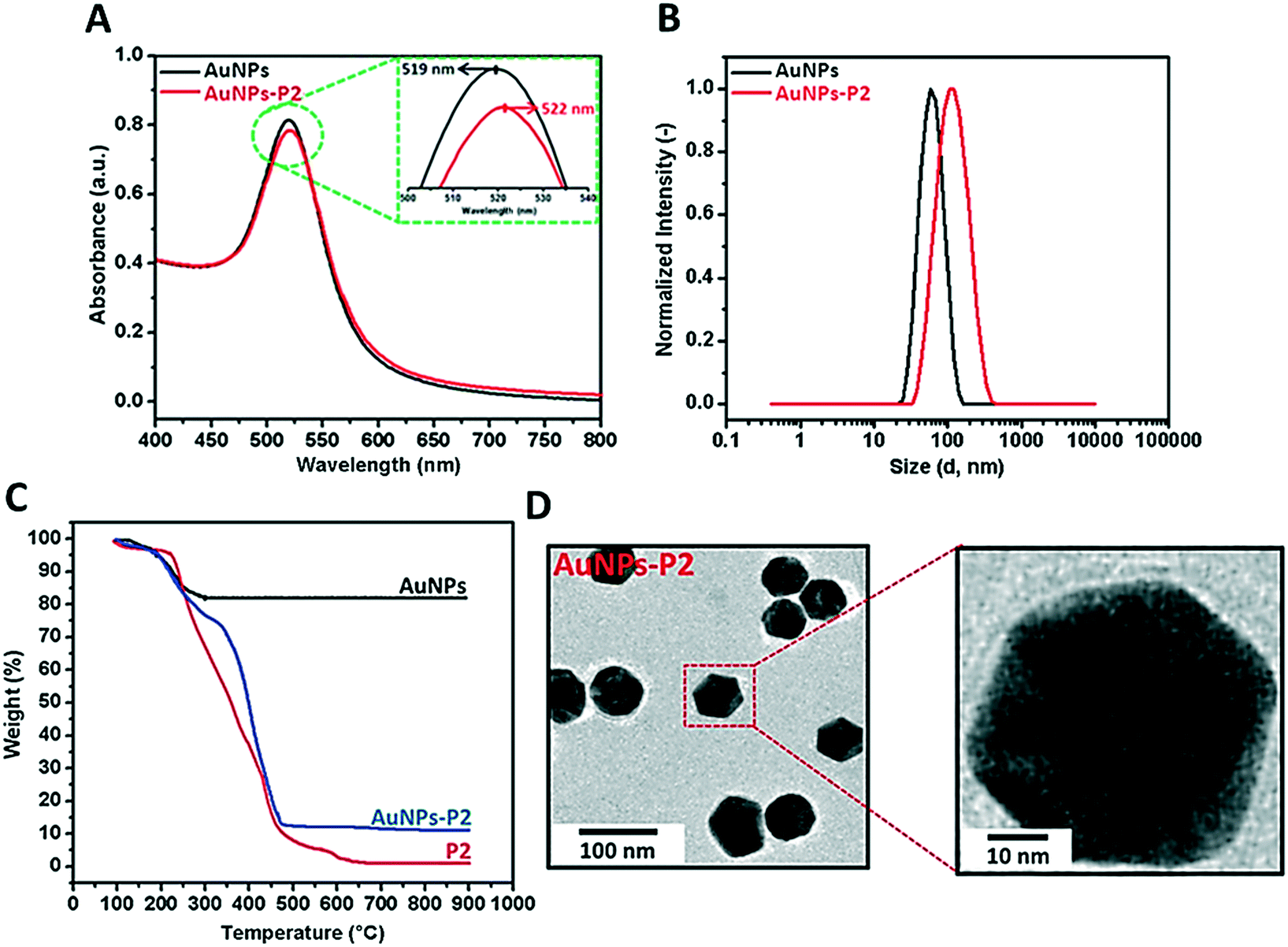 | ||
| Fig. 2 A) UV/vis spectroscopy characterization of AuNPs and the obtained AuNPs-P2 nanoparticles; B) DLS measurements of the synthesized AuNPs-P2 and AuNPs; C) TGA measurements of AuNPs and the obtained AuNPs-P2; D) TEM images of the obtained P2-coated AuNPs. | ||
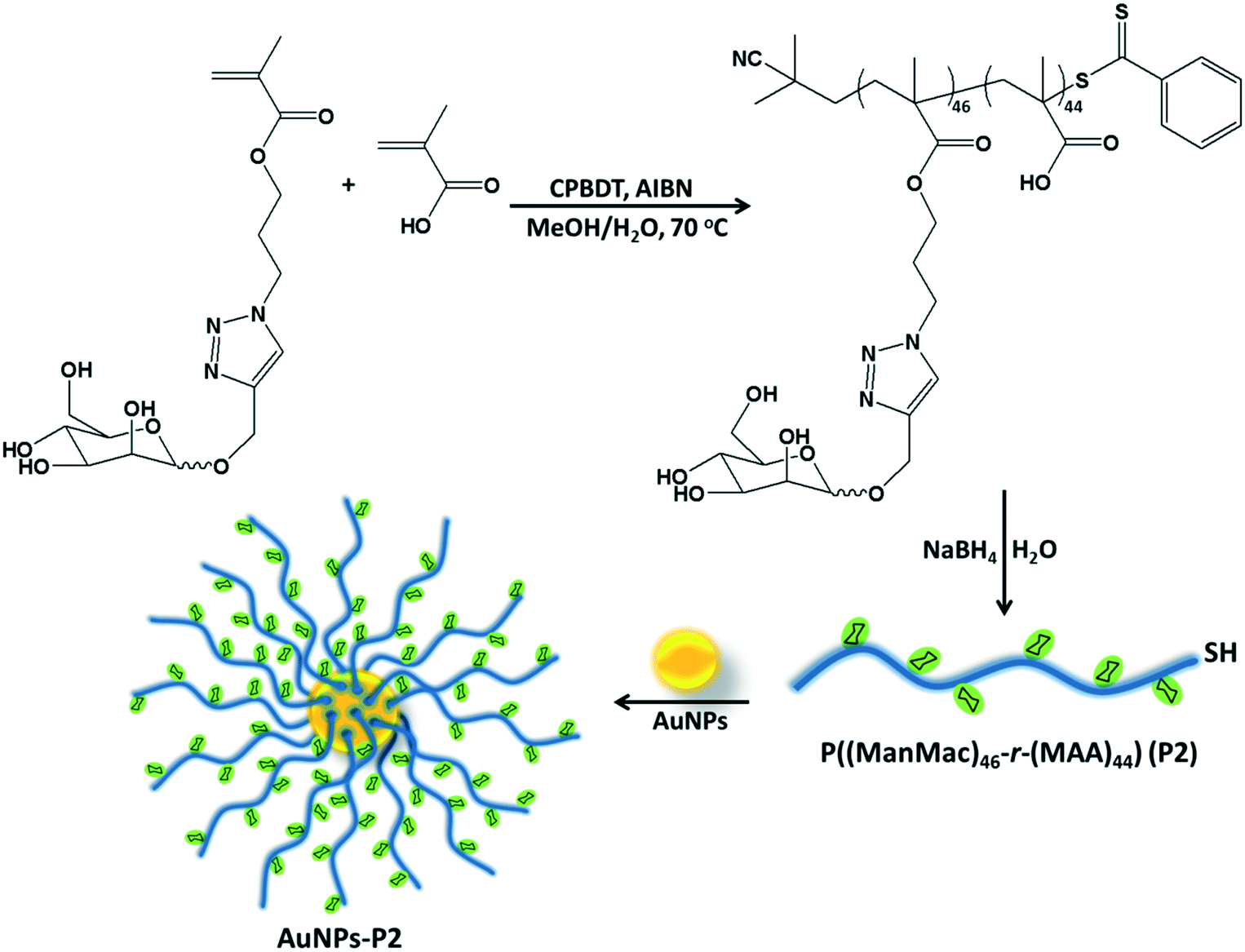 | ||
| Scheme 1 The overall synthetic strategy for the preparation of glycosylated AuNPs. | ||
Polymer-substituted AuNPs were characterized in terms of size and zeta potential by using DLS. There were significant increases in the hydrodynamic volume between AuNPs and polymer–AuNPs, indicating the successful immobilization of the polymers onto the surface. As depicted in Fig. 2 and S3,† the size of the gold nanoparticles after coating with polymers increased from 55 ± 0.2 nm to 94 ± 0.7–101 ± 0.9 nm with the dispersity index (Đ) between 0.15 and 0.22, signifying a narrow particle size distribution. Furthermore, the negative zeta potential of AuNPs has changed depending on the nature of the polymer moieties. For instance, even though the magnitude of the negative zeta potential of P(MAA)-coated AuNPs increased from −26.9 ± 0.2 mV to −41.5 ± 2.3 mV due to the deprotonation of the carboxyl group (RCOOH ↔ RCOO− + H+) in aqueous solution, the negative zeta potential of P(ManMac)-coated AuNPs decreased to −24.0 ± 1.5 mV. According to the transmission electron microscopy (TEM) images (Fig. 2 and S6†), the size of the polymer-coated gold nanoparticles ranged between 57 ± 1.8 and 61 ± 1.7 nm. The average size of the monodisperse nanoparticles is summarized in Table 2.
