
Ligand and solvent control of selectivity in the C–H activation of a pyridylimine-substituted 1-naphthalene; a combined synthetic and computational study†
 *b
Gregory A.
Solan
*a
*b
Gregory A.
Solan
*a
Abstract
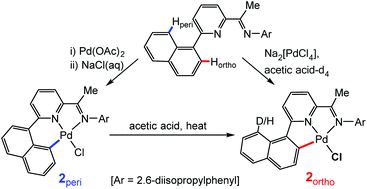
The pyridylimine-substituted 1-naphthalenes, 2-(1-C10H7)-6-{CR![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(2,6-i-Pr2C6H3)}C5H3N (R = Me HLMe, H HLH), react with Na2[PdCl4] in acetic acid at elevated temperature to afford either ortho-C–Hnaphthyl activated (LMe)PdCl (2ortho) or the unactivated adduct (HLH)PdCl2 (1b). Alternatively, 1b and its ketimine analogue (HLMe)PdCl2 (1a), can be prepared by treating (MeCN)2PdCl2 with either HLMe or HLH in chloroform at room temperature. Regio-selective ortho-C–H activation to form 2ortho can also be initiated by the thermolysis of 1a in acetic acid, while no reaction occurs under similar conditions with 1b. Interestingly, the C–H activation of HLMe to give 2ortho is found to be reversible with 100% deuteration of the peri-site occurring on reacting Na2[PdCl4] with HLMe in acetic acid-d4. By contrast, heating 1a in toluene gives a 55 : 45 mixture of 2ortho and its peri-activated isomer 2peri. Pure 2peri can, however, be obtained either from (LMe)PdOAc (3peri) by OAc/Cl exchange or by the sequential reactions of 1a with firstly silver acetate then with aqueous sodium chloride. Intriguingly, a peri to ortho interconversion occurs on heating 2peri in acetic acid to give 2ortho. DFT calculations have been used to investigate the C–H activation steps and it is found that in acetic acid ortho-C–H activation is kinetically and thermodynamically favoured but peri-CH activation is kinetically accessible (ΔΔG‡ = 2.4 kcal mol−1). By contrast in toluene, the reaction appears to be irreversible with the difference in barrier height for ortho- and peri-C–H activation being very small within the error of the method (ΔΔG‡ = 0.7 kcal mol−1), findings that are in agreement with the empirically observed product distribution for 2ortho and 2peri. Single crystal X-ray structures are reported for 1a, 1b, 2ortho and 2peri.
N(2,6-i-Pr2C6H3)}C5H3N (R = Me HLMe, H HLH), react with Na2[PdCl4] in acetic acid at elevated temperature to afford either ortho-C–Hnaphthyl activated (LMe)PdCl (2ortho) or the unactivated adduct (HLH)PdCl2 (1b). Alternatively, 1b and its ketimine analogue (HLMe)PdCl2 (1a), can be prepared by treating (MeCN)2PdCl2 with either HLMe or HLH in chloroform at room temperature. Regio-selective ortho-C–H activation to form 2ortho can also be initiated by the thermolysis of 1a in acetic acid, while no reaction occurs under similar conditions with 1b. Interestingly, the C–H activation of HLMe to give 2ortho is found to be reversible with 100% deuteration of the peri-site occurring on reacting Na2[PdCl4] with HLMe in acetic acid-d4. By contrast, heating 1a in toluene gives a 55 : 45 mixture of 2ortho and its peri-activated isomer 2peri. Pure 2peri can, however, be obtained either from (LMe)PdOAc (3peri) by OAc/Cl exchange or by the sequential reactions of 1a with firstly silver acetate then with aqueous sodium chloride. Intriguingly, a peri to ortho interconversion occurs on heating 2peri in acetic acid to give 2ortho. DFT calculations have been used to investigate the C–H activation steps and it is found that in acetic acid ortho-C–H activation is kinetically and thermodynamically favoured but peri-CH activation is kinetically accessible (ΔΔG‡ = 2.4 kcal mol−1). By contrast in toluene, the reaction appears to be irreversible with the difference in barrier height for ortho- and peri-C–H activation being very small within the error of the method (ΔΔG‡ = 0.7 kcal mol−1), findings that are in agreement with the empirically observed product distribution for 2ortho and 2peri. Single crystal X-ray structures are reported for 1a, 1b, 2ortho and 2peri.
 Please wait while we load your content...
Please wait while we load your content...

