
Comparison of interference in chemical, electrochemical and UV-photochemical generation methods of volatile Se species
E.
Nováková
 *,
M.
Rybínová
,
J.
Hraníček
,
P.
Rychlovský
and
V.
Červený
*,
M.
Rybínová
,
J.
Hraníček
,
P.
Rychlovský
and
V.
Červený
Charles University, Faculty of Science, Department of Analytical Chemistry, Albertov 6, Prague 2, CZ 128 43, Czech Republic. E-mail: eliska.novakova@natur.cuni.cz
First published on 24th November 2017
Abstract
We present a comparison of interference in three methods of generation of volatile Se species to compare their performance with regard to matrix effects. The effects of selected hydride forming elements, transition metals and electrolytes are quantified and the similarities and differences between the techniques are compared for individual interferents. The possibility that Co(II), Cr(III) and Ni(II) could be used to increase the response in UV-photochemical generation of volatile Se species is discussed. It was found that the increased sensitivity in the presence of Ni(II) is not due to the increased efficiency of generation but due to processes in the gaseous phase. In general, increased Se signals resulting from modified reactions were associated with alternative vapour generation methods. Electrochemical vapour generation suffers from permanent interference effects located on the cathode, but these were not observed with chemical and UV-photochemical vapour generation. Sb(III) interfered more severely than As(III) irrespective of the method used.
1. Introduction
Volatile species generation permits the separation of the determined element from the sample matrix, thus reducing spectral interference. However, this advantage is often offset by the effects caused by the matrix during the generation process (liquid phase interference) and during the transport of the volatile species and the atomisation process (gaseous phase interference). Liquid phase interference may be caused by reduced generation efficiency due to decomposition of the evolved hydrides on solid particles formed in the reaction medium or due to competition with other hydride forming elements.1 Gaseous phase interference is typical for other hydride forming elements and it is caused by the competition of the generated compounds during atomisation.2 Gaseous phase interference can be decreased by the choice of atomizer.3–5 Both types of interference can be caused by the direct presence of the concomitant or by memory effects. The terminology used in categorization of interference was developed by Dědina6 and it is described in detail in the monography on hydride generation atomic absorption spectrometry by Dědina and Tsalev.7Selenite can be converted to a volatile compound and subsequently determined using chemical generation of volatile species with tetrahydroborate (CVG)8 or using alkylation with tetraethylborate or tetrapropylborate.9,10 It can also be determined using electrochemical generation of volatile species (EcVG)11 or ultraviolet photochemical generation of volatile species (UV-PVG).12 All the above mentioned methods are affected by the matrix but to different degrees.
The interference mechanisms are well described only in CVG. The mechanism of interference of transition and noble metals is attributed to the adsorption or decomposition of the evolved hydride on reduced metal or metal-boride particles.1 The origins of this conclusion started with Kirkbright and Tadia, who observed the formation of a fine black precipitate during the chemical generation of AsH3 in the presence of Cu, Ni, Pt and Pd and associated this precipitate with the decrease of AsH3 formation.13 Since then other studies dealing with this topic have been published.14–17 Henden et al.18 proposed that the species of As and Sb adsorbed on Ni borides may not be the generated hydrides but rather the dissolved species of As(III), As(V) and Sb(III). They also characterized the nickel boride nanoparticles concluding that the nanoparticles produced under their reaction conditions had amorphous structures with diameters of less than 40 nm and contained Ni, Ni2B, Ni3B and Ni(OH)2.
The need for more environmentally friendly and robust analytical methods led researchers to develop alternative methods of volatile species generation aiming at lower limits of detection and better tolerance to interference. EcVG is a substantial cost-effective alternative to CVG. Laborda et al. summarized in their review the bulk of knowledge of the EcVG technique and showed that its performance is comparable to CVG.19 The most important mechanism of liquid phase interference in EcVG appears to be the reduction of transition metals on the cathode surface causing a change in the generation efficiency.20 Also, whereas metal-boride interference does not exist in this generation method, a possible interaction of the generated hydride with reduced metal particles released from the cathode cannot be ruled out. An insight into the processes taking place on the cathode was given by Bolea et al.,21 who studied the interference of low concentrations of Fe(II) and Fe(III) in the electrochemical SbH3 generation using differential pulse anodic stripping voltammetry. Their conclusion was that Fe0 is co-deposited with Sb0 on the same active sites on the cathode surface leading to decreased efficiency of SbH3 formation and release and permanent fouling of the active sites by Fe0.
UV-photochemical generation of volatile selenium species was pioneered in 2003 by Guo et al.12,22,23 The generation process is based on UV irradiation of the sample in the presence of low molecular weight organic acid. In contrast to previous techniques, almost no hydrogen is formed during UV-PVG and thus it has to be added for atomisation.12 The first papers dealing with UV-PVG reported a good tolerance of the technique to the presence of interferents and the tolerance was explained by the low reduction efficiency of the radical intermediates, which were assumed to be responsible for the formation of volatile species.12 Nevertheless, the definite mechanism of formation of the volatile species remains a matter of discussion.24
This work compares the interference in CVG, EcVG and UV-PVG methods to uncover similarities in the location and in the mechanisms of interference. Some general trends have been observed, which might even be applicable to analytes other than selenium. We also discuss the possibility of increasing the sensitivity of Se determination by UV-PVG using reaction modifiers.
2. Experimental
2.1. Reagents
Deionized water prepared by using a MilliQplus system (Millipore, USA) was used throughout the measurements. If not stated otherwise all chemicals were of analytical purity. Selenium samples were diluted from a stock solution (1000 μg mL−1 of Se) prepared from sodium selenite pentahydrate (Sigma-Aldrich, USA). Hydrochloric acid (suprapure grade, Merck, Germany) diluted to 1.0 mol L−1 served as the catholyte solution and 2.0 mol L−1 sulphuric acid (research grade, Merck, Germany) as the anolyte solution in the EcVG of selenium hydride. Formic acid diluted to 0.5 mol L−1 was used as the photochemical reagent in UV-PVG and 1.0 mol L−1 HCl and 0.5% NaBH4 in 0.4% NaOH were used in CVG. Argon of 99.998% purity (Linde, Czech Republic) was always used as the carrier gas. Hydrogen of 99.90% purity was used as an auxiliary atomisation gas in UV-PVG.The effect of concomitant ions on the volatile selenium species generation was evaluated using the solutions diluted from the standard solutions of As, Sb, Pb, Cd, Ni, Fe, Mn, Co, Cr, Cu, and Ag (prepared in HNO3, all from Analytika, Czech Republic). Pure mineral acids were used to prepare the diluted solutions of HNO3 (extrapure grade, Merck, Germany), H2SO4 (research grade, Merck, Germany), HCl (suprapure grade, Merck, Germany), and H3PO4 (Merck, Germany). The solutions of salts were prepared from NaNO3, Na2SO4, Na3PO4 (all from Lachner, Czech Republic) and NaCl (Merck, Germany).
