
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlternative binders for sustainable electrochemical energy storage – the transition to aqueous electrode processing and bio-derived polymers
Dominic
Bresser
 ab,
Daniel
Buchholz
ab,
Arianna
Moretti
ab,
Alberto
Varzi
ab and
Stefano
Passerini
*ab
ab,
Daniel
Buchholz
ab,
Arianna
Moretti
ab,
Alberto
Varzi
ab and
Stefano
Passerini
*ab
aHelmholtz Institute Ulm (HIU), Helmholtzstrasse 11, D-89081 Ulm, Germany
bKarlsruhe Institute of Technology (KIT), P.O. Box 3640, D-76021 Karlsruhe, Germany. E-mail: stefano.passerini@kit.edu; dominic.bresser@kit.edu; daniel.buchholz@kit.edu; arianna.moretti@kit.edu; alberto.varzi@kit.edu
First published on 16th October 2018
Abstract
In this review, we discuss the most recent developments in the field of green binders for batteries and supercapacitors and explain how they could decrease cost and environmental impact, and yet improve the performance of electrochemical energy devices. The different classes of green binders reported to date in the literature are firstly classified according to their processability (the solvent required for electrode manufacturing), chemical composition (F-free), and natural availability (synthetic or bio-derived). The benefits originating from their employment are analysed for different devices. The most popular lithium-ion batteries are thoroughly discussed both from the anode and the cathode side. While high capacity Si-based anodes benefit from enhanced cyclability due to the interaction between the active particles’ surface and the functional groups of, e.g., polysaccharides such as carboxymethyl cellulose and alginate, the transition to water-processable cathodes is certainly more challenging. In particular, strategies to suppress the aluminium corrosion affecting most lithiated transition metal oxides are discussed. Despite the much more limited literature available, the role of the binder is increasingly recognized in the emerging field of lithium–sulphur and sodium-ion batteries, and electrochemical double layer capacitors and, therefore, here discussed as well.
 Dominic Bresser | Dominic Bresser is presently establishing a young investigator research group at the Helmholtz Institute Ulm (HIU), affiliated with the Karlsruhe Institute of Technology (KIT), Germany, focusing on the development of alternative anode materials for lithium-ion batteries. Prior to this, he held a postdoctoral position at CEA in Grenoble, France, where he was studying new macromolecular electrolytes. Beforehand, he completed his PhD at Muenster University, Germany, on nanostructured lithium-ion active materials. He is co-author of >50 peer-reviewed publications (h-index: 24; Scopus), 3 book chapters, and 16 patents and patent applications. |
 Daniel Buchholz | Daniel Buchholz studied chemistry at the University of Muenster and completed his PhD studies at MEET battery research centre – University of Muenster with the focus on the synthesis, optimization and characterization of new and existing cathode materials for lithium- and sodium-ion batteries. He is co-author of 41 publications in peer reviewed international scientific journal (h-index: 21; Scopus). |
 Arianna Moretti | Arianna Moretti is senior scientist at the Karlsruhe Institute of Technology, Helmholtz Institute Ulm (Ulm, Germany) since April 2014. She completed her PhD at University of Camerino (Italy) on synthesis, electrochemical and structural characterization of cathodes materials for lithium ion batteries and afterward spent one year as a postdoc at the MEET battery research center at the University of Muenster (Germany). Her research activities are currently focused on vanadium oxides materials and ionic liquids based electrolytes for lithium, sodium and magnesium batteries. |
 Alberto Varzi | Alberto Varzi studied Chemistry of Materials at University of Bologna (Italy) and graduated in 2008. He continued his education in Germany and received a PhD in 2013 from the Ulm University and ZSW. After a postdoctoral period at WWU Münster/MEET, he is now a senior scientist at the Helmholtz-Institute Ulm for Electrochemical Energy Storage of the Karlsruhe Institute for Technology. Currently his research interests are focused on materials for lithiumion, lithium–sulfur batteries and supercapacitors. |
 Stefano Passerini | Stefano Passerini is Professor at the Karlsruhe Institute of Technology (KIT), Helmholtz Institute Ulm (HIU, Germany) since January 1, 2014. Formerly Professor at the University of Muenster (Germany), he co-founded the MEET battery research centre (Muenster, Germany). His research activities are focused on electrochemical energy storage in batteries and supercapacitors. He is co-author of more than 400 scientific papers (h-index: 70; Scopus), a few book chapters and several international patents. In 2012, he has been awarded the Research Award of the Electrochemical Society Battery Division. |
Broader contextCompared to other technologies, electrochemical storage offers the most energy efficient way to store electricity produced from renewable sources. This is leading to a continuously growing market of devices such as batteries and electrochemical double layer capacitors. However, it also raises concerns about the impact that their production and disposal could have on the environment. Interestingly, although simply considered as an inert component, the polymeric binder, indispensable for electrode production, is one of the factors defining the cost, environmental friendliness, and recycling/disposal of such devices. In fact, the state-of-the-art fluoropolymers, e.g., poly(vinylidene difluoride) (PVdF), are not only expensive, but require a toxic solvent (i.e., N-methyl-2-pyrrolidone, NMP) for electrode manufacturing. Greener alternatives are represented by water-processable F-free polymers, which may additionally allow for a cost reduction by a factor of 2–3 for the polymer and by a factor of about 100 for the processing solvent (NMP vs. water). |
1. Introduction and outline
Our society is presently facing the great challenge to switch from depleting energy sources like oil, coal, or gas, to renewables such as solar and wind. With regard to their inherent intermittency and commonly decentralized generation, however, efficient and sustainable energy storage is of utmost importance. Beside large-scale solutions like hydropower or compressed air, electrochemical energy storage, including secondary batteries and electrochemical double-layer capacitors (EDLCs), is currently considered to be the most suitable technology, particularly for relatively smaller applications like transportation or short- to mid-term stationary energy storage.1–4 As a matter of fact, the number of electric vehicle (EV) sales has steadily increased within the past years and the same trend is observed for the implementation of secondary batteries and EDLCs for buffering the intermittent energy supply by solar and wind.1–4 Consequently, batteries and EDLCs play a vital role for moving towards a more sustainable “energy future”. However, some concerns arise from the making of these devices. With respect to lithium-ion batteries, the cathode processing is based on the use of fluorine-containing polymers, in particular poly(vinylidene difluoride) (PVdF), as binders for the electrode preparation, and teratogen and toxic N-methyl-2-pyrrolidone (NMP)5–9 as a solvent/dispersant. Switching to aqueous electrode processing routes and non-toxic binders, as already achieved, e.g., for graphite-based lithium-ion anodes, would provide a great leap forward towards the realization of ideally fully sustainable and environmentally friendly electrochemical energy storage devices.Herein, we will initially present a summary of the general advantages for aqueous electrode processing technologies compared to those based on NMP, followed by a brief classification of the presently investigated aqueous binders with regard to their processability and biocompatibility. After this introductory part, the focus will be set on the recent developments and achievements concerning the implementation of aqueous binders for lithium-ion battery (LIB) electrodes, i.e., anodes and cathodes, and sodium-ion batteries as a promising alternative, in particular, for stationary applications, before eventually reviewing the research efforts conducted towards water-processed EDLCs as a high-power complement for efficient and effective energy storage.
1.1 General advantages of aqueous electrode processing
The major advantage of implementing large scale water-based processing is certainly related to the reduction of the environmental impact of LIB production. A comparative life cycle assessment (LCA), studying the impact of the transition from NMP to water, showed substantially reduced emissions of CO2 equivalents for the latter.5–8,10 Most importantly, NMP is an hazardous, teratogenic and irritating compound, while PVdF is mutagenic and teratogenic.8As a consequence, several countries like the USA11 or the European Union (EU)12 started to limit the use of NMP to a minimum. In fact, Annex XVII to REACH (Regulation concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals; EU) states that after May 9th 2020 “NMP shall not be used as a substance on its own or in mixtures in a concentration equal to or greater than 0.3% unless manufacturers and downstream users take the appropriate risk management measures and provide the appropriate operational conditions to ensure exposure of workers is below the Derived No-Effect Levels (DNELs) of 14.4 mg m−3for exposure by inhalation and 4.8 mg kg−1per day for dermal exposure”. Moreover, it has very recently, i.e., in April 2018, been included in the list of substances of very high concern that may have serious and often irreversible effects on human health and the environment by the EU, following a proposition of the Netherlands. Not least considering the battery manufacturing in these countries, this raises concerns about the whole electrode processing chain, from mixing of the slurry to the final solvent recovery (Fig. 1). The exposure of humans to these chemicals, as well as their release in the working environment, has therefore to be controlled carefully or ideally fully prevented.
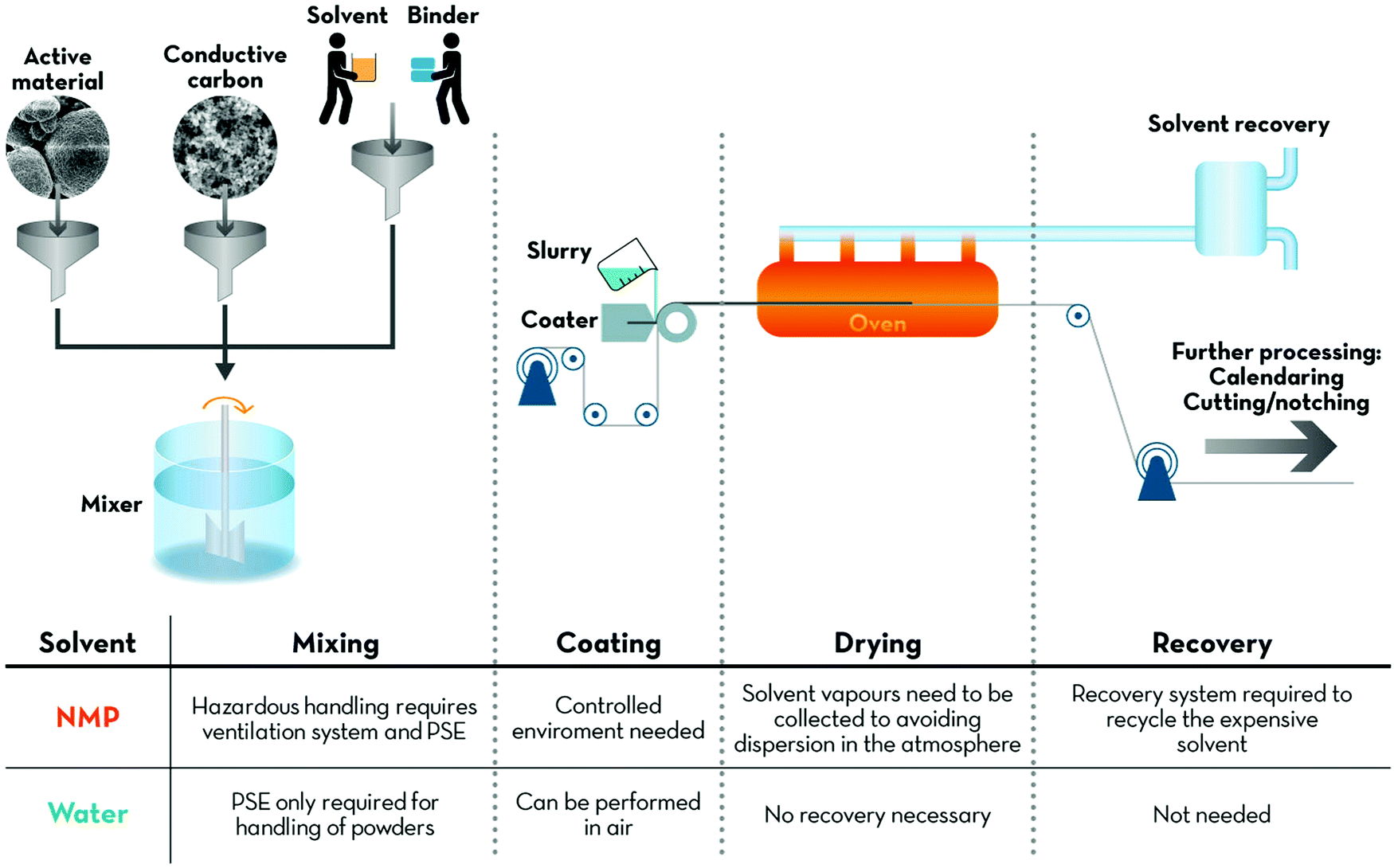 | ||
| Fig. 1 Simplified schematic description of a generic battery/EDLC electrode manufacturing process. From left to right, the main advantages of water over NMP are highlighted for each step, from the initial mixing of the slurry, its coating on the current collector, the drying of the electrode layer and, finally, the solvent recovery. | ||
Besides, the implementation of aqueous processing has also an impact on production costs and, in turn, on the overall battery price. Recent projections foresee a price of 100 $ kW h−1 for LIBs to be reached in 2030.13 A processing cost breakdown study indicates the reduction of costs associated with the use, drying and recovery of the organic solvent to be a key point to reach this ambitious target.14 A simple comparison of binder and solvent costs already indicates the convenience of water-based electrode processing. NMP is rather expensive (1–3 $ kg−1; compared to about 0.015 $ L−1 for water) and so is PVdF with a price of 8–10 $ kg−1. The most commonly used aqueous binders like carboxymethyl cellulose (CMC) are significantly cheaper at 2–5 $ kg−1 and even the rather costly alginate is only about 8 $ kg−1, i.e., still at the lower end of the cost range for PVdF.15,16 Even though such an analysis will eventually have to be carried out by industry, these numbers already indicate the potential cost reduction by transitioning to water-based electrode processing from the materials’ point of view.
The impact of the solvent on the electrode drying process is poorly investigated in the scientific literature so far – especially when it comes to a comparison of different binders. Generally, aqueous processed electrodes are reported to be more hydrophilic, but a drying temperature of 100 to 120 °C appears to be sufficient to reduce the residual water content to an acceptable level, i.e., below 50 ppm.17,18 In fact, when preparing aqueous processed electrodes on the lab-scale, one easily recognizes that the electrode drying is much faster compared to NMP-processed ones. This finding was recently confirmed by Susarla et al.,19 reporting that the equilibrium vapour pressure of water is 35-times higher compared to NMP (in line with the substantially lower boiling point of 100 °C vs. 203 °C), even though the latent heat of evaporation of water, i.e., the energy that is required to induce the liquid-to-gas phase transition, is 4-times higher than the one for NMP. This potentially results in relatively faster evaporation of water, leading to shorter drying times. A recent simulation of the corresponding cost revealed that the cost per kW h is presumably comparable for both systems, when evaluating the drying step only.17 The key for saving energy, i.e. cost, and reducing the required investment in the case of aqueous electrode processing, however, is the dispensability of recovering the NMP solvent via condensation and/or distillation to prevent release into the environment.14,17 In fact, according to the aforementioned calculation, this cost reduction may account for almost half of the current electrode processing and associated plant capital costs.17
1.2 Classification of aqueous binders
The term “green” is widely used to express the environmental friendliness of materials and processes. However, its meaning can significantly vary depending on the context of use. When talking about polymeric binders, green alternatives could be identified according to (i) processability, (ii) chemical composition and (iii) natural availability (see Fig. 2). According to the first criterion, binders soluble or dispersible in environmentally benign solvents, such as water or ethanol, could be classified as green alternatives. Poly(tetrafluoroethylene) (PTFE), also known as Teflon® (Dupont), is highly hydrophobic with strong C–C and C–F bonds providing high chemical and mechanical stability, rendering it very suitable for highly resistant coatings and membranes.20 It can be dispersed in water and stabilized by wetting agents, thus making electrode processing feasible. These wetting agents, however, are frequently environmentally and/or biologically hazardous chemicals (e.g., fluorine-containing carboxylic acids and/or ammonium lauryl sulphate). Hence, the potential aqueous processing alone does not make this binder a very green one. Recently, another water-processable binder, denoted as TRD202A (a fluorinated acrylate polymer latex), has been commercialized by JSR Corporation (Japan).21,22 This fluorinated acrylate polymer promises enhanced adhesion to the current collector, low resistance, and long cycling life. Although their water processability would reduce the environmental impact of the electrode manufacturing, fluoropolymers are, however, more difficult to dispose of at the end of the life of the battery. In fact, they are not soluble in the majority of solvents besides a few expensive, flammable, and/or toxic ones (e.g., N,N-dimethylformamide (DMF), N,N-dimethylacetamide (DMAC), NMP and dimethyl sulfoxide (DMSO)).23 Consequently, the fluorinated binding agents cannot be easily separated from the electrode active materials and would require thermal treatments for their elimination. This raises further environmental concerns, as most of the decomposition products of fluoropolymers in N2 (e.g., cycloperfluorobutane, hexafluoropropene, perfluoroisobutene, fluorophosgene, and other fluorocarbons) are extremely toxic24 and known to affect the ozone layer. Thus, F-free compounds would represent a real step towards greener energy storage devices. To date, a variety of synthetic polymers have been proposed as alternative binders for both batteries and supercapacitors. Polyacrylates are certainly the most common, with PAA being the most popular choice, often used in its neutralized water-soluble form (either with Li or Na). Other aliphatic and aromatic synthetic polymers, such as polyethylene (PE) and polystyrene (PS), have also been proposed as F-free electrode binders. Owing to their generally low glass transition temperature (Tg), these soft and rubbery materials are easy to process and provide good mechanical stability to the electrodes, which is expected to enhance cycling life. All these synthetic plastics are industrially produced in large volumes (several millions of tonnes per year) and already employed in a variety of fields, ranging from adsorbents to paint components, and food packaging. However, although such established industrial production could guarantee a smooth transition from PVdF-based electrodes; the electrode manufacturing would still rely on non-renewable materials. For this reason, bio-polymers and their derivatives would represent a more sustainable choice. Polysaccharides, for example, are naturally occurring polymers that can be extracted from a variety of non-edible sources. These generally share the same glucose monomer as the building block, while different glycosidic bonds and substituents on the pyranose ring define the peculiar properties of each polymer (e.g., cellulose, alginate, starch, etc.). As previously mentioned, CMC is the most famous bio-derived compound used in the current state-of-the-art production of LIB negative electrodes together with styrene–butadiene rubber. Although its primary role in LIB electrode processing is as a thickener, i.e., to stabilize the slurry and adjust its viscosity, its contribution to the electrode binding is now ascertained. Such a linear polysaccharide is a cellulose derivative, made water-soluble by replacing some –OH groups with carboxymethyl moieties (–CH2COOH), and is extensively used as a thickener in the food industry. In Table 1, we provide an overview of the herein reviewed and discussed binding agents, summarizing the earlier mentioned classification parameters (‘F-free’, ‘Processability’, ‘Synthesis’, ‘Sustainability’, and ‘Ease of disposal’) in order to allow for a general ranking concerning the environmental friendliness of these binders. In addition, Table 1 provides an overview of the cited references, in which the electrolyte swelling and mechanical properties of the resulting electrodes (e.g., the coating adhesion, elastic behaviour, and tensile strength) are reported. Particularly the latter two columns and the number of given references, indeed, also serve as a rough indication for the stage of development of the different binders and may serve as a guideline for future studies.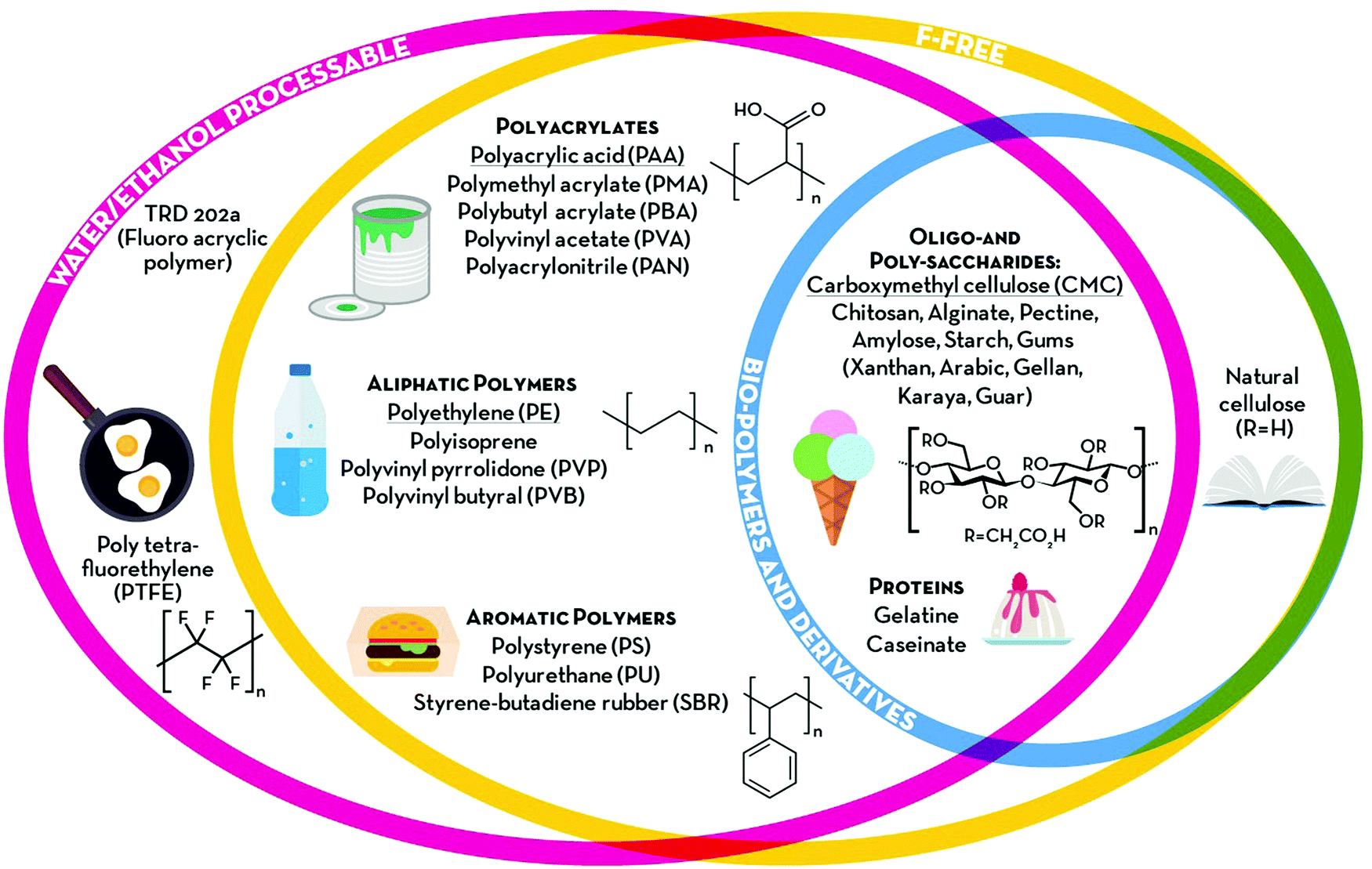 | ||
