
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and properties of MoCl4 complexes with thio- and seleno-ethers and their use for chemical vapour deposition of MoSe2 and MoS2 films†
Yao-Pang
Chang
,
Andrew L.
Hector
 ,
William
Levason
,
Gillian
Reid
* and
Joshua
Whittam
,
William
Levason
,
Gillian
Reid
* and
Joshua
Whittam
Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
First published on 29th January 2018
Abstract
Treatment of trans-[MoCl4(MeCN)2] with L (L = Me2S, Me2Se, THT, ½MeSCH2CH2SMe) in CH2Cl2 solution, or reaction of MoCl5 with excess L′ (L′ = nBu2S, nBu2Se, ½MeSCH2CH2SMe, ½iPrSCH2CH2SiPr, ½MeSCH2CH2CH2SMe, ½MeSeCH2CH2CH2SeMe) in MeCN, produces the Mo(IV) complexes, [MoCl4(L)2] and [MoCl4(L′)2], respectively, in good yield. The new complexes have been characterised by IR and UV-vis spectroscopy, elemental analysis and magnetic measurements, whilst crystal structure analyses of trans-[MoCl4(Me2S)2], cis-[MoCl4{RS(CH2)2SR}] (R = Me, iPr) and cis-[MoCl4{MeS(CH2)3SMe}] confirmed their identities and distorted octahedral geometries. The potential of [MoCl4(nBu2E)2] (E = S, Se) as the first examples of molybdenum halide derived single source CVD precursors for the growth of MoE2 thin films was first probed by TGA, which showed multi-step decomposition processes, with the masses of the final residues consistent with MoSe2 (E = Se) and MoCl4 (E = S), respectively. Low pressure CVD experiments conducted at 750 °C (E = S) and 525 °C (E = Se) gave silvery and golden yellow films, respectively. Grazing incidence and in plane XRD data confirmed these to be 2H-MoE2, whilst the texture of the MoSe2 was investigated using pole figure measurements. SEM and EDX data, optical and Raman data on the films are also reported.
Introduction
The coordination chemistry of molybdenum extends over nine formal oxidation states (−II to +VI) and has been intensively studied over the last 50 years.1–3 Over this period, in addition to exploring the basic chemistry, much effort has been devoted to studying the binding and reduction of dinitrogen by molybdenum complexes,2,4 and in modelling the active sites of molybdoenzymes.4,5 In the last 10 years the study of early d-block dichalcogenides, including MoS2 and MoSe2, has become the focus of much effort. Layered early transition metal dichalcogenides, ME2 (M = Nb, Ta, V, W, etc.; E = S, Se or Te), are inorganic analogues of graphene, and their properties can be tuned by varying the metal and the chalcogen. Production of the materials as thin films maximises the anisotropy of their magnetic or electronic properties and thus methods to deposit 2D layers are of particular interest currently.6–8 Applications of these materials in optoelectronics, spintronics, sensors, electrocatalysts and magnetic materials are being actively developed.8,9 The most common methods of forming MoS2 or MoSe2 thin films have been exfoliation from bulk samples, or heating MoO3 (or sometimes molybdenum metal) with S or Se in a hydrogen/inert gas atmosphere, to form the desired MoE2 (E = S or Se) 2D films on an appropriate substrate.10,11–13 Thermal decomposition of [NH4]2[MoS4] in a hydrogen atmosphere has also recently been reported.13Chalcogenoether complexes of the early d-block metal halides can function as single source CVD precursors for some ME2 (for example, M = Ti, V or Nb; E = S or Se).14,15 Chemical vapour deposition from molybdenum halide complexes has rarely been used to form molybdenum dichalcogenides, although MoSe2 films were produced by dual source atmospheric pressure chemical vapour deposition (CVD) using MoCl5 and either Et2Se or nBu2Se.16 The tetrathiolate complex, [Mo(StBu)4], has also been used to deposit MoS2 films by CVD.17
Although there are a few early reports of MoCl4 complexes with mono-, di- and poly-thioethers and thiamacrocycles,1,2,18 the only recent work is the detailed characterisation, including the X-ray crystal structures, of trans-[MoCl4(Et2S)2]19 and trans-[MoCl4(THT)2] (THT = tetrahydrothiophene).20
Here we report the preparation, spectroscopic and structural characterisation of a series of new complexes of molybdenum(IV) chloride with thio- and seleno-ether ligands and the evaluation of a subset of these as single source low pressure CVD reagents for the deposition of the corresponding MoE2 thin films.
Experimental
Syntheses were performed by using standard Schlenk and glove-box techniques under a dry N2 atmosphere. MoCl5 was obtained from Sigma-Aldrich and stored in a glovebox. Tetrahydrothiophene, Me2S and nBu2S were purchased from Sigma-Aldrich and Me2Se from Strem. Solvents were dried by distillation from CaH2 (CH2Cl2, MeCN), Na (Et2O) or Na/benzophenone ketyl (n-hexane). Dichalcogenoethers MeE(CH2)nEMe (E = S, Se; n = 2, 3), iPrS(CH2)2SiPr and nBu2Se were prepared via literature methods.21,22Infrared spectra were recorded using a PerkinElmer Spectrum 100 spectrometer in the range 4000–200 cm−1, and samples prepared as Nujol mulls between CsI plates. UV/visible spectra were recorded as powdered solids, using the diffuse reflectance attachment of a PerkinElmer 750S spectrometer. Microanalyses on new compounds were undertaken by London Metropolitan University. Thermogravimetric analysis (TGA) used a Netzsch TG209 F1 Libra analyser under a flow of argon at 65 mL min−1, contained within a dry, nitrogen purged glovebox. The temperature was increased at a rate of 10 °C min−1.
Single crystal X-ray experimental
Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073) rotating anode generator with VHF Varimax optics (70 micron focus) with the crystal held at 100 K (N2 cryostream). Structure solution and refinement were performed using SHELX(S/L)97 and SHELX-2014/7 and were straightforward.23–25 H atoms were added and refined using a riding model. The X-ray data collection details are in Table S1 (ESI†). CCDC reference numbers in CIF format are [MoCl4(NCMe)2] 1557208; [MoCl4(Me2S)2] 557209; [MoCl4(MeSCH2CH2SMe)] 557210; [MoCl4(iPrSCH2CH2SiPr)] 1557211; [MoCl4(MeSCH2CH2CH2SMe)] 1557212; [MoCl5(Me2S)][Me2SCH2SMe] 1557213.†Complex synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700, 19400, 16600 (sh).
