
Electrochemical deposition for the separation and recovery of metals using carbon nanotube-enabled filters†
Megan P.
O'Connor
 a,
Riley M.
Coulthard
b and
Desiree L.
Plata
*b
a,
Riley M.
Coulthard
b and
Desiree L.
Plata
*b
aDepartment of Civil and Environmental Engineering, Duke University, Durham, North Carolina 27708, USA
bDepartment of Chemical and Environmental Engineering, Yale University, New Haven, Connecticut 06520, USA. E-mail: desiree.plata@yale.edu
First published on 26th October 2017
Abstract
Rare earth and specialty elements (RESE) are functionally integral to several clean energy technologies, but there is no domestic source of virgin RESE in the United States. Manufacturing waste streams, which are relatively simple compositionally, and electronic wastes, which are chemically complex, could both serve as viable sources of secondary RESE if efficient methods existed to recover and separate these metals for reuse. Leveraging differences in RESE reduction potentials, high surface area, high conductivity carbon nanotubes (CNTs) could enable space- and solvent-efficient, selective recovery of RESE from mixed metal wastes. In this study, unaligned CNTs encapsulated in polyvinyl alcohol were used to develop an electrochemically active filter and tested for recovery of six metals or metalloids (Cu, As, Eu, Nd, Ga, and Sc) as a function of flow rate (1–5 mL min−1), pH (2–10), and voltage (0.1–3.0 V), with maximum recoveries of 86–96%, except for As, which was unretained. All metals were recovered as oxides, rather than their zero valent or reduced forms (except for Cu, which was partially reduced at low pHs). Deaeration experiments suggested electrochemical reduction of dissolved O2 and O2 derived from water splitting were jointly responsible for metal capture, where metal oxides were first formed via metal hydroxide intermediates, and this mechanism was enhanced at higher pHs. A synthetic, multi-metal waste stream of Cu and Eu was successfully separated on multiple stages with increasing voltages (97 ± 0.1% Cu and 65 ± 0.3% Eu recovery), indicating the approach might be useful for the treatment of electronic end-of-life and manufacturing derived wastes.
Water impactCarbon nanotube-enabled filters were used to electrochemically recover Cu, Eu, Nd, Sc, and Ga based on differences in reduction potential and metal oxide stabilities. The device could treat industrial, mixed metal waste streams, ultimately providing a secondary source of material for manufacturing, mitigating the need for mining primary ores, a process with substantial water use and environmental impacts. |
Introduction
The Intergovernmental Panel on Climate Change (IPCC) Fifth Assessment Report (AR5) recommended a shift away from carbon-based fuels to renewable energy sources in order to diminish anthropogenic CO2 emissions, which reached a record high of 49 ± 4.5 GtCO2 eq (in 2010).1 Resoundingly, this transition to renewable energy will rely on technologies that require rare earth and specialty elements (RESE; e.g., in permanent magnets, batteries, and catalysts for the wind, solar, and automotive industries).2 However, the U.S. Department of Energy (DOE) and European Commission have labeled some RESE as “critical” (i.e., there is a large demand for and insufficient supply of the metal).3The criticality designation is estimated based on the metal's supply risk coupled with its importance to the technical objective (e.g., the clean energy sector),3 considering demand, possible substitutions, competing technologies, and political, social, and regulatory factors. The critical status of many RESE stems from recent use of and increasing reliance on them in developing technologies and an emergent bottleneck effect, where the primary supply of these metals can no longer support demand.4 (In addition, the only domestic supply of RESE was recently reduced when Molycorp, Inc. announced closure in August 2015, promoting a transition to Estonian and Chinese source materials).5 Without intervention, the supply chain imbalance and market instabilities associated with sole or few sources could be exacerbated as clean energy technologies continue to grow.6 In order to create a secondary supply of these metals, a novel materials management strategy and supporting technologies will be needed,2 where there is enhanced reuse and recycling at the end-of-life and manufacturing stages (i.e., “closing the loop” from end-of-life to manufacturing and within the manufacturing stage itself to reduce material losses during production). Such systematic changes will not only secure the supply of metals for the requisite technologies, but also have the ancillary benefit of further reducing GHG emissions by reducing mining and refining of primary metal.
While there are no standard recovery technologies (e.g., commercialized and readily integrated onto an assembly line) available to manufacturers seeking to remove valuable materials from their own waste streams, end-of-life waste management strategies currently recycle 20% of global municipal solid waste and less than 1% of RESE (circa 2010 and 2008, respectively).1,7 These low recycling rates result from many interrelated factors including consumer behavior, government policy, and lack of infrastructure; these have been reviewed elsewhere.8–10 In addition, there are few recycling technologies able to reclaim the critical materials or separate the metals from one another for reuse.3,7 Present solid waste recycling techniques include: disassembly and targeting of valuable and hazardous components, upgrading the valuable components via magnetic and mechanical sorting processes, and finally, refining. Refining is used to recover the materials of interest, typically via pyrometallurgical and hydrometallurgical processes, which consist of either melting or dissolving the waste.11 Both hydro- and pyrometallurgical approaches work well for bulk metals (e.g., Cu, Ni, and Zn, all with recycling rates above 50%),7 but neither can capture all RESE.11
Following acid digestions, several recovery strategies for bulk metals are possible, including electrowinning, which involves applying a voltage to a cathodic material in a large tank of metal-rich waste, where the metal of interest is reduced out of solution onto the cathode surface. The cathode is generally made out of large stainless steel or aluminum sheets and the anode is an inert metal, such as titanium or lead. This electrodeposition technique yields high recovery for bulk metals (e.g., Cu, Ni, and Co11,12) but suffers high space requirements due to the large surface area of smooth cathodic material needed. This limitation makes the recycling method impractical for the low concentrations of RESE metals found in many waste streams and smaller processes important for clean energy and nanotechnologies (i.e., advanced metal-deposition techniques such as e-beam sputtering, lithography, and printing in semiconductors).2
High surface area, conductive materials could enable adaption of the basic principles of electrowinning to a broader range of elements and at lower area and volume requirements. Recently, carbon nanotube (CNT) membranes and filters with high surface area-to-mass ratios and excellent conductivity have been developed for water treatment technologies. These research endeavors are well documented, with significant efforts in anti-fouling membranes,13 virus inactivation,14 and the oxidative treatment of organic matter,15,16 but few studies explore metal recovery on CNT-enabled filters.17,18
Here, we seek to utilize the concept of electrochemical precipitation and the exceptional properties of conductive CNT filters as a cathode material to exploit differences in reduction potentials and/or solubility to recover metals of interest.16 In particular, we explore the possibility of selective recovery and separation of metals out of mixed metal streams using case studies of six metals/metalloids, each representative of different industrial materials in clean energy applications, including copper (Cu), europium (Eu), neodymium (Nd), gallium (Ga), scandium (Sc), and arsenic (As). Broadly, this work aims to develop a new method to reclaim and separate valuable materials from industrial waste streams (e.g., e-waste and manufacturing process waters), facilitating closed loop manufacturing and decreasing losses from the material supply chain at a product's end-of-life.
