
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorinated Aryl Sulfonimide Tagged (FAST) salts: modular synthesis and structure–property relationships for battery applications†
Mingjun
Huang‡
 a,
Shuting
Feng‡
b,
Wenxu
Zhang
a,
Livia
Giordano
c,
Mao
Chen
a,
Chibueze V.
Amanchukwu
b,
Robinson
Anandakathir
d,
Yang
Shao-Horn
*c and
Jeremiah A.
Johnson
*a
a,
Shuting
Feng‡
b,
Wenxu
Zhang
a,
Livia
Giordano
c,
Mao
Chen
a,
Chibueze V.
Amanchukwu
b,
Robinson
Anandakathir
d,
Yang
Shao-Horn
*c and
Jeremiah A.
Johnson
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
cDepartment of Mechanical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
dSamsung Advanced Institute of Technology (SAIT), 3 Van de Graaff Drive, Burlington, MA 01803, USA
First published on 21st March 2018
Abstract
Solid-state electrolytes are attracting great interest for their applications in potentially safe and stable high-capacity energy storage technologies. Lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) is widely used as a lithium ion source, especially in solid-state polymer electrolytes, due to its solubility and excellent chemical and electrochemical stability. Unfortunately, chemically inert LiTFSI cannot be easily modified to optimize its properties or allow for conjugation to other molecules, polymers, or substrates to prepare single-ion conducting polymer electrolytes. Chemical modifications of TFSI often erode its advantageous properties. Herein, we introduce Fluorinated Aryl Sulfonimide Tagged (FAST) salts, which are derived from successive nucleophilic aromatic substitution (SNAr) reactions. Experimental studies and density functional theory calculations were used to assess the electrochemical oxidative stabilities, chemical stabilities, and degrees of ion dissociation of FAST salts as a function of their structures. FAST salts offer a platform for accessing functional sulfonimides without sacrificing many of the advantageous properties of TFSI.
Broader contextThe increasing demands of modern electronics necessitate the development of energy storage devices that feature greater power and energy densities without compromising affordability and safety. With the advantages of broad electrochemical stability window, high thermal stability, and low vulnerability towards hydrolysis, lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) is widely used as a lithium ion source in new battery chemistries with higher theoretical energy densities beyond lithium-ion batteries, such as lithium–air and lithium–sulfur batteries. LiTFSI is also the most studied lithium salt especially in solid-state polymer electrolytes, due to its desirable solubility and excellent stability. However, chemically inert LiTFSI cannot be easily modified to optimize its properties or for conjugation to other molecules, polymers, or substrates to prepare single-ion conducting polymer electrolytes. Herein, we report the synthesis, chemical and electrochemical stability, and conductivity study of a new family of modular salts, Fluorinated Aryl Sulfonimide Tagged (FAST) salts that are derived from successive nucleophilic aromatic substitution (SNAr) reactions. The tunable chemical and electrochemical oxidative stability as well as Lewis basicity of FAST salts opens up new opportunities for the design and applications of polymer–FAST conjugates and single-ion conductors in solid-state electrolytes for safe and stable high-energy storage technologies. |
Introduction
The high energy density, reliability, and low cost of rechargeable lithium-ion batteries (LIBs) have revolutionized the consumer market for portable electronic devices.1,2 However, the increasing demands of modern electronics necessitate the development of energy storage devices that feature greater power and energy densities without compromising affordability and safety.3–5 As LIBs approach the theoretical specific energies of cathode/anode materials, extensive studies have focused on finding new battery chemistries beyond LIBs.6 Two tantalizing options are lithium–air (Li–air) batteries7–9 and lithium–sulfur (Li–S) batteries.10–12 While the gravimetric theoretical energy densities of these battery technologies are several times higher than conventional LIBs, both face numerous challenges that must be addressed before commercialization.7,10–15 For example, new electrolytes with high conductivity (>10−4 S cm−1 at room temperature), stability, and safety are needed.7,13,14,16 Most electrolyte materials that have been studied to date rely on mixtures of lithium salts such as lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) or lithium hexafluorophosphate (LiPF6) and a suitable solvent and/or polymer.16,17 Comparing LiTFSI and LiPF6, LiTFSI offers a broad electrochemical stability window,18 greater thermal stability, and higher resistance to hydrolysis,19 which lead it to be preferred in Li–air and Li–S batteries.19 Additionally, due to its high solubility in water (>21 M) and ability to form a passivation layer (mainly LiF), LiTFSI has been used in “water-in-salt” electrolytes enabling high-voltage aqueous lithium-ion batteries.20,21 Moreover, encouraging results have been reported on utilizing TFSI salts in sodium–air batteries22 and multivalent energy storage systems such as magnesium batteries.23Though great progress has been made on the development of solid polymer electrolytes wherein LiTFSI is dissolved in an aprotic polymer matrix of poly(ethylene oxide) (PEO), the transference number of Li+ in such materials is typically as low as 0.2,24,25 which leads to polarization at the battery electrodes and deleterious effects such as dendrite growth and limited power delivery.4,26–28 One strategy to improve the Li+ (or Na+ in sodium batteries) transference number involves anchoring the anions to a polymeric backbone, making the cation the only mobile ion (i.e., single-ion conducting polymer electrolytes).29 Unfortunately, the TFSI anion is not readily chemically modifiable, and attempts to attach sulfonimides to polymers via replacement of one or both of the electron withdrawing trifluoromethyl groups of TFSI with phenyl or alkyl groups often lead to materials with inferior properties compared to TFSI.25,30–33 Indeed, replacement of a trifluoromethyl group from TFSI with an electron rich group would be expected to decrease the electrochemical oxidative stability of the resulting salt, increase Li+–anion association, and potentially reduce ion conductivity.34–36
TFSI derivatives where one or both trifluoromethyl groups are replaced with functional yet still electron withdrawing substituents, such that the beneficial properties of TFSI are not comprised, would be highly desirable. We identified perfluoroarylsulfonimides A, B, and C (Scheme 1) as potential starting points to achieve this goal. The installation of perfluoroaryl substituents in these compounds could maintain the electron deficient nature of the anion and open the possibility of chemical modification via nucleophilic aromatic substitution (SNAr) reactions. Herein, we report our studies on the synthesis and characterization of this new class of sulfonimide salts, which we call “Fluorinated Aryl Sulfonimide Tagged” (FAST) salts. Successive SNAr reactions between A, B, or C and oxygen- and/or nitrogen-base nucleophiles enabled rational tuning of the electron density, electrochemical oxidative stability, chemical stability toward superoxide and peroxide anions, and Lewis basicity of FAST salts as assessed by both experimental studies and density functional theory (DFT) calculations. Our design led to several FAST salts that display electrochemical oxidative stability at 4.0 VLi, negligible chemical degradation, and reasonable ion conductivity in 1,2-dimethoxyethane (DME) and PEO. FAST salts offer a synthetically tunable platform for the identification of optimal anion structures that could replace TFSI in a variety of applications.
