
Unprecedented size-sieving ability in polybenzimidazole doped with polyprotic acids for membrane H2/CO2 separation†
Lingxiang
Zhu
 ,
Mark T.
Swihart
and
Haiqing
Lin
*
,
Mark T.
Swihart
and
Haiqing
Lin
*
Department of Chemical and Biological Engineering, University at Buffalo, The State University of New York, Buffalo, NY 14260, USA. E-mail: haiqingl@buffalo.edu
First published on 12th December 2017
Abstract
Polymers with efficient and tight chain-packing and thus strong size-sieving ability are of great interest for H2/CO2 separation. Herein, we demonstrate a new approach to manipulating polymer structure by acid doping, leading to superior H2/CO2 separation performance. We have doped polybenzimidazole (PBI) with polyprotic acids, specifically H3PO4 and H2SO4. These acids cross-link PBI chains and drastically decrease free volume, improving the material's H2/CO2 selectivity to far surpass the Robeson's 2008 upper bound for membrane performance. For example, PBI doped with H3PO4 at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 exhibits an unprecedented H2/CO2 selectivity of 140 at 150 °C, which exceeds that of previously known polymeric materials and is superior or comparable to that of state-of-the-art 2D materials with sharp size separation, such as graphene oxide, MoS2, and metal–organic frameworks. This facile approach to enhancing polymer chain-packing efficiency opens up a new avenue for designing strong size-sieving polymers for membrane gas separations.
:1 exhibits an unprecedented H2/CO2 selectivity of 140 at 150 °C, which exceeds that of previously known polymeric materials and is superior or comparable to that of state-of-the-art 2D materials with sharp size separation, such as graphene oxide, MoS2, and metal–organic frameworks. This facile approach to enhancing polymer chain-packing efficiency opens up a new avenue for designing strong size-sieving polymers for membrane gas separations.
Broader contextCO2 capture, sequestration and utilization is an important approach to mitigating CO2 emissions to the environment. A key strategy in this regard is to decarbonize fossil fuels, producing H2 and CO2 at high temperature and pressure, which must then be separated to obtain CO2 for sequestration/utilization. Membrane technology has inherent advantages for gas separation because of its high energy efficiency, small footprint and ease of scale-up. However, materials with superior H2/CO2 separation properties at 150 °C or above are lacking. Here we report, for the first time, that polybenzimidazole doped with an anhydrous polyprotic acid shows an unprecedented H2/CO2 selectivity of 140 at 150 °C. This selectivity is the highest among known polymers. The simple yet elegant approach of acid doping provides a flexible and exciting platform to enhance polymer chain-packing efficiency, and thus achieve sharp molecular size separation in polymers. |
H2 is a key chemical used in oil refining and production of ammonia and methanol, and it has been explored as a clean fuel for fuel cells and electricity generation. Currently, H2 is mostly generated by steam reforming of fossil fuels followed by the water–gas shift reaction. The produced H2 is always mixed with CO2, and it must be purified before utilization. Ideally, the CO2 is captured for utilization or sequestration to prevent its emission to the environment.1–3 Membranes that are permeable to H2 and reject CO2 at syngas processing conditions (150 °C and above) have been demonstrated as a low-cost and energy-efficient means of H2 purification and CO2 capture.4–6 The key to success of this technology is cost-effective membrane materials with superior H2/CO2 separation properties. Recently, inorganic materials have been widely explored, including graphene oxide (GO),7,8 MoS2,9 zeolites,10 and metal–organic frameworks (MOFs).11–13 2D layered materials, including GO and MoS2, were stacked to form sub-nanometer channels between layers,7–9 while MOFs and zeolites were designed with apertures between the kinetic diameters of H2 (2.89 Å) and CO2 (3.3 Å),11 leading to extremely strong size-sieving ability and thus superior H2/CO2 separation properties. However, commercial-scale production of defect-free membranes with sub-nanometer channels presents an enormous challenge. Current commercial membranes for gas separation are based on polymers, partially owing to their good processability and ease of scale-up.14 However, conventional polymers are amorphous and cannot achieve the sharp size-based separation possible with stacked GO and MoS2 layers, zeolites, or MOFs.
