
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOuterly functionalized and non-functionalized boron clusters intercalated into layered hydroxides with different modes of binding: materials for superacid storage
Klára
Melánová
a,
Josef
Holub
b,
Jan
Hynek
b,
Jindřich
Fanfrlík
 c,
Ludvík
Beneš
d,
Petr
Kutálek
d,
Anna
Krejčová
d,
Drahomír
Hnyk
*b and
Vítězslav
Zima
*a
c,
Ludvík
Beneš
d,
Petr
Kutálek
d,
Anna
Krejčová
d,
Drahomír
Hnyk
*b and
Vítězslav
Zima
*a
aInstitute of Macromolecular Chemistry of the Czech Academy of Sciences, Heyrovského nám. 2, 162 06 Prague 6, Czech Republic. E-mail: vitezslav.zima@upce.cz
bInstitute of Inorganic Chemistry of the Czech Academy of Sciences, 250 68 Husinec – Řež, Czech Republic. E-mail: hnyk@iic.cas.cz
cInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, 166 10 Prague 6, Czech Republic
dFaculty of Chemical Technology, University of Pardubice, Studentská 95, 532 10 Pardubice, Czech Republic
First published on 7th August 2018
Abstract
Two binary boron hydrides (NH4)2B10H10 and Na2B12H12 and mono- and dicarboxy p- and m-carboranes (namely, 1-(COOH)-closo-1,7-C2B10H11, 1,12-(COOH)2-closo-1,12-C2B10H10 and 1,7-(COOH)2-closo-1,7-C2B10H10) were intercalated into ZnAl-layered double hydroxides (ZnAl-LDH) and into Zn5(OH)8(NO3)2·2H2O. The formed compounds were characterized using elemental analysis, thermogravimetry analysis, X-ray powder diffraction, infrared spectroscopy and solid state NMR. All the intercalated boron compounds are present in the interlayer space of the layered hosts as anions. It is presumed that in the case of B10H102−, B12H122− and 1,12-(COO)2-closo-1,12-C2B10H102−, the guest molecules form a monolayer, whereas in the case of 1-(COO)-closo-1,7-C2B10H111− and 1,7-(COO)2-closo-1,7-C2B10H102− a bilayer arrangement is more probable. In the case of 1,7-(COO)2-closo-1,7-C2B10H102−, the guest molecules are strongly interdigitated resulting in lowering of the interlayer distance. Two different modes of binding were found. Whereas the carboxylate derivatives of p- and m-carboranes are bonded through classical hydrogen bonds, the corresponding parent borane anions interact with the host structures by mainly dihydrogen bonding. In effect, both kinds of hydrogen bonding are mainly of an electrostatic nature. The dihydrogen bond is detected, e.g. in crystal engineering, and represents a driving force for interactions of boranes with biomolecules. Since the latter dicarboxylic acids were found to be superacids, their interactions with the host structures should be stronger than in the case of the benzoic and terephthalic acid intercalates.
Introduction
Layered metal hydroxides are compounds that are interesting due to their easy synthesis and wide possible applications. Besides layered single hydroxides, wide attention is paid to layered hydroxides containing two metals in different valence states, called layered double hydroxides (LDH). The most common group of LDH may be represented by a general formula: [MII(1−x)MIIIx(OH)2][x/nAn−]·mH2O, where MII (typically Mg, Zn, Ni, and Co) and MIII (Al, Cr, or Fe) are divalent and trivalent cations, respectively, and An− are exchangeable anions of charge n, which compensates the positive charge induced by the presence of MIII in the layers. The anions are accommodated in the interlayer region, most often together with water molecules. In many cases, these anions can be exchanged for other negatively charged species, a feature that is used for the incorporation (intercalation) of functional organic or organometallic species with anionic groups (carboxylate, phosphonate, and sulfonate) into these layered hosts.1With regard to the intercalation of boron containing compounds, many papers reporting the interactions of boric acid or borates with layered double hydroxides have been published up to now. LDH or calcined LDH is used for removing boric acid or borate anions from water;2,3 LDH intercalated with borate anions were used as flame retardants,4,5 and as catalysts for the Beckmann rearrangement of cyclohexanone oxime.6–8 Less attention has been paid to the intercalation of more complicated boron containing compounds. A tetra-(8-hydroxyquinolinato) boron complex, Na[B(C9H6ON)4], was intercalated into MgAl-LDH;9 the intercalate exhibits an enhanced solid-state blue luminescence due to a more rigid and constrained environment of the host. The simplest benzoxaborole (C7H6BO(OH)) and its fluorinated analogue (C7H5FBO(OH)), a recently developed antifungal drug called tavaborole, were intercalated into MgAl-LDH and the properties of these inorganic–organic hybrids, which could be useful for the drug-delivery applications of these materials, were determined.10 The study of the intercalation of the so-called carborane† derivatives into LDH and the use of the prepared intercalates in boron neutron capture therapy has been described in the patent by Choy et al.11 The intercalates of o-carborane-1,2-dicarboxylic acid, i.e. 1,2-(COOH)2-closo-1,2-C2B10H10, p-carborane-1,12-dicarboxylic acid, i.e. 1,12-(COOH)2-closo-1,12-C2B10H10, and the mercaptoundecahydro-dodecaborate anion, i.e. 1-SH-closo-B12H112−, with ZnAl-LDH were prepared by coprecipitation and characterized using X-ray powder diffraction and infrared spectroscopy, but the composition of the intercalates was not given. Cytotoxicity and an effect of these intercalates on cancer cells after neutron irradiation was also described. Recently, Ay et al.12 have reported the intercalations of other carborane derivatives into MgAl-LDH. In the case of p-carborane-1,12-dicarboxylic acid (see above), 1-methyl-o-carborane-2-carboxylic acid, i.e. 1-CH3-2-COOH-closo-1,2-C2B10H10, and 1-mercapto-o-carborane, i.e. 1-SH-closo-1,2-C2B10H11, they observed an increase of the basal spacing indicating an insertion of the guest species into the interlayer space of the LDH host, but the content of the guests in the intercalate is rather low. In the case of 1-phenyl-o-carborane-2-carboxylic acid, i.e. 1-C6H5-2-COOH-closo-1,2-C2B10H10, the basal spacing after the reaction was nearly the same as in the case of the parent MgAl-LDH, which suggests that the guest was not intercalated.
In the presented work, a set of selected boranes and carboranes (see Fig. 1) were intercalated into zinc–aluminum layered double hydroxide and into layered zinc hydroxide nitrate. As the guests we chose mono- and dicarboxylic acids based on m-carborane, the derivatives of which were not considered in the above-mentioned studies. In addition, we describe intercalates with anions of two parent boron hydrides, which do not contain a carboxylic functional group. For them we also suggested their arrangement in the interlayer space of the hosts and their interactions with the host layers on the basis of computing the mode of hydroxide⋯boron hydride interactions.
