
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Macroscopic heat release in a molecular solar thermal energy storage system†
Zhihang
Wang
 a,
Anna
Roffey
a,
Raul
Losantos
b,
Anders
Lennartson
a,
Martyn
Jevric
a,
Anne U.
Petersen
a,
Maria
Quant
a,
Ambra
Dreos
a,
Xin
Wen
a,
Diego
Sampedro
b,
Karl
Börjesson
c and
Kasper
Moth-Poulsen
*a
a,
Anna
Roffey
a,
Raul
Losantos
b,
Anders
Lennartson
a,
Martyn
Jevric
a,
Anne U.
Petersen
a,
Maria
Quant
a,
Ambra
Dreos
a,
Xin
Wen
a,
Diego
Sampedro
b,
Karl
Börjesson
c and
Kasper
Moth-Poulsen
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 41296 Gothenburg, Sweden. E-mail: kasper.moth-poulsen@chalmers.se
bDepartment of Chemistry, Centro de Investigación en Síntesis Química (CISQ), Universidad de La Rioja, Madre de Dios 53, E-26006 Logroño, La Rioja, Spain. E-mail: diego.sampedro@unirioja.es
cDepartment of Chemistry and Molecular Biology, University of Gothenburg, Kemigården 4, 41296 Gothenburg, Sweden
First published on 20th August 2018
Abstract
The development of solar energy can potentially meet the growing requirements for a global energy system beyond fossil fuels, but necessitates new scalable technologies for solar energy storage. One approach is the development of energy storage systems based on molecular photoswitches, so-called molecular solar thermal energy storage (MOST). Here we present a novel norbornadiene derivative for this purpose, with a good solar spectral match, high robustness and an energy density of 0.4 MJ kg−1. By the use of heterogeneous catalyst cobalt phthalocyanine on a carbon support, we demonstrate a record high macroscopic heat release in a flow system using a fixed bed catalytic reactor, leading to a temperature increase of up to 63.4 °C (83.2 °C measured temperature). Successful outdoor testing shows proof of concept and illustrates that future implementation is feasible. The mechanism of the catalytic back reaction is modelled using density functional theory (DFT) calculations rationalizing the experimental observations.
Broader contextThermal energy can be used for a broad range of applications such as domestic heating, industrial process heating and in thermal power processes. One promising way to store solar thermal energy is so-called molecular solar thermal (MOST) energy storage systems, where a photoswitchable molecule absorbs sunlight and undergoes a chemical isomerization to a metastable high energy species. Here we present an optimized MOST system (providing a high energy density of up to 0.4 MJ kg−1), which can store solar energy for a month at room temperature and release the thermochemical energy “on demand” in a closed energy storage cycle. In addition to a full photophysical characterization, solar energy capture of the present system is experimentally demonstrated by flowing the MOST system through an outdoor solar collector (≈900 cm2 irradiated area). Moreover, catalyst systems were identified and integrated into an energy extraction device leading to high temperature gradients of up to 63 °C (83 °C measured temperature) with a short temperature ramp time of only a few minutes. The underlying step-by-step mechanism of the catalytic reaction is modelled in detail using quantum chemistry calculations, successfully rationalizing the experimental observations. |
Introduction
According to the 2015 United Nations Climate Change Conference (COP21) Paris agreement on climate change, global emissions must be reduced by 60% prior to 2050.1 The energy consumption is, however, predicted to double in the next 40 years due to an increasing world population.2 For this reason, it is prudent to explore other sustainable energy sources, in addition to electricity generated by either wind power or solar cells. The latter, driven from sunlight, has always been considered to be an abundant renewable energy source; in fact, the International Energy Association (IEA) has predicted that solar energy could provide over 25% of the global energy needs around the deadline of the Paris agreement.3 In recent decades, solar energy has been widely investigated, with a more than 900% increase in installed capacity, demonstrating a rapid expansion of its use.1 Nevertheless, one of the greatest challenges for mass implementation of solar energy technology is the intermittence of supply and load levelling.2,4 Effective storage of solar energy, therefore, is fundamentally important for future development of this energy source.One promising solution is the molecular solar thermal energy storage (MOST) system, where a photoswitchable parent molecule that absorbs sunlight undergoes a chemical isomerization to a metastable high energy species. This concept has been proven viable by incoporating the photoswitch into solid materials or into liquid based systems.5,6 In the case of solution based MOST, a catalyst can be used to release this stored energy “on demand” in the form of heat, and as a result regenerating the parent molecule.6 In contrast to more traditional solar thermal concepts, the MOST system operates in an entirely different manifold, by converting photons to stored chemical energy at room temperature, meaning that e.g. no insulation materials are needed for practical devices. However, many factors have to be considered for the optimal design of a photoswitchable molecule for MOST applications. Ideally, a strong absorption in the UV-visible region of the solar spectrum by the parent molecule with no absorption by the corresponding high energy isomer is preferred. Other prerequisites include a high photoisomerization quantum yield, exceptional robustness and also a low molecular weight, so as to maximize the energy density.7,8 For solutions, it's preferable that the solvent features a low heat capacity, yet can accommodate a large amount of photoswitching. Several photochromic motifs have been identified as potential candidates, such as the dihydroazulene/vinylheptafulvene couple,9 anthracene dimerization,10 azobenzene,5,11,12 azaborinine derivative Dewar isomers,13 difulvalenediruthenium complexes,14 and norbornadiene–quadricyclane.15 The latter system has gained increased attention partly due to its high energy density, but relatively little effort has been dedicated to releasing the stored energy, which is the focus of this work.