Gas transport in polymeric membranes generally follows a solution-diffusion mechanism.14,15 Gas permeability (P) for each species is the product of its solubility (S) and diffusivity (D) in the polymer. Selectivity is thus determined by a combination of solubility selectivity and diffusivity selectivity. Because H2 is smaller and less condensable than CO2, polymers usually have favorable H2/CO2 diffusivity selectivity and unfavorable solubility selectivity, leading to only moderate H2/CO2 selectivity.1 For example, commercial polymeric membranes of cellulose acetate (CA), polysulfone (PSF) and Matrimid® have H2/CO2 selectivities below 3.16
To achieve high H2/CO2 selectivity, polymers should have rigid chains with efficient chain-packing to obtain high H2/CO2 diffusivity selectivity. For example, poly[2,2′-(m-phenylene)-5,5-bibenzimidazole] (PBI) exhibits H2/CO2 selectivity of 14 to 20 at 150 °C depending on the polymer grades and processing conditions.17–21 The efficiency of polymer chain-packing can be improved by cross-linking.22 For example, PBI can be cross-linked using terephthaloyl chloride to increase its H2/CO2 selectivity by almost 100%.21 Thin films of polyimides such as 6FDA-durene and 6FDA-ODA/NDA can also be cross-linked using diamines to yield high H2/CO2 selectivity.23,24 However, these cross-linked polyimides are not stable at 150 °C or above, due to thermally induced re-imidization.25,26
Herein, we demonstrate a facile approach to improve polymer size-sieving ability by doping PBI with polyprotic acids, specifically H3PO4 and H2SO4. As shown in Fig. 1a, H3PO4 can strongly interact with PBI chains via proton transfer from the acid to imidazole rings of PBI and hydrogen bonding, and thus it cross-links the PBI.27 In comparison, a monoprotic acid such as HCl is not expected to cross-link the PBI chains. PBI doped with H3PO4 has been widely explored for use in high-temperature proton exchange membranes for fuel cell applications.28–31 However, there are no prior reports of its application for membrane gas separation. This study, for the first time, investigates PBI doped with polyprotic acids for membrane gas separation. A series of H3PO4 doped PBIs (i.e., PBI–(H3PO4)x) were prepared by immersing PBI films (with a thickness of ca. 10 μm) in solutions containing H3PO4 and methanol (Fig. 1b). Different H3PO4 doping levels (x = 0.16–1.0) were obtained by varying the H3PO4 concentration in the solutions from 0.05 to 1.0 wt% (see Table S1 in the ESI†). The maximum doping level employed in this study was x = 1.0 because H3PO4 cross-links PBI at a stoichiometric ratio of 1:1. At a sub-stoichiometric doping level (1 or less), acid doping can increase the elastic modulus of PBI due to protonation and strong hydrogen bonding between PBI and H3PO4. However, a higher doping level (particularly x > 2) deteriorates mechanical strength because the excess (non-bonded) acid can swell PBI, increasing the space between PBI backbones. For instance, increasing the doping level from 2.3 to 5.6 was shown to drastically decrease ultimate tensile strength of PBI from 160 MPa to 10 MPa at 125 °C.32 The enhanced mechanical properties of PBI–(H3PO4)x with x = 0.16–1.0 would also be advantageous for their use in membrane gas separation. Fig. 1c and d show the elemental distribution of phosphorus on the surface and cross-section of a PBI–(H3PO4)1.0 film, respectively, as measured using scanning electron microscopy/energy-dispersive X-ray spectroscopy (SEM/EDS). The even distribution of phosphorus in the analyzed area indicates that the PBI films are homogenously doped with H3PO4.
