
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable succinylation of cellulose in a CO2-based switchable solvent and subsequent Passerini 3-CR and Ugi 4-CR modification†
Z.
Söyler
 a,
K. N.
Onwukamike
ab,
S.
Grelier
b,
E.
Grau
b,
H.
Cramail
b and
M. A. R.
Meier
*a
a,
K. N.
Onwukamike
ab,
S.
Grelier
b,
E.
Grau
b,
H.
Cramail
b and
M. A. R.
Meier
*a
aKarlsruhe Institute of Technology (KIT), Institute of Organic Chemistry (IOC), Materialwissenschaftliches Zentrum MZE, Straße am Forum 7, 76131 Karlsruhe, Germany. E-mail: m.a.r.meier@kit.edu
bLaboratoire de Chimie des Polymères Organiques, Université de Bordeaux/CNRS/ Ecole Nationale Supérieure de Chimie Biologie & Physique, 16 Avenue Pey-Berland, 33607 Pessac Cedex, France
First published on 20th November 2017
Abstract
A rapid and effective dissolution and activation of cellulose was demonstrated by a reversible reaction of CO2 with the hydroxyl groups of the cellulose backbone in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). The dissolved cellulose was subsequently subjected to in situ derivatization with succinic anhydride without the need of any additional catalyst under very mild conditions. As a result of our optimization studies, cellulose was successfully converted to cellulose succinates with degrees of substitution (DS) ranging from 1.51 to 2.59, depending on the reaction conditions and the molar ratio of succinic anhydride. The optimized reaction conditions were successfully applied to different types of cellulose samples including microcrystalline cellulose (MCC) and organosolv wood pulp (WP), exhibiting similar conversions. Furthermore, the carboxylic acid moiety, introduced by the succinylation, was further converted via Passerini three-component reactions (Passerini-3CR) and Ugi four-component reactions (Ugi-4CR). 31P NMR revealed the quantitative conversion of carboxylic acid moieties on the cellulose backbone under mild conditions. All obtained products were thoroughly characterized by ATR-IR, 1H, 13C, and 31P NMR spectroscopies as well as by size exclusion chromatography (SEC). Thermal properties of the obtained products were investigated by differential scanning calorimetry (DSC) and by thermogravimetric analysis (TGA), revealing glass transitions (Tg) for all the Passerini and Ugi products between at 76–116 °C and high thermal stability between 263–290 °C. The reported methodology represents a very mild, highly efficient and sustainable route for the dissolution of cellulose and the synthesis of cellulose succinates. The subsequent modifications of the obtained cellulose succinates via multicomponent reactions resulted in materials with improved thermal properties and offers a straightforward and versatile modification strategy for cellulose.
Introduction
Most of the raw materials used in the chemical industry are petroleum derived. The increasing dependency of mankind on crude oil highlights the concern of its finite availability along with its environmental impact. In this context, renewable materials research has become a mainstream topic.1 Biomass constitutes a very promising alternative renewable resource, offering sufficient amounts of carbon with a broad range of chemical structures.2 Particularly, lignocellulosic biomass has an annual natural production of 170 × 109 tons per year.3 Cellulose, which constitutes 35–50% of the total lignocellulosic biomass, offers considerable advantages as potential sustainable alternative to petroleum-derived materials due to its positive attributes, such as high mechanical and thermal stabilities as well as biodegradability and biocompatibility. Cellulose is a high molecular weight hydrophilic homopolymer consisting of β-1,4 linked D-glucose units with 3-OH groups per anhydroglucose (AGU). However, its lack of processability and insolubility in common organic solvents as well as its insufficient physico-chemical properties constitute limiting factors for its utilization by the chemical industry. Thus, chemical modification via its accessible free hydroxyl moieties are required and investigated.4,5 Chemical modifications of cellulose, such as esterification,6 etherification7 and grafting (from or onto)8 are among the most common techniques. Such derivatizations could be achieved via heterogenous or homogenous approaches.5 Until recently, heterogenous modifications on the surface of cellulose fibres have been more common due to the solubility challenge of cellulose. Homogenous modifications of cellulose, on the other hand, are highly desirable as the latter enable to control the DS simply by adjusting the reaction conditions.9 Over the past decades, various solvent systems have been shown to be capable of solubilizing cellulose including N-methylmorpholine N-oxide (NMMO),10 mixtures of dimethylsulfoxide-tetrabutylammonium fluoride (DMSO–TBAF),11 or N,N-dimethylacetamide–lithium chloride (DMAc–LiCl).12In 2002, Swatloski et al.13 reported on the possibility to solubilise cellulose in ionic liquids (ILs). Due to their very low vapor pressure and recyclability, ILs are regarded as environmentally benign solvents for cellulose solubilisation. As shown in the review of Mäki-Arvela et al.,14 extensive studies on homogenous modifications of cellulose have been reported using ILs as solvents. However, along with the debate over their efficiencies, ILs still suffer from some limitations since they are relatively expensive and easily contaminated in the course of the reaction, thereby making their recovery relatively difficult. Moreover, their toxicity and economic production are still under investigation.15
Renewability as such, not only for the production of synthetic polymers, cannot be considered satisfactory in terms of sustainability. Thus, in order to reach fully sustainable methods and procedures, avoiding toxic chemicals and limiting waste production, or more generally considering all 12 principles of green chemistry for novel procedures, is essential.16
Recently, a more sustainable CO2-based switchable solvent system for cellulose solubilisation was proposed by Xie et al.17 as well as by Zhang et al.18 almost simultaneously. This CO2 based switchable solvent strategy can be considered a breakthrough for the solubilisation of cellulose in terms of efficiency and sustainability. Basically, two distinct strategies were reported concerning this solvent strategy, a derivative18 and a non-derivative17 approach (Scheme 1). The derivative approach includes the direct incorporation of CO2 to the hydroxyl groups of the cellulose in the presence of a superbase, such as DBU, resulting a reversible carbonate complex on the cellulose backbone, in DMSO as a solvent. The non-derivative approach employs an additional source of hydroxyl groups, such as ethylene glycol, for the in situ solubilization of cellulose. The term ‘switchable’ comes from the role of CO2 in the system. A carbonate anion is formed resulting in the solubilisation of native cellulose, which can afterwards readily be regenerated by releasing the CO2 from the system. A superbase, which is a part of the reversible complexation could be regenerated as well upon CO2 release, thus facilitating the recovery of the superbase from the system. Compared to classic ionic liquids, this solvent system is more efficient, more sustainable (i.e. no multi-step synthesis is required for IL synthesis and recovery is straightforward), and cheaper, since the only chemical to be synthesized is DBU (or another superbase). Moreover, solvent recovery is easier. Recently, several studies have been reported on the homogenous modifications of cellulose using this solvent system, including acylation and grafting from approaches. In all cases, milder reaction conditions leading to higher conversions compared to classic ionic liquids were described.19,20 Recent studies have shown that the superbase has a dual role in this system, both being part of the solvent system and acting as a catalyst in reactions.19,20 Indeed, the superbase is not fully converted into its protonated form in the solvent system. Therefore, the remaining free superbase maintains its catalytic activity, thus explaining higher conversions under mild reaction conditions.21
 | ||
| Scheme 1 Derivative and non-derivative approaches for the dissolution of cellulose using CO2 as a reagent. | ||
The acylation of cellulose using succinic anhydride is one approach to introduce carboxylic acid moieties to the cellulose backbone in a sustainable manner. Succinic anhydride can be produced from bio-based raw materials such as sugars.22 In addition, succinic anhydride does not lead to an undesired corresponding by-product, which is yet another contribution to the reduced environmental impact of the procedure. Extensive studies are available on the homogenous succinylation of cellulose using succinic anhydride leading to various DS ranging from 0.3 without any catalyst to 2.3 using catalysts such as 4-(dimethylamino)pyridine (DMAP), depending also on the reaction conditions.23–25 A variety of solvent systems and catalysts have been studied for this cellulose modification so far. For instance, Yin et al.25 studied a N,N-dimethylacetamide/LiCl solvent mixture for the succinylation of cellulose in the presence of triethylamine as catalyst, leading to DS values of up to 1.45. In addition, Sun et al.23 achieved a DS of up to 2.34 in 1-butyl-3-methylimidazolium chloride (BMIMCI) as solvent in the presence of DMAP as catalyst at 100 °C. Chen et al.26 utilized the same catalyst for homogenous succinylation of cellulose in a tetra butylammonium fluoride (TBAF)/dimethyl sulfoxide (DMSO) solvent mixture, leading to a DS up to 2.11. Recently, homogenous succinylation of cellulose was also studied under catalyst free conditions. Liu et al.27 studied a BMIMCI/DMSO solvent mixture as well as 1-allyl-3-methylimidazolium chloride (AMIMCl) for the succinylation of cellulose without catalyst, obtaining DS values of up to 0.53 and 0.22, respectively. These previous studies have shown that the presence of a catalyst is essential to obtain high conversions. Interestingly, none of these reports focused on the recovery of the solvent systems and/or the catalyst, which is of course important regarding the sustainability of the procedure.