A representation of the MOST concept and a possible domestic implementation can be seen in Fig. 1a. Sunlight is collected and stored via photochemical reaction under flow conditions. When energy is required, a solution of the metastable molecule can be passed through a catalytic bed reactor to release the energy in the form of heat, which could be used for, in this instance, heating water or creating steam. In order for MOST to be viable, the energy has to be storable for a long period of time, thus requiring a high energy barrier (ΔH‡therm) for the thermal conversion from the high energy isomer to the parent molecule (see Fig. 1b). Yet, at the same time, the heat release upon demand must be rapid and efficient. For the norbornadiene (NBD)–quadricyclane (QC) couple, it has been shown that a rapid conversion of unsubstituted QC to NBD can be effected electrochemically16 as well as through the use of a catalyst.17 Both approaches give rise to a release of the energy by lowering the activation barrier (ΔH‡cat) from the photoisomer to the parent molecule. In addition, to be suitable for use in a closed cycle operational device capable of undergoing successive cycles, heterogeneous catalysis should be employed.17 Previous research has identified many catalysts, including various transition metal salts and complexes.18–20 However, the main challenge remains to incorporate an effective catalyst into a working device based on a sunlight absorbing MOST system.21
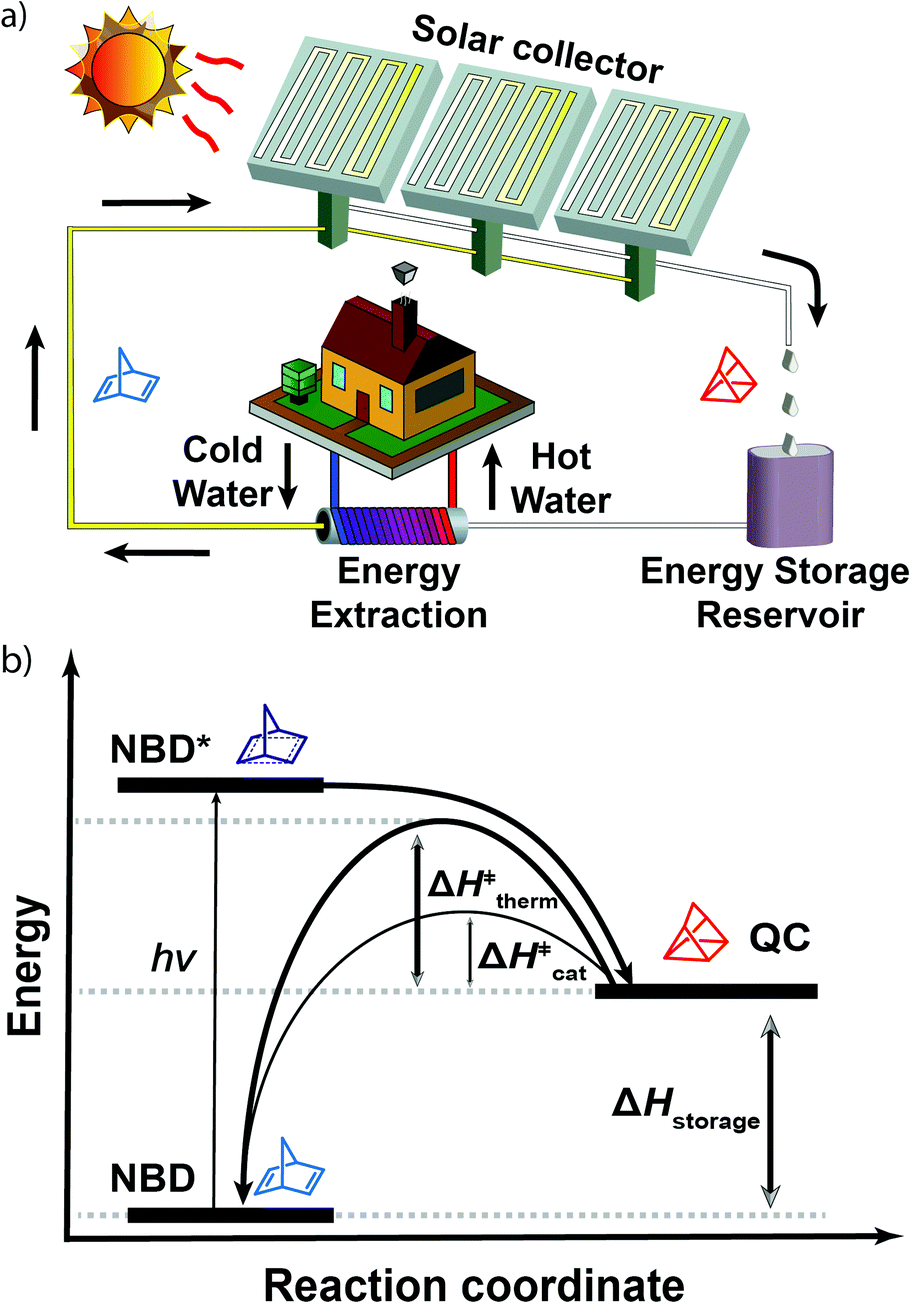 | ||
| Fig. 1 (a) Operating set up based on the MOST concept. (b) Illustration of MOST molecules. ΔH‡therm and ΔH‡cat represent the activation enthalpy for QC → NBD by heat and a catalyst, respectively. ΔHstorage corresponds to the energy storage enthalpy. | ||
In 1988, Miki et al. used a fixed bed catalyst to release heat (ΔT = 58.5 °C) from a solution of unsubstituted QC. Unfortunately, the corresponding NBD has no absorptivity over 300 nm and sunlight cannot be used to drive the forward reaction necessary for solar energy storage.17 Later, it was shown that a solution of a substituted QC, derived from an NBD having an absorption overlap with the solar spectrum (λonset ≈ 380 nm), although with a small molar absorptivity (Amax@350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm ≈ 400 M−1 cm−1), yielded a temperature rise of 50.5 °C. However, in this instance, the reaction set up was very large, consisting of a 4 cm inner diameter, 58 cm long cylindrical reactor with a large loading of a cobalt porphyrin derivative attached to an alumina surface (in total ∼0.7 g of the catalyst), seemingly impractical for real applications.20
nm ≈ 400 M−1 cm−1), yielded a temperature rise of 50.5 °C. However, in this instance, the reaction set up was very large, consisting of a 4 cm inner diameter, 58 cm long cylindrical reactor with a large loading of a cobalt porphyrin derivative attached to an alumina surface (in total ∼0.7 g of the catalyst), seemingly impractical for real applications.20
Here, we present a novel MOST study involving the implementation of a highly suitable NBD derivative (NBD1, QC1), which combines a large spectral difference between the parent molecule and the corresponding photoisomer with a high energy storage density (0.4 MJ kg−1). In addition, the metastable photoisomer exhibits a good ambient stability in toluene (t1/2 = 30 days at 25 °C), and this has allowed us to examine the heat release properties under catalytic conditions to provide rapid heat generation.