2.2. Instrumentation
The measurements were carried out using a Unicam 939 AA spectrometer (Unicam, UK). The absorption of radiation was detected at 196.0 nm using a band-pass of 0.5 nm. The atomisation of volatile species was carried out in an externally heated quartz furnace atomizer (RMI, Czech Republic) at 950 °C. Continuous flow mode was selected because it represented the simplest approach; kinetics interference (both release and transport) was eliminated because the signal height in continuous flow mode is independent of the hydride release rate.7 The experimental setup is shown in Fig. 1. A reaction coil used in CVG (Fig. 1A) was made of 0.8 m of PTFE tubing (1.0 mm i.d./2.0 mm o.d., Sigma-Aldrich, USA). The photochemical reactor (Fig. 1C) is described in detail in previous publications25,26 and it consisted of 3.5 m PTFE tubing (1.0 mm i.d./1.4 mm o.d) coiled around a 20 W low pressure Hg lamp (Ushio, Japan). A thin-layer flow-through electrochemical cell (Fig. 1B) with a lead wire cathode (Sigma-Aldrich, USA) and Pt foil anode (Goodfellow, UK) described in previous work27 was used in EcVG. A new cathode was used for each interferent. A MasterFlex L/S peristaltic pump (Cole-Parmer, USA) was used for driving the solutions. Mass flow controllers (0–50 mL min−1 and 0–100 mL min−1, Cole-Parmer, USA) were used for the control of the gas flow rates. A HR-CS-AAS spectrometer ContrAA 700 was used for the estimation of generation efficiency through the determination of unreacted Se in waste liquid. The AAS method with electrothermal atomisation employed cuvettes with an integrated platform. Temperature programs recommended for the determination of Se and IBC background correction were used. | ||
| Fig. 1 Experimental setup and reactors, A, B and C, describe in detail the reactors used and correspond to number 3 in the general diagram, purple dashed lines denote parts used only in one method, (1) peristaltic pump, (2) carrier gas (Ar), (3) reactor ((A) CVG, (B) EcVG, (C) UV-PVG), (4) gas–liquid separator, (5) heated quartz furnace atomizer, (6) waste, (1a) HCl and sample channel, (1b) NaBH4 channel, (1c) catholyte and sample channel, (1d) anolyte channel, corresponds to the dashed purple line in the general diagram, (3a) cathode compartment, (3b) anode compartment, (3c) anolyte outlet, (7) atomisation gas (H2, UV-PVG only), and (8) connection of channels during separate generation of volatile species of the analyte and interferent. | ||
2.3. Measurement procedure and evaluation of results
Each concomitant element or electrolyte was prepared in a series of solutions with growing concentrations containing the same Se concentration. The series of solutions was then measured and the attained signals were compared to the signal of selenium solution without interferents. The measured absorbance in the presence of concomitants (Asample) was divided by the absorbance of pure Se solution (ASe) and expressed as the relative Se response (RSR) in percentage units; see eqn (1). Selenium solutions without interferents were randomly inserted into the measurement sequence to show whether the concomitant caused only direct or also memory interference effects.| RSR = Asample/ASe × 100% | (1) |
Experiments employing separate generation of volatile compounds of As, Sb, Pb and possible volatile forms of Ni and Cu were carried out to classify the mechanism of interference. In these series of experiments, the possible volatile form of the interferent was generated in one channel and volatile Se species in the second channel and these channels were connected after gas–liquid separation. Pairs of generators and gas–liquid separators of the same design and dimensions were used. Experimental conditions were selected to keep the atomisation conditions unchanged; all conditions were the same as the optimum conditions only the gas flow rate was halved in each of the channels.
Due to different Se concentrations used with different volatile species generation methods (see Section 3.1); the RSR curves were plotted against the ratio of concomitant-to-Se mass concentrations rather than directly against the concentrations of concomitants.
3. Results and discussion
3.1. Optimized experimental conditions and figures of merit
Experimental conditions were optimized for each generation approach, but the optimization experiments are not discussed here. The optimum conditions for CVG were 1.0 mol L−1 HCl, 0.5% NaBH4 in 0.4% NaOH, flow rate of HCl/sample 5.0 mL min−1, NaBH4 flow rate 1.7 mL min−1, and carrier gas flow rate 20 mL min−1. The optimum conditions for EcVG were 1.0 mol L−1 HCl serving as the catholyte and 2.0 mol L−1 H2SO4 as the anolyte, generation current 1.0 A, catholyte/sample flow rate 5.5 mL min−1 and anolyte flow rate 2.0 mL min−1, and carrier gas flow rate 70 mL min−1. Similarly, for UV-PVG the conditions were 0.5 mol L−1 formic acid, formic acid/sample flow rate 6.0 mL min−1, carrier gas flow rate 15 mL min−1 and H2 flow rate 2 mL min−1. Using the apparatus and experimental conditions described in the Experimental section we achieved LODs of 58 pg mL−1, 490 pg mL−1, and 45 pg mL−1 for CVG, EcVG and UV-PVG with LDRs reaching up to 15.0, 100.0, and 12.0 ng mL−1, respectively. The attained sensitivities were 0.0408, 0.00758, and 0.0327 mL pg−1 and the repeatabilities calculated as relative standard deviations of the concentration from the middle of the calibration curve were 1.40%, 3.10%, and 1.50%, respectively. The higher LOD obtained with EcVG could be explained by higher noise associated with the deterioration of the cathode surface during longer use. Due to obtaining different LODs with the tested methods, we chose the concentration of selenium giving an absorbance of approximately 0.20 for the interference study; the concentrations of Se used were 5 ng mL−1 in CVG and UV-PVG and 25 ng mL−1 in EcVG. Higher Se concentrations were used in the determination of unreacted Se in the waste liquid after volatile species generation. In particular, 40 ng mL−1 of Se was subjected to CVG and 20 ng mL−1 to UV-PVG. These concentrations were selected on the basis of generation efficiencies published by Rybínová et al.283.2. Visual observations and memory effects
No formation of a visible reduced metal layer was observed with UV-PVG and CVG and measurements of the sample without interferents at random intervals did not attest to permanent memory effects caused by the studied interferents. Thus, we concluded that CVG and UV-PVG were resistant to permanent memory effects. In contrast, an opposite observation was made with EcVG. With EcVG, permanent memory effects were observed with all elements with the exception of Cd, Pb, Fe and Mn. A change of color of the cathode surface was visible after the measurement of copper interference. This observation is consistent with the theory of the modification of the cathode surface.293.3. Overall interference
3.3.1.1. Hydride forming elements. The cross-influence of hydride forming elements in volatile species generation has been discussed by other authors6,30–32 with the conclusion that the produced hydrides compete for hydrogen radicals during atomisation in the quartz furnace atomizer. Therefore, we restrict ourselves to the discussion of the effects of As(III), Sb(III), Pb(II) and Cd(II) that we observed in this work. It is worth noting that the 1.0 mol L−1 optimum HCl concentration used is sufficient for the formation of volatile species of even the less reactive hydride forming elements.
