
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly luminescent lanthanide complexes sensitised by tertiary amide-linked carbostyril antennae†
Daniel
Kovacs
,
Dulcie
Phipps
,
Andreas
Orthaber
 and
K. Eszter
Borbas
*
and
K. Eszter
Borbas
*
Department of Chemistry, Ångström Laboratory, Uppsala University, Box 523, 75120, Uppsala, Sweden. E-mail: eszter.borbas@kemi.uu.se
First published on 13th June 2018
Abstract
Carbostyrils are among the most widely used sensitising antennae for luminescent lanthanides; they afford bright complexes with Eu and Tb, and can also sensitise the emissions of the less commonly used Sm, Dy, Yb and Nd. Systematic studies on the effect of structural variations on the photophysical properties and lanthanide sensitising abilities of carbostyrils can therefore have a large impact. We replaced the secondary amide linker that connects the metal binding site to the antenna with a carboxymethyl-substituted tertiary amide. Eight Tb and Eu complexes were prepared. All had higher lanthanide luminescence quantum yields (ΦLn) than their secondary amide analogues; three Tb emitters had ΦTb > 40%. Eu complexes had ΦEu up to 11.6%. The antenna singlet and triplet excited states are slightly shifted, while the metal coordination sphere is unchanged by the introduction of the carboxymethyl group.
Introduction
Lanthanide (Ln)-based emitters occupy a unique niche among luminescent compounds. They have long emission lifetimes, narrow emission bands, are often highly photostable, and have negligible phototoxicities.1–6 These properties are in sharp contrast to the rapid degradation, broad emission profiles and short lifetimes of organic emitters, or the toxicity of transition metal-based phosphorescent dyes or quantum dots. Ln(III) emission results from Laporte-forbidden f–f-transitions, and direct Ln(III) excitation is inefficient. Sensitisation by a light-harvesting antenna is common, and bypasses the small extinction coefficients of the Ln(III). Energy transfer (ET) from the antenna to the Ln can be efficient, and in the most successful cases, bright luminescent complexes are obtained.2,7 The brightness of Ln(III) emitters (B = ε·Φ; ε: molar decadic absorption coefficient at λex, Φ: dye's fluorescence quantum yield) depends on several factors, e.g. the number of absorbing and emitting units,8–11 the efficiency of the antenna absorption and of the energy transfer,12,13 the intrinsic quantum yield of the Ln(III), and the quenching processes that deplete the antenna and Ln(III) excited states.14,15The development of new emitters is a lengthy and high-risk task. Therefore, there are substantial efforts directed towards the optimization of already reported luminescent Ln complexes, which encompass the understanding of the energy transfer mechanism and, if possible, elimination of quenching pathways. A well-known Ln excited state quenching pathway involves X–H overtones (X = O, N, C)16,17 but can be avoided by the saturation of the Ln inner coordination sphere with a multidentate ligand, and, in some cases, by ligand deuteration.18,19
The quenching of the antenna excited state by atmospheric oxygen21–23 and biologically relevant reductants has also been studied.4,24,25 These quenching processes could be harnessed for the construction of responsive probes, or environmentally-activated Ln-based theranostics.23,26–28
Carbostyrils (quinolin-2(1H)-ones) are among the most widely used antennae for the sensitisation of Eu and Tb, of which the most commonly used one is cs124 (Scheme 1, 2a).29–37 Some are even effective for Sm and Dy,38 as well as the near infrared (NIR) emitting Yb and Nd.39 A variety of substituted carbostyrils have been reported (Fig. 1a).38,40 Many were evaluated as antennae, even though in-depth photophysical characterizations are rare.39 Most of the structural variations were limited to the peripheral substituents, usually in the 3 and 4 (R1 and R2, respectively in Fig. 1) positions. There are also a few examples of core N-substitutions (alkylations).34 The effects of exocyclic N-alkylations on Ln sensitization have not been studied in detail, presumably because changes were expected to be small.
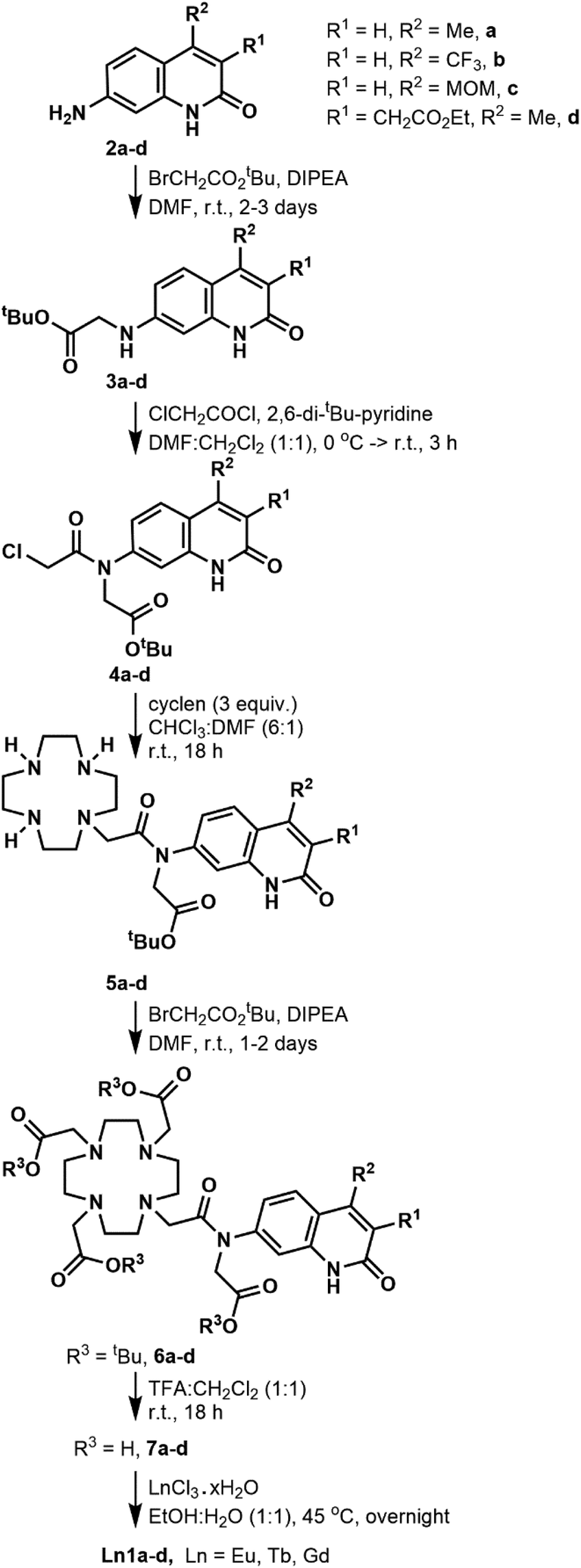 | ||
| Scheme 1 Synthesis of the Ln complexes studied here. | ||
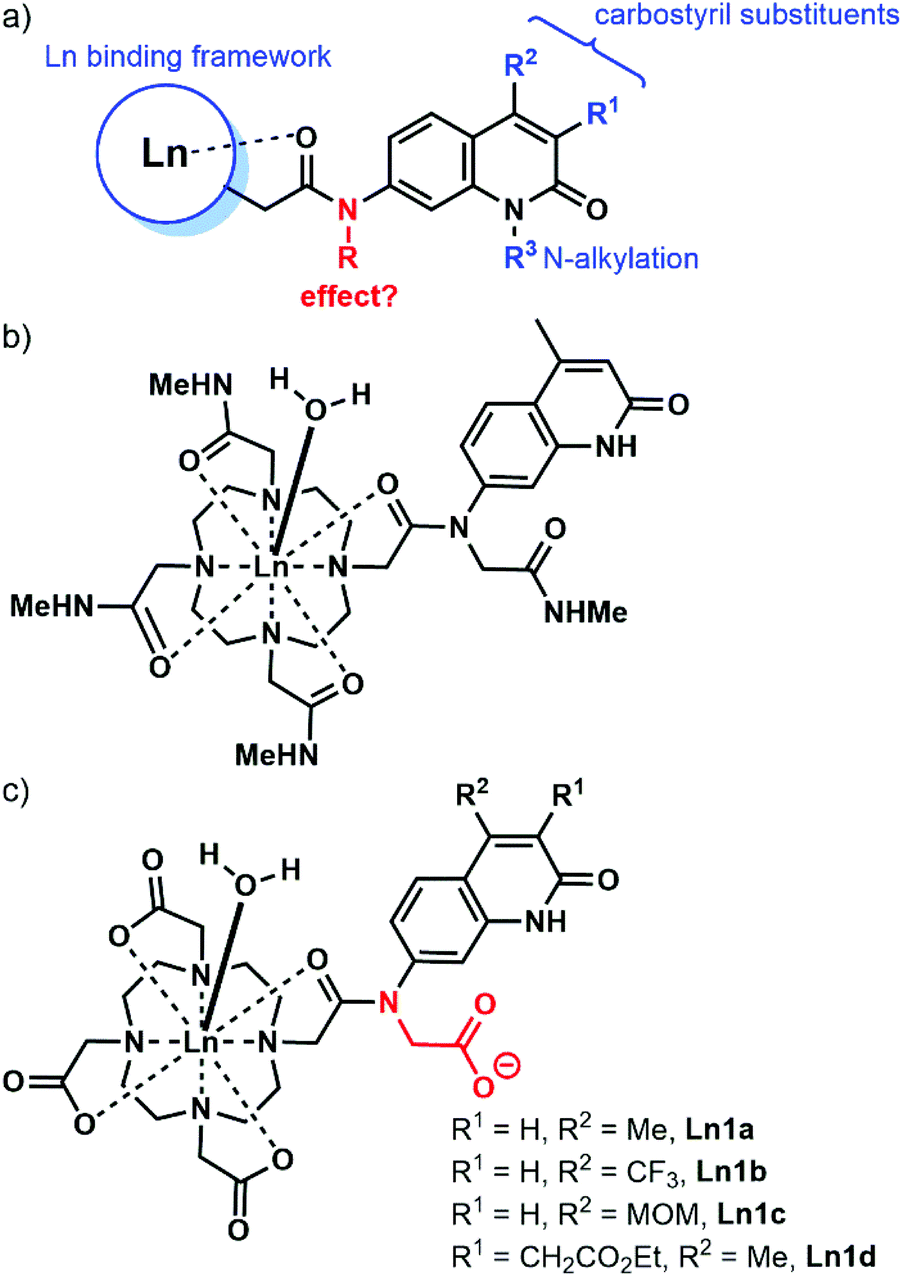 | ||
| Fig. 1 (a) Common variations shown in blue in carbostyril-appended Ln complexes. (b) Eu complex reported by Parker and Williams in ref. 20. (c) Complexes studied here. | ||
We hypothesised that the removal of the N–H bond may have a measurable effect on the Ln emission quantum yield, at least for the more sensitive Eu complexes. The majority of the reported carbostyril-appended Ln-emitters retain this N–H bond. Parker and Williams have prepared the tetraamide shown in Fig. 1b.20 However, the methylamide arms bring further N–H oscillators into the proximity of the Ln. Furthermore, the +3 charge of this complex facilitates photoinduced electron transfer (PeT) from the excited antenna to the Eu by destabilizing Eu3+. For most of the sensitised Eu(III) emitters, PeT quenches the luminescence.41 Because of the combination of detrimental processes the evaluation of the contribution of the N-alkylation to the photophysics of the complex shown in Fig. 1b difficult. Here, we investigate the role of exocyclic N-alkylation in carbostyril-sensitised DO3A-type Ln complexes (Fig. 1c, DO3A = 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid). Surprisingly, we found that N-alkylation with a carboxymethyl group afforded a dramatic increase in Ln emission for both Eu and Tb emitters. We attempt to explain these results based on spectroscopic and structural analyses.