Further, it was found that cobalt phthalocyanine (CoPc) effectively catalyses the back reaction of QC1 to NBD1. The requirement for this complex to be suitable for a working device dictates that it should be immobilised on a solid support. This was realized using a miniaturized fixed bed catalytic reactor (0.1 cm diameter, 1 cm long cylindrical tubing, in total ≈ 0.5 mg of the catalyst) containing CoPc physisorbed on an activated carbon support (CoPc@C). Using this catalyst in conjunction with the new QC derivative leads to a record macroscopic heat release of up to 63.4 °C. In addition to these experimental findings, a detailed mechanism of the catalytic cycle was proposed using DFT calculations rationalizing the experimental observations thus giving insight that can be used in the development of future high performance catalysts.
Results and discussion
Characterization of the NBD1–QC1 couple
NBD1 contains two substituents (see Fig. 2a), an electron donor and an electron acceptor across one of the olefins, giving a so called “push–pull” system.6 This leads to a red shift of the absorption and a better match with the solar spectrum. Two approaches were employed in the synthesis of NBD1, both of which gave the target material in good yield. One method utilized a palladium catalyzed Suzuki protocol to afford NBD1. Alternately, NBD1 could be generated both economically and on a large scale from commercially available 4-methoxyacetophenone, the key step being a Diels–Alder reaction of 3-(4-methoxyphenyl)propiolonitrile with cyclopentadiene (Supplementary S2, ESI†). | ||
| Fig. 2 (a) Structure of NBD1 and QC1; (b) absorption spectra of NBD1 (in blue) and its photoisomer QC1 (in red). The sample was irradiated with a λ = 310 nm LED light. The inset figure shows the kinetic back conversion of QC1 at 80 °C; (c) a DSC thermogram for QC1 conversion to NBD1 (ΔHstorage = 88.5 kJ mol−1 was based on an average of two measurements). The dashed line shows the second run after heat release; (d) cyclability experiment for a solution of 0.7 × 10−4 M in toluene showing the absorbance of NBD1 in blue dots and QC1 in red dots at λ = 325 nm. The black line indicates a degradation of 0.14% per cycle over 43 cycles. | ||
Firstly, to assess the suitability of the NBD1–QC1 couple for MOST applications, the solar spectrum match, isomerization efficiency, cycling robustness, and potential energy density were evaluated. Fig. 2b shows the contrast between absorbance profiles for toluene solutions of NBD1 and QC1. NBD1 displayed a broad absorption feature with a high maximum absorptivity. (λmax@326 = 1.3 × 104 M−1 cm−1, Supplementary S3, ESI†). The onset of this absorbance extends to 380 nm, thus leading to a calculated 4.0% absorption of all incoming photons from the solar spectrum (Supplementary S4, ESI†). Meanwhile, this feature was greatly diminished for QC1, showing a large change in absorptivity and a spectral window of 34 nm for the exclusive absorption of sunlight by NBD1. The photoisomerization quantum yield for the formation of metastable QC1 was determined to be 61%, indicating that the majority of all absorbed photons resulted in a successful photoisomerization event (Supplementary S5, ESI†). Having confirmed that the photophysical properties for NBD1 fulfilled the criteria needed for MOST, a lab-to-site transfer demonstration was necessary to prove its practical use. Much to our delight, a quantitative conversion for NBD1 to corresponding QC1 was achievable in an outdoor testing facility. This consisted of a ≈ 900 cm2 reflector directing sunlight onto a glass receiver tubing containing NBD1, demonstrating that the photoisomerization reaction can also work under real sunlight by using continuous flow in a toluene solution, thus providing a basic proof-of-principle and illustrating that future implementation is entirely feasible. A more detailed discussion can be found in ESI† (Supplementary S6).
Differential scanning calorimetry (DSC) was used to measure ΔHstorage for metastable QC1 (Supplementary S7, ESI†). Neat QC1 was prepared through the irradiation of a chloroform-d solution of NBD1, and gave an experimental storage energy ΔHstorage = 88.5 kJ mol−1 (Fig. 2C). By comparison, MP2 calculations were carried out with a theoretical ΔHstorage of 101.1 kJ mol−1 (Supplementary S8, ESI†), thus confirming an energy density of 0.4 MJ kg−1. Using the specific heat capacity found for QC,22 1.66 J g−1 K−1, the energy storage density can be translated to a possible adiabatic temperature increase of 239 °C, taking into account the melting temperature of NBD1 between 49.2 and 51.8 °C.
In order to store the converted energy for long periods of time, the half-life of the photoisomer should be as long as possible and it is important to know this value at room temperature. The thermal conversion from QC1 to NBD1 was measured at different temperatures, and an Eyring analysis was performed revealing that ΔH‡therm and ΔS‡therm equalled 104 kJ mol−1 and −22 J K−1 mol−1, respectively. By extrapolation to room temperature (25 °C), the back-conversion half-life for QC1 was calculated to be 30 days in toluene, thus exhibiting an excellent stability under ambient conditions. The life time of this metastable isomer shows that this light absorber has the potential for use in solving the intermittency of solar energy production between night and day, as well as over weekly or even monthly cycles (Supplementary S9, ESI†).
In the blueprint for the possible domestic use of MOST, such as the one described in Fig. 1a, NBD1 needs to be converted to QC1 back and forth many times. The photostability is therefore an important property in evaluating molecules for the MOST concept.6 An ideal light harvester should therefore operate for an infinite number of cycles without showing any signs of degradation. Thermogravimetric analysis (TGA) showed that the thermal decay temperature tolerance of NBD1 was 150 °C (Supplementary S10, ESI†). Due to this reason, the stability test of the NBD1/QC1 couple was conducted in an accelerated cyclability experiment at 85 °C. After 43 complete cycles of photoisomerization and thermal backconversion using a solution of NBD1, it was found that the degradation was only 0.14% per cycle (Fig. 2d and Supplementary S11, ESI†) affirming the robustness of this system.
In summary, it was found that the properties of NBD1 were more suitable for MOST compared to other NBD derivatives intensively studied in the group. Arylalkyne NBDs exhibit an absorbance profile that better matches the solar spectrum, however, this is accompanied by a low quantum yield (28–47%) and a half-life of hours (5.1–22.0 h) for the corresponding QC in toluene.7,8 In contrast, diaryl substituted NBDs, on account of the additional aromatic ring, have a lower maximum absorptivity (maximum 10100 M−1 cm−1), a blue shifted spectrum and a small spectral difference between the NBD and the QC form upon comparison with NBD1.23 In fact, the cyclability was studied for an acetylenic bridged NBD and was found to only undergo 0.2% degradation per cycle, while we are happy to report that NBD1 was more robust when subjected to multiple cycles with less degradation per cycle.
Screening of catalysts
An efficient catalytic conversion from QC1 to NBD1 in which all the accumulated energy is rapidly released is essential for a MOST system.6 Furthermore, the catalyst should be heterogeneous (i.e. a part of a solid), so as to facilitate device design and to avoid a purification step after each energy storage cycle. Different transition metal catalysts have previously been found to convert substituted QC to NBD.21 Here, 14 different salts and complexes (selected from Cu, Co and Pd inorganic salts, as well as CoPc, and cobalt porphyrin variants) were screened for their catalytic activity by evaluating the outcome using 1H NMR, upon their introduction. UV-vis analysis was used to qualitatively probe the rate of the back reaction in the instances where catalytic activity was observed. Cobalt phthalocyanine (CoPc), although with low solubility in toluene (7.2 × 10−5 M),24 was found as the most attractive candidate due to both a clean conversion and high conversion rate (measured reaction rate ≈172 s−1 M−1, Supplementary S12, ESI†). The task remained to immobilise CoPc in a form that prevents toluene from leaching it from a solid support.In order to generate a high reaction surface area in the form of a heterogeneous catalyst, CoPc was physisorbed onto activated carbon. X-ray photoelectron spectrometry showed that a loading of 13% of CoPc was obtained after this preparation and the reaction rate in toluene was measured to be some 69 times higher than the one for the untreated CoPc in solution (≈1.2 × 104 s−1 M−1). This was rationalised by the fact that pure CoPc can undergo strong intermolecular π-stacking in solution, however the dispersion of CoPc onto charcoal instead gives a larger contact surface area. The CoPc@C catalyst was then used directly in the flow experiments (Supplementary S13, ESI†).
Heat release
To practically demonstrate the compatibility of QC1 in conjunction with CoPc@C, a heat release device was constructed (see Fig. 3 and Supplementary S14, ESI†). PTFE tubing containing approximately 5 mg of CoPc@C was placed in the centre of a high vacuum chamber (up to 10−5 mbar) providing thermal insulation. Thermocouples were placed before (T1) and after (T2) the catalytic bed, which were used to measure the temperature rise resulting from the back conversion of QC1 (ΔT = T2 − T1). Using this setup, it was possible to pass a solution of QC1 through the CoPc@C site under flow conditions. The adiabatic heat release from a solution of QC1 can be calculated from (1):14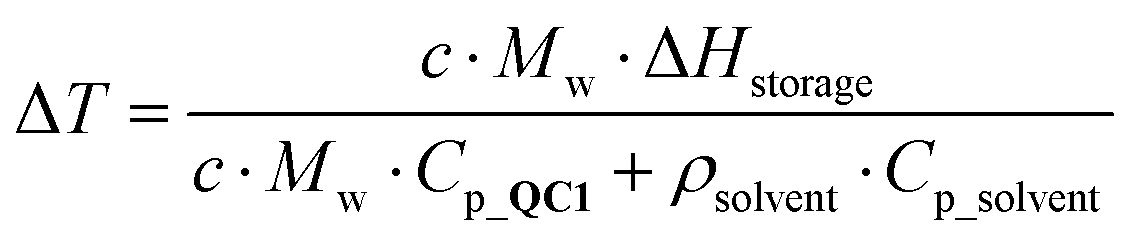 | (1) |
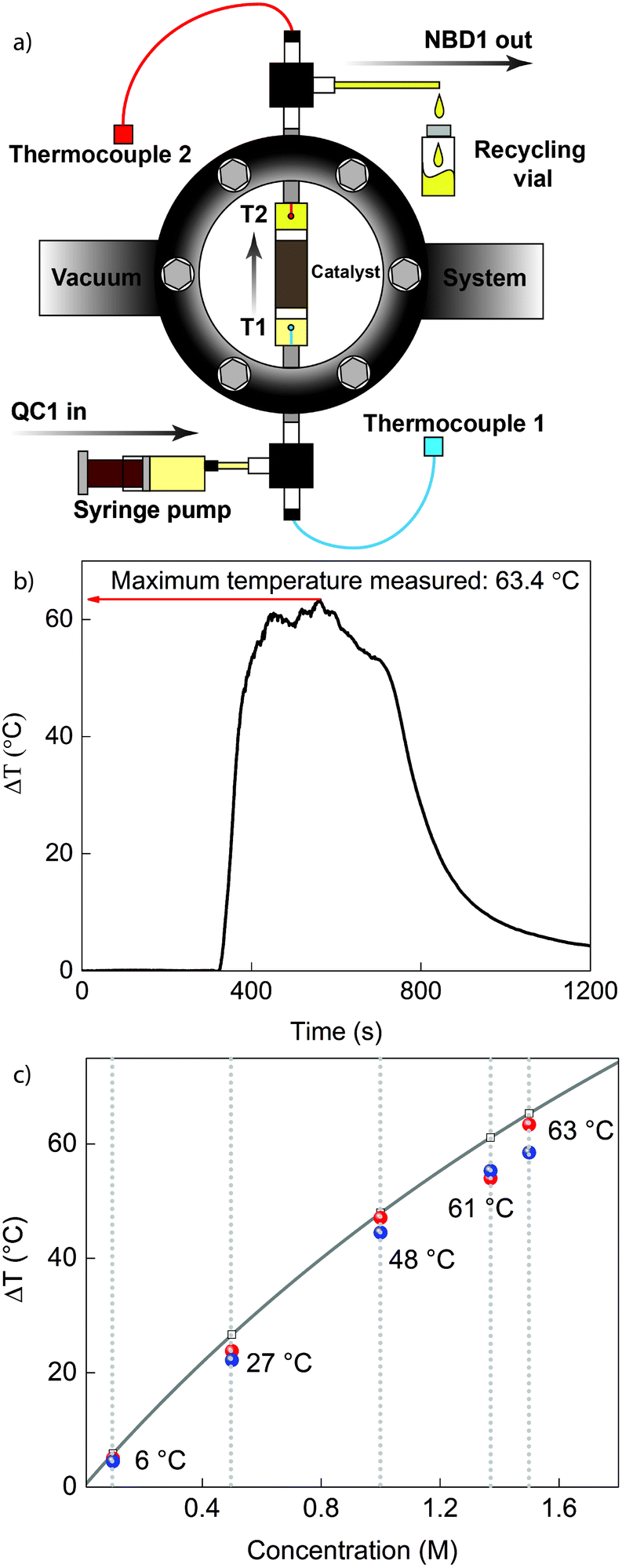 | ||
| Fig. 3 (a) Illustration depicting the design of the vacuum chamber; the picture of the actual setup is found in ESI.†T1 and T2 correspond to the temperature measured by thermocouples before and after the catalytic centre, respectively. (b) Thermogram for heat release from a 1.5 M toluene solution of QC1, where the highest temperature gradient of 63.4 °C was measured. (c) Theoretical simulation (grey line) and experimental data (blue dots for the first measurement and red dots for the second measurement) of heat release vs. concentration. | ||
Computational study
The use of several salts is known to allow the catalysed back-reaction from QC to NBD and some hypothesized mechanisms have been reported.21,25 In addition, some computational information on the photochemistry of the parent NBD26 and the substituent effect on the energy storage in NBD/QC systems27,28 is available. In contrast, the mechanism for the energy release has not been systematically explored. A detailed mechanistic picture of the back-reaction could be helpful in the design of new and more efficient catalysts. Thus, as the energy release is affected by the mechanism of the back-reaction,29 it was decided to approach the study of this process by computational means. In particular, the role of CoPc as a catalyst in the reaction from QC to NBD within the framework of density-functional theory (DFT) was explored. A set of different functionals, chemical structures and conditions were used to gather a comprehensive view of the reaction (Supplementary S16, ESI†). At our highest level of theory (PCM-M06/6-31+G*), the full QC1/NBD1 and CoPc structures were used considering the four possible orientations between QC1 and CoPc. Doing so, a good modelling of the experimental conditions could be ensured.The computed mechanism implies that one of the labile C–C bonds in QC oxidatively added to the metal centre of CoPc. The different possible orientations cause the appearance of several alternative transition structures and energy minima. Due to the development of a positive charge and the different stabilizing effects of the substituents R1–R4, these four minima (2_i to 2_iv, see Fig. 4a) have very different energies. Out of the four possible TSs, only two could be located (TS(1-2)_i and TS(1-2)_ii) with similar energies of 45.1 kJ mol−1 and 58.6 kJ mol−1 relative to the separated reagents, respectively. Due to the high energy difference (more than 54 kJ mol−1 with 2_i and 2_ii), 2_iii, 2_iv and the corresponding TSs can be further discarded as they will not be competitive reaction pathways.
