
N, S-containing MOF-derived dual-doped mesoporous carbon as a highly effective oxygen reduction reaction electrocatalyst†
Ai-Dong
Tan
a,
Kai
Wan
b,
Yi-Fang
Wang
a,
Zhi-Yong
Fu
 *ac and
Zhen-Xing
Liang
*a
*ac and
Zhen-Xing
Liang
*a
aKey Laboratory on Fuel Cell Technology of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510641, P. R. China. E-mail: zyfu@scut.edu.cn; zliang@scut.edu.cn; Tel: +86 20 87113584
bDepartment of Materials Engineering, KU Leuven, Leuven 3001, Belgium
cState Key Laboratory of Photocatalysis on Energy and Environment, Fuzhou University, Fuzhou 350002, P. R. China
First published on 23rd November 2017
Abstract
Nitrogen, sulfur-codoped carbon catalysts with enriched mesopores were, for the first time, synthesized by pyrolyzing a novel N, S-containing MOF, viz. [Zn2(TDC)2(DABCO)]·4DMF (SCUT-12, TDC = 2,5-thiophenedicarboxylic acid, DABCO = 1,4-diazabicyclo[2.2.2]octane). Single-crystal X-ray diffraction reveals that the S-containing ligand (TDC) and N-containing ligand (DABCO) are regularly crystallized into the SCUT-12 framework, which act as internal sulfur and nitrogen sources. As such, the two dopant atoms can be uniformly introduced into one precursor at the molecular level, and the codoping of two elements can be achieved in one step during carbonization, simplifying the conventional protocol. Structural characterization reveals that the resultant carbon is mesoporous and has high specific surface area, which favors the exposure of electrocatalytic active sites and mass transfer of the reactive species. As a result, the N, S-codoped carbon pyrolyzed at 900 °C (NSMC-900) shows a decent activity towards the oxygen reduction reaction (ORR) with an onset and half-wave potential up to 0.98 and 0.85 V, respectively, which are comparable with those of a 40 wt% commercial Pt/C catalyst. NSMC-900 also shows a superior long-term stability to Pt/C. Finally, it is found that the dual element-doped carbon outperforms either one of the mono-element-doped carbons, demonstrating the favorable synergistic effect of dual element-doping.
1. Introduction
Metal–organic frameworks (MOFs), which are assemblies of metal centers and organic linkers, have been developed as one type of novel organic–inorganic hybrid functional material.1 MOFs have shown a wide range of potential applications, such as gas storage and separation, optical sensing, molecular magnetism, and heterogeneous catalysis, due to their defined and controllable architectures.2–4 Recently, MOF-derived carbon has received increasing attention owing to its electrochemical applications.5–9 The advantages of using MOFs as precursors to synthesize carbon are as follows.i) All elements are distributed in MOFs at the molecular level by a specific chemical configuration, which yields a uniform dopant distribution in multi-element-doped carbon and atom-level control over composition.
ii) During pyrolysis, metal/metal oxides are uniformly generated and gaseous products (like CO2) are released, both of which favor the formation of a porous structure and high specific surface area.10–13
These advantages are acknowledged to facilitate both the reaction kinetics and mass transfer in developing a high-performance electrocatalyst.14–18 For example, nitrogen-doped porous carbon derived from MOFs has been widely investigated, which shows much better activities towards the oxygen reduction reaction (ORR) than does Pt/C in alkaline media.10,11,19–24 Dai and Lan et al.25,26 synthesized N, S-codoped porous carbon by introducing urea and dimethyl sulfoxide (DMSO) as external nitrogen and sulfur sources, respectively, into the micropores of MOF-5 precursors. The obtained N, S-codoped porous carbon shows a comparable ORR activity to the Pt/C catalyst with nitrogen and sulfur contents of 3.31 and 1.08 at%, respectively. Lin and Wang et al.27 synthesized co-encapsulated N, S-codoped carbon by pyrolyzing ZIF-67 and an external sulfur source (thiourea, NH4SCN, sublimed sulfur, and thiophene). The optimal sulfur content was found to be 1.51 at% in terms of the ORR activity. Sun and Liu et al.28 reported an effective strategy to synthesize N, S-codoped carbon by pyrolyzing a mixture of N-doped carbon derived from ZIF-8 and an external sulfur source, thiourea. This N, S-codoped carbon also shows excellent ORR activity with 0.3 at% S and 5.4 at% nitrogen. It is interesting that the above protocols have the following features in common: i) for S-doping, an external sulfur source is needed to introduce sulfur into either the precursor MOFs or MOF-derived carbon; ii) the preparation process is rather complicated, involving multiple steps to dope each hetero-element; iii) the content of dopant sulfur is not high as compared with dopant nitrogen, which is caused by the serious evaporation of the S-containing organics during pyrolysis. As a result, the promoting effect of sulfur-doping has rarely been observed in the literature and is probably underestimated. Therefore, it is of vital importance to develop novel and facile strategies to further promote sulfur doping and thus improve the electrocatalytic activity.
On the basis of the “atom-level control over composition” merit of MOFs, we synthesized a novel N, S-containing MOF, [Zn2(TDC)2(DABCO)]·4DMF (SCUT-12, TDC = 2,5-thiophenedicarboxylic acid, DABCO = 1,4-diazabicyclo[2.2.2]octane, DMF = N,N-dimethylformamide), with a classical “pillar-layer” structure. This MOF, SCUT-12, contains coordination bonds to fix S-containing linkers in its framework, inherently acting as an internal sulfur source. Hence, the adsorption procedure to introduce an external sulfur source is eliminated in synthesis. As expected, the resultant N, S-codoped mesoporous carbons (NSMCs) obtained by a one-step synthesis method show high content and uniform distribution of dopants.
To our knowledge, this is the first report on the use of a N, S-containing MOF as the precursor to synthesize N, S-codoped carbon. As an example of its application, the resultant carbon shows comparable ORR activity to that of a 40 wt% commercial Pt/C catalyst and remarkable long-term stability. Moreover, the synergistic effect of dual-element-doping is testified as compared with the mono-element-doped carbons.
2. Experimental section
2.1. Preparation of carbon
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:3:576 was treated under solvothermal conditions at 90 °C for 48 h, and then at room temperature for 16 h. The resultant colourless cubic crystals were collected by artificial isolation and washed with DMF.