| Fig. 2 Overview of the different classes of binders discussed in the present review. The polymers are divided into three main categories according to their processability (water and/or ethanol processable), chemical composition (F-free) and natural availability (bio-polymers and derivatives). | ||
| Name of Binder (incl. the corresponding derivatives) | F-free | Processability | Synthesis | Sustainability | Ease of disposal | “Green score” | Binder properties | |
|---|---|---|---|---|---|---|---|---|
| Swelling with electrolyte | Mechanical (adhesion, elastic behaviour, tensile strength, …) | |||||||
| a CMC in combination with a not specified water-soluble elastomer. b Commonly used in combination with other binders as, for instance, CMC. | ||||||||
| State of the art | ||||||||
| CMC | + | + | + | + | + | +5 | 40, 138, 158 | 12, 44, 81, 119,a 134, 136, 158, 304, 200, 201 |
| PTFE | − | + | − | − | − | −3 | n.a. | n.a. |
| PVdF | − | − | − | − | − | −5 | 40, 158, 165 | 12, 134, 136, 158, 166, 300, 303, 306 |
| Alternatives | n.a. | n.a. | ||||||
| Agar-agar | + | + | + | + | + | +5 | n.a. | n.a. |
| Alginate | + | + | + | + | + | +5 | 84, 138, 165 | 46, 86, 91, 97, 300 |
| Amylose | + | + | + | + | + | +5 | n.a. | 95 |
| Arabic gum | + | + | + | + | + | +5 | n.a. | n.a. |
| Carrageenan | + | + | + | + | + | +5 | n.a. | n.a. |
| Caseine | + | + | + | + | + | +5 | n.a. | 304 |
| Chitosan | + | + | + | + | + | +5 | n.a. | 136, 137, 166 |
| Cyclodextrines (carbonyl-beta) | + | + | + | + | + | +5 | n.a. | 90 |
| Ethylene propylene diene monomer (EPDM) rubber | + | + | 0 | − | 0 | +1 | n.a. | n.a. |
| Gelatine | + | + | + | + | + | +5 | n.a. | 46 |
| Gellan gum | + | + | + | + | + | +5 | n.a. | n.a. |
| Guar gum | + | + | + | + | + | +5 | 56 | 56, 57 |
| Karaya gum | + | + | + | + | + | +5 | n.a. | 94 |
| Cellulose (natural) | + | + | + | + | 0 | +4 | n.a. | n.a. |
| Pectine | + | + | + | + | + | +5 | n.a. | 57, 95 |
| PEDOT-PSS | + | + | + | − | + | +4 | n.a. | 100, 136 |
| Polyacrilic acid (PAA) | + | + | 0 | − | 0 | +1 | 40, 78, 69, 138, 158 | 12, 36, 44, 69, 81, 133, 158, 300 |
| Poly(methyl acrylate) (PMA) | + | + | + | − | 0 | +2 | n.a. | n.a. |
| Poly(vinyl alcohol) (PVA) | + | + | 0 | − | 0 | +1 | n.a. | 78 |
| Poly(vinyl acetate) (PVAc) | + | + | + | − | 0 | +2 | n.a. | n.a. |
| Polyacrylonitrile (PAN) | + | + | + | − | 0 | +2 | n.a. | n.a. |
| Polyisoprene (PIpr) | + | + | 0 | + | 0 | +3 | n.a. | n.a. |
| Polyaniline (PANi) | + | + | + | − | 0 | +2 | n.a. | n.a. |
| Polyethylene (PE) | + | + | 0 | − | 0 | +1 | n.a. | n.a. |
| Polyimide (PI) | + | + | 0 | − | 0 | +1 | n.a. | n.a. |
| Polystyrene (PS) | + | + | 0 | − | 0 | +1 | n.a. | n.a. |
| Polyurethane (PU) | + | + | 0 | − | + | +2 | n.a. | n.a. |
| Polyvinyl butyral (PVB) | + | + | 0 | − | 0 | +1 | n.a. | 308 |
| Polyvinyl pyrrolidone (PVP) | + | + | + | − | + | +3 | n.a. | 306, 307 |
| Starch | + | + | + | + | + | +5 | n.a. | n.a. |
| Styrene butadiene rubber (SBR)b | + | + | 0 | − | 0 | +1 | 40 | 44, 133, 200, 201 |
| Tara gum | + | + | + | + | + | +5 | 56 | 56 |
| Tragacanth gum | + | + | + | + | + | +5 | n.a. | 46 |
| TRD202A | − | + | n.a. | n.a. | − | n.a. | n.a. | n.a. |
| Xanthan gum | + | + | + | + | + | +5 | n.a. | 91, 134, 139 |
| Green score legend | |||
|---|---|---|---|
| + | 0 | − | |
| F-free | yes | no | |
| Processability | Water/ethanol-based slurry | Organic solvent-based slurry (e.g., NMP) | |
| Synthesis | No chemistry involved (natural binder) or aqueous chemistry | Non-aqueous chemistry | Non-aqueous chemistry involving fluorinated chemicals |
| Sustainability | Obtainable from renewable sources | Derived from non-renewable sources (e.g., petroleum) | |
| Ease of disposal | Recyclable by water treatment (simple washing) | Suitable for hydrometallurgical with no F emissions | Hydrometallurgical or pyrometallurgical methods required and F emissions |
2. Lithium-ion batteries
For many years, the binder has been considered to be of minor importance for battery electrodes. Indeed, PVdF was performing fairly well, and the scale of LIB production was not as high as today, rendering processing issues secondary. However, the continuously rising demand for high-performance LIBs has led to a reconsideration of these aspects. In fact – beside the advantageous introduction of aqueous electrode processing technologies, as discussed in Section 1.1 – it was found that the binder plays a decisive role in the electrode performance, despite its rather low content of a few percent only (up to 5 wt%, but usually 2 wt%) with respect to the total electrode composition (i.e., active material, binder, and conductive additives), and its chemical and electrochemical inactivity. Nonetheless, the selection of a suitable binding agent commonly follows the following general criteria: (i) ensuring suitable cohesion between the active material particles and the additional electrode components as, e.g., the conductive agent; (ii) guaranteeing strong adhesion of the electrode coating to the current collector; (iii) facile electrode processing; (iv) insolubility in the electrolyte and low electrolyte swelling; (v) high chemical, thermal, and electrochemical stability, while not detrimentally affecting the electron and ion transport in the electrode composite; and (vi) providing low additional cost, ideally being also environmentally-friendly.In light of these general considerations, we will review in the following the past and recent development and achievements in the field of lithium-ion batteries, starting from the anode side, for which aqueous electrode processing is already fairly well established and continuing with the cathode side, for which still several challenges remain to be overcome.
2.1 Anodes – active materials with limited volume variation upon lithiation
Electrodes made with CMC and CMC–SBR mixtures displayed lower 1st cycle irreversible capacity loss (ICL) than PVdF-based electrodes,26,30 which is attributed to faster SEI-formation induced by the interactions between carbon and Na-CMC.30 It is recognized that the presence of hydroxyl and carboxyl functions is the key feature which is responsible for the improved electrochemical performance, as displayed by the electrodes featuring CMC and other synthetic and natural polyelectrolytes with respect to PVdF. For example, gelatine was found to act as a surface modifier providing nucleation centres for the formation of a thinner passivation film, hence, reducing the lithium consumption associated with the solid electrolyte interphase (SEI) build-up.31–33 The reactivity of CMC with the electrolyte was studied in more detail by El Ouatani et al.34via XPS. The authors found that CMC readily reacts with the electrolyte upon simple soaking (prior to cycling), generating a protective surface layer composed of R–O–PF4 and LiF. The efficacy of this “artificial SEI” is evidenced by the better cyclability (in sulfolane-based electrolytes) of graphite electrodes made with CMC and practical loading compared to their PVdF counterparts.35
However, the role of the functional groups of the binder is better illustrated in polyacrylates (e.g., PAA or poly(methyl acrylate) (PMA)), which contain a higher number of functional groups than CMC and, thus, adsorb more strongly on the graphite surface via hydrogen bonding.36 Komaba et al.37–39 proposed that Li+ is coordinated by the polarized oxygen atoms along the polymeric chains of polyacrylates (like in poly(ethylene oxide); PEO) promoting its de-solvation from solvent molecules (ethylene carbonate (EC) or propylene carbonate (PC)). Although the amount of binder in the electrodes used for these studies was often quite high (5–15 wt%), it has been demonstrated that a homogenous coating of graphite particles with polyacrylates behaves like an “artificial SEI”, as found for Na-CMC, enabling reversible cycling in non-SEI forming solvents such as PC37 or in PC-rich electrolytes.40 Moreover, the solvent up-take of polyelectrolytes is found to be lower than that of PVdF, reducing the risk of co-intercalation of other species (e.g., PC molecules) together with Li+.40 These findings are extremely interesting, as PC has a lower melting point than EC (−49 °C and +36 °C, respectively) and its use as the main solvent or co-solvent in the electrolyte formulation would enlarge the operational temperature range of LIBs. Nevertheless, the ability of functionalised binders to act as an “artificial SEI” decays when reducing the binder amount to practical values (2–5 wt%) as a result of the incomplete particle coverage.40 Generally, a more uniform graphite particle coverage is achieved when using Li-PAA and Na-PAA rather than for non-neutralized PAA. The more extended conformation in water of dissociated Li/Na-PAA, due to electrostatic repulsion between the carboxyl groups, was proposed as the reason for the higher slurry viscosity and the better uniformity of the particle coating.38 Better first cycle coulombic efficiency, a smoother electrode surface after cycling, and prolonged cycle life when using neutralized Li-PAA and Na-PAA were also reported by Chong et al.41 These authors highlighted the importance of carefully dosing the SBR amount (used to compensate for the brittleness of polyacrylates as for Na-CMC), since a monotonous decrease of the electronic conductivity was found upon increasing the quantity of the elastomer.
When moving from laboratory to pilot-line scale studies,42 which implies practical loadings of graphite (>2 mA h cm−2) and the used binder (<5 wt%) as well as different mixing and drying conditions, the Na-CMC–SBR mixture outperforms both Na-PAA and the Na-CMC/Na-PAA mixture in terms of rheological properties of the slurry, coating adhesion, and electrochemical performance. Furthermore, it is found that Na-PAA is more sensitive than Na-CMC/SBR to different mixing methods and drying conditions. The electrodes based on the Na-CMC/SBR formulation preserved their adhesion strength upon doubling the electrode loading, while both PVdF and Na-PAA showed a significant loss of their initial mechanical properties.
Interestingly, as mentioned in the introduction, the binder may affect also the battery safety. In fact, a DSC (differential scanning calorimetry) study on lithiated graphite revealed that less heat is generated upon SEI breakdown for electrodes made with CMC or polyacrylate compared to PVdF, confirming the protective nature of the SEI formed in the presence of the former binder layers.43
The binding ability of polyacrylate, arising from the high number of functional groups, has been exploited by cross-linking PAA with CMC to prepare thick graphite electrodes (13.8 mg cm−2). The electrodes, which included also SBR in the formulation, displayed better adhesion and capacity retention and, at the same time, reduced resistance compared to those prepared with CMC/SBR.44
The extended work conducted on CMC and polyacrylate stimulated further investigation of other functionalised binders and, especially, of bio-derived polymers such alginate, chitosan, gums (e.g., guar, xanthan, karaya, tragacanth, and arabic), agar–agar, pectine, and carrageenan.45–48 All of them are stable (at least) up to 200 °C and, thus, compatible with the relatively high electrode drying temperatures. Additionally, they are electrochemically inert in the potential range from 0 to 3 V vs. Li/Li+.45 They are, therefore, potential alternatives to Na-CMC, providing the advantage of possessing functional groups, which, instead, have to be chemically introduced into cellulose. However, although the published studies are very encouraging, little is known about the processability, stability, and rheological properties of large-scale slurries made with such bio-polymers. Indeed, the viscosity of the binder solution will determine how easily the solid content of the slurry can be dispersed and, thus, the mixing conditions. The final slurry must be stable over time and no sedimentation should occur to obtain a homogenous coating. For instance, a recent study reported higher stability and viscosity for Na-alginate slurries (of different compositions) compared to Na-CMC.49 These properties are key factors to assess the feasibility of up-scaling innovative electrode formulations.
To summarize, polymers with hydroxyl and carboxyl functional groups such as CMC and PAA have been demonstrated to be suitable binders for graphite electrodes, with improved performance compared to PVdF and with “artificial SEI” formation ability.
2.2 Anodes – active materials with extensive volume variation upon de-/lithiation
While the contact between the Si particles is easily retained upon lithiation (because the resulting volume expansion of lithiated Si, up to 280%, favours the contact between all particles in the composite electrode), the volume decrease upon delithiation can lead to electrode pulverization, a loss of mechanical properties and strong capacity fading. Therefore, a binder capable of chemically binding the Si particles while maintaining a certain flexibility and homogeneously coating their surface to avoid electrolyte decomposition, is expected to enable longer cycle life. In the search for an alternative binder to PVdF, it was quickly recognized that CMC could mitigate the negative effects associated with the large Si particle volume variation upon lithiation.30,65–67 Surprisingly, however, the use of SBR together with CMC (a rather stiff and brittle polymer) did not result in improved electrochemical performance, although improving the flexibility of the electrode.65 Therefore it is the ability of CMC to chemically interact with the active materials particles (more precisely the characteristics of the formed bonds) to influence the electrochemical behaviour rather than the overall electrode flexibility.68,69 Accordingly, further studies were conducted to clarify the nature of the potential interactions between Si and CMC, which are related to the Si surface characteristics during the aqueous processing. Upon exposure of Si to air or water, siloxane Si–O–Si and silanol Si–OH surface terminations are formed.70 These are susceptible to forming both covalent and hydrogen bonds with the carboxyl groups present in the polymeric chains of many water-processable binders (e.g., CMC, alginate, pectine, gellan and xanthan gums, and carboxymethyl chitosan). Hochgatterer et al.71 demonstrated the central role of the binder carboxyl groups by replacing them with hydroxyethyl and cyanoethyl groups. In fact, the Si–C-composite anodes prepared using such modified CMCs displayed poorer electrochemical performance than those employing the original CMC. Based on this evidence, a condensation reaction occurring between the carboxylic units and the silanol groups upon drying was proposed. The improved adhesion strength of covalently bonded Si and CMC with respect to the trapped Si particles in the CMC matrix was proven by atomic force microscopy (AFM).72 Furthermore, a recent study evidenced that a thin layer of SiO2 covering the Si particles is beneficial for the formation of ester bonds, thereby improving the electrode cyclability, although increasing the first cycle ICL.73
Using different polymers and modifying the Si surface, Bridel et al.68 observed that when no Si–binder interaction can be established (i.e., when there are no accessible –OH groups on the Si surface) or if strong Si–binder covalent bonds are formed (e.g., peptide bonds between Si and the binder, e.g., chitin), the electrochemical performance of Si electrodes is rather poor. The volume change during the electrochemical process causes contact loss between carbon and Si in the first case, and the rupture of covalent bonds in the second scenario. Therefore, the authors suggest that the improved performance observed for Si–CMC electrodes must be ascribed to the formation of H-bonds, which are weak enough to break upon volume increase, but able to regenerate through the so-called “self-healing” mechanism, given the presence of residual water.68 A schematic description is presented in Fig. 3.