Sb(III) suppressed Se signals more than As(III) at higher concentrations but As(III) interfered more than Sb(III) at low concentrations (see Fig. 2). For example, Se signals in the presence of Sb(III) were not affected up to the concentration 100 ng mL−1 but the RSR at 500 ng mL−1 of Sb(III) was only 13%. In the case of As(III), good tolerance was observed in the presence of 20 ng mL−1, but 500 ng mL−1 of As(III) gave 22%.RSR.
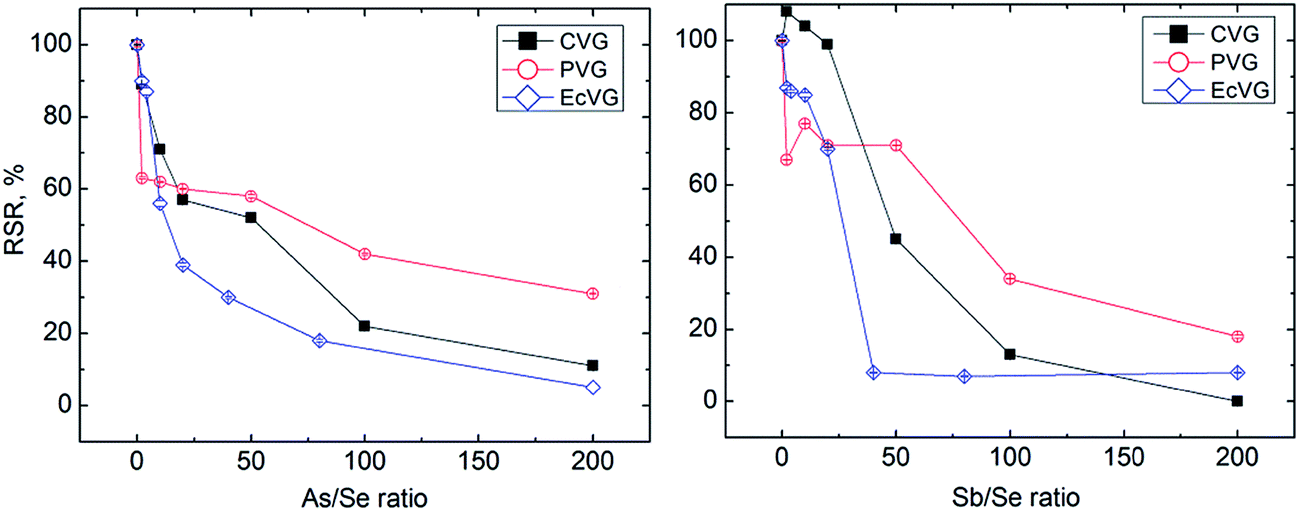 | ||
| Fig. 2 Effect of As and Sb present in the liquid matrix on CVG, EcVG and UV-PVG, with the results expressed as the relative selenium response (%) and the amount of interferent expressed as the concomitant-to-Se mass concentration ratio, see Section 2.3 for further explanation of axis labels. | ||
No interference was observed in the presence of Pb(II) in the samples even at concentrations as high as 100 μg mL−1. Cd(II) interference effects appeared at 20 μg mL−1 and Cd(II) suppressed Se signals to only 15% at 100 μg mL−1. Thus Cd(II) is a more severe interferent than Pb(II). The different effects of Pb(II) and Cd(II) arise from the fact that successful chemical generation of plumbane requires the addition of reaction modifiers,33,34 which is not necessary for CVG of CdH2. However, some authors have reported an increased generation efficiency from organized media or in the presence of thiourea and Co or Ni ions.35–39
3.3.1.2. Transition and noble metals. Transition metals (particularly Cu and Ni) are considered important interferents in CVG, because they decrease generation efficiency. We observed varied effects of transition metals on Se signals, although when a pronounced effect appeared, it was always negative. The most severe interference was observed with Cu(II), Ni(II) and Ag(I) and the influence increased in the direction Ni < Cu < Ag. Only 5 ng mL−1 of Ag led to RSR that was within the tolerable ±10% limit and complete suppression of the Se signal was observed at 250 ng mL−1, other metals had smaller effects. For example, complete suppression of the Se signal was achieved with 1000 ng mL−1 of Cu(II), whereas the same concentration of Ni(II) in the sample gave 83% RSR. The generally accepted mechanism of noble metal interference (Cu, Au, Ag, Pd) is a hydride-solid reaction on metal-boride particles confirmed by D'Ulivo et al.17,40 In addition, Pagliano et al. reported that the presence of noble metals (Au, Pd and Pt) can affect the hydride generation through modification of hydrogen transfer from NaBH4 to the analyte element. They termed this transfer of a larger amount of hydrogen derived from the solvent to the final hydride as mechanistic interference.41 In contrast, Fe(III), Mn(II), Co(II) and Cr(III) did not significantly affect the analytical signals even at high excess of the interferent over Se (see Table 1).
