
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ammonium [11C]thiocyanate: revised preparation and reactivity studies of a versatile nucleophile for carbon-11 radiolabelling†
Tom
Haywood
a,
Sara
Cesarec
 a,
Steven
Kealey
a,
Christophe
Plisson
b and
Philip W.
Miller
*a
a,
Steven
Kealey
a,
Christophe
Plisson
b and
Philip W.
Miller
*a
aDepartment of Chemistry, Imperial College London, South Kensington, SW7 2AZ, London, UK. E-mail: philip.miller@imperial.ac.uk; Tel: +44 (0)2875942847
bImanova Limited, Imperial College London, Hammersmith Hospital, Burlington Danes Building, Du Cane Road, London, W12 0NN, UK
First published on 2nd July 2018
Abstract
Herein we report the preparation of ammonium [11C]thiocyanate via the reaction of [11C]CS2 with ammonia. The [11C]SCN− ion is demonstrated as a potent nucleophile that can be used to readily generate a range of 11C-labelled thiocyanate molecules in high conversions. Furthermore, novel 11C-labelled thiazolone molecules can be easily prepared from the intermediate α-thiocyanatophenones via an acid mediated cyclisation reaction.
Positron emission tomography (PET) is now a widely used clinical and research imaging technique for the study and diagnosis of a range of neurological, oncological and cardiac conditions.1–5 The expansion of PET imaging centres worldwide over the past decade has led to an accompanying increase in demand for both new and existing PET tracers. The development of PET tracers presents a number of key chemical and biological challenges due to the short half-lives of positron-emitting nuclides, the peculiarity of tracer level chemical reactions, the complexity of biological interactions, and the strict working environment required for safe handling of radioactive substances. The synthetic chemistry hurdles are particularly apparent considering that the half-lives of the two most common PET nuclides, carbon-11 and fluorine-18, are only 20.4 min and 109.7 minutes, respectively. For example, a clinical radiopharmaceutical carbon-11 tracer production involving radiosynthesis, purification and formulation, typically needs to be complete within a 30–45 min timeframe. The development of new tracers is further confounded by the limited number of available PET precursors and methods for labelling, meaning that there are only a certain number of reactions that can be used to introduce the radionuclide.6 This limitation clearly restricts access to what can ultimately be radiolabelled and where on the target molecule the radionuclide can be introduced.
Despite these challenges, an array of novel fluorine-18 and carbon-11 labelling methods have been reported in recent years to try overcome these challenges.7–12 Carbon-11 is typically incorporated into tracers via well-established methylation protocols using [11C]CH3I or the more reactive [11C]CH3SO3CF3.6,13 Carbon-11 methylation is, however, unsuitable for labelling many interesting biological molecules, either due to the absence of a methyl group on the target molecule or due to unfavourable metabolism of the labelled methyl group in vivo. A range of other reactive small molecule carbon-11 reagents have therefore been developed to label in different positions. For example [11C]CO2,14 [11C]CO,15 [11C]HCN,11 [11C]CH2O16 and [11C]COCl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 have been used to radiolabel carbonyl and cyano functional groups. Recently, our group reported the production of a novel carbon-11 labelling reagent, [11C]carbon disulfide, and described its initial reactivity studies for the efficient labelling of organosulfur compounds such as dithiocarbamates, thioureas and other biological molecules.18–20 Herein, we report the facile production of ammonium [11C]thiocyanate, prepared via the reaction of [11C]CS2 with ammonia, and describe preliminary radiolabelling reactions of this versatile nucleophilic reagent.
17 have been used to radiolabel carbonyl and cyano functional groups. Recently, our group reported the production of a novel carbon-11 labelling reagent, [11C]carbon disulfide, and described its initial reactivity studies for the efficient labelling of organosulfur compounds such as dithiocarbamates, thioureas and other biological molecules.18–20 Herein, we report the facile production of ammonium [11C]thiocyanate, prepared via the reaction of [11C]CS2 with ammonia, and describe preliminary radiolabelling reactions of this versatile nucleophilic reagent.