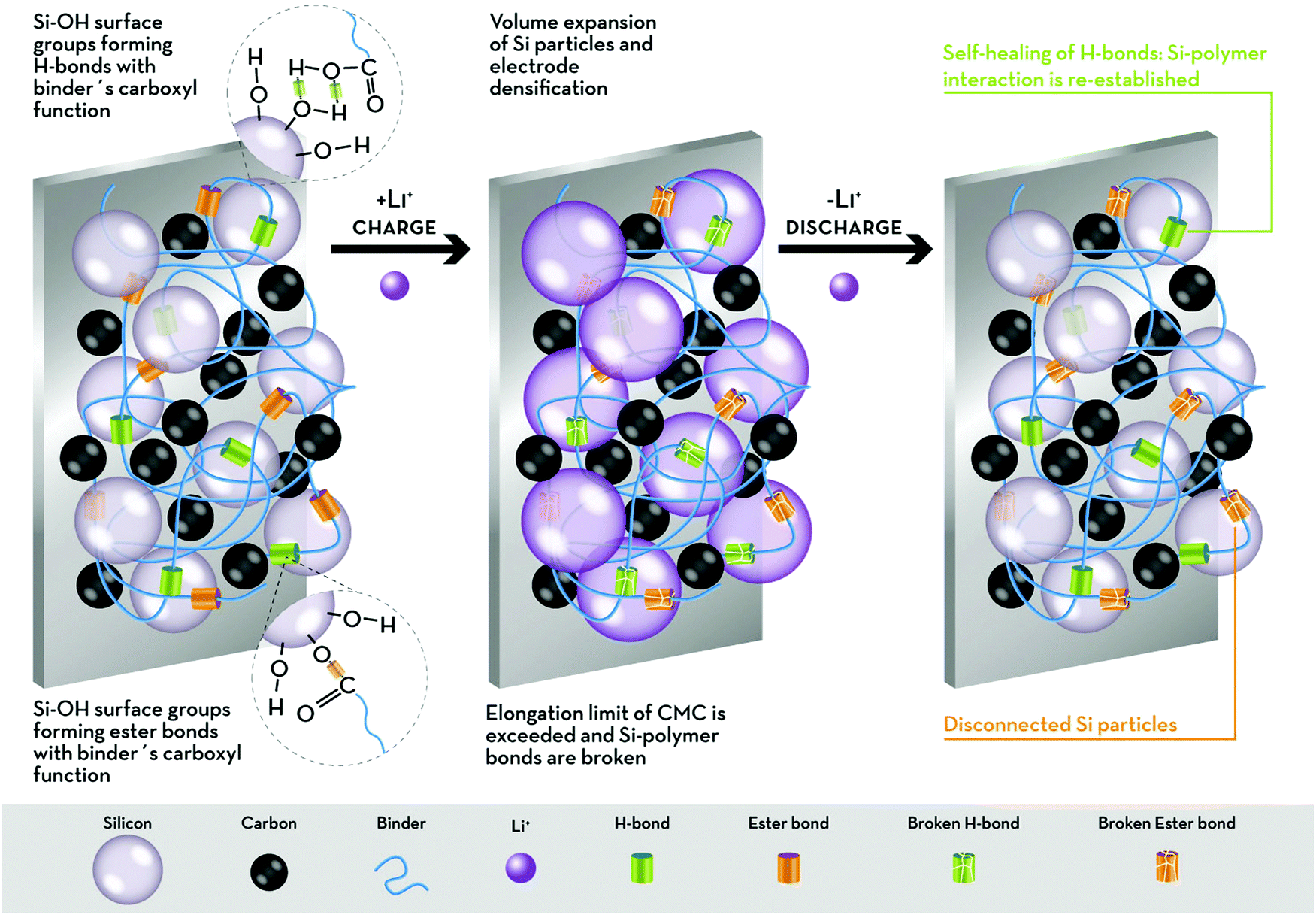 | ||
| Fig. 3 Schematic illustration of the proposed interactions occurring between the carboxylic groups of CMC and Si particles. Both H-bonds and ester bonds are established with the silanol (Si–OH) groups on the Si particle surface. The volume expansion occurring upon lithiation causes the rupture of the bonds. During the de-alloying process, the Si particles shrink, but the ester bonds are not re-established. On the other hand, the self-healing property of the weaker H-bonds permits the contact between the binder and the Si particles to be retained. | ||
Also the role of the slurry pH in the establishment of chemical bonds was investigated. In fact, adjusting the slurry pH to 3 should lead to the presence of SiOH and COOH, which are expected to promote the formation of covalent ester bonds upon electrode drying.71,74,75 For example, Bridel et al.75 compared the performance of electrodes made from slurries without adjusting the pH, to favour the formation of H-bonds in the dried electrode, and at pH = 3, to induce Si–binder ester bond formation upon drying. These authors observed that in this latter case, good long-term cyclability of the Si electrodes was achieved only by limiting the lithiation degree, i.e., when the volume expansion of the Si particles can be buffered by the electrode porosity. Indeed, lithiation degrees above 1.7 eq. of Li per Si result in exceeding the elongation limit of CMC (5–8%),65 resulting in the rupture of the CMC–Si covalent bonds, which are not reformed. On the other hand, when the slurry pH is not controlled, the formation of the weak H-bonds is favoured leading to the full capacity retention of the electrodes for over 100 cycles, since these bonds can be regenerated spontaneously.75
However, it is important to note that at pH = 3 a better dispersion of the conductive carbon and Si is reported.71 Additionally, a simulation study revealed that the CMC chains rearrange to increase the number of contact points between adjacent Si particles, giving rise to a “spring-like” conformation.76 The CMC chain rearrangement is due to the tuning of inter- and intra-chain interactions, regulating the viscosity of the slurry.74,76,77 The DS and the countercation (e.g., smaller cations lower the interactions due to the poor dissociation and less extended conformation in water) are also expected to affect inter- and intra-chain interactions.68
Further studies have proved that the cycling stability of Si anodes can be further improved by the use of polyacrylate binders.69,78 In fact, polyacrylates possess more carboxyl groups than CMC, which can graft on the Si particle surface via H-bonds. It has been reported that during the electrode's drying step, linear PAA chains bridge and cross-link together via the formation of H-bonds and carboxylic anhydride groups, giving rise to a polymeric network with improved mechanical properties and a more uniform particle coverage than linear chain polymers.78 Different from PVdF, PAA does not significantly swell in the electrolyte, thus, retaining better adhesion and protecting the surface of Si from a direct reaction with the electrolyte, acting as an “artificial SEI”.78 Porcher et al.79 carefully investigated the effect of PAA and Li-PAA on the coulombic efficiency obtained from Si electrodes with a practical loading of 2.5 mA h cm−2. The study revealed that, compared to CMC, PAA and its neutralized form, Li-PAA (that has a more elongated chain conformation in solution), grant lower ICLs during the initial cycles, exploiting the “artificial SEI” behaviour due to the more uniform binder distribution. For PAA and Li-PAA with a low degree of neutralization (i.e., a low number of Li counter cations), the “spring-like” conformation mentioned for CMC was actually visualized by transmission electron microscopy (TEM).80 Additionally, Erk et al. confirmed the optimised performance of PAA and Li-PAA as binders for Si electrodes with rather high areal capacity (5.1 mA h cm−2).80
Recently, it has been shown that the mechanical strength and the cyclability of Si-based anodes can be further improved by including a “maturation” treatment in a humid atmosphere.81,82 This study revealed that an essential parameter is the acidic pH of the slurry. Indeed, improved capacity retention and reduced first cycle coulombic efficiency can be obtained for both CMC- and PAA-based electrodes coated from acidic (pH = 3) slurries. The maturation in an acidic environment was reported to lead to Cu corrosion, resulting in the formation of Cu–binder ester bonds and, thus, improving the adhesion of the coated electrode with the current collector. It was also observed that, during the maturation, the binder migrates from the surface and concentrates in the interstices between the Si particles and in the macropores generated by the carbon agglomerates, forming reinforcing bridges.81,82
The copolymerization reaction of PAA with other monomers or polymers allows tuning of the mechanical and chemical properties of the binder. Following this approach, Koo et al.66 crosslinked PAA with CMC. The resulting Si composite electrodes outperformed those made with the single polymers. The achievement of an improved capacity retention and coulombic efficiency was attributed to the higher mechanical strength of the crosslinked PAA/CMC binder and to the reduction of the amount of free hydroxyl groups of crosslinked CMC, which can react with LiPF6.34,66 Instead, crosslinked PAA/PVA (poly(vinyl alcohol)) permits the Si anode cyclability to be improved due to the deformable properties introduced by PVA.83
Promising results have also been obtained using alginate, a natural polysaccharide.84 Similarly to CMC and PAA, alginate does not show swelling or a change in stiffness when in contact with the electrolyte. Moreover, inspired by algae cells, the authors suggested Li+ ion hopping to occur between neighbouring carboxylic groups, which could enhance the Li+ ion conduction. Following this line, the more homogeneous distribution of functionalities in alginate with respect to CMC, appears to be responsible for the better performance observed. In addition, the high polarity of alginate results in a higher viscosity of the slurry, permitting the reduction of the total amount of binder in the electrode formulation.31,84 Alginate was successfully employed to prepare electrodes with Si, which was obtained from the waste slurry of diamond-sliced solar grade crystalline Si-ingots.85 The study showed that an appropriate surface treatment to obtain a thin SiOx coating, coupled with a binder that can interact with the Si surface, enabled the reversible cycling of micro-plates (200 cycles at 0.2C in the voltage range 1.0 V to 0.01 V) without severe electrode pulverization. Recently, it has been reported that electrodes made with Al-alginate and Ba-alginate (caution, Ba salts are highly toxic) display enhanced adhesion compared to Na-alginate and higher Vickers hardness due to the crosslinked structure, induced by the multivalent cation.86 The carboxylated form of chitosan (a highly abundant natural polysaccharide), offering –NH2 groups for the interaction with the Si surface and cross-linking with other polymers, has also been successfully employed to prepare Si anodes.87–89
Similar to cross-linked polyacrylates, branched polysaccharides offer increased contact points for the binder/silicon interactions. Branched β-cyclodextrin, for example, shows higher adhesion strength than alginate and extended capacity retention due to the “self-healing” properties of the multiple H-bonds upon Si-anode delithiation.90 Jeong et al.91 observed stronger adhesion capability for native xanthan gum (and in general for charged polysaccharides rather than for neutral ones), which was attributed to the existence of ion–dipole interactions between –COO− and the Si surface, maximized by the double-helix superstructure of this polymer.
With regard to lithium conduction, a mechanism similar to that found in PEO (and polyacrylates, see graphite, Section 2.1(a)) has been proposed for guar gum (GG), another natural polysaccharide in which, however, only hydroxyl groups are present. Li+ moves along the binder chain across complexations sites offered by the lone pairs of the oxygen heteroatoms assisted by the segmental motion of the polymer.92 Another polymer from the family of natural gums, gellan, has been used to prepare Si-based anodes and the resulting electrodes showed better cyclability then CMC at both 20 and 60 °C.93 The higher number of functional groups, leading to extended self-healing, non-covalent Si–binder interactions, and the more stable SEI formed, were identified as the sources of the observed improved performance. For karaya gum, a branched natural polysaccharide, the formation of both covalent and H-bonds was observed, as well as improved adhesion.94
Inspired by biological systems, Yoon et al.95 investigated the influence of glycosidic linkages and side-chain substituents in polysaccharide binders. The α-glycosidic linkage, present in pectine and amylose, is more elastic than the β counterpart as in CMC, due to the chair-to-boat conformation transition taking place among pyranose rings upon elongation.96 The elastic modulus determination confirmed their hypothesis, i.e., a lower resistance to elongation was found for pectin with respect to CMC. The measurement also revealed the role of side groups, as amylose (with only hydroxyl groups) presents an elastic modulus similar to the β-linked, more rigid CMC. The effect was attributed to the high level of inter-chain H-bonds possible for amylose. The elastic properties of pectine resulted in better mechanical characteristics and electrochemical performance compared to CMC.
Biological systems inspired also the work of Ryou et al.,97 who introduced cathecol functionalities, responsible for the adhesion of mussels even on anti-adhesion materials,98 in alginate and PAA polymers, thus increasing their adhesive properties on Si nanoparticles.
Wu et al.99 followed a different approach, producing in situ a 3D electronically conductive binder. The reaction in water of Si nanoparticles, phytic acid, and aniline in the presence of a crosslinking agent, resulted in the formation of a viscous hydrogel, ready to be cast onto the current collector. Although the feasibility of the method to produce high loading electrodes for practical applications needs to be assessed, the in situ polymerized PANi–Si (PANi = polyaniline) showed improved cyclability and rate capability. The porous 3D matrix provides sufficient void space for Si expansion, while the in situ method grants uniform coating of the particles with the electronically conductive PANi binder. Recently poly(3,4-ethylenedioxy-thiophene):poly(styrene sulfonate) (PEDOT:PSS) conductive polymer crosslinked with polysaccharides has also been introduced as a water-soluble binder.100–102 This approach has the advantage of reducing the conductive carbon additive in the electrode formulation, to exploit the ability of the polymer to sustain large volume changes (up to 400%100) and the derived electrode shows promising performance at relatively high Si loadings (1–2 mg cm2).
In sum, a suitable binder for Si-based anodes should: (i) offer weak interaction with the electrolyte which implies no swelling, and no change in stiffness and mechanical properties; this allows direct contact with the renewed active material surface, which induces further electrolyte degradation, to be reduced/avoided; (ii) provide Li+ conduction forming a uniform “artificial SEI” layer on Si particles; Li conduction can be assisted by segmental motion/coordination for neutral molecules (as in GG) or by hopping through carboxylic groups (as for PAA); (iii) include β-glycosidic bonds (as in branched polysaccharide-based binders) and functional groups that can induce ion–dipole interactions, providing more elongation and higher adhesion strength; (iv) possess self-healing properties to regenerate the interactions with the Si surface upon delithiation; and (v) allow for the formation of “bridges” between the particles and the current collector to improve the mechanical properties of the electrode.
The beneficial effect on the Si-based electrode performance originating from numerous sites for binder interactions with the active material and the current collector has been recently studied by Cao et al.,103 using graft copolymers of glycol chitosan and Li-PAA. A systematic structure-to-property relationship study conducted by Kwon et al.104 rationalized the importance of the above-mentioned characteristics. These authors synthesized different polymers, varying the ratio of the block units, each one accounting for a particular property, such as Meldrum's acid for crosslinking and covalent interactions, styrene for the backbone stiffness, methyl methacrylate for the flexibility, and hydrolysed Meldrum's acid neutralized with Li for the “self-healing” properties. The study revealed that the stiffness provides only minor improvements. Instead, to achieve better performance, the covalent interaction ability and polymer crosslinking play a major role with respect to the flexibility induced by the methyl methacrylate unit, confirming the initial studies.65 Overall, however, the work of Kwon et al.104 highlighted the self-healing ability of the polymer as the principal factor to achieve an enhanced cycle life of the Si-based anode.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles without substantial capacity decay. A crosslinked CMC/PAA binder enabled enhancement of the cyclability of non carbon-coated Co3O4 by preserving the cohesion between the active material particles upon volume contraction/expansion.114 Improved performance when using functionalized water processable binders has also been reported for flake-like SnS2 nanoparticles115 and hierarchically porous ZnMn2O4/bio-carbon anodes.116
000 cycles without substantial capacity decay. A crosslinked CMC/PAA binder enabled enhancement of the cyclability of non carbon-coated Co3O4 by preserving the cohesion between the active material particles upon volume contraction/expansion.114 Improved performance when using functionalized water processable binders has also been reported for flake-like SnS2 nanoparticles115 and hierarchically porous ZnMn2O4/bio-carbon anodes.116
2.3 Cathodes – water sensitivity issues vs. interface stabilization
:1 mixture). The resulting electrodes revealed a high mechanical flexibility and good adhesion to the aluminium current collector. Moreover, the aqueous processed electrodes provided an increased reversible capacity of 157 mA h g−1 for the first cycle (compared to 148 mA h g−1 for those comprising PVdF as the binder), comparable cycling stability, and less irreversible capacity at an elevated temperature (60 °C). Another interesting feature is certainly the higher electrode density of CMC-based electrodes (1.7 g cm−3 compared to 1.5 g cm−3 for PVdF), generally allowing for higher volumetric energy densities. Motivated by these results, Porcher et al.120,121 investigated the stability of LFP in water, discovering that the material was not fully stable. In fact, the long-term exposure in water resulted in the formation of a few nanometres thin amorphous surface layer of Li3PO4 accompanied by the partial oxidation of Fe2+ to Fe3+. This “lithium leaching” is also reflected by a slight increase of the slurry pH to about 9.121 Nonetheless, their studies showed as well that this reaction with water leads to a loss of active material of only about 1% after 24 h immersion in water,120 indicating that the corresponding capacity loss should remain negligible upon electrode preparation. This has been also confirmed by Lux et al.122 later on, who detected solely divalent iron by means of Mössbauer spectroscopy for aqueous processed LFP electrodes. As a consequence, further studies focused in particular on the optimization of the electrode microstructure, i.e., the realization of a homogeneous dispersion of the comprised components, to enhance the electrochemical performance by ensuring a percolating network of the active material and conductive carbon. For this purpose, the proper dispersion of the (hydrophobic) carbon-coated LFP and conductive carbon particles is of utmost importance. One research strategy to achieve this is the addition of suitable surfactants. Such surfactants are commonly characterized by a (short) ionophobic part (e.g., a carbonaceous chain), which preferably interacts with the carbonaceous surface of the conductive additive as well as the carbon-coated LFP, and an ionophilic part, that allows for the homogenous dispersion in polar solvents such as, for instance, water. A schematic illustration of the underlying working principle is presented in Fig. 4, highlighting the proper dispersion of the hydrophobic electrode components in water, thus, allowing for the realization of a percolating electronically conductive network of carbon black throughout the electrode coating layer. A comparative analysis, conducted by Porcher et al.,123 of three different surfactants for which the ionophilic part was anionic (sodium dodecyl sulphate), non-ionic (isooctylphenylether of polyoxyethylene; Triton X-100; Aldrich), or cationic (hexadecyltrimethyl ammonium bromide; CTAB), revealed that the therein investigated ionic surfactants are not suitable for large-scale electrode fabrication, as they induce corrosion of the aluminium current collector. The use of Triton X-100, however, resulted in better electronic wiring of the conductive carbon throughout the electrode, i.e., a more homogeneous electrode microstructure leading to enhanced rate capability. It appears noteworthy that the authors employed as a binder either a mixture of polyvinyl alcohol and polyethylene glycol (added as a plasticizer) or butadiene-acrylonitrile copolymer rubber latex. The included CMC was considered to serve solely as a “thickener” in order to optimize the slurry viscosity. Nevertheless, as reported in another study by Porcher et al.,124 the choice of the “thickener” played an important role, since the use of hydroxypropylmethyl cellulose (HPMC) instead of CMC led to inferior electrode microstructure homogeneity and, consequently, to lower specific capacities. The authors concluded that the uncharged HPMC polymer adsorbs to a greater extent to the LFP and carbon black particle surface than the negatively charged CMC. While greater adsorption of HPMC would favour the flocculation of LFP and carbon black agglomerates, lesser adsorption, i.e., the presence of “dangling” parts of the CMC polymer surrounding the particles, may favour bridging between the LFP and conductive carbon particles, thus, eventually resulting in a very homogenous dispersion of the different components. Similarly, Li and Lin125 reported the stepwise addition of CMC and SBR to the electrode slurry was of great importance, since the more adsorbing SBR, if added first, favours the formation of particle agglomerates. On the contrary, adding CMC first, which adsorbs less to the LFP particle surface, causes a steric and electrostatic repulsion between the active material particles and, hence, results in a more homogenous electrode morphology and thus, enhanced cycling stability and de-/lithiation kinetics. These results nicely illustrate that the replacement of PVdF and NMP is not an easy task – even if the active material is essentially inert vs. water – and that all components and their corresponding interactions need to be considered (and understood) in order to obtain the best possible electrochemical performance for the final electrodes. As a matter of fact, Li et al.126,127 reported that also cationic surfactants like polyethyleneimine may be suitable to enhance the electrode microstructure (in their study used together with xanthan gum as the binder), if carefully considering the isoelectric point of the electrode components (pH = 4.3 and 3.4 for LFP and carbon black, respectively). In fact, the increased repulsive potential between the particles supports their homogenous dispersion in water and, accordingly, the realization of well-structured coated electrodes. Focusing as well on the importance of well dispersed particles in an aqueous suspension, Tsai et al.128 highlighted the importance of homogeneously coated LFP in order to prevent their agglomeration – or as they referred to it, “the gelation” of the electrode slurry – as a result of the hydrogen bonding between non-coated surfaces. As a matter of fact, such gelation may also occur for carbonaceous surfaces with a high density of hydroxyl, carboxyl, or carbonyl groups – not least due to the insulating character of sp3-hybridized carbon.129 On the contrary, the presence of these surface functional groups eventually allows for the favourable interaction with water-soluble binding agents like CMC (potentially used also as a binding agent by itself),122 PAA,130–133 xanthan gum,134 or carboxymethyl chitosan derivatives.135–137 In turn, this advantageous interaction leads to advanced cycling stability, rate capability, and apparent lithium ion diffusion coefficients as well as frequently higher specific capacities of the electrodes made using such binders rather than PVdF, as a result of the more uniform electrode microstructure and the increased adhesion to the aluminium current collector, favouring the de-/lithiation kinetics and lowering the cell polarization.122,131–134 One key feature enabling
this enhanced electrode performance is the low swellability of most aqueous binders compared to PVdF.135,138 Another highly advantageous characteristic, found by Zhang et al. for PAA, is that the binder does not only ensure the mechanical integrity of the electrode, but moreover forms a protective surface coating for the active material, which stabilizes the electrode/electrolyte interface and, thus, prevents or at least dramatically reduces detrimental side reactions like the dissolution of iron – especially for elevated operational temperatures such as, for instance, 55 °C.132
 | ||
| Fig. 4 Schematic illustration of the working principle of surfactants in aqueous electrode slurries, ensuring the homogenous dispersion of the hydrophobic carbon-coated LFP active material and carbon black particles (left panel) and, thus, allowing for the realization of a percolating network of the conductive agent throughout the electrode coating layer (right panel). | ||
To further optimize the electrochemical performance of aqueous processed lithium-ion cathodes, Li et al.139 have also considered the aluminium current collector/electrode coating interface. They found that the application of proper surface treatments of the aluminium foil such as, for instance, a corona plasma treatment and the resulting partial oxidation and functionalization of the aluminium surface (e.g., by –ROOH or –RO2R) leads to better wettability by polar aqueous dispersions and, as a result, to substantially increased specific capacities by about 20 mA h g−1 relative to the untreated samples.
With respect to these great achievements in the past few years, it does not appear surprising that also lithium-ion full-cells, incorporating exclusively aqueous processed electrodes, have been reported. Using PAA in combination with a small amount of SBR, for instance, Chong et al.133 studied LFP/graphite cells and achieved a remarkable capacity retention of 70% after about 850 cycles. Kim et al.140 reported a LFP/LTO full-cell, employing CMC as the binder for both electrodes and an ionic liquid-based electrolyte (i.e., N-butyl-N-methyl-pyrrolidinium bis(fluorosulfonyl)imide-lithium bis(trifluoromethanesulfonyl)imide; PYR14FSI-LiTFSI). These cells were stably cycling for more than 150 cycles and the observed, very minor fading was assigned to detrimental interfacial reactions at the anode rather than at the cathode.
Before continuing with the next subsection, we would like to highlight two more approaches reported for LFP to enhance the electrochemical performance and sustainability of these cathodes. The first concerns the replacement of the nanoparticulate carbon by electronically conductive polymers, which may facilitate the electrode preparation and material handling. The most prominent (water-soluble) polymer in this regard is certainly PEDOT:PSS. This conductive polymer has been combined with CMC141,142 and carboxymethyl chitosan.136 In combination with CMC, Eliseeva et al.141 reported very remarkable specific capacities at lower and higher dis-/charge rates, i.e., 148 and 126 mA h g−1 at 0.2C and 5C, respectively, as well as excellent cycling stability with only 1% decay over 100 cycles at 1C. Equally impressive, Zhong et al.136 reported prototype-like prismatic 10 A h cells, including a combination of PEDOT:PSS and carboxymethyl chitosan as an electronically conductive binder matrix, providing a capacity retention of about 90% after 1000 cycles (charge/discharge = 1C/2C) and around 98% of the 1C capacity remaining when dis-/charging the cell at 7C.
The second approach was reported by Jeong et al.,143 who introduced natural cellulose as the binder for LFP/graphite full-cells, achieving stable cycling for around 120 cycles. As cellulose is not water-soluble, they utilized an ionic liquid as the solvent/dispersant (1-ethyl-3-methylimidazolium acetate; EMIMAc). This may, at first sight, appear less “green” than simply using water, but the facile recuperation by applying a simple phase inversion process using water as a co-solvent provides the great chance to establish a completely “closed” recycling loop.
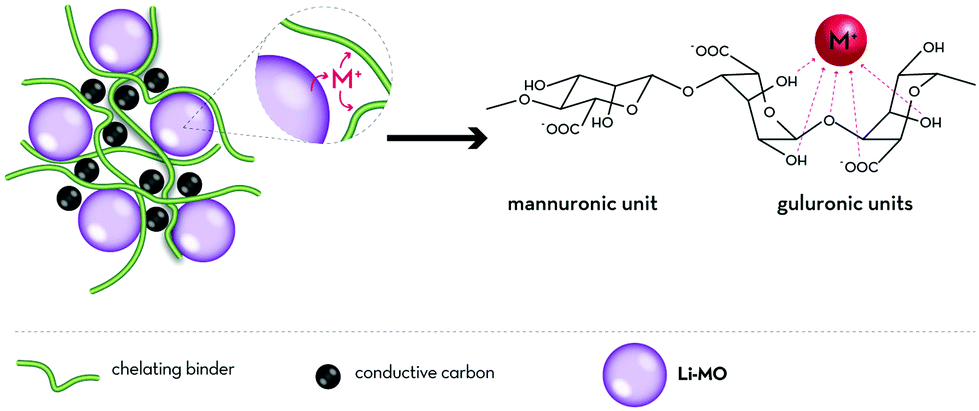 | ||
| Fig. 5 Generalized illustration of the (potential) metal cation scavenging effect of chelating polysaccharide derivatives serving as binders; as reported for sodium alginate and the scavenging of divalent manganese cations dissolved from spinel-structured LiMn2O4 by Ryou et al.155 [M = (transition) metal]. | ||
Nonetheless, the perspective for commercial applications of LMO remains limited at present, due to its comparably low energy density. Accordingly, also the number of studies towards this direction is rather low. Instead, a greater number of studies has been reported for its high voltage derivative, LNMO, initially reported by Amine et al.159 and Dahn and co-workers.160 In fact, the partial replacement of manganese by nickel allows for (theoretically) fixing the oxidation state of manganese to 4+, so that essentially only nickel is electrochemically active. By this, the aforementioned issues like Jahn–Teller distortion and Mn3+ disproportionation are ideally avoided.
We may, first, briefly review the rather performance-oriented studies now concerning aqueous electrode processing with LNMO as the active material, before focusing on the general issues, which are, as a matter of fact, of great importance also for all other lithium transition metal oxides, presently employed already or suggested to be employed soon in commercial lithium-ion batteries. To the best of our knowledge, the use of water-soluble binders for LNMO has been reported for the first time by Wang et al.161 in 2012, investigating the utilization of CMC and comparing the performance with reference electrodes comprising PVdF. They showed that the electrodes including CMC provided superior capacities over all dis-/charge rates and reduced self-discharge, while the cycling stability was roughly comparable, maybe slightly inferior to PVdF-based electrodes. This generally beneficial effect of water-soluble binders on the electrochemical performance of LNMO cathodes was supported by several studies, revealing further enhanced performance when using CMC162 and extending the findings of Wang et al.161 to CMC crosslinked via citric acid,163 sodium alginate,164,165 carboxymethyl chitosan,166,167 poly(vinyl acetate),168 PAA,169 or TRD202A.22 As for LFP, the main reasons for such improved performance were identified to be the well maintained electrode microstructure, a lower internal resistance and faster de-/lithiation kinetics, a higher peeling strength, and less swelling of the binder by the liquid organic electrolyte,162,164–166 while the proper drying of in-water-processed electrodes is apparently crucial.18,170 Once more, the use of PAA was reported to result in the formation of a very homogeneous surface coating of the active material particles,169 which is, in light of the high lithium de-/insertion potential of LNMO and the rather limited oxidative stability of common organic-carbonate-based electrolytes, of even greater relevance compared to LFP and LMO, since it turned out to stabilize the electrode/electrolyte interface, thus preventing or at least lowering the continuous electrolyte decomposition. Another very interesting aspect of the study of Pieczonka et al.169 was that the therein utilized Li-PAA may also as a Li+ reservoir, which simultaneously allows for buffering the acidity of the electrolyte due to the corresponding exchange of lithium cations (from the binder) and protons (from the electrolyte), which mitigates parasitic side reactions at the interface.
Nevertheless, the general issues when dispersing lithium transition metal oxides (including LNMO, LiCoO2, Li[Ni1−x−yCoxMny]O2 (NCM), Li[Ni1−x−zCoxAlz]O2 (NCA, with 1 − x − z ≥ 0.85), or LiMn2O3·NCM (Li-rich NCM or LR-NCM)) in water remain: (i) the leaching of lithium out of the active material host structure, (ii) the concurrent formation of LiOHaq and structural degradation of the particle surface (i.e., the outer ∼3–10 nm), (iii) the subsequent increase in pH and, eventually, (iv) the corrosion of the aluminium current collector due to the breakdown of the native alumina surface layer – as summarized in Fig. 6a.22,145,171–179 While it is still not comprehensively elucidated whether this lithium leaching is accompanied by the concomitant leaching and/or oxidation of the comprised transition metals22 or by Li+/H+ exchange180 or potentially both reactions, it is clear that the amount of electrochemically available lithium inside the active material is reduced, thus, lowering the overall reversible capacity – at least in lithium-ion full-cells, for which the cathode is the only lithium source.175,176,178
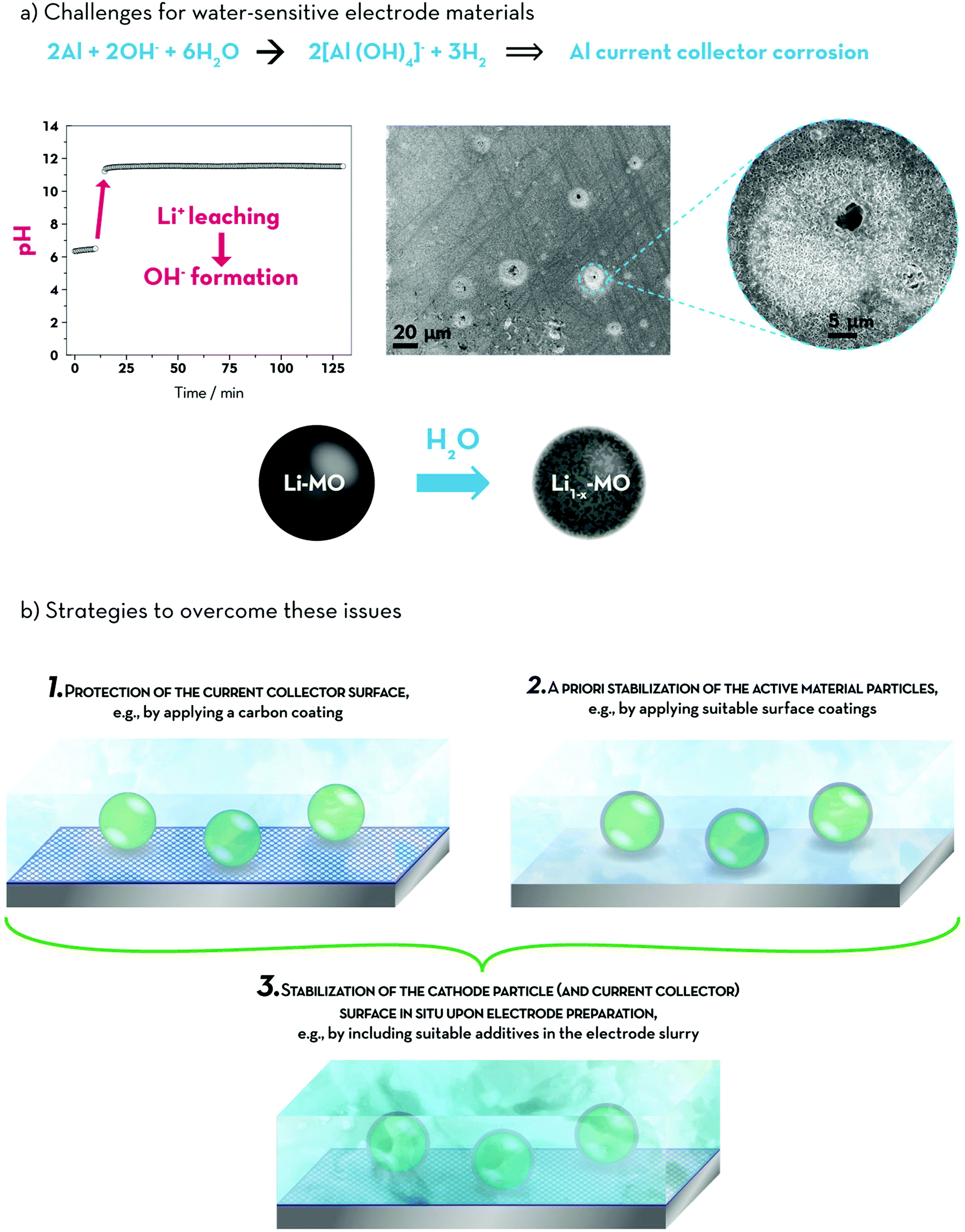 | ||
| Fig. 6 (a) General challenges for the aqueous electrode processing of water-sensitive electrode materials like NCM, LR-NCM, or LNMO and (b) potential strategies to overcome these issues, including (1) the protection of the current collector surface, (2) the a priori stabilization of the active material particle surface, and (3) the stabilization and protection of potentially both surfaces in situ upon electrode preparation. | ||
The strategies that have been so far pursued to overcome this great challenge are schematically illustrated in Fig. 6b. A rather easily implementable approach, the protection of the aluminium current collector surface by applying suitable coating layers such as, for example, carbon, in order to physically prevent the contact between the electrode coating layer and the aluminium foil (1), has led to a substantial improvement in terms of cycling stability and achievable capacity, while also leading to decreased charge transfer resistances.181,182 As a result, lab-scale lithium-ion full-cells, employing NCM and graphite – both prepared by using CMC as the binder – showed very stable cycling for more than 2000 dis-/charge cycles and a final capacity retention of around 70%.182 Nevertheless, the application of such coatings requires an additional processing step, lowers the overall energy density of the cell, and special care has to be taken to avoid detrimental defects in the coating layer, as these might still serve as nucleation sites for aluminium corrosion. Maybe even more importantly, this strategy does not really address the issue's origin, but instead its negative outcome. Similarly, the addition of mild (organic) acids (e.g., formic acid) during electrode preparation has been reported,179,183 which, however, once again, addresses the detrimental effects of the lithium leaching by keeping the pH value of the electrode slurry well within the aluminium passivation regime, but not its origin. In fact, the addition of such mild acids may even have a negative impact on the active material integrity, since the transition metal dissolution significantly increases.184 On the contrary, an approach that addresses the issue at its origin is the application of protective surface coatings for the active material itself (2). Tanabe et al.,22 for example, prepared water-resistant LNMO by coating the particles with carbon, Al2O3, or Nb2O5. Employing TRD202A as an aqueous binder, the thus prepared electrodes showed a slightly worsened rate capability, but stable cycling and no aluminium current collector corrosion was detected – even though the pH still increased to values around 10 upon long-term storage of the active material in water, indicating that the lithium leaching was not fully prevented, yet. Slightly better results were obtained by the same group185 when coating NCA with TiOx, providing a comparable rate capability and cycling stability as for the PVdF-containing reference electrodes – presumably due to the realization of a really thin coating layer with a thickness of only 2–3 nm. Also the application of a vanadium oxide coating for LR-NCM successfully prevented the aluminium current collector corrosion, while moreover allowing for substantially increased specific capacities (+30 mA h g−1) at moderate rates.186 As a matter of fact, these coatings do not necessarily have to consist of inorganic materials. Instead, the use of, for instance, PAA169 or crosslinked polyurethane in combination with CMC187 – both resulting in the formation of polymer films, which homogenously cover/encapsulate the particle surface – may be employed as well. This approach possesses the great advantage that no additional, potentially costly processing step is needed and the surface coating is obtained in situ upon electrode preparation.
A different approach, essentially combining all the aforementioned strategies, was very recently reported by Loeffler et al.188 Rather than adding a mild organic acid during the electrode fabrication process, the authors added a minor amount of phosphoric acid (PA) to buffer the otherwise dramatically increasing pH of the electrode slurry. Remarkably, the added PA spontaneously reacts at the NCM particle surface, resulting in a (very thin) metal (nickel/cobalt/manganese/lithium) phosphate surface coating, which stabilizes the active material/electrolyte interface and, thus, allows for less transition metal dissolution compared to the addition of formic acid, higher specific capacities, enhanced de-/lithiation kinetics, and advanced cycling stability. In addition, the current collector corrosion is suppressed, as the pH remains well in the aluminium passivation regime. As a result, NCM/graphite lithium-ion full-cells – both incorporating CMC as the binder – showed stable cycling for more than 300 cycles with an excellent capacity retention of 96%. The general applicability of this easily implementable approach has subsequently been confirmed by studying its effect also for the aqueous electrode preparation of NCM424 (i.e., NCM with a relatively higher nickel and manganese content),189 LNMO,163 and LR-NCM.190 The latter included moreover a comparison of three different binding agents, i.e., CMC, sodium alginate, and TRD202A, revealing slightly superior performance for CMC compared to alginate – in good agreement with an earlier study by Xu et al.191 – and inferior cycling stability for TRD202A. Concerning the addition of PA, interestingly, the beneficial effect was more pronounced for CMC and alginate compared to TRD202A and it was found that the first two binders remain chemically unaffected by the addition of PA – at least in the presence of LR-NCM. Contrarily, TRD202A reacts with PA, indicating that this reaction and the reaction of the active material with PA are competitive and, hence, highlighting that the electrode preparation may be far more complex than simply mixing an inert binder, a conductive agent, and the active material in a suitable solvent/dispersant – an aspect that is frequently overlooked in the literature.
When investigating the impact of adding PA for LNMO-based cathodes using CMC as the binding agent, Kuenzel et al.163 addressed also the issue of finding a suitable binder combination for high-voltage cathodes. While graphite anodes commonly contain a combination of CMC and SBR to ensure the flexibility and adhesion of high mass loading electrode coatings192–195 (see also Section 2.1), the latter cannot be used in this case because of its limited electrochemical stability towards oxidation.194–199 Nevertheless, it is worth mentioning in this context that two studies on LiCoO2,200,201 implementing a mixture of CMC and SBR, have reported its general suitability – even for anodic cut-off potentials as high as 4.5 V.201 In fact, it appears that the initial oxidation of SBR leads, indeed, to a higher initial irreversibility, but benefits the stability of the cathode/electrolyte interface, resulting in less polarization upon cycling and an improved capacity retention compared to PVdF-based electrodes.
Kuenzel et al.,163 however, followed another approach and crosslinked CMC using citric acid as the crosslinking agent, as this had been reported to be advantageous for low-voltage sulphur cathodes202 and phosphorus-based sodium-ion anodes.203 In combination with the addition of PA, substantially enhanced cycling stability and increased capacities were observed compared to the simple CMC reference. Beside the stabilization of the LNMO particle surface and suppressing the aluminium current collector corrosion, the addition of PA also led to the formation of a thin AlPO4 layer on the current collector surface, presumably further contributing to the advantageous impact of adding PA to the electrode slurry.
Before eventually highlighting a few studies on prototype-like lithium-ion full-cells, which nicely illustrate the advanced development stage of aqueous cathode processing technologies, we may point at one more, initially maybe surprising advantageous effect of employing water-soluble binding agents. LR-NCM is an attractive candidate for next generation, high-energy LIBs, theoretically providing energy densities as high as 1000 W h kg−1 and commonly characterized by relatively low cobalt content.204–206 Its application in commercial cells, however, is so far hindered by the continuous voltage decay upon cycling, which is suggested to be the result of ongoing structural changes – although the precise reason remains to be comprehensively understood.3,204,206 In this regard, it appears noteworthy that several recent studies investigating CMC,207 alginate,208 or guar gum209 as binders for LR-NCM-based electrodes revealed reduced voltage fading in addition to generally improved electrochemical performance. These findings may, thus, pave the way for eventually employing LR-NCM in future LIBs without the need for expensive and extensive particle coatings to overcome the detrimental surface-driven changes upon continuous de-/lithiation.210,211
Finally, as mentioned earlier, we would like to highlight two studies that have reported the successful realization of prototype LIBs including aqueous processed cathodes. The first of these two was published by Wood et al.117 in 2017. The authors assembled 1.5 A h full-cells, using NCM532 and graphite as the cathode and the anode, respectively, and CMC as the binder. Providing a capacity retention of around 80% after 900 cycles, comparable to the therein studied reference cells incorporating PVdF, these results render the completely water-based processing of lithium-ion electrodes feasible. Even larger cells, i.e., NCM111/graphite full-cells with a total capacity of 15–18 A h, were reported by Loeffler et al.212 – also in 2017. The graphite anode was based on a combination of CMC and SBR as the binder and the cathode contained a mixture of CMC and an acrylic waterborne latex. These cells showed a remarkable capacity retention of 99% after 500 dis-/charge cycles – or in other words a capacity fading of only 0.004% per cycle – rendering the realization of completely aqueous processed LIBs more than feasible, in fact.
3. Sodium-ion batteries
Non-aqueous Na-ion batteries (SIBs) are, indeed, very similar to LIBs, but promise lower prices as well as higher sustainability.238 Obviously, the use of cheap and environmentally friendly materials and processes is crucial to achieve very low costs. In light of this, the use of natural binders and aqueous electrode processing is especially appealing if not fundamental for the future success of non-aqueous SIBs. However, SIBs and LIBs show some peculiarities with respect to the binder, due to (i) the larger ionic radius of Na+, leading to different volume change upon reversible uptake and release; (ii) the use of Al instead of Cu as the negative current collector; and (iii) the somehow higher water sensitivity of the positive and negative active materials.The large ionic radius of the Na+ cation leads to higher volume changes upon sodiation and desodiation. In consequence, the careful selection and optimization of the binder is of great importance in order to maintain the mechanical integrity of the composite electrode and its adhesion to the current collector, especially for those active materials undergoing conversion and/or alloying reactions with Na+.239–242 In fact, the use of Al as the negative current collector is one of the fundamental advantages of SIB technology.243–245 However, it also represents an additional challenge to be addressed for the aqueous processing of negative electrodes because of the earlier mentioned Al corrosion (see Section 2.3).
The most prominent active material classes in SIBs are essentially similar to those in LIBs. Nevertheless, they exhibit different intrinsic properties, such as water sensitivity and volume changes upon cycling in SIBs, which strongly affects aqueous processing and binder selection.
3.1 Aqueous electrode processing of positive electrode materials for SIBs
Generally, not many studies specifically target the implementation and investigation of aqueous electrode processing for positive electrode materials in SIBs. In fact, the large majority of cathode materials are still processed with a PVdF binder.245The use of a CMC binder has been reported for the Na0.44MnO2 material, which exhibits rather low water sensitivity.246 The electrolyte (optimized: 1 M NaPF6 in EC:DEC 3:7 (v/v) + 2 wt% FEC; fluoroethylene carbonate) was found to be important to prevent the anodic dissolution of Al upon long-term cycling. Like Na0.44MnO2, polyanionic materials show typically low water sensitivity. For example, the aqueous electrode processing of carbon-coated Na2FePO4F with a polyacrylic latex binder247 and of Na3V2O2x(PO4)2F3−2x248 and Na3V2(PO4)2F3249 with a CMC binder has been reported. These electrodes showed enhanced and more stable performance in comparison to those made using PVdF resulting from better electronic conductivity,247,248 enhanced electrode adhesion,247–249 and facilitated Na+ ion diffusion.247,249 Furthermore, Zhao et al.249 reported improved electrode integrity for Na3V2(PO4)2F3 with CMC after cycling, negligible electrode swelling in contact with the liquid electrolyte, and the formation of a protective, solid and permeable interface film limiting the electrode resistance increase.
Na-Based hexacyanometalates NaxM1[M2(CN)6]y·zH2O (M1, M2 = transition metal; 0 ≤ x ≤ 2; y ≤ 1) contain interstitial water whose removal leads to improved electrochemical performance.250 Aqueous electrode processing with a CMC binder was reported for the sodium iron hexacyanoferrates Na2Fe2(CN)6·2H2O and Na1+xFe2(CN)6·3.1H2O.251,252 The effect of PVdF, Na-CMC, PTFE, and ethylene propylene diene monomer (EPDM) rubber binders on the performance of Na0.75Fe2.08(CN)6·3.4H2O was investigated very recently.253 The best electrochemical performance, especially at low current rates, was observed with a PVdF binder although electrodes with CMC delivered comparable electrochemical performance at high current rates (10C). Partial solubility and instability of the active material in water were reported to aggravate the electrode processing and adhesion of the coated electrode to the current collector.
The aqueous processing of layered oxide cathode materials is connected with several challenges. Layered oxides are typically categorized into P2- or O3-type according to the structural classification reported by Delmas et al.254 Both differ with respect to the number of transition metal layers per unit cell and the coordination of the Na-cation, being either octahedral (O) or trigonal prismatic (P). Most layered oxides are sensitive towards moisture and air, undergoing side reactions with water and carbon dioxide leading to the formation of sodium hydroxide and sodium carbonate, respectively.245,255 Hence, aqueous electrode processing of layered oxides can lead to active material degradation leading to poor electrochemical performance. As a result, increasing efforts are focusing on the development of air and moisture stable layered oxides. Air-stable and Co/Ni-free O3-type Na0.9[Cu0.22Fe0.30Mn0.48]O2 with high water stability has been reported.256 Improved air and water stability was also reported for O3-type NaNi0.45Cu0.05Mn0.4Ti0.1O2, synthesized via Cu/Ti-doping of NaNi0.5Mn0.5O2.257 Lu and Dahn258 studied the intercalation of water in different P2-Na2/3[CoxNi1/3−xMn2/3]O2 materials in 2001 and found increased water sensitivity at higher cobalt contents. They assumed the super-lattice ordering in the transition metal layer to aggravate water intercalation and to be suppressed by addition of cobalt. Interestingly, P2-type Na2/3Ni1/3Mn2/3O2 was found not to be hygroscopic and stable towards moisture. In fact, Kubota and Komaba259 reported the enhanced electrochemical performance for this material with a CMC binder in comparison to PVdF. Recently, the aqueous electrode processing with a CMC binder has also been reported for P2-type Na2/3Ni1/3Mn5/9Al1/9O2.260
3.2 Aqueous electrode processing of negative electrode materials for SIBs
In comparison to cathode materials, many studies report the aqueous processing of anode materials in SIBs, but often with copper as the current collector. In the very recent review of Bommier and Ji,261 the binders most commonly used for carbon-, alloying-, conversion- and insertion-based anode materials in SIBs are evaluated. The most commonly used binder is PVDF, but in particular for alloying- and conversion-based anode materials, i.e., those materials showing higher volume changes upon reversible Na+ storage, CMC and other aqueous binders were found to be preferentially used. In that review, the effect of the binder on the initial coulombic efficiency was statistically evaluated as well. With the exception of carbonaceous materials, all anodes made utilizing a CMC binder showed substantially higher initial coulombic efficiencies, frequently above 75%.The most commonly used anode materials in SIBs are, however, hard carbons. Several studies, focusing on the hard carbon material design and/or de-/sodiation mechanism, report aqueous processing with, e.g., CMC,262,263 Na-alginate,264,265 or sodium polyacrylate,259,266 sometimes employing Al as the current collector.263,265,266 Dahbi et al.263 demonstrated superior performance of hard carbon electrodes on Al current collectors made with CMC, rather than PVdF, in 1 M NaPF6 in PC with 2 vol% of FEC as an additive. An XPS study revealed that the hard carbon particles are initially covered by a thin layer of CMC, which contributes to the formation of a uniform and thin passivation film upon sodiation. In contrast, the PVdF-based electrodes show the formation of thick surface films, and of binder de-fluorination, resulting in the formation of NaF. In turn, the binder decomposition led to loosened composite electrodes and increasing resistance. The study by Vogt et al.267 confirmed the partial decomposition of PVdF and formation of NaF when using 1 M NaClO4 in PC as the electrolyte, indicating that these processes negatively affect the initial coulombic efficiency and the long-term cycling performance of the cell.
Fan et al.268 reported the effect of the binder on the performance of nitrogen doped hollow carbon nanotubes and found improved performance and higher initial coulombic efficiency with a sodium polyacrylate (Na-PAA) binder in comparison to PVdF. This was explained via the electrochemical activity of Na-PAA, the lower internal resistance of the electrode, and the formation of a homogeneous passivation layer covering the active material particles. The binding capability of Na-PAA was found to drop, however, with decreasing molecular weight.
For insertion-based anode materials such as titanates, e.g., Li4Ti5O12,269 Na-doped Li4Ti5O12,270 and the sodium titanate Na2Ti6O13,271 similar results like for hard carbons have been observed. In detail, a more stable long-term cycling performance has been obtained with sodium alginate and CMC in comparison to a PVdF binder on a Cu current collector. Zarrabeitia et al.272 investigated the moisture and water stability of Na2Ti3O7.
The importance of proper binder selection for alloying based materials such as Sn267,273–275 or Sb276 is illustrated by several studies, reporting improved electrochemical performance with aqueous binders in comparison to PVdF. In detail, Komaba et al.273 reported an improved electrochemical performance and enhanced initial coulombic efficiency of Sn electrodes made with PAA (polyacrylic acid) in comparison to those made with PVdF. Dai et al.274inter alia evaluated the performance of Sn nanoparticles with PVdF or CMC as the binder. The use of CMC enabled an improved performance because of its higher binding ability due to the formation of strong H-bonds between the carboxylic groups in CMC and the Sn nanoparticles. Yui et al.275 investigated the effect of PVdF, PAA, sodium polyacrylate, CMC, and polyimide on the electrochemical performance of Sn–Co based anodes. Confirming previous observations, these authors obtained very good performance with PAA and CMC in comparison to the other binders. The improved electrochemical behaviour is due to the enhanced electrode adhesion for the PAA (0.72 kN m−1) and CMC (0.62 kN m−1) based electrodes with respect to those based on PVdF (0.60 kN m−1) as well as the decreased electrode swelling after the initial cycle, i.e., 111% for PAA and 125% for CMC compared to 198% for PVdF.
The electrochemical reaction of sodium with black or red phosphorous leads to the formation of sodium phosphide (Na3P), which is associated with the strong volume expansions occurring, ranging from 292% (red phosphorous) to 390% (black phosphorous), i.e., in the same range as Sb (Na3Sb: ≈293%) and Sn (Na3.75Sn: ≈423%). Thus, P-based electrodes require binders that have elastic moduli, e.g., oxygen containing functional groups, capable of buffering the large volume changes to maintain mechanical integrity and adhesion to the current collector. In fact, electrodes with phosphorous and Na-PAA,277,278 PAA,279 or CMC280 show much better performance than those based on PVdF as the binder.
For FeP, the combination of CMC and PAA as the binder was reported to enable enhanced performance as compared to electrodes with PVdF or CMC only.281 The formation of the earlier mentioned CMC/PAA cross-linked structure, capable of buffering the mechanical stresses generated by large volume changes, was assumed as the possible origin. The results correlated well with the previous work by Sun and co-workers,282 reporting strongly improved electrochemical performance of Co3O4 and NiO–C electrodes made with CMC/PAA (5 wt% each) in comparison with PVdF as the binder. The same observation was made by Zhou et al.283 for a SnS–graphene hybrid anode material with PAA/CMC. The use of crosslinked chitosan as the binder (dissolved in 1 wt% acetic acid in aqueous solution) has been reported for Sb.284 The authors argued that crosslinked polymer binders may offer a stronger, more rigid network and, thus, better mechanical integrity for materials undergoing large volume changes during cycling in comparison to regular non-crosslinked polymers. The latter, in fact, may go through plastic deformation upon repeated extraction and uptake of sodium, eventually failing in maintaining the electrode integrity.
Recently, Luo et al.285 reported the use of a hydroxyl-rich Na-alginate binder with an organic oxygen-rich sodium rhodizonate active material and observed that the binder filled the cracks in the coated electrode, which resulted from the large volume changes of the active material because of the formation of strong H-bonds between the binder and the active material. This led to improved adhesion and mechanical stability as well as lower resistance and, in consequence, enhanced electrochemical performance. Similar observations were made for α-Fe2O3 with natural hydroxyl-rich arabic gum,286 demonstrating the importance of binder–active material bond formation for the achievement of improved performance.
4. Electrochemical double-layer capacitors
Among the different electrochemical devices covered in this review, EDLCs are those where the role of the binder has been, so far, more neglected. The quite scarce literature available on the topic reflects the limited interest of the scientific community; reasons being the relatively easy charge storage mechanism (double layer charging does not involve breaking/creation of chemical bonds, phase transitions, nor considerable volume changes) and, probably, the more limited market demand of such devices. Nevertheless, the binder may have a significant impact on the performance, cycle life, and environmental impact of EDLCs as well. An ideal binder should, indeed, provide sufficient mechanical and electrochemical stability without impeding wettability, ion mobility in the electrode pores, and electron conduction. Therefore, besides being “greener”, alternatives binders should provide physicochemical properties of the electrode layer as close as possible, if not superior, to those obtained by conventional F-containing polymers.The first attempt to replace conventional PTFE was done in 1998 by Bonnefoi et al.287 They targeted CMC as an alternative, mostly to reduce cost. As the aim of their work was to increase the volumetric capacitance of the electrode, raising the activated carbon (AC) content was mandatory. Unfortunately, though, when using only CMC, they could not exceed 80 wt% AC without greatly decreasing the mechanical properties of the electrodes. Whereas, PTFE allowed mechanically stable electrodes with an AC content up to 95 wt%, without affecting the electrode resistivity nor their frequency behaviour. As the use of CMC appeared to be incompatible with high AC loading, to accomplish their target (i.e., fabricating an economical 2 V, 300 F supercapacitor in a total volume of 100 cm−3) they decided to only partially replace PTFE with CMC. Although the composition of the two investigated PTFE/CMC ratios was not specified, in both cases a reduction of the resistivity was observed compared to the PTFE counterpart for the same electrode density (ca. 0.4 g cm−3) and composition (95 wt% AC, 5 wt% binder).
Beck and Dolata extended the exploration of greener binders for EDLCs by evaluating commercially available aqueous dispersions of PS, SBR and ethylene-acrylic acid copolymers for electrode preparation.288 In an acidic electrolyte (12 M H2SO4), however, none of the chosen binders could deliver the same capacitance as the PTFE-based electrodes. Stability over prolonged cycling also appeared to need improvements, with the loss of capacitance attributed to the low molecular weight of the F-free binders which could be transported into the pores and block them. Overall, the closest performance to PTFE could be achieved with Butonal LS 133, which is a styrene–butadiene copolymer. A similar binder was also proposed by Sun et al.289 An organic solvent-free solution/dispersion, mainly constituted by an SBR–PAN (polyacrylonitrile) mixture, was used to fabricate electrodes with 10 wt% binder content. An interesting feature provided by this novel binder is its thermal stability. In fact, it shows minimal to no mass loss upon heating up to 300 °C in an inert atmosphere (N2). Nevertheless, the fast heating rate (10 °C min−1) used for the thermogravimetric analysis may have led to an overestimation of the performance. Compatibility with both organic (Et4NBF4 in PC and acetonitrile; ACN) and IL (EMIMBF4) electrolytes was also proven by CV, galvanostatic cycling, and float voltage tests.
In an attempt to develop a green supercapacitor exclusively composed of environmentally friendly materials, Dyatkin et al.290 explored alternative binders. Unfortunately, the large majority of investigated polymers (and mixtures thereof) did not enable sufficient mechanical stability, impeding the fabrication of robust freestanding electrodes. This was attributed, mainly, to the poor elasticity, molecular mass, and number of polar bonds capable of binding the activated carbon particles.290 The only two viable alternatives resulted to be a 1:1 blend of poly(vinyl acetate) and polyisoprene (PVAc–PIpr) and a crosslinked PVA-based glue. They provided comparable malleability to PTFE, resulting in flexible, but freestanding electrodes. However, the curing step necessary to crosslink the hydroxyl groups with BO4 (in sodium borate solution) substantially affected the specific surface area and pore volume of the PVA-based electrode. The decreased available surface was also attributed to the morphological properties of the different binders – with PTFE creating a fibrous network entangling the carbon particles – while PVA shows lamellar deposits on the carbon surface (Fig. 7a). Also in terms of the obtained capacitance, none of the greener binders could outperform PTFE. Additionally, a substantial decrease of the rate capability was observed as a result of the higher resistance. Overall, the authors concluded that no readily available polymer could offer, to date, the same features as PTFE.
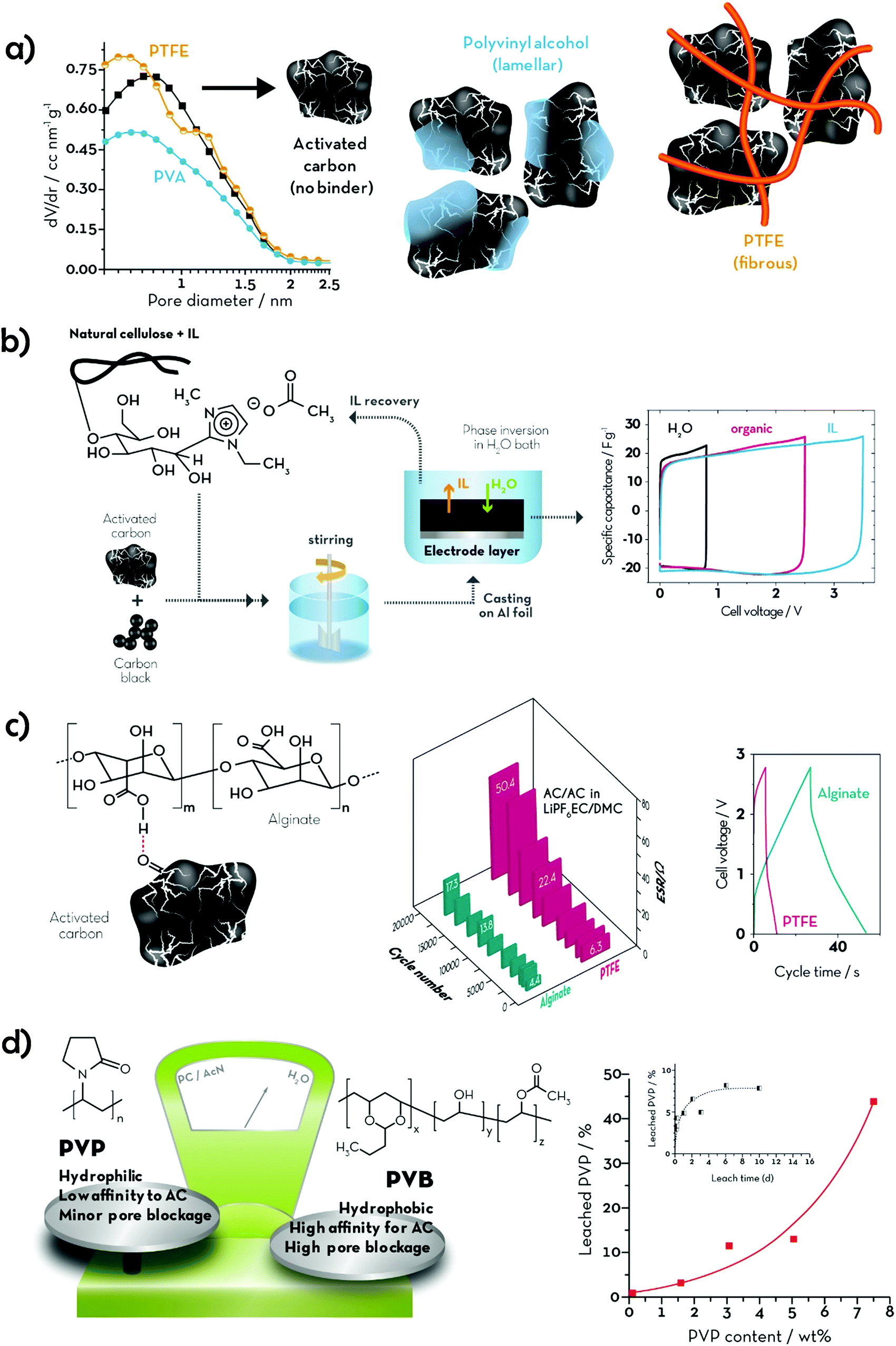 | ||
| Fig. 7 Effect of different binders on EDLC electrode properties and performance. (a) Possible pore blockage caused by a lamellar PVA binder compared to fibrous PTFE.290 (b) Method for producing universal EDLC electrodes based on natural cellulose. These can be employed, indeed, with aqueous, organic and IL-based electrolytes.295,298,314 (c) Hydrogen bonds formed by the interaction of carboxylic groups of alginate and basic moieties on the activated carbon surface, resulting in longer cycling life and reduced resistance growth.300 (d) PVP/PVB mixtures as binders for aqueous EDLCs. By tuning the ratio of the two components it is possible to tune the affinity towards activated carbon and the porosity and, at the same time, minimize the PVP leaching.307 | ||
In the early stage of binder research for EDLCs, CMC has certainly been the most popular choice, with the group of Balducci extensively reporting its use as an alternative to fluorinated polymers.291–294 Nevertheless, CMC is far from being the ideal binder for this application. From one side, it is not a “universal” binder. CMC-based electrodes can certainly operate in the majority of organic (propylene carbonate or acetonitrile) and IL-based electrolytes, but cannot be employed in aqueous environments due to the high solubility of CMC. This is a pretty restricting feature, as aqueous EDLCs would benefit from enhanced power performance enabled by the high conductivity of the water-based electrolyte. In order to overcome this issue, Böckenfeld et al.295 proposed natural cellulose (NC) as a potentially universal binder for EDLCs. NC is, in fact, insoluble in water as well as in most organic solvents. This does complicate the electrode fabrication method, but substantially decreases cost and environmental impact. NC is, indeed, cheaper than CMC (0.5–1.5 EUR kg−1vs. 1–2 EUR kg−1) and is not subject to any chemical treatment.296 Böckenfeld et al.295 exploited the solubility of NC in an imidazolium-based IL for obtaining a homogeneous slurry that could be cast on Al foil and, finally, the IL was completely recovered by phase inversion with water. Such a clever approach, which was previously proven for LIB electrodes by Jeong et al.,143 led to NC-based AC electrodes compatible with aqueous (neutral, 1 M Na2SO4), organic (1 M Et4NBF4), and IL-based electrolytes (PC-PYR14TFSI, 1:1 w/w; Fig. 7b). The same technique was adopted by Murashko et al.297 to fabricate free-standing AC electrodes for aqueous EDLCs. The AC–NC weight ratio 6:1 was found to be optimal in terms of internal resistance, rate capability, and stability upon prolonged cycling. Interestingly, Varzi et al.298 also demonstrated that NC provides enhanced stability at high voltages. Extended float voltage tests performed at 3.7 V in the pure IL electrolyte (PYR14TFSI) showed reduced capacitance fading and less pronounced ESR (equivalent series resistance) growth compared to analogous electrodes containing PVdF. The improved performance was ascribed to the higher cathodic stability of NC, which, in such demanding conditions, does not suffer from the dehydrofluorination occurring with PVdF.
A further limitation of CMC is the difficulty to obtain sufficiently flexible and mechanically stable electrodes. Indeed, CMC-based electrodes become extremely brittle after drying, resulting in easier cracking and peeling of the electrode layer.299 To address this issue, other naturally occurring polysaccharides have been proposed. Alginate, for example, a major constituent of brown algae and a copolymer of 1 → 4-linked β-D-mannuronic and α-L-guluronic acid, has been reported as an EDLC binder for the first time by Yamagata et al.299 Similarly to the initial report of Kovalenko et al.84 with Si electrodes for LIBs, they observed a strong affinity of alginate with activated carbon too. This leads to substantially reduced impedance, which is fundamental to achieve the high power density expected from EDLCs.299 As explained later by Tran et al.,300 the advantage of alginate is the high content of carboxylic moieties (from the pyranose ring of α-L-guluronic acid) that can interact with the basic groups on the AC surface and establish stable H-bonds (Fig. 7c). These guarantee superior cycling stability in a lithium containing electrolyte (1 M LiPF6 in EC:DMC) for Li-ion capacitors, despite the high residual water content due to the aqueous electrode processing (ca. 597 ppm compared to 43 and 69 ppm of PTFE and PVdF, respectively). Also, alginate-based electrodes showed a larger accessible porosity (mostly in the mesoporous region), resulting in faster energy delivery and uptake. The same group (Tran et al.300) also studied PAA and Na-PAA as water-soluble binders. Although these enable more compact electrodes and better adhesion to the current collector, higher pore blockage was observed in the case of Na-PAA (attributed to the presence of 0.5% SBR, necessary for the electrode fabrication) and increasing ESR upon cycling for PAA (probably related to the current collector corrosion due to the acidic slurry, i.e., pH = 3.3). They concluded that, overall, alginate is the most promising green binder among those investigated. Additionally, according to stress relaxation tests, alginate-based slurries possess very promising rheological properties. In fact, they can maintain a rather stable viscosity over time, which renders alginate the best candidate to obtain stable and, therefore, up-scalable slurries.300
Recently, Varzi et al.301 reported the benefits of a further polysaccharide binder, namely starch. Being highly abundant and extractable from renewable sources (e.g., non-edible potatoes growing in marginal areas), starch would be a perfect candidate for replacing the fluorinated polyolefin. Starch is mostly composed of a mixture of amylose and amylopectin. Similar to NC, CMC and alginate, amylose is a linear polymer, whereas amylopectin has moderately branched chains instead. Depending on the source, the ratio between the two components can vary, but the branched amylopectin is often the main component (>80%).302 This appears to be the key for overcoming the brittleness and shrinkage upon drying typical of CMC, which become particularly problematic when aiming at high active material loading for high areal capacitance. The use of potato starch enabled the fabrication of homogeneous and crack-free electrode layers with thicknesses as high as 240 μm and AC loadings up to 9.3 mg cm−2, whereas, the analogous slurry based on CMC showed fractures and poor adhesion to the current collector already at 88 μm and 5.1 mg cm−2. This behaviour is attributed to the branched structure of amylopectin capable of better releasing the stress upon drying compared to the linear CMC chains. Potato starch also demonstrated cycling stability and electrochemical stability comparable with those of other previously reported polysaccharides (e.g., NC) and superior to those of conventional PVdF.
The same group also proposed casein as a further green alternative binder for EDLC electrodes.303 Protein-based binders have been known since ancient times for their excellent adhesive power and have been extensively employed in mural paintings and wood crafting. However, their role in the electrochemical energy storage field was never explored. Among the possible choices (e.g., collagen, egg white, etc.), Varzi et al.303 selected casein because it is water soluble, biodegradable, and F-free. Furthermore, it can be easily extracted from food leftovers and, specifically, milk products. Interestingly, as evidenced in a recent study, a huge amount of milk is poured down the drains each year, mostly because it has passed its sell-by date or become sour.304 This does not only represent a waste of edible resources but, when released in water streams, could cause serious harm to the environment providing food for bacteria and algae, which will deplete the oxygen levels in the water, ultimately affecting the aquatic life.305 Therefore, using milk leftovers as a source for binder production would have multiple beneficial environmental impacts. Compared to PVdF, the study by Varzi et al. evidenced that casein can provide (i) one order of magnitude higher adhesion strength to the Al foil and (ii) reduced pore clogging. Its thermal stability, proven in both dynamic and static mode, showed no degradation up to at least 180 °C. Casein appears to be compatible with both PC- and acetonitrile (ACN)-based electrolytes offering capacitance values comparable with those of CMC- and PVdF-based electrodes for over 10000 cycles. Unfortunately, though, casein-based electrodes also showed relatively higher resistivity, which affected the power capability. Reducing the binder content and pressing/calendaring the electrodes was found to be beneficial to mitigate this issue.