| Concomitant element | Concomitant-to-Se ratio | CVGa | EcVGb | UV-PVGa |
|---|---|---|---|---|
| a 5 μg L−1 of Se(IV). b 25 μg L−1 of Se(IV). c n – not determined. | ||||
| As(III) | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
71 ± 3 | 56 ± 5 | 62 ± 2 |
| 100:1 |
22 ± 1 | nc | 42 ± 1 | |
| Sb(III) | 10:1 |
104 ± 2 | 85 ± 3 | 77 ± 3 |
| 100:1 |
13 ± 1 | n | 34 ± 1 | |
| Pb(II) | 10:1 |
104 ± 1 | 79 ± 5 | 83 ± 3 |
| 200:1 |
101 ± 2 | 85 ± 4 | 3 ± 1 | |
| Cr(III) | 200:1 |
100 ± 3 | 30 ± 3 | 113 ± 3 |
| Mn(II) | 10:1 |
114 ± 3 | 84 ± 4 | 101 ± 2 |
| 200:1 |
106 ± 2 | 101 ± 5 | 112 ± 2 | |
| Fe(III) | 10:1 |
102 ± 3 | 82 ± 3 | 65 ± 3 |
| 200:1 |
104 ± 2 | 97 ± 3 | 64 ± 3 | |
| Co(II) | 200:1 |
99 ± 2 | 82 ± 2 | 188 ± 5 |
| Ni(II) | 10:1 |
98 ± 2 | 87 ± 5 | 97 ± 2 |
| 200:1 |
83 ± 3 | 33 ± 1 | 121 ± 3 | |
| Cu(II) | 10:1 |
102 ± 2 | 61 ± 4 | 132 ± 4 |
| 200:1 |
2 ± 1 | 176 ± 7 | 4 ± 2 | |
| Ag(I) | 10:1 |
52 ± 2 | 74 ± 5 | 19 ± 2 |
| 200:1 |
0 ± 2 | 34 ± 3 | 0 ± 1 | |
| Cd(II) | 100:1 |
99 ± 3 | n | 79 ± 3 |
| 200:1 |
105 ± 3 | 89 ± 4 | 103 ± 4 | |
3.3.1.3. Acids and salts. We found CVG fairly resistant to the presence of inorganic acids in contrast to the alternative volatile species generation methods. No interference was observed in the presence of 1.0 mol L−1 sulphuric acid and phosphoric acid and a similar trend was observed for their salts; neither sulphates nor phosphates interfered up to 1.0 mol L−1. Nitric acid was tolerated only to 0.1 mol L−1 but 1.0 mol L−1 led to still an acceptable RSR of 65%. The presence of nitrates was tolerated up to the concentration of 0.5 mol L−1. The interference of nitric acid and nitrates is likely caused by the diminishment of the selenium form available for generation. Another possibility is the production of nitrogen oxides interfering either in the liquid or in the gaseous phase similarly to the presence of nitrites in the solution.42 The effect of hydrochloric acid was not tested, because it served as the reaction medium, but it would appear that mild concentrations of chloride might act as reaction modifiers; 0.1 mol L−1 of NaCl led to obtaining the relative Se response of 123% and 0.5 mol L−1 concentration to 160%. However, the effect was accompanied by increased deviation of measurements. This observation was surprising, because it was the only indication that we had not achieved complete conversion of selenite to its volatile compound. However, determination of the unreacted Se in the waste liquid did not prove the increased efficiency of generation of volatile Se species in the presence of NaCl (see Section 3.1). The effect of NaCl on the chemical generation of SeH2 thus remains unexplained and needs to be studied further.
3.3.2.1. Hydride forming elements. Most studies proposing EcVG as an alternative to CVG state that it is less liable to interference from other hydride forming elements;19,43 however, we observed results contrary to this assertion. The curves correlating the RSR to the ratio of As(III)-to-Se or Sb(III)-to-Se content were very similar for EcVG and CVG (see Fig. 2 and Table 1). The addition of Sb(III) led to more severe interference than the respective concentration of As(III). Pb(II) caused only small interference up to 10 μg mL−1, but the presence of higher concentrations led to a steep fall of Se signals, possibly due to a large excess of lead over selenium in the vicinity of the cathode. Separate generation of volatile lead species showed that the interference of Pb may be at least partially attributed to atomisation interference (see Section 3.4). Pb(II) is not inert to EcVG and, therefore, interference in the gaseous phase may take place.44
3.3.2.2. Ions of transition and noble metals. Ding and Sturgeon45 reported severe interference (both direct and memory effects) of nickel and copper in the determination of Se. They observed “poisoning” of the Pb cathode in the presence of 7.5 μg mL−1 Cu(II). Also the presence of 40 μg mL−1 of Ni(II) in the solution of 4 ng mL−1 selenium led to the decrease of response to 60% in contrast to solution without interferents. Laborda et al. concluded that Cu is the source of the most severe interference in EcVG.19 Conversely, Junková et al.43 reported that Cu led to less pronounced interference effects in EcVG than in CVG, which was not observed in this study. However, the different effects could be explained by the memory effects encountered in our study.
Based on the previously published data, we expected that the interference with EcVG will follow similar trends as with CVG. However, we observed two kinds of trends with transition metals. Whereas Ni, Cr and Ag exhibited only a negative influence, Mn, Fe, Co and Cu exhibited more complicated trends. At low concentrations of these concomitants the Se signals decreased; however, in certain concentration regions no or only a weak effect of the interferent on Se signals could be observed (see Table 2). Cu and Mn caused an increase of RSR in the 1–10 μg mL−1 concentration range, Fe in 1–5 μg mL−1 and Co in the 2–5 μg mL−1 range. The presence of 2 μg mL−1 of Cu(II) actually led to a response of 216%. Similar observations regarding the effect of Cu were also made for the vapour generation of other hydride forming elements. Pohl et al.46 reported increased As signals after the addition of 5 μg mL−1 Cu to the solution of 2 μg mL−1 of As. Arbab-Zavar et al.47 reported a 30% rise of Tl signals in the presence of 10 μg mL−1 Cu from the Pb–Sn cathode.
| Concomitant electrolyte | Electrolyte conc., ×10−3 mol L−1 | CVGa | EcVGb | UV-PVGa |
|---|---|---|---|---|
| a 5 μg L−1 of Se(IV). b 25 μg L−1 of Se(IV). c n – not determined. | ||||
| H2SO4 | 5 | 95 ± 2 | 104 ± 4 | 91 ± 1 |
| 50 | 101 ± 2 | 99 ± 2 | 38 ± 1 | |
| 250 | 105 ± 1 | 86 ± 4 | 7 ± 1 | |
| Na2SO4 | 5 | 91 ± 2 | 100 ± 3 | 89 ± 2 |
| 50 | 108 ± 2 | 85 ± 5 | 96 ± 3 | |
| 250 | 95 ± 2 | 72 ± 3 | 104 ± 2 | |
| HCl | 5 | nc | n | 153 ± 5 |
| 100 | n | n | 104 ± 3 | |
| 500 | n | n | 10 ± 1 | |
| NaCl | 10 | 98 ± 4 | 78 ± 7 | 105 ± 2 |
| 50 | 104 ± 4 | 49 ± 6 | 91 ± 3 | |
| 250 | 144 ± 15 | 27 ± 6 | 45 ± 1 | |
| 500 | 160 ± 20 | 9 ± 4 | n | |
| HNO3 | 5 | 107 ± 3 | 96 ± 3 | 230 ± 6 |
| 50 | 106 ± 2 | 9 ± 3 | 6 ± 2 | |
| 1000 | 65 ± 3 | n | n | |
| NaNO3 | 5 | 94 ± 2 | 111 ± 4 | 146 ± 4 |
| 50 | 92 ± 1 | 12 ± 3 | 3 ± 3 | |
| 500 | 92 ± 2 | n | n | |
| H3PO4 | 5 | 109 ± 3 | 76 ± 3 | 101 ± 2 |
| 100 | 103 ± 1 | 52 ± 1 | 77 ± 1 | |
| 1000 | 101 ± 1 | 13 ± 1 | 70 ± 3 | |
| Na3PO4 | 5 | 121 ± 2 | 106 ± 3 | 88 ± 2 |
| 50 | 120 ± 3 | 117 ± 6 | 98 ± 3 | |
| 250 | n | 45 ± 3 | n | |
It is an accepted fact that Pb has a very high hydrogen overvoltage and represents one of the cathode materials with the highest generation efficiency.20 It is, therefore, safe to assume that the cause of the increase of RSR at higher Mn, Fe and Cu concentrations is not due to increased generation efficiency. We hypothesize that it may be caused by one of the following effects: (i) some process located on the cathode surface, e.g. desorption of Se bound or adsorbed on the cathode surface caused by an excess of interferents, (ii) stabilization of the volatile Se compound in the gaseous phase on a cogenerated volatile form of interferents. The second assumption was tested experimentally, because reaction modification in the presence of various transition elements was encountered also in UV-PVG. However, the hypothesis of the stabilization of volatile Se species on the volatile form of Cu was not supported. We concluded that increased Se responses achieved with the addition of Cu(II) into the solution was caused by effects taking place on the cathode. For detailed results of the experiments with separate generation of volatile Se species and volatile forms of selected interferents see Section 3.4.