| Sample | DLS | TEM size (nm) | Zeta potential (mV) | SPRband(max) (nm) | |
|---|---|---|---|---|---|
| D h (nm) | Đ | ||||
| AuNPs | 55 ± 0.2 | 0.11 | 43 ± 1.2 | −26.9 ± 0.2 | 519 |
| AuNPs-P1 | 101 ± 0.9 | 0.15 | 57 ± 1.8 | −41.5 ± 2.3 | 521 |
| AuNPs-P2 | 94 ± 0.4 | 0.21 | 59 ± 2.1 | −19.4 ± 7.3 | 522 |
| AuNPs-P3 | 96 ± 0.8 | 0.22 | 61 ± 1.7 | −27.8 ± 2.8 | 524 |
| AuNPs-P4 | 95 ± 0.6 | 0.18 | 60 ± 2.4 | −24.0 ± 1.5 | 523 |
In order to determine the amount of the polymer immobilized onto AuNPs, thermal gravimetric analysis (TGA) was utilized to analyze each material's thermal profile. As seen in Fig. 2 and S5,† each polymer-coated AuNP exhibited mass loss until approximately 650 °C due to the decomposition of the polymer onto the surface and the remaining fraction was the Au core of the synthesized nanoparticles which was unaffected even at temperatures as high as 650 °C. It was observed that polymer-substituted AuNPs contained approximately 86.2–88.7% polymer.
Characterization of Man tagged polymer conjugates
As stated in the Introduction section, monosaccharide modified glycoconjugates as nanotools are an advancing route. These glycoconjugates can also be functionalized to metallic or polymeric NPs by active groups. In this work, Man tagged AuNP/polymer core–shell structures with different ratios of MAA, OEGMA and ManMac were investigated for drug delivery studies by using DOX as the model chemotheurapeutic anticancer agent.23 Accordingly, three different bioconjugates with DOX including P2D, P3D and P4D were prepared. These novel conjugates were also compared in reference to another polymer coated AuNP particle (PMAA coated AuNP-P1D) which was shown in our previous work.22 Within that work, it was already illustrated that these polymer covered AuNPs could be potential carriers of DOX and be pH-sensitive by utilizing the hydrazone bond. Herein, the synthesis of glyconanoparticles was performed successfully with a few modifications according to the previous work.Initially, the binding capacity of each particle was determined via a DOX standard graph between 2.5–100 μM with the equation y = 0.006x − 0.009 (R2 = 9996). The DOX binding capacities of each conjugate are as follows: 41% for P1D, 25.6% for P2D, 32% for P3D, and 33.6% for P4D. As can be seen from the binding capacities, the structural changes of the polymer-coated AuNPs affected DOX binding in relation to the conjugation strategy. P1D, which has the most DOX amount, was obtained by the well-known EDC/NHS coupling under mild conditions due to the small functional moieties coming from MAA. Concomitantly, P2D, P3D and P4D conjugations were carried out by CDI coupling due to the urethane linkages which may be more sensitive with respect to EDC/NHS coupling. Moreover, this revealed the different Man amounts with more pendant –OH groups, which can be used effectively in the CDI method.
Particularly in P3D, which showed similar binding to P4D, there is less Man in comparison to OEGMA that contains additional –OH groups by inducing more DOX binding with respect to P2D conjugation. Afterwards, the same concentrations of DOX were utilized for the rest of the work. The spectrophotometric properties were investigated by presenting the normalized absorbance of each spectrum between 350–600 nm. According to Fig. 3, small shifts to 500 nm for the newly synthesized conjugates were observed relative to the maximum absorbance peak of DOX after conjugation. Additionally, these peaks are also beneficial to the subsequent drug release studies for measuring the absorbance of the release media.
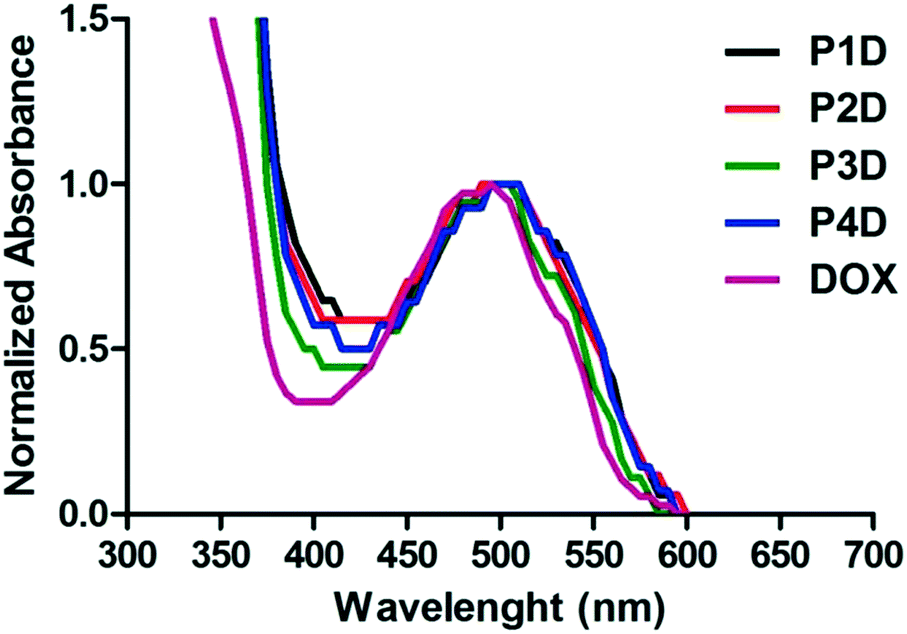 | ||
| Fig. 3 Normalized absorbance spectrum of AuNP/polymer–DOX core–shell particles and free DOX (all samples are at 10 μM DOX concentration). | ||
As the next characterization step, the hydrodynamic particle size distributions and surface charges of each bioconjugate were examined. DLS provided results in terms of RH, which is a hypothetical measure of the hydrodynamic radius of NPs in colloidal dispersions, to predict and control the fate of these NPs in cell media. Table 3 shows the differences in particle size and zeta potential between polymer coated particles and DOX-loaded bioconjugates. The DLS data confirmed that the DOX conjugated particles demonstrated the largest particle size distribution; the size of the negatively charged structure of Man tagged AuNPs decreased after bioconjugation.
