
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescence properties of mechanochemically synthesized lanthanide containing MIL-78 MOFs
Tarek
Alammar
 *a,
Ihor Z.
Hlova
b,
Shalabh
Gupta
*b,
Viktor
Balema
b,
Vitalij K.
Pecharsky
ab and
Anja-Verena
Mudring
*abc
*a,
Ihor Z.
Hlova
b,
Shalabh
Gupta
*b,
Viktor
Balema
b,
Vitalij K.
Pecharsky
ab and
Anja-Verena
Mudring
*abc
aDepartment of Materials Science and Engineering, Iowa State University, Ames, IA 50011-2300, USA. E-mail: tarek.alammar@rub.de
bAmes Laboratory, Iowa State University, Ames, IA 500011-3020, USA. E-mail: shalabh@ameslab.gov
cDepartment of Materials and Environmental Chemistry, Stockholm University, Svante Arrhenius väg 16 C, 106 91 Stockholm, Sweden. E-mail: anja-verena.mudring@mmk.su.se
First published on 27th April 2018
Abstract
Three metal–organic framework (MOF) compounds, Ln0.5Gd0.5{C6H3(COO)3}; Ln = Eu, Tb, and Dy with a MIL-78 structure, have been synthesized by a solvent-free mechanochemical method from stoichiometric mixtures of benzene 1,3,5-tricarboxylic acid, C6H3(COOH)3, also known as trimesic acid, and the respective lanthanide carbonates, Ln2(CO3)3·xH2O, Ln = Eu, Gd, Tb and Dy. MIL-78 (Ln0.5Gd0.5) shows the characteristic red, green, and yellow luminescence of Eu3+, Tb3+, and Dy3+, respectively. Efficient intramolecular energy transfer from the ligand triplet state to the excited states of Ln3+ ions can be observed. The lifetimes and quantum yields of these compounds are studied and discussed in detail. Among the three compounds, the Tb3+ containing compound shows the longest lifetime and highest quantum yield due to a smaller contribution from non-radiative decay pathways and better matching of the lowest triplet energy level of the benzenetricarboxylate ligand and the resonance level of Tb3+.
Introduction
Metal–organic frameworks (MOFs) constitute a large class of three-dimensional coordination polymers with ordered microporous structures, which have recently attracted considerable attention as highly promising materials for gas storage and separation, catalysis, magnetic cooling and as luminescent and photonic materials for sensing applications.1–5 MOFs containing lanthanides (Ln–MOFs) are of particular interest as functional luminescent materials for solid-state lighting and related applications.4,6 Typically, MOFs are prepared using solution-based techniques including conventional and solvothermal chemical syntheses,7 as well as less common microwave-assisted,8 electro-,9 sono-10 and liquid-assisted grinding11 procedures. While solvent-based methods generally produce highly crystalline MOFs, they often rely on hazardous organic solvents and generate considerable amounts of wastes when scaled-up. Among the alternatives, the solvent-free mechanochemical preparation of MOFs, also known as mechanochemistry, has proven to be a rapid, simple and efficient process12 that is readily adaptable to large-scale production. Mechanochemistry often produces materials with unique particle shapes, sizes, morphologies and structure defects, which can influence or even determine their photoluminescence properties.13 Therefore, in order to fully establish mechanochemistry as a preferred synthesis route for advanced phosphor materials, it is critical to understand the effects of mechanical processing on the luminescence properties of phosphors. For the application of Ln–MOFs in solid-state lighting, for example, in wLEDs (white light emitting diodes), aside from an efficient emission, a high photoluminescence quantum yield and excellent color purity are important prerequisites. In addition to the desired wavelength emission and high conversion efficiency, achieving enhanced stability at high temperatures while maintaining a narrow emission width is of particular interest.14 The simplicity of preparation and the cost are important factors to consider, too. In this context, a facile, scalable and reliable synthesis of Ln–MOFs like Ln–MIL-78, [Ln0.5Gd0.5{C6H3(COO)3}], is important.The luminescence properties of lanthanide-based MIL-78 MOFs were first described by Férey group.15,16 It was found that MIL-78 containing Eu, Tb and Dy exhibits excitation under UV radiation with strong red, green and blue emission, respectively.14 Other lanthanide metals, including Y, Pr, Nd, Sm, Gd and Er, form isostructural MOFs.15 However, luminescence studies on materials synthesized mechanochemically are scarce.17 Recently, several studies showed that MIL-78 can be formed by ball milling starting from lanthanide hydrides or carbonates with trimesic acid.18,19 Herein, we report a study on the luminescence properties of three MIL-78 MOFs [Ln0.5Gd0.5{C6H3(COO)3}]; Ln = Eu, Tb, and Dy synthesized using the solid-state mechanochemical approach.
Experimental
Materials
All chemicals were used without further purification. Eu2(CO3)3·4H2O, Gd2(CO3)3·4H2O, Tb2(CO3)3·6H2O, and Dy2(CO3)3·4H2O were purchased from Alfa Aesar and benzene 1,3,5-tricarboxylic acid, [C6H3(COOH)3], aka trimesic acid from Sigma-Aldrich.Synthesis of Ln{C6H3(COO)}3
Following a previously published synthesis procedure,19 lanthanide-based MIL-78 MOFs were prepared by ball milling of benzene 1,3,5-tricarboxylic acid [C6H3(COOH)3] with the respective lanthanide carbonate hydrates [Ln2(CO3)3·xH2O] (Ln = Eu, Gd, Tb, and Dy) in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. In a typical experiment, a total 1 g of the stoichiometric mixture of benzene 1,3,5-tricarboxylic acid and the Ln-metal carbonate hydrate was ball milled in a SPEX 8000 M shaker mill for 2 h in a hardened steel container together with two 12.7 mm and four 6.35 mm grinding stainless steel balls achieving a ball-to-sample ratio close to 18:1. The reaction conditions used for the synthesis of MIL-78 were optimized in our previous work.19 As noted in ref. 19, addition of anhydrous dimethylformamide and dimethyl ether prevented the progress of the reaction and, hence, the formation of any condensed species. The reactions were also carried out in the presence of 1–2 molar equivalents of additional water, but no significant changes in the reaction time or crystallinity of the products were noted. Beyond a certain limit of additional water, a plastic-like product was obtained, which was not characterized further.
:1 molar ratio. In a typical experiment, a total 1 g of the stoichiometric mixture of benzene 1,3,5-tricarboxylic acid and the Ln-metal carbonate hydrate was ball milled in a SPEX 8000 M shaker mill for 2 h in a hardened steel container together with two 12.7 mm and four 6.35 mm grinding stainless steel balls achieving a ball-to-sample ratio close to 18:1. The reaction conditions used for the synthesis of MIL-78 were optimized in our previous work.19 As noted in ref. 19, addition of anhydrous dimethylformamide and dimethyl ether prevented the progress of the reaction and, hence, the formation of any condensed species. The reactions were also carried out in the presence of 1–2 molar equivalents of additional water, but no significant changes in the reaction time or crystallinity of the products were noted. Beyond a certain limit of additional water, a plastic-like product was obtained, which was not characterized further.
Characterization
Results and discussion
The PXRD patterns of the as-prepared MOFs are shown in Fig. 1. The products obtained upon ball milling with each combination of lanthanides are isostructural, and their PXRD patterns are in good agreement with the calculated pattern of the MIL-78 (Gd) crystallizing in the monoclinic space group C2/m (no. 12).15 Because of significant line broadening of the Bragg peaks and extremely low intensities above 2θ ≅ 45°, the peak shifts expected to arise for the different compounds due to the lanthanide contraction are not discernible. No additional diffraction peaks originating from precursors or other crystalline impurities can be detected confirming the phase purity of the obtained materials. The large background is expected to arise from amorphization of a part of the materials. | ||
| Fig. 1 Powder X-ray diffraction patterns of the mechanochemically prepared MIL-78 (Ln0.5Gd0.5) Ln = Eu, Tb, and Dy and the calculated pattern of MIL-78 (Gd). | ||
The TG-DSC analysis of the as-prepared MIL-78 (Tb0.5Gd0.5) shows a two-step weight loss between room temperature and 1000 °C (Fig. 2). The first broad loss with a Tonset of ∼100 °C finishing close to 330 °C can be assigned to the loss of both physically adsorbed H2O and the water coordinated within the MIL-78 structure. A similar behaviour is observed for the as-synthesized Eu and Dy containing MIL-78 MOFs (Fig. 2). The weight losses are 10.5% (∼3.3 eq. H2O per f.u.) for the MIL-78 (Eu0.5Gd0.5), 7.3% (∼2.25 eq. H2O per f.u.) for (Tb0.5Gd0.5), and 9% (∼2.8 eq. H2O per f.u.) for (Dy0.5Gd0.5). The second weight loss at a higher temperature with Tonset ≅ 600 °C is attributed to the thermal decomposition of MIL-78 to lanthanide oxides.15 The thermal behaviours are in agreement with those reported for MIL-78 (Gd), MIL-78 (Tb0.34Dy0.33Gd0.33) and MIL-78 (Dy0.5Gd0.5).19 The absence of signals characteristic of trimesic acid and carbonates confirms that all of the precursors reacted, forming desired products.
 | ||
| Fig. 2 TG (black line) and DSC (blue line) curves of the as-prepared MIL-78 (Ln0.5Gd0.5) Ln = Tb, Eu, and Dy. | ||
The surface composition and the oxidation states of the lanthanides in the as-synthesized MIL-78 (Ln0.5Gd0.5) Ln = Eu, Tb, and Dy were investigated with X-ray photoelectron spectroscopy (XPS) and the data obtained were analyzed by a curve fitting process shown in Fig. 3. The XPS spectra illustrated in Fig. 3 contain signals that can be attributed to the electronic transitions in Eu, Gd, Tb, Dy, O and C. The Eu-3d, Tb-3d, and Dy-3d scans are presented in Fig. 3. The Eu-3d core-level XPS spectrum in MIL-78 (Eu0.5Gd0.5) shows two prominent peaks at 1165.4 eV (Eu3+ 3d3/2) and 1135.4 eV (Eu3+ 3d5/2) with three minor satellites located at 1155.4 eV (Eu2+ 3d3/2), 1143.3 eV (mult), and 1124.8 eV (Eu2+ 3d5/2).
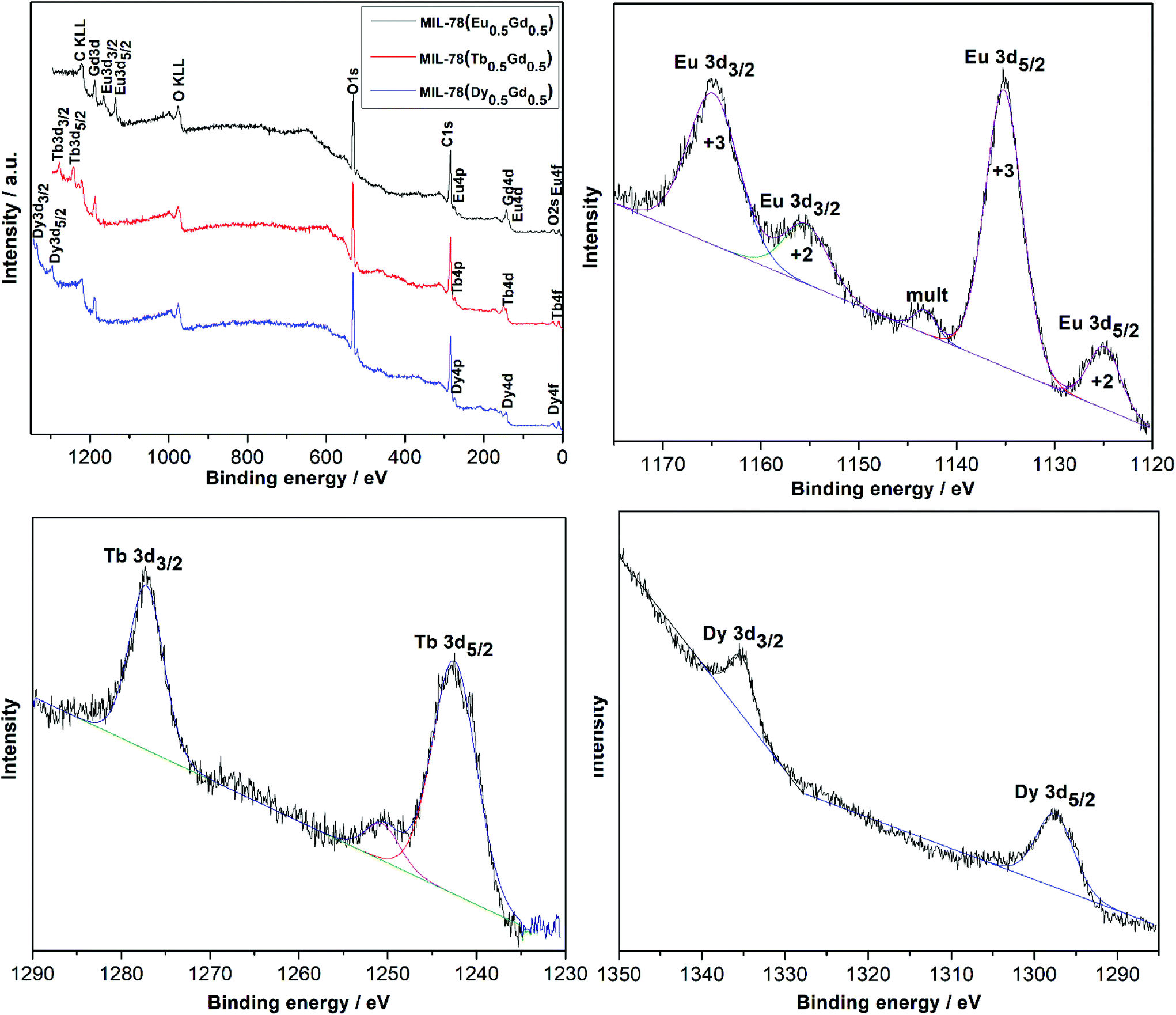 | ||
| Fig. 3 X-ray photoelectron spectrum survey and narrow scans of Eu-3d, Tb-3d, and Dy-3d regions for MIL-78 (Ln0.5Gd0.5) Ln = Eu (black), Tb (red), and Dy (blue). | ||
The presence of Eu2+ can be attributed to the reduction of Eu3+ to Eu2+ due to the charge compensation process during profiling under the ambient conditions of XPS measurements which is frequently observed for Eu3+ compounds.20–22 The XPS spectrum for the Tb-3d region of MIL-78 (Tb0.5Gd0.5) shows two peaks at 1242.5 eV (Tb3+ 3d5/2) and 1277.2 eV (Tb3+ 3d3/2). A satellite peak observed at 1250 eV is attributed to the presence of Tb4+ traces because of the oxidation of Tb3+ to Tb4+ during the electron beam irradiation, as often observed for the binary oxide and similar compounds when being investigated under similar conditions.23,24 The two peaks at 1297.5 eV and 1335.4 eV in the XPS spectrum of MIL-78 (Dy0.5Gd0.5) can be assigned to Dy3+ 3d5/2 and Dy3+ 3d3/2.25
The IR spectra of MIL-78 (Ln0.5Gd0.5) with Ln = Eu, Tb, and Dy (Fig. 4) show similar features. The characteristic bands of the protonated carboxyl groups of trimesic acid (νOH, 3085 cm−1; νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 1714 cm−1; δCO, 537 cm−1) and metal carbonates (νCO between 1500 and 300 cm−1) cannot be observed in the final products which further confirms that the starting materials are fully consumed in the reaction.26–28 Instead, in the IR spectra of MIL-78 (Ln0.5Gd0.5) Ln = Eu, Tb, and Dy, bands at 1542 cm−1 and 1357 cm−1 which originate from the asymmetric and symmetric C–O stretching vibrations of –COO−, respectively, can be noticed. The value of the difference between νas and νs is about Δν = 185 cm−1, which indicates that the carboxylate group and metal ions coordinate through a bidentate bridge in the MIL-78 structure.29 The band located at 759 cm−1 can be assigned to the ring-out-of-plane deformation vibration of the benzene core of the substituted trimesic acid. These results confirm the successful coordination of the Ln3+ ions with the trimesic acid ligands. Maiti et al. reported similar observations for the vibrational spectra of manganese 1,3,5-benzenetricarboxylate MOFs.30
O, 1714 cm−1; δCO, 537 cm−1) and metal carbonates (νCO between 1500 and 300 cm−1) cannot be observed in the final products which further confirms that the starting materials are fully consumed in the reaction.26–28 Instead, in the IR spectra of MIL-78 (Ln0.5Gd0.5) Ln = Eu, Tb, and Dy, bands at 1542 cm−1 and 1357 cm−1 which originate from the asymmetric and symmetric C–O stretching vibrations of –COO−, respectively, can be noticed. The value of the difference between νas and νs is about Δν = 185 cm−1, which indicates that the carboxylate group and metal ions coordinate through a bidentate bridge in the MIL-78 structure.29 The band located at 759 cm−1 can be assigned to the ring-out-of-plane deformation vibration of the benzene core of the substituted trimesic acid. These results confirm the successful coordination of the Ln3+ ions with the trimesic acid ligands. Maiti et al. reported similar observations for the vibrational spectra of manganese 1,3,5-benzenetricarboxylate MOFs.30
 | ||
| Fig. 4 Infra-red spectra of the as-prepared MIL-78 (Ln0.5Gd0.5); Ln = Eu, Tb, and Dy and trimesic acid. | ||
UV-Vis absorption spectra of MIL-78 (Ln0.5Gd0.5) and trimesic acid (Fig. 5) were recorded in the solid-state at room temperature. As is evident from Fig. 5, MIL-78 (Ln0.5Gd0.5) with Ln = Eu, Tb, and Dy show quite similar spectral profiles compared to trimesic acid, suggesting that the complexation of the Ln3+ ions does not significantly affect the singlet excitation state of trimesic acid. Starting from high energies, first, the π–π* type of transition in the aromatic ring and then n → π* transitions in the CO group can be observed.31,32 In comparison with the transitions in the free trimesic acid, the bands in the absorption spectrum of trimesic acid are red-shifted and additional bands beyond 300 nm are pronouncedly visible. The differences between the absorption spectrum of trimesic acid (H3BTC) and the spectra of MIL-78 (Ln0.5Gd0.5) can be attributed to the deprotonation and coordination of H3BTC (i.e. BTC3−).33 The f–f transitions of the respective lanthanide ions appear to be invisible in the absorption spectrum due to the high intensity of the transitions within the organic moiety, which have a much higher transition probability.
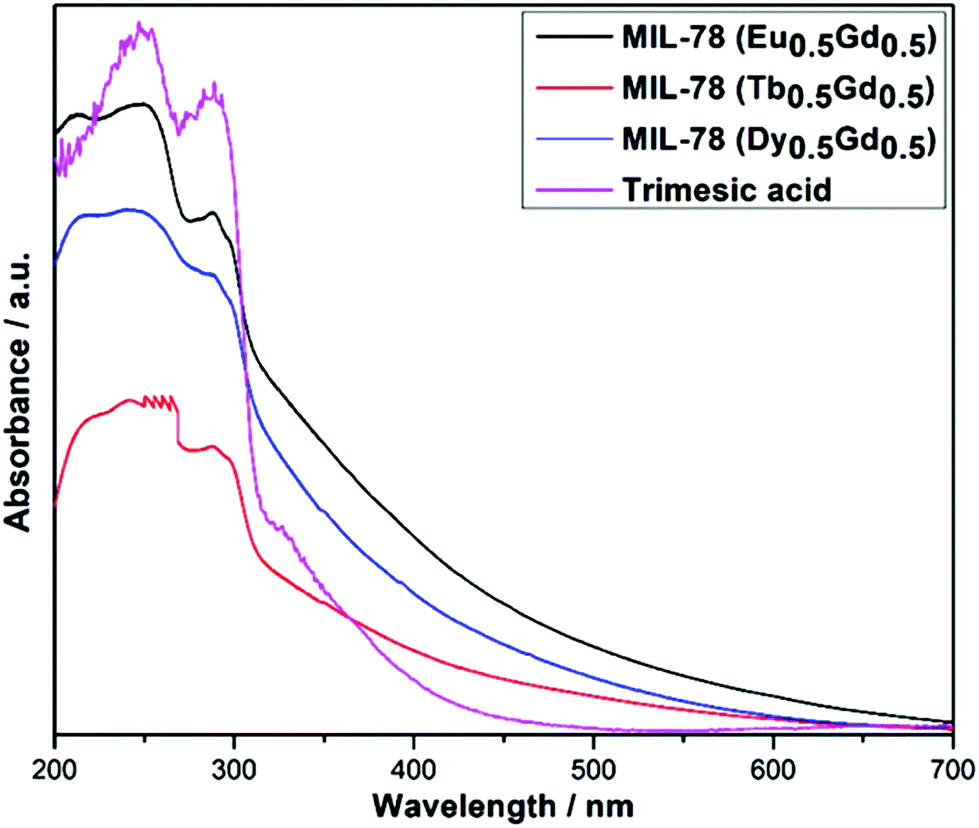 | ||
| Fig. 5 UV absorbance spectra of MIL-78 (Ln0.5Gd0.5); Ln = Eu, Tb, and Dy and trimesic acid. | ||
The luminescence properties of three compounds were determined in the solid state at room temperature. Fig. 6 shows the excitation and emission spectra of MIL-78 (Ln0.5Gd0.5); Ln = Eu, Tb, and Dy. The excitation spectra were recorded by monitoring the 5D0–7F2 transition at 613 nm for Eu3+, the 5D4–7F5 transition at 544 nm for Tb3+, and the 4F9/2–6H13/2 transition at 575 nm for Dy3+. Similar to the UV-vis absorption spectra, the excitation spectra of all the samples show broad bands below 300 nm, which can be attributed to the n–π* and π–π* transitions of the ligand, at lower and higher energies, respectively. In addition, the excitation spectrum of MIL-78 (Eu0.5Gd0.5) displays peaks with low relative intensities at 362 nm, 394 nm, and 465 nm corresponding to the transitions between the 7F0 ground state and the 5D4, 5L6 and 5D2 excited levels of Eu3+. In contrast, for MIL-78 (Tb0.5Gd0.5) no narrow bands arising from 4f–4f transitions characteristic of Tb3+ can be observed. This points to an efficient sensitization of Tb3+ by the ligand. For MIL-78 (Dy0.5Gd0.5), a series of low-intensity bands originating from 4f–4f transitions from the ground state 6H15/2 to higher excited states (325 nm (6H15/2–6P3/2 + 4M17/2), 351 nm (6H15/2–6P7/2), 365 nm (6H15/2–6P5/2 + 4I11/2), 388 nm (6H15/2–4I13/2 + 4K17/2), and 454 nm (6H15/2–4I15/2)), were observed. This hints that the 1,3,5-benzenetricaboxylate ligand can sensitize emission of the lanthanide ions, as has been reported previously for Eu- and Tb-benzene-1,3,5-tricarboxylates.29 Thus, upon excitation to ligand-centered levels below 290 nm, MIL-78 (Ln0.5Gd0.5) MOFs show the characteristic emission of Eu3+, Tb3+ and Dy3+. The emission spectrum of MIL-78 (Eu0.5Gd0.5) displays narrow peaks located at 580, 594, 613, 657.3 and 702.6 nm that can be ascribed to the 5D0–7FJ (J = 0, 1, 2, 3, and 4) transitions of Eu3+, respectively. The intensity of the hypersensitive 5D0–7F2 transition is the strongest and responsible for the red luminescence of MIL-78 (Eu0.5Gd0.5). No emission from higher excited levels such as 5D1 can be observed, suggesting the efficiency of the non-radiative relaxation to the 5D0 level.
 | ||
| Fig. 6 Excitation (left) and emission (right) spectra of MIL-78 (Ln0.5Gd0.5) with Ln = Eu, Tb, and Dy at room temperature. The insets are showing the samples under UV light. | ||
The presence of a 5D0–7F0 transition agrees with the presence of only one crystallographically independent Eu3+ located at a low-symmetry site in the crystal structure of MIL-78.15 This is also supported by the higher intensity relative to the electric dipole transition 5D0–7F2 compared to the magnetic dipole transition 5D0–7F1, aka an asymmetry ratio. The full width at half maximum of the most intense band is estimated to be 3.42 nm, which demonstrates that MIL-78 (Eu0.5Gd0.5) shows high color purity. It is worth mentioning that the absence of residual emission of the ligand in the range between 400 and 550 nm confirms the efficient intramolecular energy transfer from the ligand to the Eu3+ ion. For MIL-78 (Tb0.5Gd0.5), typical Tb3+ emission bands at 494, 544, 584, 624, 650, 671, and 683 nm are attributed to 5D4–7F6, 5D4–7F5, 5D4–7F4, 5D4–7F3, 5D4–7F2, 5D4–7F1, and 5D4–7F0 transitions, respectively. The hypersensitive 5D4–7F5 transition is the most intense among these transitions, and is responsible for its green luminescence (Fig. 6, inset). MIL-78 (Dy0.5Gd0.5) also shows typical Dy3+ emission bands at 481, 576, and 665 nm due to 4F9/2–6H15/2, 4F9/2–6H13/2, and 4F9/2–6H11/2 transitions, respectively, with the 4F9/2–6H15/2 transition (yellow emission) being the most intense.34 The color coordinates for the as-synthesized MOFs were determined to be (0.64, 0.35) for MIL-78 (Eu0.5Gd0.5), (0.30, 0.61) for MIL-78 (Tb0.5Gd0.5), and (0.37, 0.417) for MIL-78 (Dy0.5Gd0.5) and are shown in the 1931 CIE chromaticity diagram in Fig. 7, which indicates that these lanthanide-based MOFs can display good color purity for red, green, and yellow emission, respectively. The luminescence decay curves of MIL-78 (Ln0.5Gd0.5) Ln = Eu, Tb, and Dy are monitored around the respective strongest emission (5D4–7F5 for Tb3+, 5D0–7F2 for Eu3+, and 4F9/2–6H13/2 for Dy3+) at room temperature. The corresponding curves are given in Fig. 8. The curves for MIL-78 (Eu0.5Gd0.5) and MIL-78 (Tb0.5Gd0.5) are fitted by a single exponential function described using the following equation: I = I0 + A1exp(−t/τ). In turn, the curve for MIL-78 (Dy0.5Gd0.5) can only be fitted well with a bi-exponential function: I = I0 + A1exp(−t/τ1) + A2exp(−t/τ2). The lifetime values are estimated to be τ = 0.71 ± 0.007 ms for MIL-78 (Eu0.5Gd0.5), τ = 2.02 ± 0.02 ms for MIL-78 (Tb0.5Gd0.5) and τ1 = 0.008 ± 0.0001 ms and τ2 = 0.07 ± 0.002 ms for MIL-78 (Dy0.5Gd0.5).
 | ||
| Fig. 7 1931 CIE chromaticity diagram of MIL-78 (Eu0.5Gd0.5) (1), (Tb0.5Gd0.5) (2), and (Dy0.5Gd0.5) (3). | ||
 | ||
| Fig. 8 Room temperature luminescence decay curves of MIL-78 (Ln0.5Gd0.5) Ln = Eu, Tb, and Dy. | ||
The average lifetime for MIL-78 (Dy0.5Gd0.5) was calculated to be 0.0084 ms using the equation: τav = (A1τ12 + A2τ22)/(A1τ1 + A2τ2). The single exponential decay patterns of Eu3+ and Tb3+ based compounds suggest the existence of a single chemical environment around Eu3+ and Tb3+, in agreement with the crystal structure. The bi-exponential decay pattern of the Dy3+ based compound suggests the existence of two types of chemical environments around Dy3+ which may arise from local structural distortions evoked by the size difference of Dy3+ and Gd3+. The emission quantum yields were measured at room temperature using an integrating sphere under the excitation wavelength of 321 nm, yielding luminescence quantum yields of 1.24% for MIL-78 (Eu0.5Gd0.5), 19% for MIL-78 (Tb0.5Gd0.5), and 0.26% for MIL-78 (Dy0.5Gd0.5). The longer lifetime and the higher quantum yield of MIL-78 (Tb0.5Gd0.5) compared to other samples can possibly be attributed to the lower number of water molecules in the compound (as evident from the TG data) leading to a smaller contribution from non-radiative decay resulting from the OH oscillation of water molecules.
Moreover, the energy level match between the triplet state of the ligand (BTC3−) and the emissive energy level of the Ln3+ ions is an important factor which can affect the emission efficiency of the lanthanide complexes. The triplet level of the ligand needs to be higher than the emissive lanthanide cation level, ideally relatively close, but not too close that energy back transfer processes dominate. According to empirical rules established by Latva, an ideal ligand to metal transfer process occurs when the energy gaps between the ligand and the emissive level of lanthanides are larger than 2000–2500 cm−1 for Eu3+ and Tb3+.35 The triplet level of the BTC3− ligand derived from the photoluminescence of Gd-complexes has been determined to be at about 23200 cm−1.36 At 23200 cm−1 the T1 energy state of BTC3− is quite higher than that for the 5D0 level of Eu3+ (17300 cm−1), the 5D4 level of Tb3+ (20400 cm−1), and the 4F9/2 level of Dy3+ (21000 cm−1).29,34 The respective energy differences ΔE are 5900 for Eu3+, 2800 for Tb3+, and 2200 for Dy3+. So, the Tb3+ based complex shows better luminescence properties than the Eu3+ and Dy3+ complexes as a result of the good match of the lowest triplet energy level of the BTC3− ligand and the resonance level of Tb3+ leading to effective energy transfer from the ligand to the Tb3+ ions compared to other Ln3+ ions. In general, it has been reported that the ΔE that can minimize the inverse energy transition from the Ln3+ ions to the ligand is about 2500–3500 cm−1.37 The energy difference of the 4F9/2 level of Dy3+ and the BTC3− triplet state is complex and less than the optimal value, therefore it can be expected that there exists a back transfer process leading to weak luminescence properties. Furthermore, the weak luminescence of MIL-78 (Dy0.5Gd0.5) can be ascribed to a small band gap that is more prone to nonradiative deactivation,38 especially in the case of a high water content.
Conclusions
The luminescence properties of three lanthanide-containing metal–organic frameworks with a MIL-78 structure (MIL-78 (Ln0.5Gd0.5) Ln = Eu, Tb, and Dy) prepared by solvent-free ball milling from carbonates of the corresponding lanthanides and benzene 1,3,5-tricarboxylic acid were determined. The three different optically active Ln3+ ions Eu3+, Tb3+ and Dy3+ can be highly sensitized by the BTC3− ligand because the resonance levels of the Ln3+ ions are suitably located with respect to the T-level of the ligand. The lifetime varies from 0.71 ms for MIL-78 (Eu0.5Gd0.5) to 2.02 ms for MIL-78 (Tb0.5Gd0.5) to 0.0084 ms for MIL-78 (Dy0.5Gd0.5) and the quantum yields range from 0.2% for MIL-78 (Dy0.5Gd0.5) to 19% for MIL-78 (Tb0.5Gd0.5). The enhanced luminescence properties of the Tb-containing MIL-78 MOF can be attributed to a good match of the lowest triplet energy level of the BTC3− ligand with the emissive level of Tb3+.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Ames Laboratory is operated for the U.S. Department of Energy (DOE) by the Iowa State University of Science and Technology under contract no. DE-AC02-07CH11358. Research on mechanochemical synthesis and structural characterization was supported by the Division of Materials Sciences and Engineering of Basic Energy Sciences Program of the Office of Science of the U.S. Department of Energy. Research on optical properties was supported by the NSF, grant CHE-1465071.Notes and references
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef PubMed.
- S. K. Henninger, H. A. Habib and C. Janiak, J. Am. Chem. Soc., 2009, 131, 2776–2777 CrossRef PubMed.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef PubMed.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- W. P. Lustig, F. Wang, S. J. Teat, Z. Hu, Q. Gong and J. Li, Inorg. Chem., 2016, 55, 7250–7256 CrossRef PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2011, 112, 933–969 CrossRef PubMed.
- J. Klinowski, F. A. A. Paz and J. Rocha, Dalton Trans., 2011, 40, 321–330 RSC.
- U. Mueller, H. Puetter, M. Hesse and H. Wessel, BASF Aktiengesellschaft, WO2005/049892, 2005 Search PubMed.
- D. Tranchemontagne, J. Hunt and O. M. Yaghi, Tetrahedron, 2008, 64, 8553–8557 CrossRef.
- T. J. Friščić, Mater. Chem., 2010, 20, 7599–7605 RSC.
- (a) T. Friščić, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC; (b) K. Užarević, T. C. Wang, S.-Y. Moon, A. M. Fidelli, J. T. Hupp, O. K. Farha and T. Friščić, Chem. Commun., 2016, 52, 2133–2136 RSC; (c) M. Klimakow, P. Klobes, A. F. Thunemann, K. Rademann and F. Emmerling, Chem. Mater., 2010, 22, 5216–5221 CrossRef; (d) D. Prochowicz, K. Sokołowski, I. Justyniak, A. Kornowicz, D. Fairen-Jimenez, T. Friščić and J. Lewiński, Chem. Commun., 2015, 51, 4032–4035 RSC; (e) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- D. Yuan, G. S. Yi and G. M. Chow, J. Mater. Res., 2009, 24, 2042–2050 CrossRef.
- J. McKittrick and L. E. Shea-Rohwer, J. Am. Ceram. Soc., 2014, 97, 1327–1352 CrossRef.
- C. Serre, F. Millange, C. Thouvenot, N. Gardant, F. Pellé and G. Férey, J. Mater. Chem., 2004, 14, 1540–1543 RSC.
- F. Pellé, S. Surblé, C. Serre, F. Millange and G. Férey, J. Lumin., 2007, 122, 492–495 CrossRef.
- W. Liu, K. Zhu, S. J. Teat, B. J. Deibert, W. Yuan and J. Li, J. Mater. Chem. C, 2017, 5, 5962–5969 RSC.
- N. K. Singh, M. Hardi and V. P. Balema, Chem. Commun., 2013, 49, 972–974 RSC.
- N. K. Singh, S. Gupta, V. K. Pecharsky and V. P. Balema, J. Alloys Compd., 2017, 696, 118–122 CrossRef.
- R. Vercaemst, D. Poelman, L. Fiermans, R. L. Van Meishaeghe, W. H. Laflere and F. Cardon, J. Electron Spectrosc. Relat. Phenom., 1995, 74, 45–56 CrossRef.
- S. Kumar, R. Prakash, R. J. Choudhary and D. M. Phase, Mater. Res. Bull., 2015, 70, 392–396 CrossRef.
- F. Mercier, C. Alliot, L. Bion, N. Thromat and P. Toulhoat, J. Electron. Spectrosc. Relat. Phenom., 2006, 150, 21–26 CrossRef.
- W. Cartas, R. Rai, A. Sathe, A. Schaefer and J. F. Weaver, J. Phys. Chem. C, 2014, 118, 20916–20926 Search PubMed.
- K. G. Tshabalala, I. M. Nagpure, H. C. Swart, O. M. Ntwaeaborwaa, S.-H. Cho and J.-K. Park, J. Vac. Sci. Technol., B, 2012, 30, 031401 Search PubMed.
- D. Barreca, A. Gasparotto, A. Milanov, E. Tondello, A. Devi and R. A. Fischer, Surf. Sci. Spectra, 2007, 14, 52–59 CrossRef.
- L. Song and M. Rongjun, J. Cryst. Growth, 1996, 169, 190–192 CrossRef.
- B. Vallina, J. D. Rodriguez-Blanco, A. P. Brown, J. A. Blanco and L. G. Benning, J. Nanopart. Res., 2013, 15, 1438 CrossRef.
- A. Sungur and M. Kizilyalli, J. Less-Common Met., 1983, 93, 419–423 CrossRef.
- E. Souza, I. Silva, E. Teotonio, M. Felinto and H. Brito, J. Lumin., 2010, 130, 283–291 CrossRef.
- S. Maiti, A. Pramanik, U. Manju and S. Mahant, ACS Appl. Mater. Interfaces, 2015, 7, 16357–16363 Search PubMed.
- C. Chaitanya, Spectrochim. Acta, Part A, 2012, 86, 159–173 CrossRef PubMed.
- G. Jin, Z. Liu, H. Sun and Z. Tian, Luminescence, 2016, 31, 190–194 CrossRef PubMed.
- D. Yang, Y. Tian, W. Xu, X. Cao, S. Zheng, Q. Ju, W. Huang and Z. Fang, Inorg. Chem., 2017, 56, 2345–2353 CrossRef PubMed.
- H. Zhang, R. Fan, W. Chen, X. Zheng and Y. Yang, J. Lumin., 2013, 143, 611–618 CrossRef.
- M. Latva, H. Takalo, V. Mukkala, C. Matachescu, J. Rodriguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef.
- W. Yan, L. Wang, K. Yangxio, Z. Fu and T. Wu, Dalton Trans., 2016, 45, 4518–4521 RSC.
- S. Biju, D. B. Ambili Raj, M. L. P. Reddy and B. M. Kariuki, Inorg. Chem., 2006, 45, 10651–10660 CrossRef PubMed.
- V. Eliseeva and J.-C. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
| This journal is © The Royal Society of Chemistry 2018 |