Ag(I) had purely signal-depressing effects but smaller than in CVG or UV-PVG; content of 10 ng mL−1 decreased the RSR to 84% and 1 μg mL−1 to 59%. The presence of 100 ng mL−1 and 1 μg mL−1 of Ni gave responses of 95% and 66%, respectively and presence of equal concentrations of Cr(III) suppressed relative responses to 74% and 51%, respectively.
3.3.2.3. Acids and salts. One more mechanism of interference of inorganic salts and acids can be expected in EcVG compared to CVG: the formation of insoluble salts on the surface of the lead cathode.20 This mechanism was a probable source of the phosphate interference in our work. The addition of phosphoric acid caused a decrease of response even in low concentrations and 1.0 mol L−1 gave a relative Se response of only 13%. Similarly, a less pronounced trend was observed in the presence of phosphate (See Table 2). In contrast, sulphuric acid, which would be expected to act as a strong interferent due to the formation of Pb(SO4)2, did not decrease Se signals in concentrations below 0.1 mol L−1 and in the concentration of 1.0 mol L−1 led to a relative response of 68%. Again, very similar results were obtained with sulphate. Nitric acid caused loss of signals, but low concentrations of nitrate increased RSR (to 159% at 0.002 mol L−1 or to 111% at 0.005 mol L−1). At concentrations higher than 0.005 mol L−1 even nitrate strongly suppressed the generation of volatile Se species. The effect of hydrochloric acid was not studied due to its presence in the reaction medium, but chloride decreased the relative Se response. Deterioration of the Pb cathode surface in NaCl due to slow dissolution of Pb facilitated by electric current was observed and reported by our group in a previous publication.48
3.3.3.1. Hydride forming elements. The products of Se CVG and EcVG are assumed to be hydrides whereas the product of UV-PVG from formic acid should be a mixture of selenium hydride and selenium carbonyl.12,19,22,49 Despite this the observed trends of the effects of As and Sb did not differ from CVG and EcVG (see Fig. 2). It would appear that even trace contents of As(III) and Sb(III) can decrease Se signals as RSRs below 80% were observed at only 1 ng mL−1 As(III) or Sb(III). However, the decrease of Se response with a growing excess of Sb was less steep than that for CVG. Comparing the results with the experiments in the gaseous phase leads to the conclusion that the cause of interference at very low concentrations of As(III) and Sb(III) lies in the liquid phase and only a higher concentration of hydride forming elements manifests itself by gaseous interference.
The behaviour of Pb(II) and Cd(II) did not resemble those of the typical hydride forming elements. No interference was observed for Pb(II) below 20 ng mL−1, but RSR corresponding to the presence of 250 ng mL−1 (50:1 ratio) of Pb(II) was only 13%. The behaviour of Cd was even closer to the behaviour of transition metals and is discussed in Section 3.3.3.2.
3.3.3.2. Ions of transition and noble metals. Guo et al.12 stated that no interference from Ni or Co in the determination of 100 μg L−1 of Se(IV) was detected in an acetic acid medium, even for concentrations as high as 500 mg L−1 for Ni(II) and 100 mg L−1 for Co(II). Conversely, only 3 μg mL−1 of Fe(II) decreased the response by 20%. Their results also showed that Fe(II) and Fe(III) could have different effects. García et al.50 observed reaction modification in the presence of Co(II) prepared from CoCl2·6H2O in formic acid.
We observed an apparent interference or reaction modification dependent on both the concentration and the identity of the concomitant. In general, interference was observed at very high concentrations of the concomitant, whereas reaction modification was observed at lower concentrations of interferents. For example, we observed signals corresponding to 90% RSR or higher with 50–1000 ng mL−l Co(II) and 50–10000 ng mL−1 Ni(II) (see Fig. 3). We tested the possibility that addition of Ni(II), Co(II) or Cr(III) could increase the efficiency of volatile Se species generation by the determination of unreacted Se in the waste liquid. The Se content found was the same in the presence of concomitants as that without them; therefore the reaction modification is not due to increased efficiency of generation but due to some other mechanism. In the case of Ni, the reaction modification was proved to lie in the gaseous phase (see Section 3.4). Increased RSR values in the presence of Ni(II) or Ni gaseous species form a wide flat maximum, and thus Ni is suitable to be used to increase the sensitivity of UV-PVG determination of Se. Some authors have already proposed trace metal assisted photochemical generation of Pb and As utilizing reaction modification caused by transition metals.51,52
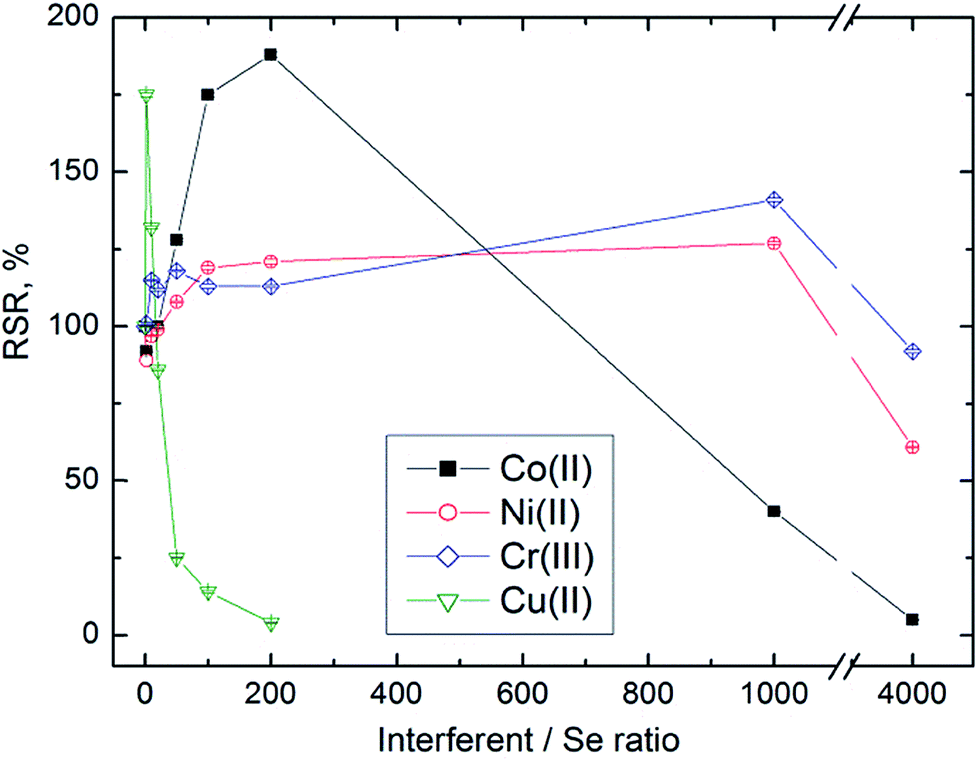 | ||
| Fig. 3 Effect of Co, Ni, Cr and Cu present in the liquid matrix on UV-PVG of Se, with the results expressed as the relative selenium response (%) and the amount of concomitant expressed as the interferent-to-Se mass concentration ratio, see Section 2.3 for explanation of axes labels. | ||
Similar to EcVG we observed a different behaviour of Cu and Ag. The presence of Ag(I) caused only negative interference effects in the form of a sharp decline of signals from 5 ng mL−1, whereas the presence of Cu(II) in concentrations 1–50 ng mL−1 resulted in increased RSR (see Fig. 3). Campanella et al.53 observed only negative effects of Cu(II) on Se UV-PVG and ascribed it to the formation of a heterophasic system in the UV-reactor due to formation of Cu(0). According to their work the interference could be alleviated only partially (by 25–30%) by varying the formic acid concentration.
Addition of Fe(III) decreased the Se response by 33% even when its concentration was 1 ng mL−1 and this decrease was constant up to 1 μg mL−1. The presence of Cd(II) led to the decrease of RSR in the region of 10–100 ng mL−1 but no interference was observed between 500 and 1000 ng mL−l.
3.3.3.3. Acids and salts. In general, UV-PVG is the most susceptible method to interference caused by the presence of acids and salts. Other authors discussed mainly the effect associated with the addition of nitric acid or nitrate to formic acid reaction medium.12,23,26,28 For example, Rybínová et al.28 observed that 0.005 mol L−1 of nitric acid increased the Se signal more than twice and that the increase of sensitivity was, at least for some experimental set-ups, accompanied by an increase of generation efficiency. The effect was ascribed to the formation of SeCO rather than a mixture of SeH2 and SeCO, which is produced when only HCOOH without NHO3 is used.22 However, any further increase of nitric acid concentration led to loss of the Se signal and at 0.05 mol L−1 of NHO3 the Se signal reached a zero value. This observation means that samples acidified with nitric acid have to be pre-treated prior to analysis to reduce the content of nitric acid. A similar observation, only less pronounced, was made with nitrate, which is in agreement with the observation of García et al.50 and Guo et al.23
The addition of hydrochloric acid and chloride led to different dependencies. Chloride did not interfere up to 0.02 mol L−1 and then the Se signals decreased reaching zero absorbance at 0.5 mol L−1 of NaCl. Hydrochloric acid caused an increase of Se absorbance with maximum 155% at 0.015 mol L−1. Assuming, that UV radiation causes production of Cl˙, we would expect similar trends for both HCl and chloride. At the same time, we do not rule out some combined acid–base and redox reactions. Sulphuric acid caused interference at concentrations higher than 0.05 mol L−1 but sulphate was tolerated up to 0.5 mol L−1. The smallest effect was observed with phosphoric acid and phosphates, thus making UV-PVG a more suitable alternative generation method for samples with higher phosphate content (e.g. biological).
3.4. Gaseous phase interference study
The effects of possible volatile forms generated from As(III), Sb(III), Pb(II), Ni(II) and Cu(II) during the atomisation process were studied, the quantified effects are summarized in Table 3. In general, copper species did not have any stabilizing effect in the gaseous phase in any of the methods. Volatile copper species were not detected and the effects caused by copper should therefore be limited to liquid phase reactions. Both As and Sb produced volatile species capable of interference during atomisation in all methods; however, the effects differed in extent. The generally lower effect during UV-PVG is more likely to be caused by lower production of volatile As and Sb species compared to CVG and EcVG. There are several arguments to support this hypothesis. First, only a low number of articles deal with UV-photochemical generation of volatile As and Sb species52,54–57 in contrast to the number of articles on selenium UV-PVG. Second, McSheehy et al.58 observed that many products formed during irradiation of inorganic As in an organic acid are water soluble species. Third, we should have observed more severe interference in UV-PVG if competing species were present in the atomizer because we used a much lower flow rate of hydrogen in UV-PVG compared to CVG or EcVG.| Concomitant element | Concomitant-to-Se ratio | CVGa | EcVGb | UV-PVGa |
|---|---|---|---|---|
| a 5 μg L−1 of Se(IV). b 25 μg L−1 of Se(IV). c n – not determined. | ||||
| As(III) | 10:1 |
97 ± 3 | 84 ± 4 | 101 ± 3 |
| 50:1 |
75 ± 2 | 53 ± 5 | 85 ± 4 | |
| 100:1 |
nc | 13 ± 3 | 75 ± 4 | |
| Sb(III) | 10:1 |
83 ± 5 | 83 ± 4 | 105 ± 7 |
| 50:1 |
67 ± 3 | 46 ± 4 | 86 ± 6 | |
| 100:1 |
14 ± 3 | 5 ± 5 | 68 ± 9 | |
| Pb(II) | 10:1 |
106 ± 4 | 98 ± 3 | 80 ± 8 |
| 50:1 |
100 ± 3 | 88 ± 2 | 70 ± 5 | |
| 100:1 |
n | 76 ± 3 | n | |
| Cu(II) | 10:1 |
100 ± 3 | 98 ± 5 | 101 ± 10 |
| 50:1 |
103 ± 3 | 96 ± 6 | 95 ± 5 | |
| 100:1 |
n | 90 ± 6 | 90 ± 6 | |
| Ni(II) | 10:1 |
104 ± 3 | 99 ± 4 | 97 ± 6 |
| 50:1 |
100 ± 4 | 98 ± 5 | 156 ± 10 | |
| 100:1 |
98 ± 4 | 99 ± 4 | 157 ± 9 | |
| 160:1 |
n | n | 160 ± 8 | |
Nevertheless, the overall interference from As and Sb was comparable for all three methods, thus leading to the conclusion that during UV-PVG As(III) and Sb(III) cause also liquid phase interference.