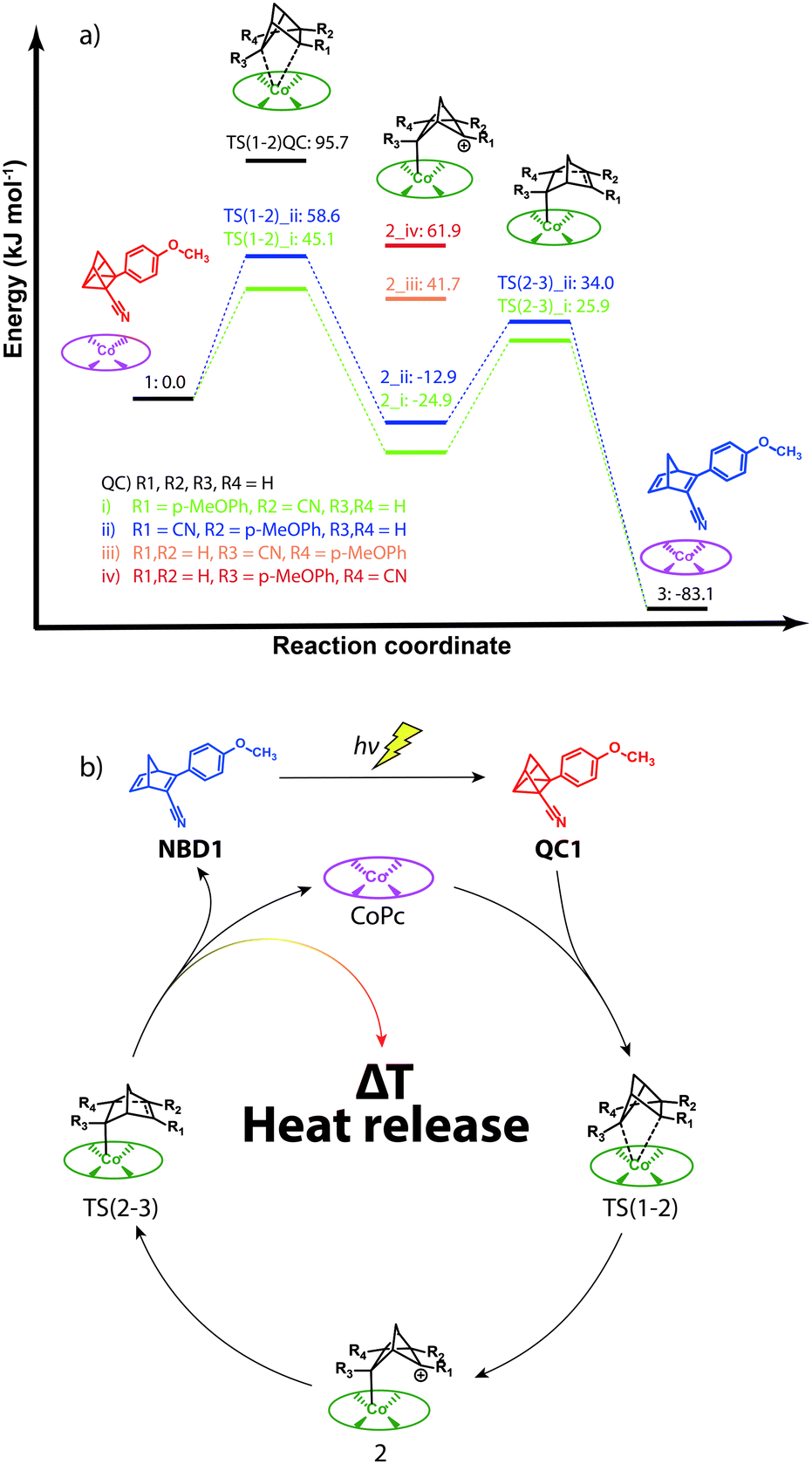 | ||
| Fig. 4 (a) Critical points along the potential energy surface for the catalytic conversion of QC1 to NBD1 using CoPc (free energies in kJ mol−1 computed in toluene with respect to a 10 Å separation of CoPc and QC1). The color coding represents molecular structures or orientations. (b) Catalytic cycle for the back-reaction. | ||
From 2_i and 2_ii, a new barrier of similar energy (50.8 kJ mol−1 for TS(2-3)_i and 46.9 kJ mol−1 for TS(2-3)_ii) has to be surmounted to finally afford NBD1 and the recovery of the catalyst (see Fig. 4b). This final product is clearly more stable than QC1 implying a favored energy release. From our computational data, the energy barriers (TS(1-2) and TS(2-3)) for both orientations i and ii seem to be involved in the control of the reaction outcome for the back-reaction. Due to the very similar maximum energy barriers (50.8 kJ mol−1 for TS(2-3)_i and 58.6 kJ mol−1 for TS(1-2)_ii), the two pathways could be acting at the same time, while the path i would be preferred. These relatively low energy barriers are caused by the substituents included in the NBD1/QC1 system, as the unsubstituted QC features an energy barrier of 95.7 kJ mol−1 for TS(1-2)QC, see Fig. 4a. These low energy barriers cause the efficient heat release as experimentally measured for NBD1 and they are due to charge stabilization in the oxidative addition adduct when cyano (ii) or p-methoxyphenyl (i) substituents are placed in R1. From the computed relative energy between 1 and 3, a heat release value of 61.7 °C could be obtained, in good agreement with the experimental data as well as the theoretical limit achieved from DSC data. Any subsequent improvement in the catalytic energy release would imply the design of new NBD/catalyst pairs to further reduce this energy barrier. From the results shown here, a new generation of MOST systems could be envisioned in which the charge generated in the rate-determining step of the back-reaction could be further stabilized to provide an improved heat release. For the design of these new systems, the whole system (QC, catalyst and solvent) should be considered as the substituents present in the QC, the metal and ligands of the catalyst and the solvent could have an impact on the energy barrier. Thus, the computational methodology presented herein will be useful in the search for new catalytic systems and the design of a new generation of MOST systems.
Conclusions
