
Confinement and enhancement of an airborne atmospheric laser-induced plasma using an ultrasonic acoustic resonator†
Andreas
Bierstedt
,
Ulrich
Panne
and
Jens
Riedel
 *
*
Bundesanstalt für Materialforschung und -prüfung, BAM, Richard-Willstätter-Straße 11, 12489, Berlin, Germany. E-mail: Jens.Riedel@bam.de; Fax: +49-30-8104-1003; Tel: +49-30-8104-1003
First published on 22nd November 2017
Abstract
Optical elemental analysis in the gas phase typically relies on electrically driven plasmas. As an alternative approach, laser-induced plasmas (LIPs) have been suggested but have so far been only scarcely used. Here, a novel signal enhancement strategy for laser-based airborne plasma optical emission spectroscopy for gas phase analytics is presented. In contrast to an electrically driven plasma, in the laser-induced analogue dynamic matter transport equilibrium builds up. The latter results in a rarefied density regime in the plasma core itself, surrounded by an area of compressed matter. The central rarefaction leads to a decrease in plasma intensity and analyte number density, both of which are detrimental for analytical purposes. Since the repetitive ignition of LIPs is a transient process, a restoration of the former gaseous medium by other dynamically equilibrated diffusion processes would be favourable. The presented combination of an airborne LIP and an ultrasonic acoustic resonator yields a fourfold signal enhancement while the background contribution of ubiquitous air is at the same time effectively suppressed. Since the entire enhancement effect occurs without contact, no additional sources for abrasive sample contamination are introduced.
Introduction
Currently, plasma-based elemental analysis of gaseous samples or gases/aerosols almost entirely relies on inductively coupled plasma (ICP) coupled with either optical emission spectroscopy (OES) or mass spectrometry (MS) detection. Despite the vast advantages of ICP, namely a high stability with respect to plasma temperature and electron number density, the technique is not without shortcomings. The major drawbacks of ICP are directly related to its consumption of energy (typically ∼10 kW) and plasma gas (10–20 L min−1 of argon). Miniaturization of ICP has often been proposed; however, resonance conditions of the radial frequency make the coil diameters scale with the plasma pressure. As a consequence, the resulting down-scaled torches can only be operated resonantly in reduced argon pressure. A promising alternative approach is the use of laser-induced plasmas (LIPs) as known in laser-induced breakdown spectroscopy (LIBS). A comprehensive comparison for liquid samples shows that LIBS has several advantages over ICP-OES, but mainly suffers from relatively high limits of detection (LODs). These higher LODs are at least partially introduced by sampling, since ICP-OES is operated with a nebulizer and the small resulting droplets show a significant pre-concentration throughout the desolvation process, whereas LIBS is typically performed in the unprocessed condensed liquid state. While for solid sample targets and bulk liquid phases LIBS is widely used, comparatively little work has been done on its use for gas phase analytes. Yalcin et al.1 showed the physical properties of LIP to be extremely robust with respect to experimental parameters such as humidity, laser fluence and even matrix gas composition. One well-studied example is the elemental mapping inside hydrocarbon flames.2,3A similar situation exists for the analysis of solid and liquid aerosols. Even though the use of nebulizers for the LIBS detection of trace metals in solution yielded much lower LODs, only a few related studies have been reported.4–7 The same holds true for the use of LIBS to detect airborne nanoparticles.8 The small amount of matter inside the nanoparticles puts the irradiated light under steady state conditions which leads to a decrease of the fluctuations in the ablation behaviour. One further step is to ignite and operate the LIP itself inside the gaseous media. Here, the micro-homogeneity of the plasma target drastically decreases pulse-to-pulse fluctuations in plasma intensities. The latter are a major contribution to the analytical matrix effect, which is known to be extremely pronounced for light–solid interactions, such as laser ablation or laser desorption. Isotope exchange experiments could show that a large amount of the actual ablation in solid target LIBS experiments does not occur by the direct laser–matter interaction but is rather driven by the plasma formed above the target and has a lifetime orders of magnitude longer than the actual laser pulse.9 This finding strongly supports an existing approach suggested by De Giacomo and co-workers,10 in which an airborne plasma is ignited in the vicinity of the sample rather than on the sample itself. All of the above sampling strategies, namely gas phase LIBS, LIBS on solid or liquid micro-/nanoparticle aerosols, and LIBS of air spark ablated material, highlight the great potential of airborne LIP-OES.