Atomisation interference of Pb was observed in EcVG, which is in concordance with the fact that unlike in CVG it is possible to generate volatile compounds from Pb(II).44,59 Slight interference of Cu and Ni was observed in EcVG, which could have been caused by volatilized nanoparticles of the reduced metals. The positive effect of Ni on Se signals in UV-PVG also manifested in the gaseous phase; we observed increased Se signals even when volatile Ni species were generated in a separate reactor. However, the assumed formation of Ni(CO)4 could not be proved. A determination of Ni at 232.0 nm wavelength via its UV photochemical volatile species generation was attempted but no Ni signals were observed. There is still a possibility that the volatile Ni species cannot be atomized under the given atomisation conditions or that it decomposes or adheres to the surfaces before reaching the atomizer.
4. Conclusions
All volatile species generation methods suffer from interference. One should have knowledge of the concomitants likely present in the matrix and the severity of their effects on the generation of a given volatile species to choose an optimum method for a particular analyte. This paper presents the comparison of overall and gaseous interferences in chemical, electrochemical and UV-photochemical generation of volatile selenium species with quartz furnace atomic absorption spectrometric detection. The concomitants were divided into groups based on similar nature – hydride forming elements, transition metals and electrolytes. Interference from As and Sb in the liquid phase showed similar trends for all three methods despite the fact that different volatile Se species may have been produced. Separate generation of interferents proved that As and Sb interfere during atomisation in all three methods; however, the atomisation interference in UV-PVG did not account for the overall interference at low concentrations of the interferent. Atomisation interference took place also in the presence of Pb at least for EcVG and PVG. No conclusion could be made for CVG because no interference was observed due to the inertness of Pb(II) to the reaction with NaBH4. Interferent effects caused by transition metals were more varied. For instance, CVG of Se species was found to be relatively immune to the presence of Fe(III) and slightly less to the presence of Ni(II), but Se signals dropped in the presence of Cu and Ag. Similarly, the UV-PVG was very sensitive to the presence of Cu and Ag, but wide ranges of Cr, Co and Ni concentrations led to a stable increase of Se signals and, thus, these elements could be used as reaction modifiers for improving the sensitivity of Se determination. Nevertheless, the increased sensitivity was not caused by increased generation efficiency; we proved that for Ni the mechanism was due to processes taking place in the gaseous phase. EcVG tolerated best the presence of Ag in solution. The effects of inorganic acids and their respective salts were similar; however some exceptions to this rule were observed in the alternative methods. Namely HCl and chloride behaved differently in UV-PVG and HNO3 and nitrate in EcVG. The presence of NaCl in the matrix during CVG led to reaction modification, whose nature was not yet explained. Thus from the three tested methods CVG would be the most suitable for analysis of samples with higher acid or salt content with the exception of NaCl.In general, the best overall resistance to interference was achieved with CVG. Interference in EcVG and UV-PVG was found comparable, but EcVG additionally suffered from memory effects. From these two alternative techniques UV-PVG is the more sensible choice.
Statement of human and animal rights
Not applicable, no experiments have been conducted on animals or human subjects.Conflicts of interest
There is no conflict of interest.Acknowledgements
This work was financially supported by the Charles University (Projects SVV 260440 and Project GA UK No. 228214) and by the Grant Agency of the Czech Republic (project GACR 14-23532).References
- A. R. Kumar and P. Riyazuddin, TrAC, Trends Anal. Chem., 2010, 29, 166–176 CrossRef CAS.
- P. Barth, V. Krivan and R. Hausbeck, Anal. Chim. Acta, 1992, 263, 111–118 CrossRef CAS.
- J. Dědina and T. Matoušek, J. Anal. At. Spectrom., 2000, 15, 301–304 RSC.
- É. M. de Moraes Flores, A. Medeiros Nunes, V. Luiz Dressler and J. Dědina, Spectrochim. Acta, Part B, 2009, 64, 173–178 CrossRef.
- J. Dědina, Spectrochim. Acta, Part B, 2007, 62, 846–872 CrossRef.
- J. Dědina, Anal. Chem., 1982, 54, 2097–2102 CrossRef.
- J. Dědina and D. L. Tsalev, Hydride Generation Atomic Absorption Spectrometry, Wiley, Chichester, 1995 Search PubMed.
- R. E. Sturgeon and Z. Mester, Appl. Spectrosc., 2002, 56, 202A–213A CrossRef CAS.
- E. Dimitrakakis, C. Haberhauer-Troyer, Y. Abe, M. Ochsenkühn-Petropoulou and E. Rosenberg, Anal. Bioanal. Chem., 2004, 379, 842–848 CrossRef CAS PubMed.
- P. Wu, L. He, C. Zheng, X. Hou and R. E. Sturgeon, J. Anal. At. Spectrom., 2010, 25, 1217–1246 RSC.
- A. Brockmann, C. Nonn and A. Golloch, J. Anal. At. Spectrom., 1993, 8, 397–401 RSC.
- X. Guo, R. E. Sturgeon, Z. Mester and G. J. Gardner, Anal. Chem., 2003, 75, 2092–2099 CrossRef CAS PubMed.
- G. F. Kirkbright and M. Taddia, Anal. Chim. Acta, 1978, 100, 145–150 CrossRef CAS.
- B. Welz and M. Melcher, Analyst, 1984, 109, 577–579 RSC.
- B. Welz and M. Melcher, Analyst, 1984, 109, 569–572 RSC.
- D. Bax, J. Agterdenbos, E. Worrell and J. B. Kolmer, Spectrochim. Acta, Part B, 1988, 43, 1349–1354 CrossRef.
- A. D'Ulivo, L. Lampugnani and R. Zamboni, J. Anal. At. Spectrom., 1991, 6, 565–571 RSC.
- E. Henden, Y. İşlek, M. Kavas, N. Aksuner, O. Yayayürük, T. D. Çiftçi and R. İlktaç, Spectrochim. Acta, Part B, 2011, 66, 793–798 CrossRef CAS.
