
Resistance-breaking profiling and gene expression analysis on an organometallic ReI–phenanthridine complex reveal parallel activation of two apoptotic pathways
Marcel
König†
b,
Daniel
Siegmund†
 a,
Lukasz J.
Raszeja
a,
Aram
Prokop
b and
Nils
Metzler-Nolte
*a
a,
Lukasz J.
Raszeja
a,
Aram
Prokop
b and
Nils
Metzler-Nolte
*a
aChair of Inorganic Chemistry I – Bioinorganic Chemistry, Faculty of Chemistry and Biochemistry, Ruhr University Bochum, Universitätsstrasse 150, D-44801 Bochum, Germany. E-mail: nils.metzler-nolte@rub.de
bDepartment of Paedriatic Oncology, Children's Hospital Cologne, Amsterdamer Strasse 59, 50735 Cologne, Germany
First published on 20th December 2017
Abstract
Emerging resistances of tumors against multiple anti-cancer agents are a major concern in the chemotherapeutical treatment of various cancers. Clearly, this raises the need for novel therapeutics with new modes of action. Herein, we report on the favorable in vitro anti-proliferative properties of a phenanthridine-containing ReI(CO)3 complex (compound 1, also abbreviated LR-166) and identify major contributions to its mode of action. The complex induces apoptosis in low micromolar concentrations even in drug-resistant Burkitt-like lymphoma (BJAB) and leukemia (Nalm-6) cell lines with known overexpression of p-glycoproteins as was confirmed by measuring the amount of hypodiploid DNA via FACS Scan analysis. Importantly, a gene expression analysis in combination with toxicity studies on a number of modified cell lines (leukemia: NALM-6, lymphoma: BJAB, melanoma: MelHO) and the reduction of mitochondrial membrane potential (determined by adding JC-1 dye, followed by FACS analysis) confirmed the activation of both, the extrinsic and the intrinsic apoptotic pathway. Finally, the mechanism of action was shown not to be influenced by overexpression of the anti-apoptotic factor Bcl-2 in Mel-HO cells which are known to be resistant to a variety of drugs. All taken together, our experiments underscore the unique opportunities inherent in this novel lead structure of Re complexes to act as an effective chemotherapeutic agent in a combination therapy to overcome documented drug resistances in tumors.
Introduction
Emerging resistances of various types of cancer towards established anti-cancer drugs, either intrinsic or acquired after initially successful treatment, seriously hamper the effectiveness of chemotherapy and frequently lead to recurrence of tumors and bad prognosis for patients.1,2 In view of the fact that chemotherapy is one of the three pillars of anti-cancer therapy, this serious drawback has triggered enormous scientific efforts and further accelerated the search for conceptually novel anti-cancer drugs.3,4 To this end, transition metal complexes have emerged as promising candidates for future treatment. Since the serendipious discovery of cisplatin which, along with its analogs carboplatin and oxaliplatin, remains one of the main components of modern chemotherapy,5 many different metal complexes and especially organometallic compounds were shown to be extremely promising candidates. Their broad structural diversity and variable ligand design in combination with unique metal-based modes of action (based e.g. on redox activity,6 ligand exchange reactions7 or phototoxicity8) offers enormous potential for the development of novel therapeutics that may help to overcome emerging resistances.9–13 Among the numerous transition metal elements considered for anti-cancer treatment, rhenium complexes received only limited attention. This is somewhat surprising as organometallic Re complexes offer potentially favorable properties for the development of anti-cancer drugs. Selected complexes, especially based on the oxidation state +I and the heavily used Re(CO)3 core structure, were shown to exhibit cytotoxic effects on a variety of cancer cell lines.14–16 Moreover, the availability of the radioactive analogues 186Re, 188Re and 99mTc provides the possibility to design complexes for diagnostic and therapeutic purposes based on the exact same coordination environment.17,18 In combination with pronounced luminescence inherent to many ReI-complexes,19 they may be regarded as versatile multimodal molecules potentially unifying diagnostic probes and therapeutic agents. Recently, we reported on a novel series of ReI(CO)3 complexes containing phenanthridine-based tridentate ligands with favorable properties for cellular imaging.20 Moreover, we demonstrated that these complexes are readily conjugated to biomolecules like peptides to influence their cellular uptake and intra-cellular localization.21 Herein, we provide the very first investigations on the potential of these ReI-phenanthridine-containing complexes to act as anti-tumor agents using the example of fac-[Re(CO)3(bis(6-phenanthridinyl-methyl)amino)acetic acid methyl ester)] bromide (compound 1, also abbreviated LR166). To investigate the cytotoxicity and selectivity of the complex, apoptosis induction is tested on the malignant BJAB (lymphocytes), Nalm-6 (leukocytes) cell lines as well as non-malignant leukocytes. Moreover, the capability of counteracting drug resistances against conventional chemotherapeutics is evaluated. By using a host of cell lines with different but know mechanisms of resistance, we could further unravel the mechanism of action, ultimately leading to the precise identification of apoptosis signaling components affected by the complex.Results and discussion
Synthesis
The Re complex 1 was synthesized as described previously (Scheme 1).21 In short, 4-[(bis(6-phenanthridinylmethyl)amino)methyl]benzoic acid methyl ester (bpm) was obtained by reacting (aminomethyl)benzoic acid methyl ester with 6-(chloromethyl)phenanthridine and subsequent purification via column chromatography. The bpm ligand was in turn reacted with [Re(CO)3(H2O)3]Br under microwave conditions to provide 1 in good yield and excellent purity after crystallization via slow diffusion of ether into a methanolic solution. | ||
| Scheme 1 Synthesis of Re(I)-complex 1: a) 2 eq. 6-(chloromethyl)phenanthridine, 5 eq. K2CO3, ACN, reflux 48 h. b) 1 eq. [Re(CO)3(H2O)3]Br, MeOH, microwave, 110 °C, 10 min. | ||
Anti-proliferative activity and apoptosis induction
The in vitro anti-proliferative activity of 1 was evaluated on the human Burkitt lymphoma cell line BJAB using a CASY Cell Counter system (Roche Applied Science). The cells were treated with serial dilutions of complex 1 and a series of cells left untreated served as negative control. Cytotoxic effects were evaluated by determining the amount of vital cells after 24 h (Fig. 1a). The overall inhibition of proliferation was assessed by the comparison between the total amount of cells treated with 1 and the control after 24 hours respectively (Fig. 1b). | ||
| Fig. 1 Cell proliferation and inhibition of BJAB cells after incubation with increasing concentrations of the rhenium complex 1 for 24 hours. The initial cell concentration was adjusted to 1 × 105 cells per mL. a) Cell proliferation after 24 h. b) Inhibition of proliferation is given in % of control. The initial concentration of 1 × 105 cells per mL represents a 100% inhibition. Depicted values are mean values ± SD of three replicates. | ||
Pronounced activity was observed in the low micromolar concentrations. A concentration of 1 μM was sufficient to inhibit the total proliferation by more than 50% (ID50). Higher concentrations starting from 2.5 μM were shown to virtually cause a G1 arrest. The ability of 1 to induce apoptosis in BJAB cells was tested by modified cell cycle analysis with a FACS Calibur flow cytometer after 72 hours of incubation with increasing concentrations of the analyte (Fig. 2). The concentration of 1 necessary to induce apoptosis in 50% of the cells (accounting for the lethal dose 50, LD50) was determined to be between 2.5 and 5 μM while the rate of apoptosis increased in a dose-dependent manner reaching a plateau at concentrations above 5 μM.