The development of diode-pumped solid-state (DPSS) lasers has brought forth several advantages for analytical chemistry. Briefly, the main improvements over traditional flashlamp-pumped lasers are higher repetition rates (and consequently higher duty cycles) and power conversion efficiency. Accordingly, it is possible to ignite an airborne LIP at a repetition rate of 26 kHz with an optical power of 3.6 W, corresponding to an overall electrical power consumption of <50 W.11 The more efficient power conversion into optical output results in lower heat dissipation, so air cooling is often sufficient. To LIP this brings several obvious advantages which directly result from the repetition rate and the accompanying better event statistics. There is, however, an upper limit for this statistical improvement, as for ever higher rates, the relaxation processes between individual laser pulses start to fall short. A closer inspection reveals that above a threshold repetition rate the pulsed character of the plasma introduces a dynamic equilibrium governed by shockwaves, in which the plasma region is depleted of reactive plasma gas.12 Within tens of microseconds, in between two subsequent plasma events, the gas pushed outside the plasma origin by a concentric shockwave cannot be replenished and from the second laser pulse on, all plasma events are less intense and less bright than the first one. Upon application of a gas stream, it could be shown that this matter rarefaction can be compensated for by replenishing the gaseous medium in the central plasma zone. However, the application of a continuous gas stream was found to introduce a wash-out effect of the analyte concentration inside the plasma region13 and upon high gas fluxes of room temperature gas even to quench the plasma. Despite the addition of a plasma gas, several other approaches to enhance the optical brightness of LIP have been introduced.14 The latter include technically expensive solutions such as double or multiple pulse LIBS and confinement strategies (such as spatial or magnetic confinement) which introduce sources for abrasive sample contamination.
Since the repetitive ignition of LIP is a transient process in dynamic equilibrium, replenishing by another dynamically equilibrated diffusion process would be favourable. Such dynamic pressure equilibria at frequencies near the repetition rate of the laser (several kHz) exist inside acoustic resonators. In contrast to a steady flow of gas, a standing wave results in little or no net gas transport over large distances. Instead, the convective flux inside the acoustic resonator gets accelerated ensuring an effective short range matter exchange. In recent years, acoustic resonators have experienced a renaissance in contactless microfluidic applications15–18 and the technique is well understood and even commercially available. In this work, the effect of an acoustical standing wave inside an ultrasonic resonator on the performance of an airborne LIP is tested and the beneficial influence of such an acoustic field is demonstrated for (i) an overall increase of sensitivity and (ii) an effective discrimination between the signal contribution of a directionally introduced sample and the ubiquitous, diffusion-controlled background gas.
Experimental
Throughout the remainder of this work, the enhancement of a LIP ignited in an open laboratory environment in combination with an ultrasonic acoustic field was visualized by using the plasma itself as a sensor. In the optimization process the brightness of the plasma acts as the loss function. Additionally, a spectrally dispersed analysis of the emitted light allows for the differentiation between individual contributions to this overall brightness.Laser-induced plasma