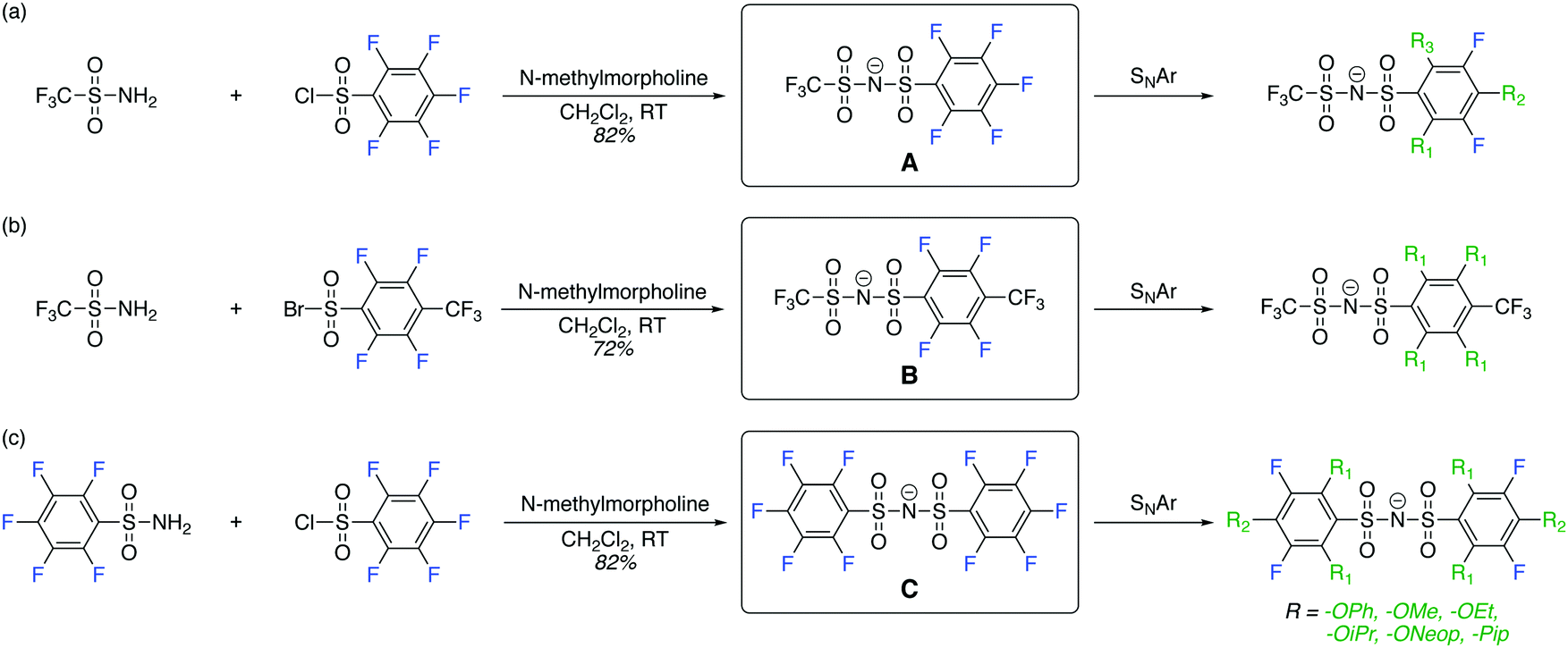 | ||
| Scheme 1 Synthesis of perfluoroarylsulfonimide anions A, B, and C and their subsequent functionalization via SNAr reactions to generate FAST anions of the general formula P-PipxORyFz. Note: the cations used in this study, Na+ and Li+, are not shown. | ||
Results and discussion
The perfluoroarylsulfonimide sodium salts A and C were prepared starting from pentafluorobenzene sulfonyl chloride in good yield (>82%), whereas salt B was prepared via condensation of 4-trifluoromethyl-2,3,5,6-tetrafluorobenzenesulfonyl bromide37 and trifluoromethanesulfonamide in 72% yield. With these compounds in hand, we embarked on the synthesis of a library of FAST salts (Fig. 1) via SNAr reactions between A, B, or C and a variety of nucleophiles selected to assess the impact of steric bulk and electronics on the salts' properties: phenoxide (OPh), alkoxides (OR: OMe, OEt, OiPr, and ONeop), and piperidine (Pip). Throughout this work, we use the general notation P-PipxORyFz to represent each FAST salt, where P is the parent salt (A, B, or C), and x, y, and z are the numbers of piperidine, alkoxide, and fluorine substituents, respectively. All FAST sodium salts were characterized by 1H, 13C, 19F NMR, MS, and in some cases, single crystal X-ray crystallography.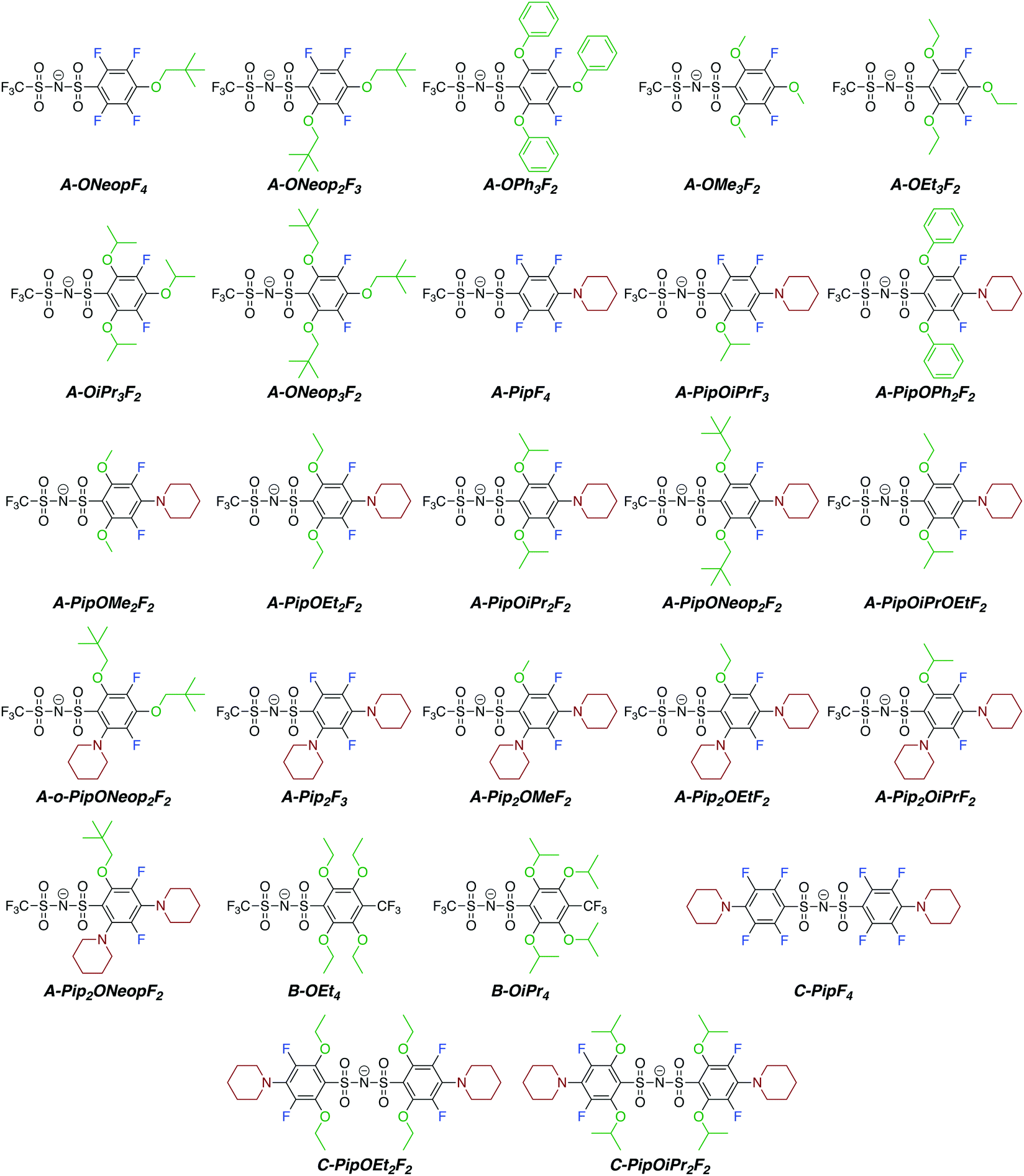 | ||
| Fig. 1 Chemical structures of FAST anions synthesized in this work via SNAr reactions between various nucleophiles and parent salts A, B, or C. | ||
We were able to exploit differences in the reactivity of the various nucleophiles in this system to control the substituent patterns in the resulting FAST salts. For example, selective SNAr of the para fluorine atom of A with Pip could be achieved to provide A-PipF4; subsequent SNAr of the remaining ortho fluorine atoms with OPh, OMe, or OEt groups provided A-PipOPh2F2, A-PipOMe2F2 and A-PipOEt2F2, respectively. The structures of the sodium salts of these compounds were confirmed by X-ray crystallography (Fig. 2). Notably, though these newly introduced N and O substituents are electron donating, this substitution pattern maintains the two electron-withdrawing meta fluorine substituents (Hammett parameters for fluorine: σmeta = 0.34 versus σpara = 0.06). As seen in Fig. 2, the sulfonimides in these structures are present as their free base; the sodium cations are not coordinated to the nitrogen but instead coordinate to the oxygen atoms from the sulfonimide groups, alkoxide groups, and/or adventitious water (Fig. 2b). When two piperidine groups are introduced onto the A scaffold the resulting FAST salts (e.g., A-Pip2F3 and its derivatives A-Pip2ORF2) are much less acidic (they are protonated during aqueous washing); the crystal structure of A-Pip2F3·H+ (Fig. S1, ESI†) reveals that the proton is coordinated by the nitrogen atom of the ortho-Pip. Therefore, FAST salts containing two Pip substituents were deprotonated with sodium hydroxide prior to further investigations.