Experimental methods
Materials
Copper chloride (CuCl2; 99.999% trace metals basis (TMB)), europium chloride (EuCl3; 99.99% TMB), scandium chloride (ScCl3; 99.99% TMB), neodymium chloride (NdCl3; ≥99.99% TMB), gallium chloride (GaCl3; ≥99.99% TMB), arsenic chloride (AsCl3; 99.99% TMB), sodium hydroxide (NaOH; 99.99% TMB), hydrochloric acid (HCl; TraceSELECT®), and sodium chloride (NaCl; ≥99%) were all purchased from Sigma-Aldrich (St. Louis, MO). Multi-walled carbon nanotube buckypaper filters encapsulated in polyvinyl alcohol were custom-made by NanoTech Labs (Yadkinville, NC; see Fig. S1†). Hydrophilic polytetrafluoroethylene (PTFE; 5 μm pore size) membranes and sodium sulfate (Na2SO4; granular ACS grade) were purchased from EMD Millipore (Darmstadt, Germany).Filter design and operation
The electrochemical filter device (adapted from Schnoor et al.16), was constructed from a modified 47 mm polypropylene filter housing (Whatman) with perforated Ti shims acting as the mechanical contact for both the anode and the cathode. Two, 47 mm CNT filters were inserted, one as the cathode and one as the anode, with PTFE insulation between (Fig. 1A). The mechanical contacts were attached to a DC power supply (Agilent E3631A) and test solutions were delivered via a peristaltic pump to maintain a constant flow (Cole-Parmer Masterflex L/S EasyLoad Pump).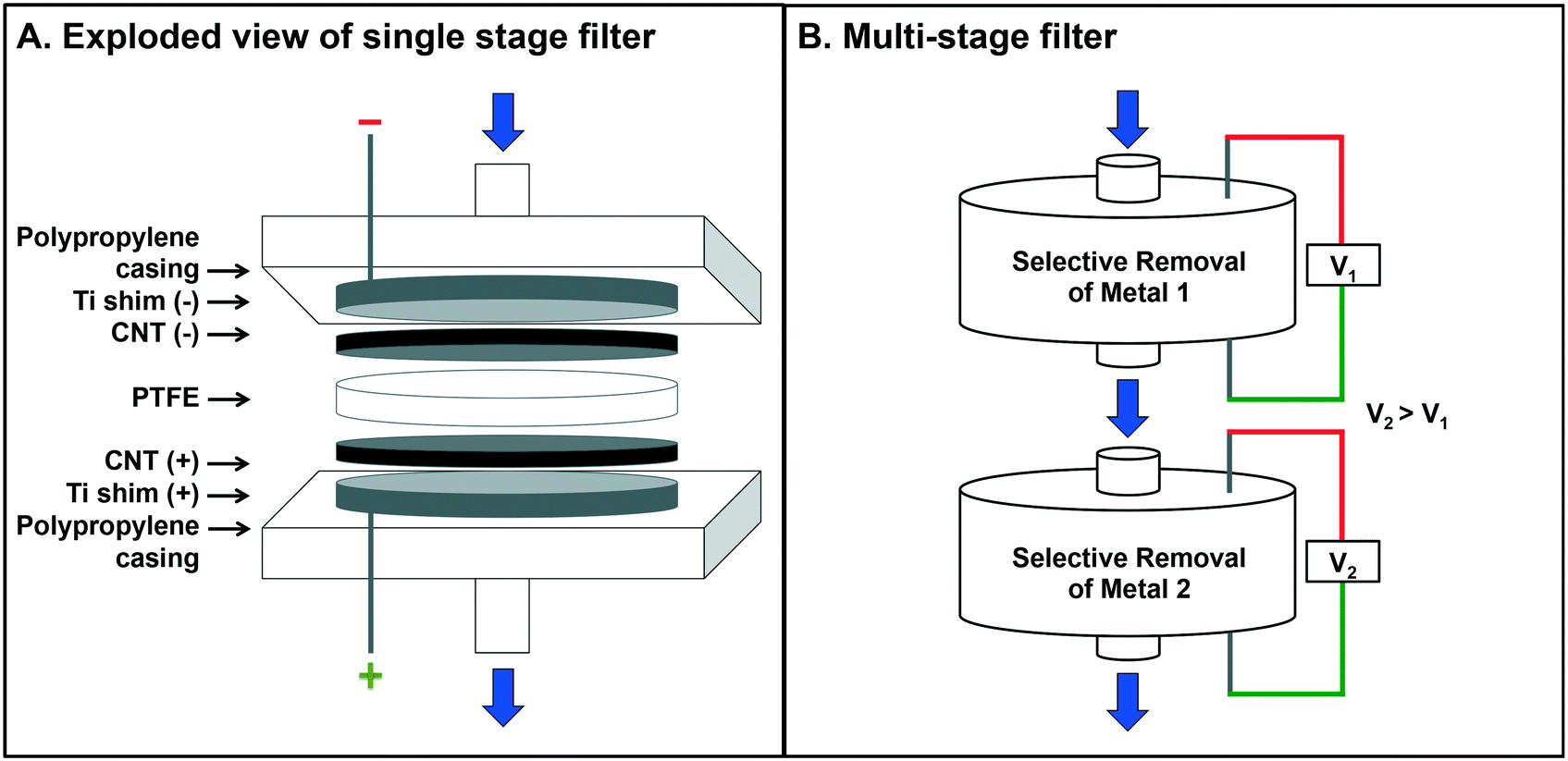 | ||
| Fig. 1 (A) Exploded view of the electrochemical filtration system. Top to bottom: Polypropylene filter casing, perforated cathodic Ti shim, cathodic CNT filter, PTFE insulating layer, anodic CNT filter, perforated anodic Ti shim, polypropylene filter casing. (B) Schematic of multiple electrochemical filters in series. A mixed metal stream is pushed through the system where the top and bottom filters are set at V1 and V2, selecting for metal 1 and metal 2, respectively, and where V1 < V2. | ||
Experimental procedures
All solutions for the electrochemical experiments were prepared in acid washed glassware (washed for at least one week in 25% v/v HCl followed by one week in 50% v/v HNO3), with 1 mM metal/metalloid and 100 mM Na2SO4 (to normalize the ionic strength); Ga was prepared with 100 mM NaCl to avoid precipitation of gallium sulfate (Ga2(SO4)3), and tested in triplicate unless otherwise noted. Effluents were collected and quantified for each metal using inductively coupled plasma mass spectrometry (ICP-MS; Perkin-Elmer ELAN DRC-e). Filters were characterized using scanning electron microscopy (SEM-EDX; Hitachi SU-70 and Hitachi SU-8230 with a BRUKER XFlash 5060FQ Annular EDS detector) and analyzed with X-ray photoelectron spectroscopy (XPS; Kratos Axis Ultra DLD) for elemental and metal speciation identification.Applied voltage ranged from 0.1 V to 3.0 V (depending on the metal) while flow rate and pH were kept constant. Keeping flow rate and voltage constant, a wide range of pH values (pH 2–10) were tested, to both ascertain the potential mechanisms of removal and define ranges of function for the device across a range of waste streams with variable pH. Finally, flow rate was tested to determine if typically slow redox kinetics were a limiting step in the recovery process (i.e., if bulk transport past the filter was too fast to allow for metal reduction or precipitation and capture). Here, flow rates ranged from 1–5 mL min−1 while pH and voltage were held constant. When relevant, experimental parameters are listed in each figure caption or image.
Results and discussion
Parameter optimization
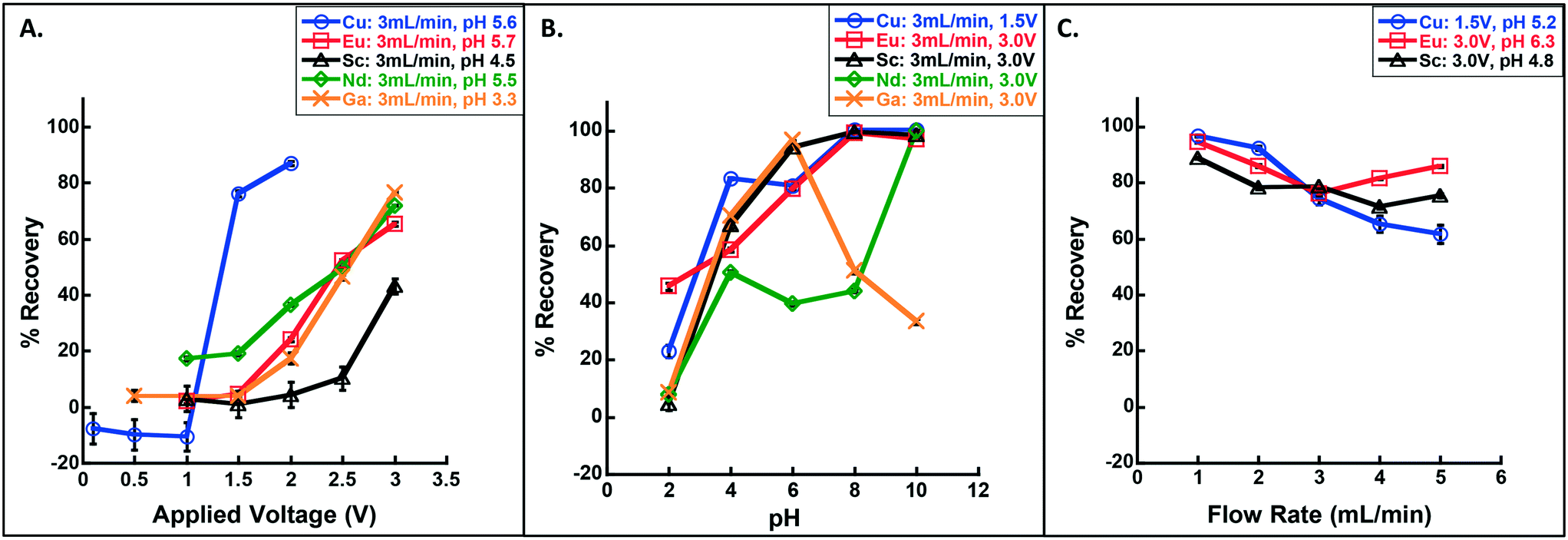 | ||