| Sample | Particle size (nm) | Zeta potential (mV) |
|---|---|---|
| P1D | 147 ± 25 | −22.7 ± 12 |
| P2D | 239 ± 7.8 | −9.5 ± 0.9 |
| P3D | 211 ± 27.1 | −12.3 ± 1.3 |
| P4D | 171 ± 34.8 | −10.9 ± 0.5 |
Over the past decade, the advanced drug carrying nanotool called “trojan horse” has been developed with different strategies.24 In particular, pH-sensitive macromolecular polymeric structures,25 vesicular systems26 or metallic NPs27 were studied. Herein, Man tagged AuNP/polymer particles linked to DOX molecules with pH-sensitive hydrazone bonds were evaluated for their in vitro release profiles, in order to investigate the potential use of these particles as delivery carriers. One of the main characteristic features of tumor sites is that they have a slightly acidic microenvironment.28
Many of the pH-sensitive bonds between the drug and the carrier, which are formed via a typical hydrazone linkage, were evaluated at pH 5.3 as a model of the extracellular pH environment of cancer cells and at pH 7.4 as a model of the environment of healthy cells.29 As shown in Fig. 4, the drug release kinetics of DOX were much faster in an acidic pH environment (pH 5.3) than in neutral pH (pH 7.4) for all samples. DOX released from AuNPs was more controllable in comparison to free DOX, which reached 100% release after 72 h. On the other hand, under slightly alkaline conditions (pH 7.4) free DOX exhibited fast release while the cumulative release of conjugated particles was slow. In particular, a negligible amount of drug release from the P4D conjugate in physiological pH (pH 7.4) supports that it could be a good candidate as a pH responsive drug carrier. As a result, the slow drug release characteristics at neutral pH and increased drug release in acidic environments ensure AuNP–DOX bioconjugates to be promising drug carriers to improve intracellular drug release in the acidic microenvironment of most tumor types.
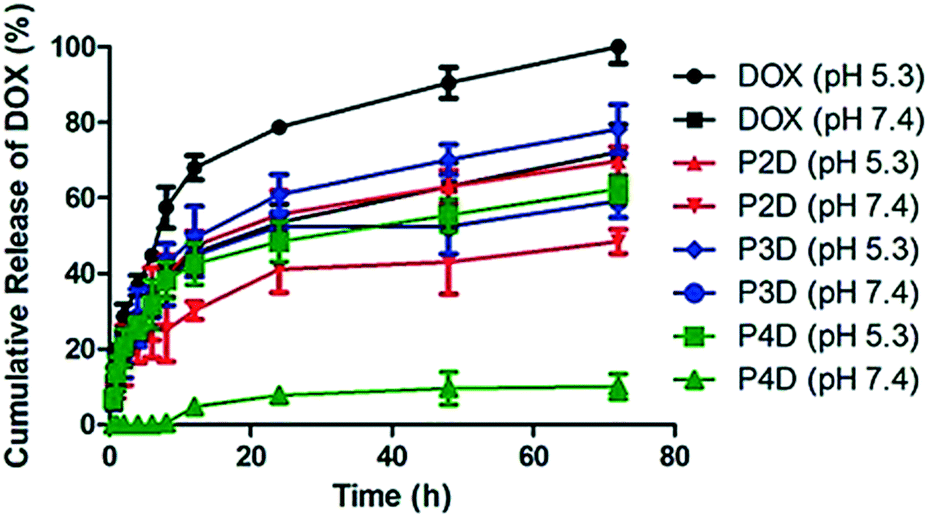 | ||
| Fig. 4 Cumulative drug release graph of free DOX at pH 5.3 and pH 7.4 and AuNP/polymer–DOX bioconjugates at pH 5.3 and pH 7.4 for 72 h at 37 °C. | ||
Effects of AuNPs on cell viability