Along with various potential applications, such as absorbents for soil in agriculture or for the removal of heavy metals from waste water,28 the carboxylic acid moieties introduced by succinylation offer unique opportunities for further modification of cellulose, for instance via multicomponent reactions (MCRs) such as the Passerini three component (Passerini 3-CR) or the Ugi four component (Ugi 4-CR) reactions. MCRs are well explored and comprehensively studied for a wide range of chemical conversions. The latter reactions are very robust, versatile and offer advantages such as high atom economy, one pot reactions and high yields.29 To the best of our knowledge, the corresponding multicomponent reactions have so far only been used for the crosslinking studies of carbohydrate polymers, including carboxymethyl cellulose and chitosan, to obtain hydrogels.30 The modification of cellulose via Passerini-3CR and Ugi-4CR, which has not been reported before, is an innovative and powerful approach for the development of cellulose-based materials with desired tunable properties by selective combination of the different components employed.
Herein, we thus report an efficient and sustainable methodology for the modification of cellulose with succinic anhydride in a CO2-based switchable solvent system (Scheme 2). This improved methodology enables higher conversions under milder conditions, if compared to previous studies (see literature discussion above).23 Moreover, we introduce the use of multicomponent reactions as versatile and mild modification strategy for cellulose. Indeed, succinylated cellulose prepared with the highest degree of substitution (SFP 6) was further modified via Passerini-3CR and Ugi-4CR with excellent conversions and yields (Scheme 3).
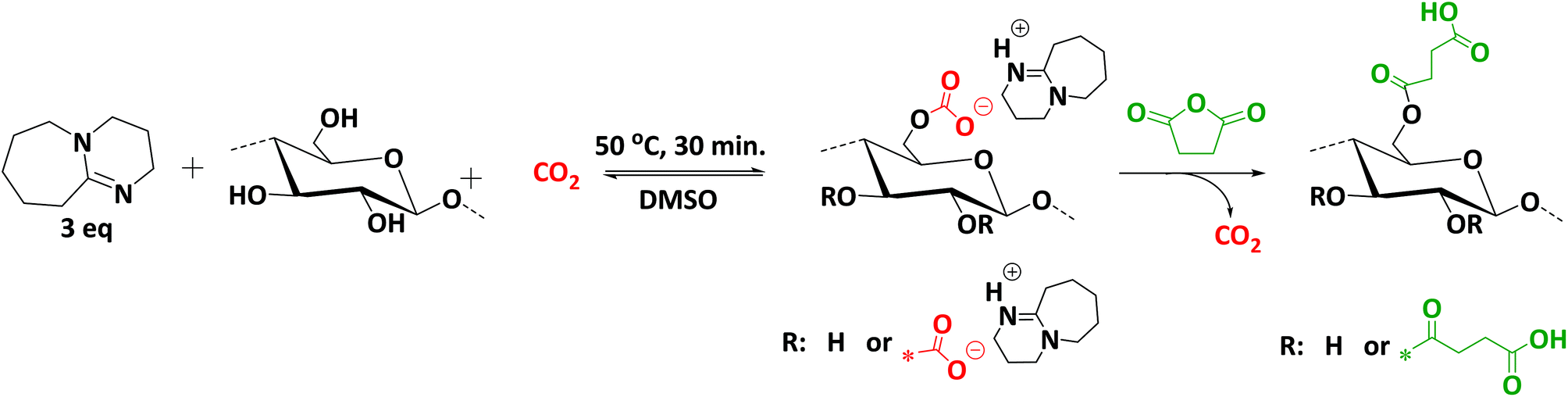 | ||
| Scheme 2 Dissolution and activation of cellulose for the subsequent derivatization with succinic anhydride using CO2-based switchable solvent. | ||
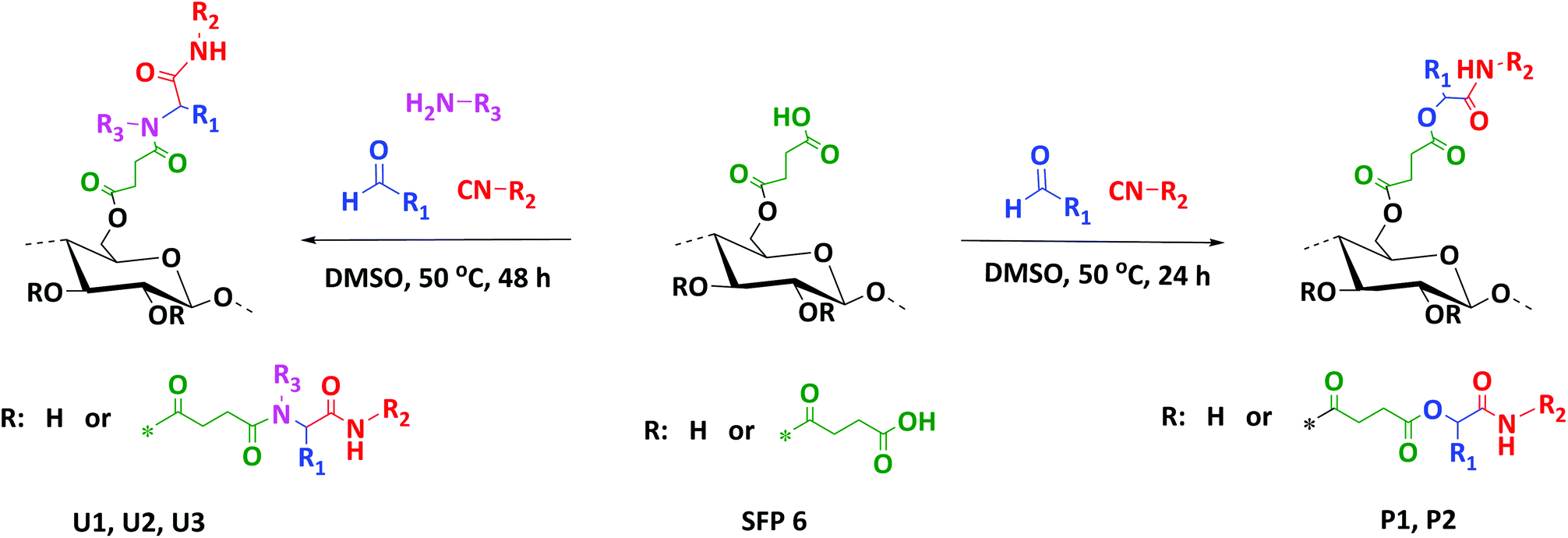 | ||
| Scheme 3 Modification of the succinylated filter paper via Passerini-3CR (right) and Ugi-4CR (left) reactions. | ||
Results and discussion
In this work, we demonstrate the rapid and efficient dissolution and activation of cellulose for the subsequent derivatization with succinic anhydride. The dissolution of cellulose was achieved at 50 °C in 30 minutes (compared to previously reported 60 °C for 3 h) by applying 2–5 bar of CO2 pressure in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 1 eq. with respect to –OH groups of cellulose) in DMSO as a solvent. Thus, applying the derivative approach (see Scheme 1 and Introduction), the hydroxyl groups on the cellulose backbone could react reversibly with CO2 to yield carbonate anions, resulting in a DMSO soluble polymeric structure (Scheme 1). Since very mild conditions were applied for the effective dissolution of cellulose, this solvent system exhibits several advantages in terms of efficiency and sustainable economy over ionic liquids, for which much longer times (6–12 h) and higher temperatures (90–120 °C) are usually required.13 The activation is reversible and under release of the CO2 cellulose can easily be regenerated. However, our observations showed that the solvent system could be preserved in a closed system under argon atmosphere for a couple of weeks, maintaining its efficiency. DMSO is often used as solvent or co-solvent for the homogenous functionalization of carbohydrates as it provides higher solubility and efficiency than its alternatives DMAc and DMF.31 Moreover, DMSO is regarded as an environmentally relatively benign solvent due to its high boiling point and low vapor pressure. Consequently, DMSO is selected as ‘greenest’ possible solvent and is thus widely employed for the functionalization of carbohydrate polymers, especially for cellulose.32 Furthermore, DBU, which is part of the solvent system in this work, is a well-known organocatalyst for various transformations, including esterification and alkylation reactions.33 This organocatalyst, along with being a part of the solvent system, maintained its catalytic activity in the course of the reactions with excellent efficiency in this work.Filter paper (whatman™ cellulose filter paper, no. 5) was utilized as a cellulose source for the optimization studies of the succinylation reaction. All the cellulose samples were dried under vacuum for 24 h at 100 °C prior to modification in order to minimize potential negative effects of residual water on the conversion. The optimization results for the succinylation of cellulose samples with succinic anhydride are summarized in Table 1. 