000, 19400, 16800 (sh). μeff: 2.22 B.M (298 K).
700, 19400, 16600 (sh).
000, 19400, 16800 (sh). μeff: 2.22 B.M (298 K).
Orange crystals of trans-[MoCl4(Me2S)2] grew by allowing a CH2Cl2 solution to evaporate slowly in a N2 atmosphere. A few orange-red crystals were also obtained by recrystallisation of crude [MoCl4(Me2S)2] from CH2Cl2; these were found to be [Me2SCH2SMe][MoCl5(Me2S)] from an X-ray structure determination.
Alternative method: MoCl5 (150 mg, 0.55 mmol) was dissolved in MeCN (10 mL) with stirring to give a dark brown solution. The solution was stirred for another 10 min and taken to dryness to produce a dark brown solid. The solid was suspended in CH2Cl2 (10 mL) before adding Me2S (3 mL). The suspended solid dissolved immediately after the Me2S was added in to give a red-brown solution. The solution was stirred for another 10 min and then pumped to dryness, leaving a dark orange solid. The product was spectroscopically identical with the product made from MoCl5 in CH2Cl2.
700, 20900, 19100, 18500 (sh).
Alternative method: MoCl5 (136 mg, 0.5 mmol) was dissolved in MeCN (10 mL) with stirring for 30 min to give a dark brown solution. The solution taken to dryness in vacuo and left a dark brown solid. CH2Cl2 (15 mL) was added, followed by a solution of nBu2S (0.4 mL, 2.0 mmol) in CH2Cl2 (2 mL). The dark solution changed to red-orange immediately. The solution was then stirred for 15 min then evaporated in vacuo to afford a dark orange oil. The product was spectroscopically identical to that made from MoCl5 in CH2Cl2.
000, 33300, 26800, 21800, 19000.
Alternative method: [MoCl4(MeCN)2] (100 mg, 0.31 mmol) was dissolved in 10 mL CH2Cl2 with stirring. A solution of Me2Se (73 mg, 0.65 mmol) and CH2Cl2 (5 mL) was then added. The solution was stirred for 1 h. resulting in a red-orange solution. Evaporation in vacuo left a dark orange powder. Yield: 99 mg, 70%. Required for C4H12Cl4MoSe2 (455.81): C, 10.54; H, 2.65. Found: C, 10.61; H, 2.54%. The product was spectroscopically identical to that made from MoCl5.
000, 32000, 28000, 22900, 21000.
500, 27300, 21000, 19600. μeff: 2.66 B.M. Orange crystals grew on allowing a CH2Cl2 solution to evaporate in a N2 atmosphere.
Alternative method: MoCl5 (136 mg, 0.5 mmol) was dissolved in MeCN (10 mL) with stirring for 10 min to give a dark brown solution. The solution taken to dryness in vacuo, CH2Cl2 (10 mL) was added, followed by a solution of MeSCH2CH2SMe (180 mg, 1.5 mmol) in CH2Cl2 (2 mL). The dark solution changed to light orange-green immediately and deposited a brown solid, which was filtered off. The brown solid was dried in vacuo and washed with Et2O (ca. 5 mL), then extracted with CH2Cl2 (ca. 5 m). The resulting solution was again taken to dryness in vacuo. Yield: 60 mg, 33%. Required for C4H10Cl4MoS2·0.1Et2O (367.4): C, 14.38; H, 3.02. Found: C, 14.38; H, 3.02% (sample contained ca. 10% Et2O from wash solvent; this was identified via1H NMR spectroscopy).
300, 26800, 25000 (sh), 21000, 18700 (sh). μeff: 2.18 B.M. Orange red crystals grew by allowing a CH2Cl2 solution to evaporate slowly under N2.