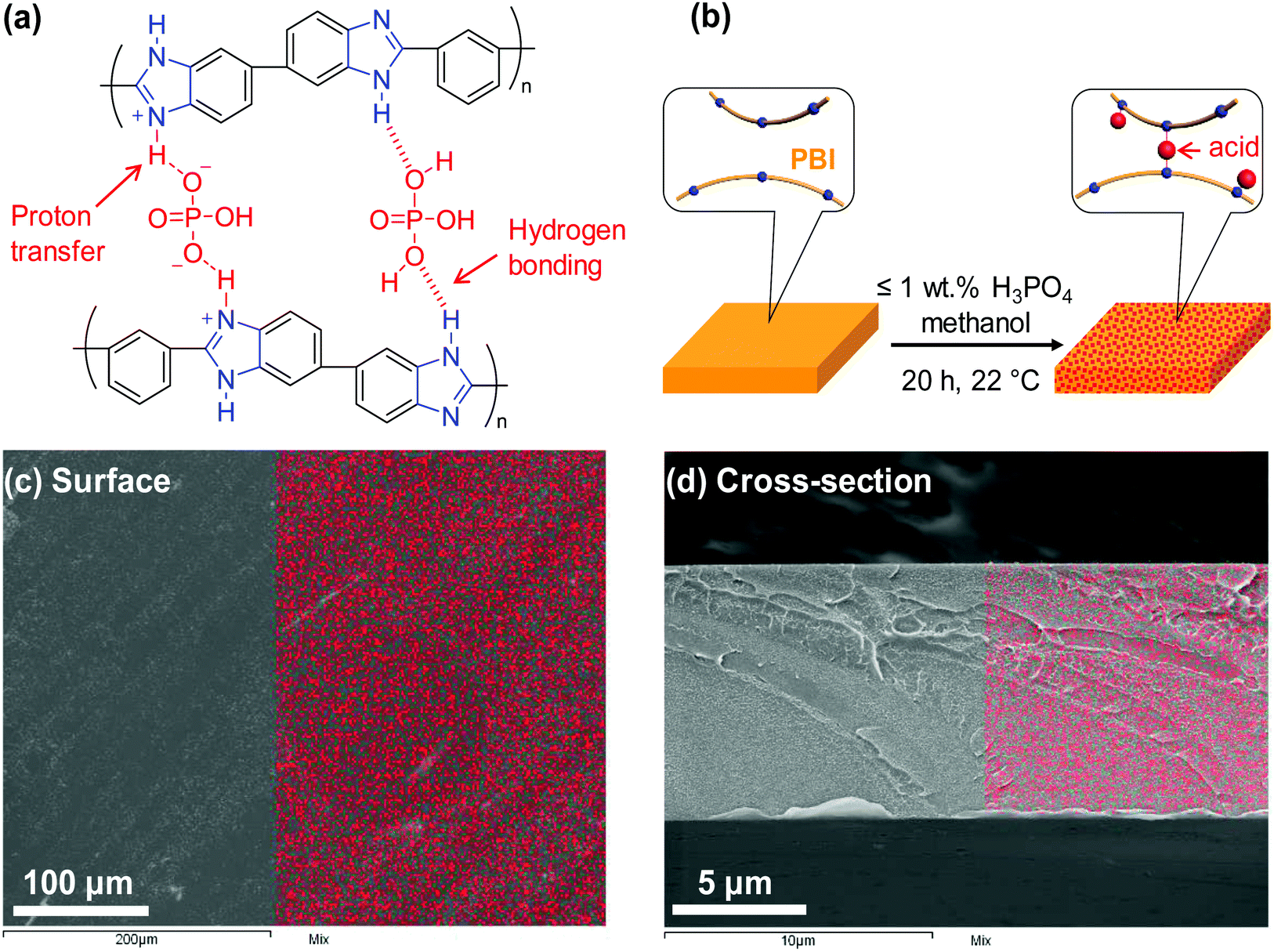 | ||
| Fig. 1 Schematic illustration of (a) proton transfer mechanism and hydrogen bonding in PBI–H3PO4 complex, and (b) preparation of H3PO4 doped PBI films with PBI backbones cross-linked by acids. SEM images with overlaid SEM/EDS mapping of phosphorus on the (c) surface and (d) cross-section of a PBI–(H3PO4)1.0 film. The red dots display the distribution of phosphorus in the polymer. | ||
Fig. 2a presents Fourier-transform infrared (FTIR) spectra of PBI–(H3PO4)x films after vacuum drying at 160 °C. The proton transfer from H3PO4 to PBI leads to the formation of H2PO4−, indicated by the characteristic peaks of PO2 (1050 cm−1) and P(OH)2 (870 cm−1 and 945 cm−1).29 The PBI–(H3PO4)x samples were also characterized by thermal gravimetric analysis (TGA, Fig. S1, ESI†) and their degradation temperature is above 200 °C, which is consistent with literature results,33 and demonstrates their promise for practical H2/CO2 separation at 150 °C. Moreover, these complexes are known to be chemically stable in the presence of CO, oxygen, water vapor, and methanol at 150–200 °C.34,35
 | ||
| Fig. 2 (a) FTIR spectra and (b) WAXD patterns of PBI and PBI–(H3PO4)x films. Curves are labelled with the values of x (defined as molar ratio of H3PO4 to the PBI repeating unit). | ||
Fig. 2b depicts the effect of H3PO4 doping on wide-angle X-ray diffraction (WAXD) patterns of the PBI–(H3PO4)x films. PBI shows a diffraction peak at 22°, which corresponds to a d-spacing (or average inter-segmental distance between polymer chains) of 4.0 Å. The H3PO4 doping shifts the diffraction peak to 25°, which corresponds to a d-spacing of 3.6 Å. This 10% decrease in interchain distance reflects the strong interaction between H3PO4 and the imidazole groups in PBI (Fig. 1a). The decrease in d-spacing inevitably increases size-sieving ability and thus H2/CO2 diffusivity selectivity.36,37
Pure-gas H2 and CO2 permeability of PBI and PBI–(H3PO4)x samples were measured at 150 °C and feed pressures ranging from 8.0 to 15 atm. The results are summarized in Table S2, ESI.† CO2 permeability for each sample was independent of feed pressure, indicating that the acid doped PBIs are resistant to CO2 plasticization. As shown in Fig. 3a, increasing the H3PO4 doping level decreases the pure-gas permeability, and drastically increases the H2/CO2 selectivity. For example, PBI shows H2/CO2 selectivity of 16, while PBI–(H3PO4)1.0 shows a remarkable selectivity of 140, which is much higher than that of any previously studied polymers. A further increase in the H3PO4 doping level to 2.5 decreased the H2 permeability to 0.65 Barrers and H2/CO2 selectivity to 100, presumably because the non-bonded H3PO4 in the PBI swells and plasticizes the polymer, decreasing its size-sieving ability.