1H NMR spectroscopy was used for the determination of the DS of the succinylated cellulose samples. For this, the obtained succinylated cellulose samples were dried under high vacuum at elevated temperatures to reduce the effect of residual water on the DS value calculation. The method used for calculation of the DS values can be found in the ESI† and relies on a large number of scans for each measurement at increased temperature, to ensure well-resolved 1H NMR spectra. The correctness of the method was later confirmed by further analysis of the obtained derivatives of the cellulose succinate (see discussion below). The effects of the concentration of cellulose, of the succinic anhydride molecular ratio as well as of the reaction time and temperature were investigated. The initial screening of the optimization studies was conducted by using ATR-IR spectroscopy. The optimization was performed by monitoring the intensity of the carbonyl peak at around 1703 cm−1 that is characteristic of the substituted ester moiety. Spectra were normalized to the intensity of the C–O stretching vibration of glycopyranose as internal reference at around 1050 cm−1 to have a relative comparison of the different degrees of modifications, since the pyranose oxygen absorption is not influenced significantly upon the succinylation process.
| Products | Eq. SAa | T (°C) | Time (min) | DSb |
|---|---|---|---|---|
| a Equivalents of succinic anhydride with respect to anhydroglucose units (AGU). b DS was calculated from 1H NMR spectroscopy. c Not determined, insoluble material. (RT: room temperature). | ||||
| SFP 1 | 2.25 | RT | 30 | 1.51 |
| SFP 2 | 2.25 | RT | 60 | 1.53 |
| SFP 3 | 2.25 | RT | 90 | 1.51 |
| SFP 4 | 3 | RT | 30 | 1.88 |
| SFP 5 | 3.75 | RT | 30 | 2.16 |
| SFP 6 | 4.5 | RT | 30 | 2.59 |
| SFP 7 | 6 | RT | 30 | 2.62 |
| SFP 8 | 4.5 | 60 | 10 | 2.22 |
| SFP 9 | 4.5 | 60 | 30 | — c |
| SFP 10 | 4.5 | 80 | 10 | 2.06 |
| SFP 11 | 4.5 | 80 | 30 | —.c |
Absorbances in the fingerprint region around 982, 896 and 606 cm−1 are associated with the anhydroglucose unit of native cellulose. The peak at around 3300 cm−1 corresponds to stretching vibrations of the OH groups of native cellulose, which is reduced and replaced after the modification by a very broad signal between 2390–3680 cm−1 characteristic of the introduced –COOH stretching vibrations. In addition, new peaks at 1704 and 1148 cm−1 were observed and attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretching vibrations of the introduced carboxyl moiety, respectively (Fig. 1). A complete overview of the optimization data can be found in the ESI.†1H and 13C NMR spectroscopies were used for a further confirmation of the structure of the succinylated cellulose. After the modification, the peaks belonging to the backbone of the cellulose and the new peaks introduced by succinylation are clearly observed (Fig. 2). The new broad peak at around 11.8 ppm corresponds to the protons of the carboxylic acid moiety. Methylene groups of the introduced succinate at around 2.5 ppm overlap with the NMR solvent DMSO-d6 and cannot be clearly observed in 1H NMR. However, 13C NMR reveals the presence of the methylene signals at around 28.36 and 28.02 ppm. Moreover, the new peaks at around 172.46 and 170.77 ppm are characteristic of the ester and acid functionalities of the introduced succinates, respectively. Moreover, peaks associated with ester groups attached to positions c6, c3 and c2 were clearly observed by 13C NMR (Fig. 2).
O and C–O stretching vibrations of the introduced carboxyl moiety, respectively (Fig. 1). A complete overview of the optimization data can be found in the ESI.†1H and 13C NMR spectroscopies were used for a further confirmation of the structure of the succinylated cellulose. After the modification, the peaks belonging to the backbone of the cellulose and the new peaks introduced by succinylation are clearly observed (Fig. 2). The new broad peak at around 11.8 ppm corresponds to the protons of the carboxylic acid moiety. Methylene groups of the introduced succinate at around 2.5 ppm overlap with the NMR solvent DMSO-d6 and cannot be clearly observed in 1H NMR. However, 13C NMR reveals the presence of the methylene signals at around 28.36 and 28.02 ppm. Moreover, the new peaks at around 172.46 and 170.77 ppm are characteristic of the ester and acid functionalities of the introduced succinates, respectively. Moreover, peaks associated with ester groups attached to positions c6, c3 and c2 were clearly observed by 13C NMR (Fig. 2).