500, 22070, 19500. μeff: 2.21 B.M. Orange crystals grew by allowing a CH2Cl2 solution to evaporate under N2.
100, 26800, 21000, 18700.
Film characterisation
X-Ray diffraction (XRD) patterns were collected in grazing incidence mode (θ1 = 1°) or in-plane mode (θ1 = 0.5°, 2θχ scan with the detector scanning in the film plane) using a Rigaku SmartLab diffractometer (Cu-Kα, λ = 1.5418 Å) with parallel X-ray beam and a DTex Ultra 250 1D detector. Phase matching, lattice parameter calculations (MoS2) and Le Bail fitting (MoSe2) used the PDXL2 software package27 and diffraction patterns from ICSD.28 Scanning electron microscopy (SEM) was performed on samples at an accelerating voltage of 10 to 15 kV using a JEOL JSM6500 or a Philips XL30 ESEM. Film thicknesses were measured by fracturing the substrate and gold sputtering the edge in order to control charging of the insulating silica surface. Energy dispersive X-ray (EDX) analysis data were obtained with an Oxford INCA x-act X-ray detector (JSM6500) or Thermofisher Ultradry NSS 3 (XL30). Raman spectra were collected using a Renishaw InVia Raman Microscope with a 100 mW He–Ne 785 nm Laser. Optical spectra were obtained in transmission mode using a PerkinElmer 750S UV-visible spectrometer.Results and discussion
Two routes were used to prepare the MoCl4 complexes (Scheme 1), either substitution of the MeCN ligands in trans-[MoCl4(MeCN)2] with the chalcogenoether ligand in CH2Cl2 solution or reaction of MoCl5 with excess ligand in CH2Cl2.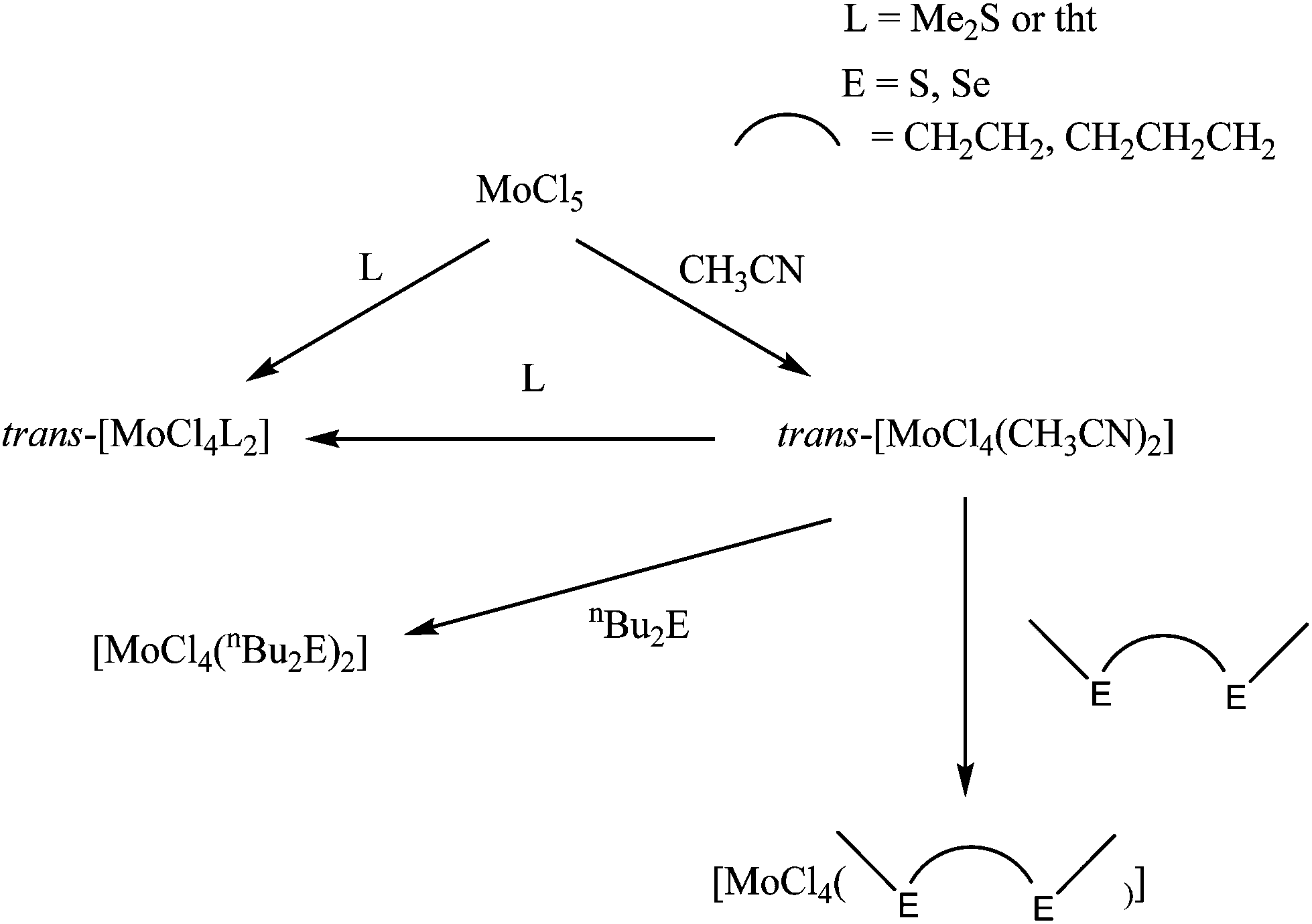 | ||
| Scheme 1 The preparative routes to the MoCl4 chalcogenoether complexes. | ||
[MoCl4(RCN)2] (R = Me, Et, nPr) are well known starting materials29–31 and an X-ray crystal structure determination of the complex with R = Me showed it to be the trans isomer (see Fig. S1, ESI†). The crystals are isomorphous with trans-[WCl4(MeCN)2].31 In the preparations using MoCl5, the chalcogen ligand reduces the molybdenum centre, being halogenated in the process, and careful washing is needed to remove the by-products. A few crystals of a minor by-product from the synthesis of [MoCl4(Me2S)2] from MoCl5 were identified by a crystal structure determination as the sulfonium salt, [Me2SCH2SMe][MoCl5(Me2S)] (Fig. 1). This cation has also been identified as a product of reaction of MoCl5 with DMSO;32 in the latter case it was obtained with an [MoOCl4]− anion.32
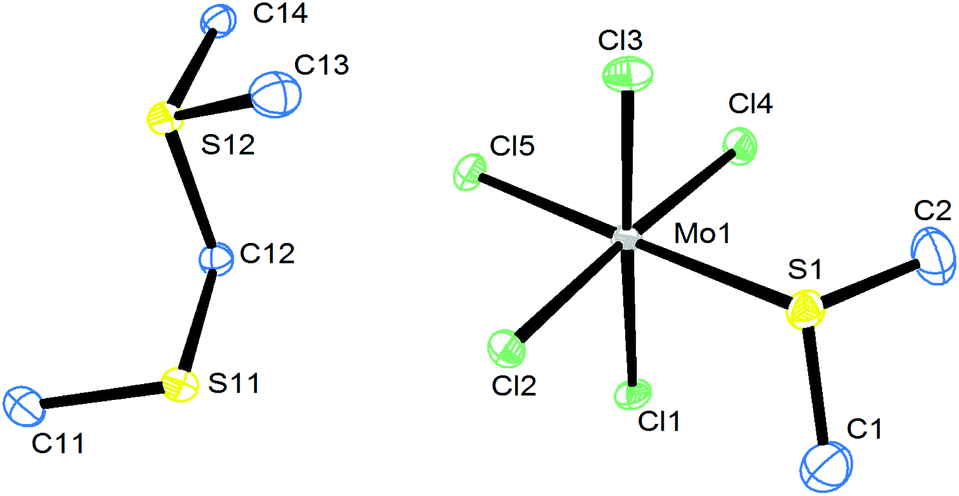 | ||
| Fig. 1 The structure of one of the two crystallographically independent cations and molybdenum anions in [Me2SCH2SMe][MoCl5(Me2S)] showing the atom numbering scheme and with ellipsoids drawn at the 50% probability level. The other cation and anion are very similar. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Mo1–Cl4 = 2.3251(17), Mo1–Cl1 = 2.3509(17), Mo1–Cl3 = 2.3633(18), Mo1–Cl5 = 2.3847(18), Mo1–Cl2 = 2.3885(17), Mo1–S1 = 2.5538(19), (cis) Cl–Mo–Cl = 88.10(6)–92.87(7), Cl–Mo1–S1 = 83.14(6)–91.74(6). | ||