The effects of the four different AuNPs were investigated using human cervix adenocarcinoma (HeLa), human lung carcinoma (A549), human neuroblastoma cells (SH-SY5Y), and monkey kidney epithelial cell line (Vero) cell lines (Fig. 5A). P1, P2, and P3 inhibited the growth of HeLa cells whereas P4 had no effect on cell viability. The inhibitory effects of P1 were similar to those of P2 and P3 at higher doses. On the other hand, at lower doses, P1 slightly decreased the cell viability. In the case of A549 cells, a dose dependent decrease in cell viability was observed for P2 and P3. There was no decline in cell viability in SH-SY5Y and Vero cells. Surprisingly, induction of cell growth was observed in the SH-SY5Y cell line. As a result, 50 μg mL−1, 10 μg mL−1, 10 μg mL−1 and 250 μg mL−1 non-toxic doses were chosen for P1, P2, P3 and P4, respectively, and used for DOX bioconjugation.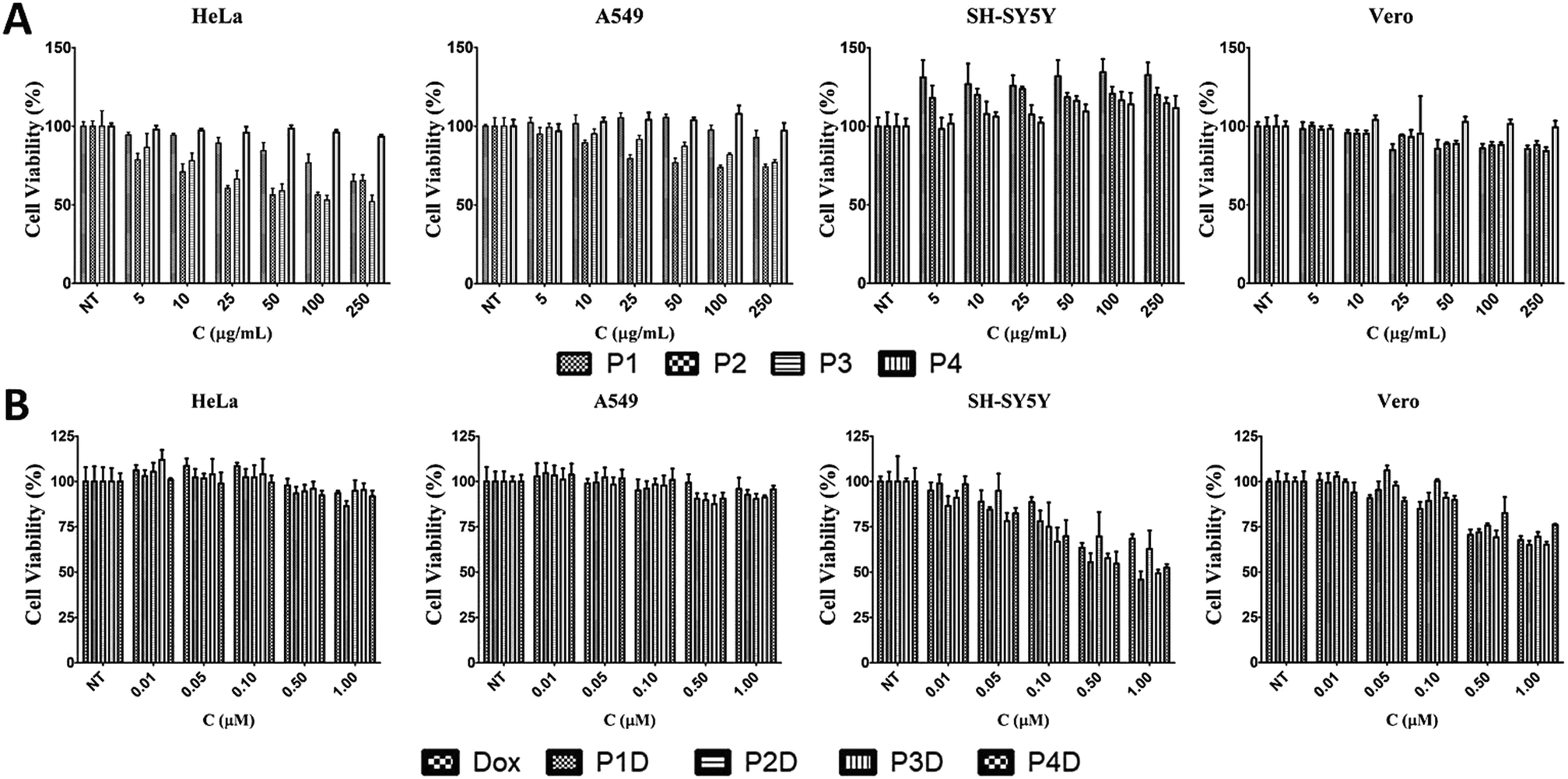 | ||
| Fig. 5 The effects of (A) AuNPs and (B) DOX conjugated AuNPs with free DOX on cell viability for 24 h [NT: non-treated cells. Bars represent ±SD, (n = 4)]. | ||
Effects of DOX conjugated AuNPs on cell viability
In this study, we selected different tumor cells which may simulate different diseased tissues in traditional 2D cell cultures as the first step before animal and preclinical studies. Literally, this novel work gives an idea of the simple characteristics of each used cell line due to the possible interaction with the Man tagged nanocarriers, since there is not a comprehensive comparison related to mannose specific cells. However, it is known that carbohydrate moieties have been beneficial labels thanks to the altered glycosylations of cancerous tissues. After determining the individual effects of AuNPs, the concentrations that have no effect on cell viability were used in the following DOX conjugation (Fig. 5B). There were no changes in cell viability for DOX-conjugated AuNPs (P1D, P2D, P3D, and P4D) and free DOX in both HeLa and A549 cells lines. In the previous work, it was also observed that HeLa cells were not affected by AuNP–DOX particles up to 1 μM concentration.22 In the case of A549 cells which contain Man receptors all over the cell surface, a study which used DOX loaded and Man tagged polymeric micelles supports our findings with an inhibitory concentration level of 5.2 μM DOX for 72 h.30 There was a dose dependent decrease in cell viability for SH-SY5Y and Vero cells. In the case of SH-SY5Y, DOX-conjugated AuNPs were more effective than free DOX except for P2D due to the high release rate of DOX outside the cells and the possibility of a lower DOX amount for each