:4:3:576 was treated under solvothermal conditions at 90 °C for 48 h, and then at room temperature for 16 h. The resultant colourless cubic crystals were collected by artificial isolation and washed with DMF.
Crystalline powders of SCUT-12 were synthesized by an amplification strategy. Zn(NO3)2·6H2O (1200.0 mg, 4.0 mmol), TDC (344.0 mg, 2.0 mmol), and DABCO (224.0 mg, 2.0 mmol) were dissolved in 50.0 mL DMF solution under stirring in a 150 mL flask. This solution was then heated at 90 °C for 36 h under vigorous stirring conditions. The resultant white powders were collected and washed twice with DMF by centrifugation at 3500 rpm for 10 min to give the yield of ca. 85.60% (750.0 mg) based on TDC.
For comparison, a mono-element-containing MOF, viz. a nitrogen-containing MOF, [Zn2(BDC)2(DABCO)] (N-MOF, BDC = 1,4-benzenedicarboxylate), and a sulfur-containing MOF, [Zn17(TDC)14(O)4(H2O)] (S-MOF), were prepared according to previous work.29,30 In short, for N-MOF, the synthesis route is similar to that for SCUT-12 powders, except that BDC was used instead of TDC. The resultant N-MOF was a white crystalline powder. For S-MOF, a mixture of Zn(NO3)2·6H2O (1200.0 mg, 4.0 mmol), TDC (412.0 mg, 2.40 mmol), and 50.0 mL DMAc (N,N-dimethylacetamide) was transferred to a 150 mL flask and heated at 90 °C for 36 h under vigorous stirring. White crystalline powders of S-MOF were collected by centrifugation at 3500 rpm for 10 min.
For N- and S-doped mesoporous carbon (NMC and SMC), the synthesis route is similar to that for NSMC-900, except that N-MOF (870 mg, ca. 1.0 mmol) and S-MOF (870 mg, ca. 0.23 mmol) were respectively used for NMC-900 and SMC-900.
For comparison, N, S-codoped carbon merely derived from SCUT-12 was synthesized in the absence of FeCl3, which is referred to as NSC-MOF.
2.2. Physicochemical characterization
Single-crystal X-ray diffraction (SXRD) measurement was performed on a Rigaku Xcalibur Eos Gemini CCD diffractometer equipped with a graphite monochromatic Mo/Kα radiation source (λ = 0.71073 Å) at 173 K. Powder X-ray diffraction (PXRD) investigations were performed on polycrystalline samples of MOFs and carbon materials in the 2θ range of 5–50° using a Bruker AXS D8-Advance diffractometer at 40 kV and 30 mA with Cu Kα radiation (λ = 1.5406 Å) with a step size of 0.1° in 2θ and a scan speed of 5° min−1. Raman spectroscopy was carried out on carbon materials using a LabRAM Aramis system. Elemental maps and energy-dispersive X-ray spectra (EDX) were obtained using a Zeiss Merlin system at 15 kV. Transmission electron microscopy (TEM) images were taken by means of a JEOL JEM-2100 system at 200 kV. X-ray photoelectron spectroscopy (XPS) investigations were performed on a Physical Electronics PHI 5600 multi-technique system using an Al monochromatic X-ray source at a power of 350 W. Nitrogen ad/desorption isotherms of the carbon materials were collected at 77 K using a Micromeritics TriStar II 3020 analyzer (Norcross, GA, USA). Before adsorption measurements, each sample was outgassed under vacuum for 12 h at 423 K. The total surface area was analyzed with the well-established Brunauer–Emmett–Teller (BET) method, and the pore size distribution was analyzed on the basis of the adsorption branches of nitrogen isotherms by the Barrett–Joyner–Halenda (BJH) method.2.3. Electrochemical characterization
The electrochemical test was performed on a Zennium (Zahner) electrochemical workstation, using a three-electrode cell at 25 °C. A double junction saturated calomel electrode (SCE) and a gold gauze electrode were used as the reference and the counter electrodes, respectively. The working electrode was a rotating ring-disk glassy carbon electrode (RRDE, disk: 5.0 mm in diameter, platinum ring: 6.5 mm inner diameter and 7.5 mm outer diameter). The thin-film electrode on the disk was prepared as follows: 10 mg of the catalyst was dispersed in 1.0 mL Nafion/ethanol (0.84 wt% Nafion) by sonication for 60 min. Then, 10.0 μL of the catalysis ink was dropped onto the disk by using a pipette to make a thin-film electrode, yielding a catalyst loading of 0.50 mg cm−2. For comparison, the ORR activity of the commercial 40 wt% Pt/C catalyst (HiSPEC4000, Johnson Matthey, London, UK) with a metal loading of 20.0 μg cm−2 was also measured.The electrolyte (0.10 M KOH) was first bubbled with Ar for 60 min. Then, a CV test was conducted at 20 mV s−1 in the potential range between 0 and 1.23 V (vs. reversible hydrogen electrode, RHE) for 20 cycles. If unspecified, the linear sweep voltammetry (LSV) curve was obtained by scanning the disk potential from 1.20 down to 0 V at 5 mV s−1 in the oxygen-saturated electrolyte solution under 1600 rpm; from this curve, the ORR polarization curve was extracted by subtracting the capacitive current. During the data collection, the potential of the ring was set to be 0.5 V (vs. RHE) in alkaline media to determine the yield of hydrogen peroxide. Herein, all potentials were referenced to the reversible hydrogen electrode (RHE): E (RHE) = E (SCE) + 1.005 V in 0.10 M KOH.
The H2O2 yield and the electron transfer number (n) were calculated using the following equations:
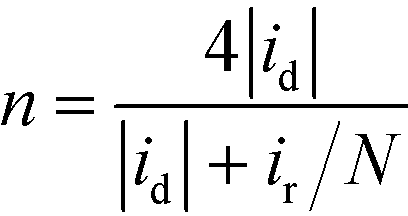
where id, ir and N are the disk current, the ring current and the current collection efficiency (21.85%) of the ring, respectively.