The thiocyanate ion is a ‘pseudohalide’ and potent nucleophile that can react either via the nitrogen atom or sulfur atom. It has been widely use to prepare organic thiocyanates and a range of other sulfur containing compounds (Scheme 1).21 The thiocyanate ion is attractive from a radiolabelling perspective because of its reactivity and potential to form a wide range of organosulfur derivatives. [11C]Thiocyanate has been previously reported, however, its synthesis relies on the production of [11C]HCN, which is not readily accessible in most PET centres, thus limiting its potential wider use. Typically, [11C]SCN− is prepared by first producing [11C]HCN, followed by conversion to a salt and then reaction with a sulfur source. The first reported method describes a two-step process where [11C]HCN is passed through a phosphorous pentoxide trap into a solution of sodium or potassium hydroxide to generate [11C]CN−, which is then reacted with elemental sulfur to give [11C]SCN− (Scheme 2, top).22,23 In an alternative approach, [11C]HCN can be converted to [11C]NaSCN via bromination in triglyme to give [11C]BrCN followed by distillation over antimony and reaction with sodium sulfide (Scheme 2, top).24 In an effort to prepare a range of sulfur based labelled compounds we sought to explore alternative methods to synthesise [11C]SCN− that circumvent the need for specialised [11C]HCN synthesis equipment (Scheme 2).
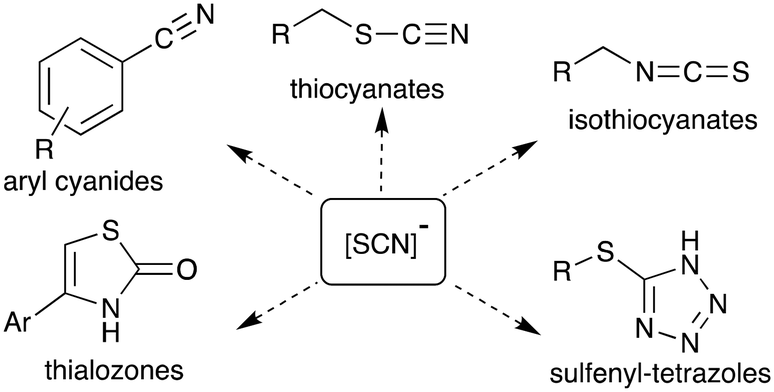 | ||
| Scheme 1 A selection of organic molecules that can be prepared using the thiocyanate ion. | ||
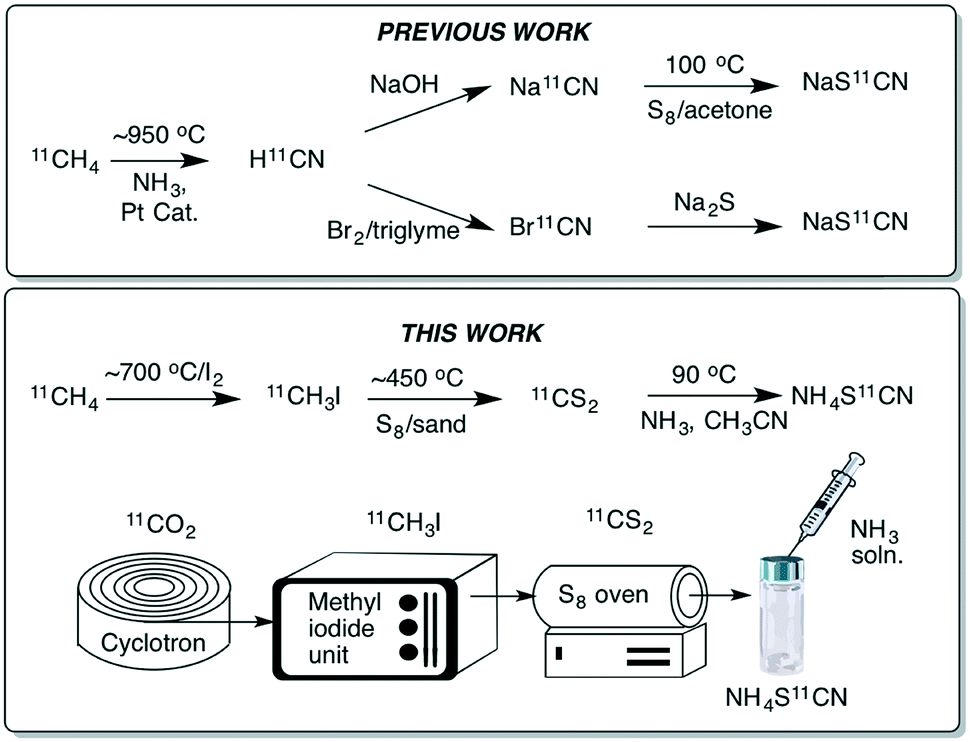 | ||
| Scheme 2 Previously reported routes to [11C]SCN− relying on generating [11C]HCN first (top); this work, which reports the synthesis of [11C]SCN−via [11C]CS2 (bottom). | ||
Our recent work on the development of [11C]CS2 led us to investigate this reagent as an alternative approach to prepare [11C]SCN−. Carbon disulfide is known to react with ammonia to give ammonium dithiocarbamate. On heating ammonium dithiocarbamate to 90 °C, ammonium thiocyanate is generated along with one equivalent of H2S. We previously reported