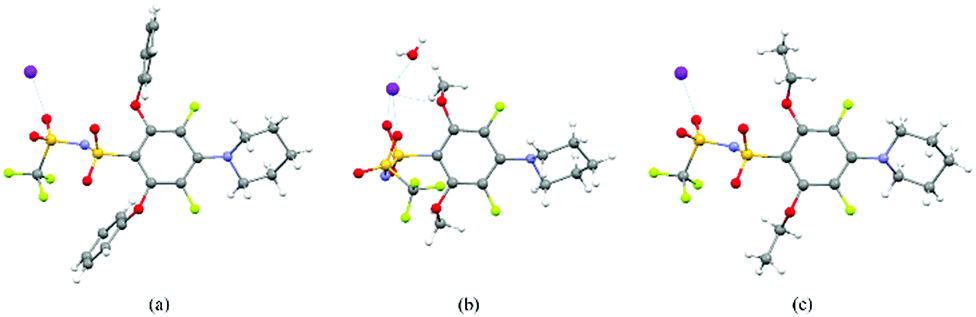 | ||
| Fig. 2 Crystal structures of FAST–Na salts (a) A-PipOPh2F2, (b) A-PipOMe2F2 and (c) A-PipOEt2F2. Atom color code: grey – carbon, white – hydrogen, red – oxygen, green – fluorine, yellow – sulfur, purple – nitrogen, and magenta – sodium. | ||
The electrochemical oxidative stability and average partial charge of aromatic carbons, c+, obtained using Natural Population Analysis (NPA)38,39 of select tri-substituted FAST salts as well as A, A-NeopF4, and A-Neop2F3 depicted in Fig. 1 were evaluated using DFT calculations (Fig. 3) following the BANE framework we developed recently.40 We observed that higher computed electrochemical oxidation potential correlated well with higher average aromatic carbon charge, c+. More specifically, FAST salts with the greatest number of electron donating Pip groups (e.g., A-Pip2ORF2) exhibit the lowest c+ and electrochemical oxidative stability. FAST derivatives with one Pip group (e.g., A-PipOR2F2) showed higher c+ and electrochemical oxidative stability than A-Pip2ORF2. As expected, the trialkoxide derivatives A-OR3F2, in turn, exhibited higher c+ and electrochemical oxidative stability than A-PipOR2F2. Finally, in the order of A-Neop2F3, A-NeopF4, and A, as the number of electron withdrawing F atoms increases, the computed c+ and electrochemical oxidative stability increase almost linearly.
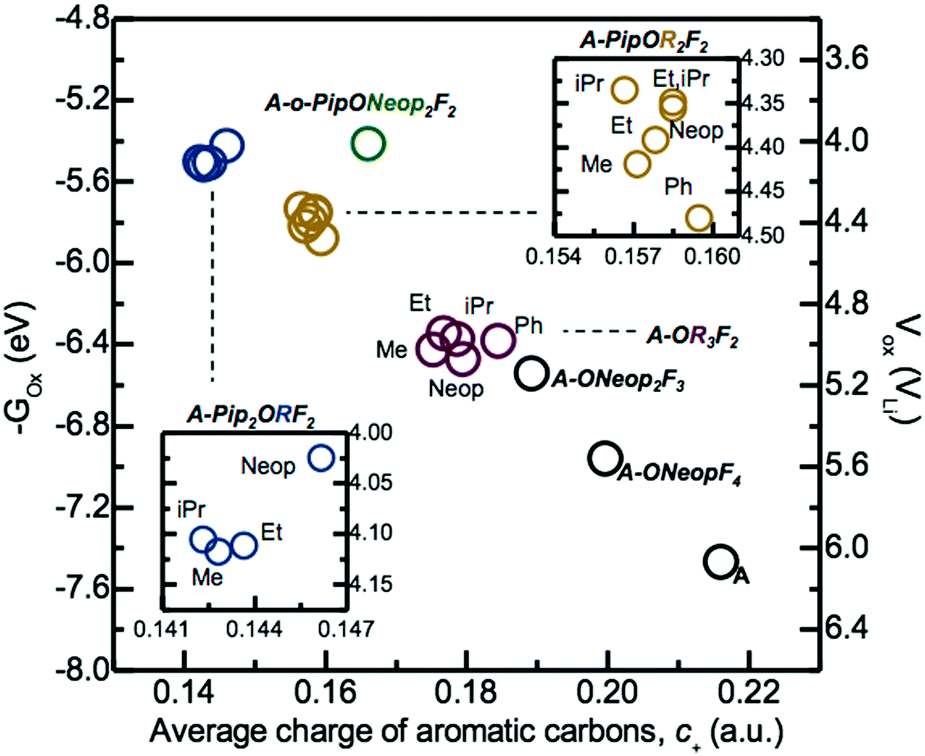 | ||
| Fig. 3 Electrochemical oxidative potentials of select FAST salts computed using B3LYP/6-311++G(d,p) with geometries fully optimized at the B3YLP/6-31G(d,p) level of theory in implicit DMSO solvent are plotted against the arithmetic average of the NPA partial charges of aromatic carbons obtained at the optimized geometries. The electrochemical oxidation potentials in experimentally measured scale versus Li/Li+, plotted on the right axis, were converted from the computed –Gox in eV by the subtraction of 1.4 V.41,42 | ||
To further understand the electrochemical oxidative stability of our FAST salts, HOMO and LUMO maps for four representative salts, A-ONeop3F2, A-PipONeop2F2, A-o-PipONeop2F2, and A-Pip2ONeopF2 were compared (Fig. 4). These salts show similar LUMOs but significantly different HOMOs: the HOMO of A-ONeop3F2 is uniformly distributed on the aromatic ring with little density on the oxygen atoms of the alkoxide substituents. FAST salts with a Pip group featured HOMOs that were heavily localized on the Pip nitrogen atom. Surprisingly, the HOMO maps of A-PipONeop2F2 and A-o-PipONeop2F2 are drastically different. The HOMO of A-PipONeop2F2 is distributed on both the benzene ring and the Pip nitrogen atom, while nearly all the HOMO is concentrated on the Pip nitrogen atom in the ortho position in both A-o-PipONeop2F2 and A-Pip2ONeopF2. These observations may explain the observed basicity of A-o-PipONeop2F2 and A-Pip2ONeopF2 that was not observed for other salts.