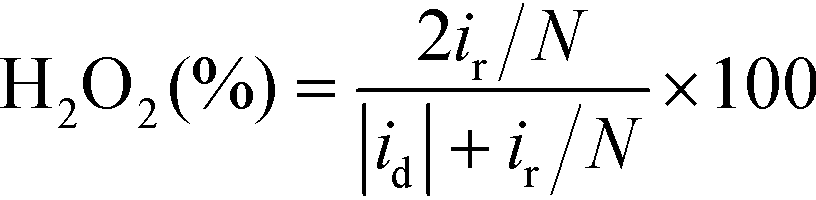
3. Results and discussion
The single-crystal X-ray diffraction analysis (see Tables S1 and S2†) reveals that SCUT-12 is isostructural with a previously reported MOF, [Zn2(BDC)2(DABCO)] (BDC = 1,4-benzenedicarboxylate, DABCO = 1,4-diazabicyclo[2.2.2]octane, abbreviated as N-MOF).29 SCUT-12 contains binuclear paddle wheel [Zn2(COO)4] structural units, which are connected by TDC2− (TDC = 2,5-thiophenedicarboxylic acid) to create a two-dimensional (2D) square-grid-like [Zn2(TDC)2] layer. The DABCO ligands are coordinated with the binuclear units in the axial orientation to extend the 2D layer into a three-dimensional (3D) “pillar-layer” cubic framework, in which DABCO acts as a pillar (see Fig. S1†). The structure analysis reveals that both nitrogen and sulfur atoms are uniformly distributed in SCUT-12, which benefits the efficient co-doping of nitrogen and sulfur into carbon upon pyrolysis (vide infra, Fig. 2g). Also, the high content of oxygen and Zn in SCUT-12 favors the formation of a porous structure upon pyrolysis and acid-leaching (vide infra).10,11The powder X-ray diffraction (PXRD) patterns of the as-synthesized MOFs were collected to analyze the purity of the crystalline phase, as shown in Fig. S2a–c.† It is seen that the experimental patterns of the as-synthesized SCUT-12, N-MOF, and S-MOF powders are well consistent with their simulated ones, revealing their high phase purity. The PXRD pattern of the precursor (a mixture of SCUT-12 and FeCl3) reveals that the presence of FeCl3 does not affect the crystalline phase of SCUT-12, and the peak marked by a rhombus should be attributed to FeCl3 (Fig. S2d†). After the thermal treatment and acid-leaching process, the obtained NSMC-900 (N, S-codoped mesoporous carbon) shows two broad peaks centered at 2θ = 25.0° and 44.5°, which correspond to the characteristic (002) and (101) crystal planes of carbon, respectively.31,32 No diffraction peaks indexed to Fe- and Zn-containing species are found in the PXRD pattern, suggesting that they are either amorphous in nature or negligible in amount.
In the Raman spectra (see Fig. S3†), all of the samples show two characteristic peaks at ca. 1340 and 1580 cm−1, which correspond to the D band and G band, respectively.33,34 The ID/IG ratios of NSMC-800, -900, and -1000 are 1.04, 1.02, and 0.99, respectively. The mono-element-doped carbon, which was calcined at 900 °C using either N-MOF or S-MOF, shows a similar ID/IG ratio to NSMC-900, which is respectively 1.01 for NMC-900 and 1.02 for SMC-900. These results are unsurprising as the graphitization degree is dependent on the pyrolysis temperature, as acknowledged in the literature.35,36
Fig. 1 and S4† show the nitrogen ad/desorption isotherms of the NSMCs, NMC-900 and SMC-900. It is found that the isotherms of the five samples are basically the same in shape and belong to typical type IV, confirming their mesoporous nature.37 The total specific surface area (SBET) of NSMC-800, -900, -1000, NMC-900 and SMC-900 are 452, 499, 492, 508 and 536 m2 g−1, respectively (see Table S3†). All of the three NSMCs show a broad mesopore size distribution in the range of 2–20 nm with an average pore diameter (DBJH) of 6.3 nm for NSMC-800, 6.7 nm for NSMC-900, and 7.2 nm for NSMC-1000. In comparison, both NMC-900 and SMC-900 show a much narrower mesopore size distribution, which is in the range of 2–10 nm with a DBJH of 5.4 nm for the former one and 2–6 nm with a DBJH of 3.4 nm for the latter one. In addition, the nitrogen ad/desorption isotherm of NSMC-900 without acid-leaching was collected to explain the pore formation mechanism in NSMCs (see Fig. S4c†). First, the pore size distribution (see Fig. S4d†) reveals the presence of mesopores with a size of <10 nm, indicating that these mesopores should be directly generated during pyrolysis. At high temperatures, the oxygen-containing functional groups in the MOF are decomposed, releasing gas (like CO, CO2 and H2O) and thus leaving these mesopores.11 It is, thus, understandable why both the total specific surface area and pore size show a slight increase with the pyrolysis temperature. Second, the SBET of NSMC-900 without acid-leaching (268 m2 g−1) is remarkably lower than that with acid-leaching, indicating that more pores are generated in the post-treatment step. The pore size distribution result confirms that more mesopores are present with the size being extended up to 30 nm. These larger mesopores should originate from the dissolution of Zn-containing particles (viz. zinc oxide/sulfide) in acid, which were formed during the pyrolysis of SCUT-12.38,39 It is expected that the mesopores with hierarchical pore size will favor both the exposure of the active sites and mass transfer of active species.16
