
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanisms of the polyol reduction of copper(II) salts depending on the anion type and diol chain length†
Johannes
Teichert
a,
Thomas
Doert
 a and
Michael
Ruck
*ab
a and
Michael
Ruck
*ab
aFaculty of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany. E-mail: michael.ruck@tu-dresden.de
bMax Planck Institute for Chemical Physics of Solids, Nöthnitzer Str. 40, 01187 Dresden, Germany
First published on 18th September 2018
Abstract
The mechanisms of the polyol reduction of copper(II) compounds were investigated by various systematic experiments. The course of the reduction in ethylene glycol strongly depends on the anion present in solution. Elemental copper can only be obtained in high yields starting from Cu(NO3)2, Cu(OAc)2, or Cu(OH)2; but not from CuCl2 or CuSO4. Intermediate compounds were observed, namely Cu2O, and the alkoxide compounds Cu(C2H4O2) and Cu3(OAc)2(C2H4O2)2. Cu3(OAc)2(C2H4O2)2 was characterised by single-crystal X-ray diffraction. It is a 2D coordination polymer with bridging bidentate acetate and bidentate deprotonated EG ligands leading to vertex- and edge-linked CuO5 polyhedra. Alkoxide compounds were also detected for higher glycol ethers. With growing glycol ether chain length the stability of alkoxide intermediates decreases, enhancing the stability range of Cu2O. Because the majority of copper species is precipitated as copper(II) alkoxide compounds or Cu2O before the reduction to elemental copper occurs, only few nuclei are formed in solution leading to microparticles instead of nanoparticles. Much smaller but strongly agglomerated particles were observed under alkaline reaction conditions.
Introduction
Polyalcohols (polyols) can act as mild reducing agents for many metal cations, yielding metallic micro- or nanoparticles from easily available metal salts. This so-called polyol process has been known for over three decades and was successfully applied to produce a variety of mono- and multimetallic materials.1–3 In these reactions, the polyol normally acts as both solvent and reducing agent. Moreover, as a surface-active substance, it can also attach to the formed particles and affect the product's particle size and morphology.4 Polyols such as ethylene glycol (EG) have good chelating properties for metal cations, which compensate the lower polarity compared to water, and therefore the solvation behaviour of metal salts in water and polyols is quite similar.4 Applying polyols as high-boiling reaction media allows synthesis temperatures up to 320 °C facilitating the formation of crystalline nanoparticles without post-treatment.4The redox potential of a polyol decreases with rising temperature.5,6 While noble metal ions are reduced at comparatively low temperatures, even at room temperature in the case of Pd2+,7 the reduction of less noble metals cations such as Co2+, Ni2+, or Cu2+ requires higher temperatures. For a given temperature the redox potential increases with increasing chain length of the polyol.8 At higher temperature the chelating properties of polyols have to be considered when planning a reduction reaction. Coordination compounds with polyol ligands that might represent intermediate species in the polyol process have been crystallised for several of the less-noble metals. During the reduction of Co(OH)2 or Ni(OH)2 in EG a solid phase precipitates before the reduction to the respective metals occurs.9 This solid is in equilibrium with solvated M2+ ions and is regarded as a metal reservoir governing the nucleation and growth of the final particles. Systematic reduction experiments of cobalt(II) salts in EG revealed the importance of the type of anion and the deprotonation of the polyol for the progress of the reduction reaction.10,11
Like cobalt and nickel, copper particles can be obtained in polyols at elevated temperature. Copper and copper oxide nanomaterials, as cheap alternatives to precious metals, have found wide attention with respect to their potential application in catalysis, photovoltaics, and optics in the last years.12 As copper(I) is fairly stable, its emergence in the course of the reaction is not unexpected: Cu2O precipitates, e.g., as an intermediate when CuO13, or CuSO4 and 3 eq. of NaOH14 are heated in EG. Increasing the temperature leads to the formation of the final Cu particles. A solid Cu–EG compound similar to the Co–EG and Ni–EG complexes was described in the literature.15 By heating copper acetate in diethylene glycol (DEG) a Cu–DEG compound was obtained.16 However, these compounds were not structurally characterised.
The knowledge about copper(II) species and their coordination modes in polyols is fragmentary. It was shown that the Jahn–Teller distorted tris-chelated complex [Cu(EG)3]2+ is formed in a solution of Cu(CF3SO3)2 in EG.17 Chloride anions have a higher coordinating affinity towards copper(II) than triflate anions, which is consistent with DFT calculations indicating the formation of the complex [CuCl2(EG)2] instead of [Cu(EG)3]2+.18 The coordination ability of other anions has not been considered yet.
To establish a general understanding of the coordination and reduction behaviour of copper(II) in polyols, we carried out a series of systematic experiments. Several copper(II) precursor salts were reacted in EG, DEG, triethylene glycol (TrEG), and tetraethylene glycol (TEG) at different temperatures. Solid reaction intermediates were isolated and structurally characterised.
Results and discussion
Reduction of copper(II) in ethylene glycol
The following reduction potentials, which are only valid for aqueous solutions, have to be considered for the reduction of Cu2+ to Cu0.19 Reduction potentials of noble metal cations were found to follow the same order in EG and water.5,6 It is reasonable to assume that the order is also the same for the potentials considered here.| Cu2+ + 2e− → Cu E° = +0.34 V | (1) |
| Cu2+ + e− → Cu+E° = +0.15 V | (2) |
| Cu+ + e− → Cu0E° = +0.52 V | (3) |
| Cu2O + H2O + 2e− → 2Cu0 + 2OH−E° = −0.36 V | (4) |
As reported before,20 the reduction of Cu2+ ions to Cu0 by polyols proceeds not in one step according to (1), but in two. Initially Cu2+ is reduced to Cu+(2). Although Cu+ ions should be unstable and readily react to Cu0 according to the more positive standard potential (3), insoluble Cu2O immediately precipitates from the reaction solution. Cu2O has a more negative reduction potential (4) and gets reduced to Cu0 at a higher temperature.
The reflux temperature which can be reached for a given reaction mixture strongly depends on the water content. Polyols are hygroscopic and therefore contain a small amount of water. In this study the water content was kept low by using freshly supplied polyols. Water was also introduced by the hydrated copper salts that were used as starting materials. When 5 vol% of water were added to reaction mixtures of EG and suitable copper(II) compounds (e.g. acetate and hydroxide), the boiling point dropped to ca. 160 °C and no elemental copper was obtained under reflux conditions. However, microwave-assisted heating of mixtures in a closed vessel to 210 °C led to the formation of elemental copper. Hence, a small quantity of water does not change the general reduction behaviour but limits the boiling temperature. The water content has to be kept low to enable a complete reduction to elemental copper under ordinary reflux conditions.
The reduction of copper(II) precursor salts in EG was monitored at different temperatures. It was observed, that the anion of the copper salt has a major influence on the reduction reaction. The reduction of Cu2+ to Cu0 does not proceed when starting from copper(II) chloride. Reacting CuCl2 in EG for 2 h at 210 °C yielded no metallic copper but a clear green solution. This can be explained by the coordination affinity of chloride ligands towards the Cu2+ ion, which leads to the formation of solution species like [CuCl2(EG)2].18 This comparatively stable complex has a lower reduction potential than [Cu(EG)3]2+, which prevents the reduction of Cu2+ to metallic copper at the given temperature. A decreasing reduction potential with increasing chloride concentration is known from copper chloride complexes in aqueous solution.21 Although elemental copper could not be produced, CuCl was obtained by reacting CuCl2 in DEG at 240 °C and TrEG at 280 °C, respectively. Powder X-ray diffraction (PXRD) showed no other crystalline phases. The light green solution turns dark greenish-brown upon heating, and above 220 °C it abruptly turns into a white suspension of CuCl.
Copper(II) sulfate can be transformed to elemental copper by refluxing in EG at 210 °C. Copper was obtained only in low yields (max. 40%), and a small amount of Cu2O was observed as a byproduct even after refluxing for 60 min (Fig. S1†). Conducting the reaction at 240 °C in DEG did not significantly change the product's purity or yield.
Obviously, CuCl2 and CuSO4 are not well suited for the precipitation of metallic copper in boiling EG without further modifying the process. Syntheses starting from these materials described in the literature always required the addition of 3–4 eq. of hydroxide.14,18
The light blue solution of copper(II) nitrate in EG does not change up to 160 °C. At 180 °C the formation of nitrous gases is observed, and after 20 min a brown suspension occurs. The precipitate isolated by centrifugation mainly consists of Cu2O (Fig. 1). The dispersion is rather stable and difficult to separate completely by centrifugation. This agrees with the previously described colloidal stability of oxide nanoparticles in polyols up to 20 wt%.22 A small reflection in the powder patterns can be assigned to copper(II) oxalate Cu(C2O4)·xH2O (x = 0–1, PDF number 21–297). IR spectra confirm the presence of carboxyl groups (Fig. S2†). The formation of the byproduct is enabled by the oxidation of EG to oxalate under simultaneous reduction of nitrate to nitrous gases according to (5).
| NO3− + 4 H+ + 3e− → NO + 2H2O E° = +0.957 V | (5) |
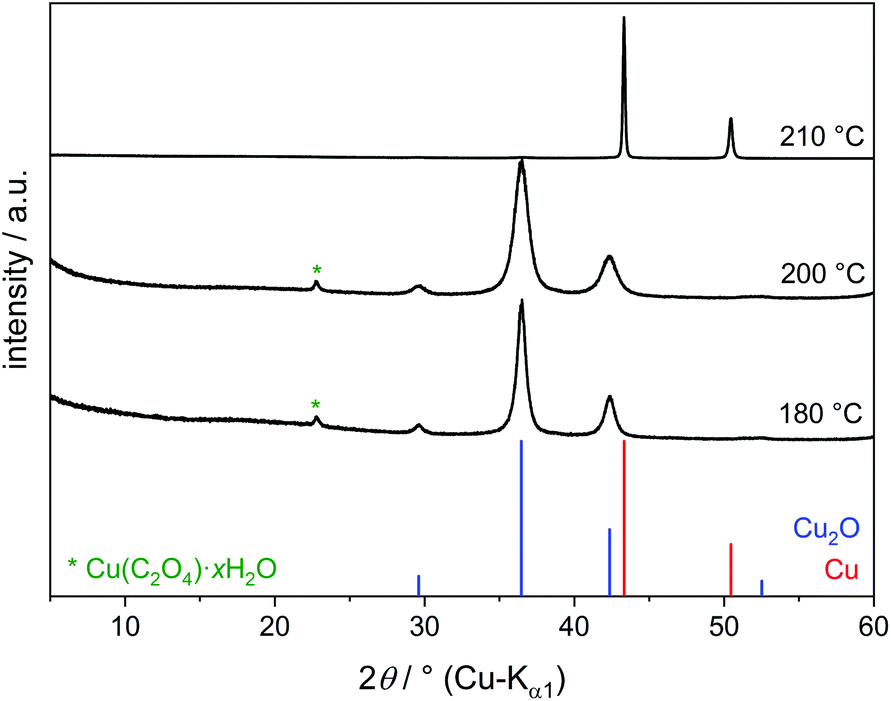 | ||
| Fig. 1 PXRD patterns of reaction products of Cu(NO3)2·2.5H2O in EG at various temperatures. | ||
Under reflux conditions at 210 °C a dark red suspension of pure elemental copper is formed after some minutes. No Cu2O was detected by PXRD (Fig. 1). However, the surface of copper particles is easily oxidised by air if no precautions are taken to prevent this.12 Such a surface layer of copper oxide is likely to be present for all the products in this study since the reactions were carried out under air and without surfactants. The amorphous surface layer is probably only a few nanometres thick. Rietveld refinements of some of the finally obtained copper powders, mixed with a known amount of silicon standard material, showed no notable amorphous content in the products.
Copper(II) hydroxide does not dissolve in EG even upon prolonged heating but transforms into an EG-containing solid. The PXRD patterns of the products obtained at 120–180 °C show primarily one intense broad reflection at 2θ = 10.8° (Fig. 2). A Cu–EG compound with a similar powder pattern was obtained from copper acetate in EG and described as a precursor for the production of nanostructured CuO. However, this compound was not further characterised.15
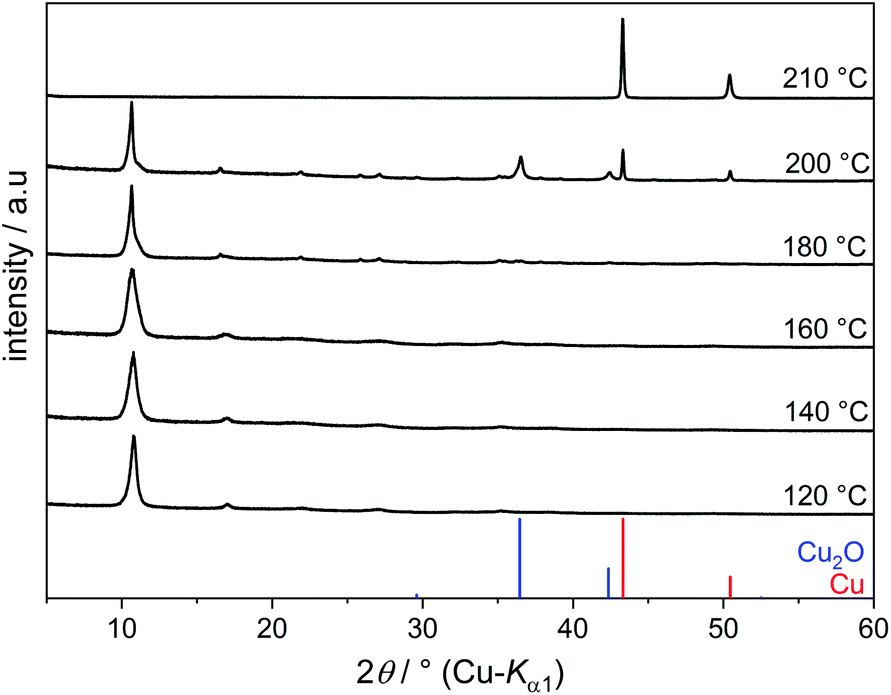 | ||
| Fig. 2 PXRD patterns of reaction products of Cu(OH)2 in EG at various temperatures. | ||
The IR spectrum of this precipitate (Fig. 3) shows some absorption bands similar to those of liquid EG: bands due to CH stretching (2950–2850 cm−1), C–O stretching (1100–1000 cm−1), C–C stretching and CH2 deformation (ca. 900 cm−1) are clearly visible in the spectrum. The absence of the broad OH stretching band at 3600–3000 cm−1 and C–OH deformation bands at 1450–1300 cm−1, on the other hand, indicates fully deprotonated EG molecules. The CH stretching vibrations are split as compared to liquid EG most probably because of a chelating coordination of the EG molecules. In combination with results from elemental analysis (measured: C 18.1 wt%, H 2.9 wt%, calculated: C 19.4 wt%, H 3.3 wt%) and the Cu content determined from the CuO residual after heating in air (determined: Cu 52.3 wt%, calculated: Cu 51.4 wt%), we propose that the solid alkoxide yielded in this study is (ethylene glycolato)-copper(II), Cu(C2H4O2).
