
Ruthenium mediated halogenation of white phosphorus: synthesis and reactivity of the unprecedented P4Cl2 moiety†
Mark
Bispinghoff
 a,
Zoltán
Benkő
b,
Hansjörg
Grützmacher
*a,
Fuencisla Delgado
Calvo
c,
Maria
Caporali
*c and
Maurizio
Peruzzini
c
a,
Zoltán
Benkő
b,
Hansjörg
Grützmacher
*a,
Fuencisla Delgado
Calvo
c,
Maria
Caporali
*c and
Maurizio
Peruzzini
c
aLaboratory of Inorganic Chemistry, ETH Zürich, Vladimir-Prelog-Weg 1, 8093 Zürich, Switzerland
bBudapest University of Technology and Economics, Szent Gellért tér 4., H-1111 Budapest, Hungary
cCNR ICCOM, Via Madonna del Piano 10, 50019 Sesto Fiorentino, Italy. E-mail: maria.caporali@iccom.cnr.it
First published on 5th November 2018
Abstract
The transition metal mediated functionalization of P4 is an approach to develop a more sustainable production of organophosphorus compounds. In this paper a ruthenium complex is presented, in which the P4 unit can be selectively converted into new P4R2 molecules in a two-step synthesis. The unsaturated 16 electron species [RuX(Cp*)(PCy3)] (Cp* = C5Me5, X = Cl, Br) reacts with a half equivalent of P4 affording a bimetallic complex bearing a planar P4X2 moiety as a ligand. The latter eliminates chloride anions under reduction with magnesium. In this process the butadiene-like P4Cl2 ligand is converted into two weakly bound P2 units which bridge the ruthenium centers. In the presence of n-BuLi, the P4Cl2 unit can be selectively alkylated, yielding the planar organophosphorus ligand P4nBu2. A detailed analysis of the electronic properties and solid state structures of the compounds combined with DFT calculations and AIM analyses demonstrate that the P4 unit in all complexes acts as an electronically highly flexible, non-innocent ligand.
Introduction
Phosphorus-containing compounds are ubiquitous in the chemical, pharmaceutical, detergent, agricultural and food industries.1 Their production starts by reducing phosphate rock to white phosphorus (P4), utilizing enormous amounts of energy, 46 MJ per kg of P4. Subsequently, P4 is oxidized to phosphorus halides, which are then converted into the desired organophosphorus compounds using organolithium or Grignard reagents. Apart from creating unnecessary salt by-products, this sequential combination of a reduction, an oxidation, and a reduction step wastes large amounts of energy. Therefore, the development of direct strategies for the functionalization of P4 is highly desirable, albeit very challenging due to the high reactivity of P4.The catalytic functionalization of P4 using transition metal complexes could be a solution to this challenge. Numerous reactions between transition metal complexes and P4 have been investigated over the last few decades. Thus, the coordination chemistry of P4 and the reactions involved in its activation seem to be well understood. It was shown that, upon exposure to a transition metal fragment, P4 can coordinate as an intact P4 tetrahedron in a single or bridging coordination mode or, upon breaking some of the P–P bonds, it can rearrange to a P4 unit of a different geometry. Alternatively, under the harsh reaction conditions used in many cases, P4 fragmentation and reaggregation to other polyphosphorus units can take place.2 However, the subsequent combination of the activated transition metal P4 species with organic substrates to yield organophosphorus compounds remains a challenge. Despite recent advances in the formation of P–C,3 P–H,4 P–N5 and P–As6 bonds in the coordination spheres of transition metals, so far no commercially viable process has been developed.
The development of such processes requires a detailed understanding of the nature of the phosphorus–metal bonds. Here, we report the formation of a dimetallic ruthenium complex with an unprecedented bridging P4Cl2 ligand. The diverse reactivity of this ligand allowed us to gain a detailed understanding of the bonding situation, which is crucial for the development of transition metal mediated P4 functionalization processes.
Results and discussion
Exposing the dark blue unsaturated 16 electron ruthenium complex [RuCl(Cp*)(PCy3)] (where Cp* = C5Me5) in n-hexane at 20 °C to P4 leads to a brown, crystalline product with the structure [RuCp*(PCy3)(μ2,η2:4-P4Cl2)RuCp*] (1) in 84% isolated yield (Scheme 1). This reaction has been described previously and the product, solely based on NMR data, incorrectly characterized as [RuCl (Cp*)(PCy3)(η2-P4)], bears an η2-coordinated P4 moiety.7 Instead, single crystal X-ray diffraction revealed a bimetallic species with a bridging P4Cl2 ligand (Fig. 1). Upon exposure to the ruthenium fragment, the P4 ligand undergoes metal-promoted activation. Formally two bonds of the P4 tetrahedron are broken and oxidatively inserted into the Ru–Cl bond, resulting in a coordinated planar P4Cl2 ligand, which has no precedence. | ||
| Scheme 1 Synthesis of the P4Cl2-bearing complex 1 and subsequent functionalization. | ||
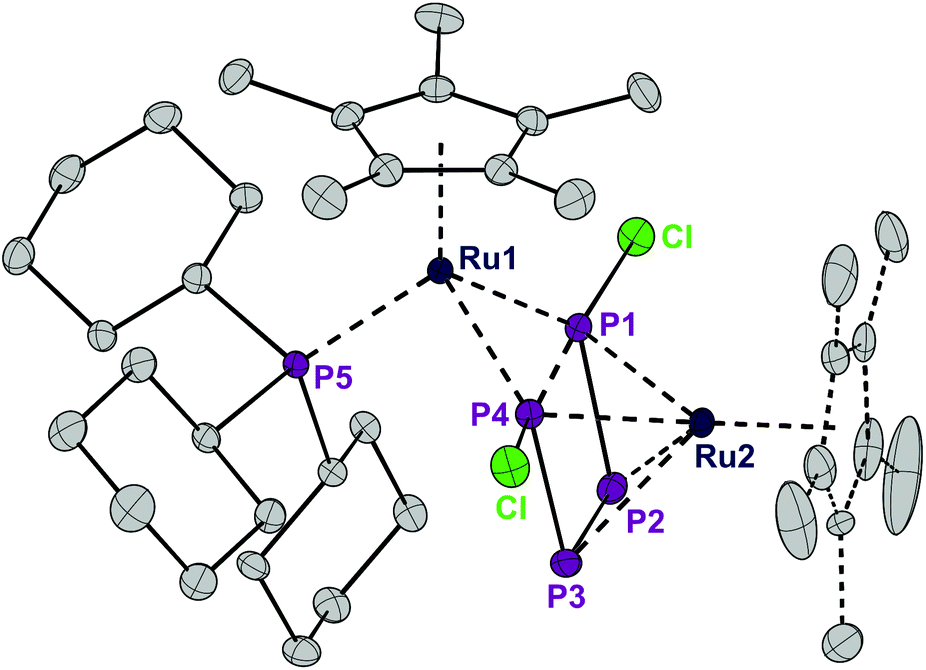 | ||
| Fig. 1 ORTEP of the P4Cl2-bearing complex 1. The dotted lines in the Cp* ligand on Ru2 correspond to the disorder. Thermal ellipsoids at 50% probability; hydrogen atoms have been omitted for clarity. | ||
The structure of 1 is highly symmetric with a mirror plane running through the two metal centers, the phosphorus center of the PCy3 group, and perpendicular to the P4Cl2 plane. Three of the four P–P distances are short and practically identical (2.16 to 2.17 Å, Fig. 2). The fourth P1–P4 distance is significantly longer (2.52 Å). The P4Cl2 ligand is best described as a Cl–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P–PP–Cl tetraphosphabutadiene in which a high degree of delocalization of the π-electrons leads to an equilibration of the bond lengths. The coordination sphere around Ru1 consists of strongly electron donating Cp* and PCy3 ligands and is completed by 4 electrons provided by two lone pairs from the terminal phosphorus centers P1 and P4. Under the assumption that the formal oxidation state at Ru1 is +2, this metal center achieves a valence electron configuration of 18. The Ru2 center is bound in an η4 fashion to the P4Cl2 ligand which accounts for an electron donation of four electrons and the second Cp* ligand as a six electron donor. Thus, under the conjecture that the formal oxidation state at Ru2 is zero, again an 18 valence electron configuration is obtained. Note that this is a very formal description and that a formulation with a dianionic [Cl2P4]2− ligand and two Ru centers in the oxidation state of +2 cannot be ruled out. DFT calculations8 on a model compound, with Cp instead of Cp* and methyl groups instead of cyclohexyl substituents, point out that the phosphabutadiene ligand donates indeed two lone pairs located at P1 and P4 towards Ru1. Indeed, the calculated Wiberg bond indices9 for Ru1–P1 and Ru1–P4 interactions are 0.843 and 0.845, respectively, very similar to the value of 0.821 found for the interaction between Ru1 and Cy3P. The calculated partial charges on both Ru centers are very similar (−1.07e for Ru1 and −0.97e for Ru2, respectively; see Table 1), underlining our suggestion that any assignment of oxidation states to the Ru centers is a formalism. The Wiberg bond indices9 of the P–P bonds (0.96 and 1.10 for the terminal and internal bonds, respectively) suggest a single bond character in the P4 butadiene-like moiety which may be a result of strong electron donation from occupied π-orbitals to Ru2 and significant metal-to-ligand electron back donation into the energetically low-lying π*-orbitals of the P4 chain.
P–PP–Cl tetraphosphabutadiene in which a high degree of delocalization of the π-electrons leads to an equilibration of the bond lengths. The coordination sphere around Ru1 consists of strongly electron donating Cp* and PCy3 ligands and is completed by 4 electrons provided by two lone pairs from the terminal phosphorus centers P1 and P4. Under the assumption that the formal oxidation state at Ru1 is +2, this metal center achieves a valence electron configuration of 18. The Ru2 center is bound in an η4 fashion to the P4Cl2 ligand which accounts for an electron donation of four electrons and the second Cp* ligand as a six electron donor. Thus, under the conjecture that the formal oxidation state at Ru2 is zero, again an 18 valence electron configuration is obtained. Note that this is a very formal description and that a formulation with a dianionic [Cl2P4]2− ligand and two Ru centers in the oxidation state of +2 cannot be ruled out. DFT calculations8 on a model compound, with Cp instead of Cp* and methyl groups instead of cyclohexyl substituents, point out that the phosphabutadiene ligand donates indeed two lone pairs located at P1 and P4 towards Ru1. Indeed, the calculated Wiberg bond indices9 for Ru1–P1 and Ru1–P4 interactions are 0.843 and 0.845, respectively, very similar to the value of 0.821 found for the interaction between Ru1 and Cy3P. The calculated partial charges on both Ru centers are very similar (−1.07e for Ru1 and −0.97e for Ru2, respectively; see Table 1), underlining our suggestion that any assignment of oxidation states to the Ru centers is a formalism. The Wiberg bond indices9 of the P–P bonds (0.96 and 1.10 for the terminal and internal bonds, respectively) suggest a single bond character in the P4 butadiene-like moiety which may be a result of strong electron donation from occupied π-orbitals to Ru2 and significant metal-to-ligand electron back donation into the energetically low-lying π*-orbitals of the P4 chain.
 | ||
| Fig. 2 Selected bond lengths within the P4 unit in complexes 1–4 in Å. | ||
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| P4 (or 2 × P2) | 1.62 | 1.58 | 1.46 | 2.26 |
| Ru2 | −2.04 | −1.96 | −1.98 | −2.17 |
| PMe3 | 1.26 | 1.26 | ||
| Cl | −0.56 | −0.19 |
The room temperature 31P NMR spectrum of 1 shows dynamic behavior with a broad doublet at δ = 250.8 ppm (w½ = 325 Hz, 1JPP = 400 Hz), a triplet at δ = 43.3 ppm (2JPP = 37 Hz) and a very broad singlet at δ = −68.8 ppm (w½ = 3000 Hz) (Fig. 3). Thus, despite the symmetry of the P4Cl2 ligand and the assumed low rotation barrier of the Cp* and cyclohexyl substituents, the four phosphorus atoms are chemically inequivalent. Low temperature NMR spectra were recorded in order to gain a deeper understanding of the dynamics of 1. At −40 °C, this dynamic behavior is frozen out and the spectrum displays five chemically inequivalent phosphorus atoms corresponding to an ABMXY spin system. The chemical inequivalence of P1/P2 and P3/P4 could stem from a chemical exchange in solution, for example dissociation of a chloride ion. However, the solid state 31P NMR spectrum likewise shows resonances corresponding to at least four inequivalent phosphorus atoms. Thus, it can only be accounted for by conformational changes, probably due to the hindered rotation of the bulky tris(cyclohexyl)phosphine. When the temperature in a solution NMR experiment is increased, the four phosphorus atoms of the P4Cl2 ligand become indeed pairwise equivalent due to enhanced molecular motion, leading to an A2MX2 spin system. Fitting of the experimental spectra and an Eyring plot revealed for the scrambling process an activation enthalpy of ΔH = 44.8(1) kJ mol−1 and ΔS = −18.5(6) Jmol−1 (see Fig. S1†).
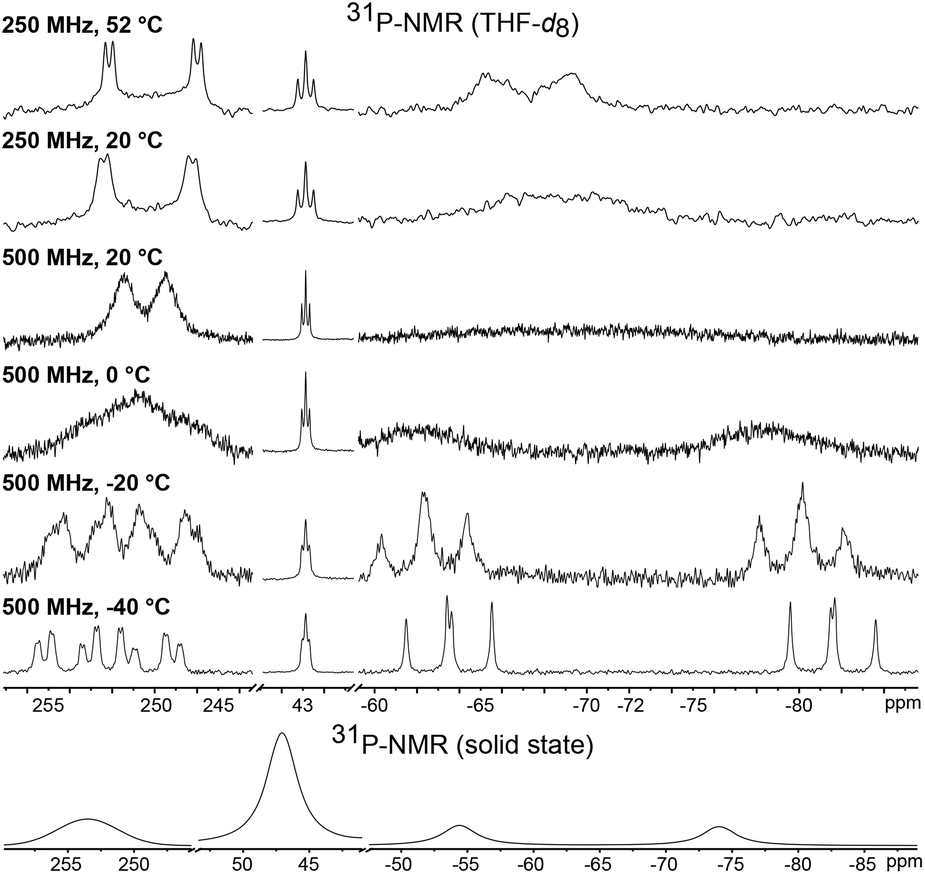 | ||
| Fig. 3 31P{1H} VT NMR spectra of 1 in THF-d8 at different field strengths (top) and in the solid state (bottom). The compound is only moderately stable at elevated temperature. Therefore, some experiments were carried out at a weaker external magnetic field. | ||
We further investigated the reactivity of the analogous ruthenium bromide complex [RuBr(Cp*)(PCy3)] towards P4. The addition of a half equivalent of white phosphorus gave the corresponding bromide complex [{Cp*Ru(PCy3)}(μ2,η2:4P4Br2){RuCp*}] (1-Br). The assignment of the same structure of the cognate chloride species to 1-Br is a consequence of the strict similarity between the 31P{1H} NMR spectra of the two compounds, which also share an identical fluxionality as is indicated by temperature dependent NMR spectra; see Fig. S2.†
Although the abovementioned possibility of dissociation of a chloride ion being a reason for the inequivalence of the 31P NMR nuclei could not be detected, the possibility of removing one or both of the chlorine centers from the P4Cl2 ligand by adding a Lewis-acid as a chloride scavenger was investigated. Indeed, exposure of 1 to gallium trichloride led to the formation of the cationic complex [Ru(Cp*)(PCy3)(μ2,η2:4-P4Cl)RuCp*]+[GaCl4]− (2). This compound is an unstable intermediate, which reacts further yielding complex 3 (see below) and could not be isolated in large quantities. However, a single crystal X-ray structure of the tetrachlorogallate salt 2 was obtained (Fig. 4). A comparison with the bond lengths of the neutral complex 1 revealed an elongation of the P1–P4 distance from 2.52 Å to 3.13 Å (Fig. 2) after chloride removal, yielding a distorted [P4Cl]+ ligand. This change makes the ruthenium centers closer; indeed examination of the X-ray structure of 2 shows that the Ru–Ru distance changes substantially from 3.709 Å in 1 to 3.102 Å in 2, which accounts for the formation of a long Ru–Ru bond. This distance is still within the expected range for the Ru–Ru single bond,10 and the elongation is likely due to the steric repulsion between Cp* ligands, and may contribute to the observed instability of 2 in solution. Thus, chloride abstraction removes an electron pair from the cluster electron count and the formation of a metal–metal bond restores the electron count to compensate.
 | ||
| Fig. 4 ORTEP of the P4Cl-bearing cation 2. Thermal ellipsoids are drawn at 50% probability; hydrogen atoms and the GaCl4− counter ion have been omitted for clarity. | ||
Complex 1 can be reduced to the complex [RuCp*(μ2,η2:2-P2)2RuCp*] (3) with elemental magnesium. Compound 3 is isolated as a light brown, crystalline solid in 82% yield. The single crystal X-ray structure shows again a planar P4 unit in a triple-decker arrangement between two RuCp* fragments (Fig. 5). The P1–P2 and P3–P4 distances (2.09 to 2.10 Å) are significantly shorter than the short P–P bonds in 1 and are in the range of PP double bonds (2.07 Å).15 But both the P2⋯P3 distance (2.59 Å) and especially the P1⋯P4 distance (3.70 Å) are significantly elongated, far above a P–P single bond length (2.21 Å).11 Consequently, the P4 ligand is cleaved in the reduction process into two P2 moieties. These bind in a μ,η2:2 coordination mode to the two Ru centers and each P2 unit is assumed to donate a total of four electrons, that is two electrons to each Ru center. A Ru–Ru distance of 2.726 Å is observed which is in between the length of a Ru–Ru single bond in metallic ruthenium (2.65 Å)11 and the distance observed in a carbonyl bridged cyclopentadienyl ruthenium complex [Ru2Cp2(CO)2(μ-CO)2] (2.73 Å).12 The Ru1–Ru2 distance in 3 is significantly shorter than the distance in the related triple-decker complexes [(RuCp*)2(μ2,η5:5-P5)] and [(triphos)Ru(μ2,η3:3-P3)Ru(triphos)]PF6, which feature a pentaphosphacyclopentadienyl (P5−) ligand (dRu–Ru = 3.73 Å)13 and a cyclotriphosphorus ligand (dRu–Ru = 4.21 Å), respectively.14 This suggests that 3 contains a Ru–Ru single bond which is further corroborated by a Bader analysis9 revealing a bond critical point (0.054 a.u.) between the two metal centers (see Fig. S4†). Thereby each Ru center reaches again a valence electron configuration of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 from Cp*−, 2 × 2 from each P2 unit, 1 from the Ru–Ru bond, and 7 from the d7-valence configuration of Ru(+1). This electron configuration may explain the remarkable stability of 3. Note that this complex is even formed when Cl− is abstracted from 1 with GaCl3 to give first the cationic intermediate 2. This seemingly decomposes according to [RuCp*(PCy3)(μ2,η2:4-P4Cl)RuCp*]+[GaCl4]− (2) → [RuCp*(μ2,η2:2-P2)2RuCp*] (3) + [PCy3Cl]+ [GaCl4]− although we could not detect the chlorophosphonium salt as an oxidized by-product and hence the exact mechanism for the formation of 3 from 1 and GaCl3 remains speculative.
:6 from Cp*−, 2 × 2 from each P2 unit, 1 from the Ru–Ru bond, and 7 from the d7-valence configuration of Ru(+1). This electron configuration may explain the remarkable stability of 3. Note that this complex is even formed when Cl− is abstracted from 1 with GaCl3 to give first the cationic intermediate 2. This seemingly decomposes according to [RuCp*(PCy3)(μ2,η2:4-P4Cl)RuCp*]+[GaCl4]− (2) → [RuCp*(μ2,η2:2-P2)2RuCp*] (3) + [PCy3Cl]+ [GaCl4]− although we could not detect the chlorophosphonium salt as an oxidized by-product and hence the exact mechanism for the formation of 3 from 1 and GaCl3 remains speculative.
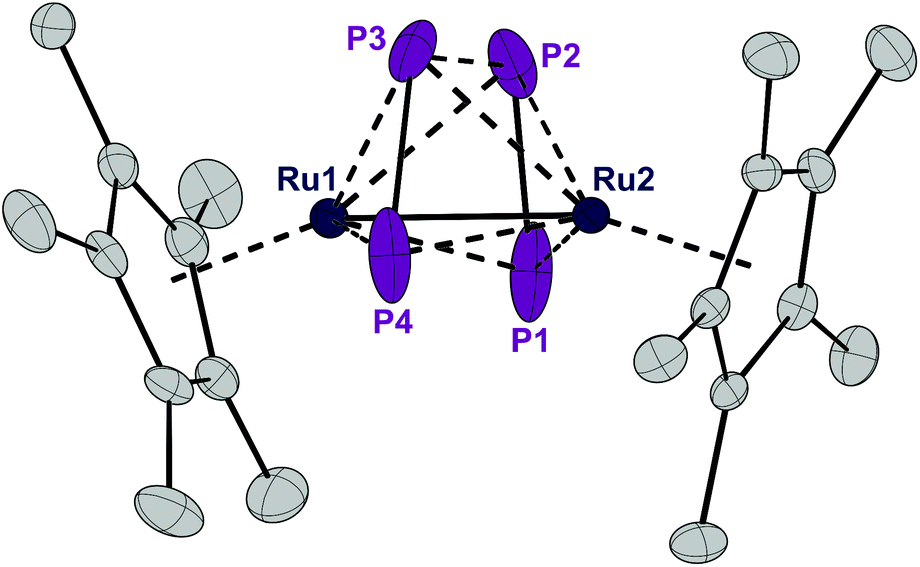 | ||
| Fig. 5 ORTEP of the P4-bearing complex 3. Thermal ellipsoids are drawn at 50% probability; hydrogen atoms have been omitted for clarity. | ||
A 31P NMR spectrum recorded in solution at 20 °C shows a single, sharp resonance at δ = 30.0 ppm which is inconsistent with the asymmetric arrangement of the phosphorus atoms as observed in the crystal structure, for which two resonances are expected. The spectra did not change upon cooling the sample to −50 °C which indicates that the compound may either possess a more symmetric structure in solution or is highly fluxional making the four P atoms chemically equivalent on the NMR time scale. The trapezoid arrangement of the two P2-units may be caused by packing effects in the solid state. We therefore employed DFT calculations in order to determine the structure of the electronic ground state of 3. The calculated structure of an isolated simplified model compound (with Cp instead of Cp*) resembles the solid state structure of 3 with two P2 units in a trapezoidal arrangement. We therefore assume that the equivalence of the 31P NMR resonances is the result of fluxionality. The 31P solid state NMR spectrum at 20 °C as well shows only a single resonance indicating that this dynamic phenomenon is also observed in the solid state at 20 °C. Unfortunately, we could not measure a solid state 31P NMR spectrum at lower temperatures for technical reasons.