The generation of a stable LIP operated in the open laboratory environment as an ionization source for ambient mass spectrometry was demonstrated recently.11 Therefore, the second harmonic output (λ = 532 nm) of a high repetition rate DPSS laser (Conqueror 3-LAMBDA, Nd:YVO4, 1–500 kHz, max. pulse energy: 300 μJ per pulse, pulse width: <10 ns, average output power: 12 W @ 50 kHz, Compact Laser Solutions GmbH, Berlin, Germany) was directed via three Nd:YAG laser mirrors (NB1-K13, Thorlabs, Dachau, Germany) consecutively on an aspheric lens with a short focal length of f = 8 mm and a high numerical aperture of NA = 0.50 (C240TME-A, Thorlabs, Dachau, Germany). Typically, an optical power consumption of 3.65 W (out of the available 12 W)11 was needed for maintaining the plasma. Microscopic photography of the active plasma revealed small plasma dimensions of only 80 μm × 25 μm.12Standing wave acoustic field generation and characterization
All experiments were performed using a homebuilt ultrasonic trap.19 Up to now, this device has been used exclusively for the acoustic levitation and subsequent contactless analysis of small sample volumes.20,21 Briefly, it consists of a USK 800 sandwich piezo transducer driven by the 200 W output signal of a MWI-400 high frequency generator at 41 kHz (MTH – Maschinenbau Technologie Herrde, Henstedt-Ulzburg, Germany), a tapered, doubly coned titanium sonotrode ending in a radiating plate and a concave shaped reflector. The sonotrode and reflector were assembled in a single axis setup and adjusted for maximum sound pressure to a resonant distance of 5/2 wavelengths (≈21 mm). Previous results indicate the resonator to be operated under acoustic saturation conditions. In contrast to levitation experiments, the trap axis was aligned parallel to the table top. Eventually, this setup was placed on an optical breadboard equipped with a 3D translational stage (3× PT1/M, Thorlabs, Dachau, Germany) allowing for individual positioning between the acoustic field and the operated LIP. Visualization of the fast pressure dynamics was performed by scanning laser Doppler vibrometry (PSV-500-3D Scanning Vibrometer, Polytec GmbH, Berlin branch, Germany).Spectral recording
The beneficial effects of acoustic confinement were evaluated by a spectral recording of the plasma species. The instrumentation used is depicted in Fig. 1. The LIP was ignited inside the gap between the sonotrode and the reflector. Single emission spectra were collected orthogonally to the beam path via an optical fibre placed at a distance of 10 mm above the plasma centre. The other end of the optical fibre was connected to a Czerny–Turner spectrograph (Shamrock SR-303i, 1800 grooves per mm grating, ANDOR Technology Ltd., Belfast, UK) coupled to a CCD camera (iDus CCD camera, −65 °C, ANDOR Technology Ltd., Belfast, UK). The entrance slit width was 150 μm and the acquisition time per spectrum was set to 0.1 s. The wavelength range between 280 and 920 nm was covered.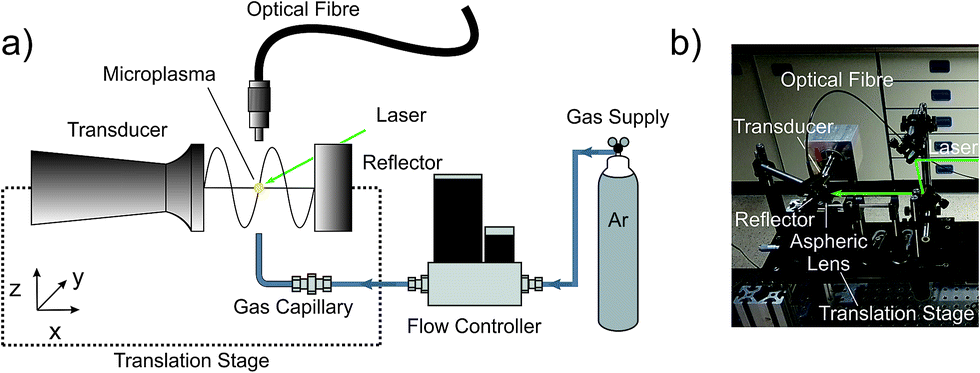 | ||
| Fig. 1 Experimental arrangement including the acoustic cavity, a 90° bent gas capillary to introduce argon, the LIP and acquisition optics. (a) Schematic representation and (b) top-view photograph of the real setup. The entire acoustic resonator comprising the driver, sonotrode and reflector, at a fixed resonant distance, was mounted on a 3D translational stage and could be displaced with respect to the other fixed components. | ||
In the second set of experiments, the position of the LIP was kept constant, while the position of the acoustic field was varied in space. Thus, a spatially resolved interrogation was achieved by conducting two individual line scans: one along the propagation of the emitted soundwave and one orthogonal to this axis. Every 250 μm a fingerprint spectrum of the plasma radiation, covering a reduced wavelength range between 700 and 900 nm, was recorded to investigate the influence of the standing wave in the acoustic field on the emission of the plasma. Eventually, to mimic an analyte introduction, the line scan experiment along the propagation axis was repeated using the LIP ignited in a gentle argon (≥99.999%, Linde, Berlin, Germany) stream. The latter was controlled by a mass flow controller (GFC17, Analyt-MTC GmbH, Müllheim, Germany) and fed from below through a 90° bent stainless steel capillary (i.d. 150 μm, length: 100 mm) fixed in a 1/16′′–1/4′′ Swagelok connector. The argon flow rate was set to 0.1 L min−1. Due to a strong increase in brightness, the acquisition time was reduced to 0.01 s and the entrance slit width of the spectrograph adjusted to 50 μm.