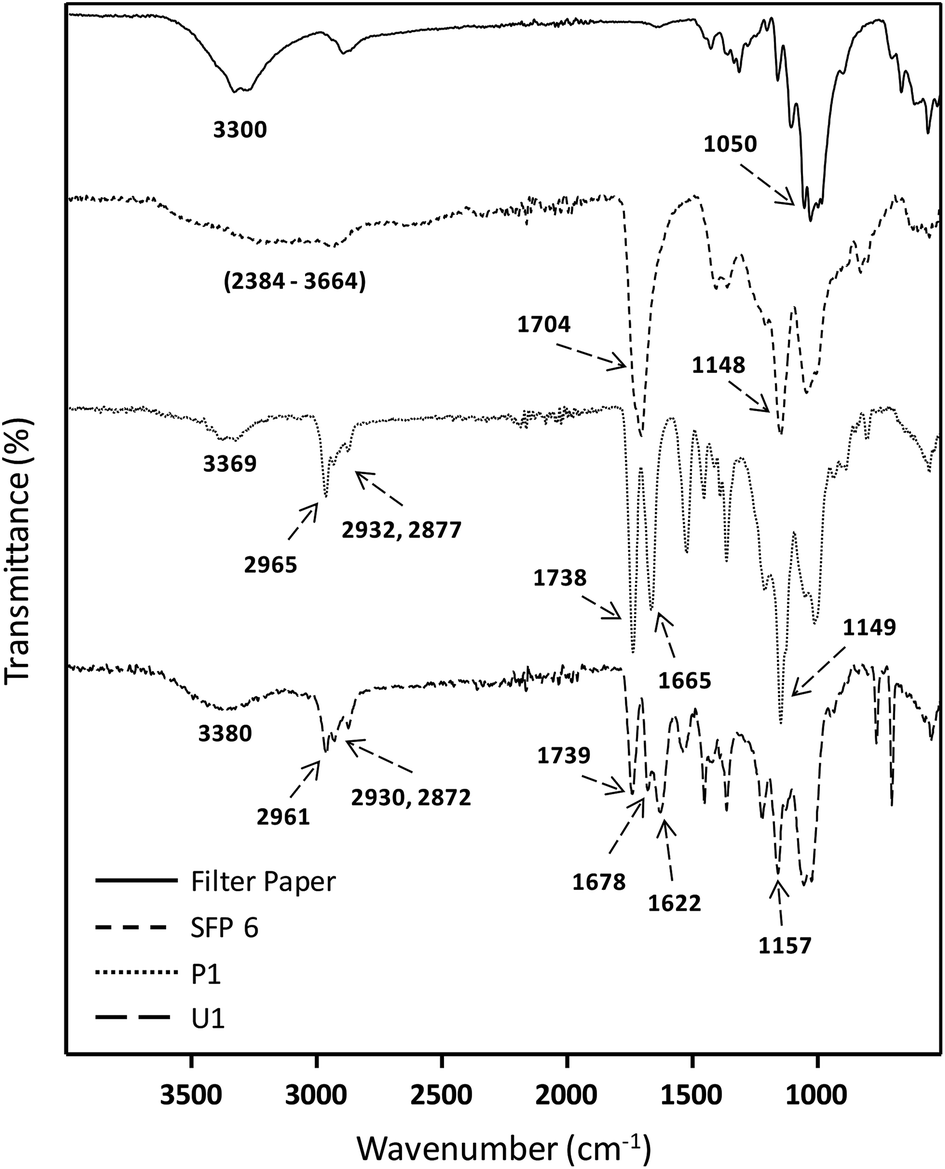 | ||
| Fig. 1 ATR-IR data of native filter paper, succinylated filter paper (SFP 6), Passerini (P1) and Ugi (U1) products of filter paper. | ||
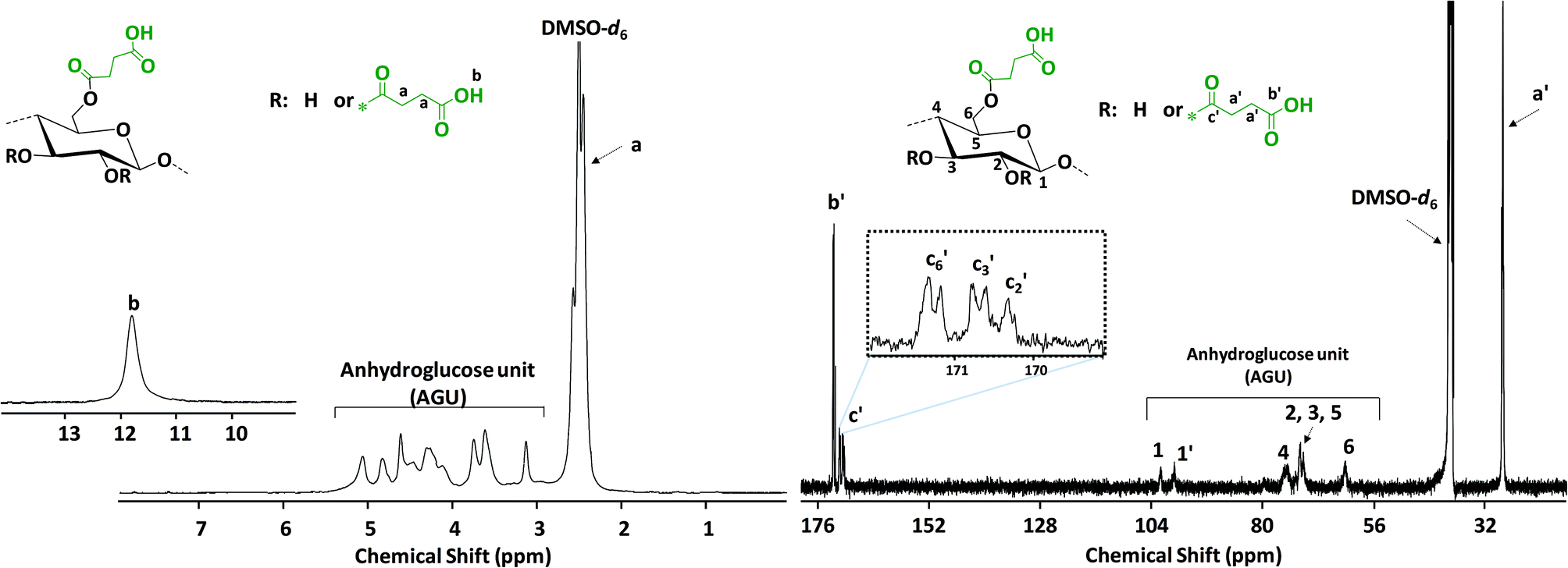 | ||
| Fig. 2 1H (left) and 13C NMR (right) of the succinylated filter paper (SFP 6) in DMSO-d6. | ||
From our observations, the concentration of cellulose constitutes a predominant factor for homogenous reactions of cellulose. Therefore, different concentrations of cellulose were tested to determine the highest possible conversion and 4% (w/w) of cellulose concentration gave the best result in terms of conversion. Lower concentrations of cellulose (i.e. 2% and 3% (w/w)) led to lower conversions (ESI Fig. S3†). Higher concentrations, above 4%, resulted in a very viscous reaction mixture during the addition of succinic anhydride. Thus, the magnetic stirring could not be maintained at these higher concentrations.
Furthermore, different reaction temperatures were tested and the highest conversions were obtained applying room temperature. Higher reaction temperatures, such as 60 and 80 °C, resulted in lower conversions and after 30 min of reaction time, the product isolated by reprecipitation was insoluble in common organic solvents, probably due to crosslinking (ESI Fig. S2, S4 and S5†). Similar results were observed in previous studies at high reaction temperatures (100 °C). Here, lower reaction temperatures (60–80 °C) resulted in crosslinked materials, presumably due to the higher reactivity of the applied solvent system in comparison to previous studies.23 The optimum reaction time for obtaining the highest conversion and yield was also investigated and found to be 30 minutes. Applying longer times did not improve the DS.
Additionally, the molar ratio of succinic anhydride has a pronounced influence on the reaction. The DS could be adjusted (from 1.51 to 2.59, the highest reported in literature yet) by changing the molar ratio of succinic anhydride (from 2.25 to 4.50 eq. per AGU), resulting in different material properties (ESI Fig. S1†). The optimized reaction conditions were applied to different types of cellulose samples, including microcrystalline cellulose (MCC) and organosolv wood pulp (WP), exhibiting similar conversions, thus also demonstrating the reproducibility and versatility of the established procedure (ESI Fig. S6†).
Size exclusion chromatography (SEC, relative calibration) of the succinylated cellulose samples revealed high molecular weights in the range of 150–270 kDa, depending on the type of the cellulose. As expected, succinylated filter paper (SFP) had the highest molecular weight with 271 kDa. Succinylated microcrystalline cellulose (SMCC) and organosolv wood pulp (SWP) exhibited similar molecular weights with 150 and 160 kDa, respectively (Table 2, Fig. 3 and ESI S25†). Moreover, although the reported values are relative to the PMMA standards used for calibration, reaching such high molecular weights for the modified cellulose samples highlights the mildness of the applied procedures and indicates that the possible degradation of the cellulose backbone was minimized. The thermal properties of the succinylated filter paper with the highest DS (SFP 6) were further characterized by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). No thermal transition was observed. Moreover, it showed low thermal stability (5% degradation temperature of 202 °C) when compared to unmodified filter paper (305 °C) (Table 2). This could be ascribed to the high acid content, introduced via succinylation, on the cellulose backbone, which presumably causes the degradation of the polymeric structure at high temperatures. After the reaction, DMSO and DBU were successfully recovered via a facile work-up. Fractional distillation was applied to recover the DMSO (90 °C, 25 mbar). The remaining mixture, composed of DBU and succinic anhydride/succinic acid or salts, was transferred into ethyl acetate and washed with a 6 M NaOH (3 × 10 mL) solution to recover DBU (56%). The high purity of recovered DBU could be shown by 1H and 13C NMR spectroscopy (ESI Fig. S30 and S31†). Indeed, the relatively low recovery yield of the DBU might be improved in the future by applying larger scale reactions by avoiding sample loss. In this work, the recovery of the unreacted succinic anhydride or succinic acid/salt was not attempted, again due to the small reaction scales used for this initial study.
 | ||
| Fig. 3 GPC traces of succinylated filter paper (SFP 6) and Passerini products P1 and P2 (left) as well as Ugi products U1–U3 (right). | ||
| Product | M n (kDa) | M w (kDa) | Đ | DSb | Conversiond (%) | Yielde (%) | T g (°C) | T d (°C) |
|---|---|---|---|---|---|---|---|---|
| a GPC measurements were carried out relative to poly(methyl methacrylate) calibration in DMAc/LiBr (1% w/w). b DS was calculated by 1H NMR. c DS value was calculated from 31P NMR of Passerini product. d Conversion of the carboxylic acid moieties were calculated by 31P NMR. e Yields were calculated from DS values (detailed calculations can be found in the ESI). f Not measured. | ||||||||
| Filter paper | — | — | — | — | — | — | — | 305 |
| SFP 6 | 271 | 714 | 2.6 | 2.59 | — | 86 | — | 202 |
| SMCC | 166 | 353 | 2.1 | 2.51 | — | 91 | — | — f |
| SWP | 167 | 593 | 3.5 | 2.57 | — | 82 | — | —f |
| P1 | 177 | 587 | 3.3 | 2.59/2.64c | 100 | 87 | 91 | 289 |
| P2 | 212 | 690 | 3.2 | 2.59/2.64c | 100 | 92 | 76 | 278 |
| U1 | 193 | 533 | 2.6 | 2.59/2.64c | 88.7 | 83 | 116 | 263 |
| U2 | 242 | 656 | 2.7 | 2.59/2.64c | 97.6 | 92 | 99 | 276 |
| U3 | 225 | 647 | 2.8 | 2.59/2.64c | 99.6 | 89 | 112 | 290 |
Finally, the obtained succinylated filter paper with the highest DS (DS: 2.59 from 1H NMR, SFP 6) was further derivatized by Passerini three-component (Passerini 3-CR) and Ugi four-component (Ugi 4-CR) reactions. Two different reactions for Passerini 3-CR and three different reactions for Ugi 4-CR were performed in order to easily tune the properties and the molecular structure of the modified cellulose. All reactions were performed under homogenous conditions using DMSO as a solvent. The concentration of the starting materials in Passerini and Ugi reactions is a key parameter. Thus, different concentrations of succinylated cellulose were tested and 50 mg mL−1 was found to be most efficient. Reactions were performed at 50 °C to ensure the homogeneity of the reaction media and also to reduce the viscosity of the reaction mixture, enabling feasible stirring. Passerini reactions were completed within the 24 hours, while 48 hours were required for Ugi reactions to achieve highest conversions of the carboxylic acid moieties. The conversion of carboxyl moieties was determined by 31P NMR spectroscopy. The unreacted carboxylic acid moieties as well as the free hydroxyl groups reacted with the phosphorylating agent, revealing a broad peak between 140–155 ppm and at around 134 ppm, respectively. Results have shown that all Passerini reactions on succinylated filter paper resulted in full conversions of carboxyl moieties, since no signal at around 134 ppm was observed in 31P NMR. Thus, the DS value of succinylation obtained from 1H NMR (DS: 2.59) could be confirmed by 31P NMR revealing very similar DS values (DS: 2.64 from 31P NMR). This consistency also verifies the reliability of our method for the determination of DS values of succinylated cellulose (Table 2 and ESI Fig. S18–S22†). The details of the calculation of the conversion of the carboxylic acid moieties of the Passerini and Ugi products by 31P NMR are described in the ESI.† Very high conversions of the carboxyl moieties (88.7–99.6%), depending on the structure of the utilized components, were observed for the Ugi reactions (Table 2). All Passerini and Ugi products of cellulose were soluble in common organic solvents including tetrahydrofuran, chloroform, dichloromethane, methanol as well as dimethyl sulfoxide and dimethyl formamide, thus significantly simplifying their analysis. The success of the MCRs was also observed by ATR-IR spectroscopy (Fig. 1 and ESI Fig. S7–S11†). In both cases, the broad signal between 2390–3680 cm−1, which represents the carboxylic acid moiety introduced via succinylation, almost completely disappeared and is replaced by less broad peaks between 3150–3700 cm−1, which are attributed to the mixtures of newly introduced ν(N–H), via MCRs, and unreacted ν(O–H) stretching vibrations from the cellulose backbone. Regarding the Passerini products, a new peak at around 1660 cm−1 corresponding to the introduced amide functionality appeared. The Ugi products exhibit an additional peak at around 1625 cm−1 associated to the CO stretching vibrations of another amide functionality, since two amide groups were introduced. Further confirmation of the multicomponent products was obtained using 1H and 13C NMR spectroscopy (Fig. 5, 6 and ESI Fig. S12–S17†). 1H NMR of the modified filter papers revealed that the broad peak at around 11.8 ppm, which is associated with hydrogen of carboxyl moiety, disappeared and new corresponding peaks of the components appeared and are clearly seen after the modification.
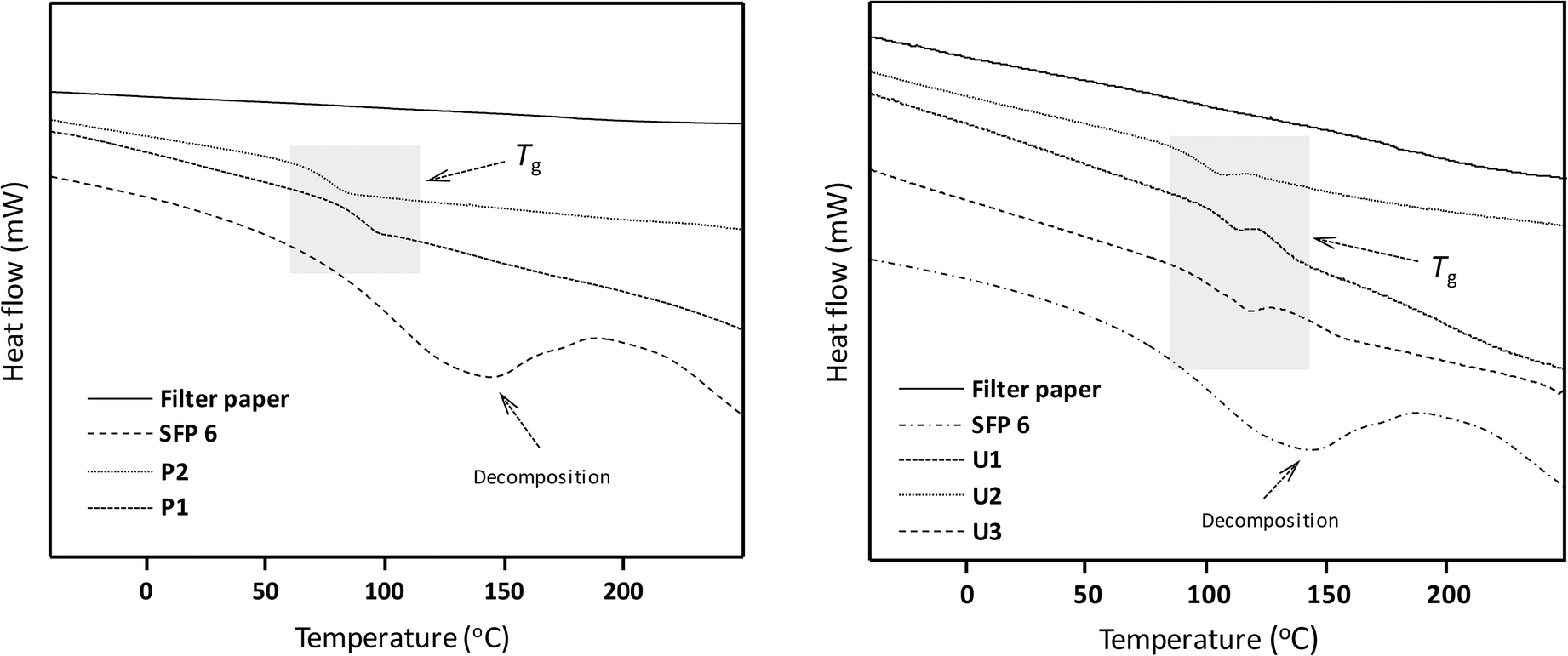 | ||
| Fig. 4 DSC data of filter paper, succinylated filter paper (SFP 6) and Passerini products P1 and P2 (left) as well as Ugi products U1–U3 (right). | ||
 | ||
| Fig. 5 1H (left) and 13C (right) NMR of the Passerini product P1. | ||
 | ||
| Fig. 6 1H (left) and 13C (right) NMR of the Ugi product U1. | ||
Moreover, the hydrogen of amide moiety, introduced via Passerini and Ugi reactions, was clearly observed at around 6.5 ppm in 1H NMR. 13C NMR provides further confirmation of the completion of the Passerini and Ugi reactions. Peaks at around 170 ppm, corresponding to carboxyl moieties of succinylated filter paper, disappeared and were replaced by peaks belonging to carbonyl groups of the amide groups at around 169 and 168 ppm for Passerini and Ugi products, respectively. The peaks associated with the cellulose backbone are barely observed by 13C NMR after the modification. The general overview of the peaks and spectra can be seen in the Experimental section and ESI,† respectively.