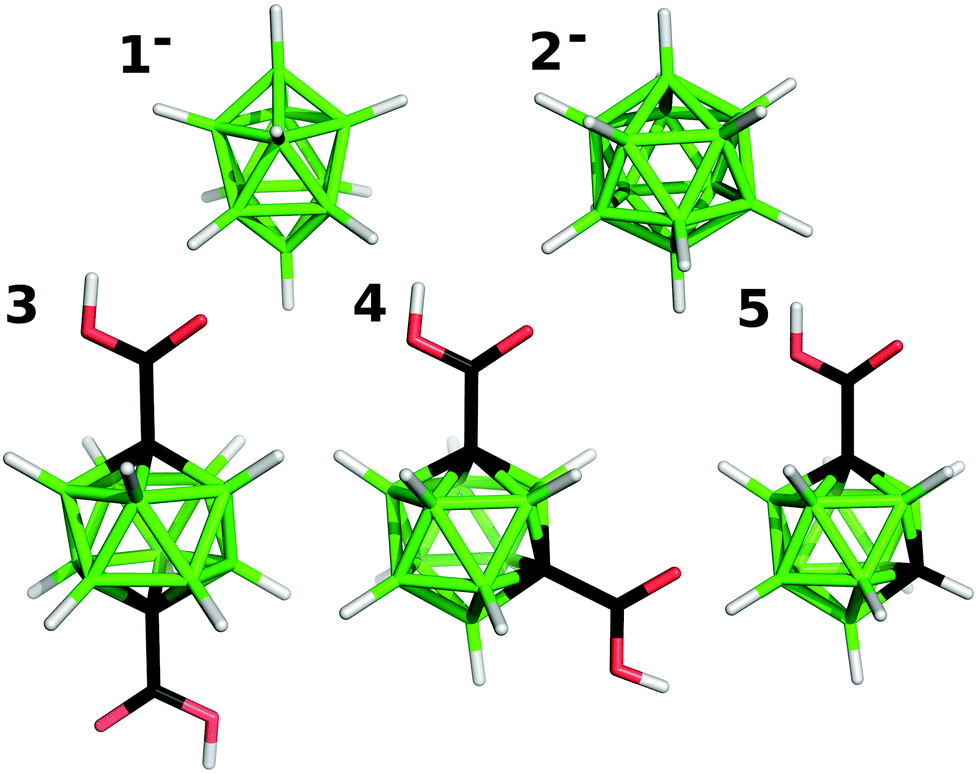 | ||
| Fig. 1 Molecular diagrams of 1−, 2−, 3, 4, and 5. | ||
Experimental section
Preparations of boranes and carboranes
Samples of (NH4)2B10H10 and Na2B12H12 were purchased from Katchem Ltd, whereas 1,12-(COOH)2-closo-1,12-C2B10H10, 1-(COOH)-closo-1,7-C2B10H11 and 1,7-(COOH)2-closo-1,7-C2B10H10 were prepared using previously described procedures.13 Their purity was checked using 11B NMR spectroscopy, and the measured data were found to be consistent with the published chemical shifts reported for both the binary boron hydrides14 and for all the carborane carboxylic acids.13Preparations of layered host materials
Layered double hydroxides with the formulae [Zn0.67Al0.33(OH)2](CO3)0.165·0.5H2O (denoted further as ZnAl-CO3) and [Zn0.66Al0.34(OH)2](NO3)0.34·0.5H2O (denoted further as ZnAl-NO3) were prepared using a urea method.15,16 Zn5(OH)8(NO3)2·2H2O was prepared by precipitation from a mixture of zinc nitrate and sodium hydroxide solutions.17Intercalations with ZnAl-NO3
The intercalates of closo-B10H102− (1−, Fig. 1) or closo-B12H122− (2−, Fig. 1) anions were prepared by the reaction of ZnAl-NO3 (0.1 g, 0.29 mmol) with solutions containing 0.14 mmol of (NH4)2B10H10 or Na2B12H12, respectively, in 25 mL of CO2-free distilled water. The reaction mixture was shaken for five days, and the solid was separated with centrifugation, washed with CO2-free distilled water and ethanol and dried in air.Intercalations with ZnAl-CO3
The intercalates of 1,12-(COOH)2-closo-1,12-C2B10H10 (3, Fig. 1) and 1,7-(COOH)2-closo-1,7-C2B10H10 (4, Fig. 1) were prepared by refluxing ZnAl-CO3 (0.1 g, 0.32 mmol) overnight in a solution of the corresponding acid (56 mg, 0.24 mmol) and dissolved in a mixture of water and ethanol (25 + 5 mL), and the carboxylic groups of the intercalated boranes were partially deprotonated with NaOH (0.16 mmol). The intercalate of 1-(HOOC)-closo-1,7-C2B10H11 (5, Fig. 1) was prepared by refluxing ZnAl-CO3 (0.1 g, 0.32 mmol) overnight in a solution of the acid (94 mg, 0.50 mmol) and partially neutralized with NaOH (0.32 mmol) in a mixture of water and ethanol (25 + 5 mL).Intercalations with Zn5(OH)8(NO3)2·2H2O
The intercalates of Zn5(OH)8(NO3)2·2H2O were prepared by shaking the host (0.1 g, 0.16 mmol) with an aqueous solution of 3 (20 mg, 0.08 mmol) or 5 (40 mg, 0.21 mmol) and neutralized with NaOH for ten days at room temperature. The solid products were separated by centrifugation and washed with ethanol and dried in air.Elemental analysis
The elemental analysis of zinc, boron and aluminum was carried out with a sequential, radially viewed ICP (Inductively Coupled Plasma) atomic emission spectrometer INTEGRA XL 2 (GBC, Dandenong, Australia), equipped with a concentric nebulizer and a glass cyclonic spray chamber (both from Glass Expansion, Australia). The contents of aluminum and zinc were confirmed using an energy-dispersive X-ray microanalyser, IXRF Systems, (detector GRESHAM Sirius 10), mounted on an electron scanning microscope, JEOL JSM-5500LV. The accelerating voltage of the primary electron beam was 20 kV.X-ray diffraction analysis
X-ray powder diffraction data were obtained with a D8 Advance diffractometer (Bruker AXE, Germany) with Bragg–Brentano θ–θ geometry (40 kV, 30 mA) using CuKα radiation with a secondary graphite monochromator. The diffraction angles were measured at room temperature from 2 to 65° (2θ) in 0.02° steps with a counting time of 10 s per step.Thermogravimetric analysis
Thermogravimetric analysis was performed using a home-made apparatus constructed of a computer-controlled oven and a Sartorius BP 210 S balance. The measurements were carried out in air between 30 and 960 °C at a heating rate of 5 °C min−1.Infrared spectroscopy
Infrared spectra in the range of 400–4000 cm−1 were recorded at 64 scans per spectrum at 2 cm−1 resolution using a fully computerized Thermo Nicolet NEXUS 870 FTIR spectrometer equipped with a DTGS TEC detector. Measurements of the powdered samples were performed ex situ in the transmission mode in KBr pellets. All spectra were corrected for the presence of moisture and carbon dioxide in the optical path.NMR
Both liquid phase (11B NMR) and solid-state 11B NMR spectra (ssNMR) for all the individual boron clusters and intercalates, respectively, were recorded on a JEOL 600 MHz NMR spectrometer. The solid-state spectra were recorded at 20 kHz MAS rate with 5 s relaxation delay. The liquid phase spectra were recorded in D2O (binary boron hydrides) and CD3CN (carborane carboxylic acids).Computational details
The electrostatic potentials of isolated molecules were computed at the HF/cc-pVDZ level using the Gaussian0918 and Molekel4.319,20 programs. As a one-electron property, electrostatic potential is correctly described by non-correlated HF wavefunctions. It has recently been shown that the double-zeta basis set size is sufficient for such purposes.21 The studied systems are quite unusual, which prevents the use of force-field based methods. On the other hand, the studied complexes are too big in reality for expensive quantum chemical computations. In order to reduce the problem to manageable dimensions, we opted for density functional theory with a small basis set and empirical dispersion correction, namely the DFT-D3/BLYP/DZVP method.22 The optimizations and energy calculations were performed using the Turbomole 6.623 and Cuby424 programs. Whereas Turbomole 6.6 was used to compute the DFT/BLYP/DZVP energy and gradients for the DFT-D3/BLYP/DZVP method, Cuby4 was used for computing both the D3 dispersion correction and geometry optimizations. The water environment was described using a COSMO implicit solvent model. The binding “free” energy (G′) is calculated as the sum of interaction energy (E) and change of solvation free energy (G) described by the implicit solvent model. Extremely demanding frequency computations were not carried out since the possible imaginary frequencies could be misleading for such a large and flexible system.Results and discussion
Choy et al.11 and Ay et al.12 prepared LDH intercalates with boranes by a coprecipitation from metal salts and the corresponding boranes in a sodium hydroxide solution, whereas we prepared the intercalates using an anion exchange method. Although the carbonate ions are strongly held in the interlayer region of the starting ZnAl-CO3, they can be replaced by desired anions used in their acidic forms.25 This reaction is favored by removing the carbonate ions from the solution. The intercalates of ZnAl-LDH with all three carboxylic acids (3, 4 and 5, see Fig. 1) were prepared by heating the ZnAl-CO3 host in a solution of the corresponding acid. If a stoichiometric amount of the acid is used, the intercalation is not complete, and the product contains an unreacted host. If an excess of acid is used, the formation of some unidentified impurities was observed. Pure intercalates were obtained when an excess of acid was neutralized with NaOH before the addition of the host. The anions 1− and 2− were intercalated into ZnAl-NO3 by an exchange of the nitrate anions for the borane anions at room temperature to obtain intercalates denoted as ZnAl-1 and ZnAl-2, respectively. The intercalates of Zn5(OH)8(NO3)2·2H2O with 3 and 5 were prepared by an exchange of the nitrate anions for the corresponding carboxylate anions by shaking the host in a solution of the corresponding acid neutralized with NaOH at room temperature, and the resulting intercalates are denoted as Zn-3 and Zn-5, respectively. The use of a pure acid or a reaction at an elevated temperature leads to a partial decomposition of the host and to products containing ZnO. The attempts to intercalate 1−, 2−, and 4 into Zn5(OH)8(NO3)2·2H2O were not successful.All the intercalates prepared were white crystalline solids. The denotation and composition of the intercalates obtained from ICP, energy-dispersive X-ray analysis, elemental analysis and TGA are given in Table 1. As can be seen from the formulas, all interlayer carbonates in ZnAl-CO3 were replaced by the carboxylate anions of 3, 4, and 5 to obtain ZnAl-3, ZnAl-4, and ZnAl-5 intercalates, which means that the content of the intercalated carborane species is significantly higher than that found in the MgAl-LDH intercalates (for example in Mg0.69Al0.31(OH)2(C4B10H10O4)0.015(NO3)0.295·2.8H2O, where C4B10H10O4 is a p-carborane-1,12-dicarboxylate anion).12 On the other hand, the ZnAl-1 intercalate also contains the carbonate anions, and the ZnAl-2 intercalate contains both carbonate and nitrate anions in the interlayer space but, as follows from the formulas, the majority of the positive charges of the layers are compensated by the borane anions. Also the Zn-3 and Zn-5 intercalates contain a significant amount of nitrate anions. For the intercalates of ZnAl-NO3 with the 1− and 2− anions, thermogravimetry was performed in the range from room temperature to 960 °C. The decrease of the weight due to the release of water was in the same range as that in the parent ZnAl-LDH. Another decrease is observed at 650 °C for the 1− intercalate and at 750° for the 2− intercalate. This decrease is due to the decomposition and the release of the borane anions, and its size corresponds to the amount of the intercalated anions determined using other methods.
| Intercalate | Formula | Basal spacing/Å |
|---|---|---|
| ZnAl-1 | Zn0.66Al0.34(OH)2(B10H10)0.12(CO3)0.05·0.5H2O | 11.38 |
| ZnAl-2 | Zn0.66Al0.34(OH)2(B12H12)0.10(CO3)0.04 (NO3)0.06·0.5H2O | 11.37 |
| ZnAl-3 | Zn0.66Al0.34(OH)2(C4B10H10O4)0.17·0.9H2O | 14.78 |
| ZnAl-4 | Zn0.66Al0.34(OH)2(C4B10H10O4)0.17·1.4H2O | 15.44 |
| ZnAl-5 | Zn0.67Al0.33(OH)2(C3B10H11O2)0.33·H2O | 20.39 |
| Zn-3 | Zn5(OH)8(C4B10H10O4)0.87(NO3)0.26·2H2O | 15.15 |
| Zn-5 | Zn5(OH)8(C3B10H11O2)1.1(NO3)0.9·2H2O | 20.69 |
Fig. 2 shows the X-ray powder diffraction patterns of the ZnAl-LDH intercalates. A series of sharp intensive (00l) diffraction lines was observed in the diffractograms together with several (hkl) lines with low intensity. The 7.60 Å or 8.90 Å reflections corresponding to the most intensive (003) lines of ZnAl-CO3 or ZnAl-NO3, respectively, were not observed in the diffractograms of the intercalates. The basal spacings of ZnAl-1 and ZnAl-2 are nearly the same (see Table 1). The basal spacing of the ZnAl-3 intercalate is very close to the value obtained by Choy et al.11 for the ZnAl-LDH intercalate of this carboxylic acid (14.88 Å) and to that obtained by Ay et al.12 for the MgAl-LDH intercalate (14.81 Å). The basal spacing of the ZnAl-4 intercalate, 15.44 Å, is comparable to that found for the ZnAl-LDH intercalate of o-carborane-1,2-dicarboxylic acid (15.56 Å).11 In the case of the ZnAl-5 intercalate, the basal spacing of 20.39 Å is slightly higher than the value found for the MgAl-LDH intercalate with 1-methyl-o-carborane-2-carboxylic acid (19.9 Å).12 The higher value of the basal spacing of these intercalates indicates that the monocarboxylic anions form a bilayer in the interlayer space. Fig. 3 shows the X-ray powder diffraction patterns of the Zn5(OH)8(NO3)2·2H2O intercalates together with the powder diffraction pattern of the pure host. Only very small peaks corresponding to the (003) reflection of the unreacted host were found in the Zn-3 and Zn-5 intercalates, despite the fact that these intercalates contain significant amounts of the nitrate ions. This means that the nitrate anions must also be present in the interlayer space of these intercalates together with the carborane anions. The basal spacing of Zn-5 (20.69 Å) is also significantly higher than that of the Zn-3 intercalate (15.15 Å).
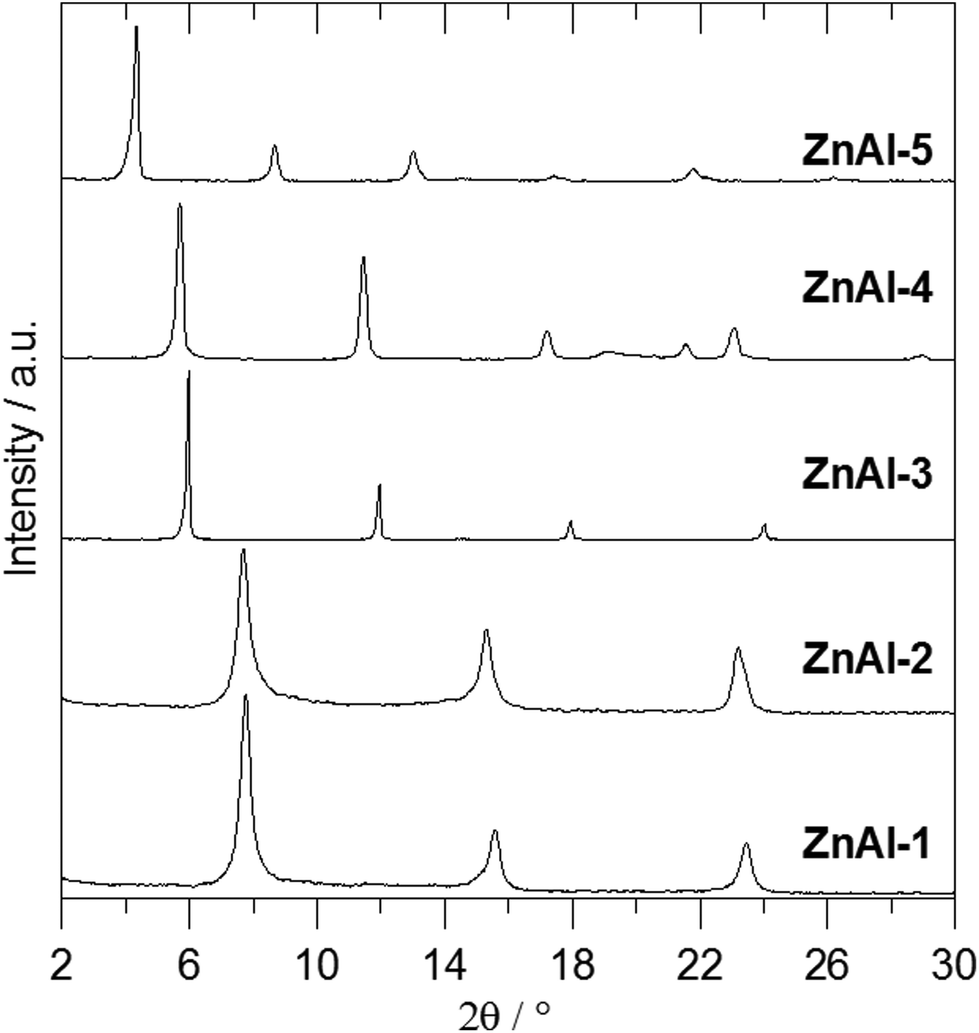 | ||
| Fig. 2 X-ray powder diffraction patterns of ZnAl-LDH intercalates. | ||
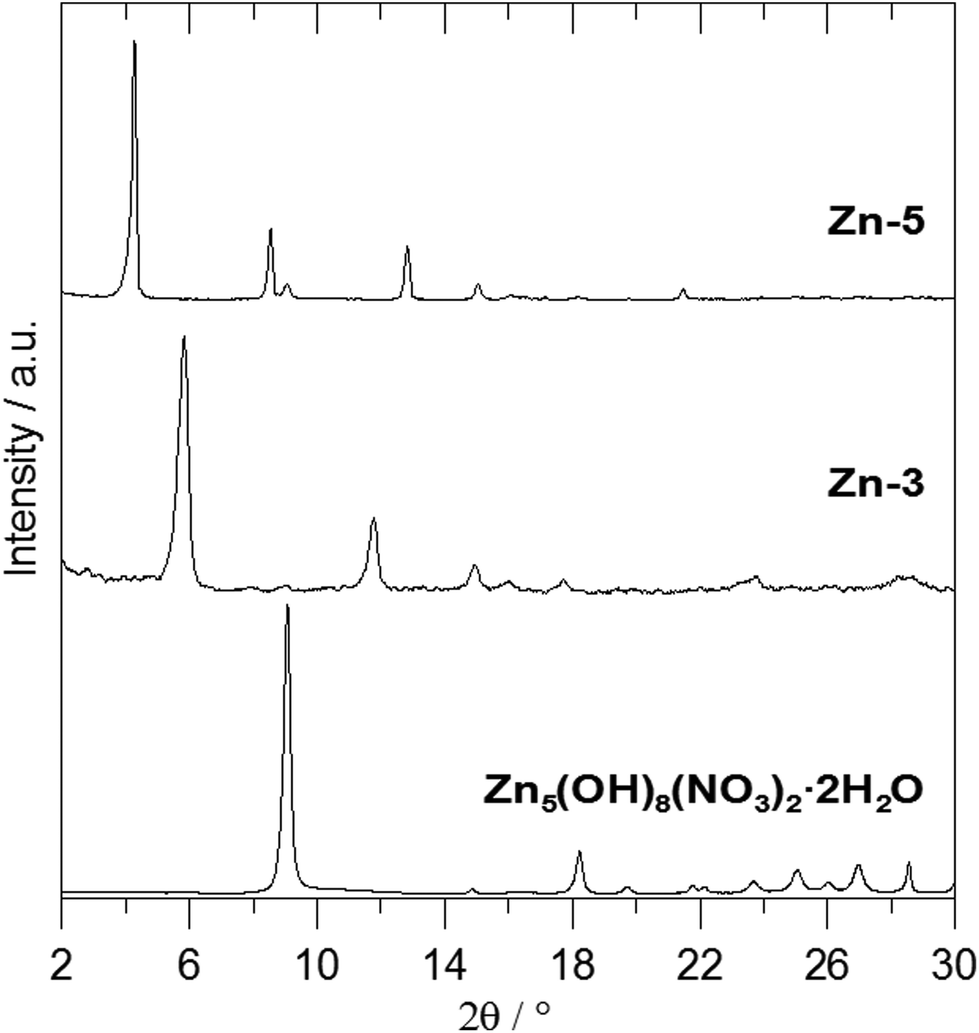 | ||
| Fig. 3 X-ray powder diffraction patterns of Zn5(OH)8(NO3)2·2H2O and its intercalates. | ||
The IR spectra of the ZnAl-1 and ZnAl-2 intercalates together with the spectra of the parent ZnAl-CO3 and ZnAl-NO3 hosts are shown in Fig. 4. A very broad and intensive band observed at 3700–2500 cm−1 in the spectra of ZnAl-CO3 and ZnAl-NO3 can be ascribed to the OH stretching vibrations of the hydroxyl groups and water molecules.26 OH bending modes from water molecules occur at 1635–1604 cm−1. A very intensive band at 1363 cm−1 and broad bands at about 880 and 680 cm−1 in the spectrum of ZnAl-CO3 correspond to the vibrations of the carbonate anion.27 A very intensive band at 1385 cm−1 and a sharp band at 825 cm−1 in the spectrum of ZnAl-NO3 are caused by vibrations of the nitrate anions.27 Bands at about 773, 621 and 426 cm−1 in the spectra of both hosts are probably caused by vibrations of the metal–oxygen bond.4 In the spectrum of the ZnAl-1 intercalate, a broad band at 3450 cm−1 also corresponds to OH stretching vibrations. A sharp band at 2483 cm−1 can be ascribed to a stretching vibration of the B–H bond, and the less intensive band at 2542 cm−1 is caused by the stretching vibration of the apical B–H bonds.28,29 A band at 1606 cm−1 can be ascribed to the deformation vibration of the co-intercalated water molecule, and an intensive band at 1360 cm−1 corresponds to the vibration of the carbonate anion. In the spectrum of the ZnAl-2 intercalate, besides the band of OH vibration at 3450 cm−1, two bands at 2520 and 2482 cm−1 in the B–H stretching region, the deformation vibration of water at 1613 cm−1, and two bands at 1082 and 1061 cm−1 corresponding to cage absorptions were observed.28,29 Bands at 1383 and 1366 cm−1 indicate the presence of both nitrate and carbonate anions in the interlayer space, which is in an agreement with the composition found by elemental analysis.