| Fig. 2 Recovery of Cu, Sc, Eu and Nd as a function of (A) applied voltage (range 0.1–3.0 V), (B) pH (range 2–10), and (C) flow rate (1–5 mL min−1). Note that only one parameter was varied at a time where others were fixed (see legends for details); for (A) the pH was measured after solution preparation rather than titrated to a controlled value. Arsenic (As) was unrecovered at all test voltages and pHs (see Fig. S3†). Error bars represent standard deviation on triplicate measure and are smaller than the symbol and/or line when not visible. | ||
The shape and speciation of the recovered material varied with metal type and, for Cu, as a function of voltage. Crystalline Cu was recovered at 1.5 and 2.0 V (consistent with Vecitis et al.'s previous finding20), as Cu2O; i.e., Cu(II) was electrochemically reduced to Cu(I) (Fig. 3; Fig. S4†). In contrast, Ga, Eu, Sc, and Nd formed large plates of amorphous material, rather than individual metal crystals (Fig. 3; Fig. S5;† X-ray diffraction (XRD) data displayed no discernable pattern (data not shown), indicating the lack of a repeating crystal structure). The RESEs were recovered in their trivalent forms as Ga2O3, Eu2O3, Sc2O3, and Nd2O3 (see further discussion below, the ESI,† and Table S1). Thus, a mechanism that promoted metal oxide deposition controlled the recovery process, and recovery efficiency would not necessarily improve with increasing standard reduction potential (i.e., that which would be expected if zero-valent metals were collected), as observed. Further, we note that metal oxide formation is inconsistent with the anticipated mechanisms for electrosorption,21 a process in which ions are electrochemically neutralized and consequently destabilized in solution (thereby promoting sorption to nearby surfaces, as in capacitive deionization).21 Instead, if electrosorption were the dominant removal mechanism, zero-valent metals/metalloids would have been recovered and the elements should be preferentially neutralized in order from highest to lowest integrated ionization energy (i.e., highest to lowest recovery would proceed Ga ≈ As > Sc > Eu > Nd ≫ Cu assuming complete neutralization; see Table S2†), which was not observed.
 | ||
| Fig. 3 Representative scanning electron micrographs of (A) Cu, (B) Sc, (C) Eu, (D) Nd, and (E) Ga at reported voltages. Flow rate was held constant at 3 mL min−1 for each metal and pH was 5.6, 4.5, 5.7, 5.5, and 3.3, respectively. SEM at all voltages for all recovered metals are available in Fig. S5.† | ||
Even in the absence of pure, zero-valent metal recovery, these data remain encouraging for selective recovery among these metals, especially between copper and the four RESE based on voltage alone. For example, at 3 mL min−1 and 1.5 V, Cu was efficiently retained (76 ± 1% recovery) while Eu recovery was minimal (5 ± 1%), indicating an ability to capture Cu on an early, low voltage stage and Eu on a later, high voltage stage. This could be very useful, especially in the electronics industry, where most metal-rich waste streams have high Cu levels, making it difficult to capture the proportionately small amount of RESE22–24 (see Mixed metal separation and recovery).
As observed with the voltage sweep, these RESEOs were recovered in the form of large platelets with no discernable morphological change from acidic to basic conditions (Fig. S6†). Interestingly, a change in Cu crystal morphology was observed and corresponded to a change in Cu speciation from Cu2O at the low end of the pH range and CuO at higher pH (Fig. S4; Fig. S6†). This reflected a shift in a partial reduction mechanism at low pH, to a metal oxide formation trapping mechanism at high pH, similar to that observed for the RESEs. Further investigation was performed to detail this mechanism of recovery (see Mechanism of recovery).
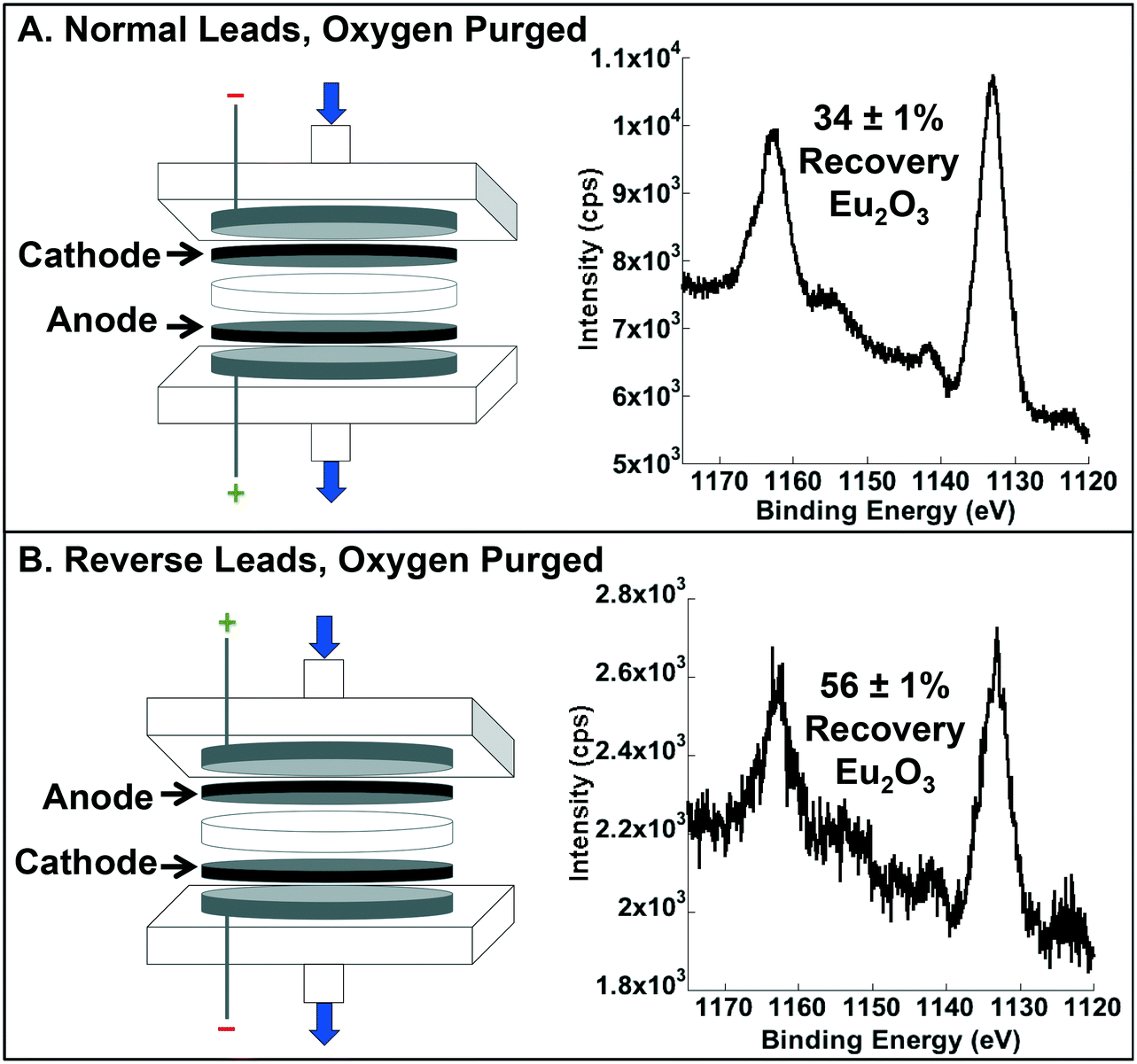 | ||
| Fig. 4 Schematic of oxygen purge lead arrangement and confirmation of Eu2O3 formation via X-ray photoelectron spectroscopy. (A) Deaerated experiment with normal leads (i.e., cathode on top) with 34 ± 1% recovery, compared to 86 ± 0.4% in the aerated system, suggesting that nearly 60% of the total recovered in an unaerated sample was due to the presence of dissolved molecular oxygen. (B) Deaerated experiment with reverse leads (i.e., anode on top; enhanced O2 transport) with 56 ± 1% recovery. Note: XPS is a surface technique, so the intensity in spectrum (B) is lower (with a correspondingly higher signal-to-noise ratio) due to metal deposition within the filter compared to surface deposition seen in (A). | ||
In the absence of O2, Eu recovery decreased from 86 ± 0.4% to 34 ± 1%, indicating that dissolved molecular oxygen is an important source of O2, and that the back diffusion of O2 derived from water splitting at the anode was substantial (i.e., 40% of the total observed in normal operation) (Fig. 4A). To confirm the importance of the latter, we qualitatively confirmed the presence of H2 (a necessary co-product of water splitting) through the observation of ignitable H2 (g) evolution and quantitatively determined there was enhanced recovery when the leads were flipped (i.e., the anode on top and the cathode on the bottom to test for enhanced oxygen transport in the direction of the bulk fluid flow; Fig. 4B). Indeed, recovery of Eu was 56 ± 1% (compared to the 34 ± 1% with normal leads), further supporting the importance of water splitting as a source of O2 for oxide formation and recovery.
In order to form the oxide, we propose the oxygen and water are being reduced in solution to hydroxide, forming metal hydroxide species and then dehydrating to the insoluble metal oxide25 (Fig. 5). Our logic follows: previous work has shown that oxygen and water can be reduced in aqueous systems at the cathode (eqn (1) and (2)),19,25 occurring at 0.19 VAg/AgCl and −1.0 VAg/AgCl, respectively.26,27
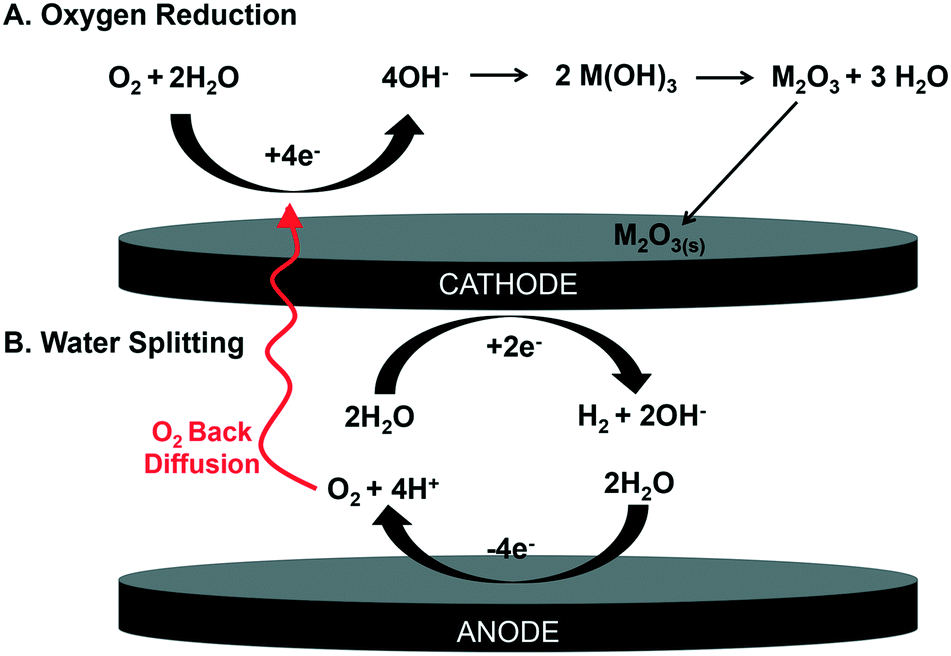 | ||
| Fig. 5 Proposed mechanism for electrochemical deposition of metal oxides (e.g., M2O3) via a metal hydroxide intermediate (e.g., M(OH)3 for trivalent RESEs). Molecular oxygen (O2) can form hydroxide at the cathode through (A) oxygen reduction or (B) water splitting. Note that water splitting contributes additional molecular oxygen (O2) via anodic water splitting. | ||
Cathodic sources of OH−:
| O2 + 2H2O + 4e− ⇌ 4OH− | (1) |
| 2H2O + 2e− ⇌ H2 + 2OH− | (2) |
Note that molecular O2 (eqn (1)) can be sourced from both dissolved O2 as well as back diffusion of O2 formed at the anode (eqn (3)).
Anodic source of O2:
| 2H2O ⇌ O2 + 4H+ + 4e− | (3) |
This O2 is sourced from the oxidation of water (−1.44 VAg/AgCl; eqn (3)). Here, we propose oxygen formed via water oxidation back diffuses to the cathode, contributing to further hydroxide formation via oxygen reduction (eqn (1)). This hydroxide could encourage the formation of insoluble metal hydroxides, which could subsequently dehydrate to the observed metal oxides (Fig. 5). Note that a similar metal oxide formation mechanism has been described superficially, but not rigorously demonstrated or detailed, in battery systems where water splitting is known to occur.28
The pH dependency supports this hypothesis: in general, RESE recovery behaved according to hydroxide solubility (e.g., Ga has high recovery at pH 6 and Eu at pH 10, where the hydroxide forms dominate; see Fig. S9†),29–31 consistent with the formation of a metal hydroxide intermediate. Arsenic, which forms no insoluble metal hydroxides (and only very soluble oxides), was not recovered at any pH or voltage. In the case of Cu, the reduction potential of Cu(II) to Cu(0) is similar to the reduction potential for O2 (eqn (1)), 0.13 VAg/AgCl and 0.19 VAg/AgCl, respectively. In this case, there are competing processes: Cu(II) to Cu(I) reduction, followed by oxide formation, prevailed at low pHs, whereas the oxygen-mediated mechanism outcompeted this reduction at high pH, where OH− was abundant and anodic water splitting would be enhanced.
Many applications utilize metal oxides as starting materials and critical components; for example, Eu2O3 is used a phosphor in electronic screen displays, Nd2O3 is used as a catalyst, and Cu2O and Ga2O3 are proposed for potential use in the semiconductor industry.32–34 Thus, the recovery of metal oxides across the entire pH range could be encouraging for direct reuse in manufacturing processes. Further, while recovery changed over the range tested, the metal speciation did not. This is uncommon in traditional metal recovery systems: pH greatly affects metal speciation and usually requires further purification processes to change the metal into a homogeneous, consistent, single form. While promising, manufacturing process streams and end-of-life waste streams are comprised of multiple metals of interest, and the selectivity of this system for individual metals (i.e., separation) remains to be shown.
Mixed metal separation and recovery
The prior optimization exercise using pure, single-metal solutions informed the possibility of selective recovery of two metals from a mixed metal solution, which is necessary for relevant process wastes, as in semiconductor manufacturing, or e-waste treatment. In this study, a mixed metal solution of Cu and Eu was prepared and tested in a stacked-filter apparatus with two filters in series (Fig. 1B), where the upstream filter was set at 1.5 V, selecting for Cu, and the downstream filter was set at 3.0 V, selecting for Eu.For these metals, separation and retention was achieved: 97 ± 0.1% Cu was retained on the first stage and 65 ± 0.3% Eu was retained on the second filter (Fig. 6). Characterization of the recovered material revealed almost full separation of the deposited Cu and Eu (with EDX mapping (Fig. 6)). Quantitative measures of Cu purity are unavailable at this time, but are qualitatively high according to the EDX color map (Fig. 6B). Note that the EDX spectra (Fig. 6C) indicate some contribution of Eu on Filter 1 and Cu on Filter 2.
 | ||
| Fig. 6 Characterization of materials collected on a dual filter stage arranged in series from a mixed metal medium containing Cu and Eu: (A) SEM, (B) SEM-EDX mapping, and (C) EDX spectra. The influent solution was prepared with 1 mM Cu, 1 mM Eu, and 100 mM Na2SO4. | ||
As observed for the high purity solutions of a single target metal, Cu crystals were deposited across the upstream filter surface and layered amorphous Eu was observed on the downstream filter (Fig. 6A). Elemental mapping (Fig. 6B) revealed almost full separation of Cu (green) and Eu (red), with only a small amount of Eu on the first filter. Counter ions were also captured on the filter (Fig. 6C), which included sulfur and chlorine (likely as sulfate and chloride, respectively) from the electrolyte and metal salts. Again, the metals were recovered as metal oxides, and the proposed mechanism of oxide formation via hydroxide intermediates prevailed in the mixed solution.
At high voltages (e.g., those needed for any metal with a high reduction potential), where both water splitting and oxygen reduction generate hydroxide ions, the application of the method will be limited to those materials that form stable, insoluble hydroxides. Since no such hydroxides are formed for arsenic (neither As(III) nor As(IV)), it is unsurprising that As was unretained at all tested pHs and voltages (Fig. S4†). Nevertheless, GaAs is commonly used in the semiconductor industry in a variety of applications including integrated circuit chips, diodes, and solar cells, and separating Ga from As could be of value for both commercial and environmental objectives.35 Ga could be effectively retained using electrochemical precipitation, but As would remain solubilized and pass with the permeate. Subsequent treatment methods to remove As could then be used for the purification of As from the waste stream.36
Implications
Here, we successfully recovered single metals out of solution with a novel electrochemical deposition method, forming micro crystals and large-area platelets, and importantly, recovered and separated two metals out of a mixed metal stream in a proof-of-concept case. Due to similarities in their reduction potentials and hydroxide solubilities, RESE are likely to be recovered as a mixed cake of oxides, unless further optimization is undertaken to exploit the subtle differences in their metal solubility. Indeed, while some information is available regarding RESE hydroxide and oxide solubility, relatively little is known regarding their thermodynamically favored speciation. Similarly, the behavior of the system with variable electrolytes (e.g., carbonate or bicarbonate) will be important if the wastewaters targeted for treatment represent some combination of natural and industrial wastes. Such studies will be critical to deployment of this technology for individual RESE separation. Nevertheless, the ability to separate easy-to-reduce bulk metals from RESE is remarkable in and of itself and presents an encouraging new route for metal recovery. This could be commercially important, particularly in nanomanufacturing waste streams where small, but valuable levels of metal are being lost with current recycling technologies.22–24 In addition, the technology might be useful for the treatment of high volume legacy wastes rich in metals, such as coal combustion residues.Under current recycling and end-of-life management practices, RESE have low to 0% recoveries.7 With many of these elements topping the critical elements list, a technology that is capable of reclaiming these metals will provide not only environmental benefit, but also enhance political and economic stability associated with abundant resource availability.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by the Environmental Protection Agency (LCNano; EPA RD-83558001) and the Department of Energy (DE-FE0026952). The authors thank Chad Vecitis and Heileen Hsu-Kim for assistance and valuable discussion, Min Li at the Yale West Campus Materials Characterization Core (MCC) and Carrie Donley at the University of North Carolina Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) for assistance with material characterization and instrumentation, as well as Robert Adams and Richard Czerw at NanoTech Labs for assistance with custom filter development.References
- M. Fischedick, J. Roy, A. Abdel-Aziz, A. Acquaye, J. M. Allwood, J.-P. Ceron, Y. Geng, H. Kheshgi, A. Lanza, D. Perczyk, L. Price, E. Santalla, C. Sheinbaum and K. Tanaka, Industry. Climate Change 2014: Mitigation of Climate Change. Contribution of Working Group III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press: Cambridge, United Kingdom and New York, NY, USA, 2014 Search PubMed.
- M. P. O'Connor, J. B. Zimmerman, P. T. Anastas and D. L. Plata, A Strategy for Material Supply Chain Sustainability: Enabling a Circular Economy in the Electronics Industry through Green Engineering, ACS Sustainable Chem. Eng., 2016, 4(11), 5879–5888 CrossRef.
- Energy, U. S. D. O., Critical Materials Strategy, 2011 Search PubMed.
- D. J. Kingsnorth, In Rare earths: facing new challenges in the new decade, 2010 Search PubMed.
- C. Jamasmie, Molycorp shuts down Mountain Pass rare earth plant, http://www.mining.com, 2015 Search PubMed.
- L. Grandell, A. Lehtilä, M. Kivinen, T. Koljonen, S. Kihlman and L. S. Lauri, Role of critical metals in the future markets of clean energy technologies, Renewable Energy, 2016, 95, 53–62 CrossRef CAS.
- B. K. Reck and T. E. Graedel, Challenges in Metal Recycling, Science, 2012, 337(6095), 690–695 CrossRef CAS PubMed.
- J. Li, X. Zeng, M. Chen, O. A. Ogunseitan and A. Stevels, Control-Alt-Delete: Rebooting Solutions for the E-Waste Problem, Environ. Sci. Technol., 2015, 49(12), 7095–7108 CrossRef CAS PubMed.
- R. Widmer, H. Oswald-Krapf, D. Sinha-Khetriwal, M. Schnellmann and H. Böni, Global perspectives on e-waste, Environ. Impact Assess. Rev., 2005, 25(5), 436–458 CrossRef.
- A. Terazono, S. Murakami, N. Abe, B. Inanc, Y. Moriguchi, S.-I. Sakai, M. Kojima, A. Yoshida, J. Li, J. Yang, M. H. Wong, A. Jain, I.-S. Kim, G. L. Peralta, C.-C. Lin, T. Mungcharoen and E. Williams, Current status and research on E-waste issues in Asia, J. Mater. Cycles Waste Manage., 2006, 8(1), 1–12 CrossRef.
- A. M. B. Hugo Marcelo Veit, Electronic Waste: Recycling Techniques, 2015 Search PubMed.
- D. A. Bertuol, F. D. R. Amado, H. Veit, J. Z. Ferreira and A. M. Bernardes, Recovery of Nickel and Cobalt from Spent NiMH Batteries by Electrowinning, Chem. Eng. Technol., 2012, 35(12), 2084–2092 CrossRef CAS.
- C.-F. de Lannoy, D. Jassby, K. Gloe, A. D. Gordon and M. R. Wiesner, Aquatic Biofouling Prevention by Electrically Charged Nanocomposite Polymer Thin Film Membranes, Environ. Sci. Technol., 2013, 47(6), 2760–2768 CrossRef CAS PubMed.
- M. S. Rahaman, C. D. Vecitis and M. Elimelech, Electrochemical Carbon-Nanotube Filter Performance toward Virus Removal and Inactivation in the Presence of Natural Organic Matter, Environ. Sci. Technol., 2012, 46(3), 1556–1564 CrossRef CAS PubMed.
- C. D. Vecitis, G. Gao and H. Liu, Electrochemical Carbon Nanotube Filter for Adsorption, Desorption, and Oxidation of Aqueous Dyes and Anions, J. Phys. Chem. C, 2011, 115(9), 3621–3629 CAS.
- M. H. Schnoor and C. D. Vecitis, Quantitative Examination of Aqueous Ferrocyanide Oxidation in a Carbon Nanotube Electrochemical Filter: Effects of Flow Rate, Ionic Strength, and Cathode Material, J. Phys. Chem. C, 2013, 117(6), 2855–2867 CAS.
- S. Arai, T. Saito and M. Endo, Cu–MWCNT Composite Films Fabricated by Electrodeposition, J. Electrochem. Soc., 2010, 157(3), D147–D153 CrossRef CAS.
- H. L. Yankun Zhan, Likun Pan, Yanping Zhang, Yiwei Chen and Zhuo Sun, Regeneration of carbon nanotube and nanofibre composite film electrode for electrical removal of cupric ions, Water Sci. Technol., 2010, 61(6), 1427–1432 CrossRef PubMed.
- A. J. Bard, R. Parsons and J. Jordan, Standard potentials in aqueous solution, CRC press, 1985, vol. 6 Search PubMed.
- C. D. Vecitis and M. H. Schnoor, Iron redox in a carbon nanotube electrochemical filter, Abstr. Pap. Am. Chem. Soc., 2013, 245, 1 Search PubMed.
- Y. Oren, Capacitive deionization (CDI) for desalination and water treatment—past, present and future (a review), Desalination, 2008, 228(1–3), 10–29 CrossRef CAS.
- H. Wang, G.-H. Gu and Y.-F. Qi, Crushing performance and resource characteristic of printed circuit board scrap, J. Cent. South Univ. Technol., 2005, 12(5), 552–555 CrossRef.
- A. Das, A. Vidyadhar and S. P. Mehrotra, A novel flowsheet for the recovery of metal values from waste printed circuit boards, Resour., Conserv. Recycl., 2009, 53(8), 464–469 CrossRef.
- W. J. Hall and P. T. Williams, Separation and recovery of materials from scrap printed circuit boards, Resour., Conserv. Recycl., 2007, 51(3), 691–709 CrossRef.
- G. H. A. Therese and P. V. Kamath, Electrochemical Synthesis of Metal Oxides and Hydroxides, Chem. Mater., 2000, 12(5), 1195–1204 CrossRef CAS.
- R. Chaim, S. Almaleh-Rockman, L. Gal-Or and H. Bestgen, Electrochemical Chromium(III) Oxide Coatings on Non-oxide Ceramic Substrates, J. Am. Ceram. Soc., 1994, 77(12), 3202–3208 CrossRef CAS.
- R. Chaim, G. Stark, L. Gal-Or and H. Bestgen, Electrochemical ZrO2 and Al2O3 coatings on SiC substrates, J. Mater. Sci., 1994, 29(23), 6241–6248 CrossRef CAS.
- C. Xia, R. Black, R. Fernandes, B. Adams and L. F. Nazar, The critical role of phase-transfer catalysis in aprotic sodium oxygen batteries, Nat. Chem., 2015, 7(6), 496–501 CrossRef CAS PubMed.
- L. Paul and C. E. Brown, Scandium, Yttrium and the Lanthanide Metals, in Hydrolysis of Metal Ions, Wiley-VCH Verlag GmbH & Co. KGaA, 2016, pp. 225–324 Search PubMed.
- L. L. Ames and D. Rai, Radionuclide interactions with soil and rock media, United States Environmental Protection Agency, Office of Radiation Programs, 1978, vol. 1 Search PubMed.
- T. W. Cheng, M. L. Lee, M. S. Ko, T. H. Ueng and S. F. Yang, The heavy metal adsorption characteristics on metakaolin-based geopolymer, Appl. Clay Sci., 2012, 56, 90–96 CrossRef CAS.
- S. I. Stepanov, V. I. Nikolaev, V. E. Bougrov and A. E. Romanov, Gallium oxide: properties and applica a review, Rev. Adv. Mater. Sci., 2016, 44, 63–86 Search PubMed.
- J. Emsley, Nature's building blocks: an AZ guide to the elements, Oxford University Press, 2011 Search PubMed.
- L.-K. Tsui and G. Zangari, Electrochemical Synthesis of Metal Oxides for Energy Applications, in Electrodeposition and Surface Finishing: Fundamentals and Applications, ed. S. S. Djokić, Springer New York, New York, NY, 2014, pp. 217–239 Search PubMed.
- R. Szweda, Gallium Arsenide, Electronics Materials and Devices. A Strategic Study of Markets, Technologies and Companies Worldwide 1999–2004, Elsevier, 3rd edn, 2000 Search PubMed.
- D. Mohan and C. U. Pittman Jr, Arsenic removal from water/wastewater using adsorbents—A critical review, J. Hazard. Mater., 2007, 142(1–2), 1–53 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ew00187h |
| This journal is © The Royal Society of Chemistry 2018 |