particle which was explained in the characterization part. The efficiency of other particles in SH-SY5Y cells indicates that this type of neuroblastoma cell is more capable of working with Man tagged formulations. However, free DOX was more effective in decreasing the cell viability in Vero cells compared to conjugated AuNPs. These valuable results indicate that these nanoparticles may also succeed in brain cancer targeting due to the mannose functionalization enabling them to cross the blood–brain barrier as shown in a previous study.31Cellular uptake
It is possible to use the intrinsic fluorescence emission of DOX to monitor drug localization in cells.29 We investigated whether DOX could enter cells when conjugated to carrier AuNPs (Fig. 6). Cellular internalization of DOX was observed in all conjugates. The incubation times of free DOX and all glycopolymer conjugates were kept at 3 h, however images were taken after 30 min and 3 h, sequentially to observe the drug release effectiveness of the conjugates by means of time without the fixation of cells.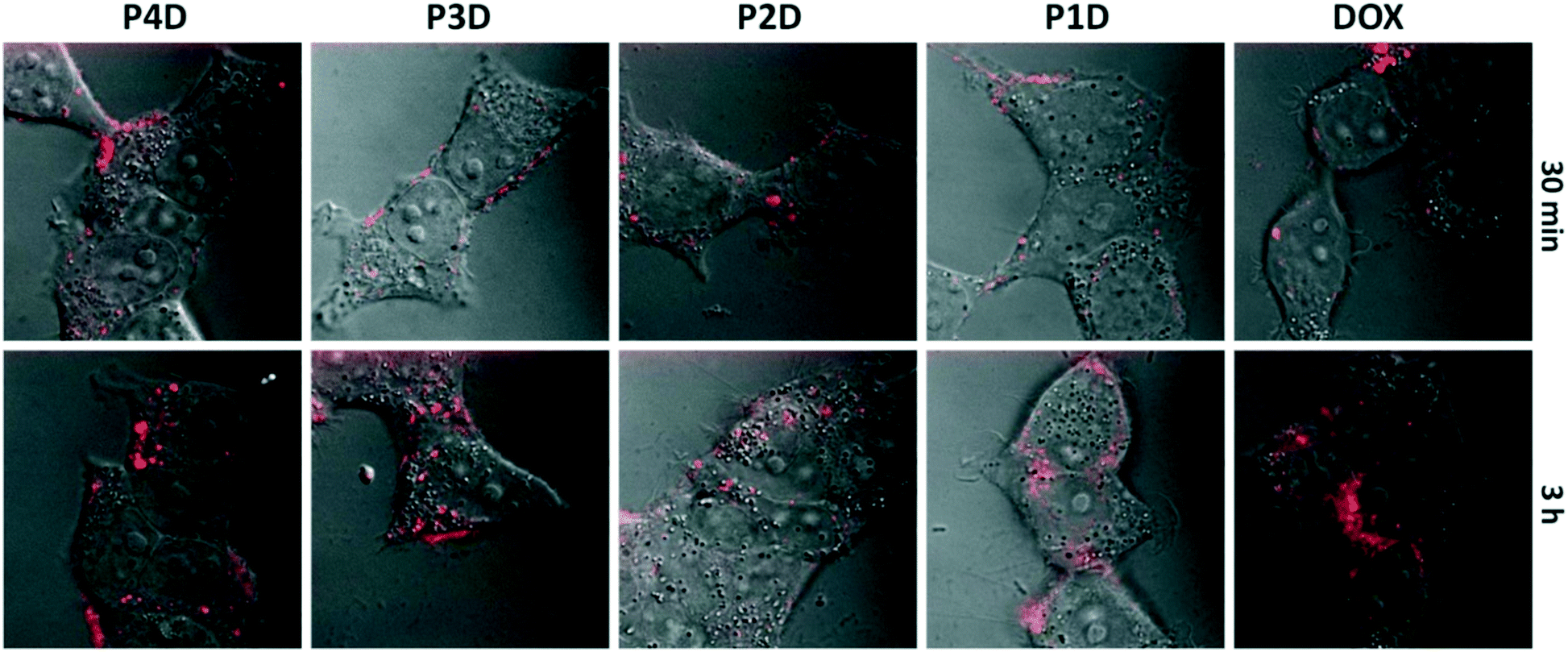 | ||
| Fig. 6 Confocal laser microscopy images of free DOX and P1D, P2D, P3D and P4D conjugations after treatment of samples for 30 min and 3 h, respectively. Bright-field images were taken in DIC mode and fluorescence images were taken by using 488 nm laser. All images were obtained with 100× magnification. | ||
By using confocal laser microscopy, the DIC images were also included to monitor the morphology of neuroblastoma cells (SHSY-5Y) and were overlaid with the fluorescence images. As can be seen from Fig. 6, all samples are near, around and/or stacked on cell membranes after 30 min. Moreover, DOX and DOX conjugated glycopolymer coated AuNPs were found in the cytoplasm and interlocalized to the environment of the nucleus. This issue can be linked to the delivery of DOX due to the breakage of linkages in cytoplasmic media which can generally occur in endosomes after endosomal uptake of nanoparticles. In accordance with the in vitro drug release study, this successful experiment indicates that free DOX in cancer cell media, which have a lower pH than that in healthy cells, was spread more effectively. These data suggest that AuNPs are membrane permeable and have no effect on DOX sub-cellular localization even in concentrations under 1.0 μM DOX as the model cancer drug.