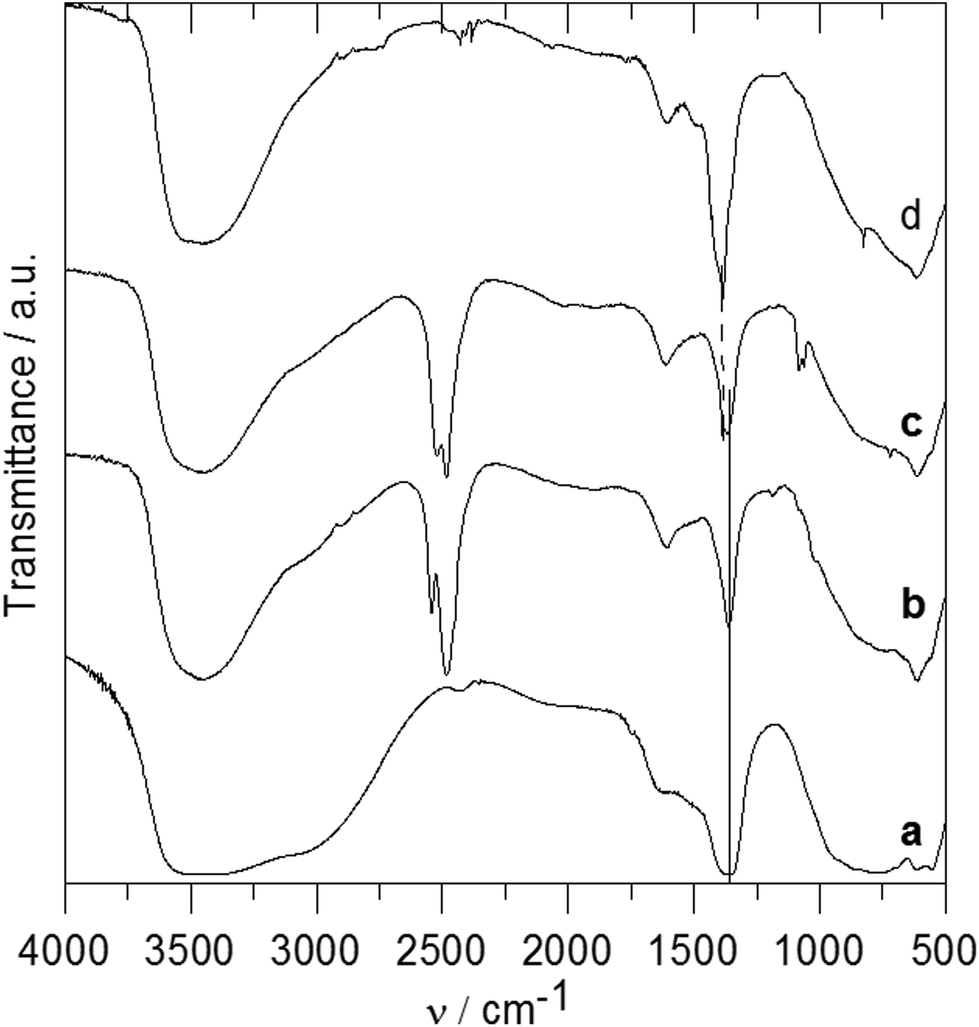 | ||
| Fig. 4 IR spectra of parent ZnAl-CO3 (a), ZnAl-1 (b), ZnAl-2 (c) and parent ZnAl-NO3 (d). | ||
In the spectrum of the ZnAl-3 intercalate (see Fig. 5a), the most intensive bands are at 2621 cm−1 corresponding to the B–H stretching vibration,30 two bands are at 1610 and 1363 cm−1 corresponding to the vibration of the carboxylate anions and a very broad band centered at 3400 cm−1 is caused by the O–H stretching vibration of the hydroxyl groups as well as of the water molecules. Bands of the B–H deformation vibrations were observed at 1133 and 992 cm−1. The infrared spectrum of the ZnAl-4 intercalate (see Fig. 5b) is very similar to that of the ZnAl-3 intercalate. In the spectrum of the ZnAl-5 intercalate (see Fig. 5c), there are strong bands at about 3400, 2609, 1608 and 1363 cm−1 that correspond to the O–H stretching, B–H stretching and carboxylate anion vibrations as in the case of the ZnAl-3 intercalate. Bands at 3061 and 2976 cm−1 can be caused by the C–H stretching vibrations.30 The 1200–400 cm−1 region in the ZnAl-5 spectrum is more complicated than that of the ZnAl-3 intercalate, and there are bands caused by the vibration of the carborane unit and metal–oxygen bonds. The spectra of all three intercalates do not contain a band at 1720–1700 cm−1, which corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration of carboxylic acid. This means that all carboxylic groups of the intercalated guests are deprotonated and the guests are present in the interlayer space as anions.
O vibration of carboxylic acid. This means that all carboxylic groups of the intercalated guests are deprotonated and the guests are present in the interlayer space as anions.
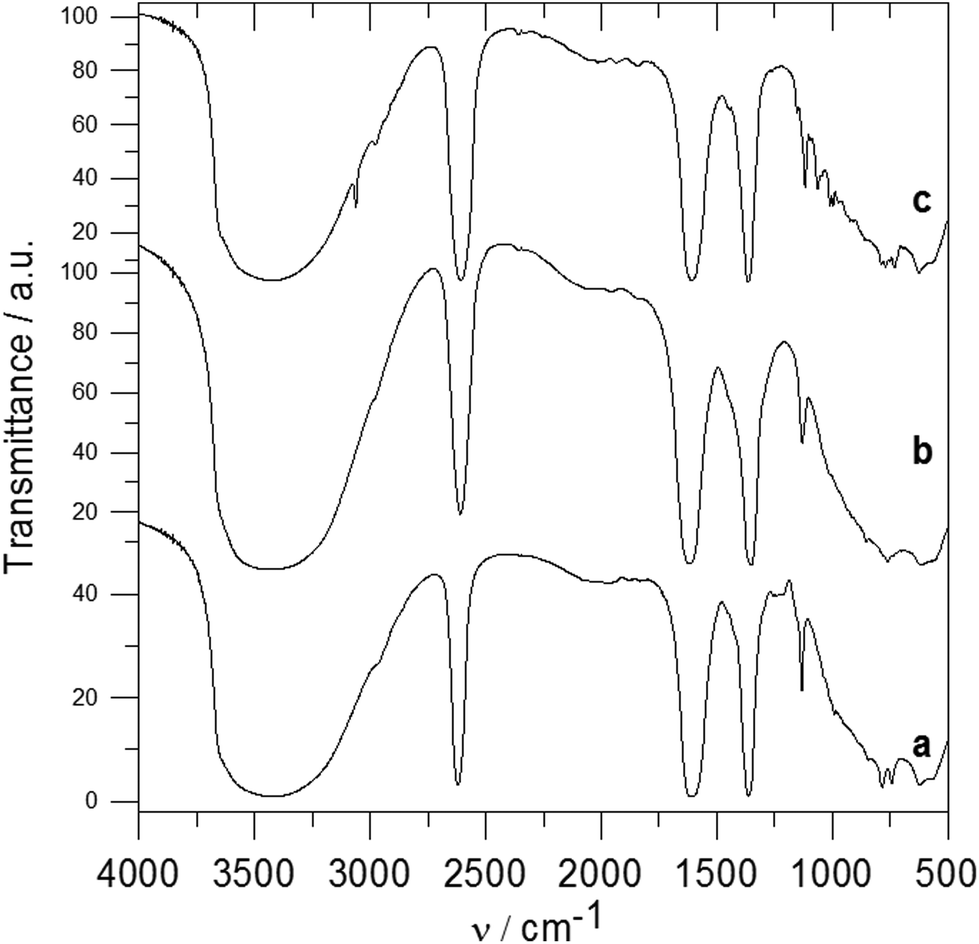 | ||
| Fig. 5 IR spectra of ZnAl-3 (a), ZnAl-4 (b) and ZnAl-5 (c). | ||
In the IR spectrum of Zn5(OH)8(NO3)2·2H2O (see Fig. 6a), bands in the region of 3600–3300 cm−1 correspond to the OH stretching modes, and a band at 1635 cm−1 arises from the H2O bending mode. Bands at 1384, 1050 and 825 cm−1 are attributed to nitrate anions.31,32 Bands between 1050 and 640 cm−1 are probably caused by the δOH bending modes of the hydroxide groups bonded to the zinc atoms. Very broad and intensive bands observed at 3800–3200 cm−1 in the spectrum of the Zn-3 intercalate (see Fig. 6b) correspond to the stretching OH vibrations. A band of B–H stretching vibration is observed at 2625 cm−1, and bands at 1140 and 993 cm−1 can be attributed to the B–H deformation vibrations. Bands at 1624 and 1367 cm−1 correspond to the vibrations of the carboxylate anions, and a shoulder at 1383 cm−1 is caused by the vibrations of non-exchanged nitrate anions. In the spectrum of the Zn-5 intercalate (see Fig. 6c), a broad band at 3700–3200 cm−1 corresponds to the OH stretching vibrations, bands at 2611 and 1121 cm−1 correspond to the B–H stretching and deformation vibrations, and bands at 1632 and 1370 cm−1 to the vibration of the carboxylate anion. The band corresponding to the non-exchanged nitrate anions at about 1384 cm−1 is in this case overlapped by a carboxylate band. In the spectra of the Zn-3 and Zn-5 intercalates no band corresponding to the CO vibration of the carboxylic acid was observed, which also means that in these intercalates all carboxylic groups are deprotonated.
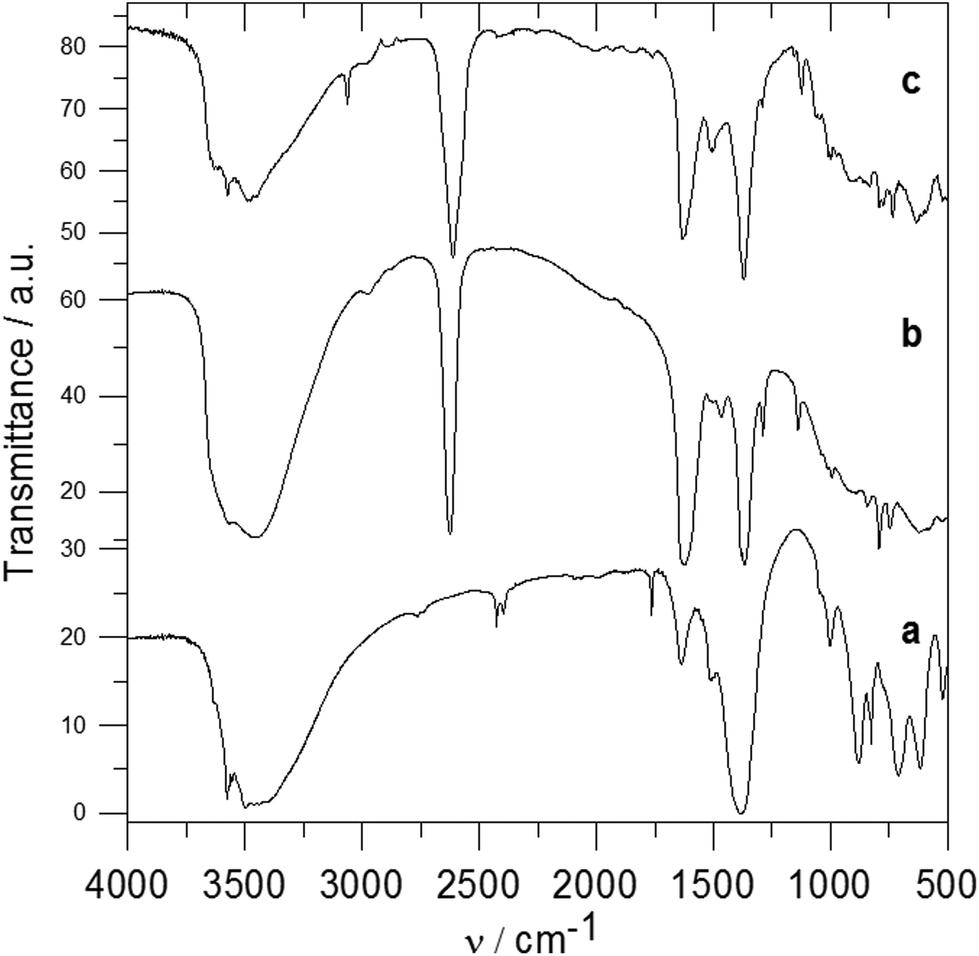 | ||
| Fig. 6 IR spectra of parent Zn5(OH)8(NO3)2·2H2O (a), Zn-3 (b) and Zn-5 (c). | ||
The ssNMR spectra of the intercalates reveal similar peaks to those of the 11B NMR spectra of the parent intercalated boron clusters. Generally speaking, the signals in the ssNMR spectra are significantly broadened (several resonances under one peak are obvious) and shifted downfield. The 11B NMR spectrum (see Fig. 7A) of 1− shows two signals at −2.0 and −31.1 ppm attributed to apical and equatorial boron atoms (a symmetry of D4h), respectively (see Fig. 1). The same pattern is retained in the corresponding ssNMR of ZnAl-1: the measured values are shifted to 2.0 and −26.7 ppm. This unambiguously indicates that 1− is intercalated without contributing any boron atom itself to such an interaction, and the position of the bicapped-square antiprism is not necessarily perpendicular to the metal-OH layers since both kinds of the boron atoms are deshielded. This usually happens due to coupling of the LUMO orbital with a suitably occupied orbital with respect to the 11B NMR spectrum before the intercalation. This coupling results in increasing negative charges on the terminal hydrogens and thus strengthening the Hδ−⋯Hδ+ bonds. The 11B ssNMR spectrum of ZnAl-2 (see Fig. 7B) reveals a single peak at −15.2 ppm, which corresponds to that at −16.3 ppm recorded in the 11B NMR of 2− (Ih symmetry). This provides a good ground for the same conclusions as for 1− in terms of the bonding type in the intercalate. Note that the body diagonal in 1− is about 10% longer (as derived from the corresponding algebra) than such diagonals in 2−, which might result in a skew arrangement of 1− between the layers, thus satisfying participation of both the equatorial and apical terminal hydrogens (see Fig. 1) in the interaction with the host layer. The 11B ssNMR spectra of ZnAl-3 and Zn-3 (see Fig. 7C) contain one signal at −13.6 ppm. This is in good agreement with the spectrum of the corresponding carboxylic acid 3 based on closo-1,12-C2B10H12, which also has one signal only due to D5d-like symmetry. The carboxylic acids 4 and 5 are based on closo-1,7-C2B10H12 that adopts a symmetry of C2v with 6 signals of intensities 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2:2:2:2. From the 11B NMR point of view both acids can be considered C2v-like symmetrical. Indeed, we detected six signals for 4 and 5 (see Fig. 7D and E). In the case of the corresponding ZnAl-4, ZnAl-5, and Zn-5 intercalates, the spectra show broad overlapped signals but the particular peaks of the intercalated borane compounds are still distinguishable. A slight de-shielding effect in the ssNMR of the intercalates with 4 and 5 with respect to their 11B NMR signals might also be attributed to the mutual interaction of the carbaborane moieties due to a relatively large dipole moment of closo-1,7-C2B10H12 (2.85 D, see ref. 33). Anyway, the de-shielding effect of the boron atoms in the intercalates containing carboxylate carbaboranes is less pronounced than in the case of intercalated parent boron hydrides, which indicates a very small interaction of the carboxylate group with the carbaborane skeleton during the intercalation.
:1:2:2:2:2. From the 11B NMR point of view both acids can be considered C2v-like symmetrical. Indeed, we detected six signals for 4 and 5 (see Fig. 7D and E). In the case of the corresponding ZnAl-4, ZnAl-5, and Zn-5 intercalates, the spectra show broad overlapped signals but the particular peaks of the intercalated borane compounds are still distinguishable. A slight de-shielding effect in the ssNMR of the intercalates with 4 and 5 with respect to their 11B NMR signals might also be attributed to the mutual interaction of the carbaborane moieties due to a relatively large dipole moment of closo-1,7-C2B10H12 (2.85 D, see ref. 33). Anyway, the de-shielding effect of the boron atoms in the intercalates containing carboxylate carbaboranes is less pronounced than in the case of intercalated parent boron hydrides, which indicates a very small interaction of the carboxylate group with the carbaborane skeleton during the intercalation.
