
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interfacial benzenethiol modification facilitates charge transfer and improves stability of cm-sized metal halide perovskite solar cells with up to 20% efficiency†
Jianfeng
Lu
 a,
Xiongfeng
Lin
b,
Xuechen
Jiao
cd,
Thomas
Gengenbach
e,
Andrew D.
Scully
e,
Liangcong
Jiang
c,
Boer
Tan
b,
Jingsong
Sun
c,
Bin
Li
c,
Narendra
Pai
a,
Udo
Bach
bef,
Alexandr N.
Simonov
*ag and
Yi-Bing
Cheng
*cfh
a,
Xiongfeng
Lin
b,
Xuechen
Jiao
cd,
Thomas
Gengenbach
e,
Andrew D.
Scully
e,
Liangcong
Jiang
c,
Boer
Tan
b,
Jingsong
Sun
c,
Bin
Li
c,
Narendra
Pai
a,
Udo
Bach
bef,
Alexandr N.
Simonov
*ag and
Yi-Bing
Cheng
*cfh
aSchool of Chemistry, Monash University, Victoria 3800, Australia. E-mail: alexandr.simonov@monash.edu
bDepartment of Chemical Engineering, Monash University, Victoria 3800, Australia
cDepartment of Materials Science and Engineering, Monash University, Victoria 3800, Australia. E-mail: yibing.cheng@monash.edu
dAustralian Synchrotron, ANSTO, 800 Blackburn Rd, Clayton, Victoria 3168, Australia
eCSIRO Manufacturing, Clayton, Victoria 3168, Australia
fARC Centre of Excellence for Exciton Science, Monash University, Victoria 3800, Australia
gARC Centre of Excellence for Electromaterials Science, Monash University, Victoria 3800, Australia
hState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China
First published on 24th April 2018
Abstract
Metal halide perovskite solar cells (PSC) exhibit outstanding power conversion efficiencies when fabricated as mm-sized devices, but creation of high-performing large-area PSCs that are stable under operating conditions on a sufficiently long timescale still presents a significant challenge. We demonstrate herein that modification of the interface between the perovskite and a spiro-OMeTAD hole-transporting material with commercially available para-substituted benzenethiol molecules facilitates fabrication of cm-sized PSCs with both improved efficiency and stability. Comprehensive analysis using specialised and conventional physical characterisation techniques has been undertaken to demonstrate that band alignment at the perovskite surface can be tuned to improve the solar cell efficiency via adsorption of benzenethiols with a significant dipole moment. Moreover, modification of the perovskite with cyano-substituted benzenethiol enhances charge extraction and reduces charge recombination in the devices. These effects enable improvements in the power conversion efficiency of PSCs from 19.0 to 20.2% and from 18.5 to 19.6% under 1 sun AM 1.5G irradiation with 0.16 and 1.00 cm2 apertures, respectively. Most importantly, benzenethiol-modified perovskite solar cells retain more than 80% of the initial performance after 185 h of continuous operation at 50% relative humidity and 50 °C device temperature under 1 sun irradiation, while devices with no interfacial modification undergo continuous deterioration down to 35% of the initial efficiency. These significant improvements are provided by a very simple and highly reproducibile modification procedure that can be readily adopted in other types of PSCs.
Broader contextPhotovoltaic technology offers a sustainable solution to the problem of soaring global energy demands. The domination of crystalline silicon solar cells is expected to ease in the foreseeable future as they approach their theoretical efficiency limit. Metal-halide perovskite solar cells have emerged as the fastest-developing new photovoltaic technology rivalling crystalline silicon, largely due to their compatibility with large-scale production using conventional printing processes. The transition from research to industrial applications of perovskite solar cells requires further advancements in both operational stability and large-area device fabrication – two factors that often display an inverse inter-dependency. The present work demonstrates that incorporation of certain commercially available benzenethiol derivatives at the interface between the perovskite light absorber and the hole-transporting material layers provides the dual benefit of improvement in the band-energy alignment and also protection of the perovskite from environmental degradation. A key feature of this work is the use of cm-sized devices rather than the mm-scale solar cells commonly employed for research purposes. The simple approach developed herein enables fabrication of larger cells with characteristics that are very similar to those of smaller devices. |
Introduction
New breakthroughs in photovoltaic technology are urgently needed to promote the global transition to solar power as a major, essentially inexhaustible source of energy. Although silicon-based photovoltaic technology that dominates the existing solar cell market has proven to be highly reliable, further improvements in its intrinsic efficiency are not straightforward.1 Instead, so-called 3rd generation solar cells have come to the forefront of photovoltaic research and have seen unprecedented progress recently.2 For example, metal halide perovskite solar cells (PSCs) technology has now reached an outstanding certified power conversion efficiency (PCE) of 22.7% after only eight years of active investigation.3 This has been achieved via numerous improvements and optimisations of the perovskite light-absorber composition,4–6 deposition procedures,7–9 selective charge transporting materials,10,11 and interface engineering.12–14 Many of these methods were developed not only to improve the efficiency but also to address a major weakness of the metal halide perovskites, viz. instability under operating conditions.15In a typical PSC, a light-absorber is confined between layers of electron-selective and hole-selective transporting materials (ETM and HTM, respectively). Diffusion lengths of photo-generated charge carriers in metal halide perovskites are on the micrometer scale under solar illumination, and hence transport losses within these materials are very low.16 Thus, the majority of losses occur due to charge recombination across the perovskite|ETM and perovskite|HTM interfaces. The properties of these interfacial regions determine both the initial photovoltaic performance and stability of the solar cells, as documented in a plethora of recent publications focusing on the optimisation of interfaces in PSCs.17,18 For example, it was reported that ionic defects at the surfaces and grain boundaries of perovskite films are detrimental to both the efficiency and stability of PSCs.19,20 Snaith and co-workers found that iodopentafluorobenzene and Lewis bases, such as pyridine or thiophene, efficiently passivate trap states on the perovskite surface via interaction with the under-coordinated halide anions and metal cations that act as charge-carrier recombination centers.17 Yang and Huang demonstrated that a layer of quaternary alkyl ammonium cations on the perovskite surface can substantially reduce the charge trap density and elongate the charge carrier lifetime, improving both device performance and stability.21,22 More recently, Seok and colleagues reported that additional iodide ions in the organic cation solution decrease the concentration of deep-level defects in perovskite films. This rational defect-engineering of the perovskite layer resulted in the highest PCE for PSCs reported to date.3
It has also been reported that the electric field at a typical perovskite|HTM or ETM|perovskite interface is not strong enough to sustain efficient electron or hole extraction.23–25 Accumulation of electrons and holes at the perovskite surface and subsurface close to the interface is currently considered to be responsible for the performance hysteresis behaviour and irreversible degradation of PSCs.18 Several groups reported that the problem can be mitigated via introduction of specifically designed interlayers between perovskite and a charge-selective material, which can additionally stabilise devices via surface passivation.26 For example, a π-conjugated Lewis base with n-type semiconductor properties 1,1-dicyanomethylene-3-indanone was used to promote charge extraction at the interface between methyl ammonium lead iodide and ETM, while also improving the stability via scavenging Lewis acid traps on the surface and at the grain boundaries of the perovskite.27 In another report, a strong interfacial dipole moment was created between a perovskite light-absorber and a C60 electron transporter by depositing a sub-nm layer of tris(2,4,6-trimethyl-3-(pyridin-3-yl)phenyl) borane to facilitate fast charge sweep-out and suppress carrier recombination at the interface.28 However, we are not aware of any previous reports on modification of the perovskite|HTM interfaces with dipoles to improve the charge-transfer as was done previously at perovskite|ETM interfaces.