Recently, polyvinylpyrrolidone (PVP) was also proposed as an alternative binder.306,307 Although synthetic, PVP is F-free, non-toxic, and water/ethanol soluble. Therefore, it might be easily implemented into casting and/or spray coating electrode manufacturing. By means of micro scratch measurements, Aslan et al.306 demonstrated that the mechanical stability of AC electrodes increases with the polymer molecular weight (MW), resulting in a maximum penetration force of 475 mN for 1300000 g mol−1 PVP. As a result, coherent films with superior mechanical stability were achieved with PVP, yet using much less binder with respect to PVdF (3.5 wt% vs. 10 wt%). Differently from PVdF, PVP appears to strongly glue the AC particles together (and to the current collector) without substantially affecting the total surface area or the pore volume of the carbon. This was ascribed, mostly, to the low affinity of the hydrophilic PVP towards the hydrophobic AC surface, thus hindering the binder easily spreading into the pores. The highly hygroscopic character of PVP (due to the tertiary amide group), however, may result in the issue that water may be easily trapped in the electrode. This has no detrimental effect in PC but, as shown by FTIR, water residues can trigger the decomposition of ACN-based electrolytes to form acetamide, H2 and CO2.306,308 Careful drying of PVP-based electrodes should therefore be considered in that case. Besides that, no degradation of PVP itself was observed. The electrochemical performance evaluated in symmetric EDLCs evidenced higher capacitance with respect to PVdF, as expectable from the previously discussed pore clogging of the latter. Comparable performance to PTFE-based electrodes was found in both PC- and ACN-based electrolytes (1 M Et4NBF4). However, most interestingly, a slightly improved rate capability could be achieved with PVP despite the much larger electrical resistance (a factor of 5–7). This result, which may appear contradictory, was attributed to the better electrode/current collector interface obtained by casting compared to rolling.
In order to extend the applicability of PVP to aqueous EDLCs, the same authors proposed a strategy involving its mixture with polyvinyl butyral (PVB),307 a hydrophobic resin extensively used for lamination of safety glasses and solar modules. PVB can be homogeneously blended with PVP in ethanol without phase separation in a wide range of compositions, accordingly tuning the hydrophobic/hydrophilic character of the electrode.307 Finding the optimal PVP/PVB ratio is fundamental to balance mechanical stability, electrode porosity, and wettability (Fig. 7d). High PVP contents enable better adhesion to the current collector, increase the wettability (owing to the hydrophilicity of PVP), and minimize pore blockage (due to the easy spreading of the hydrophobic PVB on the AC surface), while large PVB loads guarantee enhanced mechanical stability in water. The best balance was found to be a PVP/PVB ratio of 1:4 (specifically 1.5 wt% PVP and 6 wt% PVB). Interestingly, the authors determined that below 2 wt% PVP, less than 5% of its total content was leached out from the electrode into the electrolyte, as measured by high pressure liquid chromatography (HPLC). Even after prolonged storage of 10 days, a maximum of 8% PVP was lost. This was not found to negatively affect the electrochemical performance of symmetric EDLCs, employing a 1 M NaCl aqueous electrolyte. On the contrary, the capacitance and rate capability substantially improved after 78 days of operation, probably due to the improved wetting of PVB over time. A stable electrochemical response was also observed in acidic (H2SO4) and basic (KOH) electrolytes. However, the PVP/PVB mixture appears to be incompatible with a neutral Na2SO4 solution. This interesting aspect is certainly worth further investigation.
5. Summary and outlook
Although rapidly growing, the aqueous processing of battery and EDLC electrodes is still a much-unexplored field compared to, e.g., the development of new active materials. Recently though, the importance of greener battery production is being recognized and, in turn, the central role of the binder is becoming more and more relevant.With graphite anodes based on CMC/SBR mixtures already representing the state of the art, the field of LIBs is definitively the most advanced in terms of aqueous processing. Nonetheless, with the advent of high-energy density materials like silicon new challenges have to be faced – especially related to the tremendous volume changes occurring upon de-/lithiation. Focusing increasingly on the binder chemistry and its interaction with the active material rather than solely considering rather basic characteristics like stiffness or flexibility, considerable steps forward have been made in the cyclability of such alloying-type materials, particularly for silicon-based anodes. As a matter of fact, the interplay of covalent and non-covalent interactions, including inter alia the self-healing properties of hydrogen bonds, between the binder and the particle surface appears to be the key factor to achieve long-term stable cycling, while, still, further understanding of the underlying mechanisms is required to fully exploit the potential benefits of highly functional binding agents.
Similarly, the implementation of water-soluble binders for lithium-ion cathodes, especially for water-sensitive, high-energy transition metal oxides, appears to be far from being completely exploited. In fact, the first studies on this subject have been published just a few years ago. But even though being a rather unexplored field, the work reported so far has already shown great promise, as nicely highlighted by the suppressed voltage decay for LR-NCM, the scavenging effect for dissolved manganese ions, or the realization of 15–18 A h full-cells, providing a capacity retention of 99% after 500 cycles.
This great promise holds true also for sodium-ion batteries, even though this field has been opened up even more recently. Just like for lithium-ion anodes, remarkable improvements have been reported for active materials like tin, antimony, or phosphorus, which undergo severe volume variations. Specifically, crosslinked CMC and PAA have shown substantial advances in this regard and also in the case of sodium-ion batteries it appears that the impact of the binder chemistry on the electrode/electrolyte interface plays a decisive role in addition to the overall mechanical integrity of the electrode. In this context, we may anticipate that also an understanding of the interface with the aluminium current collector at the negative electrode will be crucial for further moving this technology forward.
Eventually, despite being highly underrated, the role of novel binders is slowly emerging also in the EDLC field. Interestingly though, some of the features considered beneficial for LIBs and SIBs are found to be detrimental for EDLCs. In fact, the high particle coverage observed for some aqueous binders, which provides protection towards exfoliation (graphite) and active material pulverization (silicon), can effectively block the porosity of EDLC electrodes, resulting in higher resistance and reduced capacitance. However, similarities can also be found. As a matter of fact, interactions between the basic surface group of the activated carbon and the acidic alginate chains lead to longer cycle life and a minor resistance increase upon cycling.
In general, the large majority of studies covered in this review are only lab-scale studies, usually limited to a few grams of material and/or low loading and simple electrode preparation. In order to fully assess the applicability of novel binders, these remain to be validated on a larger scale. Indeed, whenever a new binder is introduced in the electrode formulation, the rheological properties of the slurry must be fully evaluated, as well as the impact of the electrode tape post-processing (e.g., drying, calendaring) on the final electrode architecture and its mechanical properties. This evaluation ideally also includes the volumetric changes occurring on a micro- and macro-scale upon cycling, e.g., by employing techniques like in situ dilatometry,309–311 X-ray tomography,312 or in situ hydrodynamic spectroscopy,313 as such volume changes have a substantial impact on the eventual cycling performance and lifetime of the resulting electrodes.
List of abbreviations
| AC | Activated carbon |
| ACN | Acetonitrile |
| CMC | Carboxymethyl cellulose |
| EC | Ethylene carbonate |
| EDLC | Electrochemical double-layer capacitor |
| EV | Electric vehicle |
| DEC | Diethyl carbonate |
| DMAC | N,N-Dimethylacetamide |
| DMC | Dimethyl carbonate |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DS | Degree of substitution |
| DSC | Differential scanning calorimetry |
| EMIMAc | 1-Ethyl-3-methylimidazolium acetate |
| FEC | 1-Fluoroethylene carbonate |
| FSI | Bis(fluorosulfonyl)imide |
| GG | Guar gum |
| HEV | Hybrid electric vehicle |
| HPMC | Hydroxypropylmethyl cellulose |
| ICL | 1st cycle irreversible capacity loss |
| LCA | Life cycle assessment |
| LFP | Lithium iron phosphate (LiFePO4) |
| LIB | Lithium-ion battery |
| LMO | Lithium manganese oxide (LiMn2O4) |
| LR-NCM | LiMn2O3·NCM |
| LTO | Lithium titanate (Li4Ti5O12) |
| NC | Natural cellulose |
| NCA | Li[Ni1−x−zCoxAlz]O2 (with 1 − x − z ≥ 0.85) |
| NCM | Li[Ni1−x−yCoxMny]O2 |
| NMP | N-Methyl-2-pyrrolidone |
| PA | Phosphoric acid |
| PAA | Poly(acrylic acid) |
| PAN | Polyacrylonitrile |
| PANi | Polyaniline |
| PC | Propylene carbonate |
| PE | Polyethylene |
| PEDOT:PSS | Poly(3,4-ethylenedioxy-thiophene):poly(styrene sulfonate) |
| PEO | Poly(ethylene oxide) |
| Pipr | Polyisoprene |
| PMA | Poly(methyl acrylate) |
| PS | Polystyrene |
| PTFE | Poly(tetrafluoroethylene) |
| PVA | Poly(vinyl alcohol) |
| PVAc | Poly(vinyl acetate) |
| PVB | Polyvinyl butyral |
| PVdF | Poly(vinylidene difluoride) |
| PVP | Polyvinylpyrrolidone |
| PYR14 | N-Butyl-N-methyl-pyrrolidinium |
| SBR | Styrene butadiene rubber |
| SEI | Solid electrolyte interphase |
| SIB | Sodium-ion battery |
| TEM | Transmission electron microscopy |
| TFSI | Bis(trifluoromethanesulfonyl)imide |
Author contributions
D. Br. wrote Sections 1.0 and 2.3 and conceived Fig. 4–6. D. Bu. wrote Section 3. A. M. wrote Sections 1.1, 2.1, and 2.2 and conceived Fig. 3. A. V. wrote Sections 1.2 and 4 and conceived Fig. 1, 2 and 7. S. P. supervised the writing and revised the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge financial support from the German Federal Ministry of Education and Research within the Li-EcoSafe (03X4636D) and Ultimate (03ET6131E) project, from the EU under the grant agreement no. 653373 (SPICY – Silicon and polyanionic chemistries and architectures of Li-ion cell for high energy battery), as well as from the Helmholtz Association for the basic funding.References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- D. Andre, S.-J. Kim, P. Lamp, S. F. Lux, F. Maglia, O. Paschos and B. Stiaszny, J. Mater. Chem. A, 2015, 3, 6709–6732 RSC.
- D. Bresser, K. Hosoi, D. Howell, H. Li, H. Zeisel, K. Amine and S. Passerini, J. Power Sources, 2018, 382, 176–178 CrossRef CAS.
- K. P. Lee, N. C. Chromey, R. Culik, J. R. Barnes and P. W. Schneider, Fundam. Appl. Toxicol., 1987, 9, 222–235 CrossRef CAS PubMed.
- D. E. Malek, L. A. Malley, T. W. Slone, G. S. Elliott, G. L. Kennedy, W. Mellert, K. Deckardt, C. Gembardt, B. Hildebrand, S. R. Murphy, D. B. Bower and G. A. Wright, Drug Chem. Toxicol., 1997, 20, 63–77 CrossRef CAS PubMed.
- W. Zhu, D. R. Schmehl, C. A. Mullin and J. L. Frazier, PLoS One, 2014, 9, e77547 CrossRef PubMed.
- F. M. Courtel, S. Niketic, D. Duguay, Y. Abu-Lebdeh and I. J. Davidson, J. Power Sources, 2011, 196, 2128–2134 CrossRef CAS.
- T. S. Poet, C. R. Kirman, M. Bader, C. van Thriel, M. L. Gargas and P. M. Hinderliter, Toxicol. Sci., 2010, 113, 468–482 CrossRef CAS PubMed.
- V. Haufroid, V. K. Jaeger, S. Jeggli, R. Eisenegger, A. Bernard, D. Friedli, D. Lison and P. Hotz, Int. Arch. Occup. Environ. Health, 2014, 87, 663–674 CrossRef CAS PubMed.
- https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/fact-sheet-n-methylpyrrolidone-nmp#advic, accessed in June 2018, EPA, 2018.
- European Parliament and the Council concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), 2006.
- G. Berckmans, M. Messagie, J. Smekens, N. Omar, L. Vanhaverbeke and J. Van Mierlo, Energies, 2017, 10, 1314 CrossRef.
- D. L. Wood Iii, J. Li and C. Daniel, J. Power Sources, 2015, 275, 234–242 CrossRef.
- M. Valvo, A. Liivat, H. Eriksson, C.-W. Tai and K. Edström, ChemSusChem, 2017, 10, 2431–2448 CrossRef CAS PubMed.
- J. Li, Z. Du, R. E. Ruther, S. J. An, L. A. David, K. Hays, M. Wood, N. D. Phillip, Y. Sheng, C. Mao, S. Kalnaus, C. Daniel and D. L. Wood, JOM, 2017, 69, 1484–1496 CrossRef CAS.
- D. L. Wood, J. D. Quass, J. Li, S. Ahmed, D. Ventola and C. Daniel, Drying Technol., 2017, 1–11 Search PubMed.
- J. Li, C. Daniel, S. J. An and D. Wood, MRS Adv., 2016, 1, 1029–1035 CrossRef CAS.
- N. Susarla, S. Ahmed and D. W. Dees, J. Power Sources, 2018, 378, 660–670 CrossRef CAS.
- S. Feng, Z. Zhong, Y. Wang, W. Xing and E. Drioli, J. Membr. Sci., 2018, 549, 332–349 CrossRef CAS.
- K. Notake, T. Gunji, H. Kokubun, S. Kosemura, Y. Mochizuki, T. Tanabe, S. Kaneko, S. Ugawa, H. Lee and F. Matsumoto, J. Appl. Electrochem., 2016, 46, 267–278 CrossRef CAS.
- T. Tanabe, T. Gunji, Y. Honma, K. Miyamoto, T. Tsuda, Y. Mochizuki, S. Kaneko, S. Ugawa, H. Lee, T. Ohsaka and F. Matsumoto, Electrochim. Acta, 2017, 224, 429–438 CrossRef CAS.
- O. E. Bankole, C. Gonc and L. Lei, J. Environ. Ecol., 2013, 4, 14–28 CrossRef.
- G. Jiang and S. J. Pickering, Waste Manage., 2016, 48, 465–470 CrossRef CAS PubMed.
- J. Drofenik, M. Gaberscek, R. Dominko, F. W. Poulsen, M. Mogensen, S. Pejovnik and J. Jamnik, Electrochim. Acta, 2003, 48, 883–889 CrossRef CAS.
- J.-H. Lee, S. Lee, U. Paik and Y.-M. Choi, J. Power Sources, 2005, 147, 249–255 CrossRef CAS.
- C. Clasen and W. M. Kulicke, Prog. Polym. Sci., 2001, 26, 1839–1919 CrossRef CAS.
- T. Heinze and A. Koschella, Macromol. Symp., 2005, 223, 13–40 CrossRef CAS.
- J.-H. Lee, U. Paik, V. A. Hackley and Y.-M. Choi, J. Electrochem. Soc., 2005, 152, A1763 CrossRef CAS.
- H. Buqa, M. Holzapfel, F. Krumeich, C. Veit and P. Novák, J. Power Sources, 2006, 161, 617–622 CrossRef CAS.
- M. Gaberscek, M. Bele, J. Drofenik, R. Dominko and S. Pejovnik, Electrochem. Solid-State Lett., 2000, 3, 171–173 CrossRef CAS.
- M. Gaberšček, M. Bele, J. Drofenik, R. Dominko and S. Pejovnik, J. Power Sources, 2001, 97, 67–69 CrossRef.
- M. Bele, M. Gaberscek, R. Dominko, J. Drofenik, K. Zupan, P. Komac, K. Kocevar, I. Musevic and S. Pejovnik, Carbon, 2002, 40, 1117–1122 CrossRef CAS.
- L. El Ouatani, R. Dedryvère, J. B. Ledeuil, C. Siret, P. Biensan, J. Desbrières and D. Gonbeau, J. Power Sources, 2009, 189, 72–80 CrossRef CAS.
- T. Zhang, I. de Meatza, X. Qi and E. Paillard, J. Power Sources, 2017, 356, 97–102 CrossRef CAS.
- J.-H. Lee, U. Paik, V. A. Hackley and Y.-M. Choi, J. Power Sources, 2006, 161, 612–616 CrossRef CAS.
- S. Komaba, T. Ozeki and K. Okushi, J. Power Sources, 2009, 189, 197–203 CrossRef CAS.
- S. Komaba, K. Okushi, T. Ozeki, H. Yui, Y. Katayama, T. Miura, T. Saito and H. Groult, Electrochem. Solid-State Lett., 2009, 12, A107–A110 CrossRef CAS.
- S. Komaba, N. Yabuuchi, T. Ozeki, K. Okushi, H. Yui, K. Konno, Y. Katayama and T. Miura, J. Power Sources, 2010, 195, 6069–6074 CrossRef CAS.
- F. Jeschull, M. J. Lacey and D. Brandell, Electrochim. Acta, 2015, 175, 141–150 CrossRef CAS.
- J. Chong, S. Xun, H. Zheng, X. Song, G. Liu, P. Ridgway, J. Q. Wang and V. S. Battaglia, J. Power Sources, 2011, 196, 7707–7714 CrossRef CAS.
- F. Jeschull, D. Brandell, M. Wohlfahrt-Mehrens and M. Memm, Energy Technol., 2017, 5, 2108–2118 CrossRef CAS.
- Y.-S. Park, E.-S. Oh and S.-M. Lee, J. Power Sources, 2014, 248, 1191–1196 CrossRef CAS.
- D. Shin, H. Park and U. Paik, Electrochem. Commun., 2017, 77, 103–106 CrossRef CAS.
- N. Cuesta, A. Ramos, I. Cameán, C. Antuña and A. B. García, Electrochim. Acta, 2015, 155, 140–147 CrossRef CAS.
- D. Versaci, R. Nasi, U. Zubair, J. Amici, M. Sgroi, M. A. Dumitrescu, C. Francia, S. Bodoardo and N. Penazzi, J. Solid State Electrochem., 2017, 21, 3429–3435 CrossRef CAS.
- L. Zhang, L. Chai, Q. Qu, L. Zhang, M. Shen and H. Zheng, Electrochim. Acta, 2013, 105, 378–383 CrossRef.
- D. V. Carvalho, N. Loeffler, M. Hekmatfar, A. Moretti, G. T. Kim and S. Passerini, Electrochim. Acta, 2018, 265, 89–97 CrossRef CAS.
- A. García, M. Culebras, M. N. Collins and J. J. Leahy, J. Appl. Polym. Sci., 2018, 135, 46217 CrossRef.
- Z. Chen, I. Belharouak, Y. K. Sun and K. Amine, Adv. Funct. Mater., 2013, 23, 959–969 CrossRef.
- M. He, E. Castel, A. Laumann, G. Nuspl, P. Novák and E. J. Berg, J. Electrochem. Soc., 2015, 162, A870–A876 CrossRef CAS.
- N. J. J. de Klerk, A. Vasileiadis, R. B. Smith, M. Z. Bazant and M. Wagemaker, Phys. Rev. Mater., 2017, 1, 25404 CrossRef.
- G. T. Kim, S. S. Jeong, M. Joost, E. Rocca, M. Winter, S. Passerini and A. Balducci, J. Power Sources, 2011, 196, 2187–2194 CrossRef CAS.
- S.-L. Chou, J.-Z. Wang, H.-K. Liu and S.-X. Dou, J. Phys. Chem. C, 2011, 115, 16220–16227 CrossRef CAS.
- B.-R. Lee and E.-S. Oh, J. Phys. Chem. C, 2013, 117, 4404–4409 CrossRef CAS.
- B.-R. Lee, S. Kim and E.-S. Oh, J. Electrochem. Soc., 2014, 161, A2128–A2132 CrossRef CAS.
- D. Carvalho, N. Loeffler, G.-T. Kim, M. Marinaro, M. Wohlfahrt-Mehrens and S. Passerini, Polymers, 2016, 8, 276 CrossRef.
- S. J. Rezvani, M. Pasqualini, A. Witkowska, R. Gunnella, A. Birrozzi, M. Minicucci, H. Rajantie, M. Copley, F. Nobili and A. Di Cicco, Appl. Surf. Sci., 2018, 435, 1029–1036 CrossRef CAS.
- M. Mancini, F. Nobili, R. Tossici, M. Wohlfahrt-Mehrens and R. Marassi, J. Power Sources, 2011, 196, 9665–9671 CrossRef CAS.
- A. Moretti, G.-T. Kim, D. Bresser, K. Renger, E. Paillard, R. Marassi, M. Winter and S. Passerini, J. Power Sources, 2013, 221, 419–426 CrossRef CAS.
- M. Mancini, F. Nobili, R. Tossici and R. Marassi, Electrochim. Acta, 2012, 85, 566–571 CrossRef CAS.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- K. Rhodes, N. Dudney, E. Lara-Curzio and C. Daniel, J. Electrochem. Soc., 2010, 157, A1354–A1360 CrossRef CAS.
- K. Feng, M. Li, W. Liu, A. G. Kashkooli, X. Xiao, M. Cai and Z. Chen, Small, 2018, 14, 1702737 CrossRef PubMed.
- J. Li, R. B. Lewis and J. R. Dahn, Electrochem. Solid-State Lett., 2007, 10, A17 CrossRef CAS.
- B. Koo, H. Kim, Y. Cho, K. T. Lee, N.-S. Choi and J. Cho, Angew. Chem., Int. Ed., 2012, 51, 8762–8767 CrossRef CAS PubMed.
- W.-R. Liu, M.-H. Yang, H.-C. Wu, S. M. Chiao and N.-L. Wu, Electrochem. Solid-State Lett., 2005, 8, A100 CrossRef CAS.
- J. S. Bridel, T. Azaïs, M. Morcrette, J. M. Tarascon and D. Larcher, Chem. Mater., 2010, 22, 1229–1241 CrossRef CAS.
- A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C. F. Huebner, T. F. Fuller, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2010, 2, 3004–3010 CrossRef CAS PubMed.
- M. Morita, T. Ohmi, E. Hasegawa, M. Kawakami and M. Ohwada, J. Appl. Phys., 1990, 68, 1272–1281 CrossRef CAS.
- N. S. Hochgatterer, M. R. Schweiger, S. Koller, P. R. Raimann, T. Wöhrle, C. Wurm and M. Winter, Electrochem. Solid-State Lett., 2008, 11, A76–A80 CrossRef CAS.
- U. Maver, A. Znidarsic and M. Gaberscek, J. Mater. Chem., 2011, 21, 4071–4075 RSC.
- N. Delpuech, D. Mazouzi, N. Dupré, P. Moreau, M. Cerbelaud, J. S. Bridel, J. C. Badot, E. De Vito, D. Guyomard, B. Lestriez and B. Humbert, J. Phys. Chem. C, 2014, 118, 17318–17331 CrossRef CAS.
- D. Mazouzi, B. Lestriez, L. Roué and D. Guyomard, Electrochem. Solid-State Lett., 2009, 12, A215–A218 CrossRef CAS.
- J.-S. Bridel, T. Azaïs, M. Morcrette, J.-M. Tarascon and D. Larcher, J. Electrochem. Soc., 2011, 158, A750–A759 CrossRef CAS.
- M. Cerbelaud, B. Lestriez, D. Guyomard, A. Videcoq and R. Ferrando, Langmuir, 2012, 28, 10713–10724 CrossRef CAS PubMed.
- B. Lestriez, S. Bahri, I. Sandu, L. Roué and D. Guyomard, Electrochem. Commun., 2007, 9, 2801–2806 CrossRef CAS.
- S. Komaba, K. Shimomura, N. Yabuuchi, T. Ozeki, H. Yui and K. Konno, J. Phys. Chem. C, 2011, 115, 13487–13495 CrossRef CAS.
- W. Porcher, S. Chazelle, A. Boulineau, N. Mariage, J. P. Alper, T. Van Rompaey, J.-S. Bridel and C. Haon, J. Electrochem. Soc., 2017, 164, A3633–A3640 CrossRef CAS.
- C. Erk, T. Brezesinski, H. Sommer, R. Schneider and J. Janek, ACS Appl. Mater. Interfaces, 2013, 5, 7299–7307 CrossRef CAS PubMed.
- Z. Karkar, D. Guyomard, L. Roué and B. Lestriez, Electrochim. Acta, 2017, 258, 453–466 CrossRef CAS.
- C. R. Hernandez, A. Etiemble, T. Douillard, D. Mazouzi, Z. Karkar, E. Maire, D. Guyomard, B. Lestriez and L. Roué, Adv. Energy Mater., 2018, 8, 1701787 CrossRef.
- J. Song, M. Zhou, R. Yi, T. Xu, M. L. Gordin, D. Tang, Z. Yu, M. Regula and D. Wang, Adv. Funct. Mater., 2014, 24, 5904–5910 CrossRef CAS.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- L. Zhang, L. Zhang, J. Zhang, W. Hao and H. Zheng, J. Mater. Chem. A, 2015, 3, 15432–15443 RSC.
- Z.-Y. Wu, L. Deng, J.-T. Li, Q.-S. Huang, Y.-Q. Lu, J. Liu, T. Zhang, L. Huang and S.-G. Sun, Electrochim. Acta, 2017, 245, 371–378 CrossRef CAS.
- L. Yue, L. Zhang and H. Zhong, J. Power Sources, 2014, 247, 327–331 CrossRef CAS.
- C. Chen, S. H. Lee, M. Cho, J. Kim and Y. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 2658–2665 CrossRef CAS PubMed.
- Z.-H. Wu, J.-Y. Yang, B. Yu, B.-M. Shi, C.-R. Zhao and Z.-L. Yu, Rare Met. DOI:10.1007/s12598-016-0753-0.
- Y. K. Jeong, T. Kwon, I. Lee, T.-S. Kim, A. Coskun and J. W. Choi, Nano Lett., 2014, 14, 864–870 CrossRef PubMed.
- Y. K. Jeong, T. Kwon, I. Lee, T.-S. Kim, A. Coskun and J. W. Choi, Energy Environ. Sci., 2015, 9, 1224–1230 RSC.
- J. Liu, Q. Zhang, T. Zhang, J.-T. Li, L. Huang and S.-G. Sun, Adv. Funct. Mater., 2015, 25, 3599–3605 CrossRef CAS.
- S. Klamor, M. Schroder, G. Brunklaus, P. Niehoff, F. Berkemeier, F. M. Schappacher and M. Winter, Phys. Chem. Chem. Phys., 2015, 17, 5632–5641 RSC.
- Y. Bie, J. Yang, Y. Nuli and J. Wang, J. Mater. Chem. A, 2017, 5, 1919–1924 RSC.
- D.-E. Yoon, C. Hwang, N.-R. Kang, U. Lee, D. Ahn, J.-Y. Kim and H.-K. Song, ACS Appl. Mater. Interfaces, 2016, 8, 4042–4047 CrossRef CAS PubMed.
- P. E. Marszalek, A. F. Oberhauser, Y.-P. Pang and J. M. Fernandez, Nature, 1998, 396, 661 CrossRef CAS PubMed.
- M.-H. Ryou, J. Kim, I. Lee, S. Kim, Y. K. Jeong, S. Hong, J. H. Ryu, T.-S. Kim, J.-K. Park, H. Lee and J. W. Choi, Adv. Mater., 2013, 25, 1571–1576 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 CrossRef PubMed.
- L. Wang, T. Liu, X. Peng, W. Zeng, Z. Jin, W. Tian, B. Gao, Y. Zhou, P. K. Chu and K. Huo, Adv. Funct. Mater., 2018, 28, 1704858 CrossRef.
- D. Shao, H. Zhong and L. Zhang, ChemElectroChem, 2014, 1, 1679–1687 CrossRef CAS.
- T. M. Higgins, S.-H. Park, P. J. King, C. Zhang, N. McEvoy, N. C. Berner, D. Daly, A. Shmeliov, U. Khan, G. Duesberg, V. Nicolosi and J. N. Coleman, ACS Nano, 2016, 10, 3702–3713 CrossRef CAS PubMed.
- P.-F. Cao, M. Naguib, Z. Du, E. Stacy, B. Li, T. Hong, K. Xing, D. N. Voylov, J. Li, D. L. Wood, A. P. Sokolov, J. Nanda and T. Saito, ACS Appl. Mater. Interfaces, 2018, 10, 3470–3478 CrossRef CAS PubMed.
- T. Kwon, Y. K. Jeong, I. Lee, T.-S. Kim, J. W. Choi and A. Coskun, Adv. Mater., 2014, 26, 7979–7985 CrossRef CAS PubMed.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- D. Bresser, S. Passerini and B. Scrosati, Energy Environ. Sci., 2016, 9, 3348–3367 RSC.
- J. Li, D.-B. Le, P. P. Ferguson and J. R. Dahn, Electrochim. Acta, 2010, 55, 2991–2995 CrossRef CAS.
- S. Mitra, P. S. Veluri, A. Chakraborthy and R. K. Petla, ChemElectroChem, 2014, 1, 1068–1074 CrossRef CAS.
- F. Maroni, S. Gabrielli, A. Palmieri, E. Marcantoni, F. Croce and F. Nobili, J. Power Sources, 2016, 332, 79–87 CrossRef CAS.
- K. Zhou, M. Kang, X. He, Z. Hong, Z. Huang and M. Wei, J. Mater. Chem. A, 2017, 5, 18138–18147 RSC.
- C. Li, X. Wang, S. Li, Q. Li, J. Xu, X. Liu, C. Liu, Y. Xu, J. Liu, H. Li, P. Guo and X. S. Zhao, Appl. Surf. Sci., 2017, 416, 308–317 CrossRef CAS.
- D. Bresser, E. Paillard, R. Kloepsch, S. Krueger, M. Fiedler, R. Schmitz, D. Baither, M. Winter and S. Passerini, Adv. Energy Mater., 2013, 3, 513–523 CrossRef CAS.
- A. Varzi, D. Bresser, J. von Zamory, F. Müller and S. Passerini, Adv. Energy Mater., 2014, 4, 1400054 CrossRef PubMed.
- M.-H. Lee, T.-H. Kim, C. Hwang, J. Kim and H.-K. Song, Electrochim. Acta, 2017, 225, 78–85 CrossRef CAS.
- L. Yin, S. Chai, J. Ma, J. Huang, X. Kong, P. Bai and Y. Liu, J. Alloys Compd., 2017, 698, 828–834 CrossRef CAS.
- J. Chen, W. Liu, S. Liu, H. Wang, Y. Zhang and S. Chen, Chem. Eng. J., 2017, 308, 1200–1208 CrossRef CAS.
- D. L. Wood, J. D. Quass, J. Li, S. Ahmed, D. Ventola and C. Daniel, Drying Technol., 2017, 1–11 Search PubMed.
- W.-J. Zhang, J. Power Sources, 2011, 196, 2962–2970 CrossRef CAS.
- A. Guerfi, M. Kaneko, M. Petitclerc, M. Mori and K. Zaghib, J. Power Sources, 2007, 163, 1047–1052 CrossRef CAS.
- W. Porcher, P. Moreau, B. Lestriez, S. Jouanneau, F. L. Cras and D. Guyomard, Ionics, 2008, 14, 583–587 CrossRef CAS.
- W. Porcher, P. Moreau, B. Lestriez, S. Jouanneau and D. Guyomard, Electrochem. Solid-State Lett., 2008, 11, A4–A8 CrossRef CAS.
- S. F. Lux, F. Schappacher, A. Balducci, S. Passerini and M. Winter, J. Electrochem. Soc., 2010, 157, A320–A325 CrossRef CAS.
- W. Porcher, B. Lestriez, S. Jouanneau and D. Guyomard, J. Power Sources, 2010, 195, 2835–2843 CrossRef CAS.
- W. Porcher, B. Lestriez, S. Jouanneau and D. Guyomard, J. Electrochem. Soc., 2009, 156, A133–A144 CrossRef CAS.
- C.-C. Li and Y.-S. Lin, J. Power Sources, 2012, 220, 413–421 CrossRef CAS.
- J. Li, B. L. Armstrong, J. Kiggans, C. Daniel and D. L. Wood, Langmuir, 2012, 28, 3783–3790 CrossRef CAS PubMed.
- J. Li, B. L. Armstrong, J. Kiggans, C. Daniel and D. L. Wood, J. Electrochem. Soc., 2013, 160, A201–A206 CrossRef CAS.
- J.-C. Tsai, F.-Y. Tsai, C.-A. Tung, H.-W. Hsieh and C.-C. Li, J. Power Sources, 2013, 241, 400–403 CrossRef CAS.
- F.-Y. Tsai, J.-H. Jhang, H.-W. Hsieh and C.-C. Li, J. Power Sources, 2016, 310, 47–53 CrossRef CAS.
- J.-H. Lee, J.-S. Kim, Y. C. Kim, D. S. Zang and U. Paik, Ultramicroscopy, 2008, 108, 1256–1259 CrossRef CAS PubMed.
- Z. P. Cai, Y. Liang, W. S. Li, L. D. Xing and Y. H. Liao, J. Power Sources, 2009, 189, 547–551 CrossRef CAS.
- Z. Zhang, T. Zeng, H. Lu, M. Jia, J. Li and Y. Lai, ECS Electrochem. Lett., 2012, 1, A74–A76 CrossRef CAS.
- J. Chong, S. Xun, H. Zheng, X. Song, G. Liu, P. Ridgway, J. Q. Wang and V. S. Battaglia, J. Power Sources, 2011, 196, 7707–7714 CrossRef CAS.
- J. He, H. Zhong, J. Wang and L. Zhang, J. Alloys Compd., 2017, 714, 409–418 CrossRef CAS.
- M. Sun, H. Zhong, S. Jiao, H. Shao and L. Zhang, Electrochim. Acta, 2014, 127, 239–244 CrossRef CAS.
- H. Zhong, A. He, J. Lu, M. Sun, J. He and L. Zhang, J. Power Sources, 2016, 336, 107–114 CrossRef CAS.
- J. He, J. Wang, H. Zhong, J. Ding and L. Zhang, Electrochim. Acta, 2015, 182, 900–907 CrossRef CAS.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- J. Li, C. Rulison, J. Kiggans, C. Daniel and D. L. Wood, J. Electrochem. Soc., 2012, 159, A1152–A1157 CrossRef CAS.
- G. T. Kim, S. S. Jeong, M. Joost, E. Rocca, M. Winter, S. Passerini and A. Balducci, J. Power Sources, 2011, 196, 2187–2194 CrossRef CAS.
- S. N. Eliseeva, O. V. Levin, E. G. Tolstopjatova, E. V. Alekseeva, R. V. Apraksin and V. V. Kondratiev, Mater. Lett., 2015, 161, 117–119 CrossRef CAS.
- P. R. Das, L. Komsiyska, O. Osters and G. Wittstock, J. Electrochem. Soc., 2015, 162, A674–A678 CrossRef CAS.
- S. S. Jeong, N. Böckenfeld, A. Balducci, M. Winter and S. Passerini, J. Power Sources, 2012, 199, 331–335 CrossRef CAS.
- E. A. Olivetti, G. Ceder, G. G. Gaustad and X. Fu, Joule, 2017, 1, 229–243 CrossRef.
- M. M. Thackeray, J. Am. Ceram. Soc., 1999, 82, 3347–3354 CrossRef CAS.
- M. Thackeray, Nat. Mater., 2002, 1, 81 CrossRef CAS PubMed.
- D. Guyomard and J. M. Tarascon, J. Electrochem. Soc., 1992, 139, 937–948 CrossRef CAS.
- Y. Xia and M. Yoshio, J. Power Sources, 1997, 66, 129–133 CrossRef CAS.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461–472 CrossRef CAS.
- T. Ohzuku, M. Kitagawa and T. Hirai, J. Electrochem. Soc., 1990, 137, 769–775 CrossRef CAS.
- M. M. Thackeray, Prog. Solid State Chem., 1997, 25, 1–71 CrossRef CAS.
- M. M. Thackeray, Y. Shao-Horn, A. J. Kahaian, K. D. Kepler, E. Skinner, J. T. Vaughey and S. A. Hackney, Electrochem. Solid-State Lett., 1998, 1, 7–9 CrossRef CAS.
- P. Ramadass, B. Haran, R. White and B. N. Popov, J. Power Sources, 2002, 111, 210–220 CrossRef CAS.
- R. J. Gummow, A. de Kock and M. M. Thackeray, Solid State Ionics, 1994, 69, 59–67 CrossRef CAS.
- M.-H. Ryou, S. Hong, M. Winter, H. Lee and J. W. Choi, J. Mater. Chem. A, 2013, 1, 15224–15229 RSC.
- T. Gotoh, K. Matsushima and K.-I. Kikuchi, Chemosphere, 2004, 55, 57–64 CrossRef CAS PubMed.
- T. S. Pathak, J.-H. Yun, S.-J. Lee, D.-J. Baek and K.-J. Paeng, Carbohydr. Polym., 2009, 78, 717–724 CrossRef CAS.
- Z. Zhang, T. Zeng, Y. Lai, M. Jia and J. Li, J. Power Sources, 2014, 247, 1–8 CrossRef CAS.
- K. Amine, H. Tukamoto, H. Yasuda and Y. Fujita, J. Power Sources, 1997, 68, 604–608 CrossRef CAS.
- Q. Zhong, J. Electrochem. Soc., 1997, 144, 205 CrossRef CAS.
- Z. Wang, N. Dupré, A.-C. Gaillot, B. Lestriez, J.-F. Martin, L. Daniel, S. Patoux and D. Guyomard, Electrochim. Acta, 2012, 62, 77–83 CrossRef CAS.
- F. De Giorgio, N. Laszczynski, J. von Zamory, M. Mastragostino, C. Arbizzani and S. Passerini, ChemSusChem, 2017, 10, 379–386 CrossRef CAS PubMed.
- M. Kuenzel, D. Bresser, T. Diemant, D. Viera Carvalho, G.-T. Kim, R. J. Behm and S. Passerini, ChemSusChem, 2018, 11, 562–573 CrossRef CAS PubMed.
- W.-Y. Chou, Y.-C. Jin, J.-G. Duh, C.-Z. Lu and S.-C. Liao, Appl. Surf. Sci., 2015, 355, 1272–1278 CrossRef CAS.
- F. Bigoni, F. De Giorgio, F. Soavi and C. Arbizzani, J. Electrochem. Soc., 2017, 164, A6171–A6177 CrossRef CAS.
- H. Zhong, J. He and L. Zhang, Mater. Res. Bull., 2017, 93, 194–200 CrossRef CAS.
- H. Zhong, J. Lu, A. He, M. Sun, J. He and L. Zhang, J. Mater. Sci. Technol., 2017, 33, 763–767 CrossRef.
- P. P. Prosini, M. Carewska and A. Masci, Solid State Ionics, 2015, 274, 88–93 CrossRef CAS.
- N. P. W. Pieczonka, V. Borgel, B. Ziv, N. Leifer, V. Dargel, D. Aurbach, J.-H. Kim, Z. Liu, X. Huang, S. A. Krachkovskiy, G. R. Goward, I. Halalay, B. R. Powell and A. Manthiram, Adv. Energy Mater., 2016, 6, 1501010 CrossRef.
- B. P. N. Nguyen, N. Mariage, R. Fredon, E. M. Kelder and B. Lestriez, J. Electrochem. Soc., 2015, 162, A1451–A1459 CrossRef CAS.
- A. Manthiram and J. B. Goodenough, Can. J. Phys., 1987, 65, 1309–1317 CrossRef CAS.
- J. Kim, Y. Hong, K. S. Ryu, M. G. Kim and J. Cho, Electrochem. Solid-State Lett., 2006, 9, A19–A23 CrossRef CAS.
- G. V. Zhuang, G. Chen, J. Shim, X. Song, P. N. Ross and T. J. Richardson, J. Power Sources, 2004, 134, 293–297 CrossRef CAS.
- K. Matsumoto, R. Kuzuo, K. Takeya and A. Yamanaka, J. Power Sources, 1999, 81–82, 558–561 CrossRef CAS.
- X. Zhang, W. J. Jiang, X. P. Zhu, A. Mauger, Qilu and C. M. Julien, J. Power Sources, 2011, 196, 5102–5108 CrossRef CAS.
- T. Tanabe, T. Gunji, Y. Honma, K. Miyamoto, T. Tsuda, Y. Mochizuki, S. Kaneko, S. Ugawa, H. Lee, T. Ohsaka and F. Matsumoto, Electrochim. Acta, 2016, 224, 429–438 CrossRef.
- R. Moshtev, P. Zlatilova, S. Vasilev, I. Bakalova and A. Kozawa, J. Power Sources, 1999, 81–82, 434–441 CrossRef CAS.
- G. E. Brown, V. E. Henrich, W. H. Casey, D. L. Clark, C. Eggleston, A. Felmy, D. W. Goodman, M. Grätzel, G. Maciel, M. I. McCarthy, K. H. Nealson, D. A. Sverjensky, M. F. Toney and J. M. Zachara, Chem. Rev., 1999, 99, 77–174 CrossRef CAS PubMed.
- N. L. Sukiman, X. Zhou, N. Birbilis, A. E. Hughes, J. M. C. Mol, S. J. Garcia and G. E. Thompson, Aluminium Alloys – New Trends in Fabrication and Applications, Intech, 2012, pp. 47–97 Search PubMed.
- I. A. Shkrob, J. A. Gilbert, P. J. Phillips, R. Klie, R. T. Haasch, J. Bareño and D. P. Abraham, J. Electrochem. Soc., 2017, 164, A1489–A1498 CrossRef CAS.
- I. Doberdò, N. Löffler, N. Laszczynski, D. Cericola, N. Penazzi, S. Bodoardo, G. T. Kim and S. Passerini, J. Power Sources, 2014, 248, 1000–1006 CrossRef.
- N. Loeffler, J. Von Zamory, N. Laszczynski, I. Doberdo, G. T. Kim and S. Passerini, J. Power Sources, 2014, 248, 915–922 CrossRef CAS.
- A. Moretti, G. T. Kim, D. Bresser, K. Renger, E. Paillard, R. Marassi, M. Winter and S. Passerini, J. Power Sources, 2013, 221, 419–426 CrossRef CAS.
- N. Loeffler, G. T. Kim, F. Mueller, T. Diemant, J. K. Kim, R. J. Behm and S. Passerini, ChemSusChem, 2016, 9, 1112–1117 CrossRef CAS PubMed.
- T. Tanabe, Y. Liu, K. Miyamoto, Y. Irii, F. Maki, T. Gunji, S. Kaneko, S. Ugawa, H. Lee, T. Ohsaka and F. Matsumoto, Electrochim. Acta, 2017, 258, 1348–1355 CrossRef CAS.
- N. Laszczynski, J. von Zamory, J. Kalhoff, N. Loeffler, V. S. K. Chakravadhanula and S. Passerini, ChemElectroChem, 2015, 2, 1768–1773 CrossRef CAS.
- N. Loeffler, T. Kopel, G.-T. Kim and S. Passerini, J. Electrochem. Soc., 2015, 162, A2692–A2698 CrossRef CAS.
- N. Loeffler, G. T. Kim, F. Mueller, T. Diemant, J. K. Kim, R. J. Behm and S. Passerini, ChemSusChem, 2016, 9, 1112–1117 CrossRef CAS PubMed.
- Z. Chen, G.-T. Kim, D. Chao, N. Loeffler, M. Copley, J. Lin, Z. Shen and S. Passerini, J. Power Sources, 2017, 372, 180–187 CrossRef CAS.
- A. Kazzazi, D. Bresser, A. Birrozzi, J. von Zamory, M. Hekmatfar and S. Passerini, ACS Appl. Mater. Interfaces, 2018, 10, 17214–17222 CrossRef CAS PubMed.
- J. Xu, S.-L. Chou, Q. Gu, H.-K. Liu and S.-X. Dou, J. Power Sources, 2013, 225, 172–178 CrossRef CAS.
- J. H. Lee, S. Lee, U. Paik and Y. M. Choi, J. Power Sources, 2005, 147, 249–255 CrossRef CAS.
- W.-R. Liu, M.-H. Yang, H.-C. Wu, S. M. Chiao and N.-L. Wu, Electrochem. Solid-State Lett., 2005, 8, A100–A103 CrossRef CAS.
- M. Ling, J. Qiu, S. Li, H. Zhao, G. Liu and S. Zhang, J. Mater. Chem. A, 2013, 1, 11543 RSC.
- H. Buqa, M. Holzapfel, F. Krumeich, C. Veit and P. Novák, J. Power Sources, 2006, 161, 617–622 CrossRef CAS.
- S. Chou, Y. Pan, J. Wang, H. Liu and S. Dou, Phys. Chem. Chem. Phys., 2014, 16, 20347–20359 RSC.
- S. F. Lux, F. Schappacher, A. Balducci, S. Passerini and M. Winter, J. Electrochem. Soc., 2010, 157, A320 CrossRef CAS.
- Q. Wu, S. Ha, J. Prakash, D. W. Dees and W. Lu, Electrochim. Acta, 2013, 114, 1–6 CrossRef CAS.
- H. Yamamoto and H. Mori, Lithium-Ion Batteries: Science and Technologies, Springer, 2009, pp. 163–179 Search PubMed.
- C.-C. Li and Y.-W. Wang, J. Power Sources, 2013, 227, 204–210 CrossRef CAS.
- N. Yabuuchi, Y. Kinoshita, K. Misaki, T. Matsuyama and S. Komaba, J. Electrochem. Soc., 2015, 162, A538–A544 CrossRef CAS.
- Q. Pang, X. Liang, C. Y. Kwok, J. Kulisch and L. F. Nazar, Adv. Energy Mater., 2016, 1601630 Search PubMed.
- J. Song, Z. Yu, M. L. Gordin, X. Li, H. Peng and D. Wang, ACS Nano, 2015, 9, 11933–11941 CrossRef CAS PubMed.
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC.
- H. Chen, M. Chen, C. Du, Y. Cui, P. Zuo, X. Cheng and G. Yin, Mater. Chem. Phys., 2016, 171, 6–10 CrossRef CAS.
- J. Wang, X. He, E. Paillard, N. Laszczynski, J. Li and S. Passerini, Adv. Energy Mater., 2016, 6, 1600906 CrossRef.
- J. Li, R. Klöpsch, S. Nowak, M. Kunze, M. Winter and S. Passerini, J. Power Sources, 2011, 196, 7687–7691 CrossRef CAS.
- Z. Han, H. Zhan and Y. Zhou, Mater. Lett., 2014, 114, 48–51 CrossRef CAS.
- T. Zhang, J.-T. Li, J. Liu, Y.-P. Deng, Z.-G. Wu, Z.-W. Yin, D. Guo, L. Huang and S.-G. Sun, Chem. Commun., 2016, 52, 4683–4686 RSC.
- L. Baggetto, D. Mohanty, R. A. Meisner, C. A. Bridges, C. Daniel, D. L. Wood III, N. J. Dudney and G. M. Veith, RSC Adv., 2014, 4, 23364–23371 RSC.
- S. Hy, F. Felix, J. Rick, W.-N. Su and B. J. Hwang, J. Am. Chem. Soc., 2014, 136, 999–1007 CrossRef CAS PubMed.
- N. Loeffler, G.-T. Kim, S. Passerini, C. Gutierrez, I. Cendoya, I. De Meatza, F. Alessandrini and G. B. Appetecchi, ChemSusChem, 2017, 10, 3581–3587 CrossRef CAS PubMed.
- D. Bresser, S. Passerini and B. Scrosati, Chem. Commun., 2013, 49, 10545–10562 RSC.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- K. Zhang, G.-H. Lee, M. Park, W. Li and Y.-M. Kang, Adv. Energy Mater., 2016, 6, 1600811 CrossRef.
- Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- M. Wild, L. O’Neill, T. Zhang, R. Purkayastha, G. Minton, M. Marinescu and G. J. Offer, Energy Environ. Sci., 2015, 8, 3477–3494 RSC.
- D. Moy and S. R. Narayanan, J. Electrochem. Soc., 2017, 164, A560–A566 CrossRef CAS.
- J.-Y. Hwang, H. M. Kim, S. Shin and Y.-K. Sun, Adv. Funct. Mater., 2017, 28, 1704294 CrossRef.
- C. Sheng-Heng and M. Arumugam, Adv. Mater., 2014, 26, 7352–7357 CrossRef PubMed.
- X. He, J. Ren, L. Wang, W. Pu, C. Jiang and C. Wan, J. Power Sources, 2009, 190, 154–156 CrossRef CAS.
- J. Sun, Y. Huang, W. Wang, Z. Yu, A. Wang and K. Yuan, Electrochim. Acta, 2008, 53, 7084–7088 CrossRef CAS.
- Y. Huang, J. Sun, W. Wang, Y. Wang, Z. Yu, H. Zhang, A. Wang and K. Yuan, J. Electrochem. Soc., 2008, 155, A764–A767 CrossRef CAS.
- Y. Wang, Y. Huang, W. Wang, C. Huang, Z. Yu, H. Zhang, J. Sun, A. Wang and K. Yuan, Electrochim. Acta, 2009, 54, 4062–4066 CrossRef CAS.
- J. Sun, Y. Huang, W. Wang, Z. Yu, A. Wang and K. Yuan, Electrochem. Commun., 2008, 10, 930–933 CrossRef CAS.
- W. Zhang, Y. Huang, W. Wang, C. Huang, Y. Wang, Z. Yu and H. Zhang, J. Electrochem. Soc., 2010, 157, A443 CrossRef CAS.
- Q. Wang, W. Wang, Y. Huang, F. Wang, H. Zhang, Z. Yu, A. Wang and K. Yuan, J. Electrochem. Soc., 2011, 158, A775–A779 CrossRef CAS.
- M. Rao, X. Song, H. Liao and E. J. Cairns, Electrochim. Acta, 2012, 65, 228–233 CrossRef CAS.
- M. Rao, X. Song and E. J. Cairns, J. Power Sources, 2012, 205, 474–478 CrossRef CAS.
- M. He, L.-X. Yuan, W.-X. Zhang, X.-L. Hu and Y.-H. Huang, J. Phys. Chem. C, 2011, 115, 15703–15709 CrossRef CAS.
- Z. Zhang, W. Bao, H. Lu, M. Jia, K. Xie, Y. Lai and J. Li, ECS Electrochem. Lett., 2012, 1, A34–A37 CrossRef CAS.
- W. Bao, Z. Zhang, Y. Gan, X. Wang and J. Lia, J. Energy Chem., 2013, 22, 790–794 CrossRef CAS.
- S. Zhu, J. Yu, X. Yan, E. Zhao, Y. Wang, D. Sun, Y. Jin and K. Kanamura, J. Appl. Electrochem., 2016, 46, 725–733 CrossRef CAS.
- J.-T. Li, Z.-Y. Wu, Y.-Q. Lu, Y. Zhou, Q.-S. Huang, L. Huang and S.-G. Sun, Adv. Energy Mater., 2017, 7, 1701185 CrossRef.
- J. Wang, Z. Yao, C. W. Monroe, J. Yang and Y. Nuli, Adv. Funct. Mater., 2013, 23, 1194–1201 CrossRef CAS.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- H. Kang, Y. Liu, K. Cao, Y. Zhao, L. Jiao, Y. Wang and H. Yuan, J. Mater. Chem. A, 2015, 3, 17899–17913 RSC.
- S.-L. Chou, Y. Pan, J.-Z. Wang, H.-K. Liu and S.-X. Dou, Phys. Chem. Chem. Phys., 2014, 16, 20347–20359 RSC.
- M. Lao, Y. Zhang, W. Luo, Q. Yan, W. Sun and S. X. Dou, Adv. Mater., 2017, 29, 1–23 CrossRef PubMed.
- W. Zhang, M. Dahbi and S. Komaba, Curr. Opin. Chem. Eng., 2016, 13, 36–44 CrossRef.
- J. Peters, D. Buchholz, S. Passerini and M. Weil, Energy Environ. Sci., 2016, 9, 1744–1751 RSC.
- D. Buchholz, A. Moretti, R. Kloepsch, S. Nowak, V. Siozios, M. Winter and S. Passerini, Chem. Mater., 2013, 25, 142–148 CrossRef CAS.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- V. Dall’Asta, D. Buchholz, L. G. Chagas, X. Dou, C. Ferrara, E. Quartarone, C. Tealdi and S. Passerini, ACS Appl. Mater. Interfaces, 2017, 9, 34891–34899 CrossRef PubMed.
- X. Deng, W. Shi, J. Sunarso, M. Liu and Z. Shao, ACS Appl. Mater. Interfaces, 2017, 9, 16280–16287 CrossRef CAS PubMed.
- P. R. Kumar, Y. H. Jung, S. A. Ahad and D. K. Kim, RSC Adv., 2017, 7, 21820–21826 RSC.
- J. Zhao, X. Yang, Y. Yao, Y. Gao, Y. Sui, B. Zou, H. Ehrenberg, G. Chen and F. Du, Adv. Sci., 2018, 1700768 CrossRef PubMed.
- J. Song, L. Wang, Y. Lu, J. Liu, B. Guo, P. Xiao, J. J. Lee, X. Q. Yang, G. Henkelman and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 2658–2664 CrossRef CAS PubMed.
- A. Rudola, K. Du and P. Balaya, J. Electrochem. Soc., 2017, 164, A1098–A1109 CrossRef CAS.
- W. J. Li, S. L. Chou, J. Z. Wang, Y. M. Kang, J. L. Wang, Y. Liu, Q. F. Gu, H. K. Liu and S. X. Dou, Chem. Mater., 2015, 27, 1997–2003 CrossRef CAS.
- M. J. Piernas-Muñoz, E. Castillo-Martínez, J. L. Gómez-Cámer and T. Rojo, Electrochim. Acta, 2016, 200, 123–130 CrossRef.
- C. Delmas, C. Fouassier and P. Hagenmuller, Phys. B+C, 1980, 99, 81–85 CrossRef CAS.
- N. Ortiz-Vitoriano, N. E. Drewett, E. Gonzalo and T. Rojo, Energy Environ. Sci., 2017, 10, 1051–1074 RSC.
- L. Mu, S. Xu, Y. Li, Y.-S. S. Hu, H. Li, L. Chen and X. Huang, Adv. Mater., 2015, 27, 6928–6933 CrossRef CAS PubMed.
- H. R. Yao, P. F. Wang, Y. Gong, J. Zhang, X. Yu, L. Gu, C. Ouyang, Y. X. Yin, E. Hu, X. Q. Yang, E. Stavitski, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2017, 139, 8440–8443 CrossRef CAS PubMed.
- Z. Lu and J. R. Dahn, Chem. Mater., 2001, 13, 1252–1257 CrossRef CAS.
- K. Kubota and S. Komaba, J. Electrochem. Soc., 2015, 162, A2538–A2550 CrossRef CAS.
- X. H. Zhang, W. L. Pang, F. Wan, J. Z. Guo, H. Y. Lü, J. Y. Li, Y. M. Xing, J. P. Zhang and X. L. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 20650–20659 CrossRef CAS PubMed.
- C. Bommier and X. Ji, Small, 2018, 1703576 CrossRef PubMed.
- X. Dou, I. Hasa, M. Hekmatfar, T. Diemant, R. J. Behm, D. Buchholz and S. Passerini, ChemSusChem, 2017, 10, 2668–2676 CrossRef CAS PubMed.
- M. Dahbi, T. Nakano, N. Yabuuchi, T. Ishikawa, K. Kubota, M. Fukunishi, S. Shibahara, J. Y. Son, Y. T. Cui, H. Oji and S. Komaba, Electrochem. Commun., 2014, 44, 66–69 CrossRef CAS.
- Y. Li, Y. S. Hu, M. M. Titirici, L. Chen and X. Huang, Adv. Energy Mater., 2016, 6, 1–9 CrossRef.
- Y. Li, Y.-S. Hu, H. Li, L. Chen and X. Huang, J. Mater. Chem. A, 2016, 4, 96–104 RSC.
- M. Dahbi, M. Kiso, K. Kubota, T. Horiba, T. Chafik, K. Hida, T. Matsuyama and S. Komaba, J. Mater. Chem. A, 2017, 5, 9917–9928 RSC.
- L. O. Vogt, M. El Kazzi, E. Jämstorp Berg, S. Pérez Villar, P. Novák and C. Villevieille, Chem. Mater., 2015, 27, 1210–1216 CrossRef CAS.
- Q. Fan, W. Zhang, J. Duan, K. Hong, L. Xue and Y. Huang, Electrochim. Acta, 2015, 174, 970–977 CrossRef CAS.
- Y. Sun, L. Zhao, H. Pan, X. Lu, L. Gu, Y.-S. Hu, H. Li, M. Armand, Y. Ikuhara, L. Chen and X. Huang, Nat. Commun., 2013, 4, 1870 CrossRef PubMed.
- F. Zhao, P. Xue, H. Ge, L. Li and B. Wang, J. Electrochem. Soc., 2016, 163, A690–A695 CrossRef CAS.
- Y. Zhang, H. Hou, X. Yang, J. Chen, M. Jing, Z. Wu, X. Jia and X. Ji, J. Power Sources, 2016, 305, 200–208 CrossRef CAS.
- M. Zarrabeitia, E. Castillo-Martínez, J. M. López Del Amo, A. Eguía-Barrio, M. A. Muñoz-Márquez, T. Rojo and M. Casas-Cabanas, J. Power Sources, 2016, 324, 378–387 CrossRef CAS.
- S. Komaba, Y. Matsuura, T. Ishikawa, N. Yabuuchi, W. Murata and S. Kuze, Electrochem. Commun., 2012, 21, 65–68 CrossRef CAS.
- K. Dai, H. Zhao, Z. Wang, X. Song, V. Battaglia and G. Liu, J. Power Sources, 2014, 263, 276–279 CrossRef CAS.
- Y. Yui, M. Hayashi, K. Hayashi and J. Nakamura, Solid State Ionics, 2015, 288, 219–223 CrossRef.
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H.-H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS PubMed.
- N. Yabuuchi, Y. Matsuura, T. Ishikawa, S. Kuze, J. Y. Son, Y. T. Cui, H. Oji and S. Komaba, ChemElectroChem, 2014, 1, 580–589 CrossRef.
- M. Dahbi, N. Yabuuchi, M. Fukunishi, K. Kubota, K. Chihara, K. Tokiwa, X. Yu, H. Ushiyama, K. Yamashita, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, Chem. Mater., 2016, 28, 1625–1635 CrossRef CAS.
- Y. Kim, Y. Park, A. Choi, N. S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045–3049 CrossRef CAS PubMed.
- J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, Angew. Chem., Int. Ed., 2013, 52, 4633–4636 CrossRef CAS PubMed.
- W.-J. Li, S.-L. Chou, J.-Z. Wang, H.-K. Liu and S.-X. Dou, Chem. Commun., 2015, 51, 3682–3685 RSC.
- J. Ming, H. Ming, W.-J. Kwak, C. Shin, J. Zheng and Y.-K. Sun, Chem. Commun., 2014, 50, 13307–13310 RSC.
- T. Zhou, W. K. Pang, C. Zhang, J. Yang, Z. Chen, H. K. Liu and Z. Guo, ACS Nano, 2014, 8, 8323–8333 CrossRef CAS PubMed.
- H. Gao, W. Zhou, J. H. Jang and J. B. Goodenough, Adv. Energy Mater., 2016, 6, 1–7 Search PubMed.
- C. Luo, X. Fan, Z. Ma, T. Gao and C. Wang, Chem, 2017, 3, 1050–1062 CAS.
- L. Xu, H. Sitinamaluwa, H. Li, J. Qiu, Y. Wang, C. Yan, H. Li, S. Yuan and S. Zhang, J. Mater. Chem. A, 2017, 5, 2102–2109 RSC.
- L. Bonnefoi, P. Simon, J. Fauvarque, C. Sarrazin, J. Sarrau and A. Dugast, J. Power Sources, 1999, 80, 149–155 CrossRef CAS.
- F. Beck and M. Dolata, J. Appl. Electrochem., 2001, 31, 517–521 CrossRef CAS.
- X. Sun, X. Zhang, H. Zhang, B. Huang and Y. Ma, J. Solid State Electrochem., 2013, 17, 2035–2042 CrossRef CAS.
- B. Dyatkin, V. Presser, M. Heon, M. R. Lukatskaya, M. Beidaghi and Y. Gogotsi, ChemSusChem, 2013, 6, 2269–2280 CrossRef CAS PubMed.
- A. Krause and A. Balducci, Electrochem. Commun., 2011, 13, 814–817 CrossRef CAS.
- A. Krause, P. Kossyrev, M. Oljaca, S. Passerini, M. Winter and A. Balducci, J. Power Sources, 2011, 196, 8836–8842 CrossRef CAS.
- N. Böckenfeld, R.-S. Kühnel, S. Passerini, M. Winter and A. Balducci, J. Power Sources, 2011, 196, 4136–4142 CrossRef.
- A. Brandt, P. Isken, A. Lex-Balducci and A. Balducci, J. Power Sources, 2012, 204, 213–219 CrossRef CAS.
- N. Böckenfeld, S. S. Jeong, M. Winter, S. Passerini and A. Balducci, J. Power Sources, 2013, 221, 14–20 CrossRef.
- H. A. Ambjörnsson, K. Schenzel and U. Germgård, BioResources, 2013, 8, 1918–1932 Search PubMed.
- K. Murashko, D. Nevstrueva, A. Pihlajamäki, T. Koiranen and J. Pyrhönen, Energy, 2017, 119, 435–441 CrossRef CAS.
- A. Varzi, A. Balducci and S. Passerini, J. Electrochem. Soc., 2014, 161, A368–A375 CrossRef CAS.
- M. Yamagata, S. Ikebe, K. Soeda and M. Ishikawa, RSC Adv., 2013, 3, 1037–1040 RSC.
- H. Y. Tran, M. Wohlfahrt-Mehrens and S. Dsoke, J. Power Sources, 2017, 342, 301–312 CrossRef CAS.
- A. Varzi and S. Passerini, J. Power Sources, 2015, 300, 210–222 CrossRef.
- M. Murase, N. Yabuuchi, Z.-J. Han, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, ChemSusChem, 2012, 5, 2307–2311 CrossRef CAS PubMed.
- A. Varzi, R. Raccichini, M. Marinaro, M. Wohlfahrt-Mehrens and S. Passerini, J. Power Sources, 2016, 326, 672–679 CrossRef CAS.
- D. S. Reay, E. A. Davidson, K. A. Smith, P. Smith, J. M. Melillo, F. Dentener and P. J. Crutzen, Nat. Clim. Change, 2012, 2, 410–416 CrossRef CAS.
- B. S. Shete and N. P. Shinkar, Int. J. Curr. Eng. Technol., 2013, 3, 1611–1615 Search PubMed.
- M. Aslan, D. Weingarth, N. Jäckel, J. S. Atchison, I. Grobelsek and V. Presser, J. Power Sources, 2014, 266, 374–383 CrossRef CAS.
- M. Aslan, D. Weingarth, P. Herbeck-Engel, I. Grobelsek and V. Presser, J. Power Sources, 2015, 279, 323–333 CrossRef CAS.
- P. Kurzweil and M. Chwistek, J. Power Sources, 2008, 176, 555–567 CrossRef CAS.
- M. Winter, G. H. Wrodnigg, J. O. Besenhard, W. Biberacher and P. Novák, J. Electrochem. Soc., 2000, 147, 2427–2431 CrossRef CAS.
- M. M. Hantel, V. Presser, R. Kötz and Y. Gogotsi, Electrochem. Commun., 2011, 13, 1221–1224 CrossRef CAS.
- D. Sauerteig, S. Ivanov, H. Reinshagen and A. Bund, J. Power Sources, 2017, 342, 939–946 CrossRef CAS.
- M. Ebner, F. Marone, M. Stampanoni and V. Wood, Science, 2013, 342, 716–720 CrossRef CAS PubMed.
- N. Shpigel, M. D. Levi, S. Sigalov, O. Girshevitz, D. Aurbach, L. Daikhin, P. Pikma, M. Marandi, A. Jänes, E. Lust, N. Jäckel and V. Presser, Nat. Mater., 2016, 15, 570 CrossRef CAS PubMed.
- A. Brandt, S. Pohlmann, A. Varzi, A. Balducci and S. Passerini, MRS Bull., 2013, 38, 554–559 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |