 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of heat treatment atmosphere on the structure and activity of Pt3Sn nanoparticle electrocatalysts: a characterisation case study†
Haoliang
Huang
 a,
Abu Bakr Ahmed Amine
Nassr
ab,
Verónica
Celorrio
cde,
S. F. Rebecca
Taylor
f,
Vinod Kumar
Puthiyapura
fg,
Christopher
Hardacre
fg,
Dan J. L.
Brett
h and
Andrea E.
Russell
*a
a,
Abu Bakr Ahmed Amine
Nassr
ab,
Verónica
Celorrio
cde,
S. F. Rebecca
Taylor
f,
Vinod Kumar
Puthiyapura
fg,
Christopher
Hardacre
fg,
Dan J. L.
Brett
h and
Andrea E.
Russell
*a
aChemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: a.e.russell@soton.ac.uk
bElectronic Materials Research Department, Advanced Technology and New Materials Research Institute, City for Scientific Research and Technological Application (SRTA-City), New Borg Al-Arab City, Alexandria, 21934, Egypt
cSchool of Chemistry, University of Bristol, Cantocks Close, Bristol, BS8 1TS, UK
dUK Catalysis Hub, Research Complex at Harwell, RAL, Oxford, OX11 0FA, UK
eDepartment of Chemistry, University College London, Kathleen Lonsdale Building, Gordon Street, London, WC1H 0AJ, UK
fSchool of Chemical Engineering and Analytical Science, The University of Manchester, The Mill, Manchester, M13 9PL, UK
gCentre for the Theory and Application of Catalysis (CenTACat), School of Chemistry and Chemical Engineering, Queen’s University of Belfast, Belfast, BT9 5AG, UK
hDepartment of Chemical Engineering, University College London (UCL), London, WC1E 7JE, UK
First published on 16th April 2018
Abstract
Comprehensive identification of the phases and atomic configurations of bimetallic nanoparticle catalysts are critical in understanding structure–property relationships in catalysis. However, control of the structure, whilst retaining the same composition, is challenging. Here, the same carbon supported Pt3Sn catalyst is annealed under air, Ar and H2 resulting in variation of the extent of alloying of the two components. The atmosphere-induced extent of alloying is characterised using a variety of methods including TEM, XRD, XPS, XANES and EXAFS and is defined as the fraction of Sn present as Sn0 (XPS and XANES) or the ratio of the calculated composition of the bimetallic particle to the nominal composition according to the stoichiometric ratio of the preparation (TEM, XRD and EXAFS). The values obtained depend on the structural method used, but the trend air < Ar < H2 annealed samples is consistent. These results are then used to provide insights regarding the electrocatalytic activity of Pt3Sn catalysts for CO, methanol, ethanol and 1-butanol oxidation and the roles of alloyed Sn and SnO2.
Introduction
PtSn bimetallic nanoparticles (NPs) have attracted considerable attention in heterogeneous and electrocatalyst catalyst research, since the addition of Sn species increases the selectivity of hydrogenation and dehydrogenation for certain molecules1–3 and substantially lowers the activation energy for CO oxidation.4–6 The latter is key to more CO-tolerant catalysts for the anodes of reformate fuelled H2 PEM fuel cells and direct alcohol fuel cells. Fundamental studies have shown that CO oxidation occurs at reduced overpotential at Sn and SnO2 (or its hydrated form) modified Pt7–10 and at Pt3Sn11–14 compared to Pt electrodes. Similarly, Sn modified Pt surfaces and Pt3Sn catalysts have also shown enhanced activity over Pt for methanol,15–17 ethanol18–21 and butanol22 oxidation. In each case the nature of the Sn species, SnO2 or metallic Sn, and the extent of mixing of the Pt and Sn, alloy formation and the resulting Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn ratio at the surface, may play a role in determining the activities of the electrocatalysts. An understanding of these parameters is important in developing structure–property relationships. However, the simultaneous control of composition and structure is difficult for such nanoparticle bimetallic catalysts. One recent approach has been to take a parent PtxSn catalyst with a defined Pt:Sn ratio and then to subject this to heat treatment/annealing under various atmospheres, such as air, to induce surface oxidation or H2(g) to cause reduction.23–26
:Sn ratio at the surface, may play a role in determining the activities of the electrocatalysts. An understanding of these parameters is important in developing structure–property relationships. However, the simultaneous control of composition and structure is difficult for such nanoparticle bimetallic catalysts. One recent approach has been to take a parent PtxSn catalyst with a defined Pt:Sn ratio and then to subject this to heat treatment/annealing under various atmospheres, such as air, to induce surface oxidation or H2(g) to cause reduction.23–26
Structural characterisation of PtxSn catalysts seeks to determine the extent of alloy formation, any variations in lattice parameters, and the extent of surface segregation of either of the components, and to identify the presence or absence of a separate SnO2 phase. Various structural methods such as transmission electron microscopy (TEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) may each be used, but each will have its own limitations and will, therefore, provide a different perspective.
An often-noted limitation of XRD is that only the crystalline components are observed. The SnO2 in PtSn bimetallic catalysts is sometimes found to be “XRD amorphous”. For example, Colmati et al.27 investigated a range of compositions for PtxSn alloy catalysts prepared via formic acid reduction and studied their structures via XRD. The formation of Pt–Sn alloys and extent of alloy formation were suggested by using an expanded Pt lattice parameter, and no tin oxides were found. Similarly, Lim et al.28 prepared PtxSn catalysts over a wider range of atomic ratios using borohydride (a much stronger reductant than formic acid) reduction and subsequent hydrothermal treatment. These PtSn NPs exhibited expanded Pt lattice parameters similar to those measured by Colmati et al., implying a similar extent of alloy formation. However, a set of obvious diffraction peaks from SnO2 were also found by Lim et al. over the entire composition range explored. The “XRD amorphous” SnO2 implied from Colmati’s study is not a unique case in the literature and can also be found in studies of Pt–SnO2 core–shell structures29 and, more commonly, for catalysts with a low Sn content.24,30,31
Kim et al.31 applied XRD, XPS, XANES and EXAFS to investigate the influence of Sn content in carbon supported PtxSn catalysts. Like Colmati’s and Lim’s studies above, the expansion of the Pt lattice from XRD was found to be proportional to the amount of Sn used in the preparation, indicating alloy formation. In contrast, only oxidised Sn was found in the XPS spectra, and the coordination number of the Pt–Sn scattering path from EXAFS fitting did not vary systematically with composition. Such mismatches between techniques were also found in other detailed characterization studies,32,33 including our previous study,21 where we calculated the alloying degree as the proportion of Sn present in the bimetallic particles compared to the nominal composition of a Pt3Sn alloy NP (3–4 nm) to be ∼90% from the lattice parameters according to Vegard’s law,34,35 but XPS and XANES spectra indicated at least 50% of Sn atoms are oxidized.
In this work, the variation in heat treatment atmosphere approach described above is taken to provide a series of related PtSn catalysts with the same nominal composition of Pt3Sn, but with different surface compositions. A variety of structural characterisation methods were then employed to characterise the resulting nanoparticles and the results were compared. The alloy phase and SnO2 were preliminarily characterised via high-resolution transmission electron microscopy (HRTEM) and conventional XRD. XPS, XANES (X-ray absorption near edge structure) and EXAFS (extended X-ray absorption fine structure) spectra then were used to acquire additional information regarding the oxidation state and atomic environment. The CO, methanol, ethanol and 1-butanol oxidation activities of the catalysts were measured in H2SO4 solution. Activity enhancements attributed to alloyed Sn and SnO2 were then discussed.
Experimental section
Pt3Sn nanoparticles were deposited on a carbon support to yield a loading of 20 wt% metals via a conventional polyol method.36,37 Vulcan XC-72R carbon black was functionalized through refluxing in nitric acid.38 200 mg of the functionalized carbon black was dispersed into 50 mL of H2O/ethylene glycol (1:3 v/v) containing 200 mg of sodium hydroxide by 15 min of stirring and sonication. To this mixture, 110 mg of H2PtCl6·6H2O and 16 mg of SnCl2·2H2O were added, followed by stirring for 1 hour and then sonicating for 15 min. The suspension was heated to 160 °C in an oil bath and maintained at this temperature for 2 hours. The mixture was then cooled down to room temperature and 50 mL of isopropanol was added to precipitate nanoparticles. The catalyst was then collected by using a centrifuge, washed with 50% ethanol aqueous solution and dried under vacuum at 80 °C. SEM-EDX (FEI XL30 ESEM equipped with an UltraDry detector) was used to verify the composition, which was found to be Pt2.43Sn, which is close to the nominal ratio of Pt3Sn. The parent Pt3Sn/C catalyst was then divided into four parts and three parts were treated by annealing at 250 °C under either air, Ar or H2 (5%, balanced with N2). The three treated samples are denoted as the air, Ar and H2 samples. Full structural characterisation for the parent Pt3Sn/C catalyst is provided in the ESI.†
A 20 wt% Pt/C reference catalyst, supplied by Johnson Matthey, with an average particle size of ∼2.8 nm (by TEM) was used for comparison.
Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) were performed on an FEI Tecnai 12 at 80 kV and a JEOL JEM 2010 at 200 kV (Bristol University), respectively. Power X-ray diffraction (PXRD) patterns were acquired using a Bruker D2 PHASER diffractometer using Cu Kα radiation (λ = 1.5418 Å) at a voltage of 30 kV and a current of 10 mA.
X-ray photoelectron spectra (XPS) were acquired using a Kratos Axis Ultra Hybrid spectrometer (Queen’s University Belfast). XPS data deconvolution was performed using XPSPEAK 4.1. The background was corrected using the Shirley method39 and the percentage of Gaussian contribution in the peak shape was fixed in the range of 20–30%. XPS spectra were calibrated using the C 1s peak associated with adventitious carbon at 284.8 eV. In the Pt 4f spectra, the spin–orbit spitting energy and the ratio of the peak area between Pt 4f7/2 and Pt 4f5/2 are constrained to be 3.35 eV and 4:3, respectively. For the Sn 3d spectra, the spin orbit splitting energy and the ratio of the peak area between Sn 3d5/2 and Sn 3d3/2 are constrained to be 8.4 eV and 3:2, respectively.
X-ray absorption (XAS) measurements were carried out using the B18 beamline at Diamond Light Source (UK) using the Quick EXAFS mode. The samples for XAS measurements were prepared by grinding and mixing with boron nitride to form a homogeneous mixture, and then being compressed into pellets. XAS spectra of these pellets were measured in transmission mode at both the Pt L3-edge and Sn K-edges in air. Additional spectra were measured in an atmosphere of H2(g) at room temperature for the H2 annealed sample. Pt or Sn foil was measured in the reference channel along with the samples to provide energy calibration. The measured XAS spectra were processed and analysed using the Demeter software package (ATHENA and ARTEMIS).40 The edge step of each spectrum was normalized by subtracting the linear pre-edge and quadratic post edge function, and the energy shift was calibrated by choosing the zero crossing in the second derivative of all spectra to be E0 and then assigning the E0 of reference spectra to the corresponding tabulated value provided by ATHENA. The background of EXAFS was subtracted using the AUTOBK algorithm present in ATHENA. A theoretical model of SnO2 was also used as a standard to guide the background function through the XANES region at the Sn K-edge. Linear combination fitting at the Sn K-edge was also performed using ATHENA with SnO2 and Sn foil as standards.
First-shell fitting of the extended X-ray absorption fine structure (EXAFS) was performed in ARTEMIS. The Pt L3-edge and Sn K-edge spectra of each sample were co-analysed with the half scattering path (R, referred as bond length for a single-scattering path) and mean square relative displacement (σ2) of Pt–Sn and Sn–Pt first single scattering path (RPt–Sn = RSn–Pt, σPt–Sn2 = σSn–Pt2) set to be equal at both edges. Fits were carried out in an R-range of 1.1–3.3 Å at the Pt L3-edge, and 1.1–3.1 Å at the Sn K-edge. Values for amplitude reduction factor were determined to be 0.872 for Pt and 1.034 for Sn extracted from fitting the Pt foil and SnO2 standards, respectively.
The electrochemical measurements were conducted in a standard three-electrode cell connected to a PGSTAT12 potentiostat (Metrohm Autolab). A Pt mesh electrode was used as the counter electrode, and a Hg|Hg2SO4 electrode (MMS) in 1 M H2SO4 was used as the reference electrode and equipped with a Luggin capillary. The potentials reported here are with respect to the reversible hydrogen electrode (RHE) for convenience. The working electrode was prepared by drop casting 10 μL of catalyst ink on the surface of a glassy carbon electrode (5 mm diameter) and drying at room temperature. The catalyst ink was prepared by dispersing the sample powder, 2 mg cm−3, in a mixture of water, isopropyl alcohol and Nafion solution (5 wt%, Sigma Aldrich) with a volume ratio of 8:2:0.01.
All the electrochemical experiments were performed in O2-free (N2 purged) 0.1 mol dm−3 H2SO4 solution at room temperature. The electrochemical active surface area (ECSA) was measured using CO-stripping cyclic voltammetry. Before the measurement, 20 cycles between 0.05 and 0.8 V vs. RHE were performed. High-purity CO (99.9%) was then bubbled through the electrolyte for 20 min while the potential was held at 0.2 V vs. RHE. N2 was then bubbled to remove the free CO in the electrolyte. After that, the potential was swept to 1.0 V vs. RHE to oxidize the absorbed CO and then cycled between 0.05 and 1.0 V vs. RHE to confirm the complete removal of COads. The electrochemical behaviour for alcohol oxidation was evaluated using cyclic voltammetry in 0.1 mol dm−3 of the alcohol (methanol, ethanol, or 1-butanol) + 0.1 mol dm−3 H2SO4 electrolyte. The potential was cycled between 0.1 and 1 V vs. RHE for at least 20 cycles, and the peak currents for the 10th cycle were used to compare the activities.