Most of the interface modification and other strategies devised hitherto with the aim to endow perovskite solar cells with high power conversion efficiencies and robustness were applied to devices of small active areas (∼0.1 cm2), whereas examples of larger high-performing perovskite solar cell are rare.29–32 However, the performance of these small PSCs has now reached the level where demonstration of methods for fabrication of high-efficiency and stably operating cm-sized solar cells is an equally important and pressing scientific task.2 In addition, creation of high-quality interfaces to prevent recombination and degradation becomes increasingly challenging with an increase in the active area of a device.33–35
The present work explores the possibility for modification of the high-performing perovskite crystal surface with para-substituted benzenethiol molecules having different dipole moments. Such treatment was anticipated to modulate the band alignment at the perovskite|HTM interfaces, which could assist charge extraction and suppress carrier recombination. Furthermore, facilitated charge extraction can reduce the charge accumulation at the perovskite surface and thereby improve the lifetime of solar cells.18
Results and discussion
Solar cell architecture
All solar cells investigated in this work had a standard n–i–p architecture (Fig. 1a) and were fabricated by sequential deposition of components onto glass covered with fluorine-doped tin(IV) oxide, which served as a cathode in the final device. Thoroughly optimised procedures adopted from the literature36 and established in our laboratory6 were employed. The examined solar cells featured a ca. 150 nm mesoporous titania scaffold (30 nm particle size) impregnated with a high-performing Cs0.05FA0.79MA0.16PbI2.49Br0.51 perovskite (FA+ = CH(NH2)2+, MA+ = CH3NH3+) followed by a ca. 500 nm thick pure light-absorber, a spiro-OMeTAD hole-selective layer and a gold counter electrode. TiO2 and spiro-OMeTAD, which are the most widely employed electron- and hole-transporting materials in PSCs research, were employed here to facilitate comparison of our results with the ample literature. A cross-sectional scanning electron microscopic (SEM) image of a typical PSC is exemplified in Fig. 1b. The mixed perovskite was deposited by a one-step anti-solvent procedure followed by annealing at 100 °C for 60 min in an inert environment;49 formation of the target material was confirmed by X-ray diffraction (XRD) analysis (Fig. S1, ESI†). XRD patterns exhibited a set of sharp reflections expected for the cubic Cs0.05FA0.79MA0.16PbI2.49Br0.51 perovskite phase with a mean crystallite size of 142 ± 4 nm (derived from the full width at half maximum of the (100) peak). Weak diffraction peaks at 11° and 12.6° are associated with minor admixtures of yellow hexagonal δH phase and PbI2.5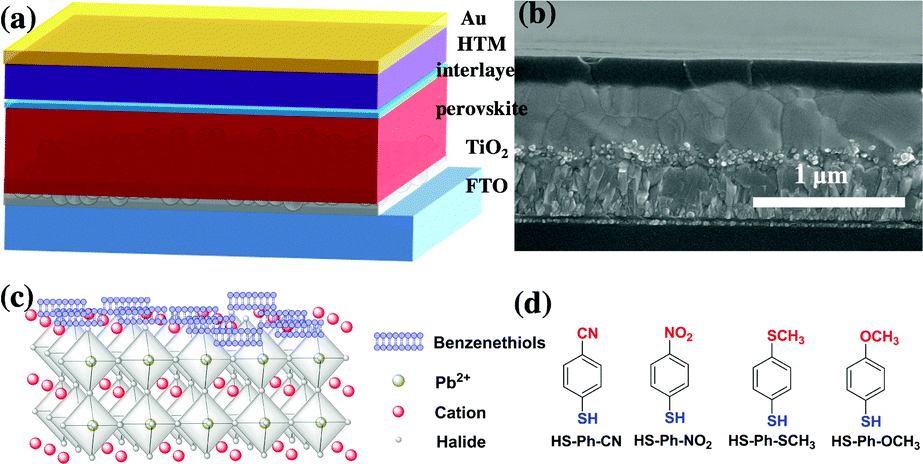 | ||
| Fig. 1 (a) Schematic presentation and (b) a typical cross-sectional SEM image of the investigated solar cells featuring a glass support covered with fluorine-doped tin(IV) oxide (FTO), a compact TiO2 coating (c-TiO2), a mesoporous TiO2 substrate filled with the Cs0.05FA0.79MA0.16PbI2.49Br0.51 perovskite (FA+ = CH(NH2)2+, MA+ = CH3NH3+), a perovskite layer, interface modification, a hole-transporting material spiro-OMeTAD and a Au counter electrode. (c) Possible binding mode of a benzenethiol molecule on the perovskite surface. (d) Chemical structures of benzenethiols used to modify the perovskite|HTM interface. | ||
Adsorption of benzenethiols onto the perovskite layer
According to the hard/soft acid/base theory, sulphur is a relatively soft (polarisable) atom. This defines the propensity of thiols to bind to other soft elements/ions, in particular lead, which motivated us to use molecules featuring –SH functional group for interfacial modification of the perovskite surface. Among other possibilities, we chose benzenethiols as they can form rigid and dense hydrophobic layers. Moreover, para-substitution in the aromatic moiety allows for tuning of the dipole moment of these molecules. Thus, benzenethiol derivatives para-substituted with –CN, –NO2, –SCH3, or –OCH3 (Fig. 1d) were examined. In some experiments, the effects of modification of the perovskite layer with HS-Ph-F having a significantly lower dipole moment were also studied.37 Importantly, the direction of the molecular dipoles for HS-Ph-NO2 and HS-Ph-CN is opposite to that for benzenethiols functionalised with –F, –SCH3 and –OCH3.37Modification of the perovskite|HTM interface was achieved by spin-coating a 20 mM benzenethiol solution in chlorobenzene onto a freshly deposited light-absorber layer prior to coating with a hole transporter. The morphology of the perovskite layer after surface treatment was examined by atomic force and scanning electron microscopy, showing no significant change as compared to the unmodified sample (Fig. S2, ESI†). A clear-cut confirmation of successful deposition of benzenethiols was derived from XPS experiments undertaken using synchrotron radiation with a photon energy of 488 eV (Fig. 2a). An incident X-ray energy close to the sulphur absorption edge significantly amplifies the photoelectron signal and thus the sensitivity of the measurements.38 More specifically, the intensity is enhanced approximately 10-fold as compared to the analysis carried out with 1486.7 eV photons. S 2p3/2 peaks within the binding energy range 164–165 eV manifesting in the spectra recorded for the benzenethiol-treated films were confidently attributed to thiol functionality.39 Importantly, no sulphur could be detected for the unmodified sample even with this highly sensitive technique (Fig. 2a). The Pb 4f spectra (recorded with a conventional laboratory-based XPS instrument; Fig. S3, ESI†) of the unmodified and HS-Ph-CN modified perovskite films exhibited one major doublet with the Pb 4f7/2 maximum at 138.4 eV, which is consistent with the Pb2+ oxidation state. Interestingly, a very weak signal with the Pb 4f7/2 peak at ca. 136.6 eV, which formally corresponds to the Pb0 state, was identified in the unmodified Cs0.05FA0.79MA0.16PbI2.49Br0.51 perovskite, but not in the benzenethiol-treated sample. Unambiguous detection of an increase in the intensity at 137.8 eV due to formation of the Pb–S bonds upon modification with HS-Ph-CN was not successful due to the very low amounts of introduced thiol. Both synchrotron-radiation and lab-based XPS also confirmed successful embedding of caesium into the perovskite structure (Fig. 2b). Independence of the Cs 3d signal intensity from the presence of benzenethiols indicates that the latter are deposited as a very thin layer. This is also supported by FTIR and UV-Vis analyses, which were unable to unambiguously detect the benzenethiol-modification of the perovskite surface under the employed conditions (Fig. S4, ESI†).
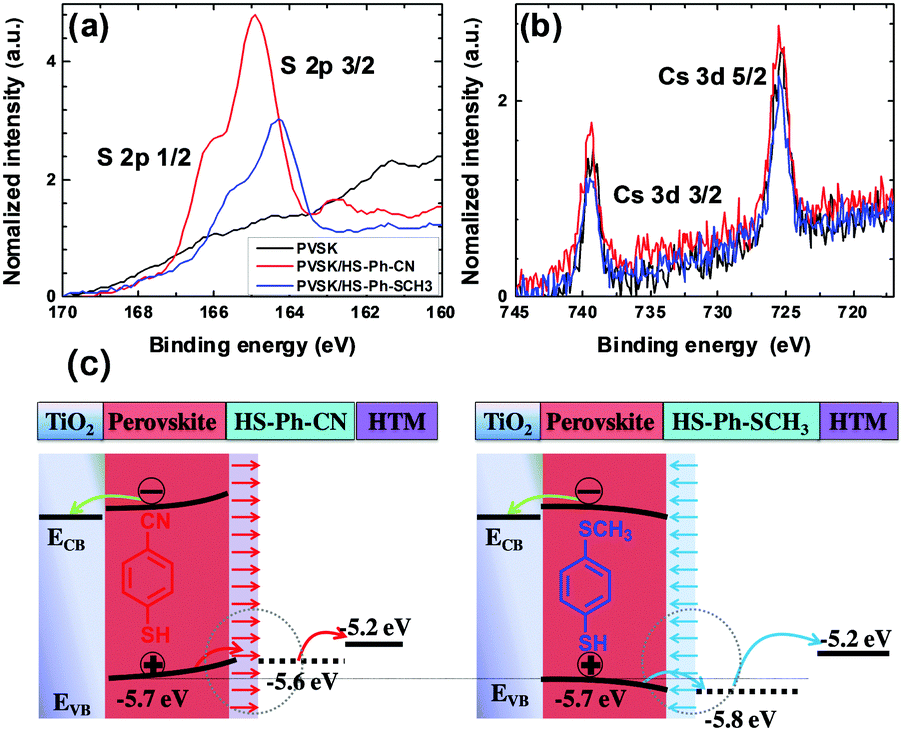 | ||
| Fig. 2 (a) S 2p and (b) Cs 3d spectra of the Cs0.05FA0.79MA0.16PbI2.49Br0.51 perovskite films that were unmodified, or treated with either HS-Ph-CN or HS-Ph-SCH3; data were derived from the synchrotron XPS analysis. (c) Hypothesised alteration of the energy levels at the perovskite|HTM interface under open-circuit conditions induced by modification with dipole benzenethiols. Valence band energy levels were derived from UPS experiments. | ||
The impact of adsorbed benzenethiols on the electronic properties of the perovskite layer was probed by ultraviolet photoelectron spectroscopy (UPS). The ionisation potential was calculated as the difference between the work function and the highest occupied molecular orbital energy cut-off (Fig. S5, ESI†) and was assumed to be equal to the valence band energy level (EVB). Unmodified Cs0.05FA0.79MA0.16PbI2.49Br0.51 perovskite films exhibited an EVB of −5.7 eV, which is consistent with the value reported in the literature.40 The adsorption of benzenethiols induced small but detectable changes in the measured valence band energy level that were consistent with the directions of the respective dipole moments of the examined molecules (Fig. 2c and Table S1, ESI†). However, the accuracy of this analysis is at a level of ±0.1 eV, and therefore the detected band shifts should not be considered as an unambiguous proof of modification of the perovskite energy level by adsorbed benzenethiols. Nevertheless, analysis undertaken on different samples and at different spots provided satisfactorily consistent results that are summarised below. Specifically, modification with HS-Ph-CN and HS-Ph-NO2 up-shifted the EVB of the perovskite to approximately −5.6 eV, while an opposite effect and valence band energy of ca. −5.8 eV was found upon treatment with HS-Ph-SCH3 and HS-Ph-OCH3 (Table S1, ESI†). The weakest effect on EVB, though still consistent with the dipole direction, was produced by HS-Ph-F having the lowest dipole moment among the investigated benzenethiols. The observed shifts can be rationalised in terms of the formation of an interfacial dipole moment that can generate a concomitant increase/decrease of the work function and an upward/downward vacuum-level shift at the interface (Fig. 2c). Further evidence for modulation of the work function in the perovskite surface layer due to modification with HS-Ph-CN was derived from the Kelvin probe measurements of a contact potential difference (Δψ) (Fig. S6, ESI†). A more negative Δψ obtained for benzenethiol-treated Cs0.05FA0.79MA0.16PbI2.49Br0.51 as compared to the intact film further supports a decrease in the work function.
Previous research on colloidal quantum dots (QD) demonstrated that their band energies can be modified by changing the nature of the capping ligand.41,42 The identity of the binding functionality and dipole moment of the ligand influence the strength of the QD-ligand surface dipole, thus shifting the valence band maximum (EVB) and conduction band minimum (ECB). For example, the EVB of PbS quantum dots treated with 1,2-ethanedithiol (EDT) is up-shifted by 0.2 eV with respect to that of the same QDs modified with tetrabutylammonium iodide. Such EDT treatment creates an electron-blocking/hole-extraction layer on the QD surface and thereby improves both the efficiency and stability of the PbS-based solar cells. Moreover, detailed investigations of this system suggested that differences in the Pb–halide and Pb–thiol interactions generate surface dipole moments, which enables a favourable band alignment shift at the interface. Based on these results, we hypothesise that the modification of the perovskite surface with HS-Ph-CN and HS-Ph-NO can also change the interfacial dipole moment and band alignment and thereby facilitate charge-transfer (Fig. 2c).
To investigate the effects of the interfacial benzenethiol on the charge transfer between the perovskite and the HTM, photoluminence (PL) measurements were undertaken. All examined films displayed qualitatively similar PL spectra with a maximum at around 770 nm, and stronger quenching was always observed for the benzenethiol-treated samples (Fig. 3a). We have further analysed the charge transfer kinetics through time-resolved photoluminescence (TRPL) measurements (Fig. 3b). The PL decay curves were fitted using a bi-exponential dependence, where the fast process is caused by bimolecular recombination of photo-generated free carriers, and the slow decay process is attributed mainly to trap-assisted recombination (Table S2, ESI†).43 The unmodified perovskite|HTM sample exhibited an average PL lifetime of 68 ns, which was notably shortened upon interfacial modification with benzenethiols down to 53 (HS-Ph-CN), 15 (HS-Ph-NO2), 29 (HS-Ph-SCH3) and 22 ns (HS-Ph-OCH3). These results suggest that the benzenethiol molecules facilitate faster charge transfer between the perovskite and the HTM, which is beneficial to the hole extraction process and therefore better device performance can be expected. Thus, our PL results are in line with the previous work on EDT-treated PbS quantum dots mentioned above,41 as well as with the reports on the improvement of the charge-transfer at the perovskite|HTM interface provided by the formation of Pb–S coordination bonds.44,45 Importantly, additional washing of the modified perovskite films with chlorobenzene induced marginal changes in the steady-state photoluminescence spectra, attesting to the firm immobilisation of the examined benzenethiols on the Cs0.05FA0.79MA0.16PbI2.49Br0.51 surface (Fig. S7, ESI†).
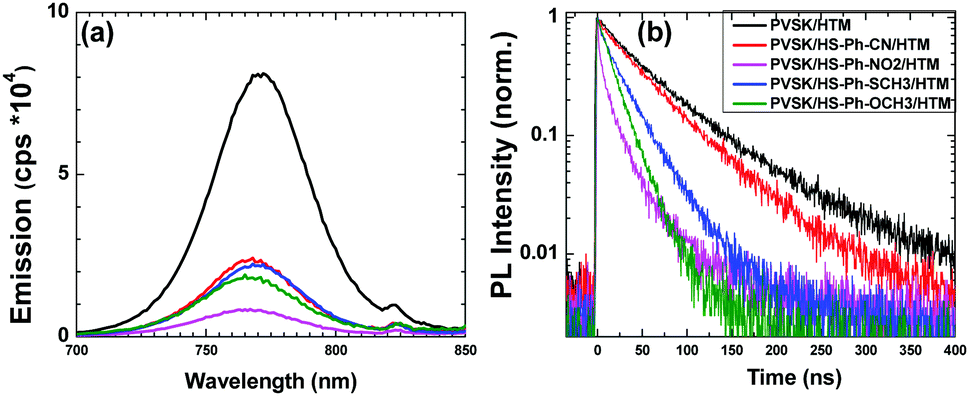 | ||
| Fig. 3 Effects of benzenethiols on the photoluminescence properties of the Cs0.05FA0.79MA0.16PbI2.49Br0.51|spiro-OMeTAD system: (a) steady-state PL spectra, (b) time-resolved PL decay curves. The perovskite layer was either unmodified, or treated with HS-Ph-CN, HS-Ph-NO2, HS-Ph-SCH3, or HS-Ph-OCH3. The excitation wavelength was 465.8 nm in all experiments. | ||
To further investigate the charge transfer between the perovskite and benzenethiol molecules, PL and TRPL measurements were carried out with no HTM present. Again, modification of Cs0.05FA0.79MA0.16PbI2.49Br0.51 induced quenching of the PL intensity by 80–95% (Fig. S8, ESI†) and lifetime from 7.6 to 0.5–1.2 μs (Table S2, ESI†). Thus, both steady-state PL and TRPL results support our hypothesis on the charge transfer from perovskite to benzenethiols. Indeed, this transfer is energetically favourable, as the HOMO levels of all examined benzenethiol molecules inferred from the photoelectron spectroscopy in air (PESA) analysis (Fig. S9, ESI†) are more positive than the valence band edge of the perovskite. It can be further suggested that the detected hole trapping can be due to a redox transformation of a benzenethiol that corresponds to an energy level situated higher than the EVB of the perovskite (Fig. 2c). In this case, trapped holes in thiol molecules would produce thyil radicals, which can undergo bimolecular recombination with the formation of disulphide species.46 When HTM is present, it can directly accept trapped holes from the benzenethiol molecules. Thus, we suggest that the benzenethiol molecules between the perovskite and the HTM function as a hole extraction interlayer, which can facilitate the charge-extraction process in a working device.
Photovoltaic performance
All photovoltaic measurements were undertaken under simulated 1 sun illumination (100 mW cm−2 intensity, AM 1.5G spectrum). In initial tests aiming to explore the effects of different benzenethiol molecules on the performance of FTO|c-TiO2|m-TiO2 + Cs0.05FA0.79MA0.16PbI2.49Br0.51|spiro-OMeTAD|Au solar cells (cross section SEM images are shown in Fig. S10, ESI†), photocurrent density (J) vs. voltage (V) characteristics and incident photon-to-charge carrier efficiency (IPCE) spectra were recorded with a small aperture providing an irradiated area of 0.16 cm2 (Fig. S11, ESI†). Major photovoltaic parameters derived from these data are summarised in Table 1. The set of unmodified devices with no interfacial modification showed comparatively high and reproducible performance.36 The best solar cell of this type produced in this work exhibited an open circuit voltage (VOC) of 1.08 V, a short circuit current density (JSC) of 22.7 mA cm−2, a fill factor (FF) of 0.78 and a PCE of 19.0%.| Device | V OC (mV) | J SC (mA cm−2) | FF | PCE (%) | SPOb (%) | |
|---|---|---|---|---|---|---|
| a Derived from the J–V curves recorded for 25 devices of each type in the forward-bias to short-circuit direction at a scan rate of 0.100 V s−1 under 1 sun simulated irradiation; VOC – open-circuit voltage; JSC – short-circuit current density (values in brackets were obtained via integration of the IPCE spectra; see discussions in the text explaining the differences between JSC derived from the J–V and IPCE data); FF – fill factor; PCE – power conversion efficiency (values in brackets were calculated using JSC derived from the IPCE spectra). b Steady-state power conversion efficiency measured for the best-performing device at a potential corresponding to the maximum power point in the J–V curve. | ||||||
| Unmodified | Best | 1076 | 22.7 (21.0) | 0.78 | 19.0 (17.6) | 18.4 |
| Average | 1081 ± 9 | 22.2 ± 0.1 | 0.77 ± 0.02 | 18.4 ± 0.4 | ||
| HS-Ph-CN | Best | 1106 | 22.8 (21.3) | 0.80 | 20.2 (18.8) | 19.6 |
| Average | 1102 ± 22 | 22.4 ± 0.6 | 0.78 ± 0.03 | 19.1 ± 0.7 | ||
| HS-Ph-NO2 | Best | 1108 | 22.9 (21.0) | 0.79 | 20.0 (18.4) | 19.3 |
| Average | 1097 ± 10 | 22.4 ± 0.3 | 0.78 ± 0.01 | 19.1 ± 0.2 | ||
| HS-Ph-SCH3 | Best | 1090 | 22.2 (20.8) | 0.79 | 19.1 (17.9) | 18.5 |
| Average | 1088 ± 11 | 21.9 ± 0.3 | 0.78 ± 0.01 | 18.6 ± 0.3 | ||
| HS-Ph-OCH3 | Best | 1087 | 22.7 (20.7) | 0.78 | 19.4 (17.6) | 18.5 |
| Average | 1085 ± 6 | 22.3 ± 0.40 | 0.77 ± 0.02 | 18.8 ± 0.4 | ||
Treatment of the perovskite surface with benzenethiols prior to deposition of a HTM layer produced solar cells that deliver photocurrent densities and FFs similar to those for unmodified devices, but their VOC was increased by ca. 5–20 mV (Table 1). Reproducibility of this improvement attests to the reliability of the effect. Inspection of the data in Table 1 and Table S1 (ESI†) suggests that HS-Ph-CN and HS-Ph-NO2 benzenethiols having their dipole moments pointing away from the perovskite surface and thereby inducing a positive shift of the valence band edge of the light-absorber (Fig. 2c) provide greater improvements in VOC and the photovoltaic efficiency. Conversely, HS-Ph-OCH3 and HS-Ph-SCH3 having dipole moments oriented in the opposite direction induce a negative shift of EVB (Fig. 2c) and increase the open-circuit voltage to a smaller extent (Table 1). The fact that methoxy- and methylsulfanyl-substituted benzenethiols also slightly enhance VOC, notwithstanding the negative shift of EVB (Table S1, ESI†), can be rationalised in terms of surface trap passivation by the self-assembled monolayers of these compounds on the perovskite surface.47 Importantly, control experiments where the perovskite surface was pre-treated with either pure chlorobenzene or HS-free anisole (chlorobenzene solution) produced essentially no improvements in VOC and other photovoltaic parameters (Fig. S12, ESI†).
Thus, the use of HS-Ph-CN to modify the perovskite surface produced the highest efficiencies, among which the best result corresponds to the following set of parameters: VOC = 1.11 V, JSC = 22.8 mA cm−2, FF = 0.80, transient PCE = 20.2% and steady-state PCE = 19.6% at 0.93 V over 500 s (Fig. S13, ESI†). The integrated JSC derived from the IPCE spectra are 21.0 and 21.3 mA cm−2 for the unmodified and modified devices, respectively, which is lower than the values obtained during the J–V measurements (Table 1). The following factors contribute to this discrepancy. Our IPCE setup is not sensitive below 350 nm, whereas the solar simulator used for J–V measurements produces the full spectrum of real sunlight, which contributes approximately 1 mA cm−2 to the difference in JSC. The relationship between JSC and illumination intensity is not strictly linear, which induces a further difference of ca. 1 mA cm−2. A minor difference of 0.5 mA cm−2 is attributed to experimental uncertainties.
Further, PSCs having an active area of more than 1.4 cm2 (aperture area 1.00 cm2) with or without a HS-PH-CN interfacial layer were fabricated and tested (Fig. 4a and Table 2). The best-performing unmodified devices (no benzenethiol interlayer) demonstrated a VOC of 1.10 V, a JSC of 23.2 mA cm−2, a FF of 0.71 and a PCE of 18.5%, which compares well with previously reported results.30 Notable improvement in all photovoltaic parameters was achieved via modification of the perovskite with HS-PH-CN, which provided devices with characteristics as high as VOC = 1.11 V, JSC = 23.8 mA cm−2, FF = 0.74 and PCE = 19.6%. The increased JSC was due to the improved incident photon-to-current conversion efficiency (IPCE), especially within the wavelength ranges 350–400 nm and 600–700 nm (Fig. 4b). However, the major contribution to the improved PCE was from the fill factor, which increased from 0.71 to 0.74 (Table 2). This improvement can be attributed to the enhanced hole extraction and reduced charge accumulation at the perovskite|HTM interface. During steady state PCE output tracking (conducted by applying a constant potential under continuous AM 1.5G for 4000 s), a drop in the PCE for the unmodified device from 18.5 to 17.2% was observed, whereas the HS-PH-CN-modified solar cell exhibited a more robust behaviour with efficiency only slightly decreasing from 19.4 to 19.0% (Fig. 4c). This PCE decrease is attributed mainly to a continuous heating of the working devices, as no cooling was provided during the measurements. This observation indicates an improved stability provided by the interfacial modification with benzenethiols, which is discussed in more detail in the next sub-section.
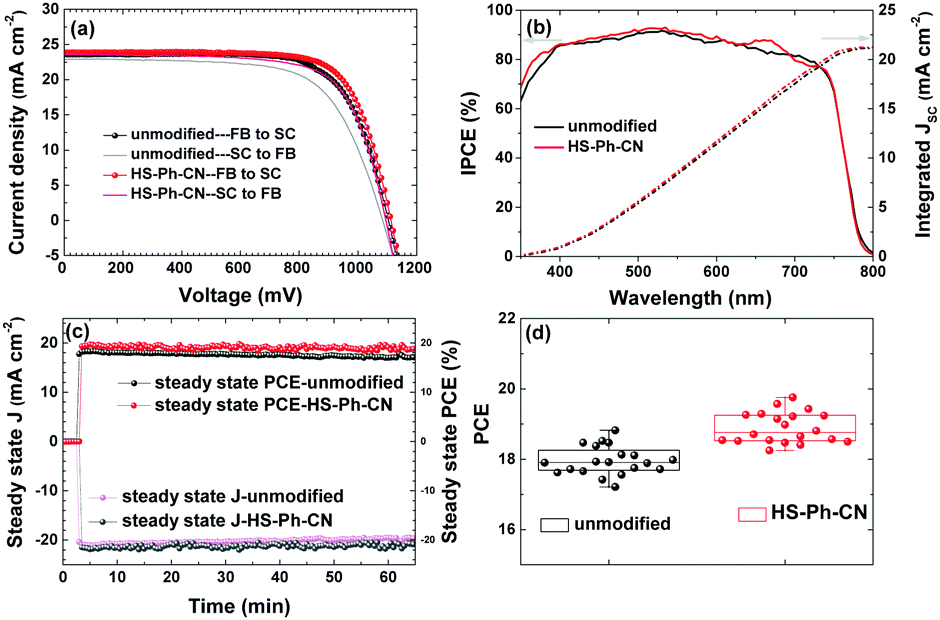 | ||
| Fig. 4 Photovoltaic performance of the large area (aperture 1.00 cm2) Cs0.05FA0.79MA0.16PbI2.49Br0.51-based solar cells without (black and grey) and with interfacial HS-Ph-CN modification (red and magenta): (a) J vs. V curves (scan rate 100 mV s−1) recorded in the forward bias (FB) to short-circuit (SC), and SC to FB directions; (b) IPCE spectra. The surface of the perovskite layer was either unmodified (black), or treated with HS-Ph-CN (red); (c) temporal evolution of the photocurrent density and the PCE at 0.87 V (unmodified) and 0.90 V (treated with HS-Ph-CN); (d) reproducibility of the major photovoltaic parameters within 20 independent devices of each type. All measurements were under AM 1.5G 1 sun irradiation. | ||
| Device | V OC (mV) | J SC (mA cm−2) | FF | PCE (%) | ||
|---|---|---|---|---|---|---|
| a Derived from the J–V curves recorded for 20 devices of each type in the forward-bias to short-circuit direction at the scan rate of 0.100 V s−1 under 1 sun simulated irradiation; VOC – open-circuit voltage; JSC – short-circuit current density; FF – fill factor; PCE – power conversion efficiency. b Steady-state power conversion efficiency measured for the best-performing device at the potential corresponding to the maximum power point in the J–V curve. | ||||||
| Unmodified | Average FB to SCb | 1096 ± 16 | 23.2 ± 0.5 | 0.71 ± 0.02 | 18.0 ± 0.4 | |
| Best | SC to FB | 1085 | 22.9 | 0.67 | 16.6 | |
| FB to SC | 1104 | 23.6 | 0.71 | 18.5 | ||
| HS-Ph-CN | Average FB to SCb | 1116 ± 10 | 23.5 ± 0.5 | 0.72 ± 0.02 | 18.9 ± 0.4 | |
| Best | SC to FB | 1095 | 23.7 | 0.70 | 18.3 | |
| FB to SC | 1113 | 23.9 | 0.74 | 19.6 | ||
Importantly, the devices displayed excellent reproducibility as concluded from testing 20 solar cells in five independent batches with and without benzenethiol modification (Fig. 4d). Moreover, J–V curves measured for five non-overlapping regions at the centre and corners of the large device using an aperture of 0.16 cm2 also demonstrated a very consistent set of photovoltaic parameters, which indicates a homogeneous distribution of the cell components over the square-centimetre scale area (Fig. S14 and Table S4, ESI†). The electroluminescence imaging technique48 also confirmed the uniformity of the perovskite and charge-transfer layers in the active area of the unmodified and HS-Ph-CN-modified devices (Fig. S15, ESI†).
To obtain deeper insights into the impact of benzenethiol modification on the charge-carrier dynamics and the enhanced performance of the PSCs, the influence of light intensity (P) on the JSC and VOC characteristics was investigated. Analysis of the obtained JSCvs. P data in bilogarithmic coordinates revealed that the photocurrent density depends linearly on the irradiation intensity for both unmodified and HS-Ph-CN-treated devices (Fig. 5a). According to the literature, this indicates that the solar cells examined herein are not significantly affected by the space charge effect.49,50 The high quality of the devices was further confirmed by the linear dependence of the VOC on the logarithm of the light intensity (Fig. 5b). This is consistent with the fundamental relationship VOC = nkBT/qln(P), where n is a constant often referred to as the ideality factor, k is Boltzmann's constant, T is the temperature and q is the elementary charge. The properties of a solar cell at open circuit are strongly dependent on recombination processes, since there is no current extraction under these conditions and all photo-generated charge carriers recombine. According to a previous study,51 the light-intensity dependence of the VOC can provide insights into the role of trap-assisted recombination versus bimolecular recombination at open-circuit. It is generally accepted that the ideality factor should be equal to 1 if Langevin recombination dominates, whereas additional involvement of the interfacial trap-assisted Shockley–Read–Hall (SRH) recombination would result in n greater than 1.52 The ideality factors obtained for unmodified (1.73) and HS-PH-CN-modified (1.64) devices imply that trap-assisted SRH recombination is present in both devices. The values of n measured here for Cs0.05FA0.79MA0.16PbI2.49Br0.51-based PSCs are consistent with the literature reports on triple cation analogues.53,54 The smaller n provided by the HS-PH-CN treatment also suggests that the benzenethiol molecules suppress SRH recombination and consequently lead to the improvement of the photovoltaic performance.
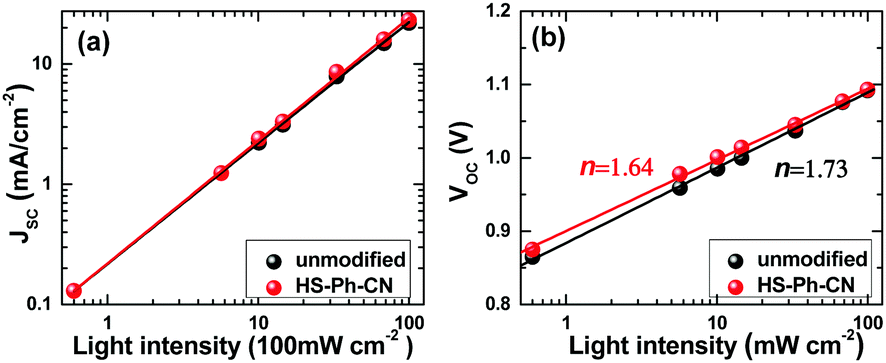 | ||
| Fig. 5 Dependence of the (a) JSC and (b) VOC of unmodified (black) and HS-Ph-CN-treated (red) Cs0.05FA0.79MA0.16PbI2.49Br0.51-based solar cells (aperture 1.00 cm2) on the light intensity. Lines show linear fits to the experimental data. | ||
To further examine the effects of interfacial HS-Ph-CN modification on recombination and charge-extraction in PSCs, transient photovoltage decays (TVD) and transient photocurrent decays (TCD) at modulated light intensity were measured. The TVD data could be well-fitted with a bi-exponential function, which indicates the presence of two distinct populations of the photogenerated carriers that recombine independently.55 The corresponding lifetimes, τ1 and τ2, were within the ranges 0.2–10 μs and 50–200 μs, respectively. According to a previous report,55 a charge population with a longer lifetime (τ2) is associated with electrons transported through the TiO2 matrix to the FTO anode, while the faster decaying component (τ1) is due to the charges confined within a perovskite layer and recombining with the holes in the HTM. Indeed, the τ2 values within the relevant VOC range for unmodified and HS-Ph-CN-treated devices were very similar (Fig. 6a), as expected for solar cells featuring identically prepared FTO|TiO2 components. However, a clear difference in the τ1-VOC profiles due to the benzenethiol modification was found, which was especially pronounced at higher light intensities (Fig. 6a). Importantly, this faster process occurring at the perovskite|HTM boundary was the predominant charge recombination pathway in the solar cells examined here, as concluded on the basis of the fitting of amplitude values (Table S4, ESI†). The latter observation is also consistent with previous studies.55,56 The transient photocurrent decay data were fitted with an exponential dependence to derive the charge-extraction times, which were always faster for the HS-Ph-CN-based devices (Fig. 6b). This result indicates that the benzenethiol treatment reduces the concentration of charge traps and allows for more efficient charge extraction.
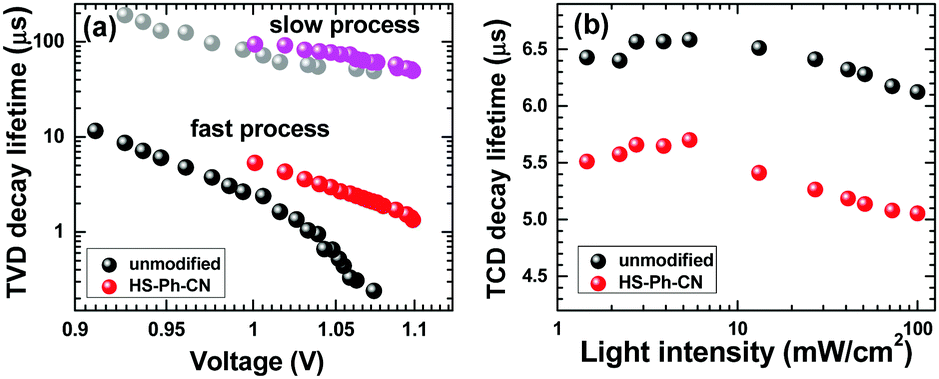 | ||
| Fig. 6 (a) Recombination lifetime versus the open-circuit voltage (derived from transient photovoltage decays at modulated light intensity), and (b) extraction lifetime versus light intensity (derived from transient photocurrent decays at modulated light intensity) for unmodified and HS-Ph-CN-treated Cs0.05FA0.79MA0.16PbI2.49Br0.51-based solar cells. | ||
Taken together, the above TVD and TCD observations along with the PL and UPS data suggest that the conduction band offset between perovskite + HS-Ph-CN and HTM can provide a barrier that prevents photogenerated electrons from flowing to the hole transporter, whereas the valence band bending can intensify the flow of photogenerated holes to the spiro-OMeTAD layer. Thus, the presented experimental evidence indicates that the HS-Ph-CN modification of the perovskite produces an interfacial hole extracting and electron blocking layer, which reduces the charge recombination and facilitates the transfer of holes from the light-harvester into the hole transport material.39 This can explain the improvements in the VOC and the FF of the HS-Ph-CN-modified devices (Fig. 4).
Device stability and long-term performance
Degradation of the Cs0.05FA0.79MA0.16PbI2.49Br0.51 films with and without benzenethiol surface modification that were directly exposed to ambient conditions (air, 45 ± 10% relative humidity, 22 ± 2 °C, continuous ambient room lighting) was monitored for 66 days. During these experiments, the unmodified perovskite film completely decomposed to PbI2, as inferred from the evolution of a sharp peak at 12.6° and the disappearance of the perovskite peak at 14.1° in the XRD patterns (Fig. S16a, ESI†). Improvement in the film stability, viz. slower evolution of the PbI2 diffraction signals, was provided by treatment of the perovskite with HS-Ph-NO2 and HS-Ph-CN (Fig. S16b and c, ESI†), but not with HS-Ph-OCH3 and HC-Ph-SCH3 (Fig. S16d and e, ESI†). Interestingly, the former two molecules exhibit a dipole moment pointing away from the perovskite, while the methoxy- and methylsulfanyl-substituted benzenethiols have dipole moments pointing towards the Cs0.05FA0.79MA0.16PbI2.49Br0.51 film. The underlying reasons for the observed phenomenon are yet to be established.Further stability tests were undertaken with 1 cm2 devices. The performance of the non-encapsulated HS-Ph-CN-modified solar cells stored at 22 °C in the dark under air with <30% relative humidity was tracked for 264 days. The devices retained 90% of the initial performance after this long-term storage test (Fig. 7ai–di and Fig. S17, ESI†).
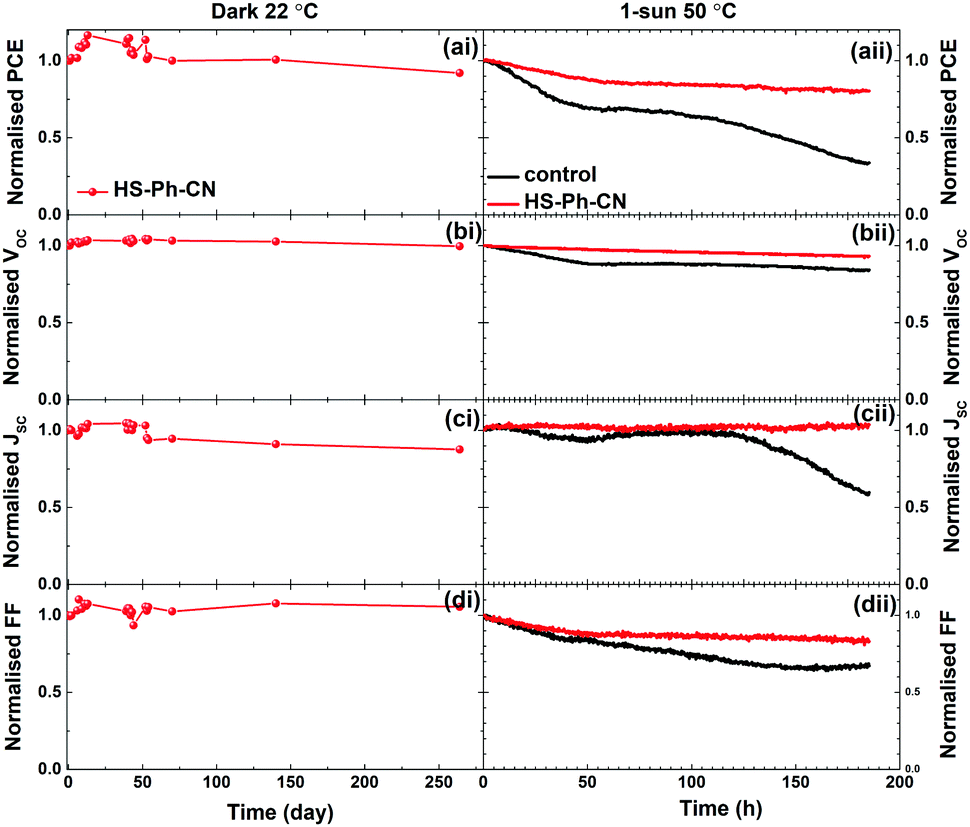 | ||
| Fig. 7 Evolution of the normalised (a) PCE, (b) VOC, (c) JSC and (d) FF for (i) non-encapsulated HS-Ph-CN-treated Cs0.05FA0.79MA0.16PbI2.49Br0.51-based PSCs (aperture 1.00 cm2) stored at 22 °C in the dark in a dry-box (RH < 30%), and (ii) encapsulated Cs0.05FA0.79MA0.16PbI2.49Br0.51-based PSCs (aperture 1.00 cm2) without (black) and with (red) interfacial HS-Ph-CN modification under 1 sun AM 1.5G irradiation at 50 °C (device temperature) in air at a relative humidity of 50%. Data were extracted from SC to FB sweeps of J–V curves recorded at 100 mV s−1. Starting parameter values were: (i) PCE = 17.0%, JSC = 22.8 mA cm−2, VOC = 1.09 V, FF = 0.69; (ii) PCE = 17.9 and 19.2%, VOC = 1.09 and 1.13 V, JSC = 22.8 and 23.6 mA cm−2, FF = 0.72 and 0.72 for the unmodified and HS-Ph-CN-modified devices, respectively. | ||
The stability of encapsulated PSCs was studied under continuous simulated 1 sun AM1.5G light soaking in an environmental chamber at a constant relative humidity of 50% and temperature of 50 °C. These conditions were chosen to mimic the real working environment of solar cells. Juxtaposition of the evolution of the normalised PCE, JSC, VOC and FF for the unmodified and HS-Ph-CN-modified devices demonstrates significant improvements in robustness provided by this benzenethiol interfacial layer (Fig. 7aii–dii). Indeed, the PCE of the benzenethiol-free solar cell degraded from 18.0% down to 6.1% over 185 hours of testing, in keeping with previous reports.57 Conversely, the performance of the HS-Ph-CN-modified devices decreased from 19.2 to 16.2% after initial 50 h and then suffered very small degradation during subsequent 135 h of the experiment (Fig. 7aii). Remarkably, only minor deterioration in VOC was observed (Fig. 7bii) and JSC did not undergo any changes (Fig. 7cii) in this experiment, which further confirms the capacity of HS-Ph-CN to stabilise Cs0.05FA0.79MA0.16PbI2.49Br0.51 perovskite. Major losses in the efficiency of the PSCs with interfacial benzenethiol modification were due to degradation of the fill factor, which, however, was much more pronounced for the unmodified devices (Fig. 7dii). This positive effect of the HS-Ph-CN modification on FF might be associated with the suppression of the corrosion reaction between inherent ionic defects in the perovskite layer and gold species migrated from the anode.58 The overall improvement in the stability of the PSCs is rationalised in terms of suppressed interfacial recombination provided by the HS-Ph-CN interlayer.
Finally, device stability was additionally assessed at 60 °C, which is expected to induce phase transition in the perovskite.59 These tests were undertaken in the dark and under continuous 1 sun irradiation. In both cases, an evident stability improvement after the HS-Ph-CN treatment was observed (Fig. S18 and S19, ESI†). However, significant deterioration in the performance of the 1 cm2 encapsulated HS-Ph-CN-modified solar cells was observed after 100 h under light and 380 h in the dark at 60 °C.
Based on the above results, we hypothesise that the stability improvement provided by the HS-Ph-CN modification is primarily due to the reduced charge accumulation60 rather than intrinsic stabilisation of the perovskite phase. Indeed, the latter effect can hardly be expected given that the benzenethiol molecules are confined to the perovskite|HTM interface and do not penetrate into the bulk of the light-absorber. Overall, the improvements in the stability found here compare well to the results reported in recent studies focused on the creation of more robust PSCs (Table S5, ESI†).
Conclusions
In summary, we developed a facile strategy to improve the power conversion efficiency and substantially enhance the stability of perovskite solar cells via modification of the interface between the light-harvester and the hole transporting layers with benzenethiol dipoles. Synchrotron X-ray photoelectron spectroscopy confirms immobilisation of the benzenethiol molecules on the perovskite film surface. Such modification facilitates hole extraction from the perovskite to the HTM and suppresses recombination of electrons from the perovskite layer with holes from the hole-transporter, as concluded from the photoluminescence and transient photocurrent/photovoltage decay data. Highest power conversion efficiencies of 20.2% for the small area devices (0.16 cm2) and 19.6% for the PSCs with an active area over 1 cm2 under 1 sun AM 1.5G irradiation were achieved with solar cells modified with HS-Ph-CN. Under simulated solar cell working conditions (1 sun AM 1.5G irradiation, 50% RH, 50 °C device temperature), such devices retained more than 80% of their initial photovoltaic performance after 50 h and operated stably over the next 135 h. In contrast, unmodified PSCs suffered continuous degradation and retained only 35% of their initial PCE after 185 h of tests. The results reported herein emphasise the critical importance of the interfacial layer at the perovskite|HTM interface for high efficiency and especially stability of larger-area perovskite solar cells, and offer an efficient method for the fabrication of better performing devices of this type.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support of this work by the Australian Centre for Advanced Photovoltaics (ACAP), the Australian Renewable Energy Agency and the ARC Centres of Excellence in Exciton Science (ACEX; CE170100026) and in Electromaterials Science (ACES; CE140100012). The authors acknowledge the use of facilities within the Monash University Centre for Electron Microscopy (MCEM), Monash X-ray Platform (funded by Australian Research Council grant LE130100072) and Australian Synchrotron. The contributions of the beamline scientists Dr Bruce Cowie, Dr Lars Thomsen and Dr Anton Tadich are gratefully appreciated.References
- M. A. Green and S. P. Bremner, Nat. Mater., 2017, 16, 23 CrossRef PubMed.
- M. A. Green, Nat. Energy, 2016, 1, 15015 CrossRef.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim and J. H. Noh, Science, 2017, 356, 1376 CrossRef PubMed.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte and B. Rech, Science, 2016, 351, 151 CrossRef PubMed.
- M. Saliba, T. Matsui, K. Domanski, J.-Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J.-P. Correa-Baena, W. R. Tress, A. Abate and A. Hagfeldt, Science, 2016, 354, 206 CrossRef PubMed.
- J. Lu, L. Jiang, W. Li, F. Li, N. K. Pai, A. D. Scully, C. M. Tsai, U. Bach, A. N. Simonov and Y. B. Cheng, Adv. Energy Mater., 2017, 7, 1700444 CrossRef.
- M. Xiao, F. Huang, W. Huang, Y. Dkhissi, Y. Zhu, J. Etheridge, A. Gray-Weale, U. Bach, Y. B. Cheng and L. Spiccia, Angew. Chem., Int. Ed., 2014, 126, 10056 CrossRef.
- F. Huang, Y. Dkhissi, W. Huang, M. Xiao, I. Benesperi, S. Rubanov, Y. Zhu, X. Lin, L. Jiang, Y. Zhou, A. Gray-Weale, J. Etheridge, C. McNeill, R. Caruso, U. Bach, L. Spiccia and Y. B. Cheng, Nano Energy, 2014, 10, 10 CrossRef.
- F. Huang, A. R. Pascoe, W. Q. Wu, Z. Ku, Y. Peng, J. Zhong, R. A. Caruso and Y. B. Cheng, Adv. Mater., 2017, 29, 1601715 CrossRef PubMed.
- W. Wu, F. Huang, D. Chen, Y. B. Cheng and R. A. Caruso, Adv. Energy Mater., 2016, 6, 1502027 CrossRef.
- T. Qin, W. Huang, J.-E. Kim, D. Vak, C. Forsyth, C. R. McNeill and Y.-B. Cheng, Nano Energy, 2017, 31, 210 CrossRef.
- H. Kim, K.-G. Lim and T.-W. Lee, Energy Environ. Sci., 2016, 9, 12 Search PubMed.
- A. Fakharuddin, L. Schmidt-Mende, G. Garcia-Belmonte, R. Jose and I. Mora-Sero, Adv. Energy Mater., 2017, 7, 1700623 CrossRef.
- A.-N. Cho and N.-G. Park, ChemSusChem, 2017, 10, 3687 CrossRef PubMed.
- M. Grätzel, Acc. Chem. Res., 2017, 50, 487 CrossRef PubMed.
- L. M. Herz, ACS Energy Lett., 2017, 2, 1539 CrossRef.
- N. K. Noel, A. Abate, S. D. Stranks, E. S. Parrott, V. M. Burlakov, A. Goriely and H. J. Snaith, ACS Nano, 2014, 8, 9815 CrossRef PubMed.
- N. Ahn, K. Kwak, M. S. Jang, H. Yoon, B. Y. Lee, J.-K. Lee, P. V. Pikhitsa, J. Byun and M. Choi, Nat. Commun., 2016, 7, 13422 CrossRef PubMed.
- D. Bryant, N. Aristidou, S. Pont, I. Sanchez-Molina, T. Chotchunangatchaval, S. Wheeler, J. R. Durrant and S. A. Haque, Energy Environ. Sci., 2016, 9, 1655 Search PubMed.
- T. Leijtens, G. E. Eperon, A. J. Barker, G. Grancini, W. Zhang, J. M. Ball, A. R. S. Kandada, H. J. Snaith and A. Petrozza, Energy Environ. Sci., 2016, 9, 3472 Search PubMed.
- S. Yang, Y. Wang, P. Liu, Y.-B. Cheng, H. J. Zhao and H. G. Yang, Nat. Energy, 2016, 1, 15016 CrossRef.
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. Zeng and J. Huang, Nat. Energy, 2017, 2, 17102 CrossRef.
- A. Abate, M. Saliba, D. J. Hollman, S. D. Stranks, K. Wojciechowski, R. Avolio, G. Grancini, A. Petrozza and H. J. Snaith, Nano Lett., 2014, 14, 3247 CrossRef PubMed.
- L. Barnea-Nehoshtan, S. Kirmayer, E. Edri, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2014, 5, 2408 CrossRef PubMed.
- J. Haruyama, K. Sodeyama, L. Han and Y. Tateyama, Acc. Chem. Res., 2016, 49, 554 CrossRef PubMed.
- L. Liu, A. Mei, T. Liu, P. Jiang, Y. Sheng, L. Zhang and H. Han, J. Am. Chem. Soc., 2015, 137, 1790 CrossRef PubMed.
- Y. Lin, L. Shen, J. Dai, Y. Deng, Y. Wu, Y. Bai, X. Zheng, J. Wang, Y. Fang, H. Wei, W. Ma, X. Zeng, X. Zhan and J. Huang, Adv. Mater., 2017, 29, 1604545 CrossRef PubMed.
- W.-H. Lee, C.-Y. Chen, C.-S. Li, S.-Y. Hsiao, W.-L. Tsai, M.-J. Huang, C.-H. Cheng, C.-I. Wu and H.-W. Lin, Nano Energy, 2017, 38, 66 CrossRef.
- W. Chen, Y. Wu, Y. Yue, J. Liu, W. Zhang, X. Yang, H. Chen, E. Bi, I. Ashraful and M. Grätzel, Science, 2015, 350, 944 CrossRef PubMed.
- X. Li, D. Bi, C. Yi, J.-D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, 353, 58 CrossRef PubMed.
- H. Chen, F. Ye, W. Tang, J. He, M. Yin, Y. Wang, F. Xie, E. Bi, X. Yang, M. Grätzel and L. Han, Nature, 2017, 550, 92 Search PubMed.
- L. K. Ono, N.-G. Park, K. Zhu, W. Huang and Y. Qi, ACS Energy Lett., 2017, 2, 1749 CrossRef.
- E. Bi, H. Chen, F. Xie, Y. Wu, W. Chen, Y. Su, A. Islam, M. Grätzel, X. Yang and L. Han, Nat. Commun., 2017, 8, 15330 CrossRef PubMed.
- W. Qiu, T. Merckx, M. Jaysankar, C. M. de la Huerta, L. Rakocevic, W. Zhang, U. Paetzold, R. Gehlhaar, L. Froyen and J. Poortmans, Energy Environ. Sci., 2016, 9, 484 Search PubMed.
- C.-H. Chiang, M. K. Nazeeruddin, M. Grätzel and C.-G. Wu, Energy Environ. Sci., 2017, 10, 808 Search PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate and A. Hagfeldt, Energy Environ. Sci., 2016, 9, 1989 Search PubMed.
- M. Shalom, S. Ruhle, I. Hod, S. Yahav and A. Zaban, J. Am. Chem. Soc., 2009, 131, 9876 CrossRef PubMed.
- B. C. C. Cowie, A. Tadich and L. Thomsen, AIP Conf. Proc., 2010, 1234, 307 CrossRef.
- Y. Cao, A. Stavrinadis, T. Lasanta, D. So and G. Konstantatos, Nat. Energy, 2016, 1, 16035 CrossRef.
- B. Philippe, M. Saliba, J.-P. Correa-Baena, U. B. Cappel, S.-H. Turren-Cruz, M. Grätzel, A. Hagfeldt and H. Rensmo, Chem. Mater., 2017, 29, 3589 CrossRef.
- C.-H. M. Chuang, P. R. Brown, V. Bulović and M. G. Bawendi, Nat. Mater., 2014, 13, 796 CrossRef PubMed.
- P. R. Brown, D. Kim, R. R. Lunt, N. Zhao, M. G. Bawendi, J. C. Grossman and V. Bulović, ACS Nano, 2014, 8, 5863 CrossRef PubMed.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano and J.-P. C. Baena, Sci. Adv., 2016, 2, e1501170 Search PubMed.
- J. Cao, J. Yin, S. Yuan, Y. Zhao, J. Li and N. Zheng, Nanoscale, 2015, 7, 9443 RSC.
- J. Cao, Y.-M. Liu, X. Jing, J. Yin, J. Li, B. Xu, Y.-Z. Tan and N. Zheng, J. Am. Chem. Soc., 2015, 137, 10914 CrossRef PubMed.
- S. F. Wuister, C. de Mello Donega and A. Meijerink, J. Phys. Chem. B, 2004, 108, 17393 CrossRef.
- L. Zuo, Q. Chen, N. De Marco, Y.-T. Hsieh, H. Chen, P. Sun, S.-Y. Chang, H. Zhao, S. Dong and Y. Yang, Nano Lett., 2016, 17, 269–275 CrossRef PubMed.
- A. M. Soufiani, Z. Hameiri, S. Meyer, S. Lim, M. J. Y. Tayebjee, J. S. Yun, A. Ho-Baillie, G. J. Conibeer, L. Spiccia and M. A. Green, Adv. Energy Mater., 2017, 7, 1602111 CrossRef.
- C. M. Proctor, C. Kim, D. Neher and T. Q. Nguyen, Adv. Funct. Mater., 2013, 23, 3584 CrossRef.
- W. Liao, D. Zhao, Y. Yu, C. R. Grice, C. Wang, A. J. Cimaroli, P. Schulz, W. Meng, K. Zhu and R. G. Xiong, Adv. Mater., 2016, 28, 9333 CrossRef PubMed.
- M. Mandoc, F. Kooistra, J. Hummelen, B. De Boer and P. Blom, Appl. Phys. Lett., 2007, 91, 263505 CrossRef.
- W. Tress, K. Leo and M. Riede, Appl. Phys. Lett., 2013, 102, 163901 CrossRef.
- T. Singh and T. Miyasaka, Adv. Energy Mater., 2017, 8, 1700677 CrossRef.
- P. Yadav, M. I. Dar, N. Arora, E. A. Alharbi, F. Giordano, S. M. Zakeeruddin and M. Grätzel, Adv. Mater., 2017, 29, 1701077 CrossRef PubMed.
- B. C. O’Regan, P. R. Barnes, X. Li, C. Law, E. Palomares and J. M. Marin-Beloqui, J. Am. Chem. Soc., 2015, 137, 5087 CrossRef PubMed.
- V. Roiati, S. Colella, G. Lerario, L. De Marco, A. Rizzo, A. Listorti and G. Gigli, Energy Environ. Sci., 2014, 7, 1889 Search PubMed.
- J. W. Lee, D. H. Kim, H. S. Kim, S. W. Seo, S. M. Cho and N. G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- K. Domanski, J.-P. Correa-Baena, N. Mine, M. K. Nazeeruddin, A. Abate, M. Saliba, W. Tress, A. Hagfeldt and M. Grätzel, ACS Nano, 2016, 10, 6306 CrossRef PubMed.
- A. Alberti, I. Deretzis, G. Mannino, E. Smecca, S. Sanzaro, Y. Numata, T. Miyasaka and A. La Magna, J. Phys. Chem. C, 2017, 121, 13577–13585 Search PubMed.
- N. Aristidou, I. Sanchez-Molina, T. Chotchuangchutchaval, M. Brown, L. Martinez, T. Rath and S. A. Haque, Angew. Chem., Int. Ed., 2015, 54, 8208–8212 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; XRD, SEM, AFM, FTIR, XPS, UPS, KPFM, photo/electroluminescence, and photovoltaic data. See DOI: 10.1039/c8ee00754c |
| This journal is © The Royal Society of Chemistry 2018 |