
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of the lanthanide contraction on the activity of a lanthanide-dependent methanol dehydrogenase – a kinetic and DFT study†
Henning
Lumpe
a,
Arjan
Pol
b,
Huub J. M.
Op den Camp
 b and
Lena J.
Daumann
*ac
b and
Lena J.
Daumann
*ac
aLudwig-Maximilians-Universität München, Department Chemie, Butenandtstr. 5-13, 81377 München, Germany. E-mail: lena.daumann@cup.lmu.de
bDepartment of Microbiology, Institute of Wetland and Water Research, Radboud University Nijmegen, The Netherlands
cCenter for Integrated Protein Science Munich (CIPSM) at the Department of Chemistry, Ludwig-Maximilians-Universität München, Germany
First published on 18th July 2018
Abstract
Interest in the bioinorganic chemistry of lanthanides is growing rapidly as more and more lanthanide-dependent bacteria are being discovered. Especially the earlier lanthanides have been shown to be preferentially utilized by bacteria that need these Lewis acids as cofactors in their alcohol dehydrogenase enzymes. Here, we investigate the impact of the lanthanide ions lanthanum(III) to lutetium(III) (excluding Pm) on the catalytic parameters (vmax, KM, kcat/KM) of a methanol dehydrogenase (MDH) isolated from Methylacidiphilum fumariolicum SolV. Kinetic experiments and DFT calculations were used to discuss why only the earlier lanthanides (La–Gd) promote high MDH activity. Impact of Lewis acidity, coordination number preferences, stability constants and other properties that are a direct result of the lanthanide contraction are discussed in light of the two proposed mechanisms for MDH.
Introduction
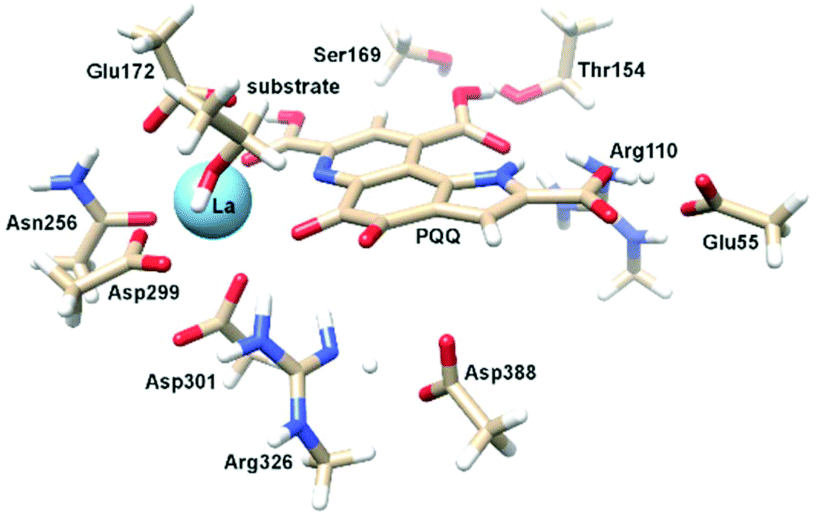
Calcium-dependent methanol dehydrogenases (MDH) have been known for over 50 years.1 However, the recently discovered lanthanide (Ln)‡-dependent MDH are also widespread in bacteria inhabiting different ecosystems and this has sparked a growing interest in lanthanide-dependent metalloenzymes.2,3 These enzymes are mostly found in methanotrophic and methylotrophic bacteria, organisms that metabolize C1 compounds such as methane or methanol. We recently reported the crystal structure of a Eu-MDH isolated from Methylacidiphilum fumariolicum SolV, a thermoacidophile that is strictly dependent on Ln for growth.4 The structure reveals, in addition to the europium ion in the active site, the presence of pyrroloquinoline quinone (PQQ), a redox cofactor that is commonly found in many alcohol dehydrogenases (Fig. 1).5 PQQ needs a Lewis acid for activation (Ca2+, Ln3+) and this cofactor is reduced to the quinol form (PQQH2) concurrent with methanol oxidation. Further, the metal ion in the active site is proposed to be involved in proper substrate orientation and binding.6 DFT calculations by Schelter and coworkers revealed that a trivalent lanthanide is superior to calcium(II) in cofactor activation.7 For Ca-MDH, cytochrome CL has been found to be the redox partner in vivo.8,9 Some bacteria possess both genes for the Ca- as well as for the Ln-dependent enzyme.10–15 In these bacteria lanthanides are also involved in gene expression and regulation and a sensing mechanism for Ln has been proposed but is still not well understood.11,16–19 Not all lanthanide ions support growth of Ln-dependent bacteria equally and not all of them are able to produce an active MDH enzyme. A preference for the lighter, more abundant Ln has been noted both in laboratory and field experiments.10,12,20,21 For example, during the Deepwater Horizon blowout in 2010, millions of moles of natural gas were released into the environment, leading to a bloom of methanotrophic bacteria.22–26 Analysis of water samples taken near the site revealed, that concurrent to the bacterial methane consumption, light lanthanides (La, Ce, Pr and Nd) were depleted during the event.21 Masuda et al. recently reported that Ho3+ and Lu3+ did not support growth of Methylobacterium aquaticum.10 In addition, the Ln-dependent alcohol dehydrogenase from Pseudomonas putida did show catalytic activity upon addition of earlier lanthanides like La3+ but not with late Ln like Yb3+.12 We recently reported that there is a link between the growth rate of strain SolV and the catalytic efficiency of the methanol dehydrogenase.4 This enzyme is pivotal to the energy metabolism of methanotrophic and methylotrophic bacteria. The early Ln like La3+ and Pr3+ led to fast growth of strain SolV and high MDH activity while cultivation with Eu3+ was slower, albeit still exponential bacterial growth occurred, and this metal ion was able to produce an active MDH enzyme. However, addition of Yb3+ to purified MDH lead to a strong decrease in activity and this element did not support exponential growth of strain SolV. These results are surprising, as the later Ln3+ should be better Lewis acids owing to their decreased ionic radius, a result from the lanthanide contraction.27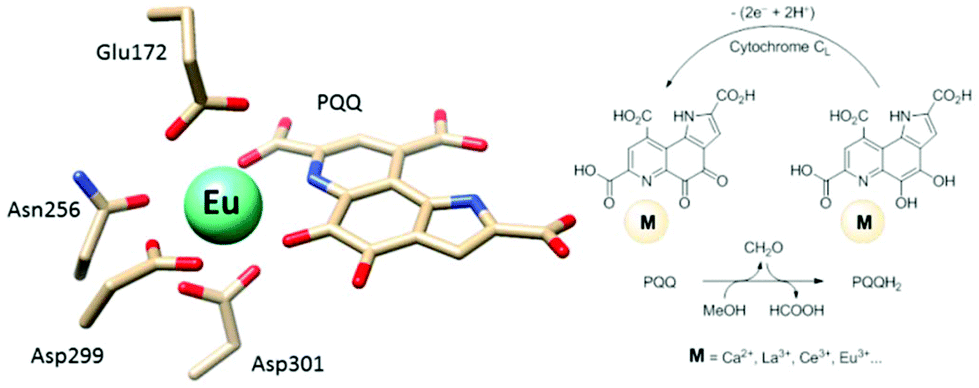 | ||
| Fig. 1 Left: Active site of Eu-MDH from strain SolV (PDB 6FKW). Image generated with the UCSF Chimera package.28 Right: Schematic overview of the MDH-methanol oxidation. MDH from strain SolV is also capable of oxidizing formaldehyde to formic acid. | ||
Why are the chemically similar lanthanides not supporting growth and MDH activity equally? This question prompted us to study the activity of MDH isolated from M. fumariolicum SolV in the presence of the entire lanthanide series (La–Lu, excluding Pm). It should be noted that strain SolV is strictly dependent on Ln, as it does not possess the gene for Ca-MDH.20 Kinetic investigations were complemented by DFT calculations of the active site and we drew from previously published studies in lanthanide coordination chemistry and the methanol dehydrogenase literature to get a full picture of the impact of the lanthanide contraction and the resulting changes such as Lewis acidity and coordination number-preference along the series on the activity of MDH.
Results and discussion
MDH assay optimization
The activity of MDH towards methanol was assessed with a dye-linked assay (Scheme 1 and Fig. S1†) using 2,6-dichlorophenolindophenol (DCPIP) as redox indicator, phenazine ethosulfate (PES) as artificial electron acceptor and PIPES buffer (20 mM, pH 7.2).1,29 The unfavorable solubility product of lanthanide phosphates precluded the use of phosphate buffer for our experiments.30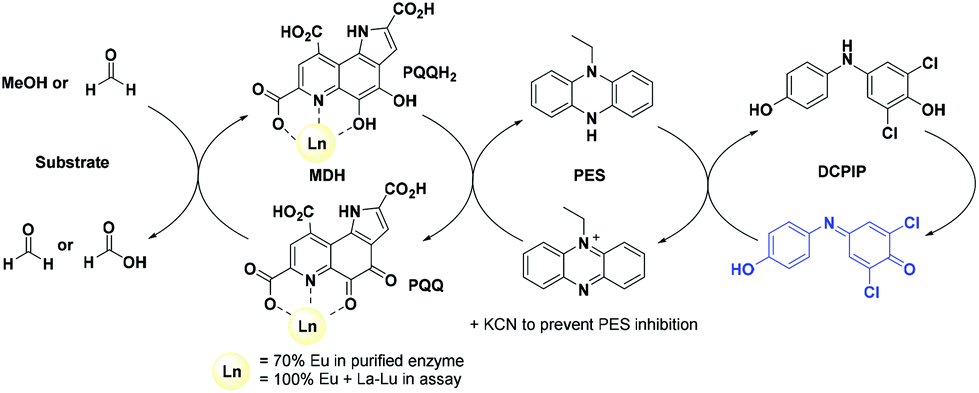 | ||
| Scheme 1 The assay components for assessing MDH activity. | ||
Further, potassium cyanide was added to the assay. While cyanide prevents inhibition of MDH by PES it is also added to suppress endogenous substrate activity in MDH.31–33 It should be noted, that under the conditions of the assay (pH 7.2), the cyanide is present mainly in its protonated form (HCN) in solution and that the solubility of HCN is temperature dependent.34 The nature of the endogenous substrate for methanol dehydrogenases has remained unresolved for the past decades but traces of alcohols and aldehydes present in the reagents used have been named.1,32 We further added a large excess of methanol (50 mM) to the assays to ensure this substrate was preferentially turned over. This assay is artificial in every way and prone to error, so it is important to investigate different systems under the same conditions and use caution when comparing results. The cultivation of strain SolV with Eu3+ and subsequent purification of Eu-MDH was reported recently.4 The enzyme is obtained in a ‘partial-apo’ form after purification where 70% of the active sites are substituted with Eu3+ and the remaining 30% are not occupied but can be reconstituted by adding additional Ln.4 The addition of increasing amounts of europium(III) to 200 nM purified ‘partial-apo’ Eu-MDH led to a gradual increase in activity until saturation around 5 to 20 μM added metal was observed.4 It is proposed that this excess of lanthanide ions will lead to a 100% occupation of the active site in MDH. The activity towards methanol oxidation upon La3+, Pr3+ and Nd3+ addition to MDH follows the same saturation kinetics as the addition with Eu3+, where, between 0 and 5 μM, a sharp increase in activity was observed, with saturation behavior for higher concentrations (Fig. 2). An investigation of a potential inhibiting effect of added lanthanide ions at higher concentrations than 20 μM was precluded by a background reaction of the assay mix (PES, KCN, DCPIP in buffer) in the absence of MDH. It is worth noting here, that the determined affinity (of ‘partial-apo’ Eu-MDH) for the chosen lanthanides was not significantly different (Kassoc(La) = 0.55 ± 0.19 μM, Kassoc(Pr) = 0.52 ± 0.20 μM, Kassoc(Nd) = 0.49 ± 0.17 μM and Kassoc(Eu) = 0.53 ± 0.21 μM) but rather the specific activity (SA) of the enzyme in the presence of these different metal ions varied (Normalized SA: La = 0.13 ± 0.01, Pr = 0.17 ± 0.01, Nd = 0.18 ± 0.01, Eu = 0.08 ± 0.01).
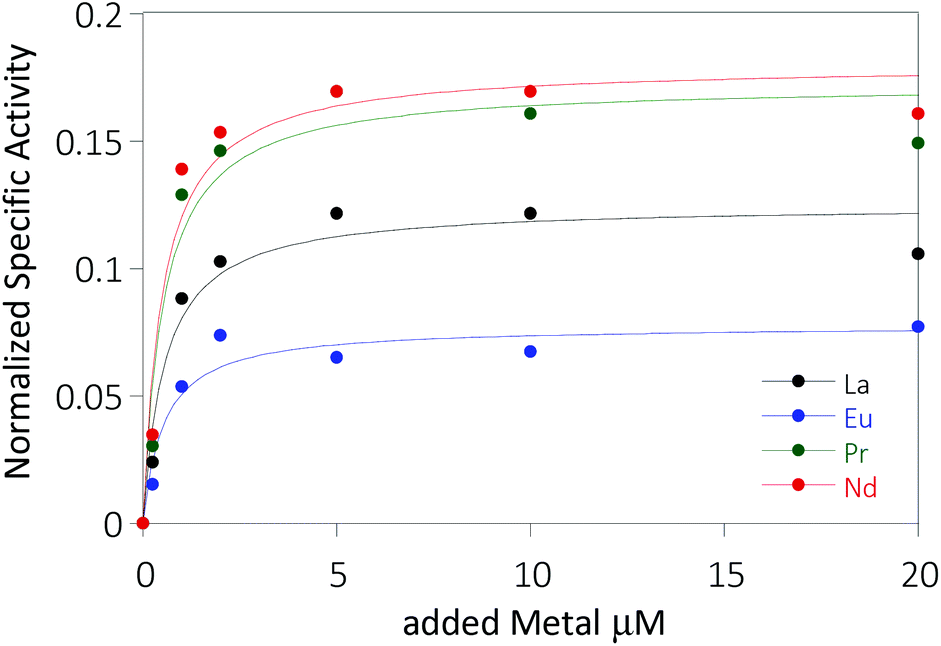 | ||
| Fig. 2 Normalized specific activity (n = 3, for 5 and 10 μM n = 1) of 200 nM ‘partial-apo’ Eu-MDH in the presence of increasing amounts of LnCl3, error bars were omitted for clarity, standard deviations from the mean were less than 10%. The data was normalized by subtracting the activity of Eu-MDH without added Ln from the data. | ||
The metal ion affinity constant for Eu differed slightly from the one reported earlier (Kassoc(Eu) = 2.6 ± 0.6 μM) as we adjusted the assay for the use of a plate-reader instrument and the resulting variations in PES, DCPIP, methanol and ‘partial-apo’ Eu-MDH concentrations and ratios are known to have an impact on the assay values.4 It is thus of importance to compare only results obtained under the exact same conditions, ideally obtained with the same stock solutions (artificial electron acceptor, redox indicator, methanol and ‘partial-apo’ Eu-MDH concentrations, temperature and pH and used buffer). We also conducted a titration with lutetium(III) under the same conditions as described in Fig. 2. However, increasing amounts of Lu3+ led to a gradual decrease of MDH activity. This indicates, that upon addition of an excess of lanthanide ions (20 μM Ln to 0.2 μM MDH), the active site metal (here Eu) can be replaced to some extent. Without the addition of MDH, the assay mixture did not show a background reaction in the presence of any of the Ln at the concentration used (20 μM).
Kinetic experiments with the entire lanthanides series
The differences in stimulated MDH activity by the early lanthanides La3+, Pr3+, Nd3+ compared to Eu3+ and Lu3+ prompted us to study the effect of equal concentrations of every lanthanide in the series to gain more insight into the differences caused by the lanthanide contraction on MDH activity. Based on the observations made in the titration experiment, we chose the same conditions for all lanthanides and added 20 μM (saturation conditions) of the respective chloride salts to 200 nM ‘partial-apo’ Eu-MDH in a subsequent assay. Fig. 3 shows the differences in activity for ‘partial-apo’ Eu-MDH upon addition of the lanthanide series (La–Lu, Pm not included). The data was normalized to 1 for the MDH activity without added lanthanides (here 10 μL H2O were added to achieve the same total volume in the sample well as in the Ln samples). Surprisingly, the addition of the late lanthanides (Tb3+ to Lu3+) decreased the activity compared to when no additional lanthanide ions were added to Eu-MDH (entry H2O in Fig. 3). Ln above Tb3+ increased activity gradually from Gd3+ to Nd3+, while Pr3+ and La3+ were slightly lower in activity than Nd3+ but still among the three best metal ions for maximum activity.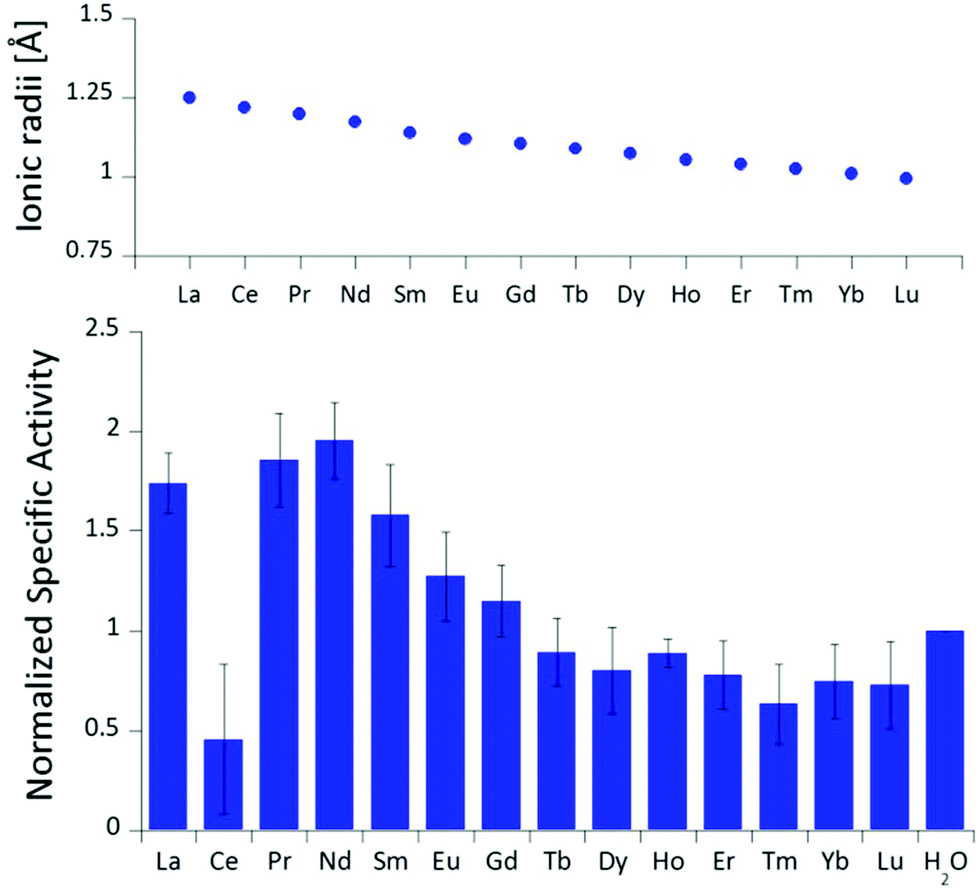 | ||
| Fig. 3 Ionic radii of the lanthanides(III) in aqueous solution as determined by D'Angelo et al.35 Normalized specific activity (n = 6) of 200 nM Eu-MDH in the presence of 20 μM of LnCl3. Conditions: 20 mM PIPES pH 7.2, 1 mM PES, 1 mM KCN, 100 μM DCPIP, 50 mM MeOH, 45 °C. | ||
The turnover point for an enhancing effect of an added Ln turning into a negative effect on catalysis occurs right after gadolinium(III). From our data, it is difficult to make a statement on whether the much discussed ‘gadolinium break’ is of relevance here.36,37 The decrease of activity in the presence of some lanthanides suggests that the later Ln are able to exchange with europium(III) in the active site. This exchange might be dependent both on the external Ln3+ concentration as well as on the properties of the Ln3+ itself. The assay with Ce3+ did not yield reproducible results, despite several attempts. While the assay mix itself (buffer, PES, DCPIP, KCN) did not show a background reaction when cerium(III) was added, the DCPIP reduction varied greatly in the presence of ‘partial-apo’ Eu-MDH. We attribute this to the property of cerium to exist not only in the trivalent form but also in its tetravalent state.27 Traces of Ce4+ present in the medium (not bound to the enzyme active site) might oxidize the reduced, colorless DCPIP dye back to the colored form and thus led to problematic results in the determination of MDH activity. This derivative was included in Fig. 3 for the sake of completeness but is not further discussed. It should be noted here again, that early (and larger) lanthanides are preferentially depleted by methanotrophic bacteria as demonstrated with samples collected during the Deepwater Horizon catastrophe.21,38 These studies could indicate that not only the activity of MDH is tuned to utilize the early lanthanides, but also the metal ion uptake mechanisms in these bacteria. This effect is most pronounced for La3+, Ce3+, Pr3+ and Nd3+, Ln3+ that also yielded highly active MDH derivatives in our experiments and in enzymatic studies by others.11 This observation is however not limited to MDH isolated from methanotrophic and methylotrophic bacteria. Klebensberger and coworkes have shown that activity of an alcohol dehydrogenase isolated from P. putida (a non-methylotrophic organism) was observed with La3+, Ce3+, Pr3+, Nd3+, Sm3+, Gd3+ and Tb3+, and was highest with Pr3+ and Nd3+. Lu3+, Tm3+, Ho3+ and Eu3+ were not investigated and Er3+ and Yb3+ as well as the two rare earth elements Sc3+ and Y3+, did not produce an active enzyme.12 These findings together with our results suggest that these alcohol dehydrogenase systems are tuned by evolution to utilize and function with the earlier, more abundant lanthanides. However, why do Pr3+ and Nd3+ stimulate the highest activity? And why does the activity gradually decrease after Nd3+ albeit increasing Lewis acidity of the central metal ion? To answer these questions, the properties of the trivalent lanthanides along the series and the different mechanisms which have been proposed for methanol dehydrogenases, have to be considered.
MDH mechanisms and possible impact of Ln size
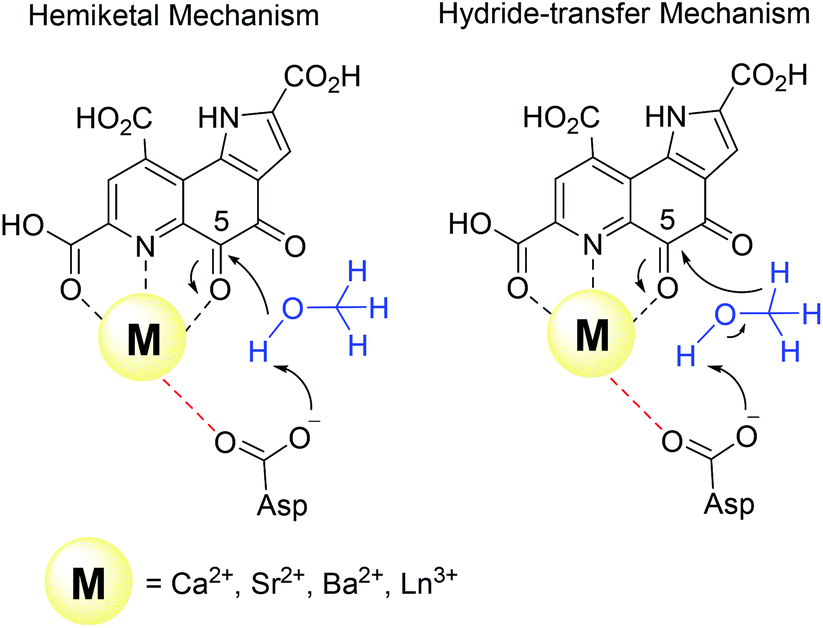 | ||
| Fig. 4 Key steps of the two proposed mechanisms in methanol dehydrogenase enzymes. | ||
Harris and Davidson reported a comparison of Ca-MDH and Sr-MDH isolated from Methylobacillus glycogenes. The authors demonstrated that Sr2+, while having a 20% larger ionic radius than Ca2+, produced the more active enzyme but with a decreased affinity for substrate (Table 1).46 Goodwin and Anthony reported the kinetic parameters of Ca-MDH, Sr-MDH and Ba-MDH from Methylobacterium extorquens.47 Here, the same trend – the larger the alkaline earth ion, the higher the vmax but the lower the substrate affinity – was observed (Table 1). Increased Lewis acidity does not seem to necessarily have a positive effect on catalysis. It is interesting that in this study the native metal ion calcium(II) does produce the slowest enzyme. However it is the derivative with the best substrate affinity. The large decrease in substrate affinity upon substitution with barium(II) suggests that this 25% larger, non-biologically relevant metal ion perturbs the active site and interferes with proper substrate binding. DFT calculations of the active site with Ca2+ or Ba2+ as the central metal were performed by Mainardi et al.48 It was shown that the activation energy for methanol oxidation of Ca2+ containing MDH was doubled compared to the Ba2+-derivative. By calculating the energy barriers for both the hemiketal- and the hydride-transfer mechanism, they further showed, that for both mechanisms almost all free-energy barriers were reduced in the presence of Ba2+.48 Within the lanthanide series, the larger derivatives are also superior to the smaller Ln and stimulate the highest activity. However, one has to keep in mind that with the lanthanide-dependent MDH, the larger Ln are the native metal ions and the later, less abundant Ln might play only a minor role in biological systems. Further, differences between Ln-MDH isolated from different bacteria have been noted.11,20 Ln-MDH isolated from strain SolV does not require activation by ammonia or methylamine in the dye-linked assay and has a pH optimum of 7 while the Ln-MDH isolated from M. extorquens does need methylamine and pH 9 to function properly (Table 1).11,13,20Table 1 also demonstrates the importance of the assay conditions such as temperature (30–60 °C were reported), the chosen buffer and pH (phosphate, PIPES or TRIS buffer, pH 7–9),4,11–13,20,46,47,49 type of electron acceptor and redox indicator (PES, PMS, Wursters Blue, cytochrome CL)12,29,33,50 and the presence of activators or competitive inhibitors (KCN, imidazole, ammonium ions, methylamine)13,32,33,46 for the kinetic parameters that are obtained. Further, some of the components e.g. PES and PMS are light sensitive and degradation during handling can impact the effective concentration of these reagents in the assay. Hence one should only compare kinetic parameters of systems that have been obtained under the same conditions.
| Enzyme system (substrate) | v max [μmol min−1 mg−1] | K M [μM] | k eff (kcat/Km) [mM−1 s−1] |
|---|---|---|---|
| a Conditions: pH 9, 30 °C, 6 mM (NH4)2SO4, 3 mM Wursters blue, 6 mM KCN, MDH from M. glycogenes. b pH 9.0, 10 mM NH4Cl. c pH 9.0, 100 mM NH4Cl, MDH from M. extorquens. d Ln = mixture of La, Ce, Pr, Nd, 60 °C, 100 nM MDH, pH 7, 2 mM PES, 40 μM DCPIP, MDH from M. fumariolicum. e pH 7.2, 45 °C, 100 nM MDH, 1 mM PES, 1 mM KCN, 80 μM DCPIP, 20 μM EuCl3, MDH from M. fumariolicum. f pH 8, 30 °C, 0.5 mM PMS (phenazine methosulfate), 150 μM DCPIP, 25 mM imidazole, 1 μM PrCl3, 1 μM PQQ, ADH = Alcohol dehydrogenase from P. Putida. g pH 9, 100 μM, LaCl3, 5 mM methylamine, 10 mM PQQ, 100 μM DCPIP, 100 μM PMS. ADH = Alcohol dehydrogenase from M. extorquens. h pH 7.2, 45 °C, 200 nM MDH, 1 mM PES, 1 mM, KCN, 100 μM DCPIP, 20 μM LnCl3, MDH from M. fumariolicum, this work. | |||
Ca-MDHa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 46 (MeOH) 46 (MeOH) |
7.3 | 15 | — |
| Sr-MDHa46(MeOH) |
21.8 | 46 | — |
| Ca-MDHb47(MeOH) |
0.81 | 3 | — |
| Sr-MDHb47(MeOH) |
1.08 | 22 | — |
| Ba-MDHc47(MeOH) |
1.61 | 3500 | — |
| Ln-MDHd20 (MeOH) |
4.4 | 0.8 | 5800 |
| Eu-MDHe4 (MeOH) | 0.189 ± 0.006 | 3.62 ± 0.44 | 55 |
| Pr-ADHf12 (EtOH) |
10.6 ± 0.4 | 177 ± 31 | 66 ± 12 |
| La-ADHg13 (MeOH) |
6.6 | 5980 | 2 |
| La-ADHg13 (EtOH) |
6.4 | 0.9 | 14500 |
| Eu/Lu-MDHh (MeOH) | 0.020 ± 0.002 | 0.82 ± 0.39 | 26 |
| Eu-MDHh (MeOH) | 0.043 ± 0.002 | 0.91 ± 0.24 | 50 |
| Eu/La-MDHh (MeOH) | 0.151 ± 0.005 | 1.30 ± 0.21 | 123 |
Ideally one could purify individual MDH derivatives with each of the different Ln in the active site and compare them under the same assay conditions. However, this endeavor is somewhat hampered by the absence of bacterial growth with the late lanthanides. In lieu of alternative purified Ln-MDH derivatives we nevertheless obtained catalytic parameters for some of the mixed Eu-MDH + Ln species in this study (Fig. 5 and Table 1). 20 μM of Eu3+, La3+ or Lu3+ were added to 200 nM Eu-MDH and increasing amounts of methanol were added (0, 1, 2, 5, 10, 20, 50 μM and 50 mM). As with the data in Fig. 2, the most striking difference among the derivatives (Eu + Lu, Eu + Eu and Eu + La) was the specific activity and the resulting difference in catalytic efficiency which increased with increasing size of the added metal ion (Table 1). Methanol affinity was in this experiment the same within error, and if there are slight differences between the derivatives they are not able to be detected due to the presence of mainly Eu3+ in the active site (Fig. 5). Previously, it was shown that MDH from SolV with a mixture of the larger Ln in the active site (La, Ce, Nd, Pr) exhibited a slightly increased affinity for substrate compared to Eu-MDH (Table 1).4,20 However it should be mentioned here that the parameters of the assay were slightly different.
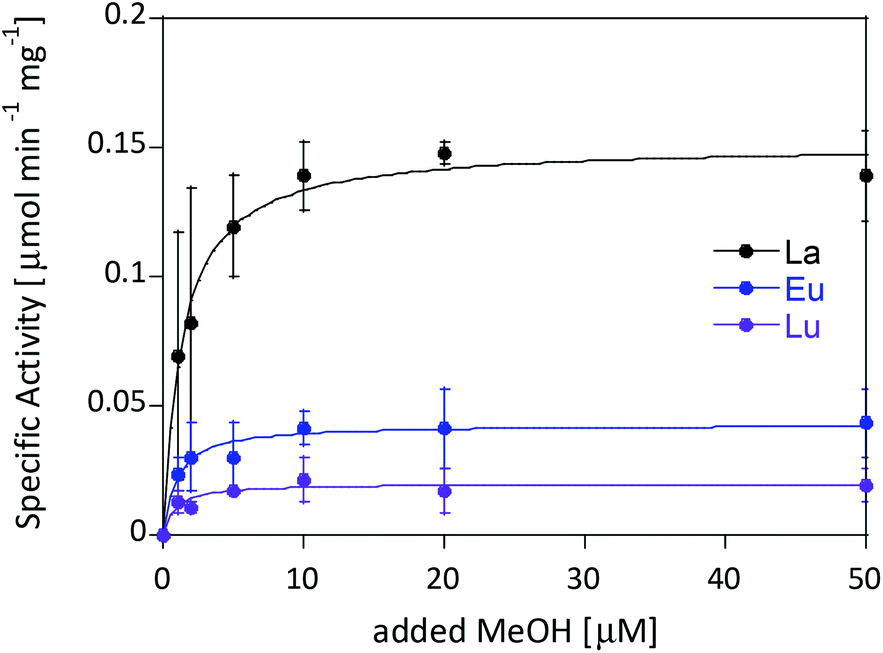 | ||
| Fig. 5 Specific activity (n = 2) of 200 nM Eu-MDH in the presence of 20 μM LnCl3 with increasing amounts of methanol. Conditions: 20 mM PIPES pH 7.2, 1 mM PES, 1 mM KCN, 100 μM DCPIP, 45 °C. | ||
We were further interested in how the specific activity would be influenced by a mixture of an early (La3+ or Nd3+) and a late (Lu3+) lanthanide. Since activity measurements included the addition of different Ln3+ to Eu-MDH which is already occupied to 70% with Eu3+, a competition among the different metals for the active site has to be taken into account. Additional activity measurements were conducted in the presence of La3+, Nd3+, or Eu3+ together with increasing amounts of Lu3+ as competing metal for the active site (Fig. 6). The total amount of additional metal was kept constant at 20 μM. Hence, 100% in Fig. 6 stands for 100% La3+ (20 μM), 75% stands for 75% La3+ and 25% Lu3+ (15 μM and 5 μM, respectively) and so forth. While the amount of Lu3+ decreased the MDH activity in the presence of La3+ and also, slightly, the one with Eu3+, the activity of MDH in the presence of Nd3+ was relatively unaffected by lutetium(III) addition. Interestingly, all measurements showed a higher activity at 50% e.g. La3+/Lu3+ (10 μM each) than the averaged values between 0 and 100% Lu3+. The following paragraphs will discuss how the central metal ion could impact catalytic parameters and explain the differences among the lanthanides.
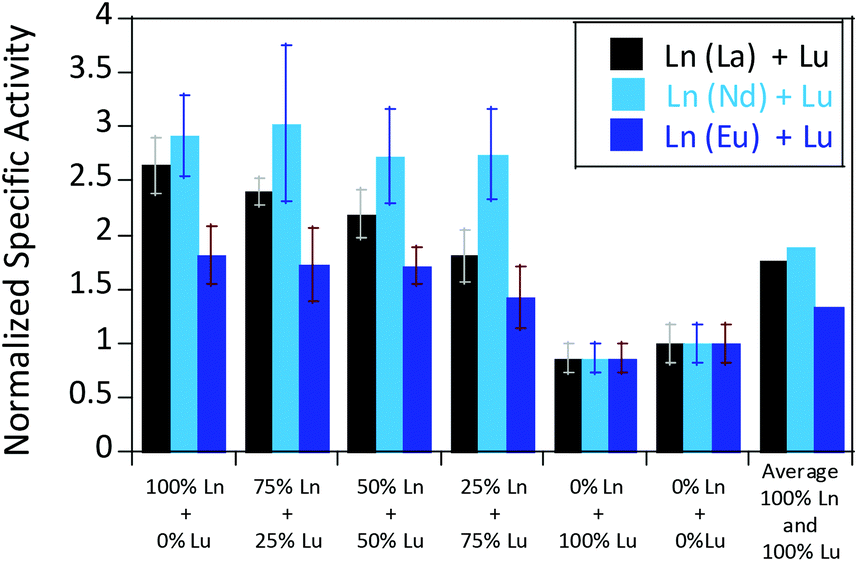 | ||
| Fig. 6 Mixed Ln normalized specific activities (n = 3) of 200 nM Eu-MDH in the presence of 20 μM LnCl3 (Ln = Lu + La/Nd/Eu). Conditions: 20 mM PIPES pH 7.2, 1 mM PES, 1 mM KCN, 100 μM DCPIP, 50 mM MeOH, 45 °C. | ||
In general, a preference for a lower coordination number in the active site, throughout the Ln series was shown by the DFT calculations in the gas phase, which would be in line with the observations made by coordination chemists studying the entire Ln-series.27,45,51,55 However, the CN in MDH is mostly set by the protein environment and the most flexible ligand would still be the substrate. In our experiments however, substrate affinity was hardly affected.
There are several other aspects to how the Lewis acid in the active site might influence methanol oxidation: (i) Substrate deprotonation. Proton abstraction from the substrate by the aspartate residue (here Asp299) is proposed to be assisted by the Lewis acid (Fig. 4).40 In crystal structures of MDH, MeOH is often found coordinating to the Lewis acid and this ligation has been proposed to be beneficial for proton abstraction by lowering the pKa value.56 In our calculations of the active quinone PQQ form with substrate, the distance of the substrate oxygen to the PQQ C5 carbon was relatively constant between 3.41–3.42 Å. However, the distance of the substrate oxygen to the central metal decreased with decreasing ionic radius as expected throughout the lanthanide-series (2.67 Å for La, 2.56 Å for Lu, Table S1†). This is a common feature found in Ln-complexes. For the complex [Ln(TREN-1,2-HOIQO)(H2O)], the sum of Ln–O bond lengths quadratically decrease throughout the Ln-series.55 This pull towards the Lewis acid could hinder the H-transfer or the nucleophilic attack of the methanolate. Further it is interesting to note, that the distance of the aspartate residue in the active site (the one proposed to act as a general base to abstract the proton from the substrate) to the Lewis acid differs greatly in the reported structures of the Ca- and Ln-MDH derivatives. In Ca-MDH from M. extorquens (PDB: 1H4I, 1.94 Å resolution) the Asp303-Ca distance (shown as red dashes in Fig. 4) was reported to be 3.56 Å or 3.61 Å (PDB: 1W6S, 1.2 Å resolution) while in Ca-MDH from Methylophilus methylotrophus W3A1 (PDB: 4AAH, 2.4 Å resolution) the Asp297-Ca distance is reported as 3.25 Å.57–59 The two available Ln-MDH structures available to date are both enzymes isolated from strain SolV and show for Ce-MDH (PDB: 4MAE, 1.6 Å resolution) a significantly shorter Asp299-Ce distance of 2.86 Å and for Eu-MDH (PDB: 6FKW, 1.4 Å resolution) a Asp299-Eu distance of 2.95 Å.4,20 While the latter two values of Ln-MDH might be the same within the error of the experiment, the difference in distance from the Asp-residue to the metal ion between Ca and Ln is significant. However in calculations by Leopoldini et al. on the mechanism of Ca-MDH, this Ca-Asp distance was found to vary between 2.38 and 2.82 Å, showing a high degree of flexibility of this amino acid during catalysis.39 We have also noted this flexibility in our calculations. The calculated distances of Ln-Asp in the active site with PQQ in the relevant resting state semiquinone form (PQQ˙−) are found to be much shorter than the experimental values (Fig. S9, S11 and Table S6†). The nature of the metal ion (Ca, La–Lu) might affect the properties of this catalytic base (Asp297/303/299) during methanol oxidation and active site regeneration. For example, a stronger Lewis acid such as Lu3+, interacting with Asp299 would hinder an efficient proton abstraction from methanol, but, in turn, once protonated, coordination of Asp by a stronger Lewis acid would lower the pKa value and thus facilitate proton abstraction from this residue and regeneration of the active site. Given the high flexibility of this residue, it is unlikely that these two effects cancel each other as the distance to the Lewis acid and its positive or negative effect might vary during catalysis. (ii) Ligand Exchange Rates and Complex Stabilities. From decades-long research into the coordination chemistry of the lanthanides it is now well established that complex stabilities increase along the series.27 Cotton et al. reported that the stability constants for EDTA lanthanide complexes significantly increase from the early to the late Ln, due to changes in charge density, coordination number and entropy.60,61 These differences among the stability constants of Ln complexes are, for example, exploited in their separation by ion exchange methods. Further, Graeppi et al. measured the water exchange rates for the late Ln (Gd–Yb) in the complexes [Ln(H2O)8]3+ and [Ln(PDTA)(H2O)2]− which decrease continuously throughout the series.62 Transferring these concepts to the methanol oxidation in MDH, this could suggest that once product is formed (formaldehyde or formic acid) the higher stability of a resulting Ln-product (formaldehyde and especially formic acid, or more specifically, formate under the conditions of the assay) complex would make product release and ligand exchange less favorable for the late Ln and therefore would hinder the regeneration of the active site. This hypothesis could be tested with inhibition experiments of different derivatives of MDH. (iii) The Redox Cycling of the Cofactor PQQ. Schelter and coworkers conducted DFT-calculations of the active site of Ce-MDH and proposed that a stronger Lewis-acid would result in lowered virtual orbitals and would therefore facilitate PQQ reduction and in turn methanol oxidation.7 Using their method, we investigated whether changing to a different lanthanide would influence this redox cycling in any way. For this we optimized the resting semiquinone PQQ˙− and the active quinone PQQ0 state for Ce3+ and the two “extremes” of the Ln series – La3+ and Lu3+ (Fig. 7) – and compared the molecular orbital (MO) energies for those derivatives. While the bond lengths, the locations of the electron density as well as the MO energies of the Ce3+ PQQ0 derivative are in excellent agreement with the previously published results by Schelter, the bond lengths of the optimized structure of the published Ce3+ PQQ˙− species deviated from our calculated coordinates (Fig. S9–S12 and Tables S5–S7†). However, as the input file and the keywords for the optimization were not given, it is difficult to comment on what caused this deviation. It should be noted that in our calculations no negative frequencies were observed.
 | ||
| Fig. 7 Calculated extended active site of xoxF MDH with La as the central metal and PQQ in its active PQQ0 form. Calculation details given in Fig. S9 and 10.† Image generated with the UCSF Chimera package.28 | ||
The results presented in Fig. 8 suggest, that for the redox cycling, the nature of the lanthanide in the active site has only a small effect. The LUMO of the active PQQ0 state is PQQ-based and would be filled during the methanol-oxidation. While the energy values for the La and Ce species differ only by 0.0016 eV, the Lu LUMO is slightly lowered by 0.0408 eV. In comparison, Schelter calculated the LUMO of the Ln-MDH active site with a Ca2+ central metal to be 0.81 eV higher than the Ce3+ species. The resting semiquinone state PQQ˙− shows the same PQQ-based MO which is now a single occupied HOMO. Also in this case, the Lu species is slightly lowered by 0.0590 eV compared to the La derivative. In both cases, the Lu species seems to be slightly favoured by means of energetic values, which is also in agreement with the conclusions made by Schelter and co-workers who stated that a better Lewis acid is beneficial for methanol oxidation. Protein electrochemistry with different MDH derivatives could experimentally verify the conclusions drawn from these calculations. (iv) Activation Energies. Lastly, the activation energies of the rate-determining step could be affected by the different lanthanides in Ln-MDH similar to the Ca/Ba-MDH that was investigated by Mainardi et al.48 This could be experimentally probed by determining kinetic parameters at different temperatures for different MDH derivatives. Efforts to purify and characterize such derivatives are currently underway.
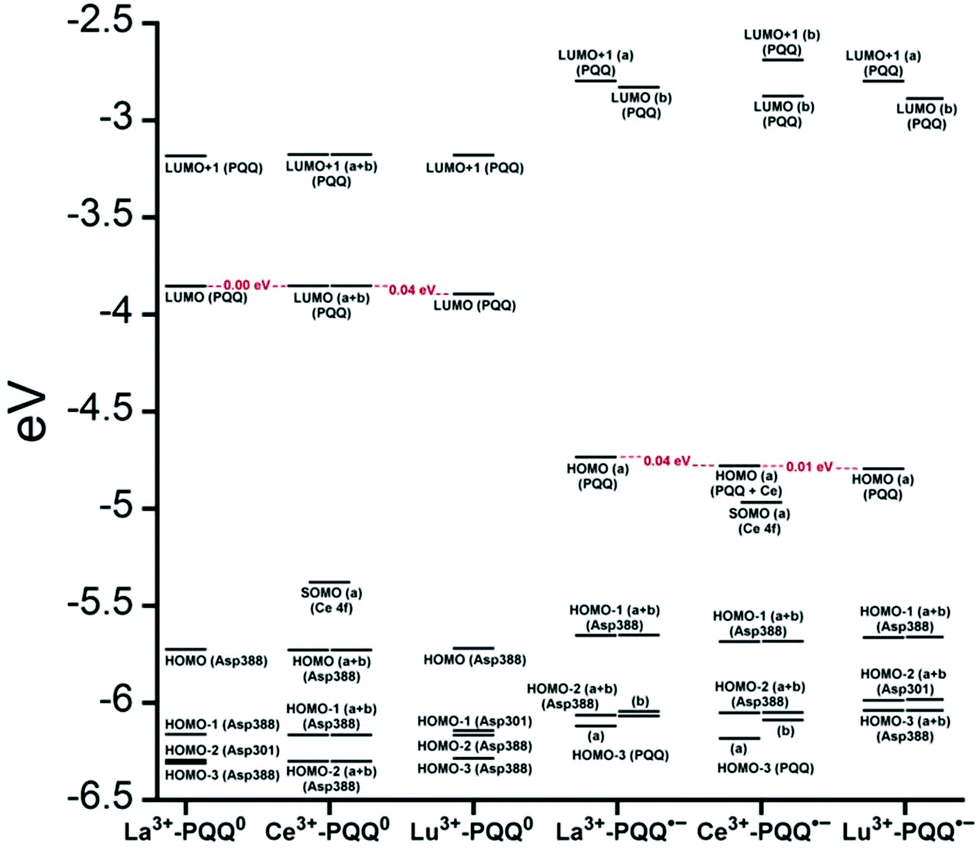 | ||
| Fig. 8 Molecular orbital diagrams of oxidized Ln PQQ0 and the semiquinone form Ln PQQ˙−. Ln = La3+, Ce3+ and Lu3+. Next to the MO description the location of electron density is shown in brackets. | ||
Materials and methods
DFT calculations
Electronic-structure calculations for determination of the redox-cycling for the Ce3+ PQQ0 and Ce3+ PQQ˙− species were performed with Gaussian 0963 as described by Schelter et al.7 Example input-files for all calculations are given in the ESI (Fig. S3, S6, S8 and S10–S12†). The B3LYP functional with the 6-31G(d) basis set for C, H, N, O was used.4 28 electron quasi relativistic effective core potentials (ECP) and segmented basis sets were used for the central metal: MWB28.64,65 Starting point of the geometry optimization was the active site of the crystal structure of Ce-MDH isolated from SolV (4MAE). The present amino acids in the active site were truncated and the terminal carbon atoms were frozen to mimic the sterics imposed by the protein. The present polyethylenglycol was truncated as well and used as a substrate-model. The conductor-like polarizable continuum model (CPCM) was used for water, with the default settings for the universal force field (UFF) and a dielectric constant of 4, to reproduce the interior of the protein and the active site. For additional calculations with alternative Ln, the central metal in the input file was exchanged with La or Lu and the used ECP was changed accordingly. Structure optimizations for the preferred coordination number were performed in a similar manner, but with several simplifications for computational savings. Again, the B3LYP functional with the 6-31G(d) basis set for C, H, N, O was used, but larger ECPs for the central metal: MWB46 + 4fn (e.g. 59 for Yb3+). Calculations were performed with 11 outer-sphere electrons, as pseudo-singlets (f-electrons included in ECP) and restricted closed-shell calculations in the gas phase. The present amino acids in the active site were reduced to the actual coordinating ones (Glu172, Asn256, Asp299, Asp301), which were truncated and the terminal carbon atoms, as well as all of the PQQ-atoms were frozen. The present polyethylenglycol was truncated as well but not frozen. The cerium atom was then exchanged with every Ln in the series. In case of Gd, the Harris functional was not able to form the initial guess, therefore the core Hamiltonian was used instead, using “guess = (core, always)”. Test calculations with solvent model (CPCM = water), with the right spin state (smaller ECP-MWB28) or unfrozen amino acids slightly influenced the overall structure, but had no influence on the coordination numbers. For additional active center calculations, the truncated polyethylene glycol residue was manually removed, and/or the PQQ-quinone part was manually converted to a quinol. For simplification, the added quinol protons were frozen as well in their position.
Kinetic experiments
Protein purification, handling and concentration determination were conducted after a previously reported protocol.4Plate-reader assay with different metal ions (as shown in Fig. 3): A phenazine ethosulfate (PES, ≥95%, Sigma-Aldrich) and 2,6-dichlorophenolindophenol (DCPIP, sodium salt dihydrate, ≥90%, Fluka) dye-coupled assay at 45 °C was used for this experiment using 96 well microtiter plates and the Epoch2 plate reader from BioTek, Winooski, VT, USA. A commonly used protocol is shown in Fig. S1.† Each well contained 185 μL assay mix (20 mM PIPES (1,4-piperazinediethanesulfonic acid, ≥99%, BioPerformance certified, Sigma) pH 7.2, 100 μM DCPIP, 1 mM PES and 1 mM KCN) and ‘partial-apo’ Eu-MDH (200 nM) and this mixture was incubated for 2 minutes at 45 °C. The assay mix without enzyme had been previously incubated at 45 °C for 15 minutes in the dark to alleviate background reactions. Then 10 μL of freshly prepared 0.4 mM stock solutions of each Ln3+ chloride salt were added to each well (LaCl3·7H2O, CeCl3·7H2O, PrCl3·7H2O, NdCl3·6H2O, SmCl3, EuCl3·6H2O, GdCl3·6H2O, TbCl3·6H2O, DyCl3, HoCl3·6H2O, ErCl3·7H2O, TmCl3·6H2O, YbCl3·6H2O, LuCl3·6H2O, all minimum of ≥99.9% purity, ABCR). Water was added to one control well in each row. The consumption of residual MeOH which had to be added for storage of the protein, was monitored at 600 nm for 2 minutes before 10 μL of a 1 M solution of MeOH (final concentration ∼50 mM, HPLC grade, Fisher Chemical) were added for a final volume of 205 μL in each well and the oxidation reaction was monitored at 600 nm. An average of six measurements (three experimental replicates with two technical replicates each) was taken to calculate specific activities (SA) and an extinction coefficient of 18.5 cm−1 mM−1 for DCPIP at pH 7.2 in PIPES buffer was used. Initial rates (the first 3 min of the slope after methanol addition) were used for the calculation of the SA. The SA of MDH without added metal ions was normalized to 1, while the SA of each of the assays with Ln was normalized by dividing through the activity of MDH without added metal ions. No background reaction of the assay mix (without enzyme) with added metal ions was observed. Metal titrations (shown in Fig. 2) with La3+, Pr3+, Nd3+ and Eu3+ were fit using the Michaelis–Menten eqn (1)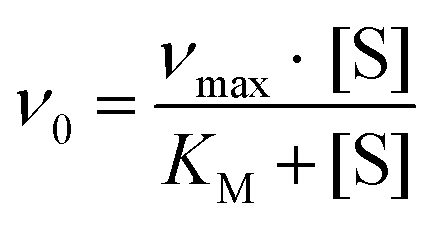 | (1) |
000 μM. Here it was important to wait until background activity from storage methanol and endogenous substrate had ceased, hence a delay of 5 minutes was introduced until methanol was added. The software KaleidaGraph 4.5 from synergy software was used to fit and plot all data.
Conclusions
Ten years ago, a biological relevance of lanthanides for living organisms was unthinkable.66–68 Now, studies of lanthanide-dependent bacterial metabolism is a quickly growing field. To answer the important questions of how these elements are made bioavailable, taken up into cells, regulate gene expression and what their advantages over calcium in the active site of enzymes are, it will take a strong collaborative effort by researchers working in the fields of microbiology, biochemistry, spectroscopy, computational and coordination chemistry. In this paper, we have investigated the methanol oxidation of a methanol dehydrogenase isolated from M. fumariolicum SolV with different lanthanides from a coordination chemists’ point of view. While activity in the presence of early lanthanides was high, the decreasing size of the lanthanides caused by the lanthanide contraction had a negative effect on catalytic activity. Lewis acidity and co-factor activation are not the only important factors that have to be considered but coordination number preference, ligand exchange rates, substrate orientation and activation and hydrogen bonding are important factors as well.Conflicts of interest
There are no conflicts to declare.Acknowledgements
LJD would like to thank the SFB 749 and the Center for Integrated Protein Science Munich for funding. HOdC is supported by the European Research Council (ERC Advanced Grant project VOLCANO 669371).Notes and references
- C. Anthony, Subcell. Biochem., 2000, 35, 73–117 CrossRef PubMed.
- A. Ramachandran and D. A. Walsh, FEMS Microbiol. Ecol., 2015, 91, fiv105 CrossRef PubMed.
- M. Taubert, C. Grob, A. M. Howat, O. J. Burns, J. L. Dixon, Y. Chen and J. C. Murrell, Environ. Microbiol., 2015, 17, 3937–3948 CrossRef PubMed.
- B. Jahn, A. Pol, H. Lumpe, T. R. M. Barends, A. Dietl, C. Hogendoorn, H. J. M. Op den Camp and L. J. Daumann, ChemBioChem, 2018, 19(11), 1147–1153 CrossRef PubMed.
- A. Jongejan, S. S. Machado and J. A. Jongejan, J. Mol. Catal. B: Enzym., 2000, 8, 121–163 CrossRef.
- C. Anthony and P. Williams, Biochim. Biophys. Acta, Proteins Proteomics, 2003, 1647, 18–23 CrossRef PubMed.
- J. A. Bogart, A. J. Lewis and E. J. Schelter, Chem. – Eur. J., 2014, 21, 1743–1748 CrossRef PubMed.
- P. R. Afolabi, F. Mohammed, K. Amaratunga, O. Majekodunmi, L. Dales, R. Gill, D. Thompson, B. Cooper, P. Wood, M. Goodwin and C. Anthony, Biochemistry, 2001, 40, 9799–9809 CrossRef PubMed.
- J. M. Cox, D. J. Day and C. Anthony, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1992, 1119, 97–106 CrossRef.
- S. Masuda, Y. Suzuki, Y. Fujitani, R. Mitsui, T. Nakagawa, M. Shintani and A. Tani, mSphere, 2018, 3(1), e00462-17 CrossRef PubMed.
- H. N. Vu, G. A. Subuyuj, S. Vijayakumar, N. M. Good, N. C. Martinez-Gomez and E. Skovran, J. Bacteriol., 2016, 198, 1250–1259 CrossRef PubMed.
- M. Wehrmann, P. Billard, A. Martin-Meriadec, A. Zegeye and J. Klebensberger, mBio, 2017, 8, e00570–e00517 CrossRef PubMed.
- N. M. Good, H. N. Vu, C. J. Suriano, G. A. Subuyuj, E. Skovran and N. C. Martinez-Gomez, J. Bacteriol., 2016, 198, 3109–3118 CrossRef PubMed.
- N. C. Martinez-Gomez, H. N. Vu and E. Skovran, Inorg. Chem., 2016, 55, 10083–10089 CrossRef PubMed.
- E. Skovran and N. C. Martinez-Gomez, Science, 2015, 348, 862–863 CrossRef PubMed.
- F. Chu, D. A. C. Beck and M. E. Lidstrom, PeerJ, 2016, 4, e2435 CrossRef PubMed.
- M. Farhan Ul Haque, B. Kalidass, N. Bandow, E. A. Turpin, A. A. Dispirito and J. D. Semrau, Appl. Environ. Microbiol., 2015, 81, 7546–7552 CrossRef PubMed.
- J. D. Semrau, A. A. DiSpirito, W. Gu and S. Yoon, Appl. Environ. Microbiol., 2018, 84(6), e02289-17 CrossRef PubMed.
- W. Gu, M. Farhan Ul Haque, A. A. DiSpirito and J. D. Semrau, FEMS Microbiol. Lett., 2016, 363, fnw129–fnw129 CrossRef PubMed.
- A. Pol, T. R. M. Barends, A. Dietl, A. F. Khadem, J. Eygensteyn, M. S. M. Jetten and H. J. M. Op den Camp, Environ. Microbiol., 2014, 16, 255–264 CrossRef PubMed.
- A. M. Shiller, E. W. Chan, D. J. Joung, M. C. Redmond and J. D. Kessler, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- B. Graham, W. K. Reilly, F. Beinecke, D. F. Boesch, T. D. Garcia, C. A. Murray and F. Ulmer, Deep Water: The Gulf Oil Disaster and the Future of Offshore Drilling, National Commission on the BP Deepwater Horizon Oil Spill and Offshore Drilling, Washington DC, 2013 Search PubMed.
- C. M. Reddy, J. S. Arey, J. S. Seewald, S. P. Sylva, K. L. Lemkau, R. K. Nelson, C. A. Carmichael, C. P. McIntyre, J. Fenwick, G. T. Ventura, B. A. S. Van Mooy and R. Camilli, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20229–20234 CrossRef PubMed.
- T. B. Ryerson, R. Camilli, J. D. Kessler, E. B. Kujawinski, C. M. Reddy, D. L. Valentine, E. Atlas, D. R. Blake, J. de Gouw, S. Meinardi, D. D. Parrish, J. Peischl, J. S. Seewald and C. Warneke, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20246–20253 CrossRef PubMed.
- M. C. Redmond and D. L. Valentine, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20292–20297 CrossRef PubMed.
- E. A. Solomon, Nat. Geosci., 2014, 7, 394 CrossRef.
- S. Cotton, Lanthanide and actinide chemistry, John Wiley & Sons, Chichester, UK, 2006 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef PubMed.
- R. Ghosh and J. R. Quayle, Anal. Biochem., 1979, 99, 112–117 CrossRef PubMed.
- F. H. Firsching and S. N. Brune, J. Chem. Eng. Data, 1991, 36, 93–95 CrossRef.
- V. Davidson, Principles and applications of quinoproteins, CRC Press, 1992 Search PubMed.
- P. Hothi, J. Basran, M. J. Sutcliffe and N. S. Scrutton, Biochemistry, 2003, 42, 3966–3978 CrossRef PubMed.
- P. Hothi, M. J. Sutcliffe and N. S. Scrutton, Biochem. J., 2005, 388, 123–133 CrossRef PubMed.
- J. Ma, P. K. Dasgupta, W. Blackledge and G. R. Boss, Environ. Sci. Technol., 2010, 44, 3028–3034 CrossRef PubMed.
- P. D'Angelo, A. Zitolo, V. Migliorati, G. Chillemi, M. Duvail, P. Vitorge, S. Abadie and R. Spezia, Inorg. Chem., 2011, 50, 4572–4579 CrossRef PubMed.
- M. Laing, J. Chem. Educ., 2009, 86, 188 CrossRef.
- N. Kaltsoyannis and P. Scott, The f elements, Oxford University Press, Oxford, 1999 Search PubMed.
- B. R. Graham, W. K. Reilly, F. G. Beinecke, D. E. Boesch, T. D. Garcia, C. A. Murray and F. Ulmer, National Comission on the BP Deepwater Horizon Oil Spill and Offshore Drilling Deep Water. The gulf oil disaster and the future of offshore drilling. Report to the President, 2011 Search PubMed.
- M. Leopoldini, N. Russo and M. Toscano, Chem. – Eur. J., 2007, 13, 2109–2117 CrossRef PubMed.
- M. Prejanò, T. Marino and N. Russo, Chem. – Eur. J., 2017, 23, 8652–8657 CrossRef PubMed.
- A. McSkimming, T. Cheisson, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 1223–1226 CrossRef PubMed.
- Y.-J. Zheng, Z.-x. Xia, Z.-w. Chen, F. S. Mathews and T. C. Bruice, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 432–434 CrossRef.
- A. Oubrie, H. J. Rozeboom, K. H. Kalk, A. J. Olsthoorn, J. A. Duine and B. W. Dijkstra, EMBO J., 1999, 18, 5187–5194 CrossRef PubMed.
- S. Itoh, H. Kawakami and S. Fukuzumi, J. Am. Chem. Soc., 1997, 119, 439–440 CrossRef.
- S. A. Cotton and P. R. Raithby, Coord. Chem. Rev., 2017, 340, 220–231 CrossRef.
- T. K. Harris and V. L. Davidson, Biochem. J., 1994, 300, 175–182 CrossRef PubMed.
- M. G. Goodwin and C. Anthony, Biochem. J., 1996, 318, 673–679 CrossRef PubMed.
- N. B. Idupulapati and D. S. Mainardi, J. Phys. Chem. A, 2010, 114, 1887–1896 CrossRef PubMed.
- O. Adachi, K. Matsushita, E. Shinagawa and M. Ameyama, Agric. Biol. Chem., 1990, 54, 2833–2837 Search PubMed.
- J. M. Armstrong, Biochim. Biophys. Acta, 1964, 86, 194–197 CrossRef.
- S. A. Cotton, C. R. Chim., 2005, 8, 129–145 CrossRef.
- S. Y. Reddy and T. C. Bruice, Protein Sci., 2004, 13, 1965–1978 CrossRef PubMed.
- X. Zhang, S. Y. Reddy and T. C. Bruice, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 745–749 CrossRef PubMed.
- A. Ramirez-Solis, J. I. Amaro-Estrada, J. Hernández-Cobos and L. Maron, Inorg. Chem., 2018, 57, 2843–2850 CrossRef PubMed.
- M. Seitz, A. G. Oliver and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 11153–11160 CrossRef PubMed.
- Y.-J. Zheng and T. C. Bruice, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 11881–11886 CrossRef.
- Z.-x. Xia, W.-w. Dai, Y.-f. Zhang, S. A. White, G. D. Boyd and S. F. Mathews, J. Mol. Biol., 1996, 259, 480–501 CrossRef PubMed.
- M. Ghosh, C. Anthony, K. Harlos, M. G. Goodwin and C. Blake, Structure, 1995, 3, 177–187 CrossRef PubMed.
- P. A. Williams, L. Coates, F. Mohammed, R. Gill, P. T. Erskine, A. Coker, S. P. Wood, C. Anthony and J. B. Cooper, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 75–79 CrossRef PubMed.
- S. A. Cotton and J. M. Harrowfield, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2011, DOI:10.1002/9781119951438.eibc2090.
- R. H. Betts and O. F. Dahlinger, Can. J. Chem., 1959, 37, 91–100 CrossRef.
- N. Graeppi, D. Hugh Powell, G. Laurenczy, L. Zékány and A. Merbach, Inorg. Chim. Acta, 1995, 235, 311–326 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, M. C. X. Li, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, Jr., F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef.
- X. Cao and M. Dolg, J. Mol. Struct.: THEOCHEM, 2002, 581, 139–147 CrossRef.
- S. A. Cotton and J. M. Harrowfield, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2011, DOI:10.1002/9781119951438.eibc2091.
- S. A. Cotton and J. M. Harrowfield, Lanthanides in Living Systems, in The Rare Earth Elements: Fundamentals and Applications, ed. D. A. Atwood, John Wiley & Sons, Chichester, UK, 2012, p. 65 Search PubMed.
- S. Lim and S. J. Franklin, CMLS, Cell. Mol. Life Sci., 2004, 61, 2184–2188 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Assay protocol for the plate reader, calculated structures, example input files, tables with calculated bond lengths and angles. See DOI: 10.1039/c8dt01238e |
| ‡ For simplicity we will use the term Lanthanide and the abbreviation Ln for the elements La–Lu throughout the document, even though lanthanum is strictly speaking not a lanthanide but has become included by common usage. |
| This journal is © The Royal Society of Chemistry 2018 |