Results
Pressure field diagnostics
Constructive interference conditions inside the acoustic resonator are greatest when the reflector opposes the sonotrode at a distance of several integer half wavelengths. These resonant conditions result in the capability of acoustic levitation, since at a given acoustic pressure the pressure gradient inside the sound field overcompensates for the gravity. Consequently, an optimal geometry was first verified by stable levitation of a 5 μL water droplet at the central pressure minimum. Maintaining the same geometry, the sound pressure distribution between the sonotrode and the reflector was mapped by 3D laser Doppler vibrometry. Fig. 2 shows a top-view photograph of the trap containing the levitated water droplet (a) and without the droplet but superimposed with a false-colour representation of the pressure field in the respective half periods of one ultrasonic cycle applied to the piezo unit of the acoustic trap in (b) and (c), respectively. The pressure distribution can be seen to develop antinodes (i.e. points of maximum displacement) of high local pressure (red and blue), intersected by pressure nodes (zones of no displacement) at the unaltered pressure of the surroundings (green). These pressure maps nicely resemble earlier simulated and recorded sound field distributions inside the used resonator.19 | ||
| Fig. 2 (a) Top-view photograph of the acoustic trap containing a levitated water droplet of 5 μL volume. (b and c) Visualized sound pressure distribution between the transducer (top) and the reflector (bottom) of the ultrasonic levitation device via laser Doppler vibrometry. The pressure maps are represented in false colour. Green indicates equilibrium pressure, whereas the red and blue areas depict zones of acoustically induced compression or rarefaction. | ||
Optical emission spectra of airborne LIP
In the first set of LIBS experiments, the LIP was ignited directly in the open laboratory gas phase, while the optical emission was spectrally analysed. The result is shown in Fig. 3a.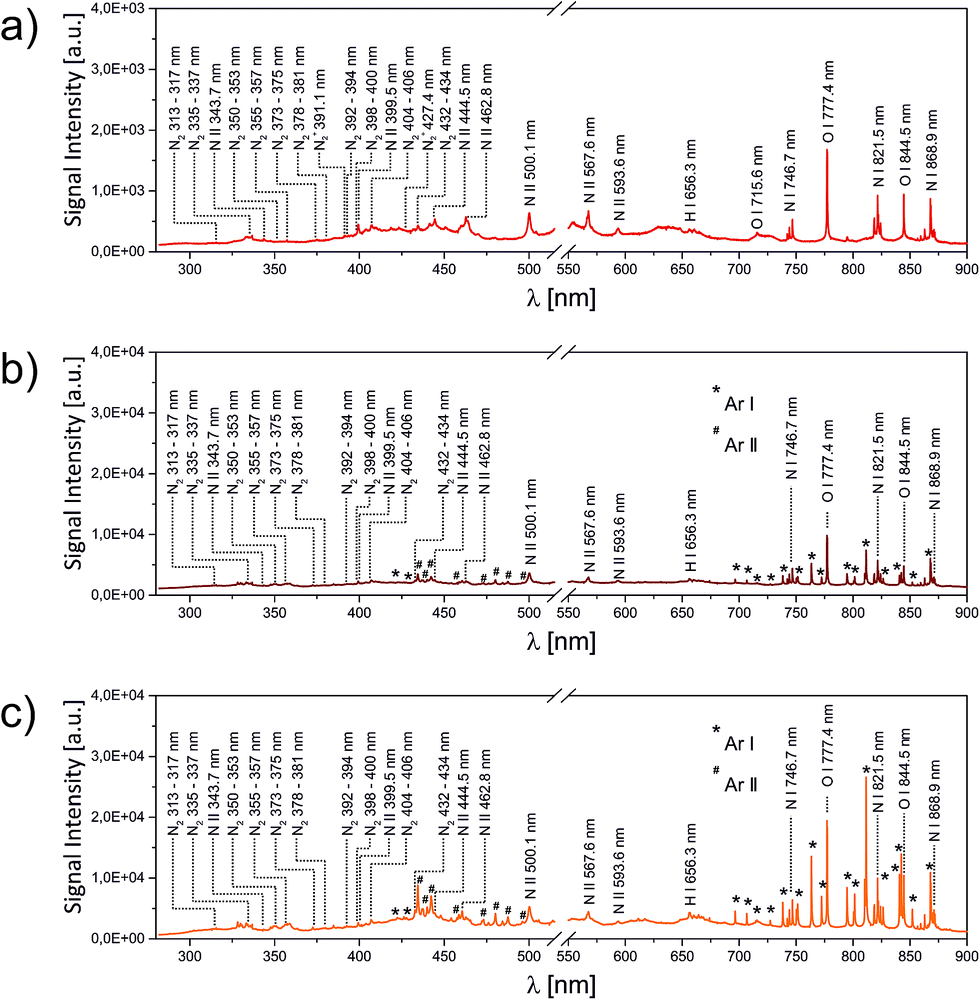 | ||
| Fig. 3 LIP emission spectrum acquired in the open laboratory atmosphere (a) and upon addition of 0.1 L min−1 argon for compensation of the rarefaction effect inside the plasma centre without applied acoustic field (b). Obtained emission spectrum of the LIP ignited inside the acoustic field and addition of 0.1 L min−1 argon (c). Please note the break in the x-axis to cut off the excitation laser wavelength. | ||
The spectrum can be seen to exhibit atomic O (715.6 nm, 777.4 nm, 855.5 nm), N (746.7 nm, 821.5 nm, 868.9 nm) and H (656.3 nm) emission lines, transitions of ionic N (343.7 nm, 399.5 nm, 444.5 nm, 462.8 nm, 500.1 nm, 567.6 nm, 593.6 nm) and progressions of weak emission lines assigned to N2 (C3Πu → B3Πg) and N2+ (B2Σu+ → X2Σg+) in the spectral range between 313–434 nm and 391–427 nm, respectively. The effect of a predominating rarefaction inside the plasma centre was observed when slightly replacing the exact plasma composition with a fresh volume of air by moving the alignment mirrors. Whenever a new spot was exposed to the laser focus, a series of brighter and louder plasma was observed, rapidly followed by a relaxation to the previous conditions.