Elemental analysis of the Passerini and Ugi products further proved the purity of the modified cellulose samples, since no residual DMSO was found (i.e. no sulphur content). Furthermore, as expected, the nitrogen content was significantly higher in the Ugi products if compared to the Passerini products (see ESI Table S1†).
Moreover, SEC traces did not reveal any major change in hydrodynamic volume upon further modification of filter paper, resulting in very high molecular weights between 177 and 225 kDa (Table 2, Fig. 3) and thus no significant degradation of the cellulosic backbone. The slight decrease of the molecular weights of the Passerini and Ugi products compared to succinylated filter paper could be ascribed to a different swelling behavior. However, comparison of the SEC data should remain within the Passerini and Ugi products, not with succinylated cellulose, due to the highly different solubility (and thus hydrodynamic volume and swelling behavior) of the samples.
The thermal properties of the Passerini and Ugi products were investigated by DSC and TGA. All Passerini and Ugi products derived from succinylated filter paper showed glass transitions (Tg) between 76–116 °C (Fig. 4). The glass transition temperatures of Ugi products were found at higher temperatures (99–116 °C) than the ones of the Passerini products (76–91 °C), presumably due to the two amide functionalities present in the Ugi products, which lead to additional hydrogen bonding and thus less mobility of the polymeric chains. Interestingly, we showed the possibility to tune the thermal properties of modified cellulose samples by simple variation of the components chosen for the MCRs, which might also lead to thermal processibility. The TGA measurements showed that all the Passerini and Ugi products possessed high thermal stability, with degradation between 263–290 °C, higher than the one of the succinylation cellulose (202 °C) and close to the native cellulose (305 °C) (ESI Fig. S26 and S27†).
Conclusions
In this work, we report a rapid and efficient dissolution and activation of cellulose for the subsequent derivatization with succinic anhydride in a CO2 based switchable solvent under very mild conditions. We demonstrated that the degree of substitutions can be adjusted (1.51 to 2.59) by changing the reactions conditions and the molar ratio of succinic anhydride. The reported solvent system is more efficient, more sustainable, and also cheaper compared to other procedures, since the only chemical to be synthesized is DBU, less toxic and easier to recover. Additionally, this solvent system did not employ any additional catalyst, since DBU is the part of the solvent system and also acted as a catalyst. Additionally, the recovery of DMSO and DBU was successful using a facile work-up. This work thus constitutes a valuable contribution towards a more sustainable polymer industry and shows several advantages compared to previous studies on the succinylation of cellulose, where more expensive solvent systems, additional catalysts (such as DMAP), higher equivalents of succinic anhydride (6 eq. per AGU) were utilized at elevated temperatures (100–120 °C). Finally, introducing thermal transitions (here Tg) to cellulose remains an important research interest. By utilizing the carboxylic acid moiety introduced in the succinylated cellulose, we successfully carried out Passerini 3-CR and Ugi 4-CR post modifications. The procedure presents a straightforward functionalization with a high atom economy leading to cellulose-based materials with adjustable thermal properties. The possibility to tune the properties of the Passerini and Ugi products by simply utilizing various aldehydes, amines and isocyanides via a one pot procedure facilitates the synthesis of a variety of cellulose-based materials that are not accessible via etherification, esterification or other commonly applied methods.Experimental part
Materials
All chemicals were used without further purification, unless otherwise stated. Filter paper was purchased from Whatman™ (filter paper, no. 5), organosolv wood pulp was kindly provided by our colleague, microcrystalline cellulose (MCC, powder), dimethyl sulfoxide (DMSO, anhydrous, ≥99.9%), 1-pentyl isocyanide (97%), butylamine (99.5%), cyclohexyl isocyanide (98%), 2-phenylpropionaldehyde (98%), benzoic acid (ACS reagent, ≥99.5%), 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP, 95%), chromium(III) acetylacetonate, (99.99% trace metals basis) were purchased from Sigma Aldrich. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU, 98%), succinic anhydride (>95) isobutyraldehyde (≥99%), tertbutyl isocyanide (98%) was purchased from TCI Europe N.V. Deuterated chloroform (CDCI3-d), deuterated dimethyl sulfoxide (DMSO-d6), deuterated tetrahydrofuran (THF-d8) was purchased from Merck. CO2 was purchased from air liquid (>99%). Native cellulose samples were dried prior to use (24 h at 100 °C) in order to reduce the amount of water to a minimum. Other solvents, such as hexane, were used in technical grade.General procedure for the solubilization and in situ synthesis of cellulose succinate
Cellulose (0.50 g, 3.08 mmol) was suspended in 12.5 mL of DMSO followed by addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (1.41 g, 9.25 mmol) and allowed to stir for 30 minutes at 50 °C in the presence of CO2 (2–5 bar) to obtain a clear solution. Afterwards, succinic anhydride (1.39 g, 13.8 mmol, 4.5 eq. per AGU) was added to the clear solution of cellulose and stirred for 30 minutes at room temperature. The resulting reaction mixture was poured to vigorously stirring water (200 mL). Then, water was slightly acidified with a diluted (0.05 M) hydrochloric acid solution to obtain white precipitates. The crude product was collected and dissolved in DMSO and reprecipitated in water (200 mL). The white product was further washed with water (200 × 3 mL) to remove DMSO, DBU and unreacted succinic anhydride, completely. The pure white product was dried under vacuum at 70 °C for further analysis. The yield was calculated from DS value. Yield: 86% ATR-IR (cm−1): 2390–3680 mixtures of ν(COOH), ν(O–H), νas(CH2), and νs(CH2), 1703 ν(CO), 1147 ν(C–O) 1H NMR (500 MHz, DMSO-d6) δH ppm: 11.8 (br, 1H), 3.0–5.3 (m, AGU, 7H), 2.46–2.57 (m, 4H). 13C NMR (500 MHz, DMSO-d6) δC ppm: 172.46, 170.77, 101.97, 99.14, 75.27, 72.03, 71.20, 62.22, 28.36, 28.02.