 | ||
| Fig. 3 IR spectra of Cu(C2H4O2) and liquid EG. | ||
Hence, copper hydroxide reacts with EG molecules to form the alkoxide upon the release of water:
| Cu(OH)2 + C2H6O2 → Cu(C2H4O2) + 2H2O | (6) |
The powder pattern of the produced alkoxide is very similar to those commonly found for layered hydroxide salts.23 In the crystal structure of Cu(OH)2, strongly Jahn–Teller distorted CuO6 octahedra are edge-linked to corrugated layers.24 These layers are stacked along the b axis with a distance of 530 pm (Fig. 4a). Corrugated layers of edge-linked CuO6 octahedra are also found in copper(II) layered hydroxide salts, e.g. Cu2(OH)3(OAc).25 Assuming a similar structure for Cu(C2H4O2), the d-spacing of the most intense reflection corresponds to an interlayer spacing of 820 pm, probably due to deprotonated EG ligands occupying the interlayer space (Fig. 4b). The EG ligands might show an intralayer–chelating or interlayer–bridging coordination mode or a mixture of both.
 | ||
| Fig. 4 Crystal structure of Cu(OH)2 (a) and a proposed structure model of Cu(C2H4O2) (b). | ||
Similar to Cu(C2H4O2), the solid alkoxides Co(C2H4O2)26 and Ni(C2H4O2)27 have been obtained by heating Co(OAc)2 and Ni(OH)2 in EG, respectively. Both compounds were described as layered brucite-like structures with turbostratic disorder. The interlayer spacing of 830 pm is close to the value for Cu(C2H4O2) determined in this study. The reported PXRD patterns and IR spectra show some similarities to those of Cu(C2H4O2).
According to DTA-TG measurements in air (Fig. S3†), Cu(C2H4O2) is mainly reduced to Cu2O in the first thermal decomposition step at about 200 °C. The oxidation of Cu2O to the final product CuO leads to a mass increase between 220 °C and 400 °C. Cu(C2H4O2) is paramagnetic. The temperature dependence of the inverse susceptibility χ−1(T) (Fig. S4†) can be modelled above 100 K by a Curie–Weiss law with the Curie constant Cmol = 0.350 cm3 K mol−1 and the Curie–Weiss temperature Θ = −61 K, indicating antiferromagnetic coupling. The effective magnetic moment per Cu atom of μeff = 1.67 μB derived from Cmol is in good agreement with the spin-only value of 1.73 expected for Cu2+ ions in a Jahn–Teller distorted octahedral environment. The χ−1(T) curve shows a steady decrease upon lowering the temperature to 2 K which might be an indication of magnetic frustration.
When reacting Cu(OH)2 in EG at 120–180 °C, the main fraction (more than 80%) of Cu2+ precipitates as Cu(C2H4O2) and only a minor fraction of Cu2+ species remains dissolved. At 200 °C, Cu2+ is reduced to Cu2O. The Bragg reflections of Cu2O are broad, indicating small domain sizes. Elemental copper is obtained under reflux conditions at 210 °C.
SEM images (Fig. 5) show drastic changes in the size and morphology of the solids collected from the reaction mixtures. The starting material Cu(OH)2 consists of prolate nanoparticles. The appearance of Cu(C2H4O2) changes between 120 °C and 200 °C from highly agglomerated almost isotropic nanoparticles (Fig. 5b) to facetted crystals up to 3 μm length (Fig. 5c). The cubic Cu2O particles with edge lengths up to 100 nm are well-stabilised in the reaction medium and rather difficult to precipitate completely by centrifugation. Due to the low Cu2+ concentration in solution prior to nucleation, the number of formed nuclei is limited and therefore copper submicron-sized particles (Fig. 5d) rather than nanoparticles are formed. These particles are poorly stabilised in EG and tend to agglomerate rapidly as the reaction time is increased.
 | ||
| Fig. 5 SEM pictures of starting material Cu(OH)2 (a), Cu(C2H4O2) (b), Cu(C2H4O2) and Cu2O (c), and copper particles (d). | ||
Combining the PXRD and IR results the reaction scheme (7) of Cu(OH)2 with EG can be deducted.
| Cu(OH)2 → Cu(C2H4O2) → Cu2O → Cu | (7) |
By adding 2 eq. of NaOH to a suspension of Cu(OH)2 in EG, a clear, dark blue solution is formed. Cu2+ might exist in this solution in the form of square-planar [Cu(OH)4]2− or [Cu(C2H4O2)2]2− complexes. The [Cu(OH)4]2− species exists in alkaline aqueous solutions.28 [Cu(C2H4O2)2]2− ions, which might be more stable in the EG solution because of the chelate effect, are known from crystals formed in alkaline mixtures of Cu2+ and group 1 or 2 metal ions in EG.29–32
A copper alkoxide intermediate precipitates from the dark blue solution by heating to 120 °C. IR spectra and TG curves of this precipitate are very similar to those of Cu(C2H4O2) obtained without NaOH, but the PXRD pattern (Fig. 6) is slightly different. Instead of one dominating reflection at 2θ = 10.8° one pronounced reflection at 10.4° and a less intense one at 11.2° are found, corresponding to interlayer distances of 850 pm and 790 pm, respectively. This suggests a structural variability of Cu(C2H4O2), which might arise from different coordination modes of the EG ligands influencing the interlayer spacing. The yield of Cu(C2H4O2) (ca. 40%) is much lower than with pure Cu(OH)2 as the starting material due to a higher solubility of copper(II) species in the alkaline solution.