The isolated, coordinatively saturated MoCl4L2 complexes are mildly hydrolytically sensitive. The crystal structures were determined for trans-[MoCl4(Me2S)], cis-[MoCl4{RS(CH2)2SR}] (R = Me, iPr) and cis-[MoCl4{MeS(CH2)3SMe}] and show the expected six-coordinate geometries (Fig. 2–4), with the chelate bite of the dithioethers responsible for the deviations from regular octahedral geometry. The bond lengths are similar to those in related Mo(IV) complexes.19,20,33
 | ||
| Fig. 2 The structure of the centrosymmetric [MoCl4(Me2S)2] showing the atom numbering scheme and with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Symmetry operation: i = −x, −y, −z + 1. Selected bond lengths (Å) and angles (°): Mo1–Cl1 = 2.3457(5), Mo1–Cl2 = 2.3323(5), Mo1–S1 = 2.5297(6); Cl1–Mo1–Cl2 = 89.39(2), Cl1i–Mo1–Cl2i = 90.61(2), Cl1–Mo1–S1i = 88.65(2), Cl1i–Mo1–S1i = 91.35(2), Cl2–Mo1–S1 = 90.51(2), Cl2i–Mo1–S1 = 89.49(2). | ||
 | ||
| Fig. 3 (a) The structure of [MoCl4(MeSCH2CH2SMe)] showing the atom numbering scheme and with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Mo1–Cl3 = 2.247(3), Mo1–Cl1 = 2.305(3), Mo1–Cl2 = 2.305(3), Mo1–Cl4 = 2.339(3), Mo1–S1 = 2.519(3), Mo1–S2 = 2.591(3), Cl3–Mo1–Cl1 = 97.61(12), Cl3–Mo1–Cl2 = 98.42(13), Cl3–Mo1–Cl4 = 96.83(11), Cl1–Mo1–Cl4 = 92.47(10), Cl2–Mo1–Cl4 = 92.95(10), Cl3–Mo1–S1 = 87.75(11), Cl1–Mo1–S1 = 82.18(10), Cl2–Mo1–S1 = 91.07(10), Cl1–Mo1–S2 = 85.34(10), Cl2–Mo1–S2 = 77.66(11), Cl4–Mo1–S2 = 92.13(10), S1–Mo1–S2 = 83.62(9). (b) The structure of [MoCl4(iPrSCH2CH2SiPr)] showing the atom numbering scheme and with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Mo1–Cl1 = 2.3203(7), Mo1–Cl2 = 2.3295(7), Mo1–Cl3 = 2.3135(7), Mo1–Cl4 = 2.3016(7), Mo–1S1 = 2.5872(7), Mo1–S2 = 2.5730(7); Cl2–Mo1–Cl4 = 93.37(3), Cl4–Mo1–Cl3 = 97.30(3), Cl4–Mo1–Cl1 = 96.60(3), Cl3–Mo1–Cl2 = 96.57(3), Cl1–Mo1–Cl2 = 96.14(3), Cl4–Mo1–S2 = 92.81(3), Cl3–Mo1–S2 = 79.16(2), Cl1–Mo1–S2 = 86.63(2), Cl3–Mo1–S1 = 87.01(2), Cl1–Mo1–S1 = 78.40(2), Cl2–Mo1–S1 = 89.61(2), S2–Mo1–S1 = 84.52(2). | ||
 | ||
| Fig. 4 The structure of [MoCl4(MeSCH2CH2CH2SMe)] showing the atom numbering scheme and with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Mo1–Cl3 = 2.2686(6), Mo1–Cl1 = 2.3143(6), Mo1–Cl4 = 2.3380(6), Mo1–Cl2 = 2.3486(6), Mo1–S1 = 2.5282(6), Mo1–S2 = 2.5390(6), Cl3–Mo1–Cl1 = 98.40(2), Cl1–Mo1–Cl4 = 97.61(2),Cl3–Mo1–Cl2 = 96.29(2), Cl1–Mo1–Cl2 = 92.82(2), Cl4–Mo1–Cl2 = 97.58(2), Cl3–Mo–1S1 = 87.41(2), Cl1–Mo1–S1 = 83.61(2), Cl4–Mo1–S1 = 79.79(2), Cl3–Mo1–S2 = 86.13(2), Cl4–Mo1–S2 = 78.20(2), Cl2–Mo1–S2 = 85.94(2), S1–Mo1–S2 = 97.370(19). | ||
The complexes are paramagnetic with μeff in the range 2.0–2.7 B.M. The values are lower than the predicted spin-only value for a d2 ion (2.82 B.M.) due to the significant spin–orbit coupling, as expected for 4d elements,34 and are similar to those observed in related complexes.18–20,29,33 The solid state UV/visible spectra exhibit two overlapping bands in the range 18000–22000 cm−1, which can be assigned in approximate Oh symmetry as the d–d transitions, 3T1g → 3T1g(P) and 3T1g → 3T2g,35 with ill-defined charge transfer transitions evident at >30000 cm−1.
CVD of molybdenum dichalcogenide films
The thermal decomposition of the [MoCl4(nBu2E)2] complexes at atmospheric pressure was first probed using thermogravimetric analysis (TGA). [MoCl4(nBu2Se)2] underwent three mass loss steps at 75–125 °C, 180–200 °C and 200–280 °C, leaving a residual mass of ca. 39.4%, which gradually reduced further as the temperature was raised to 600 °C (Fig. 5). This suggests a complex decomposition pathway with MoSe2 (40.7%) or MoCl4 (38%) as the final residue. [MoCl4(nBu2S)2] also shows three mass loss steps at 25–160 °C, 170–200 °C and 210–300 °C, leaving a residual mass of ca. 44.9%, which undergoes a further small mass loss (ca. 5%) on heating to 600 °C (Fig. 5). The residual mass is higher than that expected for MoS2 (30%), but similar to that expected for MoCl4 (44.7%); some sublimation of MoCl4 at high temperature would be expected; we note that [NbCl4(nBu2S)2] left NbCl4 upon heating in vacuo.36 It is important to note that the TGA experiments are performed at 1 atmosphere pressure under flowing argon, and hence very different from LPCVD conditions. However, TGA does provide a guide to the volatility of the precursors.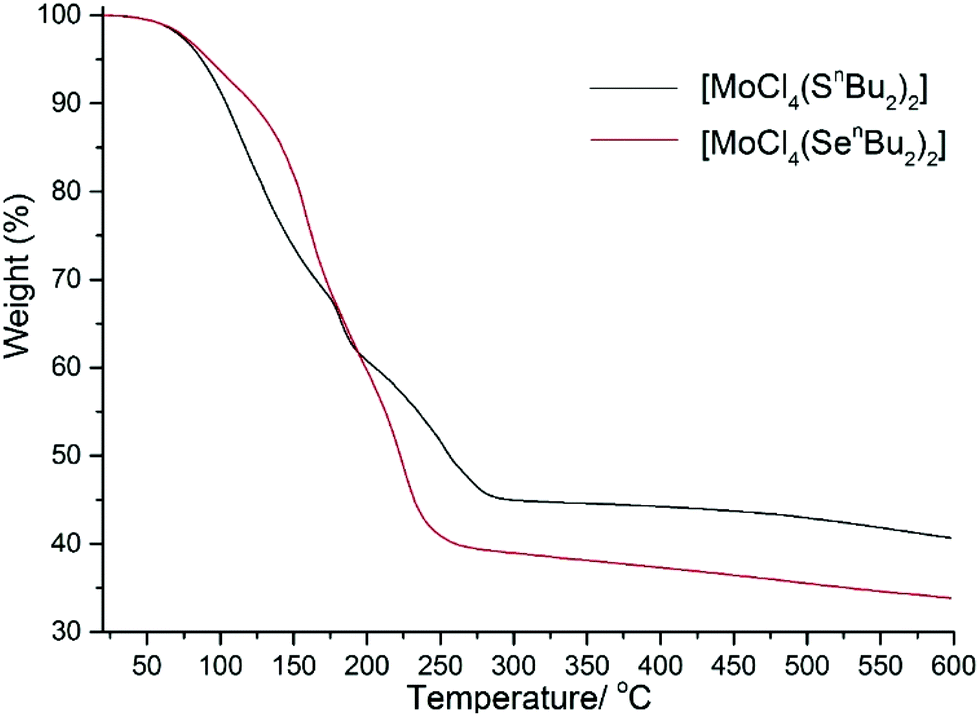 | ||
| Fig. 5 TGA of [MoCl4(nBu2E)2] (E = S or Se) obtained under flowing Ar (65 mL min−1) at ambient pressure with a heating rate of 10 °C min−1. | ||
CVD test experiments focussed on those complexes bearing nBu substituents since, unlike Me substituents, these tend to be promote elimination of the organic by-products, most likely via β-hydride elimination reactions. CVD onto silica tiles using ca. 30–70 mg of the [MoCl4(nBu2Se)2] precursor resulted in deposition of a reflective golden film on tiles positioned in the hottest part of the furnace at 400–550 °C. Grazing incidence XRD (Fig. 6) showed these films to be the 2H form of MoSe2 (P63/mmc). Lattice parameters determined from a Le Bail fit to the pattern are a = 3.2666(10) and c = 13.170(16) Å (Rwp = 1.8%, Rp = 1.3%; see ESI Fig. S2† for fit). The lattice parameters listed in ICSD for this phase are in the ranges a = 3.288–3.290 and c = 12.90–12.94 Å.28 The films are air and moisture stable (over many months), but easily scratched with a metal spatula. With deposition temperatures higher than 550 °C, MoO2 thin films were recovered, presumably resulting from deposition of molybdenum metal that oxidised post-deposition. Parkin and co-workers previously deposited films containing a mixture of the 2H- and 3R-type MoSe2 films using dual source atmospheric pressure CVD,16 but we are not aware of any other reports of single phase MoSe2 films from CVD from molybdenum chloride derived precursors.
 | ||
| Fig. 6 Grazing incidence XRD (GIXRD, incidence angle = 1°) and in-plane XRD (IPXRD, incidence angle = 0.5°) from MoSe2 thin films deposited by CVD from [MoCl4(nBu2Se)2] at temperatures as shown. The broad feature at 2θ ∼ 22° is from the SiO2 substrate. The calculated pattern for bulk polycrystalline 2H-MoSe2 (P63/mmc) with Miller indices is shown for comparison.28 | ||
The grazing incidence diffraction patterns of the MoSe2 films are dominated by the 002 reflection, suggesting <00l> alignment of the crystallites. These layered materials typically grow as platelet-shaped crystallites with the ab crystallographic axes in the plane of the crystallites, so this would suggest platelets lying flat on the substrate. An in-plane XRD measurement (Fig. 6) supressed the 002 reflection and enhanced the 100 and 110, supporting this conclusion. SEM images (Fig. 7) showed mainly the edges of platelets suggesting they were standing on edge rather than lying flat, but imaging the edge of a cross-section showed a dense, flat film with thickness of around 130 nm underlying these orthogonally oriented platelets. A pole figure measurement (Fig. 8) taken with 2θ = 13.54° (002 reflection) exhibits a single and sharp peak (FWHM ∼10°) with α = 90°, consistent with the vast majority of crystallites adopting the <00l> orientation. The pole figure with 2θ = 56.10° (110 reflection), exhibits a ring with α close to zero (the edge of the peak is cut off as the measurement cannot be made right at the substrate plane), also consistent with the <00l> preferred orientation. Unfortunately, the pole figure at the 103 reflection position was featureless. This may suggest stacking errors reducing its intensity.
 | ||
| Fig. 7 SEM images of MoSe2 films produced by CVD using ∼70 mg [MoCl4(nBu2Se)2] at 550 °C. Top view (left) and cross-section (right). | ||
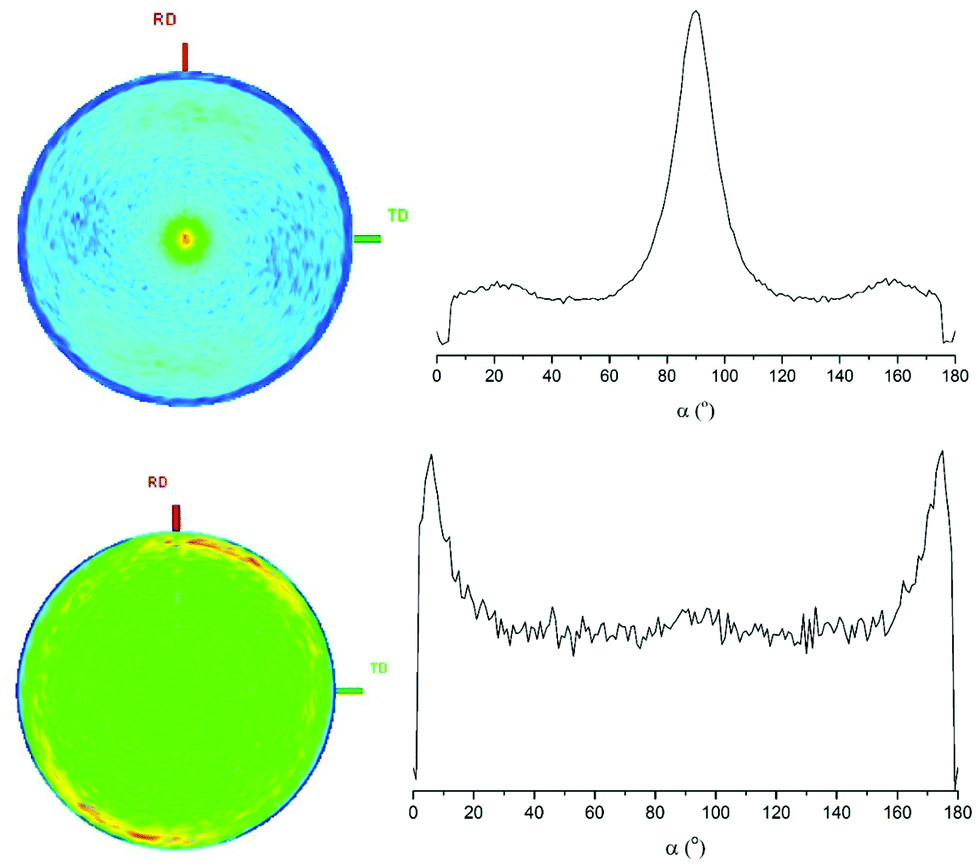 | ||
| Fig. 8 002 (top) and 110 (bottom) pole figure measurements shown as 2D plots (left) and integrated plots with α (right) for MoSe2 thin films produced by CVD using ∼70 mg [MoCl4(nBu2Se)2] at 550 °C. | ||
Energy dispersive X-ray (EDX) analysis data showed significant Si and O from the substrate, in addition to peaks due to Mo and Se, indicating that the films are thin. There was no evidence for any residual Cl in the films. The Mo:Se ratio was quantified as 1:1.9, consistent within error with the formation of MoSe2.
The crystallite size in the MoSe2 film produced at 550 °C was calculated from the grazing incidence XRD data using the Williamson–Hall method. The 6.6(9) nm size that resulted is much smaller than the largest dimensions of the platelets observed in Fig. 7 (ca. 100 nm), but the observed preferred orientation shows the bulk of the material to be contained within the dense underlying film. The diffraction crystallite size appears to be dominated by this part of the sample. Using a larger amount of precursor (ca. 200 mg) resulted in thicker films, but the optical quality of the films became worse. SEM images (ESI, Fig. S3†) showed that some crystallites continued to grow preferentially and thus the film thickness was less consistent over the area of the film.
The Raman spectrum from the MoSe2 film was collected using 785 nm excitation and shows three peaks at 140, 241 and 290 cm−1 assigned to the E1g, A1g and E2g vibration modes of 2H-MoSe2, respectively (Fig. 9),16,37–40 whilst peaks at ca. 317, 455 and 595 cm−1 can be attributed to contribution from acoustic phonons to the Raman scattering spectrum.41 The optical spectra (ESI, Fig. S4†) showed the expected excitonic transition features around 700 nm.42
 | ||
| Fig. 9 Raman spectrum of the MoSe2 film deposited by LPCVD from [MoCl4(nBu2Se)2] at 550 °C. | ||
When using similar amounts of [MoCl4(nBu2S)2] (ca. 30–70 mg) to that used in most of the MoSe2 depositions described above, CVD on SiO2 substrates at 750 °C resulted in very thin yellow films. These were too thin to generate any X-ray diffraction in grazing incidence or in plane measurement, or to image effectively in the SEM, but did produce optical spectra consistent with MoS242 (ESI, Fig. S5†). At lower temperatures no deposition occurred. However, by using larger amounts of [MoCl4(nBu2S)2] (200 mg) at 750 °C, silver films were obtained. Grazing incidence XRD (Fig. 10) showed similar features in these films to those seen for MoSe2 in Fig. 7, but with much weaker intensities. The pattern was dominated by the interlayer spacing peak (the 002 reflection), which gives no information on the exact phase of MoS2 produced. However, the in-plane XRD pattern contained stronger 100, 103 and 110 reflections, confirming that, like MoSe2, MoS2 adopted the 2H structure. The lattice parameters were refined as a = 3.13(5) and c = 13.7(8) Å, although there is significant uncertainty in this result due to the weak, broad peaks. Lattice parameters in ICSD are in the range a = 3.14–3.16 Å and c = 12.29–12.53 Å.28 The suppression of the 002 reflection in the in-plane pattern suggests <00l> preferred orientation like that observed in MoSe2, but unfortunately, the scattering from these films was not strong enough for pole figure measurements. The Raman spectrum recorded from the MoS2 film collected using 785 nm excitation showed weak bands at ca. 373 and 406 cm−1, assigned as the E2g and A1g vibrational modes in 2H-MoS2.37,40,41,43–45
 | ||
| Fig. 10 Grazing incidence (incidence angle = 1°) and in-plane (incidence angle = 0.5°) XRD patterns from the MoS2 thin film deposited from [MoCl4(nBu2S)2] at 750 °C, and the simulated XRD pattern from bulk 2H-MoS2.28 The broad feature at 2θ ∼ 22° is from the SiO2 substrate. | ||
SEM images confirm that the orthogonally oriented crystallites on the MoS2 film are very small, but that the underlying dense layer has a similar thickness to that found for MoSe2 (∼150 nm, Fig. 11). The weak and broad diffraction data suggest the crystallite sizes may also be smaller in the dense layer. Due to a close overlap between the Mo Lα (2.293 keV) and S Kα (2.307 keV) fluorescence peaks, it was not possible to obtain the Mo:S ratio from the EDX data. However, these did confirm the presence of both elements and there is no evidence for any residual Cl in the films.
 | ||
| Fig. 11 SEM images of MoS2 films produced by CVD using ∼200 mg [MoCl4(nBu2S)2] at 750 °C. | ||
Conclusions
The synthesis and characterisation of a new series of well-defined, distorted octahedral Mo(IV) chloride complexes with mono- and bi-dentate thioether and selenoether ligands has been demonstrated, with single crystal structures determined for several representative examples. TGA analysis of [MoCl4(nBu2S)2] and [MoCl4(nBu2Se)2] suggested they may be sufficiently volatile to be used as sources of MoS2 and MoSe2, respectively. This was borne out by LPCVD experiments which produced silver (2H-MoS2) and golden yellow (2H-MoSe2) thin films. The identities of the films, crystallite sizes and morphologies were determined via XRD, SEM/EDX, Raman and optical measurements. The successful identification of single source CVD precursors for their deposition is an important development given the very high level of interest in thin films of these materials currently and their range of potential applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for support (through EP/K00509X/1, EP/K009877/1 and EP/M508147/1) and the University of Southampton for a VC Scholarship (to Y.-P. C).References
- C. D. Garner and J. M. Charnock, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987, ch. 36.4 Search PubMed; E. I. Stiefel, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987 Search PubMed, ch. 36.5; G. J. Leigh and R. L. Richards, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987 Search PubMed, ch. 36.2.
- C. G. Young, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, Vol. 4, Section 4.7 Search PubMed.
- E. I. Stiefel, Prog. Inorg. Chem., 1977, 22, 1 CAS.
- M. Hidai and Y. Mizobe, in Metal Ions in Biological Systems, ed. A. Sigel and H. Sigel, Marcel Dekker, NY, 2002, vol. 39, p. 121 Search PubMed.
- Molybdenum and Tungsten; their role in biological processes, Metal ions in biological systems, ed. A. Sigel and H. Sigel, Marcel Dekker, NY, 2002, vol. 39 Search PubMed.
- M. Chhowalla, Z. Liu and H. Zhang, Chem. Soc. Rev., 2015, 44, 2584 RSC , (Eds. of themed issue on early transition metal dichalcogenides).
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef CAS PubMed; J. Liu and X.-W. Liu, Adv. Mater., 2012, 24, 4097 CrossRef PubMed.
- J. R. Brent, N. Savjani and P. O'Brien, Prog. Mater. Sci., 2017, 89, 411 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575 CrossRef CAS PubMed; K. Lee, R. Gatensby, N. McEvoy, T. Hallam and G. S. Duesberg, Adv. Mater., 2013, 25, 6699 CrossRef PubMed; Z. Yan, C. Jiang, T. R. Pope, C. F. Tsang, J. L. Stickney, P. Goli, J. Renteria, T. T. Salguero and A. A. Balandin, J. Appl. Phys., 2013, 114, 20430 Search PubMed.
- J. S. Rhyee, J. Kwon, P. Dak, J. H. Kim, S. M. Kim, J. Park, Y. K. Hong, W. G. Song, I. Omkaram, M. A. Alam and S. Kim, Adv. Mater., 2016, 28, 2316 CrossRef CAS PubMed; Y.-H. Chang, W. Zhang, Y. Zhu, Y. Han, J. Pu, J.-K. Chang, W.-T. Hsu, J.-K. Huang, C.-L. Hsu, M.-H. Chiu, T. Takenobu, H. Li, C.-I. Wu, W.-H. Chang, A. T. S. Wee and L.-J. Li, ACS Nano, 2014, 8, 8582 CrossRef PubMed; X. Wang, Y. Gong, G. Shi, W. L. Chow, K. Keyshar, G. Ye, R. Vajtai, J. Lou, Z. Liu, E. Ringe, B. K. Tay and P. M. Ajayan, ACS Nano, 2014, 8, 5125 CrossRef PubMed.
- X. Lu, M. I. Utama, J. Lin, X. Gong, J. Zhang, Y. Zhao, S. T. Pantelides, J. Wang, Z. Dong, Z. Liu, W. Zhou and Q. Xiong, Nano Lett., 2014, 14, 2419 CrossRef CAS PubMed; A. S. Pawbake, M. S. Pawar, S. R. Jadkar and D. J. Late, Nanoscale, 2016, 8, 3008 RSC.
- J. Chen, X. Zhao, S. J. Tan, H. Xu, B. Wu, B. Liu, D. Fu, W. Fu, D. Geng, Y. Liu, W. Liu, W. Tang, L. Li, W. Zhou, T. C. Sum and K. P. Loh, J. Am. Chem. Soc., 2017, 139, 1073 CrossRef CAS PubMed; Y. H. Lee, X. Q. Zhang, W. Zhang, M. T. Chang, C. T. Lin, K. D. Chang, Y. C. Yu, J. T. Wang, C. S. Chang, L. J. Li and T. W. Lin, Adv. Mater., 2012, 24, 2320 CrossRef PubMed.
- K. K. Liu, W. Zhang, Y. H. Lee, Y. C. Lin, M. T. Chang, C. Y. Su, C. S. Chang, H. Li, Y. Shi, H. Zhang, C. S. Lai and L. J. Li, Nano Lett., 2012, 12, 1538 CrossRef CAS PubMed; J. Pu, Y. Yomogida, K.-K. Liu, L.-J. Li, Y. Iwasa and T. Takenobu, Nano Lett., 2012, 12, 4013 CrossRef PubMed.
- Y.-P. Chang, W. Levason and G. Reid, Dalton Trans., 2016, 45, 18393 RSC.