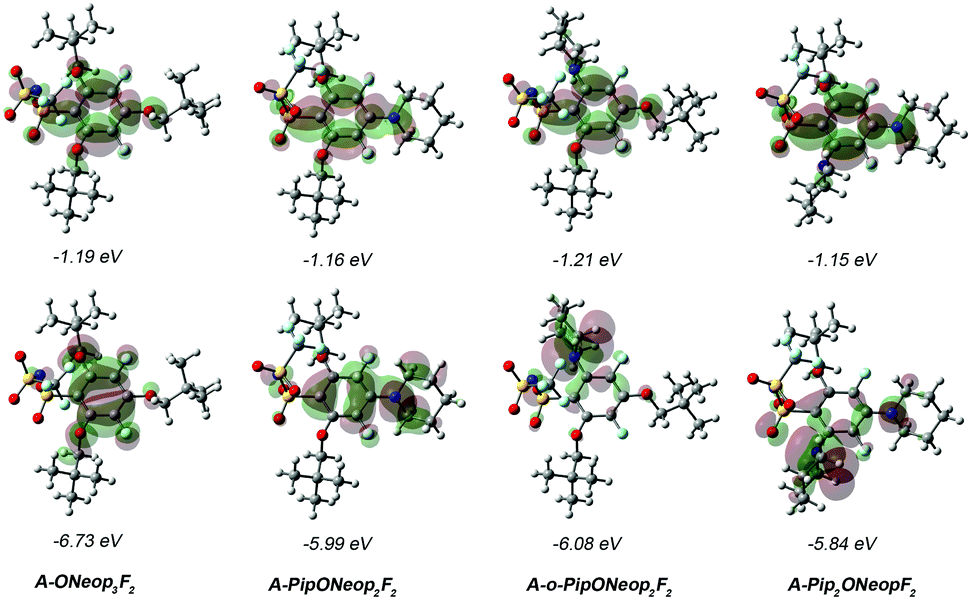 | ||
| Fig. 4 Computed LUMOs (top row) and HOMOs (bottom row) of four representative FAST anions. The HOMO/LUMO maps were generated using the optimized geometries obtained at B3LYP/6-31G(d,p). The HOMO/LUMO energy in eV were obtained at B3LYP/6-311++G(d,p) level of theory with geometries optimized using B3LYP/6-31G(d,p). Atom color code: grey – carbon, white – hydrogen, red – oxygen, aqua – fluorine, yellow – sulfur, and blue – nitrogen. | ||
We carried out experimental measurements to evaluate the electrochemical oxidative stability of several of these FAST salts under an oxygenated environment for comparison to the DFT computed trends obtained in implicit DMSO solvent. The electrochemical oxidative stability of our FAST salts was determined using potentiostatic measurements in an electrochemical cell (glass fiber separator impregnated with 0.02 M sulfonimide dissolved in propylene carbonate (PC) solution sandwiched between Li metal foil and stainless steel mesh current collector), which was pressurized with oxygen and held at potentials from 3.0 to 4.5 VLi for 3 h each. PC was chosen as the solvent due to its superior electrochemical stability,17,18 although it should be noted that its vulnerability against nucleophilic substitution makes it unsuitable as electrolyte solvent for Li–O2 battery.43,44 A relatively low concentration, 0.02 M, was employed to accommodate the low solubility of several FAST salts such as A-Pip2OEtF2 in PC. In general, A-Pip2ORF2 compounds have limited solubility in PC (<0.1 M) and 1,2-dimethoxyethane (DME, 0.1 M to 0.5 M). Nonetheless, other FAST salts shown in Fig. 1 display much higher solubility (>1 M) in common solvents used in battery systems. For example, we tested the solubility of representative sodium salts of A, A-PipF4, and A-ONeop3F2. In a typical battery system, the concentration of salt is usually 1 M. Each of these three salts have solubility larger than 1 M in PC (common solvent for aprotic LIBs) and DME (common solvent for Li-air batteries) at room temperature. The sodium salt of A has a solubility of 10 m in water. With more hydrophobic substitutions, the solubility in water would decrease. A-PipF4 has solubility larger than 1 M in water, while A-ONeop3F2 has solubility of 0.5 M in water. One key advantage of our approach is that by simple modification of the substituents, it is possible to tune solubility as needed. For example, if increased water solubility were desired, one could use hydrophilic nucleophiles in the SNAr reaction. The current response, cumulative charge, and estimated percentage of salt oxidation at each potential step from 3.6 VLi to 4.5 VLi for select salts are shown in Fig. 5. The percentage of electrochemically oxidized salt was calculated based on the assumption that the oxidation of one FAST salt molecule produces one electron. We note that deviation from this assumption and the presence of impurities can lead to overestimation of the electrochemical oxidation percentage, which can explain why several salts showed electrochemical oxidation percentages that are close to or even greater than 100%. In Fig. 5a and c, the series of A, A-ONeopF4, A-ONeop2F3, A-ONeop3F2 are compared. All four salts in this series, which feature 2 to 5 fluorine atoms and 0 to 3 ONeop substituents on the aromatic ring, were very stable towards oxidation with approximately 2% and 6% oxidized upon charging to 4.2 VLi and 4.5 VLi, respectively (Fig. 5c). Upon charging to 4.5 V, TFSI exhibited electrochemical oxidation roughly an order of magnitude lower than the four salts in this series. However, these fours salts are more stable than (phenylsulfonyl)(trifluoromethyl)sulfonimide (Ph-TFSI), a widely used TFSI alternative in battery applications.25,45 Next, the influence of the substitution pattern on the oxidative stability of triply substituted salts A-OR3F2 was measured (Fig. 5b and c). These salts showed excellent oxidative stability at voltages <4.0 VLi with the exception of A-OMe3F2, which exhibited significant oxidative current at 3.8 VLi. Generally, the FAST salts with bulkier alkoxide groups (e.g., OiPr and ONeop) exhibited superior stability than those with smaller substituents such as OMe and OEt at high voltage (>4.0 VLi). The influence of Pip on the electrochemical stability was investigated by comparing A-ONeop3F2, A-PipONeop2F2, A-o-PipONeop2F2, and A-Pip2ONeopF2 (Fig. 5d and f). It is observed that while A-ONeop3F2 is very stable at 4.2 VLi (∼2% electrochemical oxidation), A-Pip2ONeopF2 and A-o-PipONeop2F2 experienced 11% and 21% oxidation upon charging to 4.2 VLi, respectively. This stability trend matches the DFT calculations presented in Fig. 3 (A-ONeop3F2 > A-PipONeop2F2 > A-o-PipONeop2F2 ≈ A-Pip2ONeopF2). Finally, several A type FAST salts with one piperidine group A-PipOR2F2 and B type FAST salts B-OR4 were tested and compared in Fig. 5e and f. As shown by the cumulative charge and estimated oxidized percentage in Fig. 5f, A-PipOR2F2 and B-OR4 generally have higher charge accumulation and worse electrochemical oxidative stability than A-OR3F2 at >4.0 VLi. Overall, most of the salts in Fig. 2 show electrochemical oxidative stability at 4.0 VLi and thus are promising candidates in diverse battery chemistries. Notably, A-ONeop3F2 was extremely stable to electrochemical oxidation (up to 4.5 VLi with oxidative current less than 0.25 μA and oxidation percentage is less than 6%).