The situation changed upon the addition of a directed steady gas flow towards the plasma origin. In this case 0.1 L min−1 argon was applied to the LIP, resulting in a stable enhancement of the optical brightness. This enhancement was also reflected in the volume of the audible acoustic shock emitted by each LIP. Argon was chosen because it results in a speed of sound similar to that in air while being spectrally distinguishable. In contrast to LIBS experiments at higher laser fluences,22 helium introduction effectively quenched the plasma formation. This finding was rationalized by the population of the non-ionic high energy metastable Rydberg states of helium.
A closer inspection of the corresponding emission spectrum (Fig. 3b) revealed that this increase in brilliance affects the entire spectrum. A direct comparison between Fig. 3a and b shows that in accordance with a roughly fivefold increase in signal intensity almost no change in the relative peak intensities was observed. The most striking difference consists of minor contributions by atomic or ionic argon (labelled with the superscripts * or # in the figure, respectively). In other words, a restoration of the rarefied plasma region with argon resulted in a larger number of excited air species. The subtle change in relative intensities between N and O lines indicates a comparable plasma temperature. These results can be rationalized by effective energy dissipation inside the plasma. The lowest electronically excited state in argon (2P3/2) has a term energy of 11.6 eV, only slightly exceeding the 4S term of oxygen at 10.8 eV, responsible for the strongest visible emission line at 777.4 nm, while all observed argon transitions in Fig. 3b can be assigned to include at least the second lowest 2P3/2 state at 13.1 eV, justifying the existence of the strong N I line at 821.5 nm (11.9 eV), as well as the population of ionic species, but also explaining the absence of N2+ (B2Σu+ → X2Σg+) given its higher ionization potential of 14.5 eV. Interestingly, the progression of molecular nitrogen (C 3Πu → B 3Πu; upper term energy 11.1 eV) apparently also got excited. N2 can take up energy via different pathways, either as electronic energy yielding stable highly excited dimers or as vibrational/rotational energy, eventually leading to dissociation (bond energy: 9.8 eV). Since the N/N2 ratio appeared to remain unaltered, the addition of argon more likely resulted in an electronic energy transfer rather than an increase in inelastic scattering with the excitation of dissociative vibrational states.
Fig. 3c depicts the spectral emission of the LIP in the presence of the additional acoustic field. The ultrasonic resonator was aligned in a way that the LIP spatially coincided with the central pressure node. On top of an additional threefold enhancement in absolute signal intensity, now the pattern of relative intensities also changes. While upon constant argon addition, but without applied acoustic pressure, the oxygen transition at 777.4 nm resulted in the most dominant peak, within the sound field the highest spectral intensity could be assigned to the Ar I doublet at around 811.4 nm. Obviously, the enhancement goes along with a change in the number densities of the three atomic species, N, O and Ar. Two effects can potentially cause the first observation of overall higher brilliance: (1) the presence of the acoustic field induces a higher number density in the pressure antinodes (refer to Fig. 2). (2) Both the pressure gradient and the acoustically induced particle velocity yield a higher convective matter exchange inside the resonator. In consequence, the two effects result in a static (1) or dynamic (2) increase in the local density of excitable matter inside the laser focus. Since the laser matter interaction eventually forms the plasma, both effects would result in the observed behaviour. The relative enrichment of the argon signal contribution to the spectrum, however, supports the dynamic model.