Conclusions
Incorporating the biological properties of carbohydrates into nanostructured materials has attracted much attention due to their crucial role in bio-recognition processes at a molecular level and also their functional role in living systems. Hence, glycopolymer-coated gold nanoplatforms would be a great benefit as drug carriers to enhance the exposure of target sites to drugs. In this study, thiol-terminated glycopolymers have been prepared by RAFT and then grafted onto AuNPs. Subsequently, an anti-cancer drug doxorubicin was conjugated to glycopolymers by a pH-sensitive hydrazone linkage in the presence of cysteine and a cross-linker. Following the conjugation steps, it has been observed that the size and surface charge of glycopolymers changed after DOX conjugation and the drug release profiles proved the efficient sustained release of DOX from the glycopolymeric branches of the AuNPs. In terms of cellular viability, especially for SH-SY5Y neuroblastoma cells, glycopolymer conjugates (P4D) showed their therapeutic effect and a higher toxicity to cancer cell lines compared to healthy cell lines (Vero). These AuNP/glycopolymer conjugates as potential carriers with sustainable release of DOX may create a doorway to the next step of improving 3D cell cultures and/or in vivo animal studies related to a neuroblastoma type of cancer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported by the Engineering and Physical Sciences Research Council (Proposal no: EP/P009018/1).References
- L. Sun, T. Liu, H. Li, L. Yang, L. Meng, Q. Lu and J. Long, ACS Appl. Mater. Interfaces, 2015, 7(8), 4990–4997 CAS.
- S. Hörner, S. Knauer, C. Uth, M. Jöst, V. Schmidts, H. Frauendorf, C. M. Thiele, O. Avrutina and H. Kolmar, Angew. Chem., Int. Ed., 2016, 55(47), 14842–14846 CrossRef PubMed.
- C. Geyik, M. Ciftci, B. Demir, B. Guler, A. B. Ozkaya, Z. P. Gumus, F. B. Barlas, D. Odaci Demirkol, H. Coskunol, S. Timur, Y. Yagci, Z. Y. Shen, Q. Wang and H. S. Peng, Polym. Chem., 2015, 6(30), 5470–5477 RSC.
- S. V. Lale, A. Kumar, S. Prasad, A. C. Bharti and V. Koul, Biomacromolecules, 2015, 16(6), 1736–1752 CrossRef CAS PubMed.
- A. N. Shetty, R. Pautler, K. Ghagahda, D. Rendon, H. Gao, Z. Starosolski, R. Bhavane, C. Patel, A. Annapragada, C. Yallampalli and W. Lee, Sci. Rep., 2016, 6, 27863 CrossRef CAS PubMed.
- B. Demir, F. B. Barlas, E. Guler, P. Z. Gumus, M. Can, M. Yavuz, H. Coskunol, S. Timur, G. Civinskiene, S. Velziene, J. Muselik and Z. Chalupova, RSC Adv., 2014, 4(65), 34687–34695 RSC.
- R. De Coen, N. Vanparijs, M. D. P. Risseeuw, L. Lybaert, B. Louage, S. De Koker, V. Kumar, J. Grooten, L. Taylor, N. Ayres, S. Van Calenbergh, L. Nuhn and B. G. De Geest, Biomacromolecules, 2016, 17(7), 2479–2488 CrossRef CAS PubMed.
- E. Guler, F. B. Barlas, M. Yavuz, B. Demir, Z. P. Gumus, Y. Baspinar, H. Coskunol and S. Timur, Colloids Surf., B, 2014, 121, 299–306 CrossRef CAS PubMed.
- M. Panagiotopoulou, Y. Salinas, S. Beyazit, S. Kunath, L. Duma, E. Prost, A. G. Mayes, M. Resmini, B. Tse Sum Bui and K. Haupt, Angew. Chem., Int. Ed., 2016, 55(29), 8244–8248 CrossRef CAS PubMed.