a fast and easily accessible route to [11C]CS2via the reaction of sulfur vapour with [11C]CH3I. Unlike [11C]HCN, [11C]CH3I is a much more commonly used carbon-11 labelling reagent that is prevalent in many PET centres. In our initial experiments to prepare [11C]SCN−, a stream of gaseous [11C]CS2 was passed into an ammonia solution (0.1 M in MeCN) and heated to 90 °C for 5 min. Analysis of the crude reaction mixture confirmed [11C]NH4SCN formation, albeit in low radiochemical yields (RCY) (<10%, Table 1). We hypothesised that passing the [11C]CS2 gas stream directly through the ammonia solution resulted in significant loss of the volatile ammonia, hence greatly reducing its concentration and therefore limiting the reaction. To circumvent this, the order of addition was changed to avoid ammonia loss; first trapping the [11C]CS2 in acetonitrile (1 mL), then adding the ammonia solution (50 μL, 2 M NH3 in CH3OH) and heating to 90 °C for 5 min. Pleasingly, this resulted in near quantitative radiochemical conversion of [11C]CS2 to [11C]NH4SCN as observed by radio-HPLC (Table 1).
| 11CS2 + 2NH3 → NH4S11CN + H2S | ||
|---|---|---|
| Synthesis methoda | RCYb [11C]CS2 (%) | RCYb [11C]NH4SCN (%) |
| a Method A: [11C]CS2 gas stream bubbled through ammonia solution (50 μL, 2 M NH3 in CH3OH, acetonitrile 1 mL) followed by heating to 90 °C for 5 min. Method B: addition of ammonia solution (50 μL, 2 M NH3 in CH3OH) to [11C]CS2 in acetonitrile (1 mL) followed by heating to 90 °C for 5 min. b Non-isolated radiochemical yield (RCY) determined by analytical radio HPLC of the crude product based on conversion from [11C]CH3I. Average of minimum n = 3. | ||
| A | 90 | 10 |
| B | 1 | 99 |
Given its ease of synthesis and potential utility as a nucleophile, we aimed to explore the reactivity of the [11C]thiocyanate ion for substitution reactions. A simple model reaction of [11C]NH4SCN with benzyl bromide was initially investigated. In a typical labelling experiment, a solution of benzyl bromide was injected into the reaction vial containing in situ generated [11C]NH4SCN, and the mixture heated for 5 min at 90 °C (Scheme 3).