Results and discussion
The morphologies of the heat treated Pt3Sn catalysts were characterized using TEM (Fig. 1). The images show good dispersion of the nanoparticles on the carbon support, and that the diameters of the nanoparticles show some dependency on the annealing atmosphere: average particle diameters were ∼3 nm for the sample annealed under air, ∼5 nm for Ar and ∼6 nm for H2. The approximate compositions of these nanoparticles were then measured using the spacing of lattice fringes observed in HRTEM images (top inset of Fig. 1). The spacing was interpreted in Fourier-transformed space. The distance from the resulting bright spots to the centre, which is the reciprocal of the spacing, was measured and indexed to possible crystal facets with the help of centrosymmetric rings (bottom inset of Fig. 1). Whilst the (111) plane and the (200) facet (for the air sample) of face-centric cubic (fcc) Pt or the Pt3Sn alloy are observed in all samples, the (110) facet of rutile SnO2 was only found for the Ar and air samples. Pt and the Pt3Sn alloy have similar crystal structures, which cannot be distinguished from each other in HRTEM images.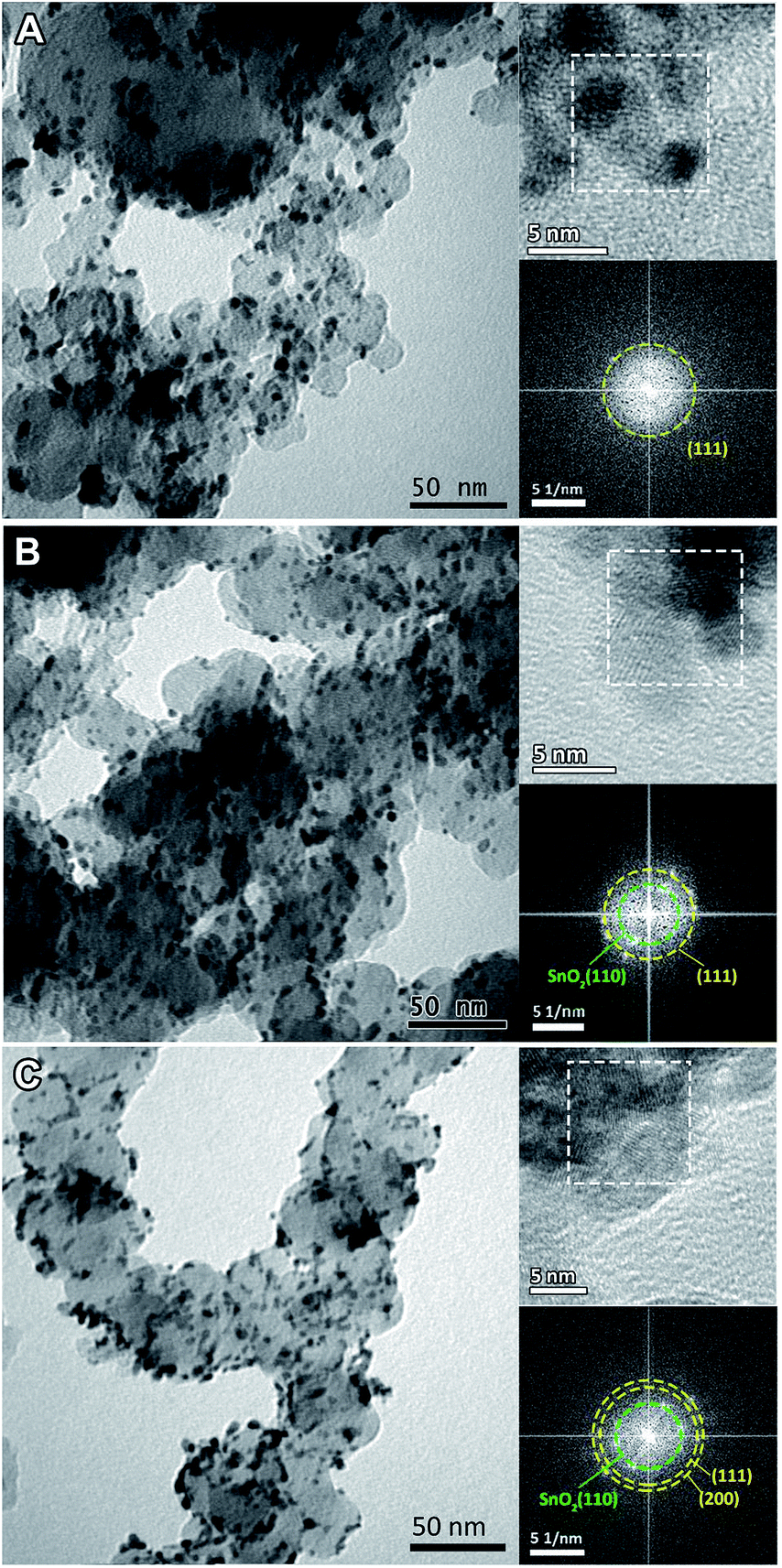 | ||
| Fig. 1 TEM images of Pt3Sn nanoparticles annealed under (A) H2, (B) Ar and (C) air. The upper insets are the corresponding HRTEM images and the bottom insets are the fast-Fourier transform patterns on the selected area (white dashed square) in the corresponding HRTEM images. The dashed rings are drawn to connect bright spots at the same distance to the centre and represent the lattice fringes of the selected areas. | ||
The powder XRD patterns for each of the treated catalysts are shown in Fig. 2. The Pt3Sn catalysts annealed under different atmospheres show features similar to the fcc structure of platinum, with strong diffraction peaks at ∼39°, ∼46°, ∼67° and ∼80°, in agreement with lattice fringes observed in HRTEM images. The XRD pattern of the air annealed sample more closely matches that of Pt standard (JCPDS no. 04-0802) with additional peaks present at ∼29° and ∼34° attributed to SnO2. The latter peak is not present in the XRD pattern for the Ar and H2 annealed samples. The peak positions for the H2 annealed sample more closely match those of the Pt3Sn standard (JCPDS no. 35-1360), with the positions of the peaks for the Ar sample lying between those of the air and H2 annealed samples (inset of Fig. 2). The lattice parameters were calculated using the peak positions of the (220) plane and found to be 3.996 Å for the H2 annealed sample, 3.971 Å for the Ar sample, and 3.951 Å for the air sample.
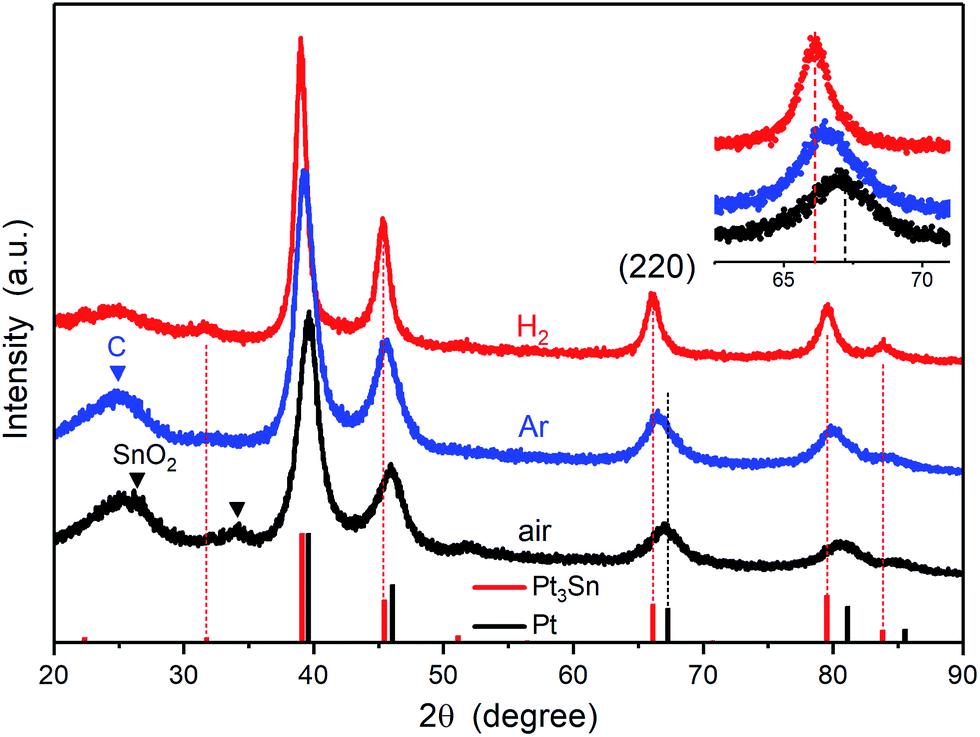 | ||
| Fig. 2 XRD patterns of Pt3Sn nanoparticles annealed under H2, Ar and air. The inset shows the magnified region of the (220) peak. The standard patterns of Pt3Sn (35-1360) and Pt (04-0802) were obtained from the JCPDS database. | ||
Combining the results from HRTEM images and XRD patterns, the existence of a SnO2 phase is still ambiguous and only clearly confirmed for the air annealed sample. The existence of SnO2 in the H2 and Ar annealed samples cannot be excluded, as Sn is relatively dilute (∼3 wt%) and amorphous SnO2 would not be detected in the XRD pattern as previously noted in the introduction.
The presence of SnO2 and speciation of the Sn at the surface of the nanoparticles was further investigated using X-ray photoelectron spectroscopy (XPS). The Sn 3d spectra (Fig. 3) show two peaks corresponding to Sn 3d3/2 and Sn 3d5/2 states. These peaks are deconvoluted into two doublets of Sn0 (green) and SnIV (blue) with some constraints (see experimental section), and a peak at the high energy side of Sn 3d3/2 (red) related to the loss feature of metallic tin. The deconvoluted peaks clearly show that the Sn species present on the surface of the nanoparticles are composed of SnIV and Sn0, with the ratio of the species dependent on the annealing conditions. The ratios of the peak areas for Sn0/SnIV are summarised in Table 1. The large value, 11:1, for the H2 annealed sample is likely to be an overestimate of the Sn0 component as SnII was not included in the deconvolution. The binding energies of SnII and SnIV are very similar and the 3d peak does not discriminate between these two peaks adequately. However, the large ratio obtained does indicate that much less of the Sn is present as a separate SnO2 phase, in agreement with both the XRD and HRTEM results.