Results and discussion
Synthesis
The DO3A-derived ligands were synthesised as shown in Scheme 1. For the Ln complex numbers see Fig. 1c. The general procedure was amenable to the preparation of all four ligands without significant adjustments in the protocols. Briefly, the carbostyrils 2a–d were N-alkylated with tert-butyl bromoacetate in the presence of DIPEA. Acylation of secondary amines of 3a–d was performed with 2,6-di-tert-butylpyridine base. Other, less hindered and more nucleophilic bases (e.g. Et3N) could not be used, as they got acylated by chloroacetyl chloride faster than the modestly nucleophilic carbostyril amines. The chloroacetylated derivatives 4a–d were obtained in at least 72% yield after column chromatography on silica gel. Monoalkylation of cyclen yielded 5a–d along with small amounts of di- and trialkylated side-products, which were readily removed upon purification. Less side-product was seen than in similar reactions of secondary amide carbostyrils, as due to the better solubilities of 4a–d in CHCl3 much less DMF co-solvent was needed, which improved the selectivity.The secondary amines in 5a–d were alkylated in DMF in the presence of DIPEA base. These conditions minimise the formation of the by-products that are N- or O-alkylated in the carbostyril core. The drawback of these conditions is that the DIPEA·HBr co-elutes with the product on silica gel in CHCl3/acetone/MeOH systems. Therefore, the protected ligands 6a–c required several chromatographic purification steps, and the purified products still contained varying amounts of DIPEA·HBr and DMF. For 6d CHCl3/acetone/EtOH eluent worked best, and an analytically pure sample was isolated after a single chromatographic step. However, a large amount of the product co-eluted with the DMF residues of the reaction mixture, which diminished the yield. Other bases (e.g. Na2CO3) afforded the N- or O-alkylated by-products. Finally, the tert-butyl esters were cleaved with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CH2Cl2 and TFA. The reactions proceeded to completion overnight at room temperature, as shown by HPLC-analysis of the reaction mixtures. The ligands were isolated after column chromatography as white (6a,c) or yellow-white (6b,d) solids in at least 81% yield.
:1 mixture of CH2Cl2 and TFA. The reactions proceeded to completion overnight at room temperature, as shown by HPLC-analysis of the reaction mixtures. The ligands were isolated after column chromatography as white (6a,c) or yellow-white (6b,d) solids in at least 81% yield.
We have explored an alternative route to these ligands by reacting the known 8c39 with tert-butyl bromoacetate in acetonitrile at 70 °C in the presence of Na2CO3 (Scheme 2). After overnight reaction no carbostyril N-alkylation was observed either in the core or the exocyclic nitrogen. Longer reaction times yielded a mixture of 10c and 13c. Attempted alkylation of 5c at 50 °C gave three observable products upon HPLC analysis of the reaction mixture. After their separation the major species was identified as 11c. This sample was contaminated by approximately 5% of a product tentatively identified as 12c, based on its HPLC-behaviour, mass spectrum and UV-Vis absorption spectrum. The desired product under these unoptimised conditions was isolated in 36% yield. Due to the observed overalkylations we did not pursue further this route.
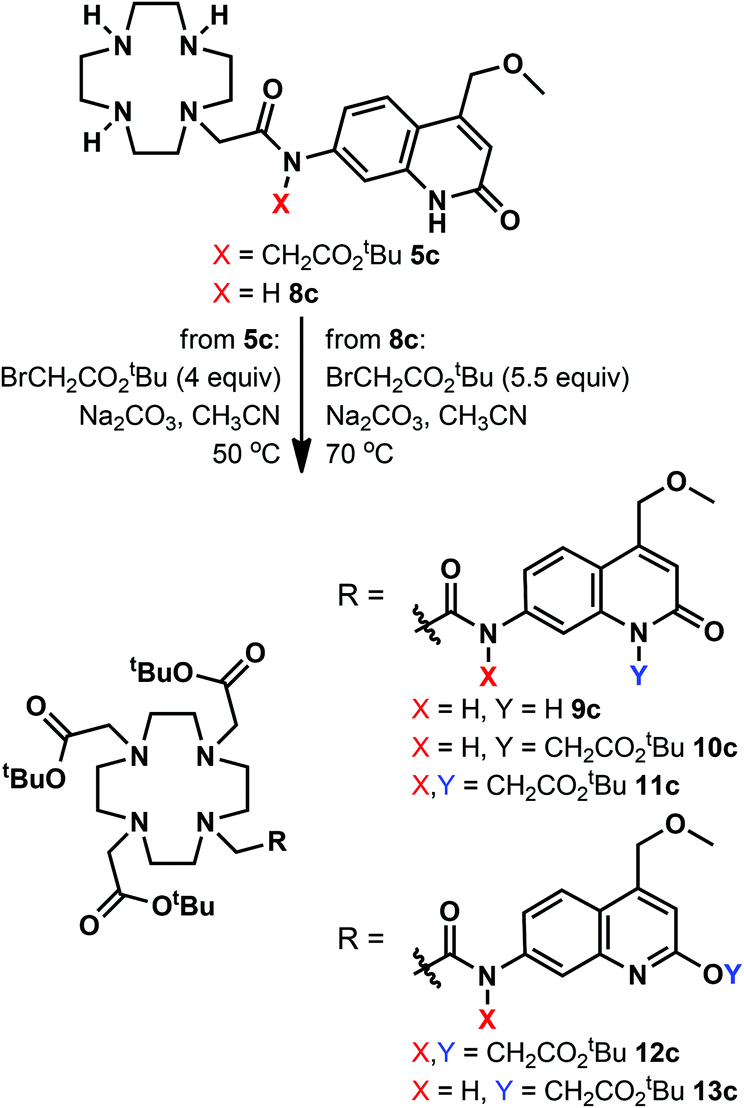 | ||
| Scheme 2 Attempted alternative syntheses of the N-alkylated ligands. | ||
Complexation with EuCl3, TbCl3 and GdCl3 was carried out in EtOH:H2O (1:1) mixture. The reaction was complete after 18 h according to HPLC-MS analysis of the reaction mixture. After completion, the crude products were isolated by extraction upon washing with Et2O (dropwise addition of the reaction mixture to Et2O). After layer separation, the aqueous phase was purified by column chromatography on silica gel. It was crucial to keep the stationary phase short. Elution from a longer column required the addition of aqueous ammonia to the eluent, which resulted in partial loss of the lanthanide ion.
Chemical characterisation
The identities of 2–7 were confirmed by 1H and 13C NMR spectroscopy and high resolution mass spectrometry (see ESI† for details). We were able to grow X-ray quality crystals from 5a (Fig. 2, S1†). The cyclen moiety is disordered over two positions in the free ligand, which was modeled as a positional disorder without any geometric constraints of the two units. The site occupation factors are 0.592 and 0.408 for the major and the minor components, respectively.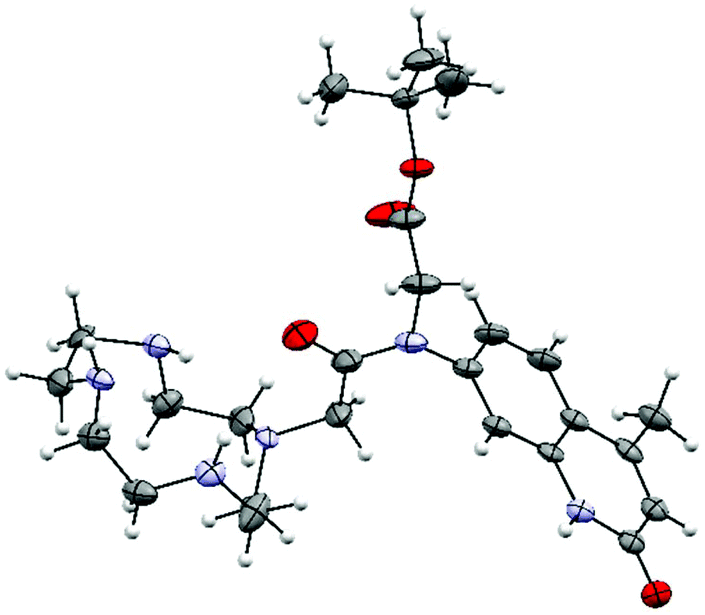 | ||
| Fig. 2 Crystal structure of 5a. Thermal ellipsoids are shown at the 30% (cyclen) probability levels. For clarity, only one of the disordered cyclen parts is shown. | ||
The complexes were shown to be pure by HPLC-MS analysis (see ESI†). High resolution mass spectrometry (HR-MS) of the Ln complexes showed the deprotonated, singly negatively charged molecule ions with the expected isotope distribution pattern. Further support for the identities of the metal complexes was provided by their photophysical properties (vide infra). Briefly, Eu and Tb complexes displayed the characteristic red and green Ln emissions, respectively, while Gd complexes only had antenna-based photophysical activities.
We could obtain crystals from a Dy complex of the non-N-alkylated analogue of the 1d ligand (Dy9d, Fig. 3). This structure shows a different configuration of the antenna-linking amide compared to that found in the current ligands, which may impact the photophysical properties (vide infra). The Dy center shows a classical monocapped square antiprismatic arrangement typical of this type of complexes. The four carboxylic oxygen and four nitrogen atoms form two near ideal planes that are almost coplanar; the angle between the least square planes is only 0.63(8)°. One additional water molecule caps the face spanned by O4 to O7. The O–Dy distances fall into two regimes: 2.300(2)–2.332(2) Å and 2.423(2)/2.433(2) Å. The two longer distances are found for the amide oxygen (O7-Dy) and capping water (O3-Dy1). The Dy–N distances are in the range from 2.600(2) to 2.657(2) Å.
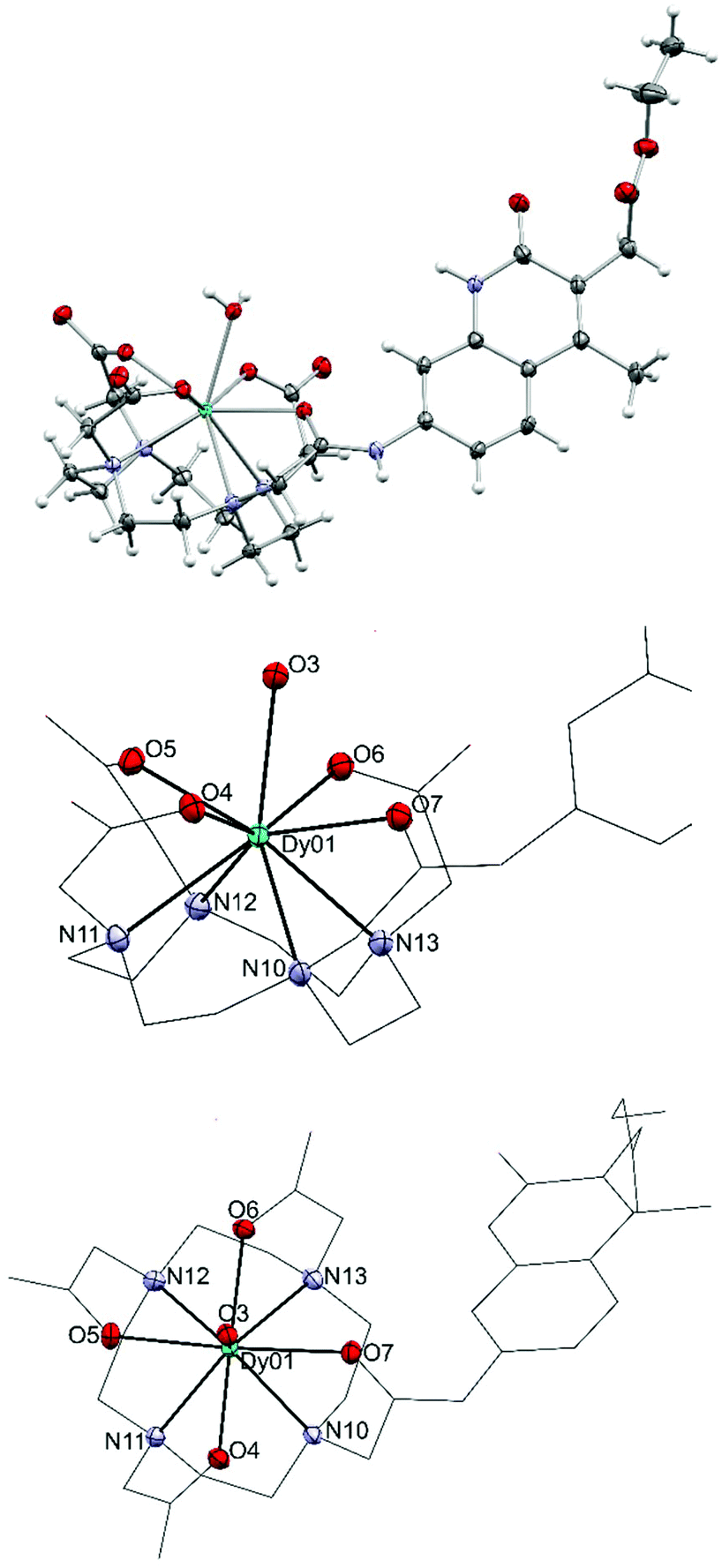 | ||
| Fig. 3 Crystal structure of Dy9d (top), and side (middle) and top views (bottom) of the metal coordination sphere. Thermal ellipsoids are shown at the 50% probability levels. | ||
In the Dy-complex, a significant void with ill-defined solvent molecules was identified. The best solution was found with ten positions with high electron density in this void. However, the diffuse nature of these contributions prompted us to treat this cavity using the solvent masking algorithm implemented in OLEX2.42 We identified a void centered on the crystallographic position −0.282 0.000 0.500 of approx. 693 Å3 containing ca. 197 electrons. In the final solution after solvent masking, only the coordinated water (O3) has been refined.
Absorption and emission spectroscopy
The photophysical characterisation of Ln1a–d was done on [Ln1a–d] = 3 × 10−5 M solutions in 0.01 M aqueous PIPES buffer at pH 6.5. These conditions were chosen because previously we observed that Ln complexes with trifluoromethylated carbostyril antennae showed a reversible loss in Ln emission at pH > 7.39 Analysis of the spectral shape of such Eu complexes showed no changes in the coordination environment, suggesting that deprotonation occurred in a non-coordinated group. As we could not exclude the loss of the core N–H proton, we have decided to do our experiments at a pH where deprotonation is not significant.The absorption and emission data are summarised in Tables 1–4. All absorption and emission spectra are given in the ESI (Fig. S3–S13†). Compared to the non-N-alkylated Ln9a–d, the new complexes had slightly blue-shifted absorption and emission maxima (by 5–6 and 1–2 nm, respectively). The exception was the emission of Ln1b, which was red-shifted by 3 nm. In all cases, the change was small. The complexes had appreciable absorptions at 337 nm, which is beneficial for laser-excitation without causing excessive damage to biomolecules.2
| Ligand | Ln | Φ L | Φ Ln |
|---|---|---|---|
| a In pH 6.5 PIPES buffer, [Ln1] = 3 × 10−5 M; λex = 336 (Ln1a), 348 (Ln1b), 338 (Ln1c,d) nm. b Using quinine sulfate as the reference. c Fold increase compared to Ln9 reference compound, calculated from data from ref. 39. Unbuffered solutions, pH 6–7, see ref. 39. Quantum yields have an error of 10%. Values given in italics were recorded in a second set of independent measurements. d In water, measured under the same conditions as reported for Tb9d. | |||
| 1a | Eu | 1.5 (×3c) | 6.0 (×1.94c) |
| Tb | 5.9 (×1.05c) | 43.4 (×1.23c) | |
| Gd | 6.8 (×0.88c) | — | |
| 1b | Eu | 2.7 (×1.6c) | 11.6 (×1.47c) |
| Tb | 3.1 (×0.69c) | 15.9 (×5.3c) | |
| Gd | 3.2 (×0.65c) | — | |
| 1c | Eu | 2.5 (×6.25c) 2.6d | 8.9 (×1.89c) 9.2d |
| Tb | 4.5 (×0.82c) 4.6d | 45.1 (×1.96c) 47.9d | |
| Gd | 5.1 (×0.74c) | — | |
| 1d | Eu | 1.7 (×4.05c), 1.9d | 5.85 (×2.1c), 5.5d |
| Tb | 7.1 (×1.11c), 7.0d | 41.7 (×4.2c), 39.9d | |
| Gd | 7.7 (×0.87c), 7.7d | — | |
| Complex | λ max/nm | λ em/nm | E 00(S1)/cm−1 | E 00(T1)/cm−1 | Δ(S1–T1)/cm−1 |
|---|---|---|---|---|---|
| a In pH 6.5 PIPES buffer, [Gd1] = 3 × 10−5 M. b In parentheses: change from Gd9a–d, calculated from ref. 39. | |||||
| Gd1a | 323 (−6)b | 364 (−2)b | 29200 (+400)b |
23900 (+400)b |
±0 |
| Gd1b | 337 (−5)b | 393 (+3)b | 27550 (+50)b |
23100 (+700)b |
−650 |
| Gd1c | 326 (−6)b | 374 (−1)b | 28700 (+400)b |
23900 (+400)b |
±0 |
| Gd1d | 326 (−5)b | 368 (−2)b | 28900 (+300)b |
23400 (+400)b |
−100 |
| Ligand | τ rad/ms | τ obs/ms | Φ EuEu | η sens | Φ EuEu ratioc | Φ EuEux ratiod | η Eux ratioe |
|---|---|---|---|---|---|---|---|
| a Calculated according to ref. 45. b Values taken from or calculated based on data reported in ref. 39. c Ratio of the intrinsic quantum yields of Eu9x and Eu1x. d Ratio of the overall quantum yields of Eu9x and Eu1x. e Ratio of the sensitisation efficiencies of Eu9x and Eu1x. | |||||||
| 1a | 5.40 | 0.65 | 12.0 | 49.9 | 1.10 | 1.94 | 1.76 |
| 9a | 5.41 | 0.59 | 10.9 | 28.4 | |||
| 1b | 5.36 | 0.66 | 12.2 | 94.7 | 1.07 | 1.47 | 1.38 |
| 9b | 5.39 | 0.615 | 11.5 | 68.8 | |||
| 1c | 5.36 | 0.66 | 12.2 | 72.8 | 1.07 | 1.89 | 1.77 |
| 9c | 5.40 | 0.613 | 11.4 | 41.1 | |||
| 1d | 5.34 | 0.65 | 12.2 | 48.4 | 1.07 | 2.09 | 1.94 |
| 9d | 5.38 | 0.60 | 11.2 | 24.9 | |||
| Ligand | Lna | τ H2O | τ D2O | q |
|---|---|---|---|---|
| a λ ex = 336 (Ln1a), 348 (Ln1b), 338 (Ln1c,d) nm; λem = 615 nm (Eu), 545 nm (Tb), initial delay: 0.05 ms; increments were adjusted between 0.2–10 μs depending on the lifetime. Lifetimes are reported as the average of three independent measurements. b Calculated as in ref. 17. | ||||
| 1a | Eu | 0.65 | 2.18 | 1.0 |
| Tb | 1.91 | 3.11 | 0.7 | |
| 1b | Eu | 0.66 | 2.16 | 1.0 |
| Tb | 0.7 | 1.34 | — | |
| 1c | Eu | 0.66 | 2.17 | 1.0 |
| Tb | 1.81 | 2.92 | 0.8 | |
| 1d | Eu | 0.65 | 2.16 | 1.0 |
| Tb | 1.56 | 2.47 | 0.9 | |
The carbostyril emissions (ΦL) in Gd1a–d were weaker than in the non-alkylated complexes Gd9a–d (Table 1). While we did not have crystals of Ln1, in its precursor 5a, which has a tertiary amide, the least squares planes (l.s.pl.) of the amide and the chromophore deviate by 86.72(14)°. This is reduced to 29.30(12)° in Dy9d, which has a secondary amide linker. Thus, there is essentially no orbital overlap between the tertiary amide and the heterocycle. In Dy9d (Fig. 3, synthesised previously, crystal structure not reported39) the amide and the heterocycle are more co-planar, which should be beneficial for the charge transfer excited state.43 The more efficiently electron-donating substituent of Ln9a–d yields a more polar emitting state in Ln9a–d than in Ln1a–d. This is consistent with the slightly higher ΦL in Ln9a–d, although the changes are small (Table 1).
All Eu and Tb complexes had robust Ln-centred emission upon antenna excitation. The Ln1 absorption spectra and the Eu and Tb excitation spectra were similar, as expected for sensitised Ln emission (Fig. 4 and S3–S10†). Ln emissions were at 490, 545, 580, 620, 650, 667 and 680 nm for Tb and at 580, 590, 615, 655 and 700 nm for Eu, corresponding to the 5D4 → 7FJ (J = 6–0) and 5D0 → 7FJ (J = 0–6) transitions, respectively (Fig. 5). In all Eu complexes the major transition was the 5D0 → 7F4 one, as in the Ln9a–d complexes. Every single one of the N-alkylated Eu and Tb complexes had higher Ln emission quantum yields than their non-alkylated analogues, Ln9 (Table 1). Tb1, with the exception of Tb1b, had ΦLn > 40%. The best result was obtained for MOM-functionalised Tb1c, ΦLn = 45%. Tb1c had a fourfold higher ΦLn than Tb9c, while for Tb1b a 5.3-fold increase was noted (to 15.9%) from non-N-alkylated Tb9b. Eu1 were less emissive than Tb1, with ΦLn in the 5.9–11.6% range; still, these values are in some cases twice as high as in the analogous Eu9 complexes.
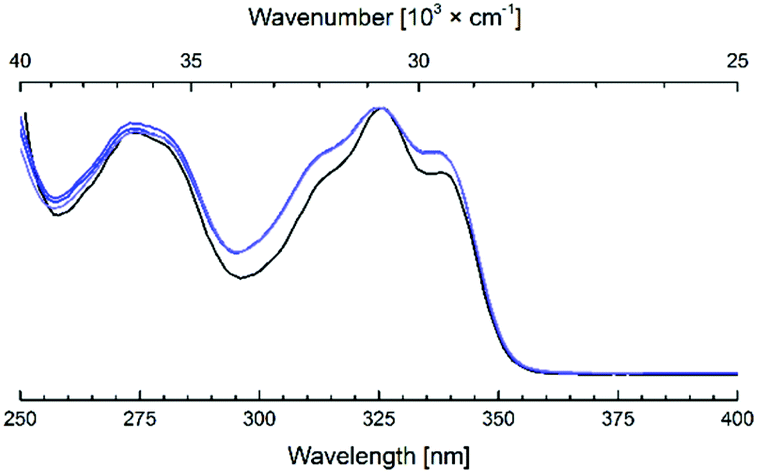 | ||
| Fig. 4 Overlaid absorption (black) and excitation (blue) spectra of Ln1d, (Ln = Eu, Tb, Gd). For Gd1d the excitation spectrum corresponds to the antenna fluorescence (λem = 374 nm), for Eu1d and Tb1d excitations of the Ln-emissions are shown (λem = 700 and 545 nm, respectively), [Ln1d] = nominally 3.0 × 10−5 M, PIPES-buffered aqueous solutions 0.01 M, pH 6.5, Ln = Eu, Tb, Gd. | ||
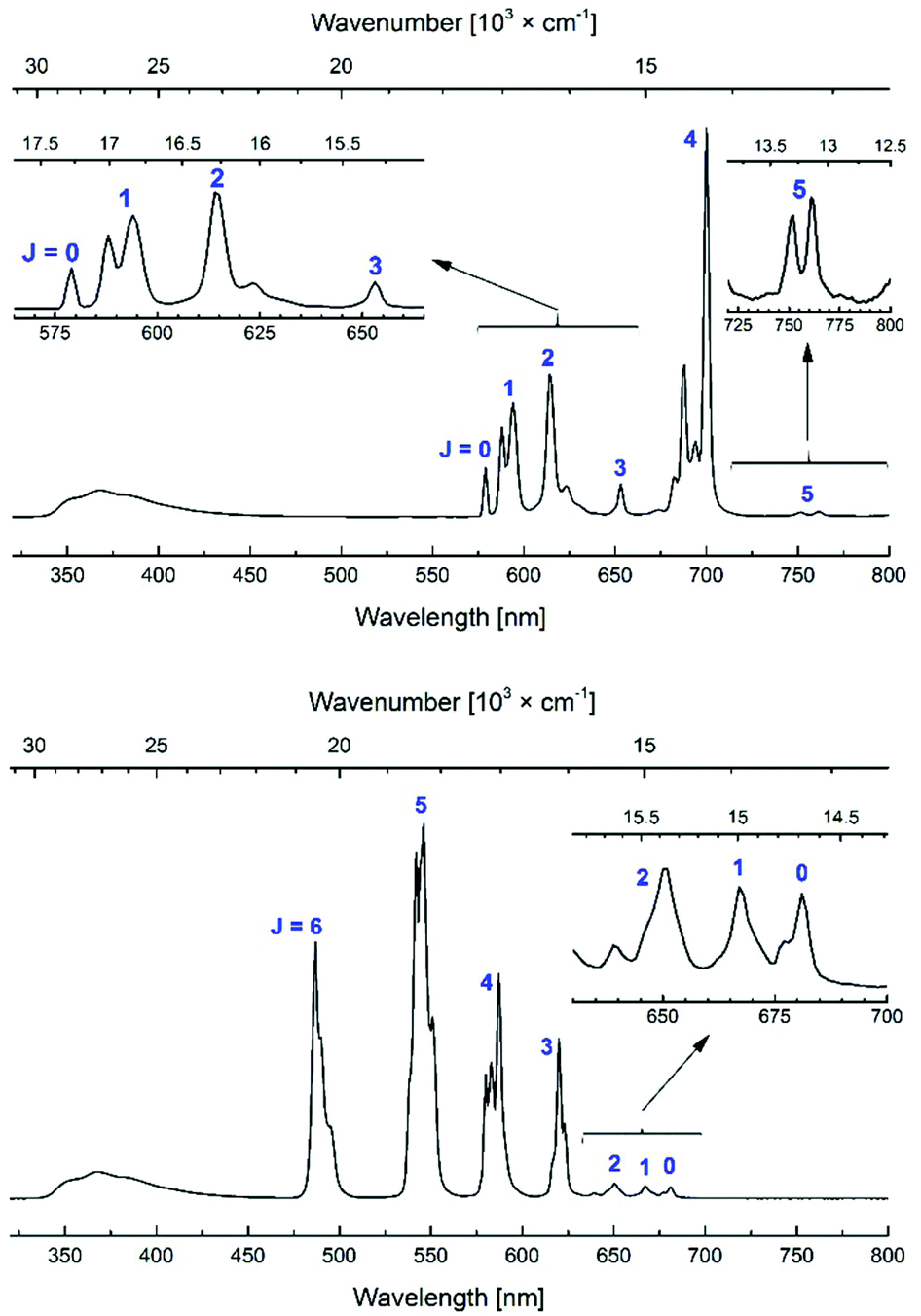 | ||
| Fig. 5 Steady-state emission spectra of Eu1d (top) and Tb1d (bottom). [Ln1d] = nominally 3.0 × 10−5 M, PIPES-buffered aqueous solutions 0.01 M, pH 6.5; λex = 336 nm. | ||
Antenna triplet states obtained from the phosphorescence bands at 77 K were located at 23100–23900 cm−1 in Gd1 (Table 2). Trifluoromethylated Gd1b had the lowest-lying antenna triplet, at 23100 cm−1. The triplet states were 400–700 cm−1 higher in energy than in Gd9a–d. Tb and Eu have excited states at 20400 (Tb), 19000 (5D1, Eu) and 17200 (5D0, Eu) cm−1.2,14 A general rule is that good triplet-mediated sensitisation requires an antenna triplet-Ln excited state energy gap of 2500–3500 cm−1.13 Previous studies have shown that a minimal energy gap of 2000–2500 cm−1 is required to avoid energy back transfer; energy transfer is then improved with an increasing energy gap until ∼24000 cm−1 for Tb.44 The presence of multiple acceptor levels in Eu makes the energy gap relation more complicated.44 Thus, 1a–d should be excellent sensitising ligands for both Eu and Tb, with the possible exception of 1b, which may be too low-lying for Tb.
In the case of the Eu complexes the increased ΦLn appears to be in large part due to improved sensitisation efficiencies for all the antennae (Table 3). Quantum yield determinations carry ∼10% relative error, and ΦEuEu should therefore be compared cautiously. Still, ΦEuEu of Eu1a–d are within experimental error (±0.1 ms) of those of Eu9a–d. This is expected based on the similarities of the coordination spheres (Fig. S11†). The observed lifetimes and the intrinsic quantum yields are identical within experimental error within the group of Eu1a–d. Interestingly, in a previous study, an Eu complex with the same Ln binding site and a tertiary amide-linked 7-amidocoumarin antenna had a very similar observed lifetime, 0.65 ms, while a non-alkylated analogue had τobs ∼ 0.6 ms.39 Thus, N-alkylation indeed increases the Eu lifetime and the intrinsic quantum yield, probably because of the removal of the N–H oscillator.
Most of the gain in overall quantum yield comes from the better sensitisation efficiency, ηsens. This is the product of the population of the feeding level (here, the antenna triplet), and the efficiency of the energy transfer. The triplet population is dependent on the efficiency of intersystem crossing, which will be affected by the S1–T1 energy gap, which was calculated in both Gd1a–d and Gd9a–d (Table 2). The differences are small, typically within experimental error, and are thus unlikely to have substantially benefited ISC; a possible exception is trifluoromethylated Ln1b. Energy transfer is dependent on the spectral overlap, on orientation factors, and on the donor–acceptor distance. In solution, the latter two are difficult to pin down, despite observations in the solid state. However, the small blue shifts of the Gd1a–d T1 states compared to Gd9a–d may allow for a better spectral overlap.
The increased Ln emission is not caused by a decrease in the number of inner sphere solvent (water) molecules, which would increase the intrinsic quantum yield (Table 4). This is not surprising. The added carboxylate is not well disposed for Ln coordination, as that would form an unfavored 8-membered ring. The shape of the Eu1 and Eu9 emission spectra are very similar, as expected for complexes with similar metal coordination environments (Fig. S11†). The q-values determined for Eu1 are the same as the values obtained for their Eu9 analogues within experimental error (1.0 vs. 1.0–1.1).39 For Tb complexes the q-values were lower than for the Eu species, which is consistent with Tb(III) being the smaller ion. The exception was Tb1b, for which an unrealistic result (q = 5) was obtained. As the antenna triplet in Tb1b is only 1800 cm−1 above the Tb excited state, this is likely due to energy back transfer. Substantial non-X–H-caused quenching makes the determination of q unreliable. In the case of back energy transfer the antenna triplet is repopulated, which in turn can be quenched by e.g. atmospheric oxygen.
The Ln complexes had modest antenna fluorescence emissions (ΦL). In Tb1a–dΦL were 87–97% of those in Gd1a–d (Table 1), which may be due to ET from the carbostyril singlet excited state to the Tb. This has been seen before in both coumarin and carbostyril sensitised species.39 Tb and Gd may also have different heavy atom effects, although these are usually assumed to be similar. PeT can be excluded for Tb and Gd complexes.
The drop in ΦL was larger in Eu1a–d than in Tb1a–d. Antennae retained only 22–84% of the ΦL of the appropriate Gd1a–d complexes. PeT and singlet ET could both contribute to this decrease. PeT from the excited carbostyril antennae to Eu3+ was found to be an efficient quenching pathway in Eu9.39 The ΦL decrease was smaller in Eu1a–d than in Eu9a–d, which may reflect decreased PeT due to the increased overall negative charge,41 or less efficient singlet ET. However, it is important to emphasize that the observed changes in ΦL in Tb1a–d and Eu1a–d compared to Gd1a–d do not support a substantial singlet mediated ET, and the contribution of the singlet state to Ln emission is small.