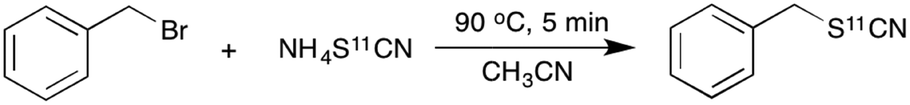 | ||
| Scheme 3 Substitution reaction of benzyl bromide with [11C]NH4SCN to generate benzyl [11C]thiocyanate. | ||
The crude product mixture was analysed by radio-HPLC and the product identity confirmed by comparison of the radioactive sample with unlabelled reference material. [11C]NH4SCN was found to react cleanly and quickly with benzyl bromide showing virtually quantitative conversion to benzyl [11C]thiocyanate (Chart 1). Encouraged by this result, we proceeded to explore this reaction with a range of α-ketobromides to generate their respective 11C-labelled α-thiocyanatophenones. We discovered that excellent conversions could be achieved for the C-11 thiocyanation of all seven α-ketobromides we tested, within a 5 min reaction time (Chart 1). Lower conversions were observed for the reaction of the diphenyl substrate (Chart 1, entry 8), most likely due to the greater steric hindrance of this substrate inhibiting nucleophilic attack of the thiocyanate ion. [11C]NH4SCN was also found to react cleanly with mannose triflate, the precursor used for 2-([18F]fluoro)-2-deoxy-D-glucose synthesis, to give the labelled sugar thiocyanate derivative (9).
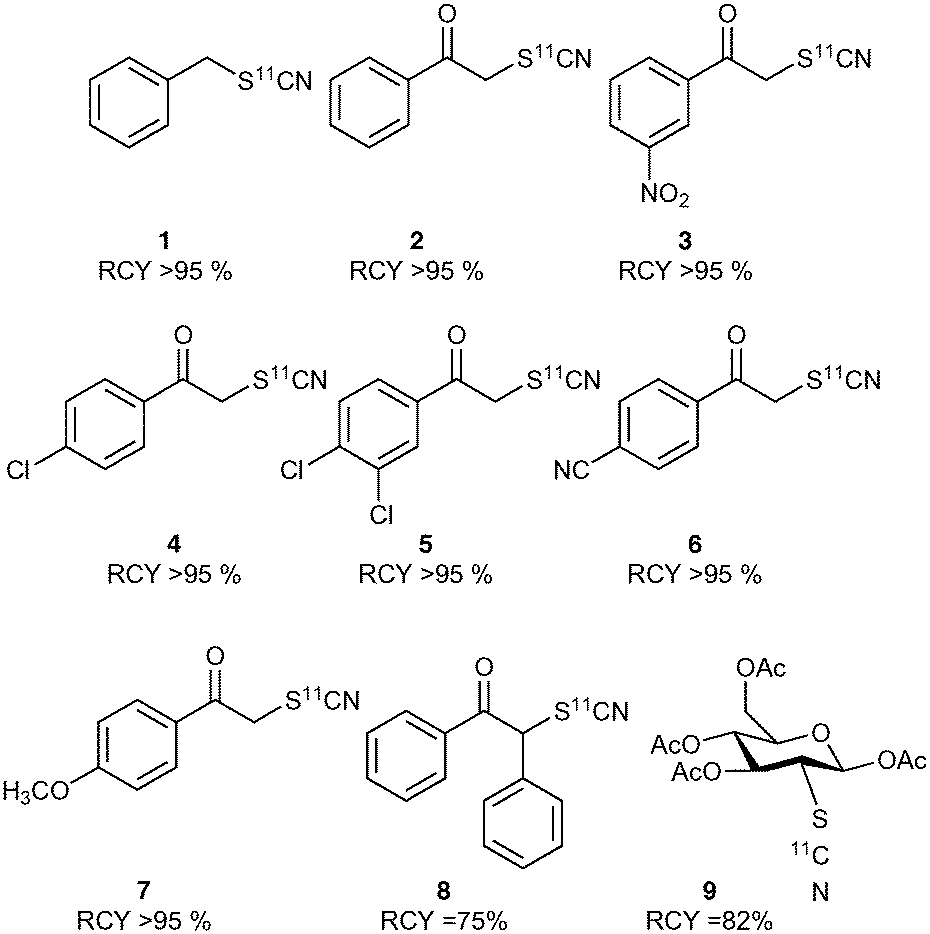 | ||
| Chart 1 Radiolabelled thiocyanate molecules. Average of minimum n = 2. | ||
Thiocyanate compounds are interesting intermediates because they can be converted into other organosulfur and heterocyclic structures.25–27 Thiazole structures are prevalent in a number of important naturally occurring compounds, for example thiamine and penicillin, and in a wide range of bioactive small molecules such as benzothiazoles, while the related thiazolones have been investigated for anti-cancer activity.28 Carbon-11 radiolabelling within the ring of such sulfur based heterocyclic structures is challenging, and to the best of our knowledge has not been achieved until now. Since thiazolones can be accessed in high yield from α-thiocyanatophenones via an acid mediated cyclisation reaction (Scheme 4), we sought to explore this pathway using our in situ generated α-[11C]thiocyanatophenones.29
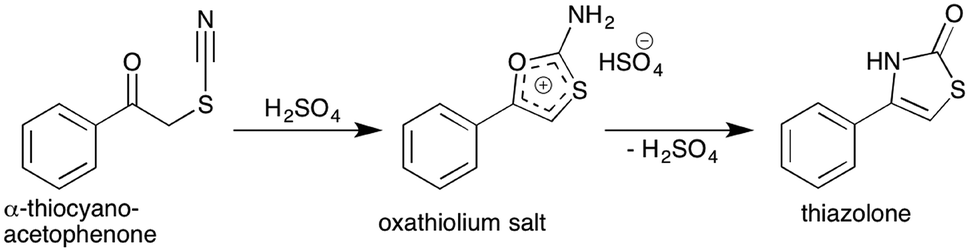 | ||
| Scheme 4 Acid mediated cyclisation of α-thiocyanatophenone which is hypothesised to proceed via the intermediate oxathiolium salt prior to thiazolone formation and elimination of sulfuric acid. | ||
To test this in the radiochemistry laboratory, a solution of 1-phenyl-2-[11C]thiocyanatoethanone (2) was added to a mixture of sulfuric and acetic acid, and heated to 90 °C for 5 min. This resulted in complete conversion of 2 to the cyclised thiazolone, 4-phenylthiazol-2-[11C]one (10) (Scheme 5, Chart 2). This cyclisation reaction was then performed with the remaining phenyl-substituted α-[11C]thiocyanatophenones, producing the corresponding 4-phenylthiazol-2-[11C]ones in high radiochemical purity (70–95%, Chart 2).
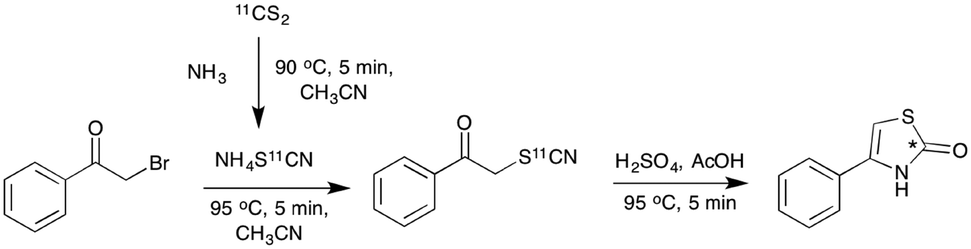 | ||
| Scheme 5 One pot radiolabelling of 4-phenylthiazol-2-[11C]one (10) via acid mediated cyclisation of the intermediate 1-phenyl-2-[11C]thiocyanatoethanone (2), (*) denotes the 11C labelling position. | ||
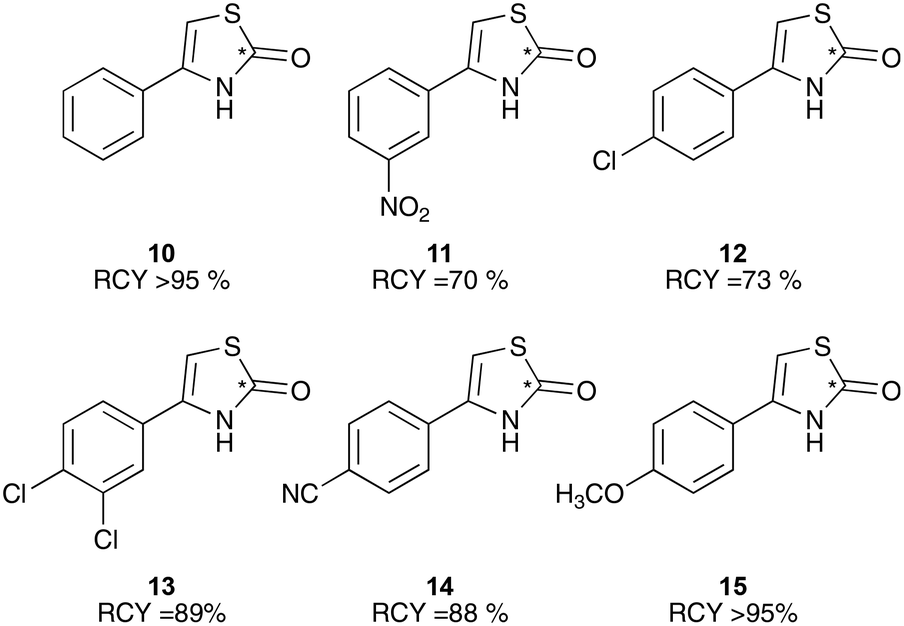 | ||
| Chart 2 11C-Radiolabelled thiazolone molecules. Average of minimum n = 2. | ||
In conclusion, we have developed a new synthetic route to ammonium [11C]thiocyanate via the reaction of ammonia with [11C]carbon disulfide. The speed and efficiency of this synthesis has enabled us to exploit the [11C]thiocyanate ion as a potent nucleophilic species for the preparation of a range of radiolabelled thiocyanate compounds in high radiochemical yields. Furthermore, we have been able to rapidly conduct cyclisation reactions with these α-[11C]thiocyanatophenones to generate unprecedented 11C-thiazolone molecules. We anticipate that this labelling route will find further applications for the preparation of carbon-11 based organosulfur tracers. Given that [11C]CS2 can be easily prepared from the widely available carbon-11 precursor [11C]CH3I we also expect that this method could be easily adopted by other PET centres with carbon-11 facilities.
We are grateful for financial support from the EPSRC (grant no. EP/L025140/1) and the Royal Society (grant no. RG110449).
Conflicts of interest
The authors declare no competing interest.Notes and references
- T. Jones and E. A. Rabiner, J. Cereb. Blood Flow Metab., 2012, 32, 1426–1454 CrossRef PubMed.
- A. Nordberg, J. O. Rinne, A. Kadir and B. Långström, Nat. Rev. Neurol., 2010, 6, 78–87 CrossRef PubMed.
- D. J. Brooks, J. Nucl. Med., 2010, 51, 596–609 CrossRef PubMed.
- E. M. Rohren, T. G. Turkington and R. E. Coleman, Radiology, 2004, 231, 305–332 CrossRef PubMed.
- L. W. Dobrucki and A. J. Sinusas, Nat. Rev. Cardiol., 2010, 7, 38–47 CrossRef PubMed.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef PubMed.
- S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef PubMed.
- T. W. Price, J. Greenman and G. J. Stasiuk, Dalton Trans., 2016, 45, 15702–15724 RSC.
- V. Bernard-Gauthier, J. J. Bailey, Z. Liu, B. Waengler, C. Waengler, K. Jurkschat, D. M. Perrin and R. Schirrmacher, Bioconjugate Chem., 2016, 27, 267–279 CrossRef PubMed.
- J.-P. Meyer, P. Adumeau, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27, 2791–2807 CrossRef PubMed.
- B. H. Rotstein, S. H. Liang, M. S. Placzek, J. M. Hooker, A. D. Gee, F. Dollé, A. A. Wilson and N. Vasdev, Chem. Soc. Rev., 2016, 45, 4708–4726 RSC.
- M. B. Haskali and V. W. Pike, Chem. – Eur. J., 2017, 1003, 8156–8160 CrossRef PubMed.
- G. Antoni, J. Labelled Compd. Radiopharm., 2015, 58, 65–72 CrossRef PubMed.
- B. H. Rotstein, S. H. Liang, J. P. Holland, T. L. Collier, J. M. Hooker, A. A. Wilson and N. Vasdev, Chem. Commun., 2013, 49, 5621–5629 RSC.
- S. Kealey, A. Gee and P. W. Miller, J. Labelled Compd. Radiopharm., 2014, 57, 195–201 CrossRef PubMed.
- J. M. Hooker, M. Schoenberger, H. Schieferstein and J. S. Fowler, Angew. Chem., Int. Ed., 2008, 47, 5989–5992 CrossRef PubMed.
- D. Roeda and F. Dolle, Curr. Top. Med. Chem., 2010, 10, 1680–1700 CrossRef PubMed.
- P. W. Miller and D. Bender, Chem. – Eur. J., 2012, 18, 433–436 CrossRef PubMed.
- T. Haywood, S. Kealey, S. Sanchez-Cabezas, J. J. Hall, L. Allott, G. Smith, C. Plisson and P. W. Miller, Chem. – Eur. J., 2015, 21, 9034–9038 CrossRef PubMed.
- S. Merchant, L. Allott, L. Carroll, V. Tittrea, S. Kealey, T. H. Witney, P. W. Miller, G. Smith and E. O. Aboagye, RSC Adv., 2016, 6, S7569–S7579 RSC.
- T. Castanheiro, J. Suffert, M. Donnard and M. Gulea, Chem. Soc. Rev., 2016, 45, 494–505 RSC.
- S. Stone-Elander, P. Roland, C. Halldin, M. Hassan and R. Seitz, Nucl. Med. Biol., 1989, 16, 741–746 Search PubMed.
- P. Johnstrom, L. Bergman, K. Varnas, J. Malmquist, C. Halldin and L. Farde, Nucl. Med. Biol., 2015, 42, 555–560 CrossRef PubMed.
- G. Westerberg and B. Langstrom, J. Labelled Compd. Radiopharm., 1994, 34, 545–548 CrossRef.
- F. R. Bisogno, A. Cuetos, I. Lavandera and V. Gotor, Green Chem., 2009, 11, 452 RSC.
- A. K. Yadav and L. D. S. Yadav, Green Chem., 2015, 17, 3515–3520 RSC.
- J. Rudolph, H. Theis, R. Hanke, R. Endermann, L. Johannsen and F. Geschke, J. Med. Chem., 2001, 44, 619–626 CrossRef PubMed.
- D. Havrylyuk, B. Zimenkovsky, O. Vasylenko, L. Zaprutko, A. Gzella and R. Lesyk, Eur. J. Med. Chem., 2009, 44, 1396–1404 CrossRef PubMed.
- K. Pihlaja, V. Ovcharenko, E. Kolehmainen, K. Laihia, W. M. F. Fabian, H. Dehne, A. Perjéssy, M. Kleist, J. Teller and Z. Šusteková, J. Chem. Soc., Perkin Trans. 2, 2002, 2, 329–336 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and HPLC traces. See DOI: 10.1039/c7md00425g |
| This journal is © The Royal Society of Chemistry 2018 |