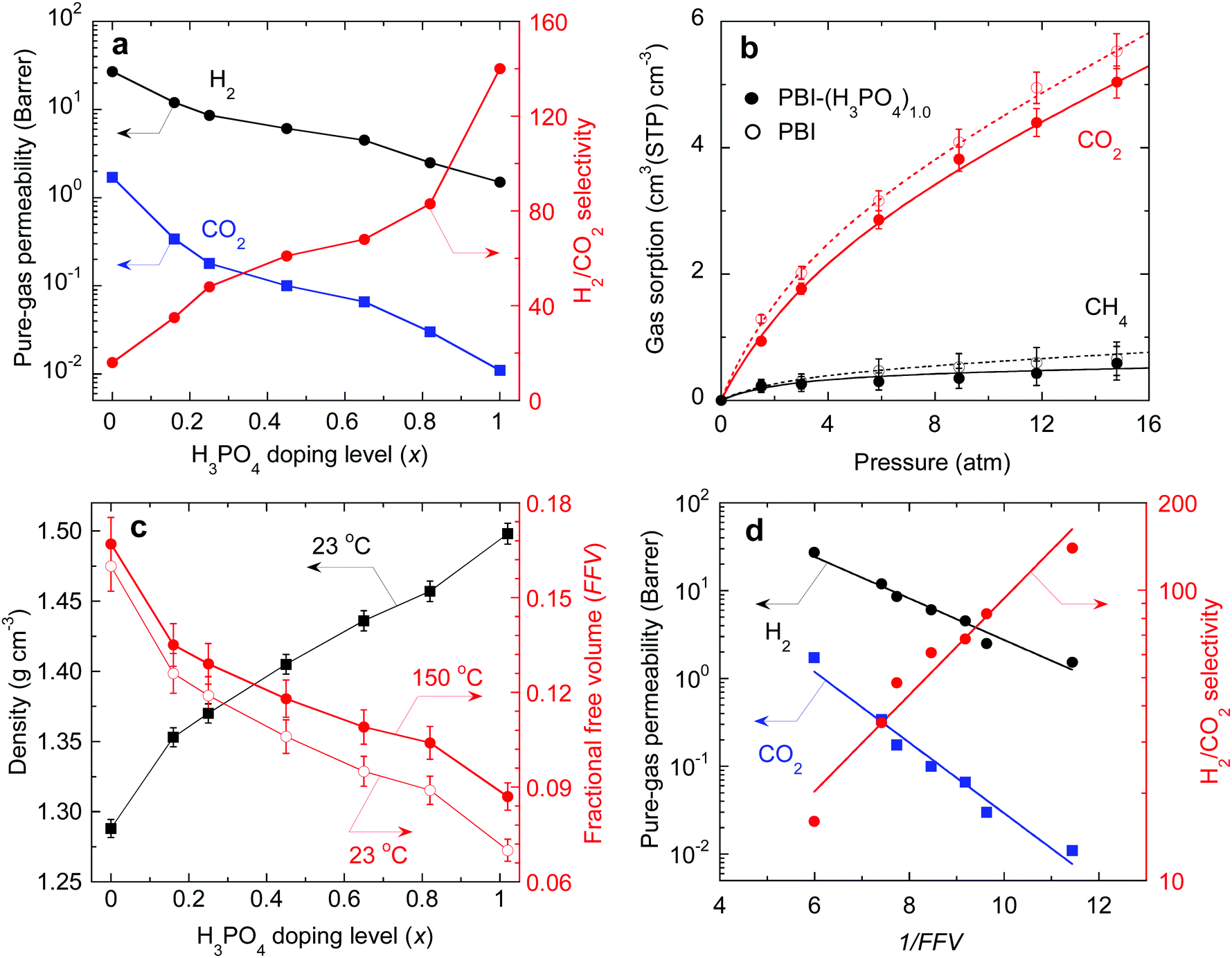 | ||
| Fig. 3 (a) Pure-gas permeability of H2 and CO2 at 150 °C as a function of H3PO4 doping level in PBI. (b) CO2 and CH4 sorption isotherms for PBI and PBI–(H3PO4)1.0 at 150 °C. CH4 is used as a surrogate for H2, and the curves are the best fits to the dual mode sorption model with fitting parameters summarized in Table S3, ESI.† (c) Effect of acid doping level on the density and FFV of PBI–(H3PO4)x at 23 °C and 150 °C. (d) Correlation of pure-gas permeability in PBI–(H3PO4)x with the FFV using the free volume model (eqn (1)) at 150 °C. | ||
To elucidate how the H3PO4 loading affects gas transport properties, pure-gas sorption of CO2 and CH4 in PBI and PBI–(H3PO4)1.0 was quantified using a gravimetric sorption analyzer at 150 °C. The resulting isotherms are shown in Fig. 3b. The experimental data can be fit well by curves based on the dual mode sorption model.38 In considering solubility selectivity, we have used CH4 as a surrogate for H2 because H2 sorption is too low to measure gravimetrically. Neither CH4 nor H2 is expected to exhibit specific interactions with polymers.38 Sorption of both CO2 and CH4 decreases slightly after acid doping, due to the reduction in accessible free volume for gas sorption. However, the CO2/CH4 solubility selectivity remains unchanged upon acid doping. For example, the CO2/CH4 solubility selectivities for PBI and PBI–(H3PO4)1.0 at 15 atm are 8.3 and 8.6, respectively. These results imply that the decrease in gas permeability and increase in H2/CO2 selectivity in the H3PO4 doped PBIs can be ascribed to changes in diffusivity and diffusivity selectivity, respectively.