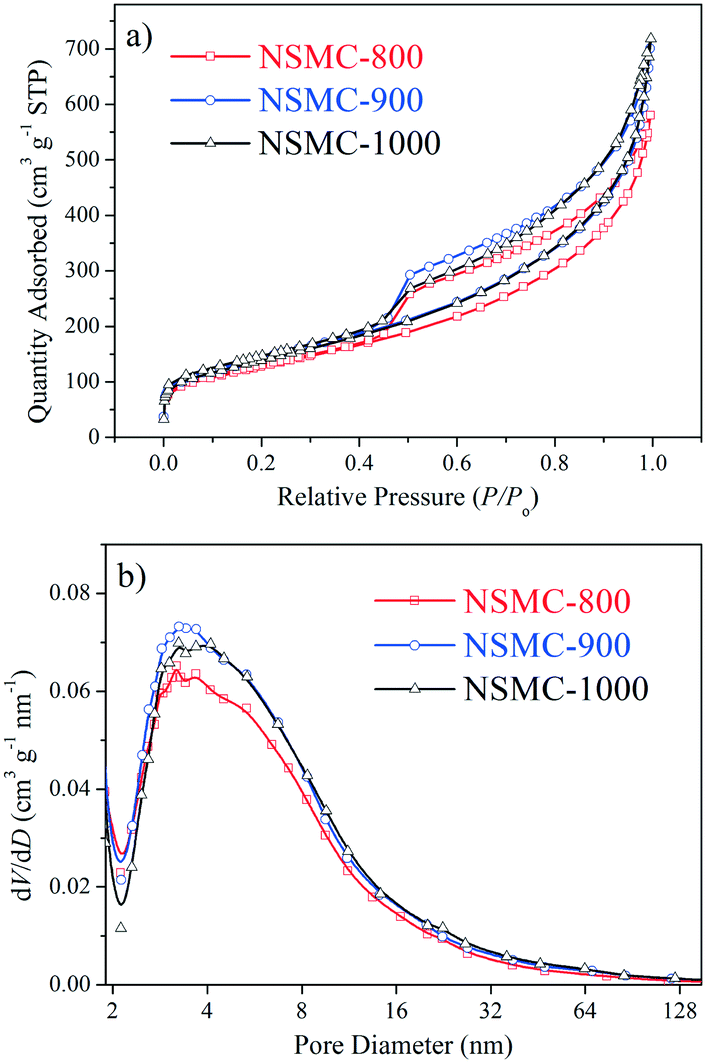 | ||
| Fig. 1 a) Nitrogen ad/desorption isotherms and b) pore size distribution of NSMCs. | ||
Fig. 2a–f show the transmission electron microscopy (TEM) images of the as-synthesized NSMCs. It is seen that abundant mesopores in the range of ca. 3–20 nm are uniformly and compactly distributed in the three carbon samples, which is consistent with the physisorption results (vide supra). Fig. 2g shows the energy-dispersive X-ray spectroscopy (EDX) elemental mapping images of NSMC-900. It is seen that both nitrogen and sulfur are uniformly doped in NSMC-900, validating the advantage of the precursor in codoping. The mapping image shows that the sulfur element generated a stronger signal than the nitrogen element, indicating a higher S content than N (see insert of Fig. 2h). Therefore, it is concluded that pyrolyzing the N, S-containing MOF can effectively promote the dual-element co-doping into carbon with uniform elemental distribution.
 | ||
| Fig. 2 TEM images of a and b) NSMC-800, c and d) NSMC-900, and e and f) NSMC-1000; g) elemental mapping of C, N, and S for NSMC-900; h) EDX spectrum of NSMC-900, the insert table shows the relative atomic content of C, N, and S. | ||
XPS was employed to further analyze the surface elemental composition and the chemical state, as shown in Fig. S6,† and the elemental content is listed in Table 1. It is found that both the N:C and S:C ratios gradually decrease with increasing pyrolysis temperature for the NSMCs. This result is understandable as the increase in pyrolysis temperature gradually deepens the decomposition of nitrogen- and sulfur-containing groups.15 Furthermore, the mono-element-doped carbons, viz. NMC-900 and SMC-900, show a similar content of dopant to NSMC-900, indicating that the content of dopant is more dependent on the pyrolysis temperature than the precursor itself. Finally, neither Fe nor Zn is detected in the five carbons, indicating that the transition metal can be effectively leached out in a strong acid.
| Sample | C | N | S | O | Fe | Zn | N:C |
S:C |
(N + S):C |
|---|---|---|---|---|---|---|---|---|---|
| NSMC-800 | 81.43 | 3.78 | 3.39 | 11.40 | 0 | 0 | 0.0464 | 0.0416 | 0.0880 |
| NSMC-900 | 86.25 | 2.19 | 2.91 | 8.65 | 0 | 0 | 0.0254 | 0.0337 | 0.0591 |
| NSMC-1000 | 91.94 | 1.12 | 2.97 | 3.97 | 0 | 0 | 0.0122 | 0.0323 | 0.0445 |
| SMC-900 | 89.86 | 0 | 2.94 | 7.20 | 0 | 0 | — | 0.0327 | 0.0327 |
| NMC-900 | 93.41 | 2.23 | 0 | 4.36 | 0 | 0 | 0.0239 | — | 0.0239 |
High-resolution N 1s, S 2p and C 1s spectra were collected to further recognize their chemical state. The N 1s peak (see Fig. 3a and S7a†) can be deconvoluted into four peaks centered at ∼398.4, ∼399.5, 400.7, and 402.6 eV, which are assigned to pyridinic-, pyrrolic-, graphitic-, and oxidic-N, respectively.40,41 The results are seen in Table 2. It is found that the pyrolysis temperature has a remarkable effect on the chemical configuration of dopant N. The content of both pyrrolic- and pyridinic-N dramatically decreases when the pyrolysis temperature increases from 800 to 1000 °C. Particularly, pyrrolic-N is only observed in NSMC-800, which is however absent in NSMCs prepared at higher temperatures. In comparison, the content of graphitic-N increases with the pyrolysis temperature. Therefore, it is concluded that edge-type (viz. pyrrolic, pyridinic) nitrogen is thermodynamically unstable, which can be converted into a more stable one at higher pyrolysis temperatures.42 Finally, it is noted that NSMC-900 and NMC-900 show similar content of the three nitrogen components, which indicates that the chemical state of the dopant depends on the pyrolysis temperature. The high-resolution S 2p spectra of NSMCs and SMC-900 can be resolved into three peaks located at 163.6, 164.8 and 167.9 eV, which correspond to S 2p3/2, S 2p1/2 and SOx (x = 2–3) (see Fig. 4 and S7b† and Table 3).43,44 The relative content of dopant S increases with the pyrolysis temperature, while the content of SOx decreases. Such a change can be attributed to the thermodynamically unstable nature of the latter species at higher temperatures. Also, NSMC-900 and NMC-900 show basically the same chemical configuration of sulfur, which is similar to the case of nitrogen (vide supra).