For an unambiguous differentiation between the separate contributions of a static or a dynamic enhancement, the 3D sound field was mapped by several individual LIP-OES experiments. The lateral distance between two subsequently recorded spectra was chosen to be 250 μm. This high resolution is possible due to the small dimensions of the LIP. First, the acoustic resonator was mapped without any additional gas stream. The resulting 3D map is represented by two central cuts through the pressure field along its principal axes in Fig. 4. As can be seen in Fig. 3, the highest informational content regarding the N/O/Ar atomic concentrations is found in the fingerprint region between 700 and 900 nm. Fig. 4a depicts the near-IR region of the OES spectrum along the resonator's axis of circular symmetry a and Fig. 4b along the orthogonal axis b. For clarity, the integral sound fields and the respective paths of the mapping experiments are depicted in an inset inside the respective subfigure. Following this path, the intensity of the individual spectra in Fig. 4a increases along the acoustic pressure (indicated in false colour in the inset). Throughout this undulating intensity distribution, the relative intensities did not change. The same trend was observed for the orthogonal case in Fig. 4b. Here, similar to the pressure, the intensity peaked at the centre and monotonically decreased towards the periphery. As a consequence, for the spectral intensity of ubiquitous static gases (as the atmospheric background signal), the static enhancement effect is responsible. The signal scaled with the equilibrated pressure distribution of the standing wave. In an analytical experiment, the positions of the pressure nodes could therefore be used for an effective background suppression. The quenching of the surrounding gas is likely to stem from the overall elevated pressure within the resonator. The latter originates from the superimposed contribution of higher order pressure terms arising from acoustic saturation. The saturated conditions are corroborated by a power dependent set of individual spectral recordings shown in the ESI.
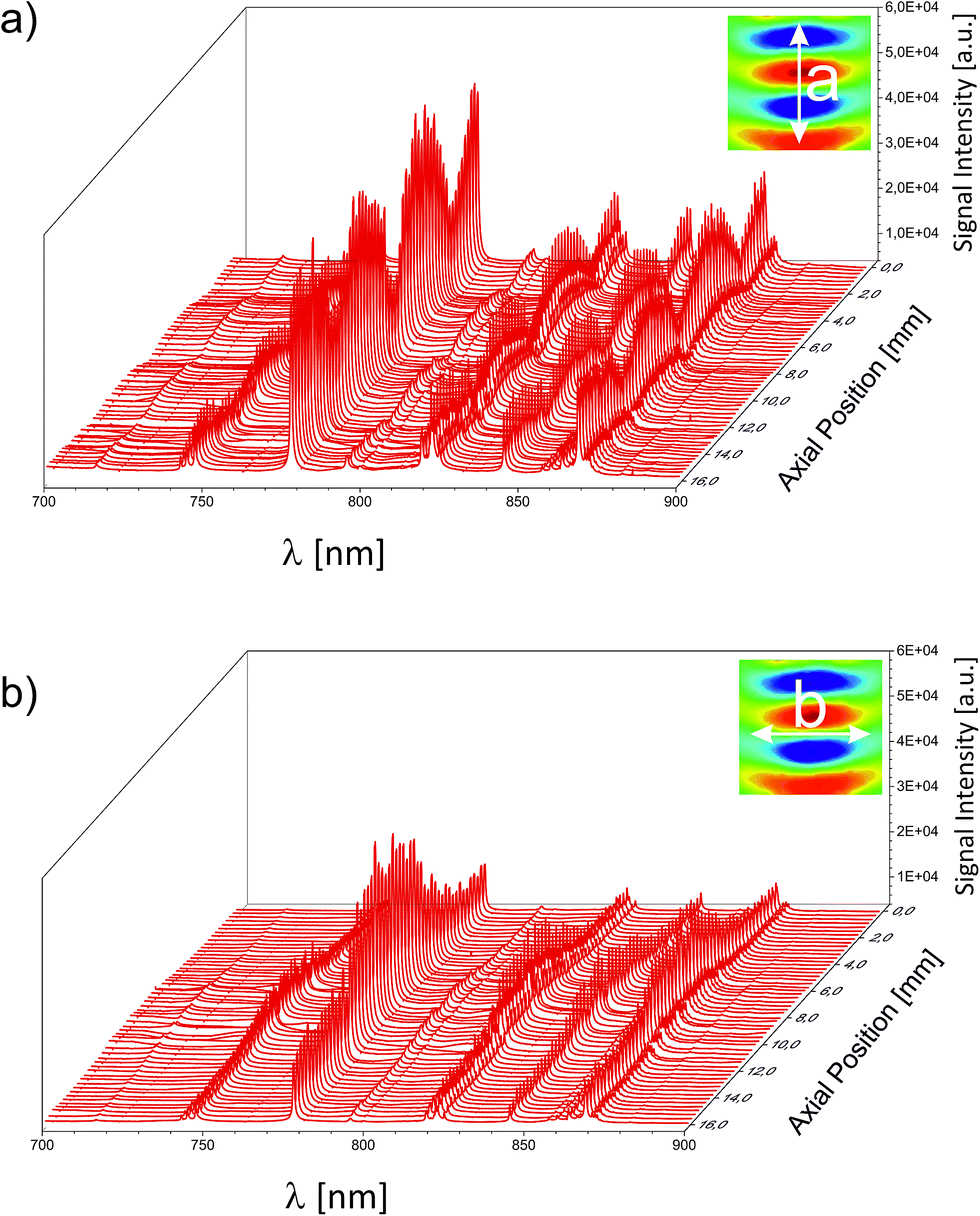 | ||
| Fig. 4 Spectrally resolved emission of the LIP inside the acoustic field screened along the two principal axes of the acoustic resonator. | ||
In contrast to the surrounding atmospheric gases, the introduction of the analyte is typically localized and directed. This analyte could be an analyte-enriched headspace vapour, a particle-laden gas stream8 or a sample ablated by the plasma itself according to the experimental proposal of De Giacomo et al.10 The latter ablation process would also occur in the vicinity of the plasma rather than inside the plasma and propagate towards the plasma centre.9 To mimic either of these sample introduction geometries, in a follow-up experiment argon was used as the model analyte. Similar to the above-mentioned sampling scenarios, the argon flow was fixed with respect to the focusing lens position. Accordingly, it was always injected from the same point of origin relative to the LIP. Using this setup, a lateral line scan along the resonator was conducted, in analogy to the scan depicted in Fig. 4a. The resulting distributions of the peak intensity of the O I spectral line at 777.4 nm and the Ar I line at 811.4 nm are plotted against the distance from the radiating plate towards the acoustic reflector. The resulting distributions are depicted in Fig. 5. For a better orientation, the pressure map is superimposed onto the figure.
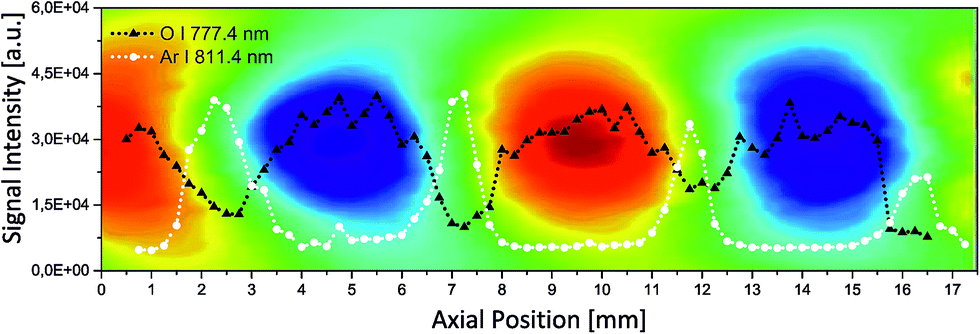 | ||
| Fig. 5 Overlay of the local pressure inside the acoustic resonator and the optical emission peak intensity along a central line scan. For the colour code of the false colour representation of the sound field refer to Fig. 2. The OES signal of O I at 777.4 nm representing the atmospheric background and the emission line at 811.4 nm of the model analyte argon are presented in black and white, respectively. | ||
The oxygen line resembles the above-discussed behaviour of a background gas, statically trapped inside the pressure antinodes with local minima at the pressure node positions. The extrinsically applied and inwardly directed argon, on the other hand, preferentially flows towards the low pressure regions avoiding areas of already elevated pressure. This local concentration leads to a fivefold increase of Ar spectral intensity. Since argon in this experiment represents the analyte, at these positions inside the resonator the acoustic confinement yields an increase in the signal-to-background ratio by a factor >20. This drastic enhancement well justifies the relatively little additional instrumental effort and potentially helps LIBS to mature to a more commonly used technique for the analysis of gaseous analytes.
Conclusions
The focused output of a high repetition rate DPSS laser can serve as a powerful source of electron-dense plasma with a high duty cycle, an efficient energy conversion and peak electronic temperatures above 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 K.12 From low repetition rate (and thus reduced duty cycles) experiments LIPs are known to yield highly reproducible plasma conditions. However, till now LIPs have hardly been used for the excitation of a gas-stream introduced sample for subsequent spectral interrogation (as in ICP-OES applications). This contribution shows that the ultrasonic field inside an acoustic resonator can serve as an intelligent tool for a synergistic combination of effective sample introduction, signal enhancement and simultaneous ambient background suppression. This approach can pave a way towards more robust and portable OES instrumentation for gas phase elemental analysis.