In this study, we successfully synthesized and comprehensively characterized a scalable new NBD/QC derivative in all aspects pertinent to the performance of a MOST system. The improved absorptivity of NBD1 (λmax@326nm = 1.3 × 104 M−1 cm−1) coupled with factors such as a large spectral difference compared to the corresponding QC1, the high photoisomerization quantum yield (61%), the long half-life (t1/2 = 30 days at 25 °C), and high solubility (cmax = 1.52 M for QC1 in toluene), makes this compound a promising candidate for future MOST applications. In addition, the robustness of a solution of NBD1 was assessed at 85 °C and is capable of withstanding 43 storage and release cycles with little degradation (0.14% per cycle). The energy storage density was confirmed to be 0.4 MJ mol−1 by both DSC measurement and MP2 calculation. A ≈900 cm2 solar collector was used to drive the NBD1 to QC1 reaction in an outdoor test facility, illustrating the possibility of a quantitative photoconversion and feasibility of lab-to-site scale up implementation. Moreover, by using the small reaction centre of the CoPc@C heterogeneous catalyst, the stored energy can be released quickly and efficiently. A macroscopic heat release of up to 63.4 °C using a 1.5 M solution of QC1 was measured, which surpasses all measured MOST systems to date. Our catalyst system in conjugation with QC1 sustained a reaction rate in toluene of 1.2 × 104 s−1 M−1 and a minimum TON of 482. In addition, the experimental work was complemented with a specific and detailed computational study on the catalytic reaction mechanism, which implies a low energy barrier in agreement with the fast conversion observed experimentally. In summary, a comprehensive proof of concept demonstration, showing both conversion under solar light and heat release with a fixed bed catalyst, has been realized. Future molecular design should focus on red shifted NBD derivatives for a better spectral overlap with the solar spectrum, without compromising other essential MOST properties.
Author contributions
Z. W. performed the TGA measurement, quantum yield measurements, cycling test, characterization of potential catalyst candidates, catalyst physisorption, outdoor test, and macroscopic heat release. A. R. performed the solubility test, outdoor test, catalyst physisorption, and macroscopic heat release. R. L. and D. S. carried out the theoretical calculation of the catalyst. A. L. and M. J. designed and contributed to the syntheses. A. U. P. contributed to the synthesis and quantum yield measurement. A. D. measured absorptivity and DSC. M. Q. performed kinetic study and measured the quantum yield and X. W. performed SEM and XPS. Z. W., A. R., K. B. and K. M. P. designed the experiments, and all authors contributed to write the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the K. & A. Wallenberg foundation and the Swedish Foundation for Strategic Research. D. S. and R. L. thank the Spanish Ministerio de Economía y Competitividad (MINECO)/Fondos Europeos para el Desarrollo Regional (FEDER) (CTQ2017-87372-P). R. L. thanks the Universidad de La Rioja for his fellowship. This work used the Beronia cluster (Universidad de La Rioja), which is supported by FEDER-MINECO grant number UNLR-094E-2C-225.References
- United Nations, Adoption of the Paris Agreement, 2015.