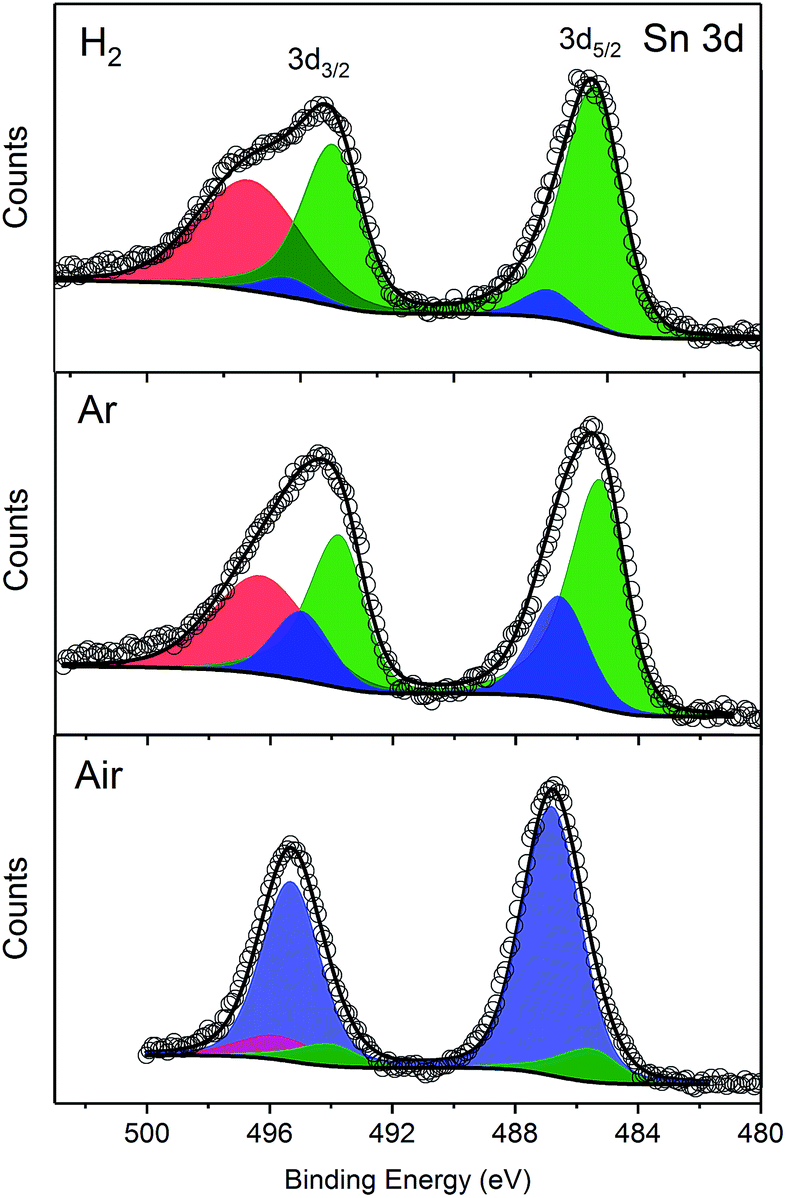 | ||
| Fig. 3 Sn 3d XPS spectra of samples synthesized under H2/N2, Ar or air. Peak deconvolution shown corresponding to Sn0 (green), SnIV (blue), and metallic Sn 3d3/2 (red). | ||
X-ray absorption near edge structure (XANES) spectra obtained at the Sn K-edge also provide speciation of the Sn, but on a per-atom basis. Fig. 4A shows the Sn K-edge spectra of the heat treated samples compared to SnO2 and Sn foil as references. The K-edge represents the transition of Sn 1s electrons to unoccupied 5p orbitals. The different electronic structures of Sn and SnO2 (Sn0: [Kr]5s25p2; Sn4+: [Kr]5s05p0) are evident as shifts in the energy required to allow the K-edge transition or edge position and a change in the intensity of the peak at the edge, which is related to the availability of suitable orbitals. The edge position, defined here as the maximum of the first derivative of XANES spectra (inset of Fig. 4A), for the Ar and air samples is similar to that for SnO2. The derivative of the XANES spectra for the H2 sample shows a broadened maximum, with a shoulder at lower energy. This suggests that more Sn atoms are alloyed with Pt following annealing in H2, in agreement with the Pt3Sn alloy phase detected by using XRD. The intensities of the peak at the edge for the heat treated samples approach that of SnO2 and follow the order, air > Ar > H2.
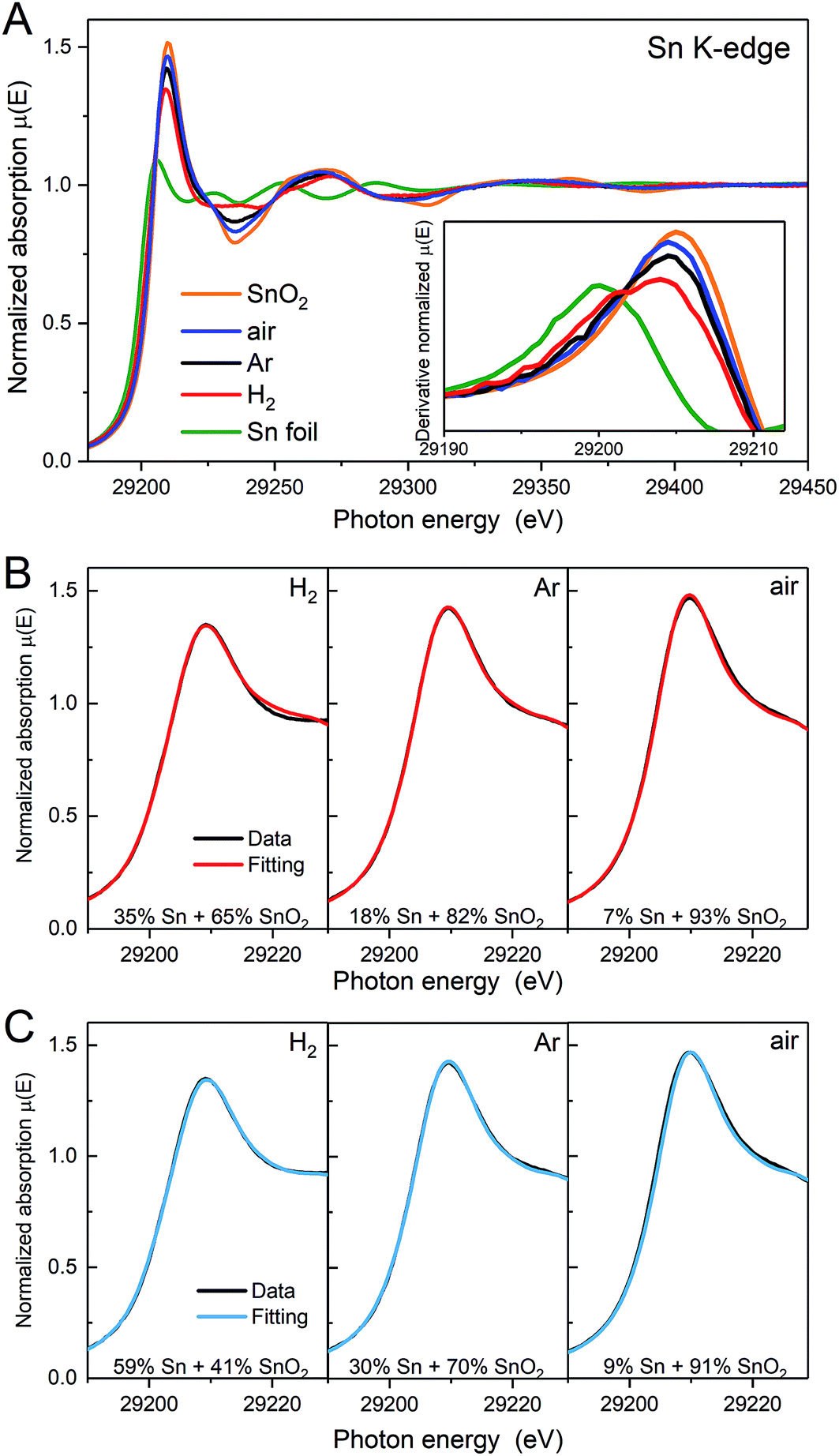 | ||
| Fig. 4 (A) XANES spectra and their first derivative (inset of A) of as-prepared samples and references on the Sn K-edge. (B and C) Linear combination fitting (LCF) of the peak. SnO2 and (B) Sn foil (red line) or (C) the Sn K-edge spectra of the H2 treated sample collected under H2 (cyan line) are used as references for the fittings. The goodness of fit (R-factors) for the fits with Sn foil and Pt3Sn are H2 treated 0.9% and 0.08%, Ar treated 0.1% and 0.2%, air treated 0.5% and 0.6%. | ||
The Sn0/SnIV atomic ratio can also be estimated by XANES linear combination fitting (LCF, Fig. 4B) using the Sn foil and SnO2 samples as the reference spectra and the results are shown in Table 1. Using Sn foil as the Sn0 reference spectrum will provide an under estimate of the Sn0. The electronic structure of Sn in the foil is not the same as in the Pt3Sn alloy as electron transfer from Sn to Pt in the alloy increases the d-band occupancy of Pt (Pt XANES data not shown) and also will decrease the occupancy of the p-orbitals in Sn. To account for these effects, a second estimate was obtained using the XANES spectrum of the H2 treated sample, collected in H2(g) to minimise the contributions of surface oxides, and the results are also shown in Fig. 4C and Table 1. As the Sn K-edge EXAFS data (discussed further below) show the presence of oxygen neighbours, this ratio is likely to be an over estimate of the Sn0.
The EXAFS data at the Pt L3 and Sn K edges provide further characterisation of the average local coordination environments of each atom in the samples. The spectra and corresponding Fourier transforms are shown in Fig. 5 along with the corresponding first shell fits. The spectra were fitted simultaneously at the Sn K-edge and Pt L3-edge using input parameters from theoretical models (rutile tin oxide and Pt3Sn for Sn, platinum oxide and Pt3Sn for Pt). The coordination parameters obtained are shown in (Table 2), all with a good R-factor <2%. Pt–O scattering paths with low coordination numbers are found for all the samples for the spectra collected in air and are attributed to the slight surface oxidation of PtSn bimetallic nanoparticles. Such surface oxidation is confirmed by comparison with the spectra obtained for the H2 annealed sample collected under H2(g) (not shown).