- A. L. Hector, M. Jura, W. Levason, S. D. Reid and G. Reid, New J. Chem., 2009, 33, 64115 RSC; Y.-P. Chang, A. L. Hector, W. Levason and G. Reid, Dalton Trans., 2017, 46, 9824 RSC; S. L. Benjamin, C. H. de Groot, C. Gurnani, A. L. Hector, R. Huang, K. Ignatyev, W. Levason, S. J. Pearce, F. Thomas and G. Reid, Chem. Mater., 2013, 25, 4719 CrossRef CAS PubMed; S. L. Benjamin, Y.-P. Chang, C. Gurnani, A. L. Hector, M. Huggon, W. Levason and G. Reid, Dalton Trans., 2014, 43, 16640 RSC; Y.-P. Chang, A. L. Hector, W. Levason and G. Reid, Dalton Trans., 2017, 46, 9824 RSC.
- N. D. Boscher, C. J. Carmalt, R. G. Palgrave, J. J. Gil-Tomas and I. P. Parkin, Chem. Vap. Deposition, 2006, 12, 692 CrossRef CAS.
- J. Cheon, J. E. Gozum and G. S. Girolami, Chem. Mater., 1997, 9, 1847 CrossRef CAS.
- A. D. Westland and U. Uzelac, Can. J. Chem., 1970, 40, 2871 CrossRef CAS; D. Sevdić and L. Fekete, Inorg. Chim. Acta, 1982, 57, 111 CrossRef; W. Levason, C. A. McAuliffe, F. P. McCullough, S. G. Murray and C. A. Rice, Inorg. Chim. Acta, 1977, 22, 227 CrossRef.
- P. Dierkes, G. Frenzen, S. Wocadlo, S. Berger, J. Pebler and K. Dehnicke, Z. Naturforsch. Teil B, 1995, 50, 159 CrossRef CAS.
- L. Favero, F. Marchetti, G. Pampaloni and S. Zacchini, Dalton Trans., 2014, 43, 495 RSC.
- F. R. Hartley, S. G. Murray, W. Levason, H. E. Soutter and C. A. McAuliffe, Inorg. Chim. Acta, 1979, 35, 265 CrossRef CAS.
- D. J. Gulliver, E. G. Hope, W. Levason, S. G. Murray, D. M. Potter and G. L. Marshall, J. Chem. Soc., Perkin Trans. 2, 1984, 429 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxforshire, England Search PubMed.
- CrystalClear-SM Expert 2.1 b29, Rigaku Corporation, Tokyo, Japan, 2012 Search PubMed.
- J. R. Dilworth and R. L. Richards, Inorg. Synth., 1990, 28, 33 CrossRef CAS.
- S. Grazulis, D. Chateigner, R. T. Downs, A. F. Yokochi, M. Quiros, L. Lutterotti, E. Manakova, J. Butkus, P. Moeck and A. Le Bail, J. Appl. Crystallogr., 2009, 42, 726 CrossRef CAS PubMed.
- ICSD: Inorganic Crystal Structure Database (ICSD), Fachinformationszentrum Karlsruhe (FIZ), accessed via the EPSRC funded National Chemical Database Service hosted by the Royal Society of Chemistry.
- F. Stoffelbach, D. Saurenz and R. Poli, Eur. J. Inorg. Chem., 2001, 2699 CrossRef CAS.
- A. V. Butcher and J. Chatt, J. Chem. Soc. A, 1970, 2652 RSC.
- A. Manteghetti, C. Belin, M. Tillard-Charbonnel, J.-L. Pascal, E. Clot and F. Favier, New J. Chem., 1999, 23, 165 RSC.
- M. Bortoluzzi, E. Ferretti, M. Hayatifar, F. Marchetti, G. Pampaloni and S. Zacchini, Eur. J. Inorg. Chem., 2016, 3838 CrossRef CAS.
- B. Modec, J. V. Brenćić and L. Golic, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 780 Search PubMed.
- B. N. Figgis and J. Lewis, Prog. Inorg. Chem., 1964, 6, 37 CAS.
- A. B. P. Lever, Inorganic electronic spectroscopy, Elsevier, NY, 2nd edn, 1984 Search PubMed.
- Y.-P. Chang, W. Levason, M. E. Light and G. Reid, Dalton Trans., 2016, 45, 16262 RSC.
- S. Sugai and T. Ueda, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 26, 6554 CrossRef CAS.
- A. Bachmatiuk, R. F. Abelin, H. T. Quang, B. Trzebicka, J. Eckert and M. H. Rummeli, Nanotechnology, 2014, 25, 365603 CrossRef CAS PubMed.
- Y.-H. Chang, W. Zhang, Y. Zhu, Y. Han, J. Pu, J.-K. Chang, W.-T. Hsu, J.-K. Huang, C.-L. Hsu, M.-H. Chiu, T. Takenobu, H. Li, C.-I. Wu, W.-H. Chang, A. T. S. Wee and L.-J. Li, ACS Nano, 2014, 8, 8582 CrossRef CAS PubMed.
- A. Roy, H. C. Movva, B. Satpati, K. Kim, R. Dey, A. Rai, T. Pramanik, S. Guchhait, E. Tutuc and S. K. Banerjee, ACS Appl. Mater. Interfaces, 2016, 8, 7396 CAS.
- P. Soubelet, A. E. Bruchhausen, A. Fainstein, K. Nogajewski and C. Faugeras, Phys. Rev. B, 2016, 93, 155407 CrossRef.
- N. Dong, Y. Li, Y. Feng, S. Zhang, X. Zhang, C. Chang, J. Fan, L. Zhang and J. Wang, Sci. Rep., 2015, 5, 14646 CrossRef CAS PubMed.
- K. K. Liu, W. Zhang, Y. H. Lee, Y. C. Lin, M. T. Chang, C. Y. Su, C. S. Chang, H. Li, Y. Shi, H. Zhang, C. S. Lai and L. J. Li, Nano Lett., 2012, 12, 1538 CrossRef CAS PubMed.
- Y. Shi, W. Zhou, A. Y. Lu, W. Fang, Y. H. Lee, A. L. Hsu, S. M. Kim, K. K. Kim, H. Y. Yang, L. J. Li, J. C. Idrobo and J. Kong, Nano Lett., 2012, 12, 2784 CrossRef CAS PubMed.
- W. Park, J. Baik, T. Y. Kim, K. Cho, W. K. Hong, H. J. Shin and T. Lee, ACS Nano, 2014, 8, 4691 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1557208–1557213. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt04352j |
| This journal is © The Royal Society of Chemistry 2018 |