- S. N. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef PubMed.
- International Energy Agency, Technology Roadmap Solar Thermal Electricity, 2014.
- K. Moth-Poulsen, Molecular Devices for Solar Energy Conversion and Storage, 2018, ch. 9 Search PubMed.
- T. J. Kucharski, N. Fettalis, A. M. Kolpak, J. O. Zheng, D. G. Nocera and J. C. Grossman, Nat. Chem., 2014, 6, 441–447 CrossRef CAS PubMed.
- Z. Yoshida, J. Photochem., 1985, 29, 27–40 CrossRef CAS.
- A. Dreos, K. Börjesson, Z. Wang, A. Roffey, Z. Norwood, D. Kushnir and K. Moth-Poulsen, Energy Environ. Sci., 2017, 10, 728–734 RSC.
- M. Quant, A. Lennartson, A. Dreos, M. Kuisma, P. Erhart and K. Moth-Poulsen, Chem. – Eur. J., 2016, 22, 13265–13274 CrossRef CAS PubMed.
- A. Vlasceanu, S. L. Broman, A. S. Hansen, A. B. Skov, M. Cacciarini, A. Kadziola, H. G. Kjaergaard, K. V. Mikkelsen and M. B. Nielsen, Chem. – Eur. J., 2016, 22, 10796–10800 CrossRef CAS PubMed.
- R. R. Islangulov and F. N. Castellano, Angew. Chem., Int. Ed., 2006, 45, 5957–5959 CrossRef CAS PubMed.
- H. Taoda, K. Hayakawa, K. Kawase and H. Yamakita, J. Chem. Inf. Comput. Sci., 1987, 20, 265–270 CAS.
- K. Masutani, M. Morikawa and N. A. Kimizuka, Chem. Commun., 2014, 50, 15803–15806 RSC.
- K. Edel, X. Yang, J. S. A. Ishibashi, A. N. Lamm, C. Maichle-Mössmer, Z. X. Giustra, S. Liu and H. F. Bettinger, Angew. Chem., Int. Ed., 2018, 57, 5296–5300 CrossRef CAS PubMed.
- K. Moth-Poulsen, D. oso, K. Börjesson, N. Vinokurov, S. K. Meier, A. Majumdar, K. P. C. Vollhardtc and R. A. Segalman, Energy Environ. Sci., 2012, 5, 8534–8537 RSC.
- A. Lennartson, A. Roffey and K. Moth-Poulsen, Tetrahedron Lett., 2015, 56, 1457–1465 CrossRef CAS.
- O. Brummel, D. Besold, T. Döpper, Y. Wu, S. Bochmann, F. Lazzari, F. Waidhas, U. Bauer, P. Bachmann, C. Papp, H. Steinrück, A. Görling, J. Libuda and J. Bachmann, ChemSusChem, 2016, 9, 1424–1432 CrossRef CAS PubMed.
- S. Miki, Y. Asako, M. Morimoto, T. Ohno, Z. Yoshida, T. Maruyama, M. Fukuoka and T. Takada, Bull. Chem. Soc. Jpn., 1988, 61, 973–981 CrossRef CAS.
- D. J. Fife, K. W. Morse and W. M. Moore, J. Am. Chem. Soc., 1983, 105, 7404–7407 CrossRef CAS.
- H. Ken-ichi, Y. Asami and Y. Osamu, Tetrahedron Lett., 1988, 29, 4109–4112 CrossRef.
- S. Miki, T. Maruyama, T. Ohno, T. Tohma, S. Toyama and Z. Yoshida, Chem. Lett., 1988, 861–864 CrossRef CAS.
- V. A. Bren’, A. D. Dubonosov, V. I. Minkin and V. A. Chernoivanov, Russ. Chem. Rev., 1991, 60, 451–469 CrossRef.
- E. J. Wucherer and A. Wilson, U.S. Air Force Research Laboratory, Edwards Air Force Base, California, 1998.
- V. Gray, A. Lennartson, P. Ratanalert, K. Börjesson and K. Moth-Poulsen, Chem. Commun., 2014, 50, 5330–5332 RSC.
- F. Ghani, J. Kristen and H. Riegler, J. Chem. Eng. Data, 2012, 57, 439–449 CrossRef CAS.
- A. L. Tchougreeff, A. M. Tokmachev and R. R. Dronskowski, Int. J. Quantum Chem., 2013, 113, 1833–1846 CrossRef CAS.
- I. Antol, J. Comput. Chem., 2013, 34, 1439–1445 CrossRef CAS PubMed.
- K. Jorner, A. Dreos, R. Emanuelsson, O. L. Bakouri, I. F. Galván, K. Börjesson, F. Feixas, R. Lindh, B. Zietz, K. Moth-Poulsen and H. Ottosson, J. Mater. Chem. A, 2017, 5, 12369–12378 RSC.
- M. Kuisma, A. Lundin, K. Moth-Poulsen, P. Hyldgård and P. Erhart, ChemSusChem, 2016, 14, 1786–1794 CrossRef PubMed.
- M. J. Kuisma, A. M. Lundin, K. Moth-Poulsen, P. Hyldgaard and P. Erhart, J. Phys. Chem. C, 2016, 120, 3635–3645 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee01011k |
| This journal is © The Royal Society of Chemistry 2019 |