General procedure for the Passerini (3-CR) on succinylated filter paper
Succinylated filter paper (0.1 g, 0.24 mmol, DS: 2.59) was suspended in 2 mL of DMSO and allowed to stir at 50 °C to obtain a clear solution. Afterwards, the (2 eq. with respect to –COOH group, DS: 2.64) corresponding aldehyde (1.26 mmol) and isocyanide (1.26 mmol) were added dropwise to the reaction mixture and allowed to stir for 24 hours at 50 °C. After the completion of the reaction, the resulting mixture was poured to vigorously stirring water (100 mL) to obtain white precipitates. The crude product was collected and dissolved in THF (3 mL) and reprecipitated in water (100 mL). Then, the precipitates were collected and dissolved in THF (3 mL) and reprecipitated in hexane (100 mL). This washing procedure with hexane was repeated two times to completely remove unreacted Passerini components. The pure white product was filtered and dried under vacuum at 50 °C, 24 h for further analysis. Yields were calculated from theoretical yields of respective Passerini products. P1: Yield: 87% ATR-IR (cm−1): 3195–3506 mixtures of ν(N–H) and ν(O–H), 2965–2877 νas(CH2), νs(CH2), 1737 ester ν(CO), 1665 amide ν(CO), 1149 ν(C–O) 1H NMR (500 MHz, THF-d8) δH ppm: 6.44 (br, 1H), 4.74 (m, 1H), 3.30–5.35 (m, AGU, 7H), 2.69 (m, 4H), 2.19 (br, 1H), 1.3 (s, 9H), 0.95 (s, 6H). 13C NMR (500 MHz, THF-d8) δC ppm: 172.72, 168.94, 79.40, 51.71, 31.47, 29.25, 19.31, 17.83. P2: Yield: 92% ATR-IR (cm−1): 3174–3509 mixtures of ν(N–H) and ν(O–H), 2872–2960 νas(CH2), νs(CH2), 1737 ester ν(CO), 1655 amide ν(CO) 1149 ν(C–O) 1H NMR (500 MHz, THF-d8) δH ppm: 7.09 (br, 1H), 4.87 (m, 1H), 3.40–5.20 (m, AGU, 7H), 3.18 (br, 2H), 2.67 (m, 4H), 2.21 (br, 1H), 1.51 (m, 2H), 1.33 (m, 4H), 0.97 (s, 6H), 0.90 (s, 3H). 13C NMR (500 MHz, THF-d8) δC ppm: 172.92, 169.54, 79.40, 39.98, 31.55, 30.17, 23.28, 19.31, 17.83, 14.45.
General procedure for the Ugi (4-CR) on succinylated filter paper
Succinylated filter paper (0.1 g, 0.24 mmol, DS: 2.64) was suspended in 2 mL of DMSO and allowed to stir at 50 °C to obtain a clear solution. Afterwards, the (2 eq. with respect to –COOH group, DS: 2.64) corresponding aldehyde (1.26 mmol), amine (1.26 mmol) and isocyanide (1.26 mmol) were added dropwise to the reaction mixture and allowed to stir for 48 hours at 60 °C. Upon completion of reaction, the mixture was precipitated in vigorously stirring distilled water (100 mL), the obtained precipitate was washed further with distilled water (100 mL) followed by dissolving in THF (3 mL) and reprecipitated in n-hexane (100 mL). This was repeated at least twice to completely remove un-reacted starting materials. The obtained white precipitate was filtered and dried under vacuum at 50 °C, 24 h for further analysis. Yields were calculated from theoretical yields of the respective Ugi products U1: Yield: 83% ATR-IR (cm−1): 3642–3132 mixtures of ν(N–H) and ν(O–H), 2961–2872 νas(CH2), νs(CH2), 1739 ester ν(CO), 1678, 1622 amide ν(CO), 1157 ν(C–O) 1H NMR (500 MHz, THF-d8) δH ppm: 7.14–7.35 (m, 5H), 6.50 (br, 1H), 4.43 (br, 1H), 3.48–3.55 (m, 3H), 3.10–5.42 (m, AGU, 7H), 2.62 (br, 4H), 1.69 (br, 2H), 1.08–1.38 (m, 14H), 0.88–0.99 (br, 3H). 13C NMR (500 MHz, THF-d8) δC ppm: 172.53, 169.42, 168.28, 143.80, 125.96–128.80, 73.30, 50.53, 50.16, 39.07, 31.85, 27.69–28.02, 20.19, 17.62, 13.08. U2: Yield: 92% ATR-IR (cm−1): 3692–3146 mixtures of ν(N–H) and ν(O–H), 2958–2856 νas(CH2), νs(CH2), 1743 ester ν(CO), 1672, 1640 amide ν(CO) 1159 (C–O) 1H NMR (500 MHz, THF-d8) δH ppm: 6.97 (br, 1H), 4.35 (br, 1H),3.65 (br, 1H), 3.38 (br, 2H), 3.02–5.40 (m, AGU, 7H), 2.67 (br, 1H), 2.45 (br, 4H), 1.22–1.31 (m, 6H), 0.92–1.08 (br, 8H), 0.79–0.92 (m, 9H). 13C NMR (500 MHz, THF-d8) δC ppm: 173.03, 169.87, 168.87, 73.78, 48.77, 45.81, 33.52, 33.34, 32.54, 28.96, 27.84, 26.51, 21.01, 20.15, 19.43, 13.93. U3: Yield: 89% ATR-IR (cm−1): 3612–3150 mixtures of ν(N–H) and ν(O–H), 2963–2872 νas(CH2), νs(CH2), 1745 ester ν(CO), 1670, 1642 amide ν(CO), 1162 ν(C–O). 1H NMR (500 MHz, THF-d8) δH ppm: 7.31 (br, 1H), 4.39 (br, 1H), 3.29 (br, 2H), 3.00–5.40 (m, AGU, 7H), 2.63–2.10 (br, 5H), 1.53 (br, 2H), 1.28 (br, 11H), 0.92(br, 6H), 0.82 (br, 3H). 13C NMR (500 MHz, THF-d8) δC ppm: 173.77, 173.02, 170.69, 73.71, 51.76, 50.99, 30.38, 29.89, 28.89, 28.94, 28.13, 27.83, 20.76, 20.27, 19.83.
Recovery of DMSO and DBU
After precipitation of the product in water, the filtrate containing DMSO and DBU was evaporated using the rotary evaporator recovering water. Then, DMSO was distilled using a distillation apparatus (90 °C, 25 mbar). The remaining mixture was transferred to ethyl acetate (50 mL) and washed with water (3 × 10 mL, 6 M, NaOH). The organic phases were collected and dried over NaSO4. Then, the mixture was filtered and ethyl acetate was evaporated using rotary evaporator to obtain DBU (recovery yield: 56%).31P NMR Method for DS determination
Degree of substitutions (DS) were determined by 31P NMR using a Bruker Ascend™ 400 MHz spectrometer with 1024 scans, a delay time d1 of 3 seconds and a spectral width of 90 ppm (190–100 ppm). Samples were prepared according to the following procedure: an exact amount of 25 mg of a sample was weighted and dissolved in 500 μL of CDCI3. Pyridine (150 μl) was added upon complete dissolution. Afterwards, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP, 100 μl, 0.63 mmol) was added to the solution and allowed to stir for 5 minutes a solution of chromium(III) acetylacetonate (50 μL, 50 mM in pyridine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CDCl3/3:2) as relaxation agent and endo-N-hydroxy-5-norbornene-2,3-dicarboximide (125 μL, 123.21 mM in Pyridine:CDCl3/3:2, 0.0154 mmol) as internal standard were added and the solution was stirred for further 5 minutes. Then, 700 μl of the solution was transferred to an NMR tube. DS values and carboxyl moiety conversions of Passerini and Ugi products were calculated according to the reported equation.34 Additionally, the calculations of the carboxyl moieties on Ugi products were performed by taking 31P NMR of the Passerini products as reference, since Passerini products showed full conversions of the carboxyl moieties.
:CDCl3/3:2) as relaxation agent and endo-N-hydroxy-5-norbornene-2,3-dicarboximide (125 μL, 123.21 mM in Pyridine:CDCl3/3:2, 0.0154 mmol) as internal standard were added and the solution was stirred for further 5 minutes. Then, 700 μl of the solution was transferred to an NMR tube. DS values and carboxyl moiety conversions of Passerini and Ugi products were calculated according to the reported equation.34 Additionally, the calculations of the carboxyl moieties on Ugi products were performed by taking 31P NMR of the Passerini products as reference, since Passerini products showed full conversions of the carboxyl moieties.
Instrumentation
000 scans and a time delay d1 of 5 seconds at 50 °C. Data are reported in ppm relative to THF-d8 at 25.37 and 67.57 ppm (for multicomponent products) and DMSO-d6 at 39.51 ppm (for succinylated cellulose samples). All products were dissolved in THF-d8 and DMSO-d6 with the concentrations of 30–50 mg mL−1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Silke Wolf for the TGA measurements. K. N. Onwukamike would like to thank the EU for PhD funding under the Horizon 2020 Marie Curie ITN Project EJD-FunMat (Project ID: 641640). We would also thank to Dr Lena Charlotte Over for providing organosolv wood pulp for these studies.References
- (a) D. E. MacArthur, D. Waughray and M. R. Stuchtey, The New Plastic Economy: Rethinking the future of plastics, World Economic Forum, 2016 Search PubMed; (b) A. Gandini, Green Chem., 2011, 13, 1061 RSC; (c) A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed; (d) R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- (a) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed; (b) R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC; (c) A. Dewaele, L. Meerten, L. Verbelen, S. Eyley, W. Thielemans, P. van Puyvelde, M. Dusselier and B. Sels, ACS Sustainable Chem. Eng., 2016, 4, 5943–5952 CrossRef CAS; (d) Z. Wang, L. Yuan and C. Tang, Acc. Chem. Res., 2017, 50, 1762–1773 CrossRef CAS PubMed; (e) Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- (a) I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed; (b) J. N. Putro, F. E. Soetaredjo, S.-Y. Lin, Y.-H. Ju and S. Ismadji, RSC Adv., 2016, 6, 46834–46852 RSC; (c) Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- M. Rose and R. Palkovits, Macromol. Rapid Commun., 2011, 32, 1299–1311 CrossRef CAS PubMed.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- J. Zhang, W. Chen, Y. Feng, J. Wu, J. Yu, J. He and J. Zhang, Polym. Int., 2015, 64, 963–970 CrossRef CAS.
- H. Hu, J. You, W. Gan, J. Zhou and L. Zhang, Polym. Chem., 2015, 6, 3543–3548 RSC.
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- (a) J. Ding, C. Li, J. Liu, Y. Lu, G. Qin, L. Gan and M. Long, Carbohydr. Polym., 2017, 157, 1785–1793 CrossRef CAS PubMed; (b) T. Heinze and T. Liebert, Prog. Polym. Sci., 2001, 26, 1689–1762 CrossRef CAS.
- H.-P. Fink, P. Weigel, H. Purz and J. Ganster, Prog. Polym. Sci., 2001, 26, 1473–1524 CrossRef CAS.
- G. T. Ciacco, Cellulose, 2003, 10, 125–132 CrossRef CAS.
- C. L. McCormick and T. R. Dawsey, Macromolecules, 1990, 23, 3606–3610 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- P. Mäki-Arvela, I. Anugwom, P. Virtanen, R. Sjöholm and J. P. Mikkola, Ind. Crops Prod., 2010, 32, 175–201 CrossRef.
- M. Gericke, P. Fardim and T. Heinze, Molecules, 2012, 17, 7458–7502 CrossRef PubMed.
- (a) A. Llevot, P.-K. Dannecker, M. von Czapiewski, L. C. Over, Z. Söyler and M. A. R. Meier, Chemistry, 2016, 22, 11510–11521 CrossRef CAS PubMed; (b) A. Llevot and M. A. R. Meier, Green Chem., 2016, 18, 4800–4803 RSC; (c) M. Eissen, J. O. Metzger, E. Schmidt and U. Schneidewind, Angew. Chem., Int. Ed., 2002, 41, 414–436 CrossRef CAS.
- H. Xie, X. Yu, Y. Yang and Z. K. Zhao, Green Chem., 2014, 16, 2422–2427 RSC.
- Q. Zhang, N. S. Oztekin, J. Barrault, K. de Oliveira Vigier and F. Jérôme, ChemSusChem, 2013, 6, 593–596 CrossRef CAS PubMed.
- L. Song, Y. Yang, H. Xie and E. Liu, ChemSusChem, 2015, 8, 3217–3221 CrossRef CAS PubMed.
- Y. Yang, H. Xie and E. Liu, Green Chem., 2014, 16, 3018–3023 RSC.
- (a) G. V. Carrera, N. Jordão, L. C. Branco and M. Nunes da Ponte, J. Supercrit. Fluids, 2015, 105, 151–157 CrossRef CAS; (b) J. Wang, Z. Xue, C. Yan, Z. Li and T. Mu, Phys. Chem. Chem. Phys., 2016, 18, 32772–32779 RSC.
- J. A. Galbis, M. d. G. García-Martín, M. V. de Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed.
- W. Y. Li, A. X. Jin, C. F. Liu, R. C. Sun, A. P. Zhang and J. F. Kennedy, Carbohydr. Polym., 2009, 78, 389–395 CrossRef CAS.
- C. F. Liu, A. P. Zhang, W. Y. Li, F. X. Yue and R. C. Sun, Ind. Crops Prod., 2010, 31, 363–369 CrossRef CAS.
- X. Yin, C. Yu, X. Zhang, J. Yang, Q. Lin, J. Wang and Q. Zhu, Polym. Bull., 2011, 67, 401–412 CrossRef CAS.
- J. Chen, M. Su, X. Zhang, R. Chen, J. Hong, L. Yang and Z. Yang, Cellulose, 2014, 21, 4081–4091 CrossRef CAS.
- (a) C. F. Liu, R. C. Sun, A. P. Zhang, J. L. Ren, X. A. Wang, M. H. Qin, Z. N. Chao and W. Luo, Carbohydr. Res., 2007, 342, 919–926 CrossRef CAS PubMed; (b) C. F. Liu, R. C. Sun, A. P. Zhang, J. L. Ren and Z. C. Geng, Polym. Degrad. Stab., 2006, 91, 3040–3047 CrossRef CAS.
- (a) T. Yoshimura, K. Matsuo and R. Fujioka, J. Appl. Polym. Sci., 2006, 99, 3251–3256 CrossRef CAS; (b) S. Hokkanen, E. Repo and M. Sillanpää, Chem. Eng. J., 2013, 223, 40–47 CrossRef CAS.
- (a) R. Kakuchi, Angew. Chem., Int. Ed., 2014, 53, 46–48 CrossRef CAS PubMed; (b) A. Sehlinger and M. A. R. Meier, in Multi-component and sequential reactions in polymer synthesis, ed. P. Theato, Springer, Cham, 2015, vol. 269, pp. 61–86 Search PubMed.
- A. E. J. de Nooy, D. Capitani, G. Masci and V. Crescenzi, Biomacromolecules, 2000, 1, 259–267 CrossRef CAS PubMed.
- (a) Z. Söyler and M. A. R. Meier, ChemSusChem, 2017, 10, 182–188 CrossRef PubMed; (b) Z. Söyler and M. A. R. Meier, Green Chem., 2017, 19, 3899–3907 RSC.
- (a) I. Soroko, Y. Bhole and A. G. Livingston, Green Chem., 2011, 13, 162–168 RSC; (b) M. Martí, L. Molina, C. Alemán and E. Armelin, ACS Sustainable Chem. Eng., 2013, 1, 1609–1618 CrossRef; (c) W. Ghezali, K. de Oliveira Vigier, R. Kessas and F. Jérôme, Green Chem., 2015, 17, 4459–4464 RSC; (d) N. García, R. Rubio-Presa, P. García-García, M. A. Fernández-Rodríguez, M. R. Pedrosa, F. J. Arnáiz and R. Sanz, Green Chem., 2016, 18, 2335–2340 RSC.
- (a) O. Kreye and M. A. R. Meier, RSC Adv., 2015, 5, 53155–53160 RSC; (b) O. Kreye, L. C. Over, T. Nitsche, R. Z. Lange and M. A. Meier, Tetrahedron, 2015, 71, 293–300 CrossRef CAS.
- A. W. T. King, J. Jalomäki, M. Granström, D. S. Argyropoulos, S. Heikkinen and I. Kilpeläinen, Anal. Methods, 2010, 2, 1499 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc02577g |
| This journal is © The Royal Society of Chemistry 2018 |