- F. Laborda, E. Bolea and J. R. Castillo, Anal. Bioanal. Chem., 2007, 388, 743–751 CrossRef CAS PubMed.
- E. Denkhaus, A. Golloch, X. M. Guo and B. Huang, J. Anal. At. Spectrom., 2001, 16, 870–878 RSC.
- E. Bolea, D. Arroyo, G. Cepriá, F. Laborda and J. R. Castillo, Spectrochim. Acta, Part B, 2006, 61, 96–103 CrossRef.
- X. Guo, R. E. Sturgeon, Z. Mester and G. J. Gardner, Appl. Organomet. Chem., 2003, 17, 575–579 CrossRef CAS.
- X. Guo, R. E. Sturgeon, Z. Mester and G. J. Gardner, Environ. Sci. Technol., 2003, 37, 5645–5650 CrossRef CAS PubMed.
- R. E. Sturgeon and P. Grinberg, J. Anal. At. Spectrom., 2012, 27, 222–231 RSC.
- M. Rybínová, V. Červený, J. Hraníček and P. Rychlovský, Microchem. J., 2016, 124, 584–593 CrossRef.
- M. Rybínová, V. Červený and P. Rychlovský, J. Anal. At. Spectrom., 2015, 30, 1752–1763 RSC.
- J. Šíma, P. Rychlovský and J. Dědina, Spectrochim. Acta, Part B, 2004, 59, 125–133 CrossRef.
- M. Rybínová, S. Musil, V. Červený, M. Vobecký and P. Rychlovský, Spectrochim. Acta, Part B, 2016, 123, 134–142 CrossRef.
- E. Bolea, F. Laborda, M. A. Belarra and J. R. Castillo, Spectrochim. Acta, Part B, 2001, 56, 2347–2360 CrossRef.
- A. D'Ulivo and J. Dědina, Spectrochim. Acta, Part B, 1996, 51, 481–498 CrossRef.
- B. Welz and P. Stauss, Spectrochim. Acta, Part B, 1993, 48, 951–976 CrossRef.
- B. Welz and M. Melcher, Anal. Chim. Acta, 1981, 131, 17–25 CrossRef CAS.
- P. Novotný and J. Kratzer, Spectrochim. Acta, Part B, 2013, 79–80, 77–81 CrossRef.
- J. Kratzer, Spectrochim. Acta, Part B, 2012, 71–72, 40–47 CrossRef CAS.
- D. Korkmaz, C. Demir, F. Aydın and Y. O. Ataman, J. Anal. At. Spectrom., 2005, 20, 46–52 RSC.
- H. Matusiewicz and M. Krawczyk, Microchem. J., 2006, 83, 17–23 CrossRef CAS.
- M. C. Valdés-Hevia y Temprano, M. R. Fernández de la Campa and A. Sanz-Medel, J. Anal. At. Spectrom., 1993, 8, 847–852 RSC.
- H. Matusiewicz, M. Kopras and R. E. Sturgeon, Analyst, 1997, 122, 331–336 RSC.
- X. Guo and X. Guo, Anal. Chim. Acta, 1995, 310, 377–385 CrossRef CAS.
- A. D'ulivo, L. Lampugnani and R. Zamboni, Spectrochim. Acta, Part B, 1992, 47, 619–631 CrossRef.
- E. Pagliano, M. Onor, J. Meija, Z. Mester, R. E. Sturgeon and A. D'Ulivo, Spectrochim. Acta, Part B, 2011, 66, 740–747 CrossRef CAS.
- D. L. Nunes, E. P. Dos Santos, J. S. Barin, S. R. Mortari, V. L. Dressler and É. M. De Moraes Flores, Spectrochim. Acta, Part B, 2005, 60, 731–736 CrossRef.
- G. Junková, J. Šíma and P. Rychlovský, Chem. Pap., 2003, 57, 192–196 Search PubMed.
- M. Sáenz, L. Fernández, J. Domínguez and J. Alvarado, Spectrochim. Acta, Part B, 2012, 71–72, 107–111 CrossRef.
- W. W. Ding and R. E. Sturgeon, J. Anal. At. Spectrom., 1996, 11, 421–425 RSC.
- P. Pohl, I. J. Zapata and N. H. Bings, Anal. Chim. Acta, 2008, 606, 9–18 CrossRef CAS PubMed.
- M. H. Arbab-Zavar, M. Chamsaz, A. Yousefi and N. Ashraf, Talanta, 2009, 79, 302–307 CrossRef CAS PubMed.
- E. Nováková, P. Rychlovský, T. Resslerová, J. Hraníček and V. Červený, Spectrochim. Acta, Part B, 2016, 117, 42–48 CrossRef.
- A. R. Kumar and P. Riyazuddin, Anal. Sci., 2005, 21, 1401–1410 CrossRef CAS PubMed.
- M. García, R. Figueroa, I. Lavilla and C. Bendicho, J. Anal. At. Spectrom., 2006, 21, 582–587 RSC.
- Y. Gao, M. Xu, R. E. Sturgeon, Z. Mester, Z. Shi, R. Galea, P. Saull and L. Yang, Anal. Chem., 2015, 87, 4495–4502 CrossRef CAS PubMed.
- O. Linhart, J. Smolejová, V. Červený, J. Hraníček, E. Nováková, T. Resslerová and P. Rychlovský, Monatsh. Chem., 2016, 147, 1447–1454 CrossRef CAS.
- B. Campanella, A. Menciassi, M. Onor, C. Ferrari, E. Bramanti and A. D'Ulivo, Spectrochim. Acta, Part B, 2016, 126, 11–16 CrossRef CAS.
- C. Zheng, Q. Ma, L. Wu, X. Hou and R. E. Sturgeon, Microchem. J., 2010, 95, 32–37 CrossRef CAS.
- X. Guo, R. E. Sturgeon, Z. Mester and G. J. Gardner, J. Anal. At. Spectrom., 2005, 20, 702–709 RSC.
- Y. Gao, R. E. Sturgeon, Z. Mester, X. Hou, L. Yang, Z. Mester, X. Hou and L. Yang, Anal. Chim. Acta, 2015, 901, 34–40 CrossRef CAS PubMed.
- Y. Gao, R. E. Sturgeon, Z. Mester, X. Hou, C. Zheng and L. Yang, Anal. Chem., 2015, 87, 7996–8004 CrossRef CAS PubMed.
- S. McSheehy, X. M. Guo, R. E. Sturgeon and Z. Mester, J. Anal. At. Spectrom., 2005, 20, 709–716 RSC.
- M. Sáenz, L. Fernández, J. Domínguez and J. Alvarado, Electroanalysis, 2010, 22, 2842–2847 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |