
Degradation of polyethylene glycols and polypropylene glycols in microcosms simulating a spill of produced water in shallow groundwater†
 ab
E. Michael
Thurman,
a
Imma
Ferrer,
a
Morgan V.
Evans,
c
Paula J.
Mouser
cd
and
Joseph N.
Ryan
*a
ab
E. Michael
Thurman,
a
Imma
Ferrer,
a
Morgan V.
Evans,
c
Paula J.
Mouser
cd
and
Joseph N.
Ryan
*a
Abstract
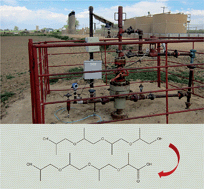
Polyethylene glycols (PEGs) and polypropylene glycols (PPGs) are frequently used in hydraulic fracturing fluids and have been detected in water returning to the surface from hydraulically fractured oil and gas wells in multiple basins. We identified degradation pathways and kinetics for PEGs and PPGs under conditions simulating a spill of produced water to shallow groundwater. Sediment-groundwater microcosm experiments were conducted using four produced water samples from two Denver-Julesburg Basin wells at early and late production. High-resolution mass spectrometry was used to identify the formation of mono- and di-carboxylated PEGs and mono-carboxylated PPGs, which are products of PEG and PPG biodegradation, respectively. Under oxic conditions, first-order half-lives were more rapid for PEGs (<0.4–1.1 d) compared to PPGs (2.5–14 d). PEG and PPG degradation corresponded to increased relative abundance of primary alcohol dehydrogenase genes predicted from metagenome analysis of the 16S rRNA gene. Further degradation was not observed under anoxic conditions. Our results provide insight into the differences between the degradation rates and pathways of PEGs and PPGs, which may be utilized to better characterize shallow groundwater contamination following a release of produced water.
- This article is part of the themed collection: The environmental geochemistry and biology of hydraulic fracturing
 Please wait while we load your content...
Please wait while we load your content...