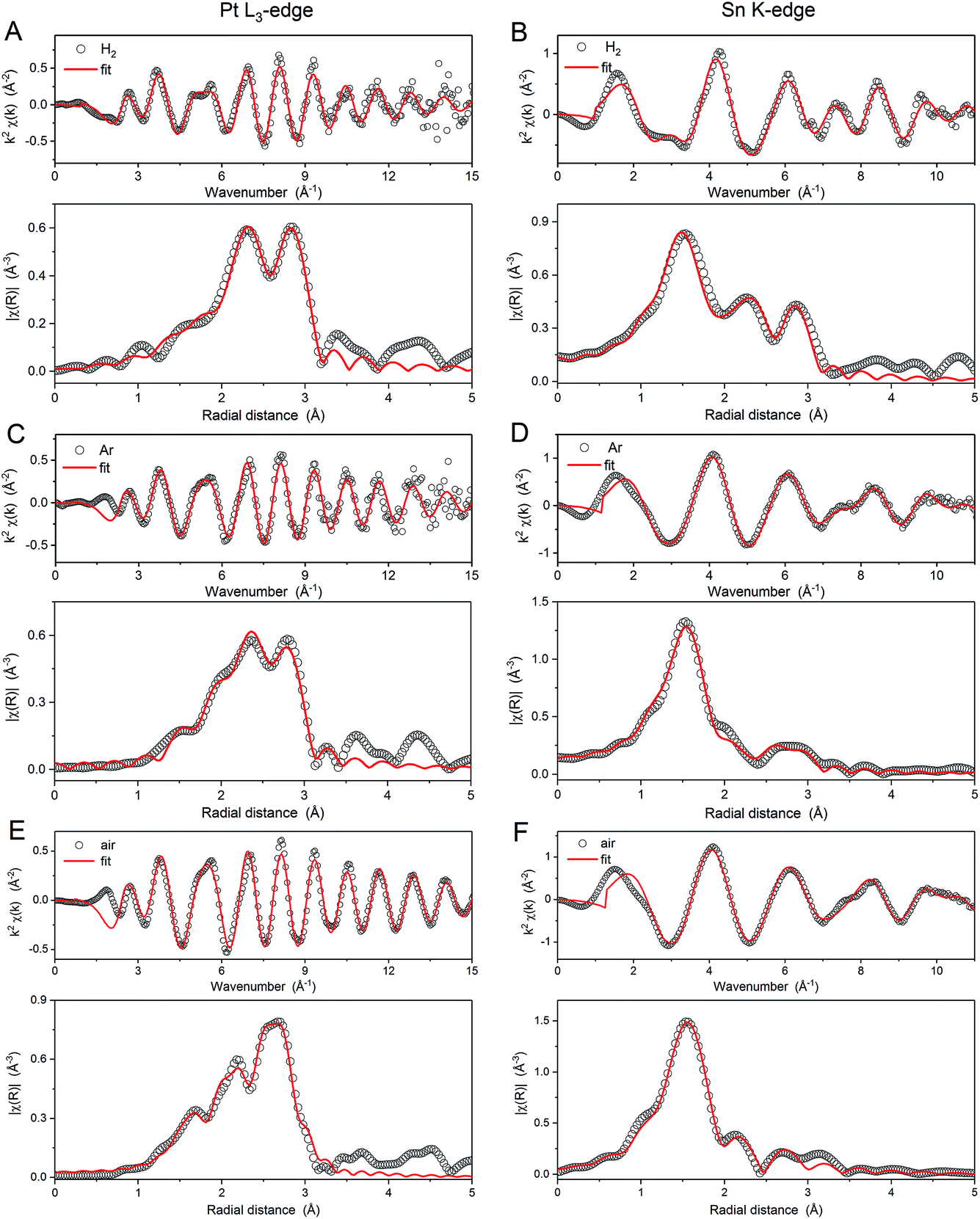 | ||
| Fig. 5 k 2-Weighted EXAFS spectra and their Fourier transforms (not phase corrected) of (A and B) Pt3Sn NPs annealed under H2, (C and D) Ar, and (E and F) air. Spectra at the Pt L3-edge and Sn K-edge are on the left and right column, respectively. The data points and the fittings are plotted as empty dots and red lines, respectively. | ||
| Samples | Shell | N | R (Å) | σ 2 × 102 (Å) | J Pt–Sn (%) | R-factor (%) |
|---|---|---|---|---|---|---|
| a N, the coordination number for the absorber-backscatterer pair; R, the average absorber-backscatterer distance; σ2, the mean square relative displacement. | ||||||
| Pt foil | Pt–Pt | 12.0 | 2.764(1) | 0.50(1) | N. A. | 0.75 |
| H2 collected in H2(g) | Pt–Pt | 7.8(2) | 2.779(2) | 0.68(2) | 60 | 0.56 |
| Pt–Sn | 2.0(2) | 2.788(5) | 1.07(9) | |||
| Sn–O | 1.2(3) | 2.03(1) | 0.6(4) | |||
| Sn–Pt | 8.1(8) | 2.788(5) | 1.07(9) | |||
| H2 | Pt–O | 0.4(4) | 1.99(6) | 0(1) | 55 | 1.19 |
| Pt–Pt | 8(2) | 2.78(1) | 0.9(2) | |||
| Pt–Sn | 1.7(7) | 2.812(9) | 1.0(1) | |||
| Sn–O | 3.0(3) | 2.025(8) | 0.7(2) | |||
| Sn–Pt | 6.9(9) | 2.812(9) | 1.0(1) | |||
| Ar | Pt–O | 0.3(3) | 1.99(6) | 0(1) | 15 | 1.40 |
| Pt–Pt | 8(2) | 2.75(1) | 0.9(2) | |||
| Pt–Sn | 0.4(6) | 2.83(1) | 0.7(2) | |||
| Sn–O | 4.5(3) | 2.061(8) | 0.6(1) | |||
| Sn–Pt | 2.6(7) | 2.83(1) | 0.7(2) | |||
| Air | Pt–O | 0.55(9) | 1.974(9) | 0.3(2) | 9 | 1.11 |
| Pt–Pt | 9.2(6) | 2.739(4) | 0.99(9) | |||
| Pt–Sn | 0.3(1) | 2.856(8) | 0.08(8) | |||
| Sn–O | 5.0(4) | 2.06(1) | 0.5(1) | |||
| Sn–Pt | 0.8(3) | 2.856(8) | 0.08(8) | |||
Comparison of the parameters obtained from the EXAFS fitting provides further insight into the structure of the nanoparticles and the effects of heat treatment conditions. The coordination numbers NPt–Sn, NSn–Pt and NSn–O (where the first element represents the edge and the second the neighbouring atom) and the Pt–Pt bond length (RPt–Pt) are in agreement with the XANES analysis, NSn–O decreases as the fraction of Sn present as SnO2 decreases in the order, air > Ar > H2. Additionally, NPt–Sn and NSn–Pt are largest for the H2 annealed sample, indicating greater mixing of the Pt and Sn in the nanoparticles. These results, consistent with XANES measurements, indicate that heat treatment under H2 promotes the formation of an alloy, but heat treatment under air leads to de-alloying and phase-segregation of SnO2. This conclusion is supported by examination of RPt–Pt, as there is a slight increase from the air sample (2.74 Å) to the Ar sample (2.75 Å), with the longest distance observed for the H2-treated sample (2.78 Å). RPt–Pt is expected to increase from 2.77 Å for metallic Pt (ICSD collection code 52250) to 2.83 Å for Pt3Sn (ICSD collection code 105796). The shorter distances observed for all the heat treated samples, compared to these two reference points, are attributed to effects of the nanoparticle size of the catalysts.41,42
The alloying degree may be quantified from the fits of the EXAFS data by using the alloying parameter (JPt–Sn) proposed by Hwang et al.,43
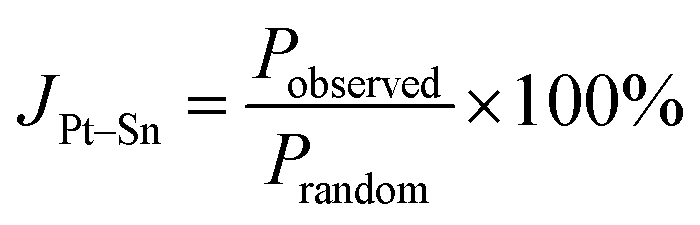
The extent of alloying was then also calculated from the perspective of each of the other various structural characterisation techniques described above. For the XPS and XANES data the extent of alloying is defined as the fraction of Sn present as Sn0, whilst for the XRD and EXAFS data it is obtained using Vegard’s law to estimate the fractional composition of alloyed Sn (xalloyed Sn) first,
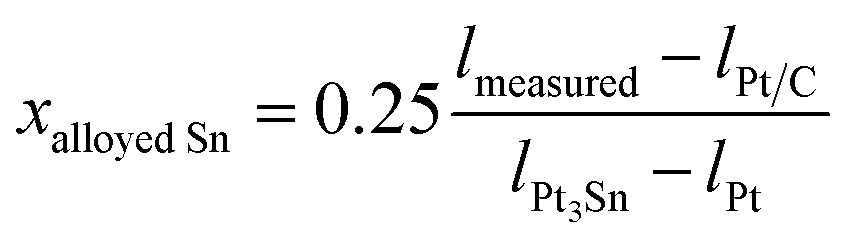
The effects of the extent of alloying on the electrocatalytic activities of the series of heat treated Pt3Sn nanoparticles were then investigated taking the oxidation of a monolayer of CO, which is a known contaminant present in H2(g) produced by reforming hydrocarbons, and methanol, which is the simplest alcohol used as a fuel in direct alcohol fuel cells. The data are compared to those obtained for a 20 wt% Pt/C reference catalyst supplied by Johnson Matthey.