Because PBI–(H3PO4)x samples show similar gas solubility, gas permeability can be correlated with the fractional free volume (FFV) using the following equation:39
| PA = AAexp(−BA/FFV) | (1) |
| FFV = (V − V0)/V | (2) |
As shown in Fig. 3d, pure-gas permeability can be satisfactorily described using eqn (1) with BA values of 0.54 and 0.93 for H2 and CO2, respectively. The larger BA value for CO2 is consistent with its larger molecular size. As the FFV decreases, certain free volume elements initially become inaccessible to the larger CO2 and only with further decrease of FFV become inaccessible to the smaller H2. Consequently, the CO2 permeability decreases more rapidly than H2 permeability. This leads to an exponential increase in H2/CO2 selectivity, as indicated by the data and best-fit line in Fig. 3d. PBI–(H3PO4)1.0 has an extremely low FFV value of 0.087 and thus exceptionally sharp molecular separation of H2/CO2 (with a selectivity of 140).
Membrane-based H2/CO2 separation has been proposed as an efficient technology to purify H2 from coal-derived shifted syngas, which is typically comprised of 56% H2, 41% CO2, and small amounts of other components, such as H2O, CO, N2 and H2S, at 150 °C or above.4 We investigated the mixed-gas separation performance of PBI–(H3PO4)x using a binary gas mixture of 50% H2 and 50% CO2 at 120–180 °C with a feed pressure of 14 atm. PBI–(H3PO4)0.16 was selected as a model sample because of its balanced H2 permeability and H2/CO2 selectivity. As shown in Table S5, ESI,† it exhibits a mixed-gas H2 permeability of 12 Barrers and a H2/CO2 selectivity of 34 at 150 °C. These values are very close to the pure-gas properties, suggesting the absence of competitive sorption and plasticization at 150 °C.41,42 This behavior is very different from conventional studies of polymers for CO2/CH4 separation at 35 °C, where CO2 can plasticize the polymer matrix, decreasing the size-sieving ability and thus CO2/CH4 selectivity.43–45 This contrast also demonstrates an advantage of operating the membrane H2/CO2 separation at high temperatures, where CO2 sorption is low, and the adverse effect of plasticization can be avoided. As shown in Fig. 4a, increasing temperature dramatically increases both H2 and CO2 permeability. On the other hand, the temperature has a negligible effect on H2/CO2 selectivity (at 33 ± 2). This behavior can be ascribed to two opposing factors. Increasing temperature decreases the size-sieving ability in polymers (or H2/CO2 diffusivity selectivity), whereas it may increase H2/CO2 solubility selectivity, resulting in almost constant H2/CO2 selectivity.21
 | ||
| Fig. 4 (a) Mixed-gas H2/CO2 separation performance of PBI–(H3PO4)0.16 in gas mixture (50%H2/50%CO2) at temperatures ranging from 120 to 180 °C. The lines are to guide the eye. (b) Long-term stability test of PBI–(H3PO4)0.16 in both dry and humidified conditions at 150 °C for 120 h. 0.3 mol% water moisture was introduced in feed gas (50%H2/50%CO2) at 11 h. (c) Pure-gas H2/CO2 separation performance of PBI–(H3PO4)x with different doping levels (x = 0.16–1.0) and PBI–(H2SO4)0.24 at 150 °C versus Robeson's 2008 upper bound (the blue line) at 35 °C.47 The orange line is the upper bound predicted for 150 °C.48 (d) Comparison of H2/CO2 separation performance of PBI–(H3PO4)x with state-of-the-art membrane materials: green squares (1 to 6) represent polymeric membranes and mixed matrix membranes (MMMs);6,50–54 black triangles (7 to 9) represent inorganic materials including GO,7 MoS2,9 and zeolite;10 and orange diamonds (10 to 14) represent MOF membranes and their composites.11,13,55–57 The box on the right explains the data points in detail. | ||
We also investigated the effect of water vapor on mixed-gas separation properties and long-term stability of PBI–(H3PO4)0.16 at 150 °C. As shown in Fig. 4b, PBI–(H3PO4)0.16 was initially tested using a dry gas mixture (50% H2/50% CO2) at a pressure of 14 atm for 10 hours. Then 0.3 mol% water vapor was introduced in the mixed-gas feed, and the mixed-gas permeability was tested over a period of 110 hours. The presence of water slightly decreased mixed-gas H2 permeability from 12 Barrers (dry state) to 11 Barrers (humidified state), while the H2/CO2 selectivity gradually increased from 34 (dry state) to 38 (humidified state). PBI is quite hydrophilic and the dry polymer can absorb up to 20 wt% water.46 The absorbed water occupies some free volume elements, which decreases the accessible free volume for gas diffusion and increases size-sieving ability. Due to the strong affinity between PBI and H3PO4, absorbed water does not displace the cross-linking acid, and no performance deterioration was observed upon exposing the PBI–(H3PO4)0.16 film to moisture for over 100 hours. This demonstrates the material's remarkable reliability under these simulated shifted syngas processing conditions and supports its potential for practical application.
Pure-gas H2/CO2 separation properties of PBI–(H3PO4)x are benchmarked in a Robeson's plot (Fig. 4c).47 The 2008 upper bound represents a tradeoff between H2 permeability and H2/CO2 selectivity, and the highest H2/CO2 selectivity achievable for any given H2 permeability in polymers at around 35 °C. For example, the separation performance of commercial membrane polymers such as CA, PSF, Matrimid and poly(2,6-dimethyl-1,4-phenylene oxide) (PPO) is below this upper bound.16 Meanwhile, our PBI–(H3PO4)x samples, operating at 150 °C, are significantly above the 2008 upper bound empirically drawn for 35 °C and another upper bound predicted for 150 °C,48 indicating their superior H2/CO2 separation performance. Interestingly, a trend line drawn over the data points of PBI–(H3PO4)x is almost parallel to the upper bound, which is consistent with the notion that decreasing free volume decreases permeability and increases selectivity.49 For comparison, gas separation properties of PBI and PBI–(H3PO4)0.16 at 35 °C are also included in Fig. 4c. They are much closer to the 2008 upper bound than the commercial membrane polymers.
Fig. 4d compares H2/CO2 separation performance of PBI–(H3PO4)x samples with other state-of-the-art materials including polymers,50 mixed matrix membranes (MMMs) containing MOFs or palladium nanoparticles,6,51–54 inorganic materials (e.g., GO, MoS2 and zeolites),7,9,10 and layered MOF nanosheets.11,13,55–57 The PBI–(H3PO4)x samples exhibit higher H2/CO2 selectivity than these leading materials with superior size-sieving ability, except for the layered MOF nanosheets.11 More importantly, in contrast to the layered 2D materials (such as MOFs, GOs, and MoS2) which present enormous challenges in membrane manufacturing and scale-up, the PBI–(H3PO4)x can be easily incorporated into current manufacturing practices for polymer-based membranes.
To demonstrate the generality of the concept of polyprotic acid doping to increase polymer size-sieving ability, PBI was also doped with H2SO4 at a level of 0.24, i.e., PBI–(H2SO4)0.24, following the same procedure used for preparing PBI–(H3PO4)x. The successful doping of H2SO4 was confirmed by the FTIR spectrum (cf. Fig. S2, ESI†). Similar to the H3PO4 doping, H2SO4 doping also increases polymer density, decreases FFV (cf., Table S4, ESI†) and reduces d-spacing (cf., Fig. S3, ESI†). As shown in Fig. 4c, PBI–(H2SO4)0.24 exhibits H2/CO2 separation performance above the upper bound. With a similar doping level, PBI–(H2SO4)0.24 and PBI–(H3PO4)0.25 exhibit almost the same H2 permeability and H2/CO2 selectivity, presumably due to the similar effect of acids in enhancing the chain-packing efficiency for PBI.
Conclusions
We have demonstrated, for the first time, the enhancement of polymer chain-packing efficiency by doping with polyprotic acids to improve the size-sieving ability and H2/CO2 separation properties in PBI at 120–180 °C. The polyprotic acids (H3PO4 or H2SO4) form complexes with PBI, which are stable up to 200 °C and show great promise for H2 purification and CO2 capture. The acid doping has a negligible effect on gas sorption, but it significantly reduces FFV and d-spacing, thereby drastically improving H2/CO2 diffusivity selectivity. For example, PBI–(H3PO4)1.0 exhibits an unprecedented H2/CO2 selectivity of 140, which is much higher than that of any known polymers, and superior or comparable to that of emerging 2D materials such as GO, MoS2, and MOFs. The free volume model was used to correlate the gas permeability and structural changes induced by acid doping.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Fossil Energy, under Award Number DE-FE0026463. This work was also partially supported by the U.S. National Science Foundation (NSF) under CAREER award number 1554236.Notes and references
- H. Lin, E. Van Wagner, B. D. Freeman, L. G. Toy and R. P. Gupta, Science, 2006, 311, 639 CrossRef CAS PubMed.
- S. Wang, X. Li, H. Wu, Z. Tian, Q. Xin, G. He, D. Peng, S. Chen, Y. Yin, Z. Jiang and M. D. Guiver, Energy Environ. Sci., 2016, 9, 1863 CAS.
- Z. Kang, M. Xue, L. Fan, L. Huang, L. Guo, G. Wei, B. Chen and S. Qiu, Energy Environ. Sci., 2014, 7, 4053 CAS.
- T. C. Merkel, M. Zhou and R. W. Baker, J. Membr. Sci., 2012, 389, 441 CrossRef CAS.
- H. Lin, Curr. Opin. Chem. Eng., 2014, 4, 54 CrossRef.
- S. Japip, K. S. Liao and T. S. Chung, Adv. Mater., 2017, 29, 1603833 CrossRef PubMed.
- H. W. Kim, H. W. Yoon, S. M. Yoon, B. M. Yoo, B. K. Ahn, Y. H. Cho, H. J. Shin, H. Yang, U. Paik, S. Kwon, J. Y. Choi and H. B. Park, Science, 2013, 342, 91 CrossRef CAS PubMed.
- H. Li, Z. N. Song, X. J. Zhang, Y. Huang, S. G. Li, Y. T. Mao, H. J. Ploehn, Y. Bao and M. Yu, Science, 2013, 342, 95 CrossRef CAS PubMed.
- A. Achari, S. Sahana and M. Eswaramoorthy, Energy Environ. Sci., 2016, 9, 1224 CAS.
- M. Yu, H. H. Funke, R. D. Noble and J. L. Falconer, J. Am. Chem. Soc., 2011, 133, 1748 CrossRef CAS PubMed.
- Y. Peng, Y. Li, Y. Ban, H. Jin, W. Jiao, X. Liu and W. Yang, Science, 2014, 346, 1356 CrossRef CAS PubMed.
- W. Yang, Y. Peng, Y. Li and Y. Ban, Angew. Chem., Int. Ed., 2017, 56, 9757 CrossRef PubMed.
- C. Kong, H. Du, L. Chen and B. Chen, Energy Environ. Sci., 2017, 10, 1812 CAS.
- H. B. Park, J. Kamcev, L. M. Robeson, M. Elimelech and B. D. Freeman, Science, 2017, 356, eaab0530 CrossRef PubMed.
- W. J. Koros and C. Zhang, Nat. Mater., 2017, 16, 289 CrossRef CAS PubMed.
- D. F. Sanders, Z. P. Smith, R. Guo, L. M. Robeson, J. E. McGrath, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 4729 CrossRef CAS.
- K. A. Berchtold, R. P. Singh, J. S. Young and K. W. Dudeck, J. Membr. Sci., 2012, 415–416, 265 CrossRef CAS.
- S. C. Kumbharkar and K. Li, J. Membr. Sci., 2012, 415-416, 793 CrossRef CAS.
- X. Li, R. P. Singh, K. W. Dudeck, K. A. Berchtold and B. C. Benicewicz, J. Membr. Sci., 2014, 461, 59 CrossRef CAS.
- R. P. Singh, X. Li, K. W. Dudeck, B. C. Benicewicz and K. A. Berchtold, Polymer, 2017, 119, 134 CrossRef CAS.
- L. Zhu, M. T. Swihart and H. Lin, J. Mater. Chem. A, 2017, 5, 19914 CAS.
- L. Zhu, M. Omidvar and H. Lin, in Membranes for Gas Separations, ed. M. A. Carreon, World Scientific, 2017, p. 243 Search PubMed.
- B. T. Low, Y. Xiao, T. S. Chung and Y. Liu, Macromolecules, 2008, 41, 1297 CrossRef CAS.
- T.-S. Chung, L. Shao and P. S. Tin, Macromol. Rapid Commun., 2006, 27, 998 CrossRef CAS.
- L. Shao, T. S. Chung, S. H. Goh and K. P. Pramoda, J. Membr. Sci., 2005, 256, 46 CAS.
- L. Shao, T. S. Chung, S. H. Goh and K. P. Pramoda, J. Membr. Sci., 2005, 267, 78 CrossRef CAS.
- R. He, Q. Li, J. O. Jensen and N. J. Bjerrum, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2989 CrossRef CAS.
- J. Wainright, J. Wang, D. Weng, R. Savinell and M. Litt, J. Electrochem. Soc., 1995, 142, 121 CrossRef.
- X. Glipa, B. Bonnet, B. Mula, D. J. Jones and J. Roziere, J. Mater. Chem., 1999, 9, 3045 RSC.
- J. Weber, K. D. Kreuer, J. Maier and A. Thomas, Adv. Mater., 2008, 20, 2595 CrossRef CAS.
- E. Quartarone and P. Mustarelli, Energy Environ. Sci., 2012, 5, 6436 CAS.
- R. He, Q. Li, A. Bach, J. O. Jensen and N. J. Bjerrum, J. Membr. Sci., 2006, 277, 38 CrossRef CAS.
- S. Samms, S. Wasmus and R. Savinell, J. Electrochem. Soc., 1996, 143, 1225 CrossRef CAS.
- Q. Li, J. O. Jensen, R. F. Savinell and N. J. Bjerrum, Prog. Polym. Sci., 2009, 34, 449 CrossRef CAS.
- J. P. Melchior, G. Majer and K. D. Kreuer, Phys. Chem. Chem. Phys., 2017, 19, 601 RSC.
- H. B. Park, C. H. Jung, Y. M. Lee, A. J. Hill, S. J. Pas, S. T. Mudie, E. Van Wagner, B. D. Freeman and D. J. Cookson, Science, 2007, 318, 254 CrossRef CAS PubMed.
- S. Luo, J. R. Wiegand, B. Kazanowska, C. M. Doherty, K. Konstas, A. J. Hill and R. Guo, Macromolecules, 2016, 49, 3395 CrossRef CAS.
- Z. P. Smith, R. R. Tiwari, T. M. Murphy, D. F. Sanders, K. L. Gleason, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 3026 CrossRef CAS.
- J. Y. Park and D. R. Paul, J. Membr. Sci., 1997, 125, 23 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- M. R. Coleman and W. J. Koros, Macromolecules, 1999, 32, 3106 CrossRef CAS.
- S. Japip, K. S. Liao, Y. Xiao and T. S. Chung, J. Membr. Sci., 2016, 497, 248 CrossRef CAS.
- C. Staudt-Bickel and W. J. Koros, J. Membr. Sci., 1999, 155, 145 CrossRef CAS.
- H. Lin, E. Van Wagner, R. Raharjo, B. D. Freeman and I. Roman, Adv. Mater., 2006, 18, 39 CrossRef CAS.
- G. Liu, N. Li, S. J. Miller, D. Kim, S. Yi, Y. Labreche and W. J. Koros, Angew. Chem., Int. Ed., 2016, 128, 13958 CrossRef.
- Q. Li, R. He, R. W. Berg, H. A. Hjuler and N. J. Bjerrum, Solid State Ionics, 2004, 168, 177 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390 CrossRef CAS.
- B. W. Rowe, L. M. Robeson, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2010, 360, 58 CrossRef CAS.
- H. Lin and M. Yavari, J. Membr. Sci., 2015, 475, 101 CrossRef CAS.
- M. F. Jimenez-Solomon, Q. Song, K. E. Jelfs, M. Munoz-Ibanez and A. G. Livingston, Nat. Mater., 2016, 15, 760 CrossRef CAS PubMed.
- T. Yang, Y. Xiao and T. S. Chung, Energy Environ. Sci., 2011, 4, 4171 CAS.
- T. Yang, G. M. Shi and T. S. Chung, Adv. Energy Mater., 2012, 2, 1358 CrossRef CAS.
- Z. Wang, D. Wang, S. Zhang, L. Hu and J. Jin, Adv. Mater., 2016, 28, 3399 CrossRef CAS PubMed.
- L. F. Villalobos, R. Hilke, F. H. Akhtar and K. V. Peinemann, Adv. Energy Mater., 2017, 1701567 CrossRef.
- Y. S. Li, F. Y. Liang, H. Bux, A. Feldhoff, W. S. Yang and J. Caro, Angew. Chem., Int. Ed., 2010, 122, 558 CrossRef.
- A. Huang, Q. Liu, N. Wang, Y. Zhu and J. Caro, J. Am. Chem. Soc., 2014, 136, 14686 CrossRef CAS PubMed.
- Y. Hu, J. Wei, Y. Liang, H. Zhang, X. Zhang, W. Shen and H. Wang, Angew. Chem., Int. Ed., 2016, 55, 2048 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c7ee02865b |
| This journal is © The Royal Society of Chemistry 2018 |