A comparison of the electron density plots from the X-ray structures at 100 K and 300 K further supports the assumption of dynamic behavior also in the solid state (see Fig. S3†). The plot at 300 K shows an overlap of the electron densities of all four phosphorus atoms, suggesting molecular movement, which renders them chemically equivalent. A Bader analysis reveals bond critical points between P1 and P2 as well as P3 and P4 (0.120 a.u.). A weaker bond critical point is observed between P2 and P3 (0.044 a.u.) and no bond critical point is found between P1 and P4 (see Fig. S4†). Thus, the ligand is best described as two very weakly bound P2 units which have a π–π* interaction between P2 and P3.
Similar compounds with planar P4 ligands between two metal centers have been prepared previously, albeit in very tedious and low yielding syntheses.16 We compared the structures and 31P NMR signals of these compounds to those of 3 (Fig. 6). The Rh-complex 5a shows a parallel arrangement of two P2 units and accordingly, a single resonance in the 31P NMR spectrum.16a The distortion observed in 3 could be caused by electric repulsion generated by the metal–metal bond, which is necessary to fulfill the 18 valence electron count in the Ru-complex 3 but not in the Rh-complex 5a. The Fe-complex 5b shows two parallel P2 units and a single, albeit broad singlet 31P NMR resonance.16b A single bonding Fe–Fe distance of 2.59 Å is reported but does not lead to any distortion in the phosphorus ligand. Similarly, the bimetallic Ru-complex 5c also possesses two parallel P2-units, in which the P atoms are magnetically equivalent.16c On the other hand, the Fe-complex 5d, which differs from 5b only in the more sterically demanding Cp-ligand, possesses a butadienyl-like P4 chain.16d The structural characteristics of 3 lie between those of 5c and 5d.
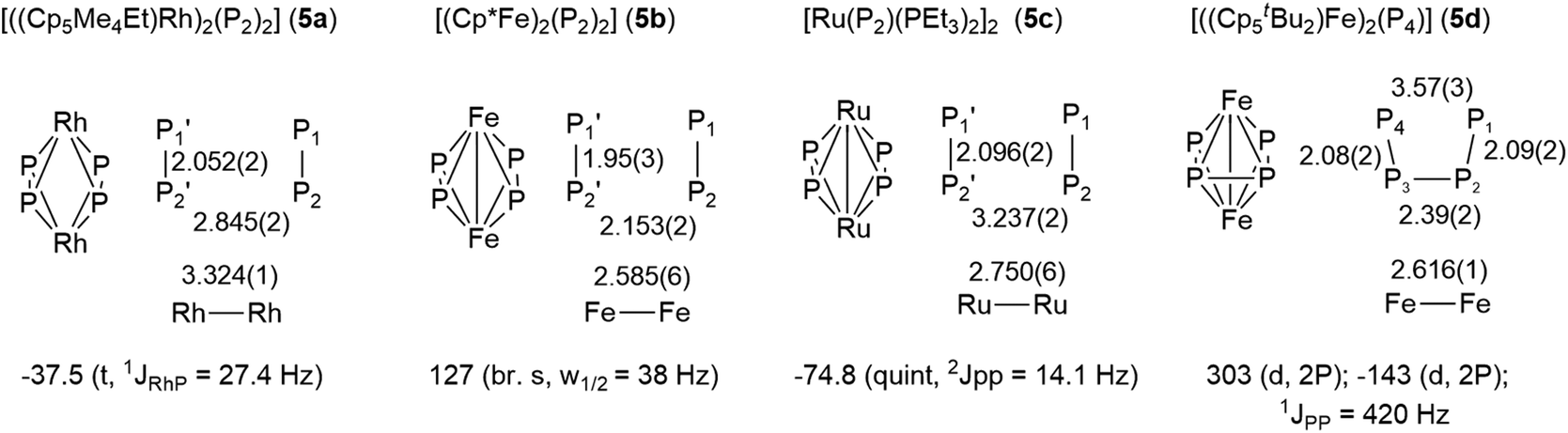 | ||
| Fig. 6 Structures, bond lengths and 31P NMR signals of the P4 unit of known compounds with a planar P4 ligand. | ||
The presence of chloro substituents in the P4Cl2 moiety of 1 led us to explore the possibility of exchanging the chloride substituents with organic groups using common synthetic protocols employing Grignard or alkyl lithium reagents. Indeed, a salt metathesis reaction with n-butyllithium leads to the selective formation of a new complex 4 with a planar organophosphorus ligand, P4nBu2, which was characterized by an X-ray diffraction study using single crystals. The structure is shown in Fig. 7 and reveals a tetranuclear compound [(RuCp*)4(μ3,η2:2:4-P4nBu2)2] (4) in which the P4nBu2 moiety serves as a bridging ligand between the Ru centers. We assume that the displacement of tricyclohexylphosphine during the reaction leads to a free coordination site at Ru1 and thus promotes dimerization, yielding a tetrametallic compound bearing two coplanar [P4nBu2] moieties.
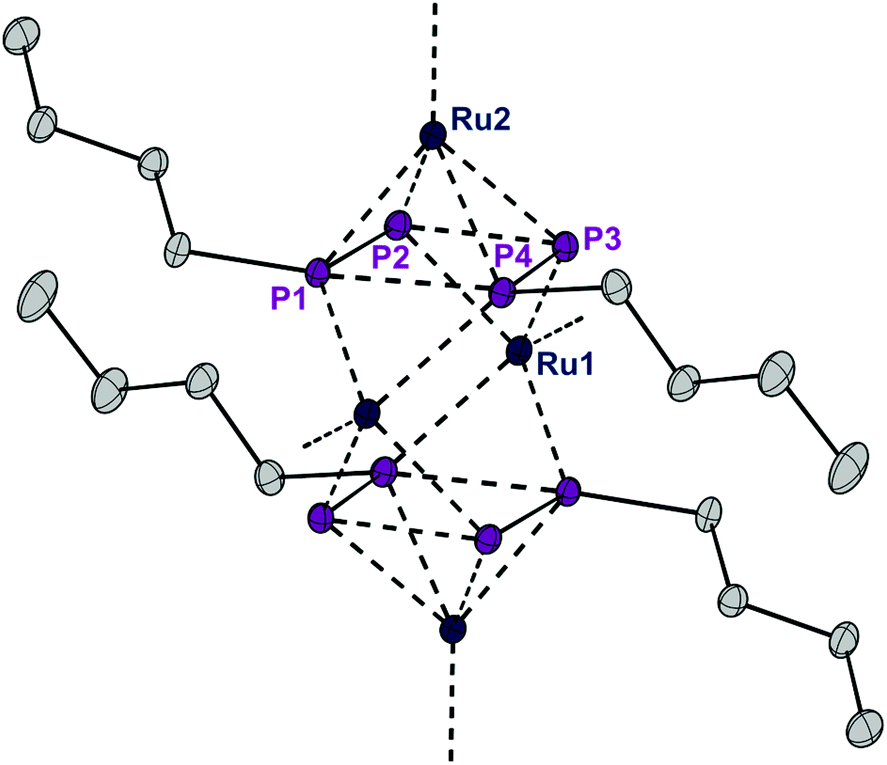 | ||
| Fig. 7 ORTEP of complex 4. Thermal ellipsoids are drawn at 50% probability; hydrogen atoms and the Cp* rings have been omitted for clarity. | ||
The atomic charges of model complexes of all complexes 1–4 (Cp instead of Cp* and Me instead of Cy) were determined by natural population analysis (Table 1).9 They reveal that the ruthenium centers, despite having formal oxidation states of +1 and +2, carry a negative charge in all cases. This suggests that the “P4” units – either as such or as two P2 groups – together with the Cp and PMe3 ligands are strongly electron donating. Furthermore, the charges of the cationic complex 2 do not differ significantly from those of the neutral complex 1, implying that the resulting positive charge of 2 is delocalized over the entire molecule. The individual charges in complex 3 which contains two P2 units as ligands do not differ significantly from the charges of the P4Cl2-complex 1.
The reduction of complex 1 by two electrons leads to a remarkable reorganization of the structure of the central P4 unit. In this process, PCy3 is lost as a two electron donor together with two chloride ions. Curiously, the formal loss of two electrons in this reduction process is compensated by the formation of a Ru–Ru bond and the two Ru centers retain on average their formal oxidation states (Ru+2/Ru0 in 1; 2 × Ru+1 in 3) while the P4 units in 1 (P4Cl2) and 3 (2 × P2) serve in each case as donors of a total of eight electrons to both metal centers. In summary, this work in combination with the previous results displayed in Fig. 6 shows that a P4 unit can act as an electronically highly flexible, non-innocent ligand rendering the assignment of oxidation states to the metal centers and charges to the phosphorus ligand almost meaningless.
Conclusions
The reaction between two equivalents of the reactive 16 valence electron species [RuX(Cp*)(PCy3)] (X = Cl, Br) and white phosphorus probably gives a first intermediate, in which the P4 tetrahedron is simply coordinated to two Ru centers. This reaction is rapidly followed by an unexpected insertion of the P4 unit into the Ru–X bond, which leads to an unprecedented planar P4X2 ligand bridging the two Ru centers in a η2- and η4-fashion, respectively. This reaction may be viewed as a metal mediated nucleophilic attack of two chloride anions on the P4 cage and electronic re-organization within the product to give a formally dinuclear Ru+2/Ru0 complex. A strong reducing agent like elemental magnesium is able to remove the chloro substituents as chloride anions and the P4Cl2 unit is converted into two P2 units within the coordination sphere of the Ru centers. Formally, the latter product 3 contains two Ru(I) centers which are connected by a Ru–Ru single bond and bridged by two weakly interacting P2 units. Furthermore, the chloro substituents in the P4Cl2 ligand can be exchanged for alkyl substituents using a classical organometallic reagent, such as nBuLi. In this transformation [RuCl(Cp*)(PCy3)] serves as a platform for a controlled activation and functionalization of white phosphorus yielding a new P4R2 organophosphorus ligand. This reaction can be formally written as 2 [RuCl(Cp*)(PCy3)] + P4 + 2 nBuLi → ½ [Ru2(Cp*)2(P4nBu2)]2 + 2 LiCl + 2 PCy3 which proceeds under mild conditions without any degradation or re-aggregation of the P4 units. DFT calculations show that neither the Ru centers, which carry a negative charge, nor the phosphorus centers, which carry a positive charge, change significantly their charges throughout all transformations reported in this paper. The P4 units serve in all instances as strong electron donors to the Ru centers. The observations reported here remain to a certain extent puzzling. Given that the d7-RuCp* fragment is isolobal to BH+ and each P center isolobal to BH−, a regular octahedral Ru2P4 structure as in the closo-borane dianion [B6H6]2− would have been expected according to the Wade–Mingos-rules. However, attempts to optimize such a structure by DFT methods failed and led to the irregular structure observed in 3. In combination with the previous results shown in Fig. 6, this study demonstrates again the remarkable electronic flexibility of P4 ligands. In the long term, it may contribute to develop selective, and eventually even catalytic, ways to convert white phosphorus into new organophosphorus species in an efficient manner.5. Experimental section
General details
All reactions were carried out under argon using either standard Schlenk techniques or an argon-filled glove box. The glassware was stored in an oven at 130 °C for at least 16 hours and cooled under vacuum prior to use. Solvents were purified using an Innovative Technology PureSolv MD 7 solvent purification system. Deuterated solvents were purchased from Cambridge Isotope Laboratories. THF-d8 and C6D6 were distilled from sodium benzophenone, whereas CD2Cl2 was dried over 4 Å molecular sieves before use. All reagents were used as received from commercial suppliers. White phosphorus was distilled prior to use and stored and weighed under argon. [RuCp*Cl]4 and [RuCp*Br]4 were synthesized according to a literature procedure.17Solution NMR spectra were recorded on Bruker 250 MHz, 300 MHz, 400 MHz and 500 MHz spectrometers. All 1H and 13C chemical shifts are reported in ppm relative to SiMe4 using the 1H and 13C shifts of the solvent as an internal standard. 31P NMR shifts are reported relative to 85% H3PO4. Solid state NMR spectra were recorded on Bruker 400 MHz and 700 MHz spectrometers equipped with 2.5 mm and 4 mm two-channel solid-state probe heads. The sample was contained in a 2.5 or 4 mm rotor and spun at 10 or 20 kHz.
31P{1H} NMR (THF-d8, 203 MHz, 233 K): δ = 253.8 (ddd, 1JPP = 427 Hz, 1JPP = 137 Hz, 2JPP = 32 Hz, 1P, P1), 246.8 (ddd, 1JPP = 429 Hz, 1JPP = 137 Hz, 2JPP = 34 Hz, 1P, P4), 43.3 (t, 2JPP = 32 Hz, 2JPP = 34 Hz, 1P, PPCy3), −62.5 (dd, 1JPP = 429 Hz, 2JPP = 378 Hz, 1P, P3), −80.1 (dd, 1JPP = 427 Hz, 1JPP = 378 Hz, 1P, P2) ppm.
1H-NMR (300 MHz, C6D6, 298 K): δ = 1.87 (m, 6H, CH2 p-, PCy3), 1.68 (m, 12H, CH2 m-, PCy3), 1.46 (m, 18H, CH, PCy3 + Cp*), 1.18 (m, 12H, CH, CH2 o-, PCy3) ppm.
31P{1H} NMR (121 MHz, C6D6, 295 K): δ = 41.9 (s) ppm. 13C NMR (75.5 MHz, C6D6, 298 K) = 74.6 (Cp*), 34.6 (d, 1JCP = 16 Hz, CH), 30.9 (CH2 m-) 28.2 (d, 2JCP = 8.5 Hz, CH2 o-), 26.8 (CH2 p-), 11.6 (CH, CH3) ppm.
1H-NMR (THF-d8, 300 MHz, 20 °C): δ = 1.90 (s, 30 H, CH3), 1.84–1.26 (m, 33 H, PCy3) ppm. 31P{1H} NMR (THF-d8, 121 MHz, 20 °C): δ = 261.0 (br. d, w½ = 255 Hz, 1JPP = 387 Hz, 2P, P1–P4), 39.9 (t, 37 Hz, 1P, PPCy3), −58.0 (br s, w½ = 1300 Hz, 2P, P2–P3) ppm.
31P{1H} NMR (THF-d8, 121 MHz, 233 K): δ = 253.8 (ddd, 1JPP = 415 Hz, 1JPP = 160 Hz, 2JPP = 35 Hz, 1P, P1), 246.8 (ddd, 1JPP = 417 Hz, 1JPP = 160 Hz, 2JPP = 34 Hz, 1P, P4), 39.9 (t, 2JPP = 35.1 Hz, 1P, PPCy3), −54.1 (dd, 1JPP = 417 Hz, 1JPP = 383 Hz, 1P, P3) −72.4 (dd, 1JPP = 415 Hz, 1JPP = 378 Hz, 1P, P2) ppm.
1H NMR (CD2Cl2, 300 MHz): δ = 1.92 (s, CH3) ppm. 13C{1H}NMR (CD2Cl2, 75 MHz): δ = 95.9 (C5), 12.6 (CH3) ppm. 31P{1H} NMR (CD2Cl2, 121 MHz): δ = 30.0 ppm.
Solution instability prevented the collection of NMR data for compound 4.
X-Ray crystallographic data collection and refinement of the structures
Single crystals suitable for X-ray diffraction were coated with polybutene oil (Mn ≈ 920 g mol−1) in a glovebox and transferred to a nylon loop. Diffraction studies were performed on a Rigaku Oxford Diffraction XCalibur S or a Bruker X8 APEX2 diffractometer, both equipped with a molybdenum X-ray tube (λ = 0.7107 Å). Preliminary data were collected to determine the crystal system. These data were processed using the corresponding diffractometer software (CrysAlisPro v38.41 and Apex2 v.2014.1)18 and corrected for absorption with a multi-scan method.The structures were solved using direct methods (ShelXS)19 or intrinsic phasing (ShelXT) and refined using least squares procedures (ShelXL) with Olex2 v1.2.8.20
CCDC reference numbers are as follows: 1: 1815071, 2: 1815069, 3: 1815070 and 4: 1815072. Crystallographic data are reported in Table S1.†
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the European Union (Marie Curie ITN SusPhos, Grant Agreement No. 317404) for financing a doctoral grant to F. Delgado Calvo and to M. Bispinghoff as well as NKFIH (PD 116329). MIUR (Italian Ministero dell'Istruzione, dell'Università e della Ricerca) is acknowledged for financing the project PRIN 2015 “Towards a Sustainable Chemistry: Design of Innovative Metal–Ligand Systems for Catalysis and Energy Applications” (contract no. 20154X9ATP). Michael Wörle (XRD), René Verel (NMR) and Carlo Bartoli are acknowledged for technical support.References
- D. E. C. Corbridge, Phosphorus, an Outline of its Chemistry, Biochemistry and Technology, Elsevier, Amsterdam, 5th edn., 1995 Search PubMed.
- (a) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed; (b) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed; (c) M. Peruzzini, L. Gonsalvi and A. Romerosa, Chem. Soc. Rev., 2005, 34, 1038–1047 RSC; (d) M. Peruzzini, R. R. Abdreimova, Y. Budnikova, A. Romerosa, O. J. Scherer and H. Sitzmann, J. Organomet. Chem., 2004, 689, 4319–4331 CrossRef CAS.
- (a) J. E. Borger, M. S. Bakker, A. W. Ehlers, M. Lutz, J. Chris Slootweg and K. Lammertsma, Chem. Commun., 2016, 52, 3284–3287 RSC; (b) B. M. Cossairt and C. C. Cummins, Angew. Chem., Int. Ed., 2010, 49, 1595–1598 CrossRef CAS PubMed; (c) B. M. Cossairt and C. C. Cummins, Angew. Chem., Int. Ed., 2008, 47, 8863–8866 CrossRef CAS PubMed; (d) P. Barbaro, M. Peruzzini, J. A. Ramirez and F. Vizza, Organometallics, 1999, 18, 4237–4240 CrossRef CAS.
- (a) P. Barbaro, C. Bazzicalupi, M. Peruzzini, S. Seniori Costantini and P. Stoppioni, Angew. Chem., Int. Ed., 2012, 51, 8628–8631 CrossRef CAS PubMed; (b) P. Barbaro, M. Di Vaira, M. Peruzzini, S. Seniori Costantini and P. Stoppioni, Angew. Chem., Int. Ed., 2008, 47, 4425–4427 CrossRef CAS PubMed; (c) M. Di Vaira, P. Frediani, S. S. Costantini, M. Peruzzini and P. Stoppioni, Dalton Trans., 2005, 2234–2236 RSC; (d) P. Barbaro, A. Ienco, C. Mealli, M. Peruzzini, O. J. Scherer, G. Schmitt, F. Vizza and G. Wolmershäuser, Chem. – Eur. J., 2003, 9, 5195–5210 CrossRef CAS PubMed.
- E. Mädl, M. V. Butovskii, G. Balázs, E. V. Peresypkina, A. V. Virovets, M. Seidl and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 7643–7646 CrossRef PubMed.
- B. M. Cossairt, M.-C. Diawara and C. C. Cummins, Science, 2009, 323, 602 CrossRef CAS PubMed.
- D. N. Akbayeva, Russ. J. Coord. Chem., 2006, 32, 329–334 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- The Wiberg bond indices and the NPA charges were obtained with NBO 3.1., see: E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.10, TCI, University of Wisconsin, Madison, 1998 Search PubMed.
- J.-I. Ito, T. Shima and H. Suzuki, Organometallics, 2004, 23, 2447–2460 CrossRef CAS.
- Tables of Interatomic Distances and Configuration in Molecules and Ions Interatomic Distances, Interatomic Distances Supplement - Special Publication No. 18, ed. L. E. Sutton, Chemical Society, London, 1965 Search PubMed.
- O. S. Mills and J. P. Nice, J. Organomet. Chem., 1967, 9, 339–344 CrossRef CAS.
- A. R. Kudinov, D. A. Loginov, Z. A. Starikova, P. V. Petrovskii, M. Corsini and P. Zanello, Eur. J. Inorg. Chem., 2002, 3018–3027 CrossRef CAS.
- F. Fabrizi de Biani, M. Corsini, A. Ienco, M. Peruzzini and F. Zanobini, Inorg. Chim. Acta, 2018, 470, 428–432 CrossRef CAS.
- H. Borrmann, I. Kovacs and G. Fritz, Z. Anorg. Allg. Chem., 1994, 620, 1818–1820 CrossRef.
- (a) O. J. Scherer, M. Swarowsky and G. Wolmershäuser, Angew. Chem., Int. Ed. Engl., 1988, 27, 405–406 CrossRef; (b) M. E. Barr and L. F. Dahl, Organometallics, 1991, 10, 3991–3996 CrossRef CAS; (c) Ł. Ponikiewski, T. Kruczyński, M. Caporali, M. Peruzzini, D. Gudat, M. Walaszkowska and J. Pikies, Eur. J. Inorg. Chem., 2016, 4241–4249 CrossRef; (d) O. J. Scherer, G. Schwarz and G. Wolmershäuser, Z. Anorg. Allg. Chem., 1996, 622, 951–957 CrossRef CAS; (e) V. A. Miluykov, O. G. Sinyashin, P. Lönnecke and E. Hey-Hawkins, Mendeleev Commun., 2003, 13, 212–213 CrossRef; (f) F. Dielmann, M. Sierka, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2010, 49, 6860–6864 CrossRef CAS PubMed.
- P. J. Fagan, M. D. Ward and J. C. Calabrese, J. Am. Chem. Soc., 1989, 111, 1698–1719 CrossRef CAS.
- CrysAlis CCD, Oxford Diffraction Ltd., Yarnton, Oxford, UK, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC range 1815069–1815072. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt01840e |
| This journal is © The Royal Society of Chemistry 2019 |