The CO oxidation voltammograms are shown in Fig. 6. Blocking of the Hads sites and additional current associated with oxidation of the adsorbed CO monolayer are observed for all of the catalysts. One measure of the enhanced activity of the bimetallic catalysts is the reduction in the onset potential for CO oxidation. This is defined in the data shown as the crossing of the first cycle (red line) with the second cycle (black line). As shown in Fig. 6, the onset potentials shift dramatically from 0.69 V vs. RHE for Pt/C to 0.39 V vs. RHE for the air annealed sample, 0.36 V vs. RHE for the Ar sample, and 0.35 V vs. RHE for the H2 sample.
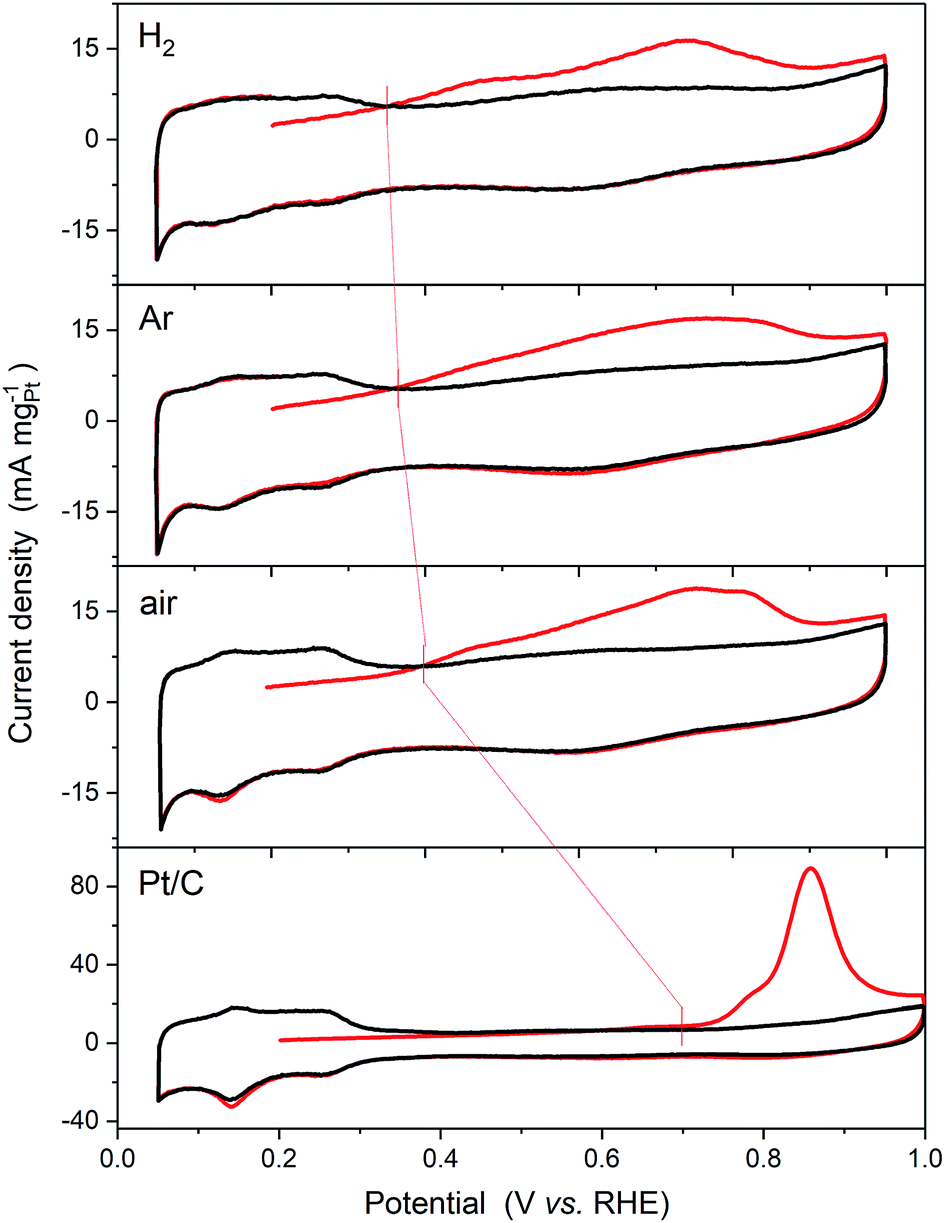 | ||
| Fig. 6 CO stripping voltammograms of the as-prepared PtSn nanoparticles and commercial Pt/C (20 wt%, JM). A monolayer of CO is absorbed at 0.2 V vs. RHE. The first cycles, starting at 0.2 V, are shown in red and the third cycle is shown in black. Measurements were carried out in 1 mol dm−3 H2SO4 at 25 °C with a scan rate of 20 mV s−1. The onset potentials of CO oxidation as defined in the text are marked by red bars. | ||
The CO stripping voltammograms are commonly used to determine the electrochemically accessible surface areas (ECSA) of Pt based electrocatalysts using a charge density of 420 μC cmPt−2.44 Similarly, the ECSA may be calculated from the Hupd peaks using a charge density of 210 μC cmPt−2. The ECSA values obtained using both methods are shown in Table 4 along with the ratio of the two. If the two methods are providing a measurement of the same surface area, the ratio of the two values should be 1 within the error of the measurement, and indeed this is the case for the Pt/C reference catalyst. Significant deviations are observed, however, for all of the PtSn catalysts, and these increase with increasing the extent of alloying, in agreement with H2(g) and CO(g) absorption on Sn surface-enriched Pt–Sn nanoparticles,45 the origins of which are two-fold. Firstly, as shown in Fig. 6, formation of the alloy results in a decrease in the Hupd peaks, which would result in an underestimate of the ECSA. This decrease may be attributed to blocking of the H adsorption sites by Sn at the surface of the nanoparticles. Rizo et al.9 have recently shown that each Sn atom adsorbed on the surface of Pt(111) single crystal electrodes blocks the H adsorption sites for three Pt atoms, suggesting that the Sn adsorbs in three-fold hollow sites on the Pt(111) surface in agreement with previous work.15 Secondly, CO may either adsorb on the Sn sites on the surface of the nanoparticles or not be blocked at the Pt sites by the formation of the alloy as found for H adsorption. DFT calculations by Zheng et al.46 strongly support the latter, as adsorbed CO was not found to be stable at any of the adsorption sites involving Sn atoms. The smaller ECSA-CO values obtained for all of the PtSn catalysts compared to the Pt/C reference catalyst are attributed to a combination of the larger average size of the nanoparticles and the proportional decrease in the Pt sites on the surface of the catalyst nanoparticles with increased incorporation of Sn.
| Samples | ECSA-CO (m2 gPt−1) | ECSA-H (m2 gPt−1) | ECSA-CO/ECSA-H |
|---|---|---|---|
| H2 | 25.1 | 7.4 | 3.4 |
| Ar | 26.1 | 8.4 | 3.1 |
| Air | 29.5 | 12.8 | 2.3 |
| Pt/C | 67.2 | 60.7 | 1.1 |
The broad CO oxidation peaks observed on all the Pt3Sn catalysts may be compared to the recent study by Rizo et al.,9 of Sn modified Pt(111) surfaces, who reported two distinct CO stripping peaks, a pre-peak below 0.5 V vs. RHE and a larger main peak at approximately 0.7 V. They attributed the pre-peak to the oxidation of CO adsorbed on Pt with adjacent Sn sites at which water is activated, whilst that at higher potentials to the promotion of CO oxidation by OH adsorbed on Sn that are not in direct contact with Pt. Closer examination of the CVs in Fig. 6 show a more distinct pre-peak between 0.4 and 0.5 V for the H2 annealed sample. As this sample has the greatest extent of alloying there should be a greater number of Sn sites at which water can be activated adjacent to Pt sites on the surface of the nanoparticles.