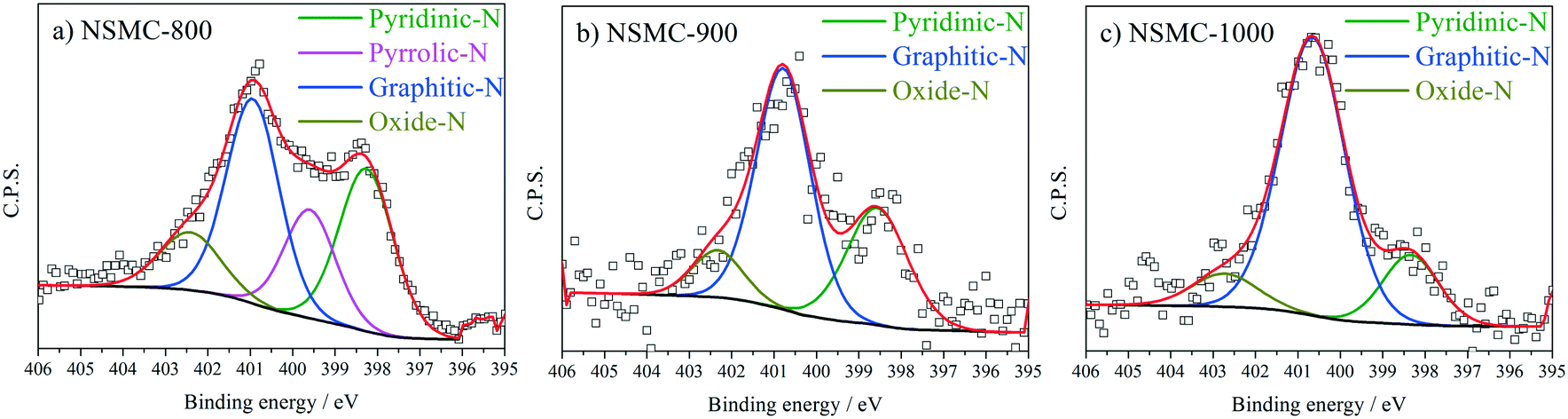 | ||
| Fig. 3 N 1s peak and the peak fitting results for a) NSMC-800, b) NSMC-900, and c) NSMC-1000. | ||
| Sample | Pyridinic-N | Pyrrolic-N | Graphitic-N | Oxidic-N |
|---|---|---|---|---|
| Note: The binding energy of each type of nitrogen is 398.4, 399.5, 400.7, and 402.6 ± 0.2 eV, respectively. The full-width at half-maximum is set to be 1.6 ± 0.2 eV. | ||||
| NSMC-800 | 30.69 | 19.30 | 37.70 | 12.31 |
| NSMC-900 | 31.66 | — | 57.81 | 10.53 |
| NSMC-1000 | 16.22 | — | 74.82 | 8.96 |
| NMC-900 | 38.48 | — | 51.54 | 9.98 |
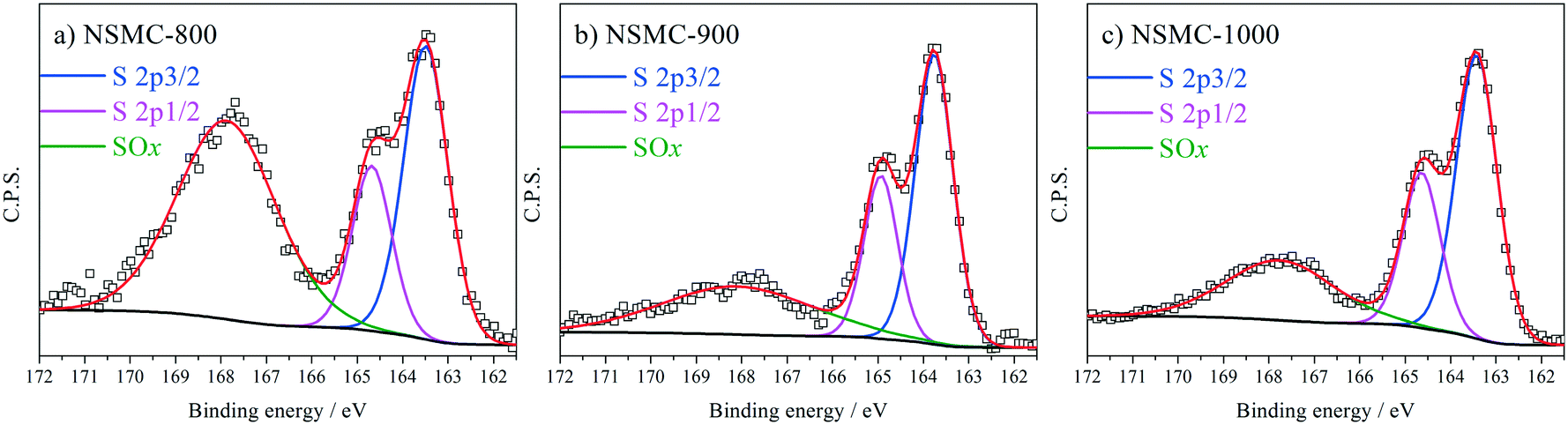 | ||
| Fig. 4 S 2p peak and the peak fitting results for a) NSMC-800, b) NSMC-900, and c) NSMC-1000. | ||
| Sample | S 2p3/2 | S 2p1/2 | SOx (x = 2–3) |
|---|---|---|---|
| Note: The binding energy of each type of sulfur is 163.6, 164.8 and 167.9 ± 0.2 eV, respectively. The full-width at half-maximum is set to be 1.0 ± 0.2 eV except for SOx. | |||
| NSMC-800 | 33.55 | 32.85 | 33.60 |
| NSMC-900 | 41.35 | 40.47 | 18.18 |
| NSMC-1000 | 42.57 | 41.68 | 15.75 |
| SMC-900 | 41.64 | 40.75 | 17.61 |
The heteroatom-activated carbon is claimed to be the active site for the ORR,15,45 of which the content is obtained by fitting the C 1s spectra (see Fig. S8 and Table S4†).46–48 It is seen that the content of the activated carbon atoms (referred to as C–N/C–S) follows a volcano-shaped relationship with the pyrolysis temperature, which first increases from 11.97 to 16.16 at% and finally drops to 14.06 at%. As such, NSMC-900 has the highest content of activated carbon atoms, which is expected to yield the best electrocatalytic activity towards the ORR (vide infra).