Experimental
Materials and methods
Compounds 1a,461b,461c,471d,40 and 8c,39 were synthesised following literature methods. All other chemicals were from commercial sources and used as received.
HPLC-analysis was performed on a RP-HPLC was performed on a Dionex UltiMate 3000 system using a Phenomenex Gemini® C18 TMS end-capped 150 mm × 4.6 mm HPLC column with water (0.05% formic acid):CH3CN (0.05% formic acid) eluent system using the methods: 0–10 min: 10% → 90% CH3CN, 0–12 min: 10% → 50% CH3CN, 0–8 min: 10 → 20% & 8–12 min: 20% iso CH3CN. Flow rate: 0.5 mL min−1, UV- (UltiMate 3000 Photodiode Array Detector) and ESI-MS detections (LCQ DECA XP MAX) were used.
Quantum yields were measured at room temperature and relative to quinine sulfate (QS) in H2SO4 0.05 M, ΦQS = 0.59(1). Quantum yields were calculated according to (1), with Φs the quantum yield of the sample, Φref the quantum yield of the reference, I the integrated corrected emission intensity of the sample (s) and of the reference (ref), fA the absorption factor of the sample (s) and of the reference (ref) at the excitation wavelength and n the refractive indexes of the sample (s) and of the reference (ref). The concentration of the complexes was adjusted to obtain an absorbance around the maxima of the antennae matching that of the QS fluorescent standard. The excitation wavelength where the absorption factors of the samples and of the reference were the same was chosen (i.e. where the absorptions are identical). The corrected emission spectra of the sample and reference standard were then measured under the same conditions over the 330–800 nm spectral range as well as blank samples containing only the solvent (i.e. water or PIPES buffered aqueous solutions). The appropriate blanks were subtracted from their respective spectra, and the antenna fluorescence and lanthanide luminescence were separated by fitting the section of the antenna emission overlapping the lanthanide emission with an exponential decay or with a scaled emission spectrum from the corresponding gadolinium complexes. The quantum yields were then calculated according to (1). The given relative error on the quantum yields (δΦ = ΔΦ/Φ, where ΔΦ is the absolute error) take into account the accuracy of the spectrometer and of the integration procedure [δ(Is/Iref) < 2%], an error of 0.59 ± 0.01 on the quantum yield of the reference QS [δ(Φref) < 2%], an error on the ratio of the absorption factors [δ(fAref/fAs) < 5%, relative to the fixed absorption factor of the reference QS] and an error on the ratio of the squared refractive indexes [δ(ns2/nref2) < 1%, <0.25% around 1.333 on each individual refractive index], which sums to a total estimated relative error that should be δΦs < 10%. A limit value of 10% is thus chosen.
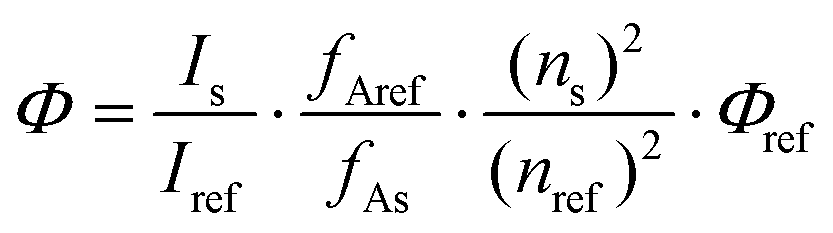 | (1) |
Low temperature measurements were done in quartz capillaries at 77 K by immersion in a liquid N2-filled quartz Dewar and with addition of glycerol (1 drop) to the solutions (9 drops) measured at room temperature.
Lifetimes were recorded 0.05 ms after pulsed excitations at the excitation maxima (λex) between 300–400 nm by measuring the decay of the lanthanide main emission peak (i.e. Eu 615 nm, Tb 545 nm). The increments after the initial delay were adjusted between 0.2–10 μs depending on the lifetime in order to have a good sampling of the decay. The obtained data were fitted by mono and double exponential decay models in OriginPro 9, and the most reliable value was chosen according to the adjusted R2 value and the shape of the residuals. A relative error of 10% is typically found among a series of measurements on the same sample.
Hydration numbers q were obtained by measuring the lifetimes of the same quantity of complex in an unbuffered solution in H2O and in D2O and fitting the difference according to the model of Horrocks et al.,16 and Beeby et al.17
Specific for 5a. NH protons are located on the difference map or placed at idealised positions. The cyclen ring shows a positional disorder which is modelled with an occupancy of 0.53 and 0.47 of the two different orientations, respectively.
Specific for Dy9d. Solvent accessible voids were treated using the solvent masking algorithm implemented in OLEX2 accounting for a 197 electrons in a 693 Å3 large void. In addition a structure refinement prior to applying solvent masking is attached.
CCDC 1832851 and 1833918 contain the supplementary crystallographic data for this paper.
Synthetic procedures
:AcOEt:iPrOH (6:4:0 → 6:3:1) for 3a, CH2Cl2:Et2O:acetone (8:2:0 → 7:3:0 → 7:1.5:1.5) for 3b, CH2Cl2:Et2O (3:2 iso) for 3c, CH2Cl2:Et2O:acetone (8:2:0 → 5:5:0 → 5:3:2) for 3d.
3a. 1.057 g (61% → 41% after recrystallization +20% after col. chrom. on the filtrate); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.43 (3, 9H), 2.29 (s, 3H), 3.79 (d, J = 6.3 Hz, 2H), 5.99 (s, 1H), 6.25 (d, J = 2.1 Hz, 1H), 6.52 (dd, J = 8.8 Hz, 2.2 Hz, 1H), 6.64 (t, J = 6.1 Hz, 1H), 7.39 (d, J = 8.8 Hz, 1H), 11.22 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 18.4 (CH3), 27.7 (CH3), 45.0 (CH2), 80.8 (Cq), 94.8, 109.4, 110.8, 115.2, 125.4, 140.7, 147.9, 162.3, 169.9; RP-HPLC tR = 6.13 min (10 min method 10% → 90%); ESI-MS obsd 289.02; HR-ESI-MS obsd 311.1373, calcd 311.1366 [(M + Na)+, M = C16H20N2O3].
3b. 1.302 g, (43% → 34% after recrystallization +9% after col. chrom. on the filtrate); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.43 (s, 9H), 3.83 (d, J = 4.7 Hz, 2H), 6.34 (d, J = 1.9 Hz, 1H), 6.49 (s, 1H), 6.65 (dd, J = 9.0, 2.2 Hz, 1H), 7.04 (brd, 1H), 11.92 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 27.7 (CH3), 44.8 (CH2), 81.0 (Cq), 94.8 (CHAr), 104.0 (CHAr), 111.1 (CHAr), 114.2 (CH), 122.9 (Cq), 124.2 (CAr), 136.7 (Cq) 142.1 (CAr), 151.0 (CAr), 160.8 (Cq), 169.5 (Cq); 19F NMR (376 MHz, DMSO-d6) δ ppm −62.3; RP-HPLC tR = 6.97 min (10 min method 10% → 90%); ESI-MS obsd 342.99; HR-ESI-MS obsd 365.1075, calcd 365.1083 [(M + Na)+, M = C16H17NF3N2O3].
3c. 1.954 g (84% → 49% after recrystallization +35% after col. chrom. on the filtrate); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.42 (s, 9H), 3.36 (s, 3H), 3.79 (d, J = 6.4, 2H), 4.54 (s, 2H), 6.13 (s, 1H), 6.27 (d, J = 2.2 Hz, 1H), 6.51 (dd, J = 8.8, 2.2 Hz, 1H), 6.67 (t, J = 6.4 Hz, 1H), 7.37 (d, J = 8.8 Hz, 1H), 11.35 (s, 1H); 13C NMR (101 MHz, DMSOd6) δ ppm 28.2 (CH3), 45.0 (CH2), 58.0 (CH3), 70.5 (CH2), 80.8 (Cq), 94.8 (CHAr), 108.4 (CHAr), 109.6 (CHAr), 113.3 (CH), 124.9 (CHAr), 141.0 (Cq), 147.3 (CAr), 150.2 (CAr), 162.4 (Cq), 169.9 (Cq); RP-HPLC tR = 6.03 min (10 min method 10% → 90%); ESI-MS obsd 318.65; HR-ESI-MS obsd 341.1469, calcd 341.1472 [(M + Na)+, M = C17H22N2O4].
3d. 3.72 g (76%); 1H NMR (400 MHz, CDCl3) δ ppm 1.25 (t, J = 7.1 Hz, 3H), 1.51 (s, 9H), 2.43 (s, 3H), 3.80 (s, 2H), 3.88 (s, 2H), 4.17 (q, J = 7.1 Hz, 2H), 6.38 (d, J = 2.3 Hz, 1H), 6.62 (d, J = 8.9, 2.3 Hz, 1H), 7.54 (d, J = 8.9 Hz, 1H), 11.59 (s, 1H); 13C NMR (101 MHz, CDCl3) δ ppm 14.3 (CH3), 15.5 (CH3), 28.2 (CH3), 32.5 (CH2), 46.0 (CH2), 60.8 (CH2) 82.4 (Cq), 96.6 (CHAr), 110.3 (CAr), 112.9 (CHAr), 119.2 (Cq), 125.9 (CHAr), 139.3 (Cq), 146.1 (CAr), 148.8 (CAr), 163.7 (Cq), 170.0 (Cq), 171.8 (Cq); RP-HPLC tR = 6.73 min (10 min method 10% → 90%); ESI-MS obsd 374.71; HR-ESI-MS obsd 397.1730, calcd 397.1724 [(M + Na)+, M = C20H26N2O5].
:1 mixture of DMF and distilled CH2Cl2 (125 mM). The solutions were cooled to 0 °C and 2,6-di-tert-butyl-pyridine (3.0 equiv.) was added followed by the addition of chloroacetyl chloride (1.2 equiv.). The reaction mixtures were then allowed to warm to room temperature. When TLC analysis indicated the completion of the reaction, the mixture was diluted with H2O and EtOAc. The phases were separated, and the aqueous layer was extracted with EtOAc. The combined organic phases were dried over MgSO4, filtered, and the filtrate was concentrated at reduced pressure. The crude products were purified by column chromatography on silica gel using the following eluent mixtures: CH2Cl2:Et2O:iPrOH (8:2:0 → 8:1:1) for 4a, CHCl3:Et2O:EtOH (9.5:0.5:0 → 8:2:0 → 8:1.6:0.4) for 4b, CHCl3:Et2O:iPrOH (9:1:0 → 8.5:1.5:0 → 8.5:1:0.5) for 4c, CH2Cl2:Et2O:EtOH (9.5:0.5:0 → 8:2:0 → 8:1.6:0.4) for 4d.
4a. 0.818 g (83%); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.42 (s, 9H), 2.43 (s, 3H), 4.13 (s, 2H), 4.29 (s, 2H), 6.44 (s, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.32 (s, 1H), 7.79 (d, J = 8.5 Hz, 1H), 11.75 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 18.5 (CH3), 27.7 (CH3), 41.8 (CH2), 52.4 (CH2), 81.5 (Cq), 114.1 (Cq), 119.4 (CAr), 120.9 (CHAr), 121.5 (CH), 126.3 (CHAr), 139.4 (Cq), 142.8 (CAr), 147.5 (CAr), 161.7 (Cq), 165.7 (Cq), 167.6 (Cq); RP-HPLC tR = 6.23 min (10 min method 10% → 90%); ESI-MS obsd 364.97; HR-ESI-MS obsd 387.1081, calcd 387.1082 [(M + Na)+, M = C18H21ClN2O4].
4b. 1.80 g (quant.); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.42 (s, 9H), 4.19 (s, 2H), 4.32 (s, 2H), 7.03 (s, 1H), 7.35 (s, J = 8.5 Hz, 1H), 7.46 (s, 1H), 7.77 (d, J = 8.5 Hz, 1H), 12.48 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 27.7 (CH3), 41.9 (CH2), 52.2 (CH2), 81.6 (Cq), 112.6 (CAr), 115.0 (CHAr), 121.0 (CHAr), 122.3 (CH), 122.4 (q, J = 275 Hz, Cq), 125.7 (CHAr), 136.2 (q, J = 32.2 Hz, Cq), 140.6 (CAr), 144.0 (CAr), 160.2 (Cq), 165.7 (Cq), 167.6 (Cq); 19F NMR (376 MHz, DMSO-d6) δ ppm −62.5; RP-HPLC tR = 7.03 min (10 min method 10% → 90%); ESI-MS obsd 419.94; HR-ESI-MS obsd 441.0795, calcd 441.0799 [(M + Na)+, M = C18H18ClF3N2O4].
4c. 1.78 g (87% → 68% after recrystallization (from EtOAc twice before chromatography) + 19% after col. chrom. on the filtrate); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.42 (s, 9H), 3.39 (s, 3H), 4.14 (s, 2H), 4.29 (s, 2H), 4.66 (s, 2H), 6.53 (s, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.35 (s, 1H), 7.75 (d, J = 8.6 Hz, 1H), 11.88 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 27.7 (CH3), 40.2 (CH2), 52.3 (CH2), 58.6 (CH3), 70.2 (CH2), 81.5 (Cq), 114.3 (CHAr), 117.2 (CAr), 119.9 (CHAr), 120.9 (CH), 125.8 (CHAr), 139.7 (Cq), 142.9 (CAr), 146.8 (CAr), 161.6 (Cq), 165.7 (Cq), 167.6 (Cq); RP-HPLC tR = 6.20 min (10 min method 10% → 90%); ESI-MS obsd 394.97; HR-ESI-MS obsd 417.1183, calcd 417.1188 [(M + Na)+, M = C19H23ClN2O5].
4d. 2.59 g (72%) (recrystallised from EtOAc after col. chrom.); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.18 (t, J = 7.1 Hz, 3H), 1.42 (s, 9H), 2.40 (s, 3H), 3.72 (s, 2H), 4.07 (q, J = 7.1 Hz, 2H), 4.14 (s, 2H), 4.30 (s, 2H), 7.25 (d, J = 8.1 Hz, 1H), 7.33 (s, 1H), 7.86 (d, J = 8.6 Hz, 1H), 11.92 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 14.1 (CH3), 15.3 (CH3), 27.7 (CH3), 32.2 (CH2), 40.2 (CH2), 52.4 (CH2), 60.2 (CH2), 81.4 (Cq), 113.9 (CAr), 119.5 (CHAr), 121.0 (Cq), 125.4 (CHAr), 126.6 (CHAr), 138.1 (Cq), 142.4 (CAr), 144.1 (CAr), 161.4 (Cq), 165.7 (Cq), 167.6 (Cq), 170.4 (Cq); RP-HPLC tR = 6.83 min (10 min method 10% → 90%); ESI-MS obsd 451.03; HR-ESI-MS obsd 473.1446, calcd 473.1450 [(M + Na)+, M = C22H27ClN2O6].
:DMF (3:1) (158 mM). The reaction mixture was stirred for 1–2 days, when TLC analysis showed full conversion of the limiting starting material. The CHCl3 was evaporated under reduced pressure, and the residue was diluted with a 1:1 mixture of CH2Cl2 and MeOH (few mL). This solution was loaded onto a silica column that had been conditioned with CH2Cl2:MeOH (1:1). Elution with CH2Cl2:MeOH:NH4OH (10:9:1) yielded the products as white (5a), off-white (5c), yellowish-white (5b, 5d) solids.
5a. 0.627 g (91%); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.41 (s, 9H), 2.10–3.75 (m, 24H (21H product, 3.36H DMSO/H2O/EtOH)), 4.22 (s, 2H), 6.40 (s, 1H), 7.17 (d, J = 7.9 Hz, 1H), 7.31 (s, 1H), 7.75 (d, J = 8.5 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 18.5 (CH3), 27.7 (CH3), 44.9 (CH2), 45.5 (CH2), 46.6 (CH2), 51.1 (CH2), 52.0 (CH2), 55.3 (CH2) 81.2 (Cq), 113.8 (CHAr), 118.9 (CAr), 120.7 (CHAr), 121.2 (CH), 126.2 (CHAr), 139.4 (Cq), 143.5 (CAr), 147.6 (CAr), 161.8 (Cq), 168.0 (Cq), 170.0 (Cq); RP-HPLC tR = 1.71, 4.05 min (16 min method: 0–12 min 10% → 50%); ESI-MS obsd 501.23; HR-ESI-MS obsd 501.3181, calcd 501.3184 [(M + H)+, M = C26H40N6O4].
5b. 1.822 g (94%); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.34–1.48 (s, 9H), 2.18–2.86 (m, 17.17H (16H product, 1.17H DMSO)), 3.29 (s, 2H), 4.25 (s, 2H), 4.99 (br., 3H), 6.93 (s, 1H), 7.21 (dd, J = 8.7, 1.2 Hz, 1H), 7.46 (d, J = 1.2 Hz, 1H), 7.70 (d, J = 7.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 27.7 (CH3), 45.0 (CH2), 45.4 (CH2), 46.7 (CH2), 51.2 (CH2), 51.9 (CH2), 55.6 (CH2), 81.2 (Cq), 112.3 (CAr), 116.1 (CHAr), 121.0 (CHAr), 121.5 (CH), 122.7 (q, J = 275.0 Hz, Cq), 125.3 (CHAr), 135.4 (q, J = 31.0 Hz, Cq), 142.7 (CAr), 144.2 (CAr), 161.7 (Cq), 168.0 (Cq), 170.2 (Cq); 19F NMR (376 MHz, DMSO-d6) δ ppm −62.3 RP-HPLC tR = 5.37 min (16 min method: 0–12 min 10% → 50%); ESI-MS obsd 555.19; HR-ESI-MS obsd 555.2919, calcd 555.2901 [(M + H)+, M = C26H37F3N6O4].
5c. 1.681 g (81%); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.34–1.50 (s, 9H), 2.11–2.99 (m, 17.76H (16H product, 1.76H DMSO/DMF)), 3.25 (br, 2H), 3.39 (s, 3H), 4.22 (s, 2H), 4.66 (s, 2H), 6.50 (s, 1H), 7.16 (d, J = 7.5 Hz, 1H), 7.35 (s, 1H), 7.71 (d, J = 8.5 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 27.6 (CH3), 44.9 (CH2), 45.5 (CH2), 46.6 (CH2), 51.1 (CH2), 51.9 (CH2), 55.3 (CH2), 58.2 (CH3), 70.2 (CH2), 81.2 (Cq), 114.0 (CHAr), 116.8 (CAr), 119.5 (CHAr), 120.7 (CH), 125.7 (CHAr), 139.7 (Cq), 143.6 (CAr), 146.8 (CAr), 161.7 (Cq), 168.0 (Cq), 170.0 (Cq); RP-HPLC tR = 1.75, 2.08, 4.12 min; (16 min method: 0–12 min 10% → 50%); ESI-MS obsd 531.24; HR-ESI-MS obsd 531.3292 calcd 531.3289 [(M + H)+, M = C27H42N6O5].
5d. 2.84 g (89%); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.18 (t, J = 7.1 Hz, 3H), 1.42 (s, 9H), 2.19–2.93 (m, 22H (19H product, 3H DMSO)), 3.24 (s, 2H), 3.72 (s, 2H), 4.06 (q, J = 7.1 Hz, 2H), 4.23 (s, 2H), 7.19 (d, J = 7.8 Hz, 1H), 7.31 (s, 1H), 7.83 (d, J = 8.6 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ ppm 14.1 (CH3), 15.3 (CH3), 27.7 (CH3), 32.2 (CH2), 45.0 (CH2), 45.5 (CH2), 46.7 (CH2), 51.2 (CH2), 52.0 (CH2), 60.2 (CH2), 81.2 (Cq), 113.6 (CHAr), 119.1 (CAr), 120.8 (CHAr), 125.0 (Cq), 126.5 (CHAr), 138.1 (CAr), 143.1 (CAr), 161.4 (Cq), 168.0 (Cq), 170.0 (Cq), 170.5 (Cq); RP-HPLC tR = 1.88, 4.63, 5.50 min (16 min method: 0–12 min 10% → 50%); ESI-MS obsd 587.27; HR-ESI-MS obsd 587.3561, calcd 587.3552 [(M + H)+, M = C30H46N6O6].
:1:0 → 8:2:0 → 8:1:1). For analytically pure products, up to 3 columns were needed.
6a. 0.429 g (81%); 1H NMR (400 MHz, CDCl3) δ ppm 1.12–1.77 (m, 67H (36H product, 31H DIPEA·HBr)), 1.79–3.86 (m, 35.5 (27H product, 8.3H DIPEA·HBr, 0.2 solvent residues)), 4.05–4.40 (br, 2H), 6.46 (s, 1H), 7.09 (dd, J = 8.5 Hz, 1.9 Hz, 1H), 7.49 (d, J = 1.6 Hz, 1H), 7.65 (d, J = 8.6 Hz), 12.43 (s, 1H); 13C NMR (101 MHz, CDCl3) δ ppm 19.3 (CH3), 27.9 (CH3), 28.0 (CH3), 48.0–49.0 (CH2), 51.8–53.0 (CH2), 52.8 (CH2), 55.6 (CH2), 55.7 (CH2), 56.0 (CH2), 81.9 (Cq), 82.0 (Cq), 114.9 (CHAr), 120.3 (CAr), 121.4 (CH), 121.4 (CHAr), 126.3 (CHAr), 139.5 (Cq), 143.0 (CAr), 148.4 (CAr), 163.8 (Cq), 167.4 (Cq), 171.6 (Cq), 172.6 (Cq); RP-HPLC tR = 4.80 min (10 min method 10% → 90%); ESI-MS obsd 843.40; HR-ESI-MS obsd 865.5041, calcd 865.5046 [(M + Na)+, M = C44H70N6O10].
6b. 0.759 g (66%); 1H NMR (400 MHz, CDCl3) δ ppm 1.14–1.74 (m, 45.9H (36H product, 9.9H DIPEA·HBr)), 1.77–3.90 (m, 30.66H (24H product, 0.96H DMF, 2.64H DIPEA·HBr)), 4.12–4.40 (br, 2H), 6.95 (s, 1H), 7.13 (dd, J = 8.7 Hz, 1.6 Hz, 1H), 7.68 (d, J = 8.0 Hz, 1H), 8.14 (d, J = 1.4 Hz, 1H), 12.64 (s, 1H); 13C NMR (101 MHz, CDCl3) δ ppm 27.9 (CH3), 28.1 (CH3), 48.0–49.3 (CH2), 51.5–53.0 (CH2), 52.1 (CH2), 55.7 (CH2), 55.8 (CH2), 56.1 (CH2), 81.7 (Cq), 81.9 (Cq), 113.8 (CAr), 116.2 (CHAr), 121.7 (CHAr), 122.4 (CH), 122.5 (q, J = 275 Hz, Cq), 125.9 (CHAr), 137.4 (q, J = 31.7 Hz, Cq), 141.2 (CAr), 143.6 (CAr), 161.0 (Cq), 167.5 (Cq), 171.6 (Cq), 172.7 (Cq); 19F NMR (376 MHz, CDCl3) δ ppm −63.5 RP-HPLC tR = 4.97 min (10 min method 10% → 90%); ESI-MS obsd 897.36; HR-ESI-MS obsd 919.4767, calcd 919.4763 [(M + Na)+, M = C44H67N6O10F3].
6c. 0.505 g (53%); 1H NMR (400 MHz, CDCl3) δ ppm 1.30–1.74 (m, 41H (36H product, 5H DIPEA·HBr)), 1.78–4.49 (m, 32H (29H product, 1.33 H DIPEA·HBr, 1.67 H solvent residues)), 4.66 (s, 2H), 6.71 (s, 1H), 7.09 (dd, J = 8.6 Hz, 1.7 Hz, 1H), 7.50 (m, 1H), 7.67 (d, J = 8.6 Hz, 1H), 12.74 (s, 1H); 13C NMR (101 MHz, CDCl3) δ ppm 27.9 (CH3), 28.1 (CH3), 48.0–49.3 (CH2), 52.0–53.5 (CH2), 71.3 (CH2), 81.8 (Cq), 82.0 (Cq), 115.1 (CHAr), 118.2 (CAr), 120.2 (CHAr), 121.6 (CH), 125.8 (CHAr), 139.9 (Cq), 147.5 (CAr), 164.0 (Cq), 167.5 (Cq), 171.7 (Cq), 172.7 (Cq); RP-HPLC tR = 10.07 min (16 min method: 0–12 min 10% → 50%); ESI-MS obsd 873.37; HR-ESI-MS obsd 895.5151, calcd 895.5151 [(M + Na)+, M = C45H72N6O11].
6d. 1.35 g (34%; yield of the analytically pure compound after the first column. A significant amount of 6d was washed away by the residual DMF and came off with the front. This part of the batch was kept in storage. From this sample crystals grew that were suitable for X-ray analysis, and has thus not been worked up); 1H NMR (400 MHz, CDCl3) δ ppm 1.20 (t, J = 7.2 Hz, 3H), 1.24–1.73 (m, 36H), 1.82–3.74 (m, 27H), 3.81 (s, 2H), 4.10 (q, J = 7.1 Hz, 2H), 4.15–4.35 (br, 2H), 7.11 (dd, J = 8.6 Hz, 1.8 Hz, 1H), 7.45 (d, J = 1.6 Hz, 1H), 7.70 (d, J = 8.6 Hz, 1H), 11.90 (s, 1H); 13C NMR(101 MHz, CDCl3) δ ppm 14.3 (CH3), 15.9 (CH3), 27.9 (CH3), 28.0 (CH3), 28.1 (CH3), 32.5 (CH2), 48.0–49.0 (CH2), 51.5–53.0 (CH2), 55.7 (CH2), 55.8 (CH2), 56.0 (CH2), 60.9 (CH2), 81.8 (Cq), 114.9 (CAr), 120.6 (CAr), 121.8 (CHAr), 125.4 (Cq), 126.5 (CHAr), 138.4 (CAr), 142.5 (CAr), 145.2 (Cq), 162.9 (Cq), 167.4 (Cq), 170.8 (Cq), 171.7 (Cq), 172.3 (Cq); RP-HPLC tR = 4.93 min (10 min method 10% → 90%) ESI-MS obsd 929.47; HR-ESI-MS obsd 951.5411, calcd 951.5413 [(M + Na)+, M = C48H76N6O12].
:H2O (9:1). Elution with acetonitrile:H2O (9:1 → 7:3) yielded the ligands as white (6a,c) and yellowish-white (6b,d) solids.
7a. 208 mg (81%); 1H NMR (400 MHz, D2O) δ ppm 1.25–1.35 (m, 1.02H DIPEA), 1.84–4.65 (m, 29.3H (29H product, 0.3H DIPEA)), 6.31 (s, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.37 (s, 1H), 7.71 (d, J = 8.4 Hz, 1H); 13C NMR (101 MHz, D2O) δ ppm 16.3 (CH3), 47.5–48.7 (CH2), 52.2–53.2 (CH2), 54.1 (CH2), 56.5 (CH2), 58.7 (CH2), 58.9 (CH2), 114.3 (CHAr), 119.4 (CAr), 120.3 (CHAr), 121.8 (CH), 126.8 (CHAr), 137.7 (Cq), 143.0 (CAr), 151.0 (CAr), 164.0 (Cq), 174.0 (Cq), 174.7 (Cq), 180.2 (Cq); RP-HPLC tR = 3.02–7.12 min (16 min method: 0–12 min 10% → 50%); ESI-MS obsd 619.27; HR-ESI-MS obsd 655.2068, calcd 655.2046 [(M + Ca − 3H), M = C28H38N6O10].
7b. 434 mg (92%); 1H NMR (400 MHz, D2O) δ ppm 1.85–4.68 (m, 26H), 6.94 (m, 1H), 7.38 (d, J = 8.7 Hz, 1H), 7.51 (m, 1H), 7.87 (d, J = 7.4 Hz, 1H); 13C NMR (101 MHz, D2O) δ ppm 51.4 (CH2), 56.6 (CH2), 58.8 (CH2), 58.9 (CH2), 114.2 (CAr), 114.9 (CHAr), 120.8 (CH), 121.6 (CHAr), 122.0 (q, J = 275 Hz, Cq), 126.8 (CHAr), 138.5 (q, J = 32.2 Hz, Cq), 139.3 (CAr), 143.9 (CAr), 162.6 (Cq), 174.1 (Cq), 174.6 (Cq), 180.2 (Cq); 19F NMR (376 MHz, CDCl3) δ ppm −63.4 RP-HPLC tR = 5.60–9.72 min (16 min method: 0–12 min 10% → 50%); ESI-MS 673.32; HR-ESI-MS obsd 709.1779, 731.1559, calcd 709.1763, 731.1583, [(M + Ca − 3H), M = C28H32N6O10F3; (M + Ca + Na − 4H), M = C28H35N6O10F3].
7c. 477 mg (quant.); 1H NMR (400 MHz, D2O) δ ppm 1.80–4.71 (m, 31H), 7.31 (d, J = 8.6 Hz, 1H), 7.43 (s, 1H), 7.73 (d, J = 8.6 Hz); 13C NMR (101 MHz, D2O) δ ppm 54.2 (CH2), 56.6 (CH2), 58.4 (CH2), 58.8 (CH2), 58.9 (CH2), 70.3 (CH2), 114.5 (CHAr), 118.0 (CAr), 118.3 (CHAr), 122.0 (CH), 126.0 (CHAr), 138.3 (Cq), 143.2 (CAr), 148.8 (CAr), 164.0 (Cq), 174.1 (Cq), 174.9 (Cq), 180.1 (Cq), 180.2 (Cq); RP-HPLC tR = 2.95–7.02 min (16 min method: 0–12 min 10% → 50%); ESI-MS obsd 649.29; HR-ESI-MS obsd 685.2175, 707.1980, calcd 685.2152, 707.1971, [(M + Ca − 3H), M = C29H37N6O11; (M + Ca + Na − 4H), M = C29H340N6O11].
7d. 345 mg (quant.); 1H NMR (400 MHz, D2O) δ ppm 1.11–1.25 (m, 4.19H (3H product, 1.19H EtOH)), 1.73–4.67 (m, 33.8H (33H product, 0.8H EtOH)), 7.32 (d, J = 8.8 Hz, 1H), 7.38–7.45 (m, 1H), 7.83 (d, J = 8.7 Hz, 1H); 13C NMR (101 MHz, D2O) δ ppm 13.4 (CH3), 15.1 (CH3), 32.6 (CH2), 47.7–48.6 (CH2), 52.3–53.1 (CH2), 54.1 (CH2), 56.6 (CH2), 58.8 (CH2), 58.9 (CH2), 62.3 (CH2), 114.1 (CHAr), 120.4 (CAr), 121.7 (CHAr), 123.9 (Cq), 137.0 (CAr), 142.7 (CAr), 147.7 (Cq), 163.0 (Cq), 173.5 (Cq), 174.0 (Cq), 174.7 (Cq), 180.1 (Cq), 180.2 (Cq); RP-HPLC tR = 5.70–9.42 min (16 min method: 0–12 min 10% → 50%); ESI-MS obsd 705.32; HR-ESI-MS obsd 741.2431, 763.2234, calcd 741.2414, 763.2223, [(M + Ca − 3H), M = C32H41N6O12; (M + Ca + Na − 4H), M = C32H44N6O12].
:1 mixture of H2O and EtOH was added (c = 0.05 M) into the vial using a micropipette, followed by the appropriate (2.4 equiv.) lanthanide salt (EuCl3·6H2O, TbCl3 (anhydrous), or GdCl3 (anhydrous)). The vials were sealed with a screw-cap and parafilm. The mixtures were sonicated to ensure full dissolution. The reaction mixtures were stirred overnight at 45 °C in an alumina bath. The following day the mixture was sonicated again, and then it was transferred dropwise to a 20 mL vial filled with Et2O. The phases were separated, (the organic phase was removed from the top), and the aqueous layer was loaded onto a silica gel chromatography column (Ø 1 cm, h = 3 cm). Elution with acetonitrile:H2O (8:2 → 6:4) yielded the Ln complexes as yellowish-white (ivory) solids. The final products contain a small amount of silica because of the polar conditions applied on the silica column. Most of the residual silica can be removed through membrane filtration (0.2 μm) of the concentrated aqueous solution of the complexes using a syringe. It is important to leave the solution standing for about a day (or at least for several hours) before filtration to allow the silica to precipitate out.
Eu1a. 8 mg (43%); RP-HPLC tR = 5.85 min (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 769.20; HR-ESI-MS obsd 767.15435, calcd 767.15568, [(M − H)−, M = C28H35N6O10Eu].
Gd1a. 10 mg (53%); RP-HPLC tR = 5.67 min (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 774.18; HR-ESI-MS obsd 772.15725, calcd 772.15896, [(M − H)−, M = C28H35N6O10Gd].
Tb1a. 7 mg (37%); RP-HPLC tR = 5.87 min (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 775.30; HR-ESI-MS obsd 773.15826, calcd 773.15953, [(M − H)−, M = C28H35N6O10Tb].
Eu1b. 36 mg (59%); RP-HPLC tR = 10.80 min (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 823.17; HR-ESI-MS obsd 821.12580, calcd 821.12879, [(M − H)−, M = C28H32N6O10F3Eu].
Gd1b. 35 mg (57%); RP-HPLC tR = 10.73 min (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 828.16; HR-ESI-MS obsd 826.12879, calcd 826.13069, [(M − H)−, M = C28H32N6O10F3Gd].
Tb1b. 44 mg (71%); RP-HPLC tR = 10.75 mind (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 829.26; HR-ESI-MS obsd 827.12989, calcd 827.13127, [(M − H)−, M = C28H32N6O10F3Tb].
Eu1c. 52 mg (85%); RP-HPLC tR = 5.78 min (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 799.18; HR-ESI-MS obsd 797.16485, calcd 797.16626, [(M − H)−, M = C29H37N6O11Eu].
Gd1c. 33 mg (53%); RP-HPLC tR = 5.45 min (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 804.26; HR-ESI-MS obsd 802.16777, calcd 802.16956, [(M − H)−, M = C29H37N6O11Gd].
Tb1c. 54 mg (87%); RP-HPLC tR = 5.43 min (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 805.32; HR-ESI-MS obsd 803.16896, calcd 803.17010, [(M − H)−, M = C29H37N6O11Tb].
Eu1d. 35 mg (58%); RP-HPLC tR = 10.92 min (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 855.15; HR-ESI-MS obsd 853.19070, calcd 853.19253, [(M − H)−, M = C32H41N6O12Eu].
Gd1d. 42 mg (69%); RP-HPLC tR = 10.90 min (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 859.76; HR-ESI-MS obsd 802.16777, calcd 802.16956, [(M − H)−, M = C32H41N6O12Gd].
Tb1d. 42 mg (69%); RP-HPLC tR = 10.87 min (16 min method: 0–8 min: 10 → 20% & 8–12 min: 20% iso); ESI-MS obsd 861.36; HR-ESI-MS obsd 803.16896, calcd 803.17010, [(M − H)−, M = C32H41N6O12Tb].
Conclusions
In conclusion, four new ligands and their Tb, Eu and Gd complexes were synthesised and characterised. The ligands have carbostyril sensitising antennae decorated with 4-Me, 4-CF3, 4-MOM or 3-CH2CO2Et and 4-Me substituents. Antennae are attached to the ligand-binding DOTA framework through a tertiary amide linker, which carries a negatively charged carboxymethyl group. The Tb and Eu complexes had greatly increased quantum yields compared to analogous species wherein the linker was a secondary amide. The increased ΦLn is due to an enhanced sensitisation efficiency, based on the analysis of the Eu spectra, and, to a much smaller extent, due to a slightly increased intrinsic quantum yield possibly caused by the removal of the amide N–H oscillator from the proximity of the Ln.The reasons for the improved photophysical properties are likely to be multiple. The blue-shifted antenna triplets should allow for better overlap with the Tb excited states, and thus allow for a more efficient energy transfer. In the case of Eu, a reduction in PeT may contribute; this effect would be smallest for the electron-poor trifluoromethylated antenna. Factors that are difficult to evaluate are: better ISC due to the larger heavy atom effect of the N-alkyl group, and the removal of the NH oscillator that may quench the triplet as well as the Ln excited state. Finally, the Ln-antenna distance and orientation may differ in complexes with secondary and with tertiary amide linkers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council (project grants 2013-4655 and 2017-04077 to K. E. B.). D. P. is an Erasmus student from the University of Glasgow. We thank Dr Julien Andres for discussions and critical reading of the manuscript, Michele Bedin for help with the HPLC analysis, and Ashleigh Castner for proofreading.Notes and references
- A. de Bettencourt-Dias, in Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials, John Wiley & Sons Ltd, 2014, pp. 1–48, DOI:10.1002/9781118682760.ch01.
- S. J. Butler, M. Delbianco, L. Lamarque, B. K. McMahon, E. R. Neil, R. Pal, D. Parker, J. W. Walton and J. M. Zwier, Dalton Trans., 2015, 44, 4791–4803 RSC.
- J. M. Zwier, H. Bazin, L. Lamarque and G. Mathis, Inorg. Chem., 2014, 53, 1854–1866 CrossRef PubMed.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef PubMed.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef PubMed.
- M. Sy, A. Nonat, N. Hildebrandt and L. J. Charbonniere, Chem. Commun., 2016, 52, 5080–5095 RSC.
- S. Petoud, S. M. Cohen, J.-C. G. Buenzli and K. N. Raymond, J. Am. Chem. Soc., 2003, 125, 13324–13325 CrossRef PubMed.
- C. Y. Chow, S. V. Eliseeva, E. R. Trivedi, T. N. Nguyen, J. W. Kampf, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2016, 138, 5100–5109 CrossRef PubMed.
- A. Foucault-Collet, C. M. Shade, I. Nazarenko, S. Petoud and S. V. Eliseeva, Angew. Chem., Int. Ed., 2014, 53, 2927–2930 CrossRef PubMed.
- E. R. Trivedi, S. V. Eliseeva, J. Jankolovits, M. M. Olmstead, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2014, 136, 1526–1534 CrossRef PubMed.
- A. Foucault-Collet, K. A. Gogick, K. A. White, S. Villette, A. Pallier, G. Collet, C. Kieda, T. Li, S. J. Geib, N. L. Rosi and S. Petoud, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17199–17204 CrossRef PubMed.
- A. T. Bui, A. Roux, A. Grichine, A. Duperray, C. Andraud and O. Maury, Chem. – Eur. J., 2018, 24, 3408–3412 CrossRef PubMed.
- J.-C. G. Bünzli and S. V. Eliseeva, in Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects, ed. P. Hänninen and H. Härmä, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 1–45, DOI:10.1007/4243_2010_3.
- D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Chem. Rev., 2002, 102, 1977–2010 CrossRef PubMed.
- A. Bourdolle, M. Allali, J.-C. Mulatier, B. Le Guennic, J. M. Zwier, P. L. Baldeck, J.-C. G. Bunzli, C. Andraud, L. Lamarque and O. Maury, Inorg. Chem., 2011, 50, 4987–4999 CrossRef PubMed.
- R. M. Supkowski and W. D. Horrocks Jr., Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, S. A. S. de, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504, 10.1039/a808692c.
- C. Doffek, N. Alzakhem, M. Molon and M. Seitz, Inorg. Chem., 2012, 51, 4539–4545 CrossRef PubMed.
- C. Doffek, N. Alzakhem, C. Bischof, J. Wahsner, T. Gueden-Silber, J. Luegger, C. Platas-Iglesias and M. Seitz, J. Am. Chem. Soc., 2012, 134, 16413–16423 CrossRef PubMed.
- D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1996, 1581–1586, 10.1039/p29960001581.
- D. Parker, P. K. Senanayake and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1998, 2129–2139 RSC.
- D. Parker, Coord. Chem. Rev., 2000, 205, 109–130 CrossRef.
- G.-L. Law, R. Pal, L. O. Palsson, D. Parker and K.-L. Wong, Chem. Commun., 2009, 7321–7323, 10.1039/B920222F.
- R. A. Poole, C. P. Montgomery, E. J. New, A. Congreve, D. Parker and M. Botta, Org. Biomol. Chem., 2007, 5, 2055–2062 RSC.
- F. Kielar, C. P. Montgomery, E. J. New, D. Parker, R. A. Poole, S. L. Richardson and P. A. Stenson, Org. Biomol. Chem., 2007, 5, 2975–2982 RSC.
- T. J. Soerensen, A. M. Kenwright and S. Faulkner, Chem. Sci., 2015, 6, 2054–2059 RSC.
- A. Watkis, R. Hueting, T. J. Soerensen, M. Tropiano and S. Faulkner, Chem. Commun., 2015, 51, 15633–15636 RSC.
- J. Lehr, M. Tropiano, P. D. Beer, S. Faulkner and J. J. Davis, Chem. Commun., 2015, 51, 15944–15947 RSC.
- W. J. Evans, J. L. Shreeve, J. W. Ziller and R. J. Doedens, Inorg. Chem., 1995, 34, 576–585 CrossRef.
- P. R. Selvin and J. E. Hearst, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10024–10028 CrossRef.
- A. Cha, G. E. Snyder, P. R. Selvin and F. Bezanilla, Nature, 1999, 402, 809–813 CrossRef PubMed.
- P. Ge and P. R. Selvin, Bioconjugate Chem., 2004, 15, 1088–1094 CrossRef PubMed.
- H. E. Rajapakse, D. R. Reddy, S. Mohandessi, N. G. Butlin and L. W. Miller, Angew. Chem., Int. Ed., 2009, 48, 4990–4992 CrossRef PubMed.
- A. Mohamadi and L. W. Miller, Bioconjugate Chem., 2016, 27, 2540–2548 CrossRef PubMed.
- M. Rajendran, E. Yapici and L. W. Miller, Inorg. Chem., 2014, 53, 1839–1853 CrossRef PubMed.
- D. R. Reddy, L. E. Pedro Rosa and L. W. Miller, Bioconjugate Chem., 2011, 22, 1402–1409 CrossRef PubMed.
- A. M. Reynolds, B. R. Sculimbrene and B. Imperiali, Bioconjugate Chem., 2008, 19, 588–591 CrossRef PubMed.
- M. S. Tremblay, M. Halim and D. Sames, J. Am. Chem. Soc., 2007, 129, 7570–7577 CrossRef PubMed.
- D. Kovacs, X. Lu, L. S. Mészáros, M. Ott, J. Andres and K. E. Borbas, J. Am. Chem. Soc., 2017, 139, 5756–5767 CrossRef PubMed.
- H.-K. Lee, H. Cao and T. M. Rana, J. Comb. Chem., 2005, 7, 279–284 CrossRef PubMed.
- D. Kovacs and K. E. Borbas, Coord. Chem. Rev., 2018, 364, 1–9 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- G. Saroja, N. B. Sankaran and A. Samanta, Chem. Phys. Lett., 1996, 249, 392–398 CrossRef.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodriguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542–1548 RSC.
- B. M. Reddy, B. Thirupathi and M. K. Patil, Open Catal. J., 2009, 2, 33–39 CrossRef.
- I. Sosic, M. Gobec, B. Brus, D. Knez, M. Zivec, J. Konc, S. Lesnik, M. Ogrizek, A. Obreza, D. Zigon, D. Janezic, I. Mlinaric-Rascan and S. Gobec, Angew. Chem., Int. Ed., 2016, 55, 5745–5748 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and LC-MS traces for new compounds, and absorption, excitation and emission spectra of Ln complexes, crystallographic characterisation. CCDC 1832851 and 1833918. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt01270a |
| This journal is © The Royal Society of Chemistry 2018 |