The methanol oxidation activity is similarly evaluated using cyclic voltammetry in 0.1 M methanol with 0.1 M sulfuric acid as the electrolyte. The current densities have been normalised to the equivalent area of Pt using the ECSA-CO values in Table 4 in Fig. 7A and the mass of Pt in Fig. 7B, representing the specific and Pt mass activities of the catalysts, respectively. The cyclic voltammograms of the heat treated Pt3Sn NPs share the same features as Pt/C, with oxidation peaks appearing in both the forward sweep and the reverse sweep. Across the potential range of these peaks, the Pt3Sn NPs, particularly the H2 sample, show larger specific activities (Fig. 7A) than that for Pt/C. The specific activity enhancement would appear to be even greater if the ECSA-H values had been used, but we suggest that such a comparison is inappropriate, as H adsorption is unlikely to be a good model of the methanol adsorption sites. In contrast, when the Pt mass activities are compared (Fig. 7B), only the H2 annealed sample shows enhanced activity.
 | ||
| Fig. 7 Cyclic voltammograms illustrating the specific activities (A, C and E) and mass activities (B, D and E) of the heat treated Pt3Sn catalysts and the commercial Pt/C reference catalyst in 0.1 mol dm−3 H2SO4 and 0.1 mol dm−3 (A and B) methanol, (C and D) ethanol, and (E and F) 1-butanol at 25 °C with a scan rate of 50 mV s−1. | ||
The ethanol and butanol specific and mass activities of the Pt3Sn catalysts are also shown in Fig. 7. The specific activities of all of the catalysts, assessed by comparison of the maximum current density, for ethanol oxidation are approximately half and those for 1-butanol oxidation are approximately one quarter of those observed for methanol oxidation. Only the H2 annealed catalyst shows enhanced specific activity for ethanol and 1-butanol oxidation compared to that for the Pt/C reference catalyst. When the mass activities are compared, none of the Pt3Sn catalysts show enhanced current densities at the maximum.
The relative activities of the catalysts for oxidation of the three alcohols may also be assessed by comparison of the onset potential, which is the potential at which the current density starts to increase significantly, with the more active catalyst having the lowest onset potential. This is more difficult to define for the alcohol oxidation CVs than for the case of CO shown above and should only be used to provide a ranking of catalyst activity. No improvement in onset potential with alloy formation is observed for methanol oxidation. A slight improvement, by approximately −0.1 V, is seen for the air annealed catalyst for ethanol oxidation, and all of the Pt3Sn catalysts show a similar −0.1 V improvement for 1-butanol oxidation.
Enhancements of the activity of bimetallic Pt-based catalysts for CO oxidation have long been attributed to a bifunctional effect in which OH adsorbed on Sn provides active O species which facilitate the oxidation of CO adsorbed on Pt sites.47 The role of this mechanism in the oxidation of small organic molecules was reviewed 30 years ago by Parsons and Van der Noot48 and more recently for CO and methanol oxidation on Pt–Sn catalysts by Antolini and Gonzalez.49 The structural characterisation data presented above indicates that a proportion of the Sn is present as SnO2, with SnO2 present as a separate phase for the air annealed catalyst. The adsorption of CO and alcohols requires available Pt sites on the surface of the catalyst nanoparticles, whilst the removal of CO and any partial oxidation products of the alcohols requires nearby Sn sites.
For CO oxidation, the lowest onset potential was obtained for the H2 annealed sample, which has the greatest extent of alloying of the three. As discussed above and in agreement with Rizo et al.9 this result suggests that the onset of the reaction is determined by the availability of Sn atoms at which water is activated, i.e. those not present as a separate SnO2 phase, adjacent to Pt atoms on which CO is adsorbed.
For methanol oxidation, no variation in the onset potential was observed, although the H2 annealed sample exhibited the largest specific activity. This result is in agreement with those of Haner and Ross15 for methanol oxidation, who showed enhanced current density at Pt3Sn(111) compared to Pt(111), but no shift in the onset or peak potentials, and in contrast to the conclusions of the review by Antolini and Gonzalez, who found that the best methanol oxidation catalyst was a mixture of Pt and SnO2, suggesting that the air annealed sample should have been best. The latter has been interpreted as the need to provide Pt sites for methanol adsorption, whilst minimising facilitating CO oxidation by the availability of active Sn sites. Our results suggest that the active Sn sites need to be associated with the Pt sites of the nanoparticles and are not easily accessible if present as a separate SnO2 phase.
For ethanol oxidation, the air annealed catalyst exhibited the lowest onset potential, whilst the H2 annealed sample exhibits the largest specific activity. These results are in good agreement with the previously reported work by Jiang et al.50 who show that the less well alloyed Pt3Sn catalyst is more active at low current densities or potentials and Godoi et al.24 who show that the oxidation current increases with alloying. This suggests that, as in the case of methanol, a balance must be struck between availability of the adsorption site, defining the onset potential, and electronic or bifunctional effects that are important to sustain higher current densities.
Finally, for 1-butanol oxidation, all of the Pt3Sn catalysts exhibited similar lower onset potentials compared to the Pt/C reference catalyst, suggesting that the availability of either adjacent Sn sites at which water activation takes place or adsorbed OH on non-adjacent Sn sites facilitates the oxidation. The observation that only the H2 annealed Pt3Sn catalyst exhibits an increase in the specific activity, suggests that the former is more important in determining 1-butanol oxidation activity.
Conclusions
The annealing of carbon supported Pt3Sn nanoparticles under air, Ar and H2 atmospheres has provided a means of varying the extent of alloying without changing the overall composition of the catalyst. The structures of the nanoparticles were then characterised using TEM, XRD, XPS, XANES and EXAFS at both the Pt L3 and Sn K edges. The extent of alloying was characterised using each method and, as shown in Table 3, the values obtained are technique dependent. TEM and XRD are biased towards the more crystalline components of the samples, XPS is biased towards the surface composition, and the XANES and EXAFS provide per-atom averages. However, the trend is the same for each of the methods with the extent of alloying increasing as air < Ar < H2 annealed, in agreement with previously published studies,23,24,50 although none of these previous studies provide as detailed characterisation as reported in this study.The activity enhancements, compared to the activity of the 20 wt% Pt/C reference catalyst, observed for CO, methanol, ethanol and 1-butanol oxidation can be understood in terms of the relative effects of active Sn atoms that are close to (alloyed with) the Pt atoms on the surface of the catalyst nanoparticles and other Sn species such as SnO2. For CO oxidation, the lowest onset potential was observed for the H2 annealed catalyst, which is also the most well alloyed catalyst, with the smallest fraction of Sn present as SnO2, supporting the view that water activation at Sn sites provides the enhancement. For methanol oxidation, no shift in the onset potential was observed with variation in the extent of alloying, but the enhanced specific activity of the H2 annealed catalyst once again suggests that the mechanism of enhancement is similar to that for CO oxidation. For ethanol oxidation, the onset potential was lowest for the air annealed catalyst, which has more Pt rich nanoparticles as more of the Sn is present as SnO2, but the largest specific activity was obtained for the well-alloyed H2 annealed sample. These conflicting results indicate that a balance must be struck between the availability of Pt sites (onset) and active Sn atoms to assist in more complete oxidation. In contrast, for 1-butanol oxidation, the onset potentials of all the heat treated Pt3Sn catalysts were similar, with only the H2 annealed sample exhibiting any significant increase in specific activity. This result suggests that, whilst both types of Sn sites enhance activity, as observed for CO, methanol and ethanol, water activation at Sn atoms adjacent to the Pt atoms on the surface of the catalyst nanoparticles is the dominant enhancement mechanism.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC (Portfolio Grants EP/K014706/2, EP/K014668/1, EP/K014854/1, EP/K014714/1, and EP/I019693/1). Open access data can be found via the University of Southampton research portal. HH acknowledges Fellowship support from the China Scholarship Council (201608440295). TEM studies were carried out at the University of Bristol Chemistry Imaging Facility with equipment funded by UoB and EPSRC (EP/K035746/1 and EP/M028216/1). XAS data were collected at Diamond on B18 as part of the UK Catalysis Hub BAG allocation (SP10306). David Inwood (Southampton) is thanked for assistance in fitting the EXAFS data.References
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef PubMed.