Fig. 5a shows the cyclic voltammograms (CVs) of the three NSMCs in 0.10 M KOH solution. It is seen that the three curves are basically the same in shape with large capacitive current. A pair of broad symmetrical peaks is observed in the potential range of 0–0.9 V, which can be attributed to the reversible electrochemical ad/desorption of OH− on/from the carbon surface.49 The capacitive current follows the order of NSMC-800 > NSMC-900 > NSMC-1000. Such a change agrees well with that in the content of the dopant, highlighting the dopant's key role in generating the electrochemically active functional groups. The corresponding ORR polarization curves (see Fig. 5b) show that the electrocatalytic activity follows the order of NSMC-900 > NSMC-1000 > NSMC-800. NSMC-900 has the highest electrocatalytic activity with an onset potential of ca. 0.98 V (vs. RHE) and a half-wave potential of ca. 0.85 V, which are comparable to those of the 40 wt% Pt/C catalyst and better than most data found in literature (see Table S5†). As mentioned above, the electrocatalytic activity is determined by the content of the activated carbon atoms.15,45 To make the correlation clearer, the fraction of the activated carbon and the current density at 0.9 V are respectively plotted vs. the pyrolysis temperature (see Fig. S9†), and the trend curves are basically overlapping.
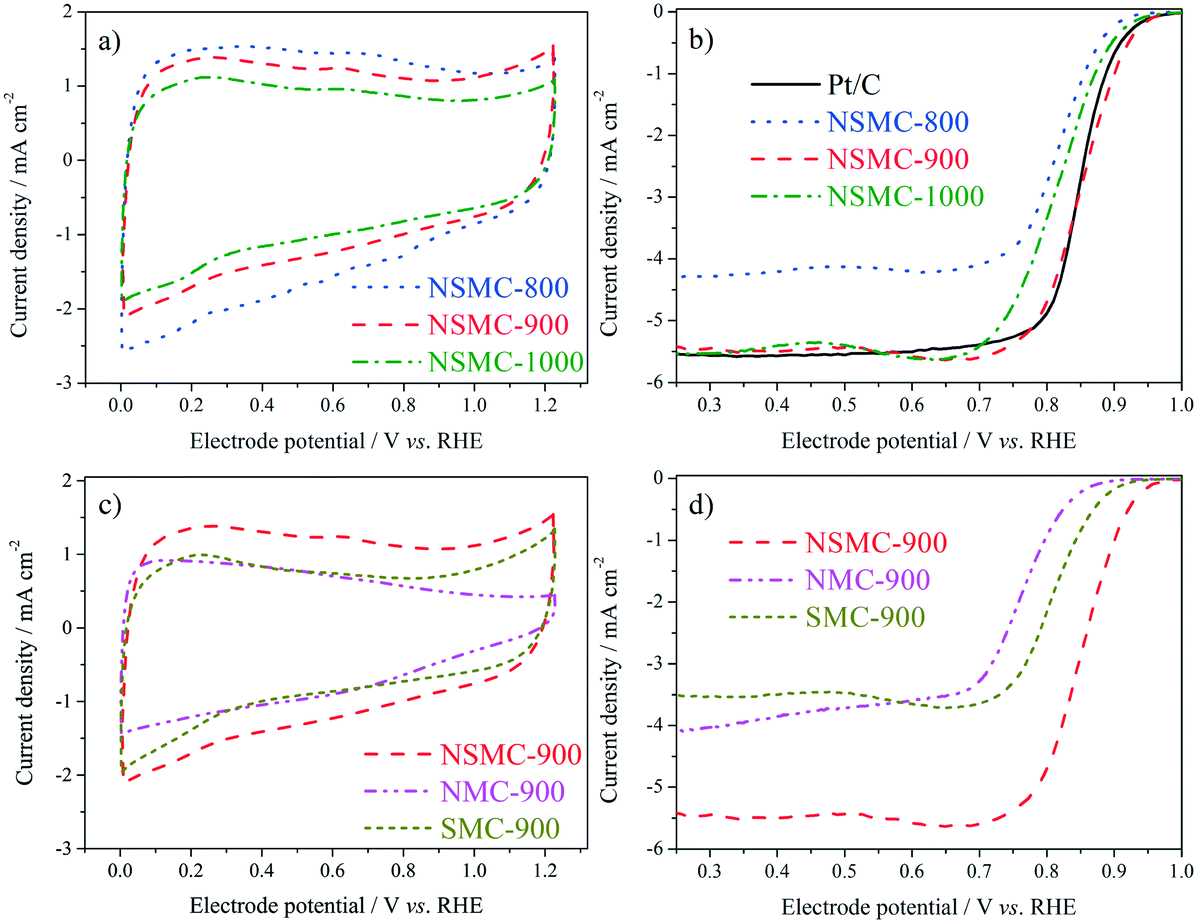 | ||
| Fig. 5 CV curves of a) NSMCs and c) N(S)MC-900 at a scan rate of 20 mV s−1 in Ar-saturated 0.10 M KOH; ORR polarization curves of b) NSMCs and 40 wt% Pt/C and d) N(S)MC-900 at a scan rate of 5 mV s−1 under rotating speeds of 1600 rpm in O2-saturated 0.10 M KOH. | ||
The CVs of NSMC, NMC, and SMC were obtained to further clarify the effect of element doping, as shown in Fig. 5c. It is unsurprising that NSMC, which has the highest content of dopant, yields the largest capacitive current. It is also noteworthy that the NSMC shows a unique redox peak at ca. 0.6 V, as compared with the other two mono-element-doped carbons. The presence of this redox couple should originate from the synergistic effect of the dual-element-doping into carbon. In line with this finding, the NSMC yields a much better electrocatalytic activity than do the other two mono-element-doped carbons, as shown in Fig. 5d. In addition, RRDE results reveal that all of the carbons are effective in catalysing the reduction of oxygen by a 4-electron transfer route (see Fig. S10†).