 | ||
| Fig. 2 Apoptosis measured by flow cytometric analysis after 72 h. BJAB cells were incubated with increasing concentrations of 1. Depicted values are mean values ± SD of three replicates. | ||
Selectivity
The induction of apoptosis in malignant cells by an anti-cancer agent is considered a very favorable event if it is not accompanied with high rates of necrosis which may result in the fatal release of cell components to extracellular space ultimately triggering inflammatory responses and preventing phagocytes from eliminating dead cells per cell debris. To estimate the degree of necrosis caused by a treatment of BJAB cells with 1, the release of cell components into extracellular space was quantified by an LDH-release assay after one hour of incubation using increasing concentrations of the complex (Fig. 3). Within the biological active concentration range no unspecific necrotic effects were detected. | ||
| Fig. 3 Cell viability assay. Unspecific necrosis was determined by LDH-release of BJAB cells incubated with increasing concentrations of 1 for 1 h. Depicted values are mean values ± SD of three replicates. | ||
These results are further supported by flow-cytometric analysis based on annexin V and propidium iodide (PI) which allows for the differentiation between apoptosis (early apoptosis and late apoptosis) and undesirable unspecific necrosis (Fig. 4). After an incubation time of 48 hours over 75% of the cells treated with concentrations between 2,5 and 10 μM were found to be late apoptotic, indicating a fast mechanism of apoptosis induction. In full accordance to the cell viability assay, the fraction of necrotic cells was found to be below 10% in each case.
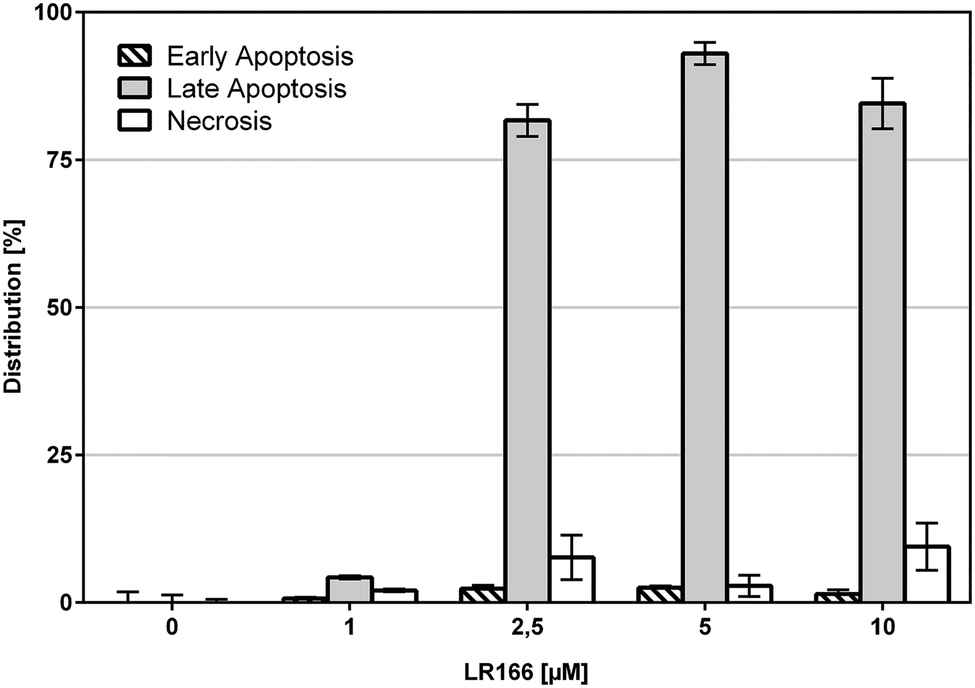 | ||
| Fig. 4 Annexin-V/PI double staining. BJAB cells were incubated for 48 h with increasing concentrations of 1. Distribution of counted cells according to cell cycle states. Vital cells, representing the remaining fraction, are not shown. Depicted values are mean values ± SD of three replicates. | ||
To investigate the selectivity towards malignant cells, the cell lines Nalm-6 (leukemia) and BJAB (lymphoma) as well as non-malignant leukocytes were treated with 1 in concentrations up to 20 μM for 72 hours. Both, BJAB and Nalm-6 cells showed significantly higher rates of apoptosis reaching an LD50 of about 5 μM and a plateau of around 75% apoptotic cells at approx. 10 μM. As opposed to the malignant cell lines, healthy leukocytes showed an apoptosis induction of 16% at 5 μM and 26% at 10 μM (Fig. 5). Thus, 1 shows significantly lower effects on healthy cells. Although the cytotoxic potential on healthy leukocytes is not negligible, the observed apparent selectivity renders 1 a promising candidate compound for further investigations.