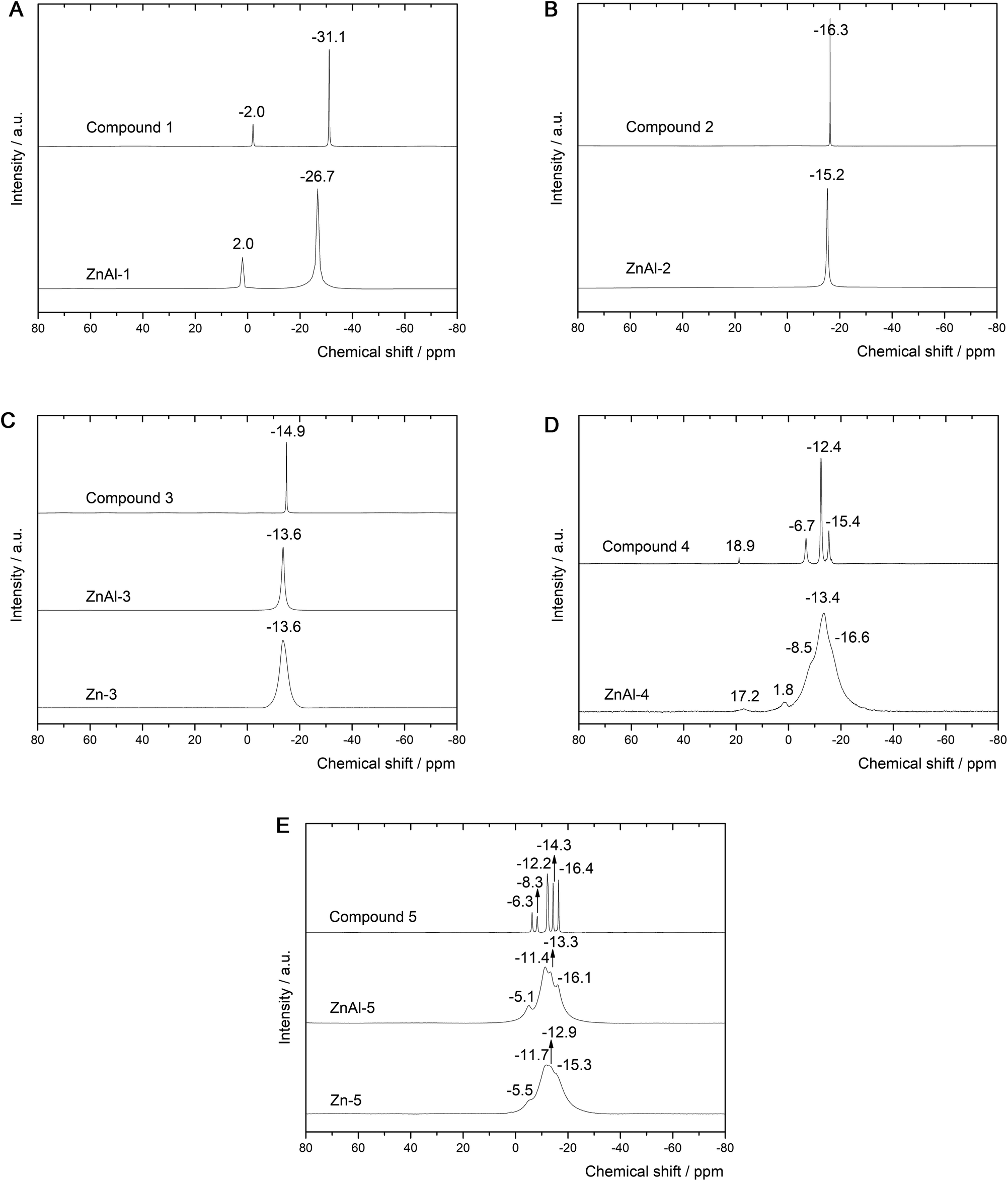 | ||
| Fig. 7 (A) Comparison of the 11B NMR spectra of 1− and ZnAl-1. (B) Comparison of the 11B NMR spectra of 2− and ZnAl-2. (C) Comparison of the 11B NMR spectra of 3, ZnAl-3 and Zn-3. (D) Comparison of the 11B NMR spectra of 4 and ZnAl-4. (E) Comparison of the 11B NMR spectra of 5, ZnAl-5 and Zn-5. | ||
Both the binary boron hydride anions, 1− and 2−, form a monolayer in the interlayer space (depicted schematically in Fig. 8a), as is evidenced by the value of the interlayer distance, which is roughly 11.4 Å in both ZnAl-LDH intercalates, see Table 1. Taking into account that the thickness of the ZnAl-LDH layer is about 4.8 Å,11 the height of the interlayer space is found to be about 6.6 Å, a value big enough to accommodate the mentioned guest molecules, whose size is about 6 Å in one layer. In contrast to the substituted icosahedral carboranes, which are likely to interact with the host layers via the charged COO− functional group, in the case of the ZnAl intercalates with 1− and 2− an interesting interaction between the host layer and the borane anion is expected to occur. Hence, we built a [Zn13Al6(OH)36](B12H12)4 model with all possible mutual interactions, which was analyzed by QM methods at the DFT-D3/BLYP/DZVP level and compared to a [Zn13Al6(OH)36](NO3)8 model. In the first step, we computed the electrostatic potential of the isolated anions of 2− and NO3−. The whole surface of 2− is highly negative (Vmax and Vmin of −151.1 and −177.7 kcal mol−1). It is more negative than the ESP surface of NO3− (Vmax and Vmin of −111.3 and −150.1 kcal mol−1, respectively; see Fig. 9). The Vmax of 2− is located on top of the H atoms, while the Vmin is in the B–B–B triangles. The Vmax of NO3− is located on the N atom on the C3 axis of the molecule, while the Vmin is between the O atoms (on the plane of the molecule).
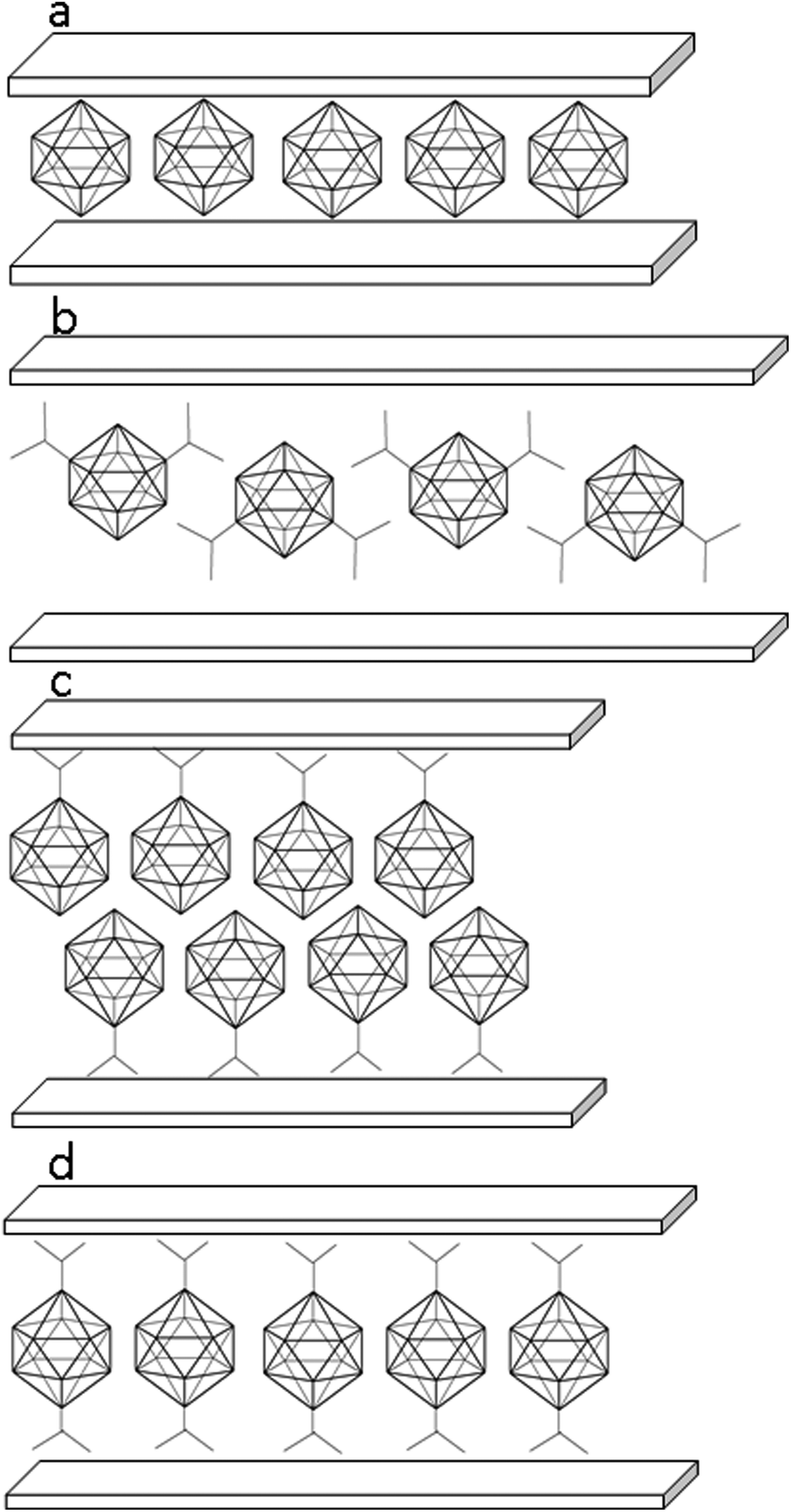 | ||
| Fig. 8 Presumed arrangement of the guest molecules in the interlayer space of the hosts: ZnAl-2 (a), ZnAl-4 (b), ZnAl-5 and Zn-5 (c), and ZnAl-3 and Zn-3 (d). | ||
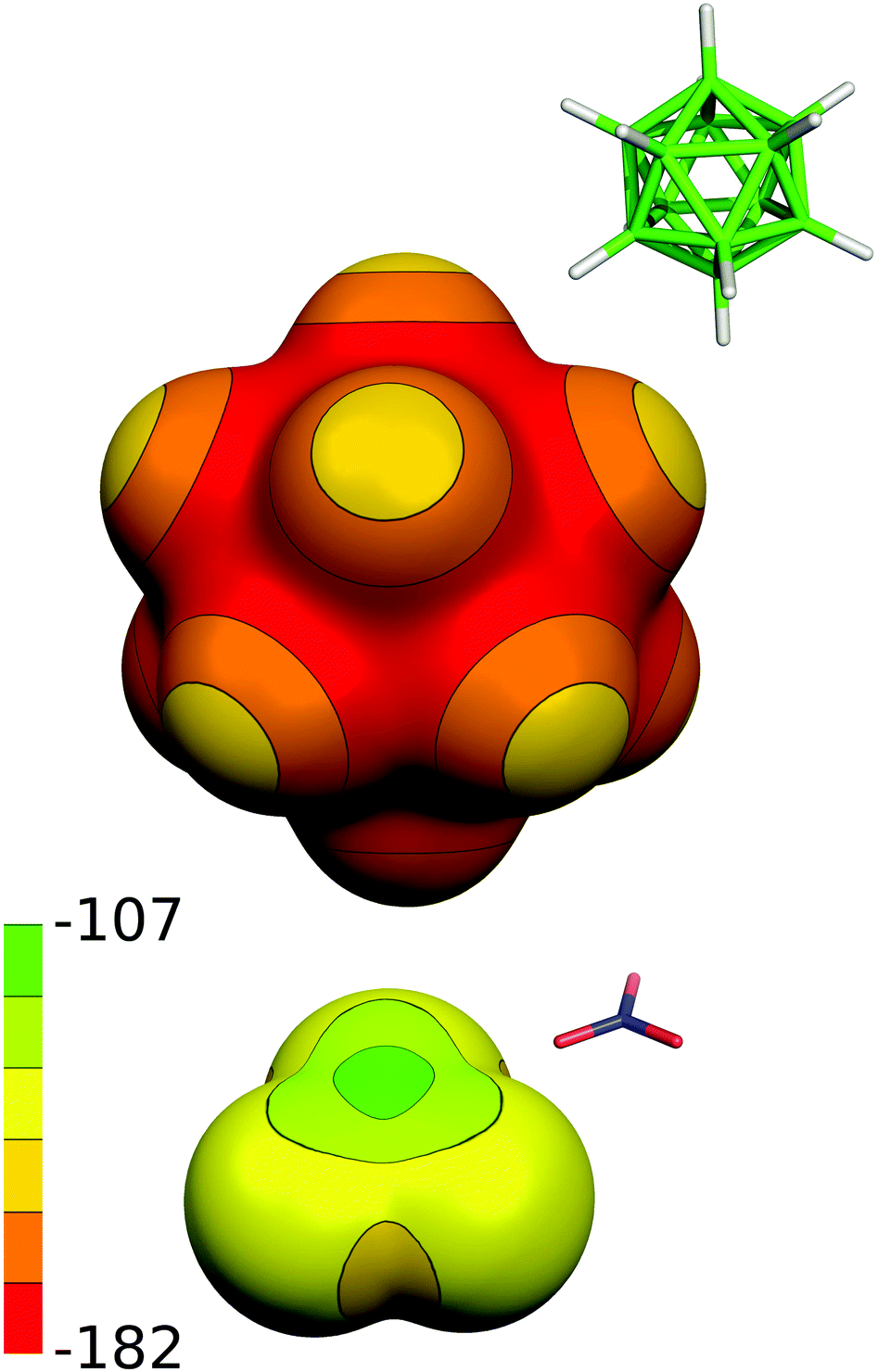 | ||
| Fig. 9 The computed electrostatic potential (ESP) surface for 2− and NO3−. The ESP color range is in kcal mol−1. | ||
The constructed [Zn13Al6(OH)36](B12H12)4 model was optimized and the computed interaction energy for each B12H122− cage is summarized in Table 2. A very strong electrostatic interaction between the dianionic B12H122− cages and the hydroxide resulted in the strong interaction energies in the gas phase, which are compensated by a large desolvation penalty. All four identical B12H122− cages in the model (denoted as molecules A to D in Table 2) had comparable binding “free” energies of about −30 kcal mol−1, which was considerably more than the binding “free” energy of NO3− (ΔG′ of about −20 kcal mol−1). The B12H122− cages interact with the hydroxide groups of the host mainly via dihydrogen bonds. The dihydrogen bonds, also called proton-hydride bonds, were formed between the positively charged hydrogen of the OH group and a σ-bond of a BH proton acceptor. Besides the dihydrogen bonds, the B and C molecules interact directly with the Zn atom (H⋯Zn distances of 1.85 and 2.04 Å, respectively). Fig. 9 illustrates the electrostatic potentials. Fig. 10 shows the structures of these models. Since the basal spacing of the intercalate with 1 (ZnAl-1) is the same as for ZnAl-2, we suppose that the model for ZnAl-1 will give similar results as the model for ZnAl-2.
 | ||
| Fig. 10 The modeled structure of ZnAl LDH with (a) 2− and (b) NO3−. | ||
| ΔG′ | ΔE | ΔΔGsolv | |
|---|---|---|---|
| [Zn13Al6(OH)36](B12H12)3⋯molecule A | −29.7 | −324.8 | 295.1 |
| [Zn13Al6(OH)36](B12H12)3⋯molecule B | −31.4 | −311.5 | 280.1 |
| [Zn13Al6(OH)36](B12H12)3⋯molecule C | −29.6 | −321.3 | 291.7 |
| [Zn13Al6(OH)36](B12H12)3⋯molecule D | −30.4 | −321.8 | 291.4 |
| [Zn13Al6(OH)36](NO3)7⋯NO3− (parallel) | −18.7 | −134.2 | 115.5 |
| [Zn13Al6(OH)36](NO3)7⋯NO3− (perpendicular) | −20.0 | −113.5 | 93.5 |
A different arrangement of the 4− anions is expected in ZnAl-4, see Fig. 8b. The molecule of this guest is anchored by both its carboxylate groups to one host layer. The guest molecules thus form a kind of a bilayer in which the molecules of the guest are bonded alternately to the upper and lower host layers and are strongly interdigitated.
The 5− anions form in the interlayer space of the host a bilayer arrangement (Fig. 8c), as follows from the interlayer distances, 20.4 and 20.7 Å, observed for the ZnAl-5 and Zn-5 intercalates, respectively. This means that the intercalate of 5− with Zn5(OH)8 has about 0.3 Å higher interlayer distance than the intercalate with ZnAl-LDH.
A monolayer arrangement is presumed for the intercalate of 3− with ZnAl-LDH, whose interlayer distance is 14.8 Å (Fig. 8d), in the way analogous to that suggested by Choy et al.11 The body diagonal of the 3− anion is 9 Å (the distance between the oxygen atoms of the carboxylic groups), i.e. 3 Å longer than that of 2. The anion of 3− can be therefore presumed to be arranged as a pillar between the layers with the carboxylic groups close to the host layers. The same arrangement can be expected for the Zn-3 intercalate whose interlayer distance is about 15.2 Å, which is about 0.4 Å higher than that of the ZnAl-LDH intercalate. Such a pillared arrangement can be thus excluded for the 4− dianions intercalated into ZnAl-LDH, because it would lead to a basal spacing of ZnAl-4 lower than that found in ZnAl-3, which is not true.
Conclusions
Five borane compounds were intercalated into two layered compounds, namely a zinc aluminum layered double hydroxide with the formula [Zn0.67Al0.33(OH)2](A)x·0.5H2O (where A represents an anion) and into Zn5(OH)8(NO3)2·2H2O by an ion exchange. The intercalated borane compounds were closo-B10H102− and closo-B12H122− borane anions and 1,12-(COO)2-closo-1,12-C2B10H102−, 1,7-(COO)2-closo-1,7-C2B10H102− and 1-(COO)-closo-1,7-C2B10H111− carborane anions. The presence of these borane compounds in their anionic form in the interlayer space of the intercalates was proved by the infrared spectra of the synthesized products. The original anions are fully exchanged in the intercalates of LDH with carboxyl group containing carboranes, whereas in the zinc hydroxide nitrate intercalates part of the nitrate anions remain unchanged. The original nitrate or carbonate anions are also present in the intercalates with borane anions. The 1, 2, and 3 guests form a monolayer in the interlayer space, with 3 forming a pillared structure thanks to its two carboxyl groups present in a para position of the carborane skeleton. The 4 and 5 guests form a bilayer, and in the case of the 4 intercalate the carborane molecules are strongly interdigitated.Compound 5 was found to be very acidic in water (measured pKa = 2 and computed pKa = 1.88),34 which suggests that this monocarboxylic acid is much stronger than benzoic acid (pKa = 4.19). The corresponding gas-phase acidity (GA) is found to be 1325 kJ mol−1 (computed GA is 1321 kJ mol−1).34 To make matters more interesting, the GA values of both dicarboxylic acids 3 and 4 could not be calculated using standard approaches as they are beyond the H2SO4 (GA = 1265 kJ mol−1) scale as revealed by computing GA.35 We may therefore consider 3 and 4 as superacids, much stronger acids than the comparable terephthalic and isophthalic acids, respectively. On that basis, the intercalates of 3 and 4 should be of much stronger nature than those in which organic aromatic acids are intercalated. Since the protic acids derived from 1− and 2−, i.e. (H3O)2B10H10 and (H3O)2B12H12,36 with an unusual intercalation bonding scheme, i.e., Hδ−⋯Hδ+ bonds (so far observed in boron-cluster-interactions with biomolecules37,38 and as one of the driving forces in the crystal packings39), are also considered to be superacids, we may consider the reported intercalates as means of potential storage of superacidic materials for delivering into various chemical reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Czech Science Foundation (projects 17-10639S (KM, LB, and VZ) and 17-08045S (DH, JH, and JF)) for financial support.References
- X. Duan and D. G. Evans, Layered Double Hydroxides, Springer Verlag, Berlin, 2006 Search PubMed.
- Y. Xu and J. Q. Jiang, Ind. Eng. Chem. Res., 2008, 47, 16–24 CrossRef.
- Z. M. Guan, J. F. Lv, P. Bai and X. H. Guo, Desalination, 2016, 383, 29–37 CrossRef.
- C. Nyambo and C. A. Wilkie, Polym. Degrad. Stab., 2009, 94, 506–512 CrossRef.
- Q. Wang, J. P. Undrell, Y. S. Gao, G. P. Cai, J. C. Buffet, C. A. Wilkie and D. O'Hare, Macromolecules, 2013, 46, 6145–6150 CrossRef.
- J. T. Lin, S. J. Tsai and S. F. Cheng, J. Chin. Chem. Soc., 1999, 46, 779–787 CrossRef.
- R. Bechara, A. D'Huysser, M. Fournier, L. Forni, G. Fornasari, F. Trifiro and A. Vaccari, Catal. Lett., 2002, 82, 59–67 CrossRef.
- G. Varga, S. Murath, A. Bajcsi, A. Kukovecz, Z. Konya, P. Sipos and I. Palinko, React. Kinet., Mech. Catal., 2017, 121, 241–254 CrossRef.
- Z. L. Wang, Z. H. Kang, E. B. Wang, Z. M. Su and L. Xu, Inorg. Chem., 2006, 45, 4364–4371 CrossRef PubMed.
- S. Sene, S. Begu, C. Gervais, G. Renaudin, A. Mesbah, M. E. Smith, P. H. Mutin, A. van der Lee, J. M. Nedelec, C. Bonhomme and D. Laurencin, Chem. Mater., 2015, 27, 1242–1254 CrossRef.
- J. H. Choy, I. R. Jeon, S. J. Choi and J. M. Oh, Boron compound-layered double hydroxide nanohybrid, method of preparing the boron compound-LDH nanohybrid, and pharmaceutical composition comprising the boron compound-LDH nanohybrid, US7572458B2, 2009 Search PubMed.
- A. N. Ay, H. Akar, A. Zaulet, C. Vinas, F. Teixidor and B. Zumreoglu-Karan, Dalton Trans., 2017, 46, 3303–3310 RSC.
- R. N. Grimes, Carboranes, Elsevier Inc., London, Amsterdam, 2011 Search PubMed.
- S. Heřmánek, Chem. Rev., 1992, 92, 325–362 CrossRef.
- U. Costantino, F. Marmottini, M. Nocchetti and R. Vivani, Eur. J. Inorg. Chem., 1998, 1439–1446 CrossRef.
- K. Melánová, L. Beneš, V. Zima and M. Vlček, Sci. Pap. Univ. Pardubice, Ser. A, 2002, 8, 103–109 Search PubMed.
- S. P. Newman and W. Jones, J. Solid State Chem., 1999, 148, 26–40 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2009.
- P. Flükiger, H. P. Lüthi, S. Portmann and J. Weber, MOLEKEL 4.3, Swiss Center for Scientific Computing, Manno, Switzerland, 2000 Search PubMed.
- S. Portmann and H. P. Lüthi, Molekel: Chimia, 2007, 28, 555–569 Search PubMed.
- K. E. Riley, K. A. Tran, P. Lane, J. S. Murray and P. Politzer, J. Comput. Sci., 2016, 17, 273–284 CrossRef.
- J. Fanfrlík, J. Holub, Z. Růžičková, J. Řezáč, P. D. Lane, D. A. Wann, D. Hnyk, A. Růžička and P. Hobza, ChemPhysChem, 2016, 17, 3373–3376 CrossRef PubMed.
- R. Ahlrichs et al., TURBOMOLE V6.2 2010, a development of the University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com. Turbomole V6, Karlsruhe, Deutschland, 2008.
- J. Řezáč, J. Comput. Chem., 2016, 37, 1230–1237 CrossRef PubMed.
- K. Melánová, L. Beneš, V. Zima and J. Svoboda, J. Inclusion Phenom. Macrocyclic Chem., 2005, 51, 97–101 CrossRef.
- A. C. Vieira, R. L. Moreira and A. Dias, J. Phys. Chem. C, 2009, 113, 13358–13368 CrossRef.
- M. J. Hernandez-Moreno, M. A. Ulibarri, J. L. Rendon and C. J. Serna, Phys. Chem. Miner., 1985, 12, 34–38 Search PubMed.
- E. L. Muetterties, J. H. Balthis, H. C. Miller, Y. T. Chia and W. H. Knoth, Inorg. Chem., 1964, 3, 444–451 CrossRef.
- E. L. Muetterties, R. E. Merrifield, W. H. Knoth, J. R. Downing and H. C. Miller, J. Am. Chem. Soc., 1962, 84, 2506–2508 CrossRef.
- L. A. Leites, Chem. Rev., 1992, 92, 279–323 CrossRef.
- W. Stahlin and H. R. Oswald, J. Solid State Chem., 1971, 3, 252–255 CrossRef.
- C. Chouillet, J. M. Krafft, C. Louis and H. Lauron-Pernot, Spectrochim. Acta, Part A, 2004, 60, 505–511 CrossRef.
- O. Exner, Dipole Moments in Organic Chemistry, Thieme Stuttgart, 1975 Search PubMed.
- J. Z. Davalos, J. Gonzalez, R. Ramos, D. Hnyk, J. Holub, J. A. Santaballa, M. Canle and J. M. Oliva, J. Phys. Chem. A, 2014, 118, 2788–2793 CrossRef PubMed.
- J. M. Oliva-Enrich, S. Humbel, J. A. Santaballa, I. Alkorta, R. Notario, J. Z. Dávalos, M. Canle-L., E. Bernhardt, J. Holub and D. Hnyk, ChemistrySelect, 2018, 3, 4344–4353 CrossRef.
- A. Avelar, F. S. Tham and C. A. Reed, Angew. Chem., Int. Ed., 2009, 48, 3491–3493 CrossRef PubMed.
- R. Sedlak, J. Fanfrlík, A. Pecina, D. Hnyk, P. Hobza and M. Lepšík, in Boron – the Fifth Element, ed. D. Hnyk and M. McKee, Springer, Heidelberg, New York, Dordrecht and London, 2015, vol. 20 Search PubMed.
- S. M. Eyrilmez, E. Bernhardt, J. Z. Davalos, M. Lepsik, P. Hobza, K. I. Assaf, W. M. Nau, J. Holub, J. M. Oliva-Enrich, J. Fanfrlik and D. Hnyk, Phys. Chem. Chem. Phys., 2017, 19, 11748–11752 RSC.
- J. Holub, P. Melichar, Z. Růžičková, J. Vrána, D. A. Wann, J. Fanfrlík, D. Hnyk and A. Růžička, Dalton Trans., 2017, 46, 13714–13719 RSC.
- J. Hostaš and J. Řezáč, J. Chem. Theory Comput., 2017, 13, 3575–3585 CrossRef PubMed.
Footnote |
| † Icosahedral and bicapped-square antiprismatic cages very frequently appear in boron cluster chemistry and are represented by closo-B12H122− and by closo-B10H102− in their parent forms with symmetries of Ih and D4h, respectively. Since the BH− vertex is formally isoelectronic with CH, replacement of BH− with CH results in the so-called carboranes; when replacing two such vertices, neutral dicarbaboranes (closo-C2BnHn+2, n = 8, 10) are obtained with different C-positions. Hence, in the case of the icosahedral shape closo-1,2-, 1,7- and 1,12-dicarbaboranes are obtained, frequently denoted as o-, m-, and p-carboranes. See e.g. D. Hnyk and D. A. Wann, Chapter 2, Challenges and Advances in Computational Chemistry and Physics, in Boron – the Fifth Element, ed. D. Hnyk and M. McKee, Springer, Heidelberg, New York, Dordrecht and London, 2015, vol. 20. |
| This journal is © The Royal Society of Chemistry 2018 |