The stability was studied by intermittently collecting the polarization curve during an accelerated durability test (ADT) with cycling potential in the range of 0.6–1.0 V at 50 mV s−1. The chronoamperometric curve (i–t) was also collected for the evaluation (see Fig. 6). The ADT results in Fig. 6a and b show that NSMC-900 is far more stable than Pt/C in catalysing the ORR. Fig. 6c shows that after 20000 s of continuous operation, Pt/C shows a remarkable loss of ca. 50% in current, while NSMC-900 yields a much slower decay of <9%.
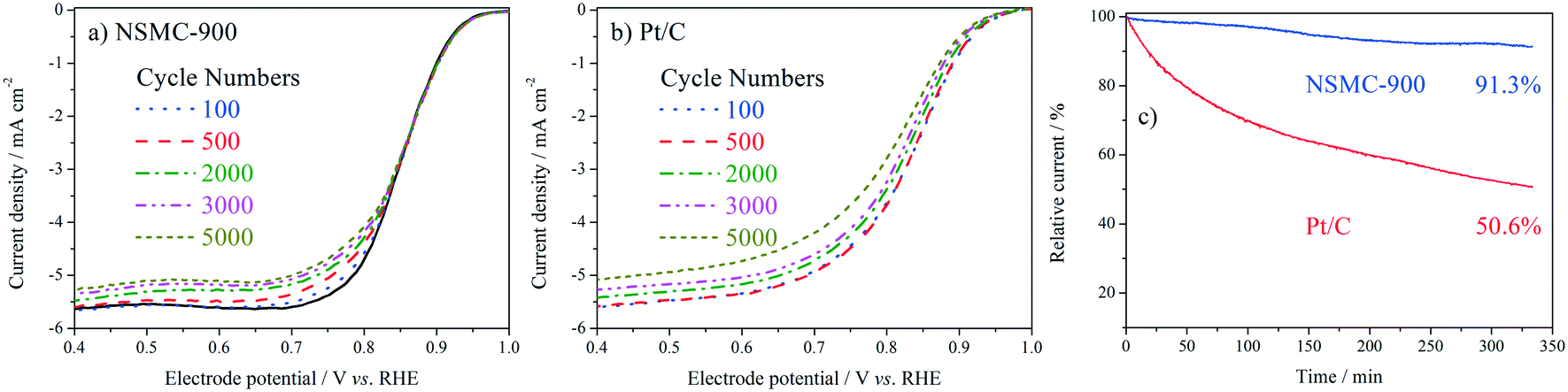 | ||
| Fig. 6 ORR polarization curves of the ADT-based stability test in 0.10 M KOH solution for a) NSMC-900 and b) 40 wt% Pt/C; c) chronoamperometric curves of NSMC-900 and 40 wt% Pt/C at 0.90 V (vs. RHE) in O2-saturated 0.10 M KOH. | ||
Conclusions
We present a new method to prepare efficient nitrogen, sulfur-codoped carbon catalysts by pyrolyzing a novel N, S-containing MOF (SCUT-12). It is found that the N- and S-containing organic linkers are regularly crystallized in SCUT-12, which can act as internal nitrogen and sulfur sources for the subsequent carbonization step. The homogeneous distribution of the two dopant elements in the precursor yields uniform dual-element-doping in the resultant carbon. In addition, zinc cation and oxygen-containing groups in the precursor act as pore-forming agents to generate hierarchical mesopores. The above features, as revealed by the electrochemical results, benefit both the formation and utilization of the active sites for the oxygen reduction reaction (ORR). As a result, the as-obtained carbon shows superior ORR electrocatalytic activity and long-term stability, demonstrating the favorable synergistic effect of dual-element-doping.Conflicts of interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.Acknowledgements
The work described in this paper was jointly supported by the National Natural Science Foundation of China (No. 21676106, 21476087, 21576101), the Science and Technology Program of Guangzhou (201704030065), the Science and Technology Program of Guangdong (2017A050506015), the National Key R&D Program of China (No. 2016YFB0101200 (2016YFB0101204)), and the CAS Key Laboratory on Fuel Cells and Composite Electrochemical Power Sources.Notes and references
- K. Biradha, A. Ramana and J. J. Vittal, Cryst. Growth Des., 2009, 9, 2969–2970 CAS.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- O. Toma, M. Allain, F. Meinardi, A. Forni, C. Botta and N. Mercier, Angew. Chem., Int. Ed., 2016, 55, 7998–8002 CrossRef CAS PubMed.
- P. Pachfule, D. Shinde, M. Majumder and Q. Xu, Nat. Chem., 2016, 8, 718–724 CrossRef CAS PubMed.
- A. Aijaz, N. Fujiwara and Q. Xu, J. Am. Chem. Soc., 2014, 136, 6790–6793 CrossRef CAS PubMed.
- Y. Z. Chen, C. M. Wang, Z. Y. Wu, Y. J. Xiong, Q. Xu, S. H. Yu and H. L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed.
- L. Jiao, Y. X. Zhou and H. L. Jiang, Chem. Sci., 2016, 7, 1690–1695 RSC.
- X. Zhang, J. Luo, P. Tang, X. Ye, X. Peng, H. Tang, S. G. Sun and J. Fransaer, Nano Energy, 2017, 31, 311–321 CrossRef CAS.
- A. D. Tan, Y. F. Wang, Z. Y. Fu, P. Tsiakaras and Z. X. Liang, Appl. Catal., B, 2017, 218, 260–266 CrossRef CAS.
- L. Ye, G. L. Chai and Z. H. Wen, Adv. Funct. Mater., 2017, 27, 1606190 CrossRef.
- A. J. Amali, J. K. Sun and Q. Xu, Chem. Commun., 2014, 50, 1519–1522 RSC.
- S. Liu, H. Zhang, Q. Zhao, X. Zhang, R. Liu, X. Ge, G. Wang, H. Zhao and W. Cai, Carbon, 2016, 106, 74–83 CrossRef CAS.
- L. Tan, Y. D. Yang, N. Li, S. Chen and Z. Q. Liu, Catal. Sci. Technol., 2017, 7, 1315–1323 CAS.
- K. Wan, G. F. Long, M. Y. Liu, L. Du, Z. X. Liang and P. Tsiakaras, Appl. Catal., B, 2015, 165, 566–571 CrossRef CAS.