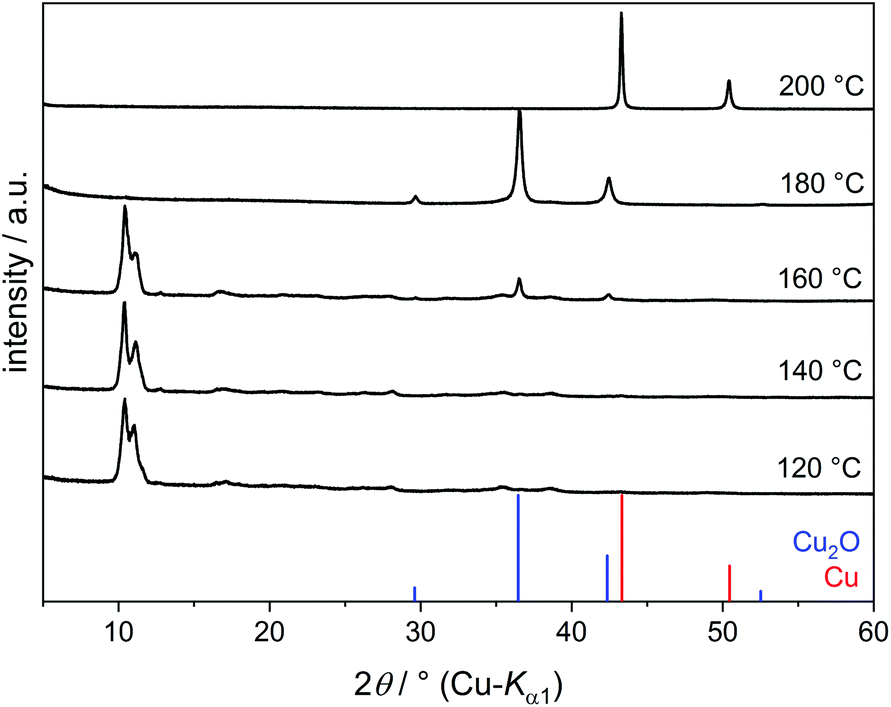 | ||
| Fig. 6 PXRD patterns of reaction products of Cu(OH)2 and 2 eq. of NaOH in EG at various temperatures. | ||
The reduction of [Cu(C2H4O2)2]2− to elemental copper proceeds according to the reaction scheme (7) shown above, but at a significantly lower temperature compared to pure Cu(OH)2. According to PXRD results, Cu2O begins to form at 160 °C and quantitatively transforms to Cu at 200 °C. This could be due to the solubility of the precursor as well as the increased pH value. Hydroxide ions are assumed to initiate the reaction by deprotonation of the α-carbon atom of the (C2H4O2)2− ligand, thus leaving two electrons for the reduction of the metal cation.18
The majority of the finally obtained, severely agglomerated Cu particles (Fig. 7) have a significantly smaller size compared to those obtained from pure Cu(OH)2 due to a higher concentration of Cu2+ in the reaction solution prior to nucleation.
 | ||
| Fig. 7 SEM picture of copper particles obtained from Cu(OH)2 and 2 eq. of NaOH in EG at 200 °C. | ||
Copper(II) acetate completely dissolves in EG at ca. 80 °C yielding a bright turquoise solution. Further heating to 120 °C leads to the precipitation of a dark green crystalline compound (Fig. 8). The IR spectrum of this compound (Fig. 9) shows CH stretching (2950–2850 cm−1), C–O stretching (1100–1000 cm−1), C–C stretching and CH2 deformation (ca. 900 cm−1), but no OH stretching band, indicating fully deprotonated EG groups. Bands in the range 1600–1300 cm−1 are due to COO stretching and CH3 deformation because of the presence of acetate ions.
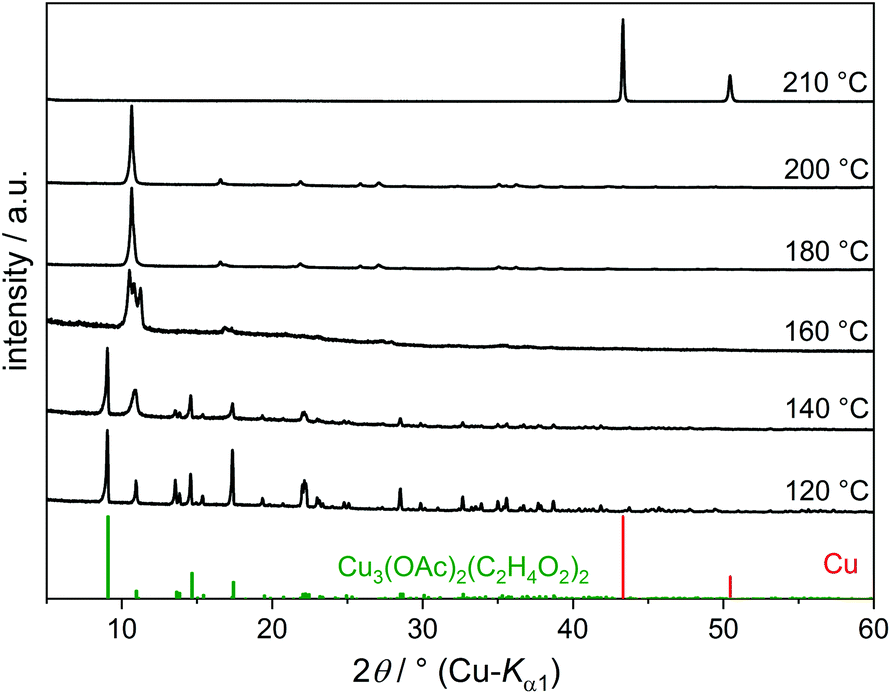 | ||
| Fig. 8 PXRD patterns of reaction products of Cu(OAc)2·H2O in EG obtained at various temperatures. The reference pattern for Cu3(OAc)2(C2H4O2)2 was simulated from the single-crystal structure data. | ||
 | ||
| Fig. 9 IR spectra of reaction products from Cu(OAc)2·H2O in EG obtained at different temperatures. | ||
The structure determination of the obtained product by single-crystal X-ray diffraction revealed the coordination polymer Cu3(OAc)2(C2H4O2)2. It crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and shows a 2D network structure generated by bridging bidentate acetate and deprotonated EG ligands (Fig. 10). The three crystallographically independent Cu atoms have approximately square-pyramidal coordination environments of O atoms. Four O atoms are located in distances of 192–200 pm forming a distorted square-planar array. The apical O atoms are more remote with distances between 224.6 pm and 251.4 pm. Valence-bond sums33 of 2.11 (Cu1) and 2.04 (Cu2, Cu3) unambiguously indicate copper(II). The CuO5 pyramids are linked via their edges and vertices forming bands along [100] that consist of alternating Cu6O6 and Cu4O4 rings. The longest Cu–O distance of 251.4 pm (at Cu2) links the chains to a strongly corrugated layer parallel (001). The layers are stacked along the c axis and not linked via hydrogen bonds.
and shows a 2D network structure generated by bridging bidentate acetate and deprotonated EG ligands (Fig. 10). The three crystallographically independent Cu atoms have approximately square-pyramidal coordination environments of O atoms. Four O atoms are located in distances of 192–200 pm forming a distorted square-planar array. The apical O atoms are more remote with distances between 224.6 pm and 251.4 pm. Valence-bond sums33 of 2.11 (Cu1) and 2.04 (Cu2, Cu3) unambiguously indicate copper(II). The CuO5 pyramids are linked via their edges and vertices forming bands along [100] that consist of alternating Cu6O6 and Cu4O4 rings. The longest Cu–O distance of 251.4 pm (at Cu2) links the chains to a strongly corrugated layer parallel (001). The layers are stacked along the c axis and not linked via hydrogen bonds.
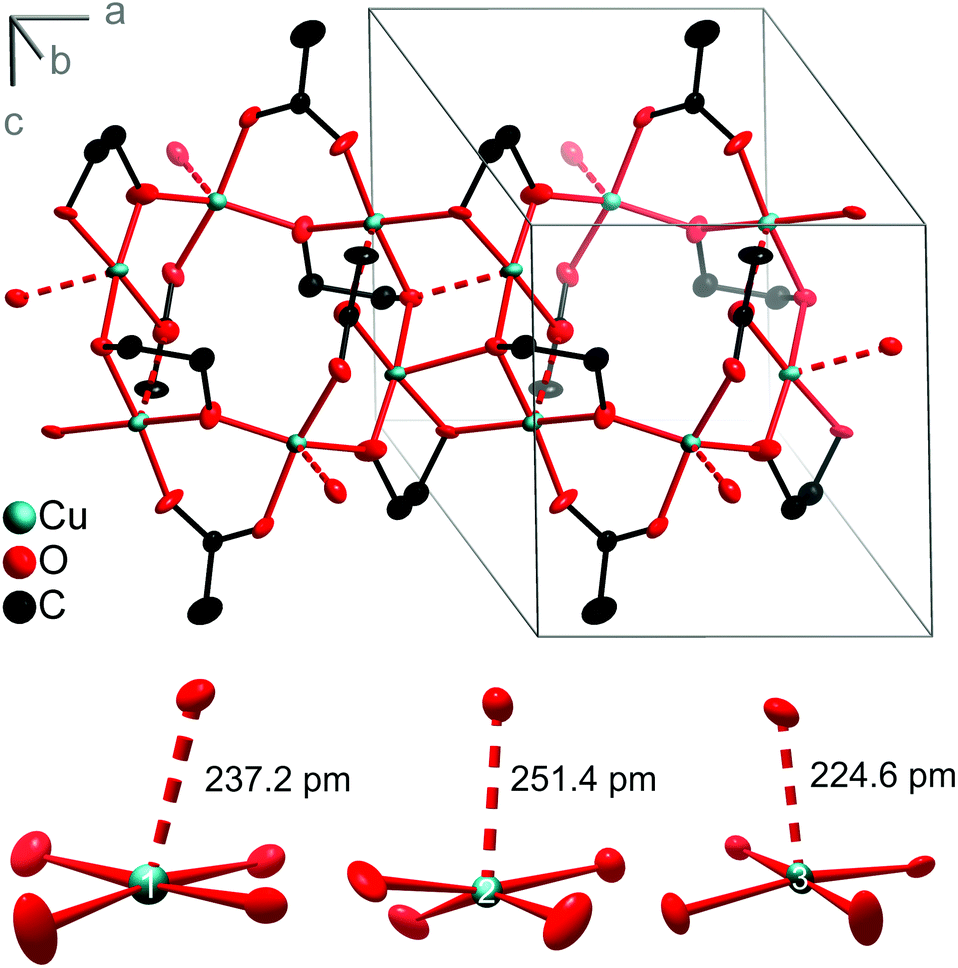 | ||
| Fig. 10 Crystal structure of Cu3(OAc)2(C2H4O2)2 and coordination environment of the three independent Cu atoms. The longest Cu–O distances are represented by dashed lines. Hydrogen atoms are omitted for clarity. Ellipsoids represent a probability density of 90%. | ||
The mixed coordination of metal cations to solely double-deprotonated EG and acetate ligands, which occurs in Cu3(OAc)2(C2H4O2)2, was also reported for the compound Sb(OAc)(C2H4O2).34 Mono-deprotonated EG molecules were found in Zn2(OAc)3(C2H5O2).35 The occurrence of deprotonated EG is rather unusual, especially regarding the fact that no base was added to the reaction mixture. Acetic acid (pKa = 4.75) is a much stronger acid than EG (pKa = 15.1),19 and therefore the deprotonation of EG by acetate anions is an unlikely process. However, the stability of the product compound or the evaporation of acetic acid from the reaction solution may drive the reaction. The yield of the precipitated alkoxide is ca. 50%.
DTA-TG measurements of dry Cu3(OAc)2(C2H4O2)2 in air (Fig. S5†) showed that the compound is stable up to 200 °C and decomposes in an exothermic reaction to Cu2O and then to the final product CuO. Cu3(OAc)2(C2H4O2)2 shows paramagnetic behaviour. The χ−1 plot (Fig. S6†) can be modelled above 200 K by a Curie–Weiss fit. The obtained Curie constant Cmol = 0.412 cm3 K mol−1 corresponds to an effective magnetic moment μeff = 1.81 μB, which is slightly higher than the spin-only value of 1.73 μB expected for Cu2+ ions.
The acetate–alkoxide coordination polymer is not stable at reaction temperatures of 160 °C and higher. Products isolated after 30 min at 180 °C and 200 °C show PXRD patterns (Fig. 8) and IR spectra (Fig. 9) characteristic for the already described compound Cu(C2H4O2). The solid alkoxide is formed in high yields and only a small amount of Cu2+ remains in solution. Elemental copper is obtained under reflux conditions at 210 °C. Cu2O could not be detected after 30 min because of its small stability range in this particular reaction system, which is accompanied by the high stability of Cu(C2H4O2).
By combining the PXRD and IR results, reaction scheme (8) can be deducted for the reduction of copper acetate in EG.
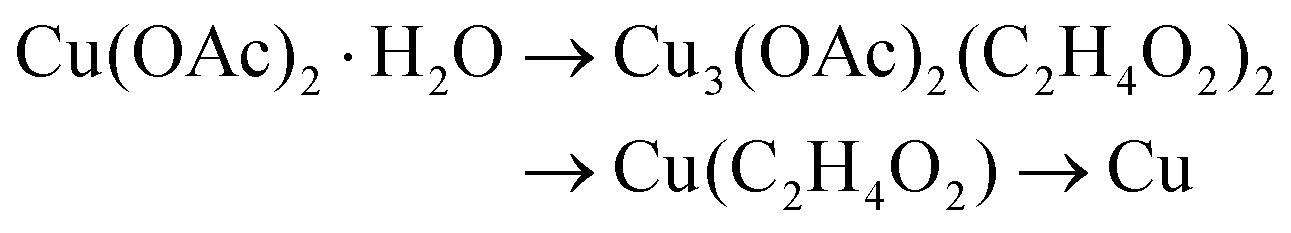 | (8) |
Reduction of copper(II) in higher glycol ethers
Besides EG, the reduction of copper(II) hydroxide and acetate was also monitored in the longer-chain diols DEG, TrEG, and TEG. The resulting solid phases starting from copper hydroxide and acetate are summarised in Tables 1 and 2, respectively.| Polyol | Cu(OH)2 | Alkoxide | Cu2O | Cu |
|---|---|---|---|---|
| EG | 120–200 | 200 | ≥210 | |
| DEG | 120–200 | 180–200 | ≥220 | |
| TrEG | 120 | 120–180 | 180–200 | ≥220 |
| TEG | 120–160 | 140–220 | ≥200 |
| Polyol | Acetate–alkoxide | Alkoxide | Cu2O | Cu |
|---|---|---|---|---|
| EG | 120–140 | 160–200 | ≥210 | |
| DEG | 120 | 140–200 | 200–220 | ≥220 |
| TrEG | 140–240 | ≥240 | ||
| TEG | 120–240 | ≥240 |
On reacting Cu(OH)2 with DEG, an alkoxide compound is obtained between 120 °C and 200 °C. PXRD patterns (Fig. 11) show an intense reflection at 2θ = 11.0° and weaker reflections at 16.2° (d = 550 pm) and 32.7° (d = 270 pm). IR spectra indicate the absence of acetate in this compound as they only show bands associated with the DEG molecules. No OH stretching band can be observed, although in some samples there is a broad signal around 3000 cm−1 arising from adsorbed water or DEG molecules. The formula deducted for this alkoxide compound is Cu(C4H8O3) which is in agreement with results from elemental analysis (measured: C 28.7 wt%, H 4.8 wt%, calculated: C 28.7 wt%, H 4.8 wt%) and the Cu content calculated from the CuO residual after heating in air (determined: 36.8 wt%, calculated: 37.9 wt%). The compound described here had already been synthesised and two structural models, containing either mono-deprotonated or a mixture of double-deprotonated and neutral DEG molecules, had been proposed.16 However, no or only very few OH groups might be present in the structure according to our results.
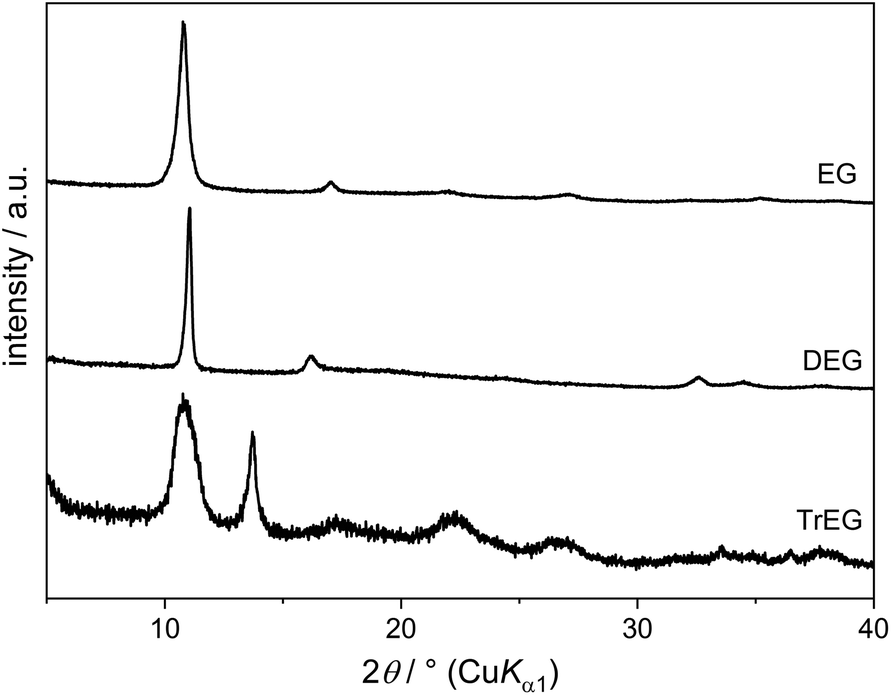 | ||
| Fig. 11 PXRD patterns of alkoxides synthesised from the reaction of Cu(OH)2 with EG, DEG, and TrEG. | ||
The starting compound Cu(OH)2 can still be found in the solid products obtained with TrEG and TEG up to 140 °C and 160 °C, respectively. Similar to EG and DEG, an alkoxide compound is formed with TrEG. The PXRD pattern (Fig. 11) shows intense broad reflections at 2θ = 10.8° (d = 820 pm) and 13.7° (d = 645 pm).
In contrast to the previously discussed polyols TEG does not form an alkoxide compound with Cu2+. Only Cu(OH)2, Cu2O and copper can be detected in the obtained solid by PXRD. IR spectra show weak CH and C–O stretching bands, possibly due to TEG molecules adsorbed to the particle surface.
With copper acetate as precursor material (Table 2), only with EG and DEG alkoxides are formed. Similar to the reaction sequence in EG, an acetate–alkoxide compound is formed when copper acetate is heated in DEG to 120 °C. Both DEG and carboxylate vibration bands are visible in the IR spectrum of the precipitate. The powder diffractogram shows one intense and sharp reflection at 2θ = 9.4° (d = 940 pm). At 140 °C desert-rose shaped particles of Cu(C4H8O3) are obtained (Fig. S11†). Cu(C4H8O3) transforms to Cu2O and finally to copper. No alkoxide compounds could be detected for TrEG and TEG. However, the reduction to Cu+ starts at a much lower temperature and the stability range of Cu2O is significantly enhanced. There is a small amount of a byproduct at 180 °C for TrEG and at 160 °C for TEG (Fig. S8†). IR spectra of the samples show intense carbonyl bands at 1650 cm−1 (Fig. S14†). Elemental copper is produced at higher temperatures when compared to copper acetate as the starting material. This is due to the stronger alkalinity of Cu(OH)2 which leads to deprotonation of EG molecules.
Coordination affinity of anions to copper(II) in ethylene glycol
Experiments including more than one anion in the solution showed some trends from which a general sequence of affinity towards Cu2+ could be deducted. Addition of NaOH to mixtures of CuCl2, CuSO4, Cu(NO3)2, or Cu(OAc)2 in EG first yields a light blue precipitate (2 eq. of NaOH), which subsequently dissolves resulting in a dark blue solution (4 eq. of NaOH). The reaction behaviour of these solutions is very similar to that of Cu(OH)2 or [Cu(OH)4]2−/[Cu(C2H4O2)2]2−. With 4 eq. of NaOH, the complete reduction of the starting material to elemental copper can be achieved at 180 °C. Seemingly, anions other than hydroxide do not affect the reduction. However, with sulfate anions Na2SO4 was formed as byproduct during the reaction (Fig. S7†). Na2SO4 is only sparingly soluble in EG and has to be removed from the solid product by multiple washing cycles.Addition of 2 eq. of NaOAc to solutions of CuCl2, CuSO4, and Cu(NO3)2 produces bright turquoise solutions which show the same reaction behaviour as those of pure Cu(OAc)2. Remarkably, CuCl2 can be reduced to elemental copper at 210 °C (Fig. S7†). Because of its chelating ability, acetate seems to be a stronger ligand for Cu2+ than chloride.
The addition of 2 eq. of NaCl prevents the reduction of Cu(NO3)2 or CuSO4 to elemental copper. Clear solutions without any solid products were obtained after 30 min at 210 °C. Chloride acts as a Lewis base and directly binds to Cu2+, probably forming the solution species [CuCl2(EG)2], which leads to the same reaction behaviour as for CuCl2.
Based on these results, we propose a sequence of decreasing affinity towards Cu2+ ions in EG solutions: hydroxide > acetate > chloride > nitrate/sulfate.
Conclusions
Systematic experiments were carried out to study the anion-dependent course of the reduction of copper(II) salts in polyols. Elemental copper can only be obtained in high yields starting from Cu(NO3)2, Cu(OAc)2, or Cu(OH)2; but not with CuCl2 or CuSO4. With EG, the intermediate compounds Cu3(OAc)2(C2H4O2)2, Cu(C2H4O2), and Cu2O were observed during the reaction. Experiments with higher glycol ethers show that increasing the polyol chain length decreases the stability of alkoxides und therefore enhances the stability range of Cu2O. In the case of TEG, no alkoxide compound but phase-pure Cu2O was detected in a very wide temperature range. The formation of Cu2O or alkoxide compounds reduces the Cu2+ concentration in the reaction solution and therefore leads to a small number of nuclei resulting in micro- rather than nano-sized copper particles. Under alkaline reaction conditions, e.g. by addition of NaOH, the lowest reduction temperature and the smallest particle sizes are achieved due to a higher Cu2+ concentration prior to nucleation.This study shows, that the anion-dependent reduction behaviour of copper in polyols is similar to the less noble metals cobalt and nickel, although the occurrence of the stable oxidation state +I is a peculiarity of the copper system. Less noble metals should be treated under alkaline conditions to ensure a complete reduction at the lowest possible temperature.
Experimental
Chemicals
Freshly supplied polyols were used as received: ethylene glycol (Fluka, 99.5%), diethylene glycol (Aldrich, 99%), triethylene glycol (Alfa Aesar, 99%), and tetraethylene glycol (Acros, 99.5%). Hydrated copper(II) salts were used for all syntheses: CuCl2·2H2O (Aldrich, 99%), CuSO4·5H2O (Aldrich, 99%), Cu(NO3)2·2.5H2O (ABCR, 99.5%), Cu(OAc)2·H2O (Aldrich, 99%). Cu(OH)2 was prepared by precipitation of an aqueous solution of CuSO4·5H2O with 2 eq. of NaOH. The light blue powder was filtrated, washed several times with water and ethanol, and dried under vacuum at ambient temperature overnight. For some experiments, solutions of NaOH (Grüssing, 99%), NaOAc·3H2O (Grüssing, 99.5%), or NaCl (VWR Prolabo, 99.9%) in EG with a concentration of 1 mol l−1 were used.Synthesis
For each experiment, the respective copper(II) compound (0.5 mmol) was mixed with 5 ml of the polyol in a flask equipped with a magnetic stirring bar and a reflux condenser. In some experiments a specified amount of NaOH, NaOAc·3H2O, or NaCl solution was added, maintaining the total volume of 5 ml. Heating was performed with a CEM Discover-SP microwave device under open vessel conditions. The reaction temperature was controlled with the in-built infrared sensor. The mixture was heated to the desired temperature in 3 min, annealed for 30 min, and rapidly cooled down to 60 °C with compressed air. The solid products were collected by centrifugation, repeatedly washed with ethanol, and finally dried under vacuum at ambient temperature.Powder characterisation
Powder X-ray diffraction (PXRD) patterns were collected with a transmission-geometry diffractometer Stadi P (Stoe) equipped with a Mythen 1K detector (Dectris) using Ge(111)-monochromated Cu-Kα1 radiation (λ = 154.056 pm). The powder samples were placed between two layers of adhesive tape. Crystallographic data from the Inorganic Crystal Structure Database was used to calculate reference powder patterns for Cu (CSD 627114), Cu2O (CSD 180846) and Cu(OH)2 (CSD 68459). Fourier-transform infrared (FT-IR) spectra were measured with a Vertex 80 (Bruker) spectrometer in attenuated total reflectance mode in the wavelength range 4000–500 cm−1. Scanning electron microscopy (SEM) was performed using a SU8020 electron microscope (Hitachi) equipped with a triple-detector system for secondary and low-energy backscattered electrons with an acceleration voltage of 3 kV. DTA-TG curves were recorded with a STA 409 thermal analyser (Netzsch). The samples were heated under air in an alumina crucible with a rate of 5 K min−1 up to 600 °C. Elemental analysis was carried out with a Vario MICRO cube CHNS analyser (Elementar). Magnetisation measurements were conducted with a Helium-cooled vibrating sample magnetometer (Cryogenic) in DC mode in the range 2–300 K.Single-crystal X-ray crystallography
A suitable single crystal of Cu3(OAc)2(C2H4O2)2 was mounted on the tip of a glass fibre and cooled to 100 K under flowing nitrogen gas. Intensity data were collected with a four-circle diffractometer Kappa Apex2 (Bruker) equipped with a CCD-detector using graphite-monochromated Mo-Kα radiation (λ = 71.073 pm). Data were corrected for Lorentz and polarisation factors,36 and multi-scan absorption correction37 was applied. The structure was solved using SHELXT intrinsic phasing38 and refined with full-matrix least-squares based on Fo2 with SHELXL39 using the OLEX240 program package. Refinement included anisotropic displacement parameters for all non-hydrogen atoms. Hydrogen atoms were refined with riding coordinates and displacement parameters. Graphics were developed with Diamond.41 The crystal structure data have been deposited at the Cambridge Crystallographic Data Centre, deposit number 1857785.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Philipp Lange for elemental analysis, Dr Ilka Kuhnert for DTA-TG measurements, Dr Martin Kaiser for assistance with the single-crystal measurement, and Dr Jens Hunger for measuring magnetic data and helpful discussions. Open Access funding provided by the Max Planck Society.Notes and references
- M. Figlarz, F. Fiévet and J.-P. Lagier, Process for the Reduction of Metallic Compounds by Polyols, and Metallic Powders Obtained by This Process, US4539041A, 1985 Search PubMed.
- F. Fiévet, J. P. Lagier, B. Blin, B. Beaudoin and M. Figlarz, Solid State Ionics, 1989, 32–33, 198–205 CrossRef.
- F. Fiévet, J.-P. Lagier and M. Figlarz, MRS Bull., 1989, 14, 29–34 CrossRef.
- H. Dong, Y.-C. Chen and C. Feldmann, Green Chem., 2015, 17, 4107–4132 RSC.
- F. Bonet, C. Guéry, D. Guyomard, R. Herrera Urbina, K. Tekaia-Elhsissen and J.-M. Tarascon, Int. J. Inorg. Mater., 1999, 1, 47–51 CrossRef.
- F. Bonet, C. Guéry, D. Guyomard, R. Herrera Urbina, K. Tekaia-Elhsissen and J.-M. Tarascon, Solid State Ionics, 1999, 126, 337–348 CrossRef.
- M. Heise, J.-H. Chang, R. Schönemann, T. Herrmannsdörfer, J. Wosnitza and M. Ruck, Chem. Mater., 2014, 26, 5640–5646 CrossRef.
- A. J. Biacchi and R. E. Schaak, ACS Nano, 2011, 5, 8089–8099 CrossRef PubMed.
- G. Viau, F. Fiévet-Vincent and F. Fiévet, Solid State Ionics, 1996, 84, 259–270 CrossRef.
- T. Matsumoto, K. Takahashi, K. Kitagishi, K. Shinoda, J. L. C. Huaman, J.-Y. Piquemal and B. Jeyadevan, New J. Chem., 2015, 39, 5008–5018 RSC.
- K. Takahashi, S. Yokoyama, T. Matsumoto, J. L. C. Huaman, H. Kaneko, J.-Y. Piquemal, H. Miyamura and J. Balachandran, New J. Chem., 2016, 40, 8632–8642 RSC.
- P. Lignier, R. Bellabarba and R. P. Tooze, Chem. Soc. Rev., 2012, 41, 1708–1720 RSC.
- F. Fiévet, F. Fiévet-Vincent, J.-P. Lagier, B. Dumont and M. Figlarz, J. Mater. Chem., 1993, 3, 627–632 RSC.
- J. Sun, Y. Jing, Y. Jia, M. Tillard and C. Belin, Mater. Lett., 2005, 59, 3933–3936 CrossRef.
- A. Cao, J. D. Monnell, C. Matranga, J. Wu, L. Cao and D. Gao, J. Phys. Chem. C, 2007, 111, 18624–18628 CrossRef.
- A. Anžlovar, Z. C. Orel and M. Žigon, J. Nanosci. Nanotechnol., 2008, 8, 3516–3525 CrossRef.
- S. E. Okan and P. S. Salmon, Mol. Phys., 1995, 85, 981–998 CrossRef.
- K. J. Carroll, J. U. Reveles, M. D. Shultz, S. N. Khanna and E. E. Carpenter, J. Phys. Chem. C, 2011, 115, 2656–2664 CrossRef.
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press, Boca Raton, Florida, USA, 2016 Search PubMed.
- Z. C. Orel, E. Matijević and D. V. Goia, J. Mater. Res., 2003, 18, 1017–1022 CrossRef.
- Y. Meng and A. J. Bard, Anal. Chem., 2015, 87, 3498–3504 CrossRef PubMed.
- C. Feldmann and H.-O. Jungk, Angew. Chem., Int. Ed., 2001, 40, 359–362 CrossRef PubMed.
- G. G. C. Arizaga, K. G. Satyanarayana and F. Wypych, Solid State Ionics, 2007, 178, 1143–1162 CrossRef.
- H. R. Oswald, A. Reller, H. W. Schmalle and E. Dubler, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 2279–2284 CrossRef.
- S. Švarcová, M. Klementová, P. Bezdička, W. Łasocha, M. Dušek and D. Hradil, Cryst. Res. Technol., 2011, 46, 1051–1057 CrossRef.
- N. Chakroune, G. Viau, S. Ammar, N. Jouini, P. Gredin, M. J. Vaulay and F. Fiévet, New J. Chem., 2005, 29, 355–361 RSC.
- K. Tekaia-Elhsissen, A. Delahaye-Vidal, G. Nowogrocki and M. Figlarz, C. R. Acad. Sci., Ser. II, 1989, 309, 469–472 Search PubMed.
- Y.-Y. H. Chao and D. R. Kearns, J. Phys. Chem., 1977, 81, 666–668 CrossRef.
- N. Habermann, G. Jung, M. Klaassen and P. Klüfers, Chem. Ber., 1992, 125, 809–814 CrossRef.
- C. P. Love, C. C. Torardi and C. J. Page, Inorg. Chem., 1992, 31, 1784–1788 CrossRef.
- A. R. Pico, C. S. Houk, T. J. R. Weakley and C. J. Page, Inorg. Chim. Acta, 1997, 258, 155–160 CrossRef.
- J. H. Rivers, K. J. Carroll, R. A. Jones and E. E. Carpenter, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, m83–m85 CrossRef PubMed.
- O. C. Gagné and F. C. Hawthorne, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 562–578 CrossRef PubMed.
- S. M. Biros, B. M. Bridgewater, A. Villeges-Estrada, J. M. Tanski and G. Parkin, Inorg. Chem., 2002, 41, 4051–4057 CrossRef PubMed.
- L. Poul, N. Jouini and F. Fiévet, Z. Kristallogr., 1998, 213, 416–418 Search PubMed.
- SAINT+: Area-Detector Integration Software, Version 8.34A, Bruker AXS Inc., Madison, Wisconsin, USA, 2013 Search PubMed.
- SADABS: Area-Detector Absorption Correction, Version 2014/4, Bruker AXS Inc., Madison, Wisconsin, USA, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- K. Brandenburg, Diamond 4: Crystal and Molecular Structure Visualization, Crystal Impact GbR, Bonn, Germany, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns and IR spectra of the solid reaction products, DTA-TG measurements and magnetic susceptibility data of alkoxide compounds. CCDC 1857785. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt03034k |
| This journal is © The Royal Society of Chemistry 2018 |