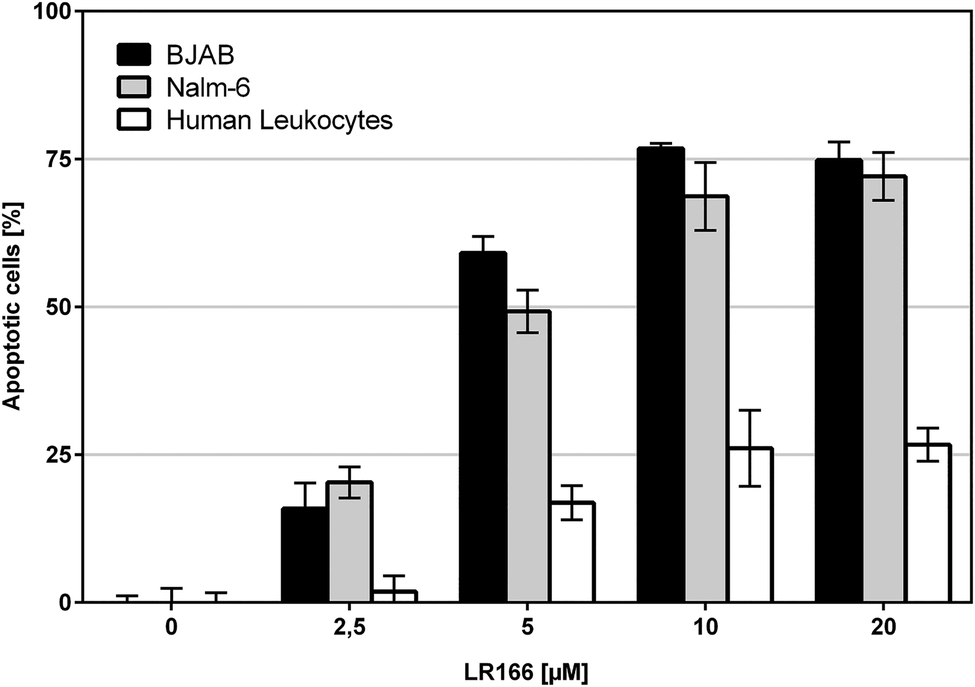 | ||
| Fig. 5 Apoptosis measured by flow cytometric analysis after 72 h. BJAB, Nalm-6 and human leukocytes were incubated with increasing concentrations of the rhenium complex. Depicted values are mean values ± SD of three replicates. | ||
In vitro drug resistance overcome
During cancer therapy, one of the most threatening complications is the development of resistances against the applied drugs. Because therapy itself can lead to an evolutionary selection of resistant cells,22 it is favorable to use multiple drugs in a combined therapy to strike different targets. The development of novel drugs which are not affected by resistances against established chemotherapeutics by exploiting different targets in the cell is a major goal in bioorganometallic chemistry. To investigate this property, complex 1 was tested on special cell lines with known resistances against different established drugs. The resistant and control cell line were both incubated with increasing concentrations of the rhenium complex. Resistance was proven by applying the respective drug on control batches.Comparison of apoptosis induction in BJAB cells and vincristine (VCR) resistant BJAB BiBo cells with overexpression of p-glycoprotein (Fig. 6) showed that toxicity of 1 was not affected by p-glycoprotein overexpression and induced comparable or even higher rates of apoptosis in the BJAB BiBo cells over the tested dosage spectrum from 2.5 μM to 10 μM.
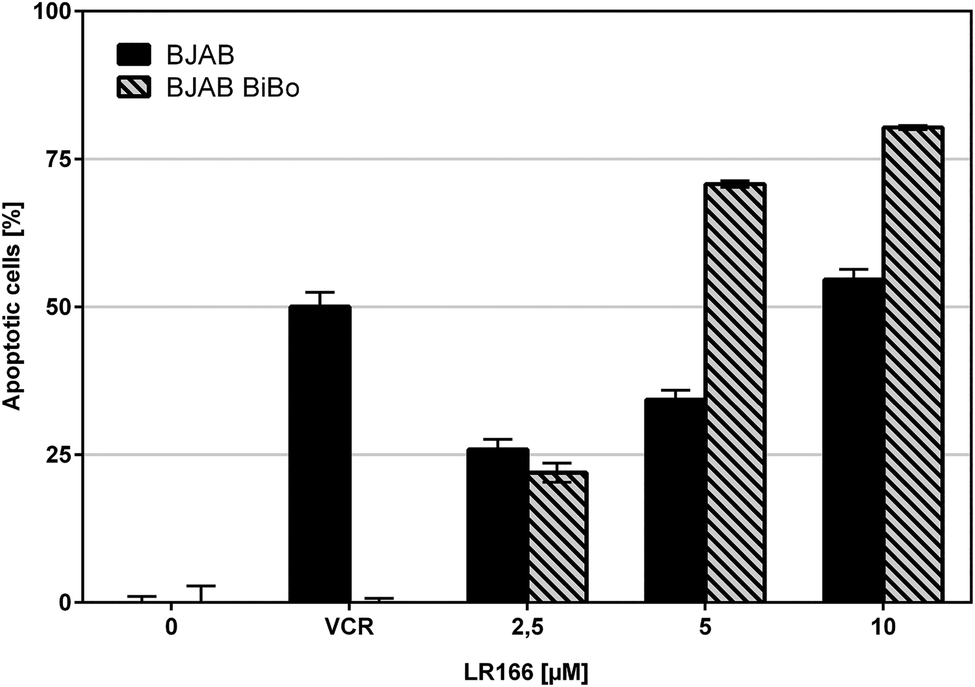 | ||
| Fig. 6 Apoptosis measured by flow cytometric analysis after 72 h using the dye propidium iodide. Values are given in % cells with hypodiploid DNA. BJAB and vincristin-resistant BJAB BiBo cells were incubated with increasing concentrations of the rhenium complex 1 and VCR as control. Depicted are mean values ± SD of three replicates. | ||
The comparison of unmodified Nalm-6 cells with multiple drug-resistant Nalm-6/DAU (overexpression of p-glycoprotein) and Nalm-6 NaKu (resistance to steroids prednisone and dexamethasone) cells showed that apoptosis induction by 1 was not influenced by the resistance mechanisms of both cell lines. In Nalm-6/DAU an apoptosis induction of 64%, comparable to the daunorubicin control was achieved at 2.5 μM (Fig. 7). In Nalm-6 NaKu cells an apoptosis rate of approximately 50% comparable to the prednisolon control was achieved at the same concentration (Fig. 8).
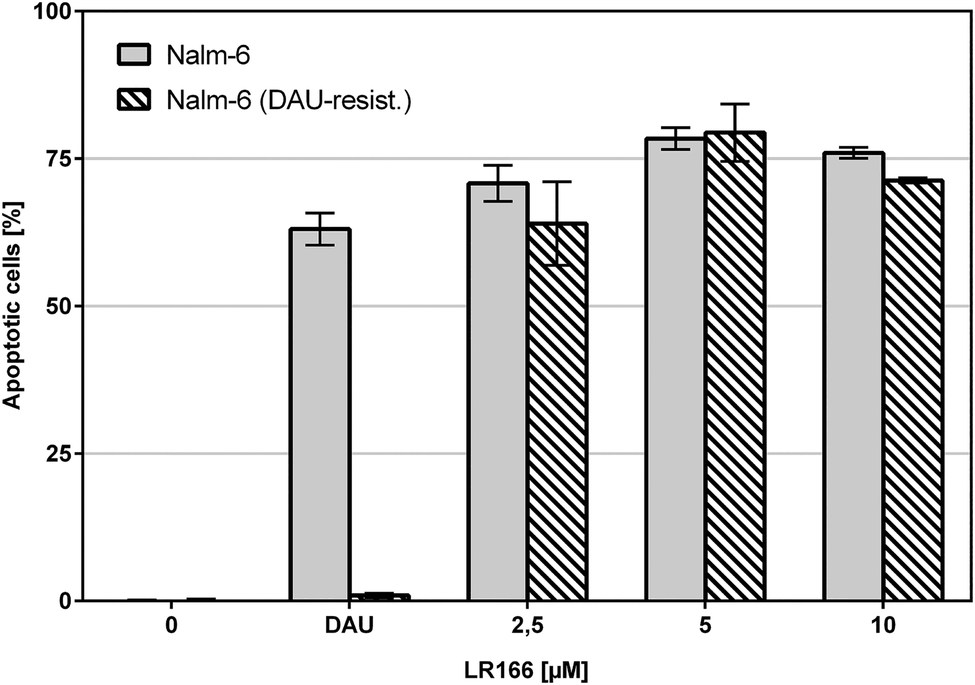 | ||
| Fig. 7 Apoptosis measured by flow cytometric analysis after 72 h using propidium iodide. Values are given in % cells with hypodiploid DNA. Nalm-6 and daunorubicin-resistant Nalm-6 cells were incubated with increasing concentrations of 1 and daunorubicin as control. Depicted are mean values ± SD of three replicates. | ||
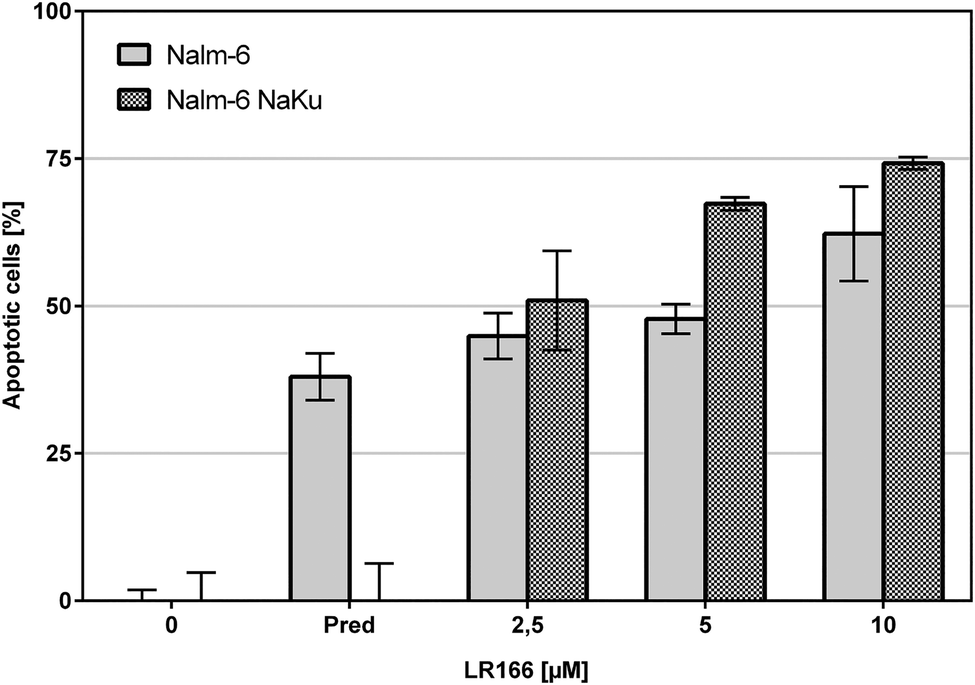 | ||
| Fig. 8 Apoptosis measured by flow cytometric analysis after 72 h using propidium iodide. Values are given in % cells with hypodiploid DNA. Nalm-6 and prednisolon-resistant Nalm-6 NaKu cells were incubated with increasing concentrations of 1 and prednisolon as control. Depicted are mean values ± SD of three replicates. | ||
Taken together, complex 1 provides the ability to overcome resistances of several types of tumors against established anti-cancer drugs. In our opinion, this study clearly outlines the major potential of this compound class to act as novel anti-cancer agent preferentially in a combination therapy. Likewise, the above experiments also demonstrate that complex 1 follows a mode of action different from the above compounds.
Mechanism of action
To further elucidate the mechanism of action of 1, the expression of a total of 84 genes involved in apoptosis was determined using the commercially available RT2 Profiler PCR Array Human Apoptosis kit. The rate of gene-expression is given in relation to untreated control cells (Table 1). Cells treated with 1 show a more than ninefold upregulation of TNFRSF1A and a fourfold upregulation of the TNFRSF1B gene respectively. Both genes encode for ligand-bound tumor necrosis factor (TNF) receptors, which are part of the membrane associated FAS receptor pathway and responsible for the activation of FADD and other death domains. Moreover, an increase in expression of APAF1 is observed, accounting for an activation of caspase-3 in a cytochrome c dependent manner. These findings suggest that a combination of the two main activation mechanisms is responsible for initiating apoptosis, namely the receptor-initiated pathway (extrinsic pathway) as well as the mitochondria dependent pathway (intrinsic pathway).The involvement of TNF receptors in the extrinsic induction of apoptosis by 1 was further investigated by co-incubation with the anti-tumor necrosis factor α monoclonal antibody infliximab. This binding inhibits TNF-α from binding the cell receptors. While infliximab induced no detectable apoptosis alone it successfully inhibited the in vitro apoptosis induction capabilities of 1 in BJAB cells. Rates of apoptotic cells after 72 hours decreased from 44% without infliximab to 34% for cells co-incubated with a concentration of 100 mg L−1 (Fig. 9). This confirms the involvement of TNF receptors in apoptosis induction by complex 1 at least partly which was implied by the gene expression analysis (Table 1).
 | ||
| Fig. 9 Apoptosis measured by flow cytometric analysis after 72 h using propidium iodide. Values are given in % cells with hypodiploid DNA. BJAB cells were incubated with the rhenium complex 1 and co-incubated with the TNF-α binding antibody infliximab. Depicted are mean values ± SD of three replicates. | ||
The extrinsic or death receptor activated apoptosis pathway is known to activate caspase-8. The dependency of the LR166-induced mechanism on caspase-8 was tested by co-incubation of BJAB cells with the caspase-8 inhibitor Z-IETD-FMK. BJAB cells treated with doxorubicin (known to cause caspase-8 dependent apoptosis) served as positive control. Cells treated with 1 and an increasing amount of the inhibitor showed a decreasing rate of apoptosis, indicating at least a partial mechanistic dependency on caspase-8 (Fig. 10).
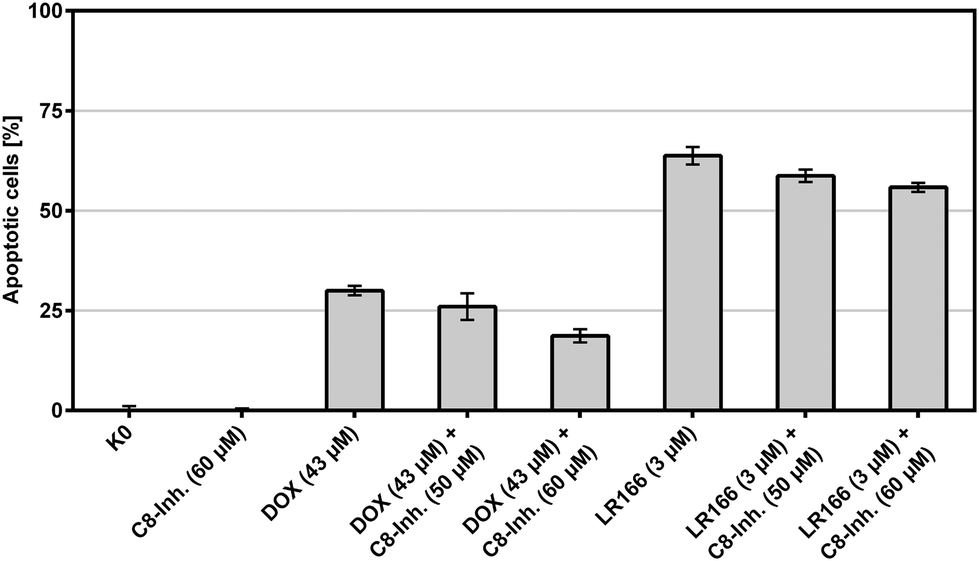 | ||
| Fig. 10 Apoptosis measured by flow cytometric analysis after 72 h using propidium iodide. Values are given in % cells with hypodiploid DNA. BJAB cells were incubated with increasing concentrations of the rhenium complex LR166 and the capase-8 inhibitor Z-IETD-FMK. Depicted are mean values ± SD of three replicates. | ||
Upon mitochondrial involvement in the apoptosis inducing signaling pathway a drop in mitochondrial membrane potential is commonly associated with the release of cytochrome c into the cytosol. This breakdown of membrane potential can be detected by the application of the dye JC-1 which can cross the mitochondrial membrane only when the membrane potential is impaired.
The tested BJAB cells treated with increasing concentrations of the rhenium complex 1 showed a dose-dependent increase of cells with impaired membrane potential (Fig. 11), thus indicating an involvement of the mitochondria as part of the apoptosis pathway.
 | ||
| Fig. 11 Change in mitochondrial membrane potential. BJAB cells were incubated with increasing concentrations of 1 for 48 hours. Depicted are mean values ± SD of three replicates. | ||
The modified BJAB cell line 7CCA carries a vector with a down-regulated capase-3 gene. Compared with an unmodified BJAB cell line a lower apoptosis induction leads to the conclusion that the compound induces apoptosis in a capase-3 dependent manner. The experiment with complex 1 showed a more than 40% lower induction of apoptosis and therefore suggests the involvement of capase-3 (Fig. 12).
 | ||
| Fig. 12 Apoptosis measured by flow cytometric analysis after 72 h using propidium iodide. Values are given in % cells with hypodiploid DNA. BJAB and caspase-3 downregulated BJAB 7CCA cells were incubated with increasing concentrations of the rhenium complex. Depicted are mean values ± SD of three replicates. | ||
The Bcl-2 protein is an important anti-apoptotic factor and proto-onkogene. The ability to induce apoptosis independent of Bcl-2 may provide a novel therapeutic option in resistant cells. Incubated in MelHO cells transfected only with the pires vector and with Bcl-2 over-regulated vector, complex 1 was able to induce comparable rates of apoptosis (Fig. 13) therefore ignoring the anti-apoptotic effect and underlining the potential of 1 to induce apoptosis even in resistant cell lines.
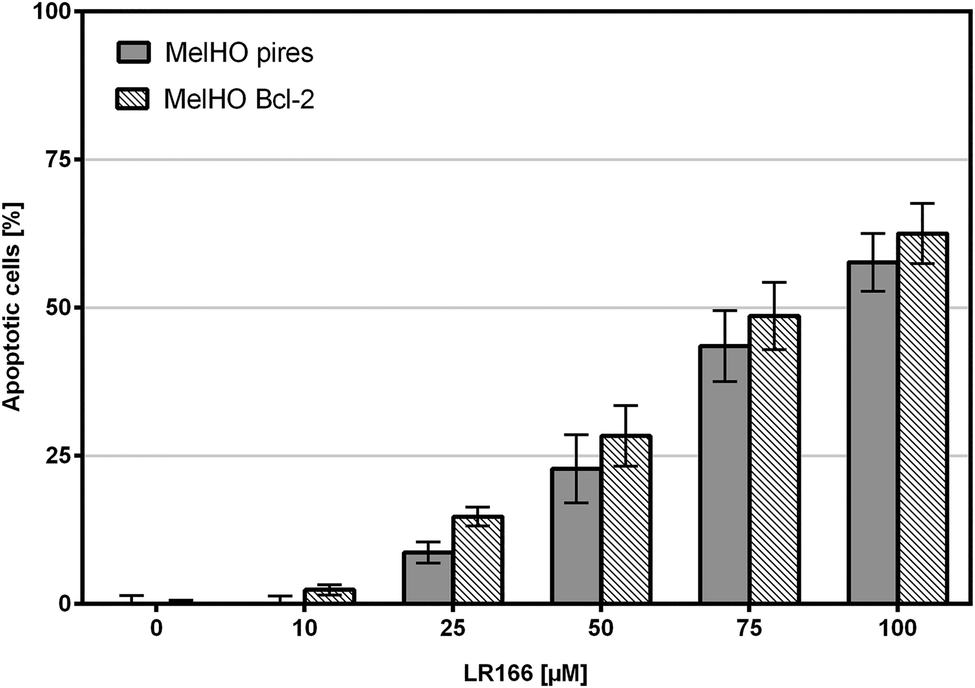 | ||
| Fig. 13 Apoptosis measured by flow cytometric analysis after 72 h using propidium iodide. Values are given in % cells with hypodiploid DNA. MelHO (pires) cells and MelHO (Bcl-2) were incubated with increasing concentrations of LR166. Depicted are mean values ± SD of three replicates. | ||
Conclusion
In conclusion, the rhenium complex 1 (LR166) was found to provide very promising antiproliferative activity in low micro-molar concentrations on a variety of cancer cells. Notably, being a methyl ester 1 might be a prodrug, with significant accumulation inside the cancer cells following hydrolysis of the ester group. A comparison with healthy human leukocytes confirmed a pronounced selectivity with much greater impact on malignant cells. Moreover, 1 was able to overcome acquired resistances based on p-glycoprotein overexpression which is a major mechanism in multi-drug resistant tumors. The complex induces apoptosis involving mitochondrial pathways including processing of caspase 3. Additionally, the application of a TNF-mediated pathway subsequently activating caspase 8 was identified as another key factor in its mechanism of action. Thus, complex 1 invokes both, the extrinsic and the intrinsic apoptotic pathway providing an additional option for therapeutic applications in resistant cell lines. Furthermore, the mechanism of action was shown not to be influenced by an overexpression of the anti-apoptotic factor Bcl-2 in MelHO cells. Remarkably, no indication of unfavorable necrosis was observed during the course of our experiments. Complexes similar to 1, but with simpler ligands derived from N,N′-bis[(quinolin-2-yl)methyl]amine15,24 were investigated for their mechanism of cytotoxicity by Gasser and coworkers both under irradiation24 and in the dark. Both classes of compounds localize in the mitochondria and cause a change in the mitochondrial potential at low concentrations. However it appears that 1, which has the phenanthridine ligands (larger than quinoline), has a higher selectivity for cancer over non-malignant cells. Moreover, the quinoline-derived complexes also illicit a significant necrotic response both in the dark and under irradiation,24 which is not the case for 1 as shown above. In terms of structure–activity relationship (SAR), there are hence beneficial effects associated with the larger, and possibly more lipophilic phenanthridine-derived ligands.Taken together complex 1, as a first member of aphenanthridine-containing class of rhenium(I) complexes, appears to be a very promising lead structure for the development of novel metal-based anti-cancer drugs. Its key properties in overcoming drug resistances and invoking multiple apoptotic pathways may be exploited in a combined chemotherapy alongside other therapeutic agents to counteract the development of tumor resistances.
Experimental section
Annexin-V/propidium iodide double staining
In early phases of apoptosis, morphological changes occur to the plasma membrane leading to the translocation of the phospholipid phosphatidyl-1-serine from inside of the cell to the outside. Annexin-V binds calcium dependent to those phospholipids which make it possible to differentiate between cells in early apoptosis. Because annexin-V is conjugated covalently to the fluorescence dye fluorescein isothiocyanate (FITC) cells can be detected in FACScan using a counter stain with propidium iodide. This enables to differentiate between early apoptosis (annexin-V positive, propidium iodide negative) and late apoptosis or necrosis (annexin-V positive, propidium iodide positive).Cell culture
Cell cultures were maintained in humidified atmosphere of 5% CO2 and air at 37 °C in an incubator. Cells were passaged twice a week; a vitality control was performed for each passage. A mycoplasma test was performed each month. Suspension cells were incubated in completed RPMI medium while adherent cells were cultivated in DMEM. Suspension cell lines were adjusted to a concentration of 3 × 105 cells per mL 24 h in advance of all experiments to maintain controlled growth conditions. Directly in advance of analyte substance exposition, the cells were diluted to 1 × 105 cells per mL and 2 mL was transferred to six well plates according to the experimental setup. Adherent cells were harvested 24 h in advance of experiments, adjusted to 1 × 105 cells per mL and 2 mL was transferred to six well plates. Prior to treatment with the analyte substance, the cells were allowed to adhere to the dishes for 24 h in humidified atmosphere of 5% CO2 and air at 37 °C.MelHO (human melanoma cells) pIres and MelHO bcl-2 (transfected MelHO cells with the vector pIres and the bcl-2 gene, overexpressing the anti-apoptotic protein Bcl-2) were kindly provided by Jürgen Eberle, Charité, Berlin, Germany; Nalm-6 (human B cell precursor leukemia) cells were kindly provided by Karlheinz Seeger, Charité, Berlin. BJAB (human Burkitt lymphoma) cells were kindly provided by Peter Daniel, Charité, Berlin, Germany. Daunorubicin- and prednisolone-resistant Nalm-6 cells (NaKu) and also vincristine-resistant (BiBo) and Bcl-2 overexpressing BJAB cells (7CCA) were generated by our group in Cologne.
Cell viability
To determine the inhibitory effects of the agent on culture cells, a cell viability measurement is performed after 24 h of exposure. The viability is determined by Casy® Cell Counter from Schaerfe System GmbH. The increase in cell amount of exposed cells is determined and compared to the cell amount of non-exposed control cells. Difference in cell amount gives the direct inhibition at given concentration. Cells were seeded at a density of 1 × 105 cells per mL and treated with different concentrations of 1; non-treated cells served as controls. After a 24 h incubation period, cells were resuspended and 100 μl of each well was diluted in 10 mL CASYton (ready-to-use isotonic saline solution) for immediate automated counting.Cytotoxicity by LDH-release
The lysis of cells caused by a necrotic cell death can be quantified by the detection of lactate-dehydrogenase release from the cytosols to the medium. The LDH activity is determined by a linked enzymatic test. In a first step NAD+ is reduced to NADH/H+ during the LDH catalyzed conversion of lactate to pyruvate. Subsequently a catalyst transfers a H/H+ equivalent to the pale yellow tetrazolium salt forming a red formazan salt which is detected at 490–492 nm.The release of lactate-dehydrogenase is accompanied with the rupture of cell membrane caused by a necrotic cell death. It is known that necrotic cell death occurs directly after exposure to the causing agent while apoptosis is a much slower process involving complex reconfiguration of cell DNA and cell compartments in advance of cell blebbing. Therefore, necrosis and apoptosis can be differentiated via the rapid quantification of LDH activity in extracellular medium after exposure with a cytotoxic agent. The lactate-dehydrogenase is quantified using the Cytotoxicity Detection Kit from Roche. The supernatants were centrifuged at 1500 rpm for 5 min; 20 μl of cell-free supernatants were diluted with 80 μl phosphatebuffered saline (PBS), and 100 μl reaction mixture containing 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-phenyltetrazolium chloride (INT), sodium lactate, NAD+ and diaphorase were added. Then, time-dependent formation of the reaction product was quantified photometrically at 490 nm. The maximum amount of LDH release was determined after lysis of the cells using 0.1% Triton X-100 in culture medium and set to represent 100% cell death.
DNA-fragmentation
Quantifying the DNA fragmentation of exposed cells gives direct relation to apoptosis rate and therefore the lethality of the tested analyte on culture cells. The measurement is performed using the modified cell cycle analysis postulated by Nicoletti.23 DNA of cells is stained with propidium iodide after cell harvest and breakup of cell membrane. Cells were seeded at a density of 1 × 105 cells per mL and treated with different concentrations of 1. After a 72 h incubation period at a temperature of 37 °C, cells were collected by centrifugation at 1500 rpm for 5 min, washed with PBS at 4 °C, and fixed in PBS/2% (v/v) formaldehyde on ice for 30 min. After fixation, cells were pelleted, incubated with ethanol/PBS (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) for 15 min, pelleted and resuspended in PBS containing 40 μg ml−1 RNase. RNA was digested for 30 min at a temperature of 37 °C, after which the cells were pelleted again and finally resuspended in PBS containing 50 μg mL−1 propidium iodide. Nuclear DNA fragmentation was quantified by flow cytometric determination of hypodiploid DNA. Data was collected and analyzed using a flow cytometry analysis by FACScan (Becton–Dickinson, Heidelberg, Germany) equipped with CELL Quest software. Data is given in percent hypoploidy (subG1), which reflects the number of apoptotic cells.
:1, v/v) for 15 min, pelleted and resuspended in PBS containing 40 μg ml−1 RNase. RNA was digested for 30 min at a temperature of 37 °C, after which the cells were pelleted again and finally resuspended in PBS containing 50 μg mL−1 propidium iodide. Nuclear DNA fragmentation was quantified by flow cytometric determination of hypodiploid DNA. Data was collected and analyzed using a flow cytometry analysis by FACScan (Becton–Dickinson, Heidelberg, Germany) equipped with CELL Quest software. Data is given in percent hypoploidy (subG1), which reflects the number of apoptotic cells.
Mitochondrial membrane permeabilization
After an incubation period of 48 h with different concentrations of 1, the cells were collected by centrifugation at 1500 rpm, 4 °C for 5 min. The mitochondrial permeability transition was then determined by staining the cells with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine iodide (JC-1; Molecular Probes, Leiden, The Netherlands). Then 1 × 105 cells were resuspended in 500 μL phenol red-free RPMI 1640 without supplements, and JC-1 was added to give a final concentration of 2.5 μg mL−1. The cells were incubated for 30 min at 37 °C with moderate shaking. Control cells were likewise incubated in the absence of JC-1 dye. The cells were harvested by centrifugation at 1500 rpm, 4 °C for 5 min, washed with ice-cold PBS, and resuspended in 200 μl PBS at 4 °C. Mitochondrial permeability transition was then quantified by flow cytometric determination of the cells with decreased fluorescence, i.e. with mitochondria displaying a lower membrane potential. Data was collected and analyzed using a FACScan (Becton–Dickinson, Heidelberg, Germany) apparatus equipped with the CELL Quest software. Data are given in % cells with low ΔΨm, which reflects the number of cells undergoing mitochondrial apoptosis.Author contributions
A. P. and N. M.-N. designed the work and oversaw the workflow of experiments. L. R. and D. S. performed the synthesis of LR166. M. K. performed the biological experiments. D. S. analyzed all data in detail and wrote the first draft of the manuscript. The manuscript was refined through contributions of all authors. All authors have given approval to the final version of the manuscript.Funding sources
We gratefully thank the Dr. Kleist-Stiftung in Berlin (AP) for the financial support of this project.Conflicts of interest
The authors declare no competing interest.Acknowledgements
Prof. S. Hahn (Ruhr University Bochum) is gratefully acknowledged for insightful discussions.References
- G. Housman, S. Byler, S. Heerboth, K. Lapinska, M. Longacre, N. Snyder and S. Sarkar, Cancers, 2014, 6, 1769–1792 CrossRef CAS PubMed.
- H. Zahreddine and K. L. B. Borden, Front. Pharmacol., 2013, 4(28), 1–8 CAS.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS PubMed.
- K. D. Miller, R. L. Siegel, C. C. Lin, A. B. Mariotto, J. L. Kramer, J. H. Rowland, K. D. Stein, R. Alteri and A. Jemal, CA. Cancer J. Clin., 2016, 66, 271–289 CrossRef PubMed.
- Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS PubMed.
- P. Zhang and P. J. Sadler, Eur. J. Inorg. Chem., 2017, 2017, 1541–1548 CrossRef CAS.
- A. M. Pizarro, A. Habtemariam and P. J. Sadler, Top. Organomet. Chem., 2010, 32, 21–56 CrossRef CAS.
- A. Leonidova, V. Pierroz, R. Rubbiani, J. Heier, S. Ferrari and G. Gasser, Dalton Trans., 2014, 43, 4287–4294 RSC.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- G. Gasser and N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 16, 84–91 CrossRef CAS PubMed.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- P. Zhang and P. J. Sadler, J. Organomet. Chem., 2017, 839, 5–14 CrossRef CAS.
- M. M. Pedro Martins, M. Marques, L. Coito, A. J. L. Pombeiro, P. V. Baptista and A. R. Fernandes, Anti-Cancer Agents Med. Chem., 2014, 14, 1199–1212 CrossRef.
- A. Leonidova and G. Gasser, ACS Chem. Biol., 2014, 9, 2180–2193 CrossRef CAS PubMed.
- I. Kitanovic, S. Can, H. Alborzinia, A. Kitanovic, V. Pierroz, A. Leonidova, A. Pinto, B. Spingler, S. Ferrari, R. Molteni, A. Steffen, N. Metzler-Nolte, S. Wölfl and G. Gasser, Chem. – Eur. J., 2014, 20, 2496–2507 CrossRef CAS PubMed.
- R.-R. Ye, C.-P. Tan, M.-H. Chen, L. Hao, L.-N. Ji and Z.-W. Mao, Chem. – Eur. J., 2016, 22, 7800–7809 CrossRef CAS PubMed.
- S. Jürgens, W. A. Herrmann and F. E. Kühn, J. Organomet. Chem., 2014, 751, 83–89 CrossRef.
- J. R. Dilworth and S. J. Parrott, Chem. Soc. Rev., 1998, 27, 43–55 RSC.
- K. K.-W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS PubMed.
- L. J. Raszeja, D. Siegmund, A. L. Cordes, J. Güldenhaupt, K. Gerwert, S. Hahn and N. Metzler-Nolte, Chem. Commun., 2017, 53, 905–908 RSC.
- L. Raszeja, A. Maghnouj, S. Hahn and N. Metzler-Nolte, ChemBioChem, 2011, 12, 371–376 CrossRef CAS PubMed.
- A. Persidis, Nat. Biotechnol., 1999, 17, 94–95 CrossRef CAS PubMed.
- C. Riccardi and I. Nicoletti, Nat. Protoc., 2006, 1, 1458–1461 CrossRef CAS PubMed.
- A. Leonidova, V. Pierroz, R. Rubbiani, Y. Lan, A. G. Schmitz, A. Kaech, R. K. O. Sigel, S. Ferrari and G. Gasser, Chem. Sci., 2015, 5, 4044–4056 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |