
Microfluidic affinity separation chip for selective capture and release of label-free ovarian cancer exosomes†

Abstract
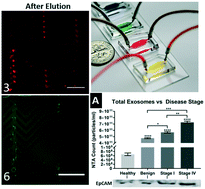
Exosomes are nanoscale vesicles found in many bodily fluids which play a significant role in cell-to-cell signaling and contain biomolecules indicative of their cells of origin. Recently, microfluidic devices have provided the ability to efficiently capture exosomes based on specific membrane biomarkers, but releasing the captured exosomes intact and label-free for downstream characterization and experimentation remains a challenge. We present a herringbone-grooved microfluidic device which is covalently functionalized with antibodies against general and cancer exosome membrane biomarkers (CD9 and EpCAM) to isolate exosomes from small volumes of high-grade serous ovarian cancer (HGSOC) serum. Following capture, intact exosomes are released label-free using a low pH buffer and immediately neutralized downstream to ensure their stability. Characterization of captured and released exosomes was performed using fluorescence microscopy, nanoparticle tracking analysis, flow-cytometry, and SEM. Our results demonstrate the successful isolation of intact and label-free exosomes, indicate that the amount of both total and EpCAM+ exosomes increases with HGSOC disease progression, and demonstrate the downstream internalization of isolated exosomes by OVCAR8 cells. This device and approach can be utilized for a nearly limitless range of downstream exosome analytical and experimental techniques, both on and off-chip.
- This article is part of the themed collection: Personalised Medicine: Liquid Biopsy
 Please wait while we load your content...
Please wait while we load your content...