000 K.12 From low repetition rate (and thus reduced duty cycles) experiments LIPs are known to yield highly reproducible plasma conditions. However, till now LIPs have hardly been used for the excitation of a gas-stream introduced sample for subsequent spectral interrogation (as in ICP-OES applications). This contribution shows that the ultrasonic field inside an acoustic resonator can serve as an intelligent tool for a synergistic combination of effective sample introduction, signal enhancement and simultaneous ambient background suppression. This approach can pave a way towards more robust and portable OES instrumentation for gas phase elemental analysis.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
Financial support was granted by the German Federal Ministry of Education and Research (BMBF) as part of the Konjunkturpaket II. The authors are indebted to Polytec GmbH, Berlin branch, for acquiring the pressure maps in their application lab.Notes and references
- S. Yalcin, D. R. Crosley, G. P. Smith and G. W. Faris, Appl. Phys. B: Lasers Opt., 1999, 68, 121–130 CrossRef CAS.
- M. Kotzagianni and S. Couris, Chem. Phys. Lett., 2013, 561, 36–41 CrossRef.
- P. Stavropoulos, A. Michalakou, G. Skevis and S. Couris, Spectrochim. Acta, Part B, 2005, 60, 1092–1097 CrossRef.
- H. A. Archontaki and S. R. Crouch, Appl. Spectrosc., 1988, 42, 741–746 CrossRef CAS.
- M. Essien, L. J. Radziemski and J. Sneddon, J. Anal. At. Spectrom., 1988, 3, 985–988 RSC.
- S. T. Jarvinen, J. Saarela and J. Toivonen, Spectrochim. Acta, Part B, 2013, 86, 55–59 CrossRef CAS.
- A. Kumar, F. Y. Yueh, T. Miller and J. P. Singh, Appl. Opt., 2003, 42, 6040–6046 CrossRef CAS PubMed.
- P. K. Diwakar, K. H. Loper, A.-M. Matiaske and D. W. Hahn, J. Anal. At. Spectrom., 2012, 27, 1110–1119 RSC.
- R. Glaus, J. Riedel and I. Gornushkin, Anal. Chem., 2015, 87, 10131–10137 CrossRef CAS PubMed.
- A. De Giacomo and O. De Pascale, Thin Solid Films, 2004, 453, 328–333 CrossRef.
- A. Bierstedt and J. Riedel, Eur. J. Mass Spectrom., 2016, 22, 105–114 CrossRef CAS PubMed.
- A. Bierstedt, H. Kersten, R. Glaus, I. Gornushkin, U. Panne and J. Riedel, Anal. Chem., 2017, 89, 3437–3444 CrossRef CAS PubMed.
- V. Hohreiter and D. W. Hahn, Anal. Chem., 2005, 77, 1118–1124 CrossRef CAS PubMed.
- G. Galbacs, Anal. Bioanal. Chem., 2015, 407, 7537–7562 CrossRef CAS PubMed.
- D. Foresti, M. Nabavi, M. Klingauf, A. Ferrari and D. Poulikakos, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12549–12554 CrossRef CAS PubMed.
- A. Marzo, A. Barnes and B. W. Drinkwater, Rev. Sci. Instrum., 2017, 88, 085105 CrossRef PubMed.
- A. Marzo, S. A. Seah, B. W. Drinkwater, D. R. Sahoo, B. Long and S. Subramanian, Nat. Commun., 2015, 6, 8661 CrossRef CAS PubMed.
- S. Santesson and S. Nilsson, Anal. Bioanal. Chem., 2004, 378, 1704–1709 CrossRef CAS PubMed.
- A. Stindt, M. A. B. Andrade, M. Albrecht, J. C. Adamowski, U. Panne and J. Riedel, Rev. Sci. Instrum., 2014, 85, 015110 CAS.
- C. Warschat, A. Stindt, U. Panne and J. Riedel, Anal. Chem., 2015, 87, 8323–8327 CrossRef CAS PubMed.
- A. Stindt, M. Albrecht, U. Panne and J. Riedel, Anal. Bioanal. Chem., 2013, 405, 7005–7010 CrossRef CAS PubMed.
- C. A. Henry, P. K. Diwakar and D. W. Hahn, Spectrochim. Acta, Part B, 2007, 62, 1390–1398 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ja00297a |
| This journal is © The Royal Society of Chemistry 2018 |