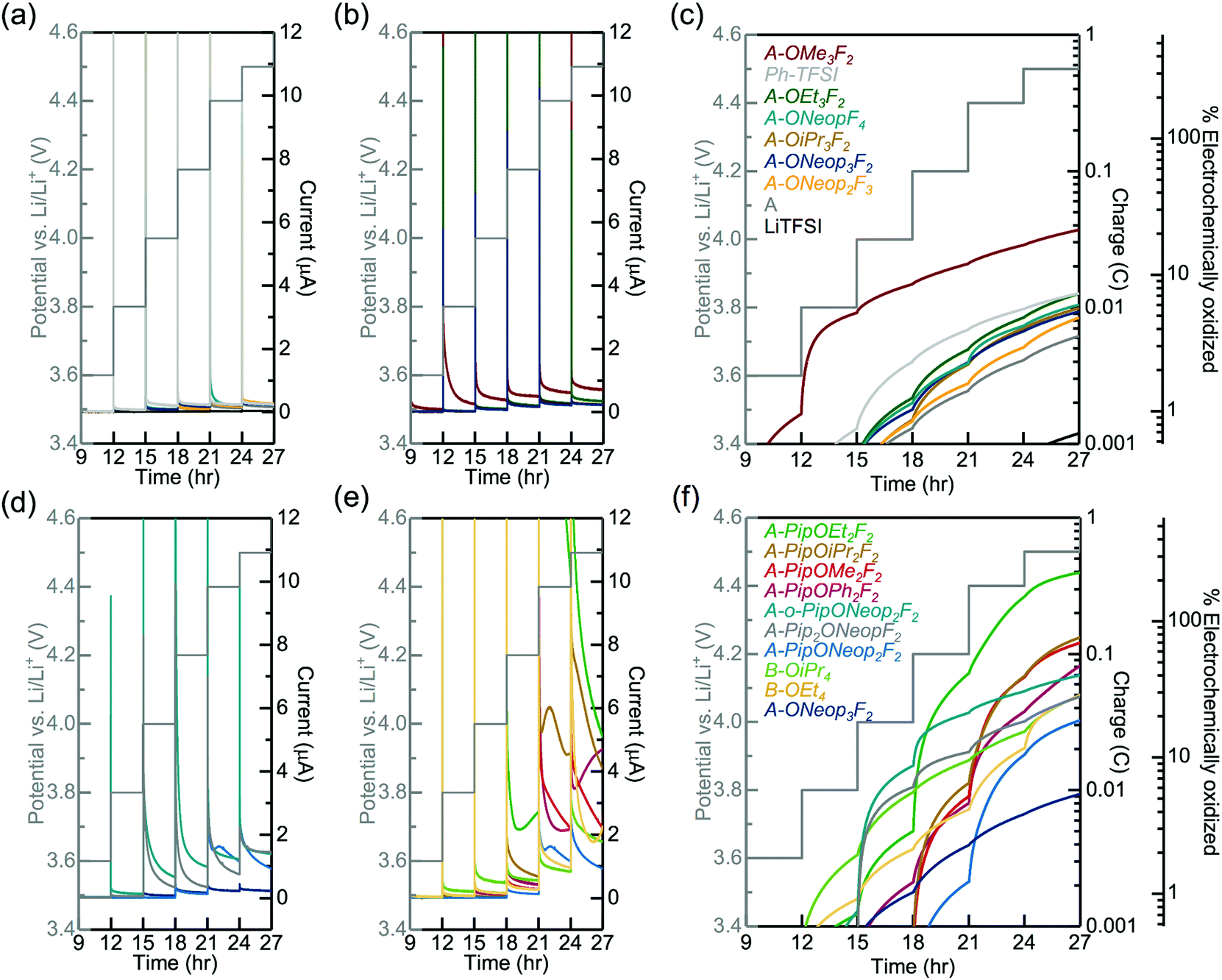 | ||
| Fig. 5 The influence of SNAr substitutions on the electrochemical oxidative stability of representative FAST salts in potentiostatic tests. The current response of A-ORyFz as functions of time show very low current up to 4.5 VLi (a). The current response of different types of alkoxide substitutions in A-OR3F2 were also compared in (b). The cumulative charge and estimated oxidized percentage of A-ORyFz and A-OR3F2 were calculated in (c). The current response of salts with different numbers of piperidine type substitutions, A-PipxORyFz, and those in the A-PipOR2F2 series were recorded in (d) and (e), respectively; their cumulative charge and estimated oxidized percentage are shown in (f). Measurements were performed using an electrochemical cell pressurized with oxygen and consisting of a stainless steel mesh current collector, 90 μL 0.02 M FAST-PC solution, one glass fiber separator, and Li metal. | ||
Next, the chemical stability of various FAST salts was investigated under solution conditions designed to mimic the oxygen electrode of a typical aprotic Li–air battery.40,46,47 Each FAST salt was dissolved in DMF (20 mg mL−1) and mixed with 12.5 equivalents of Li2O2, KO2, and 1 equivalent of 4-methoxybiphenyl as internal standard (for quantitative NMR analysis); the mixture was stirred at 80 °C for 3 days. The supernatant of the mixture was characterized by 1H, 19F-NMR, and liquid chromatography-mass spectrometry (LC-MS). Generally, FAST salts with a greater number of aryl fluoride groups displayed lower chemical stability: for salts derived from A and C, only those with two meta fluorine atoms have negligible degradation, whereas in salts derived from B no aryl fluorides were tolerated due to the strong electron withdrawing effect of –CF3 group (σpara = 0.54). For tri-substituted salts derived from A (Scheme 1 and Fig. 1), the chemical stability was strongly affected by the identity and pattern of the substituents. Fig. 6 provides a comparison of the percentage of degradation (obtained by quantitative 1H NMR) of each salt. For salts derived from A, we observed that bulkier –OR substituents improved the stability against chemical degradation. A-OMe3F2 was observed to degrade almost completely (91%) while no degradation was detected for A-ONeop3F2. FAST salts with Pip groups exhibited greater chemical stability than those with –OR substitutions: the degraded percentage decreased from 65% in A-OPh3F2 and 91% in A-OMe3F2 to 16% in A-PipOPh2F2 and 15% in A-PipOMe2F2. When two Pip groups were introduced (A-Pip2ORF2), less than 4% degradation was observed regardless of the identity of R. These experimental results for chemical stability are inversely correlated with the calculated average carbon atomic charges on the aromatic ring: A-Pip2ORF2 > A-PipOR2F2 > A-OR3F2, which supports the expectation that more electron rich FAST salts should be less susceptible to nucleophilic attack. Furthermore, we computed the Gibbs free energy for nucleophilic substitution by superoxide, ΔGnuc, at select carbon sites (i.e., O–CH3) in A-OMe3F2, A-PipOMe2F2, and A-Pip2OMeF2 (Fig. S2, ESI†); the computed trend of ΔGnuc (A-OMe3F2 < A-PipOMe2F2 < A-Pip2OMeF2) follows the trend in the chemical stability of these salts determined experimentally. Finally, we plotted the computed ΔGnuc against the increase in the NPA partial charge of the attacking oxygen in superoxide (partial charge of the oxygen after the substitution reaction minus its partial charge before the reaction; Fig. S2, ESI†). It is observed that a larger increase in the oxygen partial charge corresponds to a more favourable ΔGnuc; this correlation was also observed in our recent study on the nucleophilic substitution of small organic molecules such as carbonates and ethers by superoxide.40 This trend suggests that a larger increase in the attacking oxygen partial charge indicates stronger electron-donating strength of superoxide at the carbon site, which gives rise to more favourable ΔGnuc.
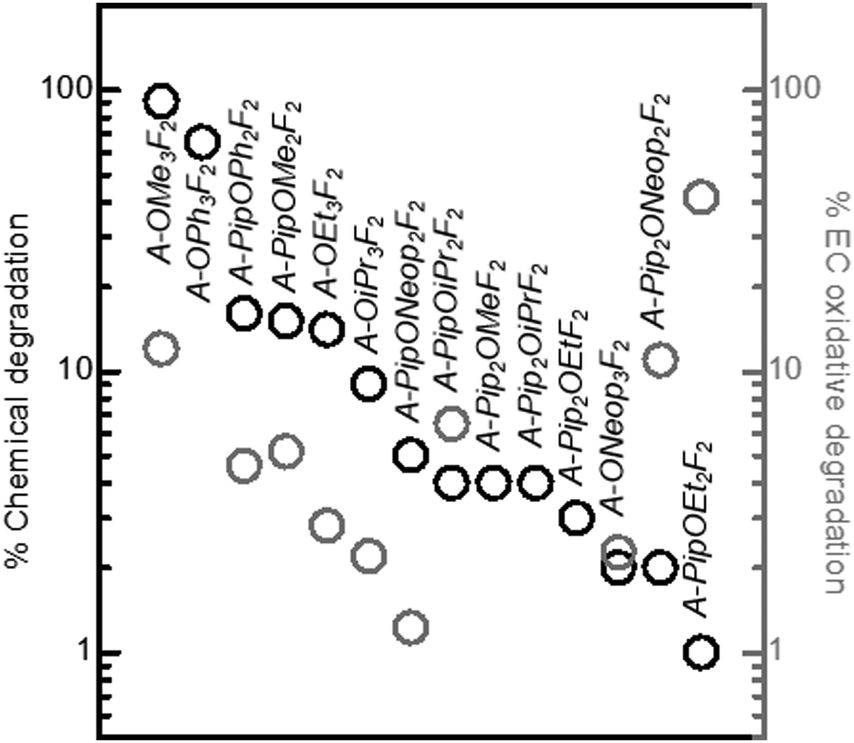 | ||
| Fig. 6 Chemical and electrochemical oxidative stability test results for FAST salts. The black circles indicate the degradation percentages of the sulfonimide salts after incubation with 12.5 equiv. of Li2O2 and KO2 in DMF for three days at 80 °C. Percent degradation was quantified by 1H NMR using 4-methoxybiphenyl as a reference standard. The electrochemical oxidation percentages of the corresponding FAST salts in the potentiostatic tests at the end of the 4.2 VLi step are shown by the grey circles. | ||
Ion conductivity in liquid electrolytes depends upon two factors: charge carrier concentration and mobility. With the same concentration of salts, the extent to which the salt is dissociated determines the charge carrier concentration. Generally, salt anions with higher Lewis basicity interact more strongly with alkali metal cations, and thus increase the extent of anion–cation association.48–50 The TFSI anion is well known for being an “innocent” anion with weak interactions with metal ions.50 To compare our FAST salts with TFSI and evaluate the extent of ion dissociation, the anion–cation interaction strengths for our FAST salts were determined by 23Na NMR.50,51 The sodium salts were prepared as 0.1 M solutions in nitromethane with 0.25 M NaClO4 in DMSO as the internal standard. The 23Na chemical shifts of FAST salts relative to NaTFSI are shown in Fig. 7a. It is immediately obvious that the nature of the anion plays an important role in the resulting chemical shift. For example, A-OR3F2 and A-PipOR2F2 have 23Na signals that are shifted downfield, indicating stronger anion–Na+ interactions than the parent type A and B salts. A considerable amount of line broadening was also observed and can be attributed to the formation of more ion pairs.51 We propose that as the FAST salt anions become more electron rich (i.e., as the number of F atoms decreases), they become more Lewis basic, display stronger interactions with Na+, and produce more ion pairs. To validate this hypothesis and study the effect of different substitution groups on anion–Na+ interaction and ion conductivity, we plotted the calculated Gibbs free energy of ion pair association, ΔGassoc, versus the relative 23Na chemical shifts as well as the ion conductivities of DME solutions containing several FAST salts at 0.1 M experimentally obtained at 25 °C (Fig. 7b). Implicit diethylether solvent with dielectric constant set at 7.2 was used to mimic the solvent used for conductivity studies (DME). As expected, salts with more negative calculated ΔGassoc values (more favorable anion–Na+ association) have more down-field shift in 23Na NMR spectrum and displayed lower conductivity. More specifically, we observed that solutions containing type A and B salts have ion conductivities that are a factor of 2 and 1.5, respectively, lower than that of NaTFSI while A-ONeop3F2, A-PipF4 and A-PipONeop2F2 exhibit conductivities that are 4 to 7 times lower. The conductivities of solutions containing these salts are inversely related to the salt anion Lewis basicity and anion–cation interaction strength. Furthermore, 23Na NMR chemical shift of other B type and C type salts were also measured shown in Fig. S3a (ESI†). Here acetonitrile was chosen as solvent due to low solubility of B and C type salts in nitromethane. Acetonitrile solvates Na+ better than nitromethane and thus decreases the anion–Na+ interaction strength difference.51 Nevertheless, we observed that salts with more SNAr substitutions have 23Na signal shift toward down field and line broadening comparing with B and C parent salts (Fig. S3a, ESI†); these salts also exhibit more favorable ΔGassoc for anion–Na+ association and lower ion conductivity in DME solution (Fig. S3b, ESI†). Overall, salts with more SNAr substitutions among all three types have greater Lewis basicity, which leads to stronger interaction with Na+ and more negative ΔGassoc values. These results highlight the balance of factors that must be considered in the design of functional FAST salts.
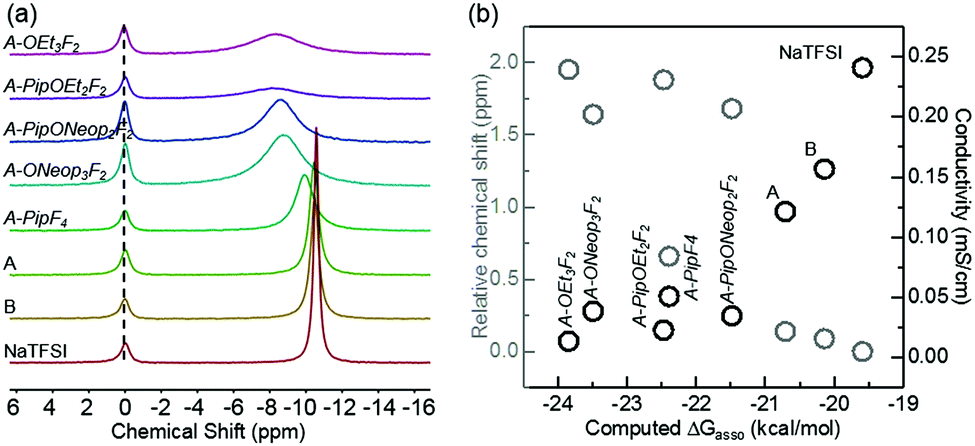 | ||
| Fig. 7 (a) The measured chemical shift of 23Na NMR signal for representative FAST salts (the 23Na signal from the inner standard, NaClO4, is set to 0 ppm). (b) The 23Na NMR chemical shift (relative to NaTFSI) and ion conductivity of 0.1 M DME solution at 25 °C versus the computed anion–Na+ association free energy, ΔGassoc, in implicit diethylether solvent with dielectric constant set at 7.2 for representative FAST salts. | ||
The Li FAST salts could be readily acquired by ion exchange of the Na salts. We prepared four Li salts from A, A-PipF4, A-ONeop3F2, and A-PipONeop2F2. The 7Li and 23Na NMR spectra show nearly complete replacement of Na+ by Li+ in A (Fig. S4, ESI†). The ionic conductivities of solid-state polymer electrolytes prepared by blending these Li salts with PEO (10 kDa; molar ratio of PEO repeat unit and Li+ [EO]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Li+] = 15:1) are shown in Fig. 8a. These FAST–PEO blends exhibited a similar ionic conductivity trend to their corresponding sodium salts in liquid electrolytes: blends with Li salts A and A-PipF4 have ionic conductivities that are 2 and 4 times lower, respectively, than that of LiTFSI. Polymer electrolytes containing Li salts of A-PipONeop2F2 and A-ONeop3F2 showed conductivities in the range from 60 °C to 80 °C that were approximately one order of magnitude lower than that of LiTFSI. As in liquid electrolyte, the lower ion conductivity of FAST–PEO blends can be attributed to higher Lewis basicity of these salts and the formation of more ion pairs (Fig. 8b), which leads to a lower concentration of charge carriers. Another reason for the observed lower ion conductivity could simply be size, as the mobility of larger FAST anions may be decreased, which would reduce overall ion conductivity. Nevertheless, in light of the all of the results discussed above, A-ONeop3F2 shows chemical and oxidative stability on par with TFSI and reasonable conductivity, thus suggesting the potential use of this and other trisubstituted FAST salts as functional TFSI replacements.
:[Li+] = 15:1) are shown in Fig. 8a. These FAST–PEO blends exhibited a similar ionic conductivity trend to their corresponding sodium salts in liquid electrolytes: blends with Li salts A and A-PipF4 have ionic conductivities that are 2 and 4 times lower, respectively, than that of LiTFSI. Polymer electrolytes containing Li salts of A-PipONeop2F2 and A-ONeop3F2 showed conductivities in the range from 60 °C to 80 °C that were approximately one order of magnitude lower than that of LiTFSI. As in liquid electrolyte, the lower ion conductivity of FAST–PEO blends can be attributed to higher Lewis basicity of these salts and the formation of more ion pairs (Fig. 8b), which leads to a lower concentration of charge carriers. Another reason for the observed lower ion conductivity could simply be size, as the mobility of larger FAST anions may be decreased, which would reduce overall ion conductivity. Nevertheless, in light of the all of the results discussed above, A-ONeop3F2 shows chemical and oxidative stability on par with TFSI and reasonable conductivity, thus suggesting the potential use of this and other trisubstituted FAST salts as functional TFSI replacements.
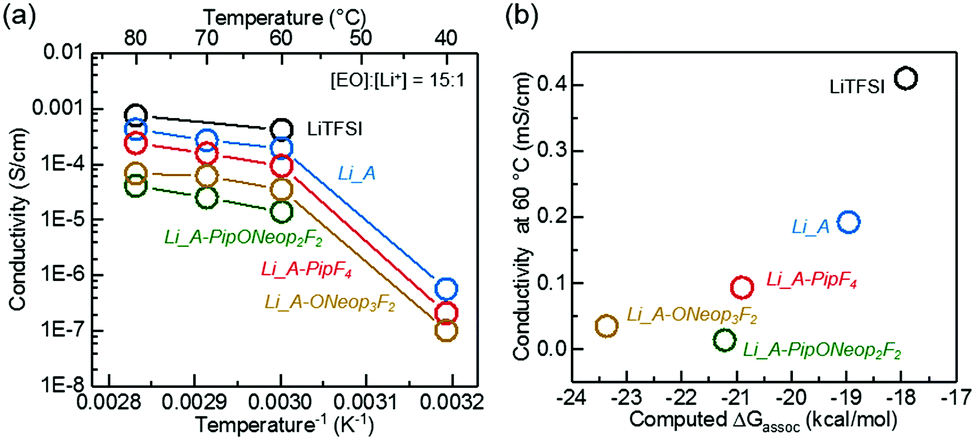 | ||
| Fig. 8 (a) The ion conductivities of solid state FAST–PEO electrolytes prepared via blending representative LiFAST salts and PEO (molar ration of PEO repeat units and lithium ion [EO]:[Li+] = 15:1) at various temperatures. (b) The conductivity of FAST–PEO electrolytes at 60 °C versus computed free energy of anion–Li+ association, ΔGassoc, in implicit diethylether solvent with dielectric constant set at 7.2. | ||
Conclusions
In summary, we have developed a novel class of sulfonimide salts that are based on successive SNAr reactions of fluorinated phenyl sulfonimides: Fluorinated Aryl Sulfonimide Tagged (FAST) salts. Using DFT calculations and experimental measurements, we demonstrated that the chemical and electrochemical oxidative stability of FAST salts are inversely correlated with each other as the number of fluorine atoms present on the aromatic ring is varied. FAST salts with strongly electron donating Pip substituents generally showed better chemical stability compared to those with ether substituents; however, the sterically hindered salt A-ONeop3F2 was also highly resistant to chemical degradation. FAST salts with Pip groups were more vulnerable to electrochemical oxidation than those containing only ether substituents; here again, ONeop3F2 displayed outstanding stability. Other properties like solubility, Lewis basicity, and conductivity could also be tuned by introducing different numbers and types of nucleophilic functional groups to the FAST salt scaffold. FAST salts provide a new anion design strategy, enabling alternatives to TFSI with properties that can be rationally varied in a highly modular fashion. In particular, the ability to readily control the pattern of functionalization on the FAST scaffold and predict the resulting chemical and electrochemical oxidative stability as well as basicity opens up new opportunities for the design of polymer–FAST conjugates and single-ion conductors, meeting the growing interest of solid-state electrolytes as potentially safe and stable high-energy storage technologies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Samsung Advanced Institute of Technology (SAIT) for funding this research. This research used resources of the National Energy Research Scientific Computing (NERSC) Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-5CH11231, and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562.Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- O. Schmidt, A. Hawkes, A. Gambhir and I. Staffell, Nat. Energy, 2017, 6, 17110 CrossRef.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed.
- Y.-C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 750–768 CAS.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- A. Manthiram, S. H. Chung and C. Zu, Adv. Mater., 2015, 27, 1980–2006 CrossRef CAS PubMed.
- J. Yi, S. Guo, P. He and H. Zhou, Energy Environ. Sci., 2017, 10, 860–884 CAS.
- S. Zhang, K. Ueno, K. Dokko and M. Watanabe, Adv. Energy Mater., 2015, 5, 1500117 CrossRef.
- D. G. Kwabi, N. Ortiz-Vitoriano, S. A. Freunberger, Y. Chen, N. Imanishi, P. G. Bruce and Y. Shao-Horn, MRS Bull., 2014, 39, 443–452 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- M. Ue, M. Takeda, M. Takehara and S. Mori, J. Electrochem. Soc., 1997, 144, 2684–2688 CrossRef CAS.
- R. Younesi, G. M. Veith, P. Johansson, K. Edstrom and T. Vegge, Energy Environ. Sci., 2015, 8, 1905–1922 CAS.
- L. Suo, F. Han, X. Fan, H. Liu, K. Xu and C. Wang, J. Mater. Chem. A, 2016, 4, 6639–6644 CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- M. He, K. C. Lau, X. Ren, N. Xiao, W. D. McCulloch, L. A. Curtiss and Y. Wu, Angew. Chem., Int. Ed., 2016, 55, 15310–15314 CrossRef CAS PubMed.
- X. Qu, Y. Zhang, N. N. Rajput, A. Jain, E. Maginn and K. A. Persson, J. Phys. Chem. C, 2017, 121, 16126–16136 CAS.
- K. Timachova, H. Watanabe and N. P. Balsara, Macromolecules, 2015, 48, 7882–7888 CrossRef CAS.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J.-P. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes, D. Devaux, R. Denoyel and M. Armand, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 CAS.
- W. H. Meyer, Adv. Mater., 1998, 10, 439–448 CrossRef CAS PubMed.
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, ACS Energy Lett., 2017, 2, 2563–2575 CrossRef CAS.
- H. T. Ho, A. Tintaru, M. Rollet, D. Gigmes and T. N. T. Phan, Polym. Chem., 2017, 8, 5660–5665 RSC.
- L. Porcarelli, A. S. Shaplov, F. Bella, J. R. Nair, D. Mecerreyes and C. Gerbaldi, ACS Energy Lett., 2016, 1, 678–682 CrossRef CAS.
- Q. Ma, H. Zhang, C. Zhou, L. Zheng, P. Cheng, J. Nie, W. Feng, Y. S. Hu, H. Li, X. Huang, L. Chen, M. Armand and Z. Zhou, Angew. Chem., Int. Ed., 2016, 55, 2521–2525 CrossRef CAS PubMed.
- P. Murmann, P. Niehoff, R. Schmitz, S. Nowak, H. Gores, N. Ignatiev, P. Sartori, M. Winter and R. Schmitz, Electrochim. Acta, 2013, 114, 658–666 CrossRef CAS.
- S. Ladouceur, S. Paillet, A. Vijh, A. Guerfi, M. Dontigny and K. Zaghib, J. Power Sources, 2015, 293, 78–88 CrossRef CAS.
- V. Morizur, S. Olivero, J. R. Desmurs, P. Knauth and E. Duñach, New J. Chem., 2014, 38, 6193–6197 RSC.
- V. Morizur, M. Braglia, S. Olivero, J.-R. Desmurs, P. Knauth and E. Duñach, New J. Chem., 2016, 40, 7840–7845 RSC.
- V. E. Platonov, A. M. Roman, A. Bredikhin and V. V. K. Maksimov, J. Fluorine Chem., 2010, 131, 13–16 CrossRef CAS.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- S. Feng, M. Chen, L. Giordano, M. Huang, W. Zhang, C. V. Amanchukwu, R. Anandakathir, Y. Shao-Horn and J. A. Johnson, J. Mater. Chem. A, 2017, 5, 23987–23998 CAS.
- S. Trasatti, Pure Appl. Chem., 1986, 58, 955–966 CrossRef CAS.
- L. Xing, O. Borodin, G. D. Smith and W. Li, J. Phys. Chem. A, 2011, 115, 13896–13905 CrossRef CAS PubMed.
- D. Aurbach, M. Daroux, P. Faguy and E. Yeager, J. Electroanal. Chem., 1991, 297, 225–244 CrossRef CAS.
- S. A. Freunberger, Y. Chen, Z. Peng, J. M. Griffin, L. J. Hardwick, F. Bardé, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040–8047 CrossRef CAS PubMed.
- A. A. Rojas, K. Thakker, K. D. McEntush, S. Inceoglu, G. M. Stone and N. P. Balsara, Macromolecules, 2017, 50, 8765–8776 CrossRef CAS.
- J. R. Harding, C. V. Amanchukwu, P. T. Hammond and Y. Shao-Horn, J. Phys. Chem. C, 2015, 119, 6947–6955 CAS.
- C. V. Amanchukwu, J. R. Harding, Y. Shao-Horn and P. T. Hammond, Chem. Mater., 2015, 27, 550–561 CrossRef CAS.
- S. S. Sekhon, N. Arora and H. P. Singh, Solid State Ionics, 2003, 160, 301–307 CrossRef CAS.
- C. M. Burke, V. Pande, A. Khetan, V. Viswanathan and B. D. McCloskey, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9293–9298 CrossRef CAS PubMed.
- M. Schmeisser, P. Illner, R. Puchta, A. Zahl and R. van Eldik, Chemistry, 2012, 18, 10969–10982 CrossRef CAS PubMed.
- R. H. Erlich and A. I. Popov, J. Am. Chem. Soc., 1971, 93, 5620–5623 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1590310–1590313. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ee03509h |
| ‡ M. H. and S. F. contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |