
Studies on enantioselectivity of chiral 4-acetylamino-6-alkyloxy-2-alkylthiopyrimidines acting as antagonists of the human A3 adenosine receptor†
Barbara
Cosimelli
 a,
Giovanni
Greco
*a,
Sonia
Laneri
a,
Ettore
Novellino
a,
Antonia
Sacchi
a,
Simona
Collina
b,
Daniela
Rossi
b,
Sandro
Cosconati
c,
Elisabetta
Barresi
d,
Sabrina
Taliani
d,
Maria Letizia
Trincavelli
d and
Claudia
Martini
d
a,
Giovanni
Greco
*a,
Sonia
Laneri
a,
Ettore
Novellino
a,
Antonia
Sacchi
a,
Simona
Collina
b,
Daniela
Rossi
b,
Sandro
Cosconati
c,
Elisabetta
Barresi
d,
Sabrina
Taliani
d,
Maria Letizia
Trincavelli
d and
Claudia
Martini
d
aDipartimento di Farmacia, Università di Napoli “Federico II”, Via Domenico Montesano 49, 80131 Naples, Italy. E-mail: giovanni.greco@unina.it; Fax: +39 081 678107; Tel: +39 081678645
bDipartimento del Farmaco, Università di Pavia, Viale Torquato Taramelli 12, 27100 Pavia, Italy
cDipartimento di Scienze e Tecnologie Ambientali Biologiche e Farmaceutiche, Università degli Studi della Campania “Luigi Vanvitelli”, Via Antonio Vivaldi 43, 81100 Caserta, Italy
dDipartimento di Farmacia, Università di Pisa, Via Bonanno Pisano 6, 56126 Pisa, Italy
First published on 9th November 2017
Abstract
Three A3 adenosine receptor (AR) antagonists (1–3) selected from 4-acylamino-6-alkyloxy-2-alkylthiopyrimidines previously investigated by us were modified by inserting a methyl group on their ether or thioether side chains. These compounds gave us the chance to evaluate whether their higher lipophilicity, reduced conformational freedom and chirality might improve the potency towards the A3 AR. Racemic mixtures of 1–3 were resolved using chiral HPLC methods and the absolute configurations of the enantiomers were assigned by chiroptical spectroscopy and density functional theory calculations. We measured the affinity for human A1, A2A, A2B and A3 ARs of the racemic mixtures and the pure enantiomers of 1–3 by radioligand competition binding experiments. Cell-based assays of the most potent enantiomers confirmed their A3 AR antagonist profiles. Our research led to the identification of (S)-1 with high potency (0.5 nM) and selectivity as an A3 AR antagonist. Moreover we built a docking-model useful to design new pyrimidine derivatives.
Introduction
Adenosine is ubiquitously located in mammalian organisms, where it acts as an agonist of four subtypes of G-protein-coupled-receptors (GPCRs), namely the A1, A2A, A2B and the A3 adenosine receptors (ARs).1,2 Activation of ARs leads to different intracellular events related to inhibition (A1 and A3) or stimulation (A2A and A2B) of adenylate cyclase, stimulation of phospholipase C (A1, A2B, and A3), activation of potassium channels and inhibition of calcium channels (A1).2 ARs are considered to be promising pharmacological targets given their key role in important physiological and pathological processes. The A3 AR is involved in cell cycle regulation and cell growth.3 Borea P. A. et al.4 have recently outlined the current and potential clinical applications of selective agonists and antagonists of this receptor. As detailed in the above quoted review, agonists can be employed as cardioprotective, cerebroprotective and anti-inflammatory agents, whereas antagonists might be useful for the treatment of stroke, glaucoma, asthma and allergies.The pyrimidine ring represents one of the heterocyclic scaffolds featured by potent antagonists of the human A3 AR,5–8 including some 4-acylamino-6-alkyloxy-2-alkylthiopyrimidines of general formula I (Chart 1, where R, R′ and R′′ are alkyl groups) reported by us some years ago.7
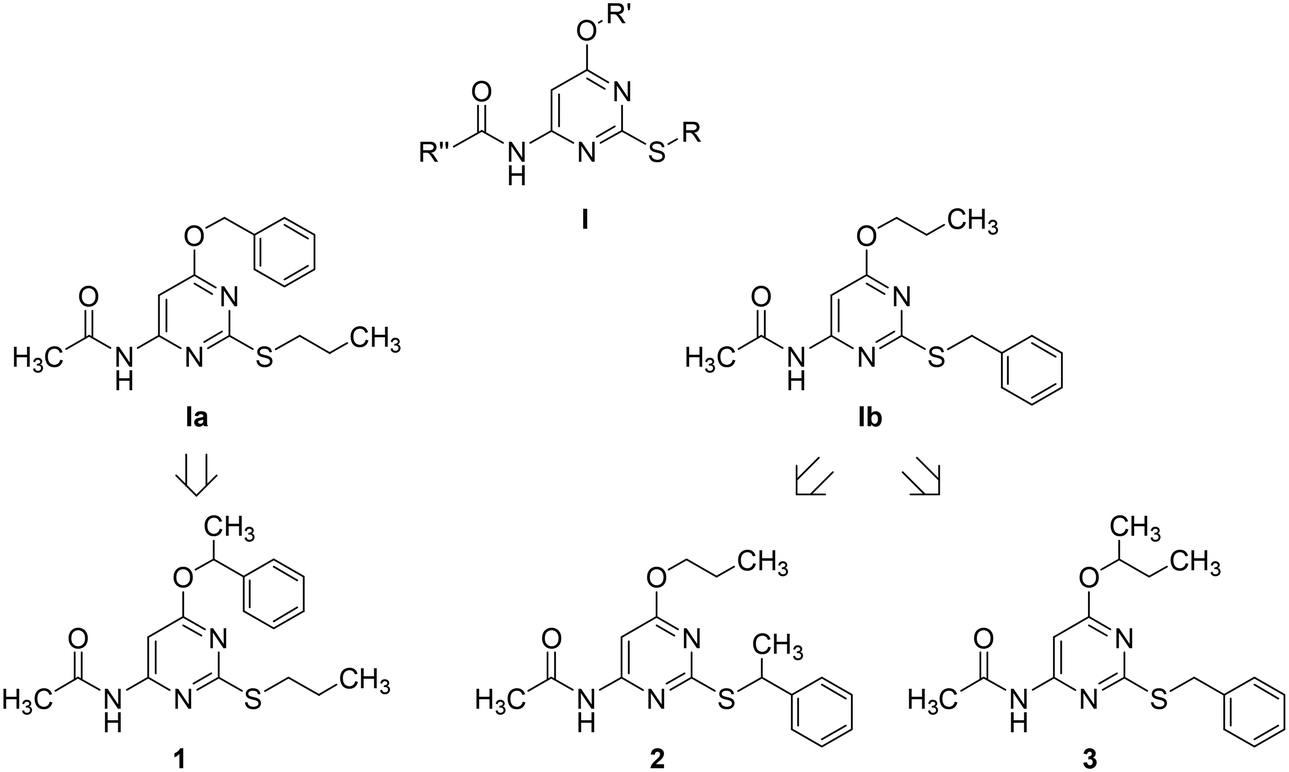 | ||
| Chart 1 Design of chiral pyrimidines 1–3 by formal methylation of Ia and Ib. | ||
Recently, we have focused our attention on compounds Ia and Ib of series I (Chart 1) as suitable structures to continue our research in this field. Particularly, we decided to prepare and test 1 as an α-methyl derivative of Ia together with 2 and 3 as α-methyl derivatives of Ib to assess whether the designed compounds, being more lipophilic and less flexible than their desmethyl counterparts, might exhibit improved potency as A3 AR antagonists. Moreover, as α-methylation of Ia and Ib introduces a stereogenic center on the ether (1 and 3) or the thioether (2) side chain, we had a chance to study the potential influence of chirality on the interaction of these pyrimidine derivatives with the target receptor. To our knowledge, only a few investigations on chiral A3 AR antagonists have been so far reported in the literature. Specifically, chiral 1,4-dihydropyridines have been investigated by van Rhee, A. M. et al.9 as well as by Jiang J.-L. et al.,10 whereas Jacobson, K. A. et al. described carbocyclic adenosine analogues characterized as A3 AR adenosine antagonists and partial agonists.11
In our work the racemic mixtures of the novel pyrimidine derivatives were resolved using chiral HPLC methods and the absolute configurations of the resulting pure enantiomers were assigned by a combined approach of chiroptical spectroscopy and density functional theory (DFT) calculations. The results of these studies have been recently published.12
In the present work, racemic mixtures and the pure enantiomers of 1–3 were assayed in competition binding experiments to measure their affinities for the human A3 AR and their selectivity trend towards the other AR subtypes. Cell-based functional experiments were carried out on the most potent enantiomers to confirm the expected antagonist efficacy profile. Finally, in silico docking studies using a 3D model of the human A3 AR were performed to hypothesize the interactions of the most potent enantiomers with the receptor binding site at the molecular level. This paper describes the results of our studies.
Results
Chemistry
Derivatives 1–3 were obtained in three steps starting from the commercially available 4-amino-6-hydroxy-2-mercaptopyrimidine monohydrate (4) as shown in Scheme 1 (additional experimental details for each step including characterization of the intermediates can be found in the ESI† of this publication). Alkylation of the sulfur atom at the 2-position by reaction with the opportune bromoalkane in 1 M NaOH gave derivatives 5–7 which were collected by filtration and resulted in sufficiently pure compounds to be subsequently used without further purification (5 and 7 were previously published).7 As a second step, the 4-oxygen atom of 5–7 was alkylated to give 8–10. When the reactant was 6 the reaction gave a mixture of isomers 9 and 9a. As a last step, the 4-amino group of 8–10 was converted into the amide group of 1–3 by reaction with acetic anhydride and concentrated H2SO4 as a catalyst.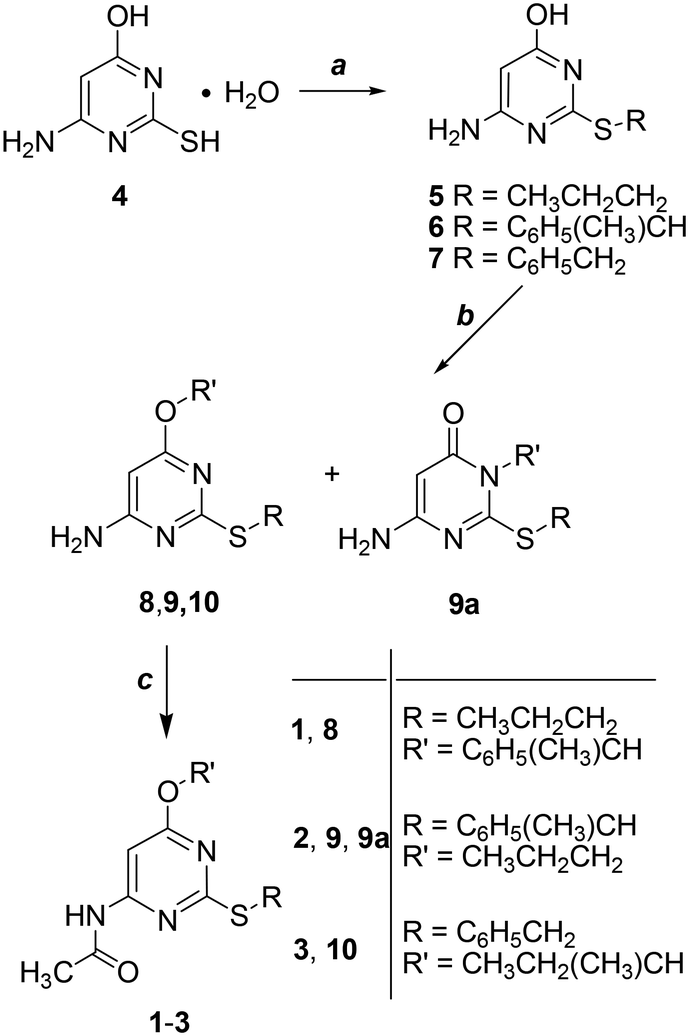 | ||
| Scheme 1 Reagents: (a) bromopropane, (1-bromoethyl)benzene or benzylbromide, NaOH 1 M, 50–55 °C, 15–90%. (b) (1-Bromoethyl)benzene, bromopropane or 2-bromobutane, K2CO3, CH3CN, reflux, 60–75%. (c) (CH3CO)2O, H2SO4 conc. (cat.), 50–60 °C, 41% (1) or 52% (2) or 70% (3). | ||
As recently reported,12 the resolution of racemic 1–3 by semi-preparative HPLC using chiral stationary phases was performed to isolate each enantiomer in small amounts suitable for configurational assignment and biological investigations.
Briefly, a standard screening protocol was firstly applied by using an analytical Chiralcel OJ-H column (4.6 mm × 150 mm, 5 μm), whose chiral selector is cellulose tris-(4-methylbenzoate) coated on a silica gel substrate. Baseline separation of 2 and 3 was achieved by eluting with pure methanol.
As regards 1, high enantioselectivity and resolution were achieved by using heptane/ethanol 95/5 (v/v) as the eluent. These experimental conditions were adopted for the chromatographic scale-up so as to afford enantiomeric 1–3 in good yields (82%, 93% and 97%, respectively) and with an optical purity above 99% suitable for both configurational studies and biological investigations.
Absolute configurations of enantiomeric 1–3 were assigned by comparing experimental and calculated optical rotatory dispersion curves and, in the case of enantiomeric 1 and 2, also comparing the electronic circular dichroism and vibrational circular dichroism spectra, with DFT calculations. According to our studies, the S absolute configuration was assigned to the levo isomers of 1–3 and the R absolute configuration to the corresponding dextro isomers (Chart 2).
 | ||
| Chart 2 Structures of the enantiomers of 1–3 represented with their absolute configurations. | ||
Biology
The affinities of 1–3 for human ARs, tested as racemic mixtures, were evaluated by radioligand competition binding experiments which assessed their ability to displace [3H]8-cyclopentyl-1,3-dipropylxanthine ([3H]DPCPX) from the human A1 AR or [3H]5′-N-ethylcarboxamidoadenosine ([3H]NECA) from the human A2A, A2B and A3 ARs expressed in Chinese hamster ovary (CHO) cells.13 In all binding assays, compounds 1–3, similarly to their desmethyl counterparts Ia and Ib, exhibited high or moderate potency for the A3 AR, whereas they lacked appreciable affinity for A1, A2A and A2B ARs (Ki values greater than 1000 nM; their percentages of inhibition of specific radioligand binding determined at 10 μM are listed in Table 1S of the ESI†). On the basis of these results, enantiomeric 1–3 were tested for their binding affinities exclusively for the A3 AR following the same procedures employed for the racemic mixtures.The antagonist efficacies of the enantiomers exhibiting the highest affinities for the A3 AR, namely eutomers (S)-1, (S)-2 and (S)-3, were evaluated by assessing their abilities to counteract 2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide (Cl-IB-MECA)-mediated inhibition of cAMP accumulation produced by forskolin (FK). Cl-IB-MECA is a selective A3 AR agonist whereas FK is a non-selective activator of adenylate cyclase. The obtained data are expressed as IC50 values (concentrations inhibiting the effect of the selective Cl-IB-MECA by 50%).14 Intracellular cAMP levels were measured as previously described.15 (Methods are detailed in the ESI†).
Table 1 lists the binding affinities of 1–3, tested as racemic mixtures or enantiomers, for the human A3 AR expressed as Ki values. The binding affinities for the same receptor of the previously described pyrimidine derivatives Ia and Ib7 are also given for comparison purposes. As mentioned, the affinities of the new compounds for the A3 AR range from subnanomolar to micromolar values. The concentration–response curves of the tested compounds are depicted in Fig. 1, whereas their antagonist potencies, expressed as IC50 values, are listed in Table 1.
|
|
||||
|---|---|---|---|---|
| No | R | R′ | K i [nM]a | IC50 [nM]a |
| hA3b | hA3c | |||
| a The Ki and IC50 values are means ± SEM of at least three determinations derived from an iterative curve-fitting procedure (Prism program, GraphPad, San Diego, CA). b Displacement of specific [3H]NECA binding in membranes obtained from CHO cells stably expressing the human A3 AR. c The IC50 values correspond to the concentrations inhibiting the effect of the selective A3 AR agonist Cl-IB-MECA by 50%. d Data from ref. 7, assayed with a purity of 96%. | ||||
| Ia | CH3CH2CH2 | C6H5CH2 | 7.5 ± 0.22 | |
| Ib | C6H5CH2 | CH3CH2CH2 | 525 ± 21 | |
| (R/S)-1 | CH3CH2CH2 | C6H5(CH3)CH | 0.6 ± 0.04 | |
| (R)-(+)-1 | CH3CH2CH2 | C6H5(CH3)CH | 1.7 ± 0.4 | |
| (S)-(−)-1 | CH3CH2CH2 | C6H5(CH3)CH | 0.5 ± 0.2 | 7.9 ± 0.9 |
| (R/S)-2 | C6H5(CH3)CH | CH3CH2CH2 | 580.1 ± 70.3 | |
| (R)-(+)-2 | C6H5(CH3)CH | CH3CH2CH2 | 737.5 ± 28.5 | |
| (S)-(−)-2 | C6H5(CH3)CH | CH3CH2CH2 | 440.2 ± 55.2 | 428.2 ± 36.9 |
| (R/S)-3 | C6H5CH2 | CH3CH2(CH3)CH | 195.1 ± 32.4 | |
| (R)-(+)-3 | C6H5CH2 | CH3CH2(CH3)CH | 1390 ± 187 | |
| (S)-(−)-3 | C6H5CH2 | CH3CH2(CH3)CH | 183.2 ± 44.1 | 152.9 ± 14.3 |
| DPCPX | >1000 | |||
| NECA | 75.5 ± 5.2 | |||
| Cl-IB-MECA | 0.2 ± 0.1 | |||
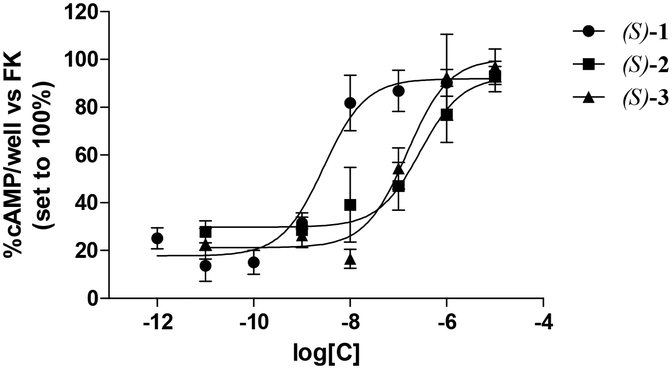 | ||
| Fig. 1 Effects of (S)-1, (S)-2 and (S)-3 on Cl-IB-MECA-mediated inhibition of cAMP accumulation in human A3 AR-transfected CHO cells. CHO cells were treated with 1 μM FK and 100 nM Cl-IB-MECA in the absence or presence of different concentrations of the tested compound (0.01 nM–10 μM). After 15 min incubation, the reaction was stopped and the intracellular cAMP levels were quantified. Data are expressed as the percentage of the cAMP intracellular levels with respect to FK, which was set to 100%, and represent the mean ± SEM of at least three different experiments. Each experiment was performed in duplicate. | ||
All the three selected compounds acted as antagonists of the A3 AR with potencies comparable to the affinities obtained in radioligand binding experiments.
Discussion
Insertion of an α-methyl group on the benzyloxy side chain of Ia to yield 1 increases potency. In fact, (S)-1 and (R)-1 are more potent than their desmethyl counterparts, by 15-fold and 4.4-fold, respectively. These findings suggest that the α-methyl group helps the PhCH(CH3)O substituent of both (S)-1 and (R)-1 to optimize their interactions within a specific region of the human A3 AR binding site. This result might be achieved by each of the two ligands through conformational effects (i.e. reduced torsional freedom) and/or a direct hydrophobic interaction with the lipophilic side chain residues of the receptor.Conversely, α-methylation of the benzylthio side chain of Ib to give 2 does not significantly affect the potency, the racemic mixture and the corresponding enantiomers being nearly equipotent with the desmethyl parent structure.
Such data indicate that this type of α-methylation somehow leaves the interactions between Ib and the A3 AR almost intact.
When a methyl group is inserted onto the α-carbon of the n-propyloxy side chain of Ib to yield 3, we observe contrasting effects on the potency depending on the chirality. Specifically, compared with the desmethyl parent compound, (S)-3 is more potent by 2.9-fold, whereas (R)-3 is less potent by 2.6-fold. In this case, α-methylation modifies the conformational properties of the s-butyloxy side chain of the eutomer and of the distomer so as to enhance or, respectively, reduce ligand–receptor attractive interactions.
Docking studies (see the ESI† for methods) carried out on the most potent enantiomers, namely, (S)-1 and (R)-1, suggest that these compounds form a double-bridged H-bond between the ligand N3–C4–NH fragment and the receptor N250 side chain (Fig. 2). Such an interaction is considered to be fundamental for tight ligand binding to both A3 and A2A ARs.16,17 By looking at Fig. 2, it can also be noticed that the pyrimidine scaffold of (S)-1 and (R)-1 engages in π-stacking and van der Waals interactions with the F168 phenyl ring on one side and with the Leu264 isobutyl moiety on the opposite side. The above described interactions seem to provide anchoring points for the two ligands and allow the placement of the PhCH(CH3)O and CH3(CH2)2S substituents in two different lipophilic clefts of the receptor. Specifically, the n-propylthio chain points towards the inner part of the receptor making van der Waals contacts with the I186, L91, W243, and L246 side chains. On the opposite side, the PhCH(CH3)O group extends towards the crevice formed by the TM2 and TM3 helices. As depicted in Fig. 2, it is clear that (S)-1 maximizes the interactions between the terminal phenyl ring and the surrounding residues (namely Y15, V65, A69, V72, L89, and L90). The same substituent in (R)-1 still nicely fits within the same cleft. However, its phenyl ring is less close to the above listed residues, thus giving rise to less favourable interactions. Our docking models seem to be consistent with the slightly higher affinity (3.4-fold) of (S)-1 compared with that of (R)-1.
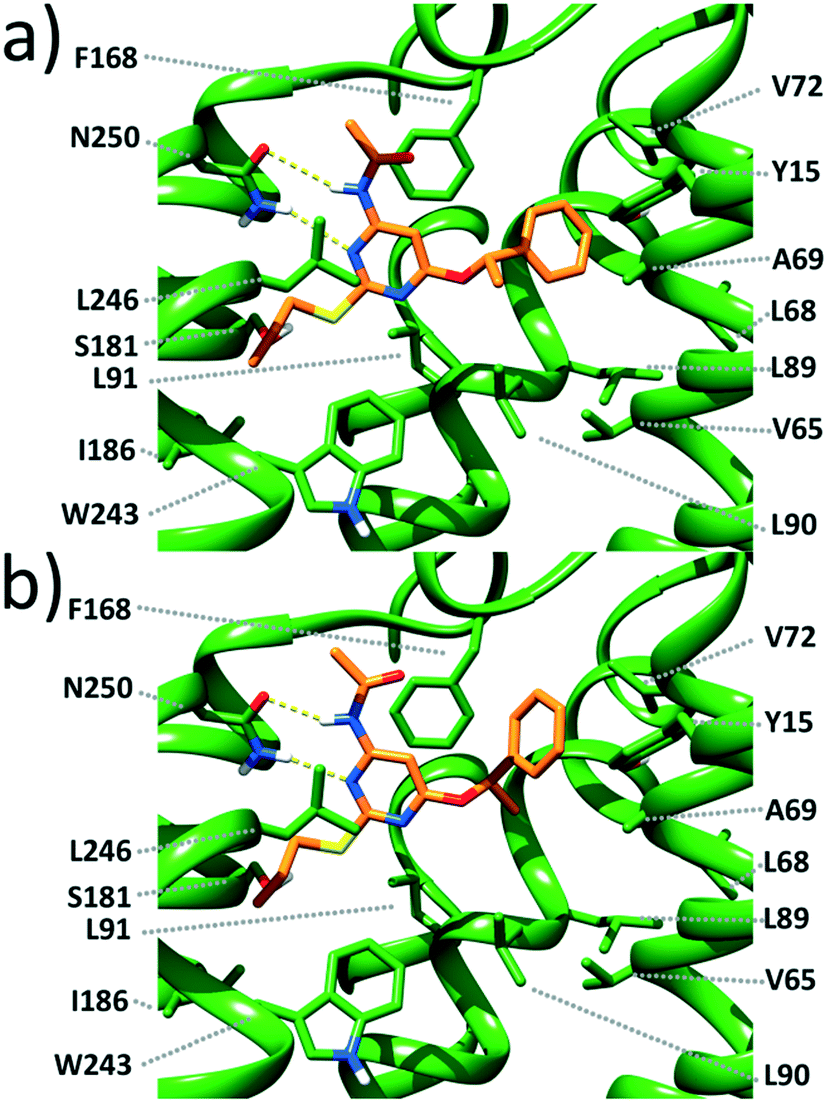 | ||
| Fig. 2 Close view of the calculated binding pose for (S)-1 (a) and (R)-1 (b) within a published 3D model of the human A3 AR.18 The ligand and the receptor are depicted as orange and green sticks, respectively. H-bonds are depicted as dashed yellow lines. | ||
A close inspection of Fig. 2 reveals that the α-methyl group of both ligands does not fit into any specific hydrophobic clefts of the receptor binding site. According to our theoretical models, the α-methyl group of (S)-1 and (R)-1 acts by “freezing” the two enantiomers in conformations where the phenyl and the methyl groups point towards opposite directions (see Fig. 3), with each conformation capable of optimizing the favourable interactions that the PhCH(CH3)O moiety within the receptor cavity is engaged in.
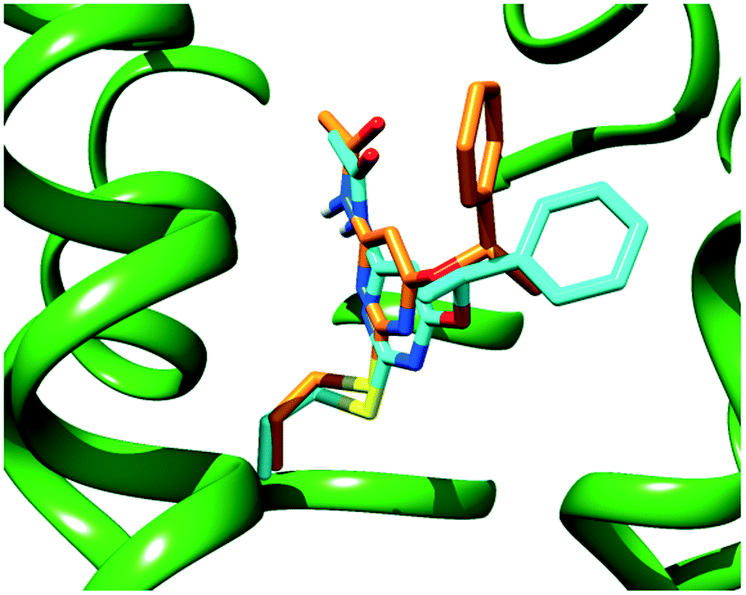 | ||
| Fig. 3 Superimposition of the calculated binding poses for (S)-1 (cyan sticks) and (R)-1 (orange sticks) within a model of the human A3 AR binding site (green ribbons). | ||
It is worth outlining that the binding modes of (S)-1 and (R)-1 hypothesized by us are very similar to those proposed by Yaziji, V. et al. for a number of 4,6-diaryl-2-amidopyrimidines fitted into the same receptor.8 Specifically, the amidopyrimidine moiety in common with their series and ours is oriented in the same way within the A3 AR binding site by establishing identical ligand–receptor interactions. Moreover, the two aryl rings of Yaziji's set fit into the same lipophilic clefts occupied by the PhCH(CH3)O and CH3(CH2)2S substituents of (S)-1 and (R)-1. The agreement between the two docking models mutually supports their robustness.
The best achievement of our lead-optimization efforts is represented by (S)-1 which stands out due to its subnanomolar affinity for the human A3 AR (0.5 nM) together with an excellent selectivity profile (no appreciable binding to human A1, A2A and A2B ARs). These properties make (S)-1 the most valuable pyrimidine derivative among those investigated so far by us as A3 AR antagonists.
The results described in the present paper will be exploited for continuing the design and synthesis of novel pyrimidine derivatives as A3 AR antagonists. Particularly, by taking 1 as a reference structure, the α-methyl will not be replaced by larger groups. Moreover, small substituents on the phenyl ring will be evaluated in docking simulations for their capability of establishing favourable interactions with the surrounding amino acids of the A3 AR binding site.
Authorship
The authors have contributed to the work as detailed below.B. Cosimelli, S. Laneri and A. Sacchi: synthesis. G. Greco and S. Taliani: design. S. Collina and D. Rossi: HPLC purification and analysis. E. Barresi, M. L. Trincavelli and C. Martini: biological experiments. E. Novellino and S. Cosconati: docking simulations.
Conflicts of interest
The authors declare no competing interest.Acknowledgements
The authors thank Dr. Rita Nasti for support of analytical work.References
- C. E. Müller and K. A. Jacobson, Biochim. Biophys. Acta, 2011, 1808, 1290–1308 CrossRef PubMed.
- B. B. Fredholm, A. P. IJzerman, K. A. Jacobson, J. Linden and C. E. Müller, Pharmacol. Rev., 2011, 63, 1–34 CrossRef PubMed.
- R. Brambilla, F. Cattabeni, S. Ceruti, D. Barbieri, C. Franceschi, Y.-C. Kim, K. A. Jacobson, K.-N. Klotz, M. J. Lohse and M. P. Abbracchio, Naunyn Schmiedebergs Arch. Pharmacol., 2000, 361, 225–234 CrossRef PubMed.
- P. A. Borea, K. Varani, F. Vincenzi, P. G. Baraldi, M. A. Tabrizi, S. Merighi and S. Gessi, Pharmacol. Rev., 2015, 67, 74–102 CrossRef PubMed.
- L. C. W. Chang, R. F. Spanjersberg, J. K. von Frijtag Drabbe Künzel, T. Mulder-Krieger, G. van den Hout, M. W. Beukers, J. Brussee and A. P. IJzerman, J. Med. Chem., 2004, 47, 6529–6540 CrossRef PubMed.
- J. P. D. van Veldhoven, L. C. W. Chang, J. K. von Frijtag Drabbe Künzel, T. Mulder-Krieger, R. Struensee-Link, M. W. Beukers, J. Brussee and A. P. IJzerman, Bioorg. Med. Chem., 2008, 16, 2741–2752 CrossRef PubMed.
- B. Cosimelli, G. Greco, M. Ehlardo, E. Novellino, F. Da Settimo, S. Taliani, C. La Motta, M. Bellandi, T. Tuccinardi, A. Martinelli, O. Ciampi, M. L. Trincavelli and C. Martini, J. Med. Chem., 2008, 51, 1764–1770 CrossRef PubMed.
- V. Yaziji, D. Rodríguez, H. Gutiérrez-de-Terán, A. Coelho, O. Caamaño, X. García-Mera, J. Brea, M. I. Loza, M. I. Cadavid and E. Sotelo, J. Med. Chem., 2011, 54, 3954–3963 CrossRef PubMed.
- A. M. van Rhee, J.-L. Jiang, N. Melman, M. E. Olah, G. L. Stiles and K. A. Jacobson, J. Med. Chem., 1996, 39, 2980–2989 CrossRef PubMed.
- J.-L. Jiang, A.-H. Li, S.-Y. Jang, L. Chang, N. Melman, S. Moro, X.-D. Ji, E. B. Lobkovsky, J. C. Clardy and K. A. Jacobson, J. Med. Chem., 1999, 42, 3055–3065 CrossRef PubMed.
- K. A. Jacobson, A. Melman and B. B.-R. Wang, Aust. Pat. Appl., AU 2009276411 A1 20100204, 2010.
- D. Rossi, R. Nasti, A. Marra, S. Meneghini, G. Mazzeo, G. Longhi, M. Memo, B. Cosimelli, G. Greco, E. Novellino, F. Da Settimo, C. Martini, S. Taliani, S. Abbate and S. Collina, Chirality, 2016, 28, 434–440 CrossRef PubMed.
- M. L. Trincavelli, C. Giacomelli, S. Daniele, S. Taliani, B. Cosimelli, S. Laneri, E. Severi, E. Barresi, I. Pugliesi, G. Greco, E. Novellino, F. Da Settimo and C. Martini, Biochim. Biophys. Acta, 2014, 1840, 1194–1203 CrossRef PubMed.
- H. O. Kim, X.-D. Ji, S. M. Siddiqi, M. E. Olah, G. L. Stiles and K. A. Jacobson, J. Med. Chem., 1994, 37, 3614–3621 CrossRef PubMed.
- C. Nordstedt and B. B. Fredholm, Anal. Biochem., 1990, 189, 231–234 CrossRef PubMed.
- Z.-G. Gao, A. Chen, D. Barak, S.-K. Kim, C. E. Müller and K. A. Jacobson, J. Biol. Chem., 2002, 277, 19056–19063 CrossRef PubMed.
- J. Kim, J. Wess, A. M. van Rhee, T. Schöneberg and K. A. Jacobson, J. Biol. Chem., 1995, 270, 13987–13997 CrossRef PubMed.
- S. Taliani, C. La Motta, L. Mugnaini, F. Simorini, S. Salerno, A. M. Marini, F. Da Settimo, S. Cosconati, B. Cosimelli, G. Greco, V. Limongelli, L. Marinelli, E. Novellino, O. Ciampi, S. Daniele, M. L. Trincavelli and C. Martini, J. Med. Chem., 2010, 53, 3954–3963 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemistry and biology experimental sections for the described compounds. See DOI: 10.1039/c7md00375g |
| This journal is © The Royal Society of Chemistry 2018 |