- Y.-S. Yang, R. P. Carney, F. Stellacci and D. J. Irvine, ACS Nano, 2014, 8(9), 8992–9002 CrossRef CAS PubMed.
- L. Dykman, N. Khlebtsov, L. L. del Mercato, D. Sasse, W. J. Parak, G. Schmid, W. Brandau, U. Simon, W. Jahnen-Dechent, N. A. Kotov, L. M. Liz-Marzán, J. M. Reid, M. M. Ames, P. Mukherjee, D. Mukhopadhyay and B. W. Gibson, Chem. Soc. Rev., 2012, 41(6), 2256–2282 RSC.
- M. Marradi, F. Chiodo, I. García, S. Penadés, G. M. Whitesides, D. Y. Lee, H. Shin, R. J. Pieters, J. M. de la Fuente, S.-H. Nishimura, P. Arosio, A. Lascialfari, D. Gatteschi and C. Sangregorio, Chem. Soc. Rev., 2013, 42(11), 4728 RSC.
- X. Li, M. Takashima, E. Yuba, A. Harada and K. Kono, Biomaterials, 2014, 35(24), 6576–6584 CrossRef CAS PubMed.
- A. M. Alkilany and C. J. Murphy, Langmuir, 2009, 25(24), 13874–13879 CrossRef CAS PubMed.
- A. Quintanilla, M. Valvo, U. Lafont, E. M. Kelder, M. T. Kreutzer and F. Kapteijn, Chem. Mater., 2010, 22(5), 1656–1663 CrossRef CAS.
- E. Boisselier, A. K. Diallo, L. Salmon, C. Ornelas, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2010, 132(8), 2729–2742 CrossRef CAS PubMed.
- C.-U. Lee, D. Roy, B. S. Sumerlin and M. D. Dadmun, Polymer, 2010, 51(6), 1244–1251 CrossRef CAS.
- E. Gullotti and Y. Yeo, Mol. Pharmaceutics, 2009, 6(4), 1041–1051 CrossRef CAS PubMed.
- T. Kitaoka, S. Yokota, M. Opietnik and T. Rosenau, Mater. Sci. Eng., C, 2011, 31(6), 1221–1229 CrossRef CAS.
- F. C. Telli, B. Demir, F. B. Barlas, E. Guler, S. Timur, Y. Salman, S. Timur, S. Kowarik, M. Weinelt, C. Wang, J. Liu, X. Fan, J. Gu, X. Chen, Y. Zhang and N. Gu, RSC Adv., 2016, 6(107), 105806–105813 RSC.
- A. Pettenuzzo, R. Pigot and L. Ronconi, Metallodrugs, 2016, 1(1), 36–61 CrossRef.
- G. Yilmaz, B. Demir, S. Timur and C. R. Becer, Biomacromolecules, 2016, 17(9), 2901–2911 CrossRef CAS PubMed.
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandez-Boussard, H. McLeod, T. E. Klein and R. B. Altman, Pharmacogenet. Genomics, 2011, 21(7), 440–446 CrossRef CAS PubMed.
- M. Ferrari, S. C. Onuoha and C. Pitzalis, Nat. Rev. Rheumatol., 2015, 11(6), 328–337 CrossRef CAS PubMed.
- B. Guler, H. Akbulut, F. B. Barlas, C. Geyik, D. O. Demirkol, A. M. Senisik, H. A. Arican, H. Coskunol, S. Timur and Y. Yagci, Macromol. Biosci., 2016, 16(5), 730–737 CrossRef CAS PubMed.
- M. Chang, S. Lu, T. Zuo, Y. Guan, T. Wei, W. Shao and G. Lin, Colloids Surf., B, 2015, 129, 175–182 CrossRef CAS PubMed.
- S. Jung, J. Nam, S. Hwang, J. Park, J. Hur, K. Im, N. Park and S. Kim, Anal. Chem., 2013, 85(16), 7674–7681 CrossRef CAS PubMed.
- L. Wong, M. Kavallaris, V. Bulmus, R. Mendichi, L. Andersson, A. Tsirk, J. Ford, G. Wu, S. Kneller, J. Davies, R. Duncan, I. S. Kim, K. Kim and I. C. Kwon, Polym. Chem., 2011, 2(2), 385–393 RSC.
- Y. Qin, W. Fan, H. Chen, N. Yao, W. Tang, J. Tang, W. Yuan, R. Kuai, Z. Zhang, Y. Wu and Q. He, J. Drug Target., 2010, 18(7), 536–549 CrossRef CAS PubMed.
- L. Yin, Y. Chen, Q. Yin, Z. Zhang, Q. Yin, N. Zheng and J. Cheng, Macromol. Rapid Commun., 2015, 36(5), 483–489 CrossRef CAS PubMed.
- A. Strambi and A. De Milito, In Tumor Cell Metabolism, Springer Vienna, Vienna, 2015, pp. 173–196 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7me00086c |
| This journal is © The Royal Society of Chemistry 2018 |