- F. Humblot, J. P. Candy, F. Le Peltier, B. Didillon and J. M. Basset, J. Catal., 1998, 179, 459–468 CrossRef.
- J. Shi, M. Zhang, W. Du, W. Ning and Z. Hou, Catal. Sci. Technol., 2015, 5, 3108–3112 Search PubMed.
- W. D. Michalak, J. M. Krier, S. Alayoglu, J.-Y. Shin, K. An, K. Komvopoulos, Z. Liu and G. A. Somorjai, J. Catal., 2014, 312, 17–25 CrossRef.
- M. Vandichel, A. Moscu and H. Grönbeck, ACS Catal., 2017, 7, 7431–7441 CrossRef.
- C. Dupont, Y. Jugnet and D. Loffreda, J. Am. Chem. Soc., 2006, 128, 9129–9136 CrossRef PubMed.
- W.-P. Zhou, W. An, D. Su, R. Palomino, P. Liu, M. G. White and R. R. Adzic, J. Phys. Chem. Lett., 2012, 3, 3286–3290 CrossRef.
- M. B. de Oliveira, L. P. R. Profeti and P. Olivi, Electrochem. Commun., 2005, 7, 703–709 CrossRef.
- R. Rizo, E. Pastor and M. T. M. Koper, J. Electroanal. Chem., 2017, 800, 32–38 CrossRef.
- B. E. Hayden, M. E. Rendall and O. South, J. Mol. Catal. A: Chem., 2005, 228, 55–65 CrossRef.
- K. Wang, H. A. Gasteiger, N. M. Markovic and P. N. Ross, Electrochim. Acta, 1996, 41, 2587–2593 CrossRef.
- B. E. Hayden, M. E. Rendall and O. South, J. Am. Chem. Soc., 2003, 125, 7738–7742 CrossRef PubMed.
- V. R. Stamenkovic, M. Arenz, C. A. Lucas, M. E. Gallagher, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2003, 125, 2736–2745 CrossRef PubMed.
- V. Stamenkovic, M. Arenz, B. B. Blizanac, K. J. J. Mayrhofer, P. N. Ross and N. M. Markovic, Surf. Sci., 2005, 576, 145–157 CrossRef.
- A. N. Haner and P. N. Ross, J. Phys. Chem., 1991, 95, 3740–3746 CrossRef.
- G. Stalnionis, L. Tamašauskaitė-Tamašiūnaitė, V. Pautienienė and Z. Jusys, J. Solid State Electrochem., 2004, 8, 900–907 CrossRef.
- X. Lu, Z. Deng, C. Guo, W. Wang, S. Wei, S. P. Ng, X. Chen, N. Ding, W. Guo and C. M. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 12194–12204 Search PubMed.
- H. Wang, Z. Jusys and R. J. Behm, J. Power Sources, 2006, 154, 351–359 CrossRef.
- T. Herranz, M. Ibáñez, J. L. Gómez de la Fuente, F. J. Pérez-Alonso, M. A. Peña, A. Cabot and S. Rojas, ChemElectroChem, 2014, 1, 885–895 CrossRef.
- A. Velázquez-Palenzuela, F. Centellas, E. Brillas, J. A. Garrido, C. Arias, R. M. Rodríguez and P.-L. Cabot, Int. J. Hydrogen Energy, 2013, 38, 16418–16426 CrossRef.
- L. Calvillo, L. Mendez De Leo, S. J. Thompson, S. W. T. Price, E. J. Calvo and A. E. Russell, J. Electroanal. Chem., 2018, 819, 136–144 CrossRef.
- V. K. Puthiyapura, D. J. Brett, A. E. Russell, W. F. Lin and C. Hardacre, ACS Appl. Mater. Interfaces, 2016, 8, 12859–12870 Search PubMed.
- G. Wang, T. Takeguchi, T. Yamanaka, E. N. Muhamad, M. Mastuda and W. Ueda, Appl. Catal., B, 2010, 98, 86–93 CrossRef.
- D. R. M. Godoi, J. Perez and H. M. Villullas, J. Power Sources, 2010, 195, 3394–3401 CrossRef.
- Y. Uemura, Y. Inada, K. K. Bando, T. Sasaki, N. Kamiuchi, K. Eguchi, A. Yagishita, M. Nomura, M. Tada and Y. Iwasawa, J. Phys. Chem. C, 2011, 115, 5823–5833 Search PubMed.
- Y. Uemura, Y. Inada, K. K. Bando, T. Sasaki, N. Kamiuchi, K. Eguchi, A. Yagishita, M. Nomura, M. Tada and Y. Iwasawa, Phys. Chem. Chem. Phys., 2011, 13, 15833–15844 RSC.
- F. Colmati, E. Antolini and E. R. Gonzalez, Electrochim. Acta, 2005, 50, 5496–5503 CrossRef.
- D.-H. Lim, D.-H. Choi, W.-D. Lee and H.-I. Lee, Appl. Catal., B, 2009, 89, 484–493 CrossRef.
- W. Du, G. Yang, E. Wong, N. A. Deskins, A. I. Frenkel, D. Su and X. Teng, J. Am. Chem. Soc., 2014, 136, 10862–10865 CrossRef PubMed.
- D.-H. Kwak, Y.-W. Lee, S.-B. Han, E.-T. Hwang, H.-C. Park, M.-C. Kim and K.-W. Park, J. Power Sources, 2015, 275, 557–562 CrossRef.
- J. H. Kim, S. M. Choi, S. H. Nam, M. H. Seo, S. H. Choi and W. B. Kim, Appl. Catal., B, 2008, 82, 89–102 CrossRef.
- M. Zhu, G. Sun and Q. Xin, Electrochim. Acta, 2009, 54, 1511–1518 CrossRef.
- H. Zhu, D. H. Anjum, Q. Wang, E. Abou-Hamad, L. Emsley, H. Dong, P. Laveille, L. Li, A. K. Samal and J.-M. Basset, J. Catal., 2014, 320, 52–62 CrossRef.
- L. Vegard, Z. Phys., 1921, 5, 17–26 CrossRef.
- L. Vegard, Z. für Kristallogr.–Cryst. Mater., 1928, 67, 239–259 Search PubMed.
- B. Liu, Z. W. Chia, Z. Y. Lee, C. H. Cheng, J. Y. Lee and Z. L. Liu, Fuel Cells, 2012, 12, 670–676 CrossRef.
- C. Bock, C. Paquet, M. Couillard, G. A. Botton and B. R. MacDougall, J. Am. Chem. Soc., 2004, 126, 8028–8037 CrossRef PubMed.
- A. B. A. A. Nassr, I. Sinev, W. Grünert and M. Bron, Appl. Catal., B, 2013, 142–143, 849–860 CrossRef.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef PubMed.
- J. T. Miller, A. J. Kropf, Y. Zha, J. R. Regalbuto, L. Delannoy, C. Louis, E. Bus and J. A. van Bokhoven, J. Catal., 2006, 240, 222–234 CrossRef.
- J. R. Gallagher, T. Li, H. Zhao, J. Liu, Y. Lei, X. Zhang, Y. Ren, J. W. Elam, R. J. Meyer, R. E. Winans and J. T. Miller, Catal. Sci. Technol., 2014, 4, 3053–3063 Search PubMed.
- B. J. Hwang, L. S. Sarma, J. M. Chen, C. H. Chen, S. C. Shih, G. R. Wang, D. G. Liu, J. F. Lee and M. T. Tang, J. Am. Chem. Soc., 2005, 127, 11140–11145 CrossRef PubMed.
- Q.-S. Chen, J. Solla-Gullón, S.-G. Sun and J. M. Feliu, Electrochim. Acta, 2010, 55, 7982–7994 CrossRef.
- T. Inoue, K. Tomishige and Y. Iwasawa, J. Chem. Soc., Faraday Trans., 1996, 92, 461–467 RSC.
- J. Zheng, M. Busch, L. Artiglia, T. Skála, J. Rossmeisl and S. Agnoli, J. Phys. Chem. C, 2016, 120, 25306–25316 Search PubMed.
- M. Watanabe, M. Shibata and S. Motoo, J. Electroanal. Chem. Interfacial Electrochem., 1985, 187, 161–174 CrossRef.
- R. Parsons and T. Van der Noot, J. Electroanal. Chem., 1988, 257, 9–45 CrossRef.
- E. Antolini and E. R. Gonzalez, Electrochim. Acta, 2010, 56, 1–14 CrossRef.
- L. Jiang, Z. Zhou, W. Li, W. Zhou, S. Song, H. Li, G. Sun and Q. Xin, Energy Fuels, 2004, 18, 866–871 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7fd00221a |
| This journal is © The Royal Society of Chemistry 2018 |