- K. Wan, A. D. Tan, Z. P. Yu, Z. X. Liang, J. H. Piao and P. Tsiakaras, Appl. Catal., B, 2017, 209, 447–454 CrossRef CAS.
- X. H. Li, K. Wan, Q. B. Liu, J. H. Piao, Y. Y. Zheng and Z. X. Liang, Chin. J. Catal., 2016, 37, 1562–1568 CrossRef CAS.
- C. Du, X. H. Gao and W. Chen, Chin. J. Catal., 2016, 37, 1049–1061 CrossRef CAS.
- Z. Li, M. Shao, L. Zhou, Q. Yang, C. Zhang, M. Wei, D. G. Evans and X. Duan, Nano Energy, 2016, 25, 100–109 CrossRef.
- S. L. Zhao, H. J. Yin, L. Du, L. C. He, K. Zhao, L. Chang, G. P. Yin, H. J. Zhao, S. Q. Liu and Z. Y. Tang, ACS Nano, 2014, 8, 12660–12668 CrossRef CAS PubMed.
- H. L. Tang, S. C. Cai, S. L. Xie, Z. B. Wang, Y. X. Tong, M. Pan and X. H. Lu, Adv. Sci., 2016, 3, 1500265–1500272 CrossRef PubMed.
- S. Pandiaraj, H. B. Aiyappa, R. Banerjee and S. Kurungot, Chem. Commun., 2014, 50, 3363–3366 RSC.
- H. L. Wang, Q. L. Zhu, R. Q. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS.
- L. J. Li, P. C. Dai, X. Gu, Y. Wang, L. T. Yan and X. B. Zhao, J. Mater. Chem. A, 2017, 5, 789–795 CAS.
- J. S. Li, S. L. Li, Y. J. Tang, K. Li, L. Zhou, N. Kong, Y. Q. Lan, J. C. Bao and Z. H. Dai, Sci. Rep., 2014, 4, 5130 CrossRef CAS PubMed.
- J. S. Li, Y. Y. Chen, Y. J. Tang, S. L. Li, H. Q. Dong, K. Li, M. Han, Y. Q. Lan, J. C. Bao and Z. H. Dai, J. Mater. Chem. A, 2014, 2, 6316–6319 CAS.
- C. Zhang, B. An, L. Yang, B. B. Wu, W. Shi, Y. C. Wang, L. S. Long, C. Wang and W. B. Lin, J. Mater. Chem. A, 2016, 4, 4457–4463 CAS.
- Z. Song, W. Liu, N. Cheng, M. Norouzi Banis, X. Li, Q. Sun, B. Xiao, Y. Liu, A. Lushington, R. Li, L. Liu and X. Sun, Mater. Horiz., 2017, 4, 900–907 RSC.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033–5036 CrossRef CAS PubMed.
- Y. X. Tan, Y. P. He and J. Zhang, Chem. Commun., 2014, 50, 6153–6156 RSC.
- J. Liu, P. Song, M. B. Ruan and W. L. Xu, Chin. J. Catal., 2016, 37, 1119–1126 CrossRef CAS.
- J. X. Zhang, D. D. Liu, H. Y. Song, Z. X. Liang, X. F. Guo, L. Du and S. J. Liao, RSC Adv., 2016, 6, 19515–19521 RSC.
- L. L. Geng, J. L. Song, B. Zheng, S. J. Wu, W. X. Zhang, M. J. Jia and G. Liu, Chin. J. Catal., 2016, 37, 1451–1460 CrossRef CAS.
- B. Frank, Z. L. Xie, K. Friedel Ortega, M. Scherzer, R. Schlogl and A. Trunschke, Catal. Sci. Technol., 2016, 6, 3468–3475 CAS.
- W. H. Niu, L. G. Li, X. J. Liu, N. Wang, J. Liu, W. J. Zhou, Z. H. Tang and S. W. Chen, J. Am. Chem. Soc., 2015, 137, 5555–5562 CrossRef CAS PubMed.
- F. J. Maldonado-Hodar, C. Moreno-Castilla, J. Rivera-Utrilla, Y. Hanzawa and Y. Yamada, Langmuir, 2000, 16, 4367–4373 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051 CrossRef CAS.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed.
- H. L. Jiang, B. Liu, Y. Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Q. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS PubMed.
- G. F. Long, X. H. Li, K. Wan, Z. X. Liang, J. H. Piao and P. Tsiakaras, Appl. Catal., B, 2017, 203, 541–548 CrossRef CAS.
- Y. W. Lee, G. H. An, S. Lee, J. Hong, B. S. Kim, J. Lee, D. H. Kwak, H. J. Ahn, W. Huh, S. N. Cha, K. W. Park, J. I. Sohn and J. M. Kim, Catal. Sci. Technol., 2016, 6, 2085–2091 CAS.
- T. Sharifi, G. Hu, X. E. Jia and T. Wagberg, ACS Nano, 2012, 6, 8904–8912 CrossRef CAS PubMed.
- L. Zhang, Y. Wang, K. Wan, J. H. Piao and Z. X. Liang, Electrochem. Commun., 2018, 86, 53–56 CrossRef CAS.
- S. Liu, G. Li, Y. Gao, Z. Xiao, J. Zhang, Q. Wang, X. Zhang and L. Wang, Catal. Sci. Technol., 2017, 7, 4007–4016 CAS.
- M. Wu, J. Wang, Z. X. Wu, H. L. L. Xin and D. L. Wang, J. Mater. Chem. A, 2015, 3, 7727–7731 CAS.
- K. Hu, L. Tao, D. D. Liu, J. Huo and S. Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 19379–19385 CAS.
- S. Bag, B. Mondal, A. K. Das and C. R. Raj, Electrochim. Acta, 2015, 163, 16–23 CrossRef CAS.
- W. Kicinski and A. Dziura, Carbon, 2014, 75, 56–67 CrossRef CAS.
- K. Wan, Z. P. Yu, X. H. Li, M. Y. Liu, G. Yang, J. H. Piao and Z. X. Liang, ACS Catal., 2015, 5, 4325–4332 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1571241. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cy02265d |
| This journal is © The Royal Society of Chemistry 2018 |