
Less hindered ligands give improved catalysts for the nickel catalysed Grignard cross-coupling of aromatic ethers†
Gavin J.
Harkness
 and
Matthew L.
Clarke
*
and
Matthew L.
Clarke
*
School of Chemistry, University of St Andrews, EaStCHEM, St Andrews, Fife, UK. E-mail: mc28@st-andrews.ac.uk; Fax: +44(0) 1334 463808; Tel: +44(0) 1334 463850
First published on 1st December 2017
Abstract
The challenging reaction of unactivated ortho-substituted aromatic ethers with Grignard reagents has been found to be most effectively catalysed using nickel complexes of less sterically hindered ligands. Air stable, cheap, commercially available [NiCl2(PnBu3)2] stands out as an improved catalyst for this type of transformation. The improved results with this catalyst even extend to some couplings of a more activated substrate when examined at higher temperatures and at catalyst loadings down to 0.1 mol%. Unusual induction periods in these latter reactions have been related to the by-product magnesium salts acting as co-catalysts.
Introduction
A key area of modern research is the cross-coupling of organometallics with aromatic electrophiles via carbon–oxygen bond cleavage. In particular, intensive research has focused on expanding this chemistry away from expensive trifluoromethanesulfonates to cheaper leaving groups that also deliver less harmful waste streams.1 The use of simple aromatic ethers is especially desired, but this is a challenging reaction: naphthyl ethers frequently deliver good results (Scheme 1a), but removing the second aromatic ring causes a large fall in reactivity and successful examples are rare and require very specific nucleophile/electrophile combinations.2,3 With the exception of aromatic ethers with ortho-directing oxazoline4,5 groups, examples of coupling ortho-substituted unactivated anisoles are even more scarce.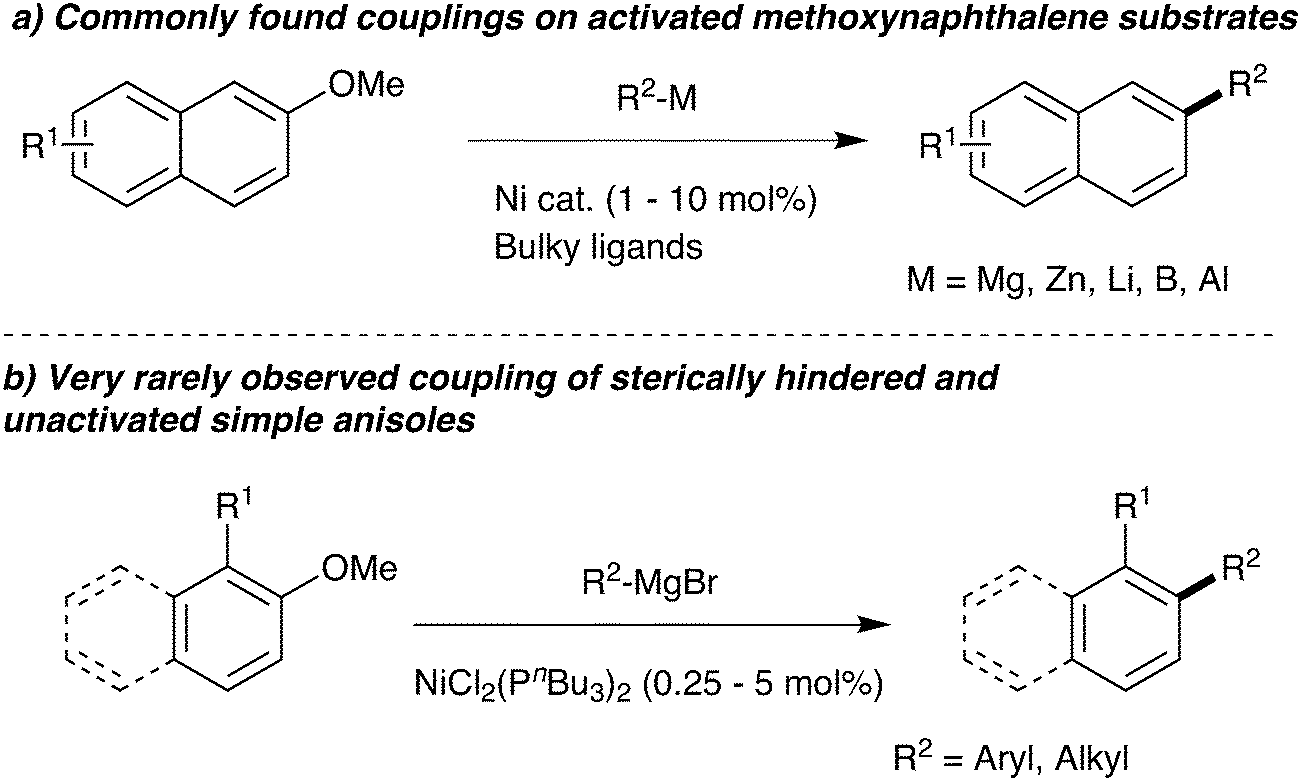 | ||
| Scheme 1 Nickel-catalysed cross-coupling using aryl ethers. | ||
Among the most common catalyst systems used in the cross-coupling of aromatic ethers are Ni(cod)2/PCy3 and [NiCl2(PCy3)2].2e,h,n,t,w,x One of the early key papers reported that using bulky tricyclohexylphosphine as ligand was substantially superior to triethylphosphine,2x which perhaps led to smaller cone angle phosphines being neglected.
During a project seeking to convert lignin-derived 2-methoxyphenol into useful products,5–7 we needed to constructively deoxygenate several ortho-substituted anisoles, leading us to study the Ni-catalysed Grignard coupling of aryl ethers. Here we show that, in contrast to most cross-couplings, less bulky catalysts such as [NiCl2(PnBu3)2] give improved performance in the Grignard cross-coupling of aromatic ethers (Scheme 1b).
Results and discussion
The model substrate used to search for ligand steric effects was 2-phenyl anisole. This could be prepared from the imidazole sulfonate of guaiacol by a Grignard cross-coupling catalysed by palladium complexes (but unfortunately not the nickel catalysts used for ether cleavage) as described in the ESI.† Our initial tests of a range of Ni pre-catalysts derived from bidentate, hemilabile and monodentate ligands did not yield useful amounts of product in this model coupling; catalysts derived from [Ni(cod)2] and Cy3P or bis-dicyclohexylphosphino-ethane were ineffective. One of the state-of-the-art catalysts for ether cleavage in the literature uses the NHC ligand IPr,2l but this was also ineffective (Scheme 2 and see ESI†). Given the hindered nature of the C–O bond in this substrate, we considered that less sterically hindered phosphines were worthy of investigation. A range of nickel complexes of electron donating phosphines with relatively small cone angles8,9 were therefore investigated as catalysts for this especially difficult Grignard cross-coupling reaction (Scheme 2). All of the catalysts derived from ligands with smaller cone angles than Cy3P outperformed benchmark catalyst, 1, the only exception being the catalyst with the least sterically hindered phosphine [NiCl2(PMe3)2], 6. Commercially available, cheap, air stable, and previously untested [NiCl2(PnBu3)2] stands out.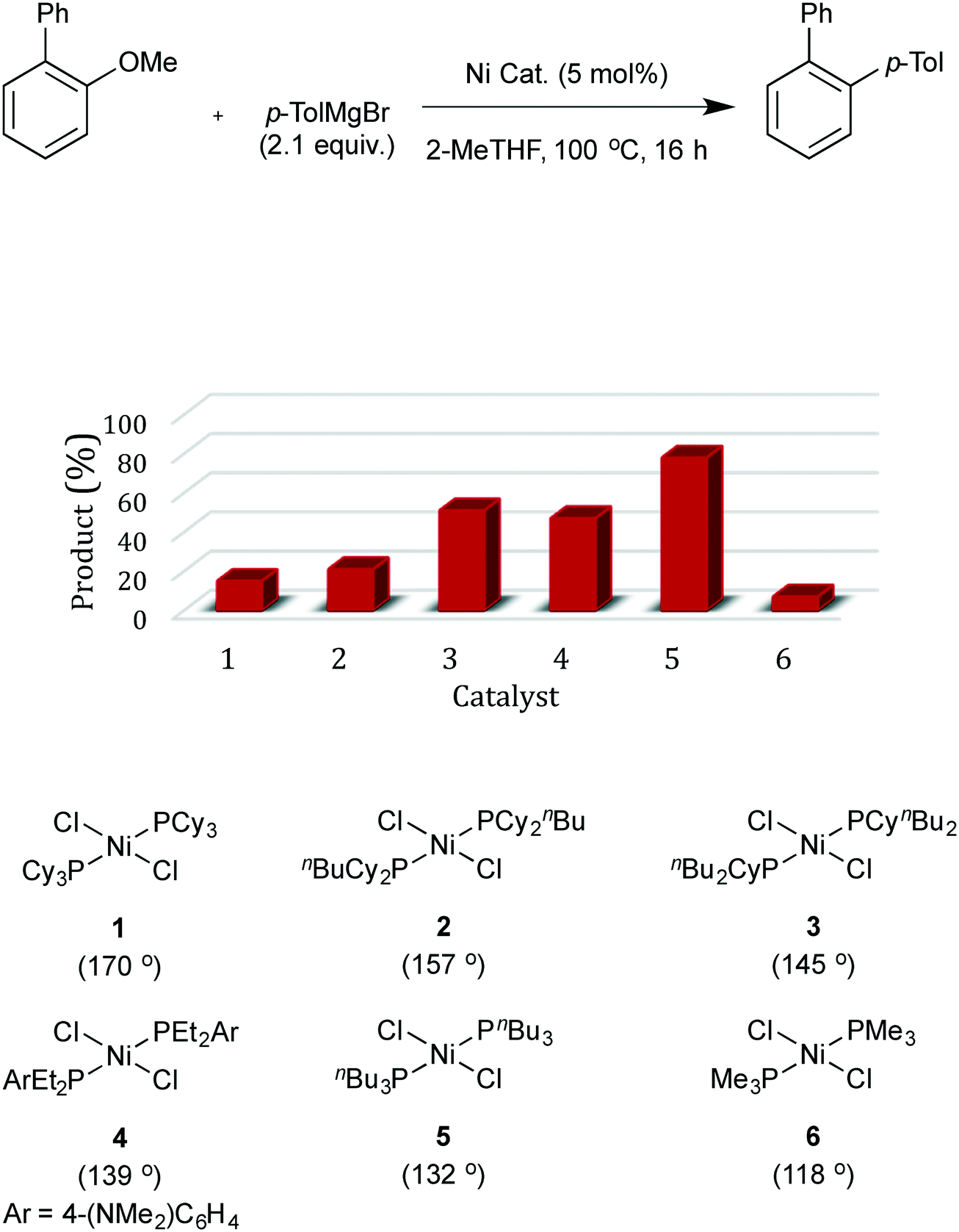 | ||
| Scheme 2 Effect of ligand cone angle on the challenging nickel-catalysed coupling of 2-methoxybiphenyl. | ||
The reactions give good results using reasonably practical conditions. For example, while excess phosphine ligand, as well as large excesses of Grignard are often required to gain satisfactory yields in this type of reaction,2x [NiCl2(PnBu3)2] works effectively with no additives and actually works better using 1.5 equivalents of Grignard rather than a large excess (Table 1, entry 3 and ESI†). While this project was not target focussed, we felt it important to establish if this catalyst gave improved results for some other combinations of ether and Grignard reagent. In fact, other Grignard reagents of varying steric properties were tested with this substrate and the same trend was apparent, with catalyst 5 outperforming 1. PhMgBr couples extremely well (Table 1, entry 7). However, the increased sterics imposed by o-TolMgBr gave lower yields (Table 1, entries 5–6). This trend for [NiCl2(PnBu3)2] to outperform bulkier ligands extends to other substrates of varying electronic and steric properties. Other biphenyl methyl ether substrates (Table 1, entries 10 and 14) gave high yields. Coupling of 2-methylanisole required 10 mol% catalyst, (Table 1, entry 12) but also gave best results with catalyst 5. In the coupling of ortho-substituted 1-phenyl-2-methoxynaphthalene at 100 °C, [NiCl2(PnBu3)2] is a better catalyst than catalyst 1 for coupling either p-TolMgBr or o-TolMgBr (Table 1, entries 19 and 21). At lower temperature, for this more activated aromatic, catalyst 1 was actually the faster catalyst. The simpler substrate methoxynaphthalene, even using a hindered Grignard reagent, ortho-tolyl magnesium bromide, can be coupled well with either catalyst (entries 22 and 23).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | ArOMe | R2MgBrb | Catalyst (mol%) | Temp (°C) | Time (h) | Conversionc (%) | Productc (%) |
| a Reactions were carried out on the scale of aryl ether (0.50 mmol), Grignard reagent (0.75 mmol in Et2O, 0.5 M), 2-MeTHF (2.25 mL) in sealed vessels unless otherwise noted. b Grignard molarity in brackets. c Conversions and yields were determined by 1H NMR using 1-methylnaphthalene as an internal standard [yield of isolated product in square brackets]. d Reaction performed in a Schlenk flask. e 2.1 equiv. Grignard added. f PhMe as reaction solvent. g 27% yield isolated with 1-methylnaphthalene impurity; total product obtained = 24%. h 40% yield isolated with 1-methylnaphthalene impurity; total product obtained = 38%. | |||||||
| 1 |
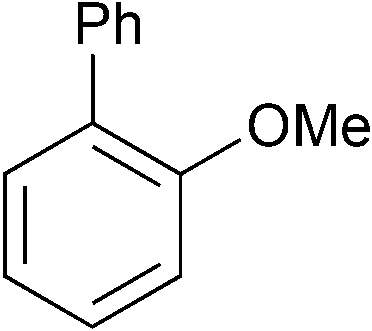
|
p-Tol | 1 (5) | 100 | 19 | 36 | 15 |
| 2e | p-Tol | 1 (5)/L (10) | 100 | 19 | 37 | 30 | |
| 3e | p-Tol | 5 (5) | 100 | 16 | 85 | 79 | |
| 4d | p-Tol | 5 (5)/L (10) | 100 | 45 | 98 | 84 [84] | |
| 5 | As above | o-Tol (2.0) | 1 (5) | 100 | 16 | 23 | 8 |
| 6 | o-Tol (2.0) | 5 (5) | 100 | 16 | 25 | 21 [21] | |
| 7 | As above | Ph (1.7) | 5 (5) | 100 | 16 | 97 | 96 [86] |
| 8e,f | As above | Me (2.4) | 1 (5) | 100 | 16 | 9 | 3 |
| 9e,f | Me (2.4) | 5 (5) | 100 | 16 | 48 | 46 [27]g | |
| 10 |
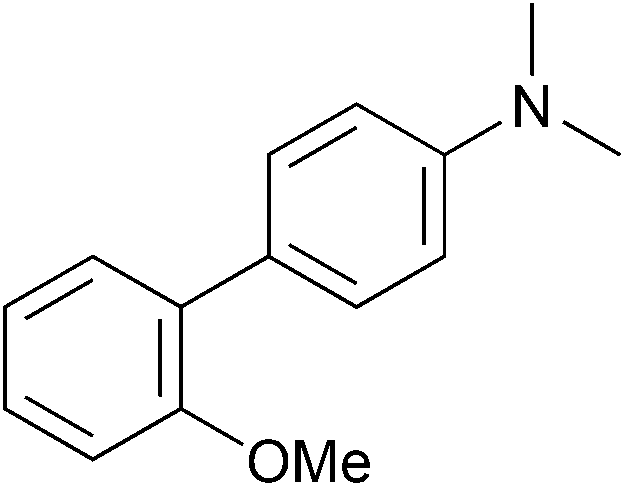
|
p-Tol | 5 (5) | 100 | 40 | 79 | 55 [40] |
| 11e |
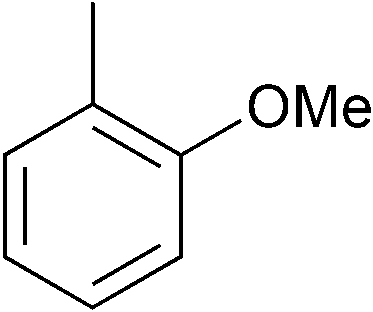
|
p-Tol | 1 (10) | 100 | 64 | 18 | 1 |
| 12e | p-Tol | 5 (10) | 100 | 64 | 76 | 56 [40]h | |
| 13 |
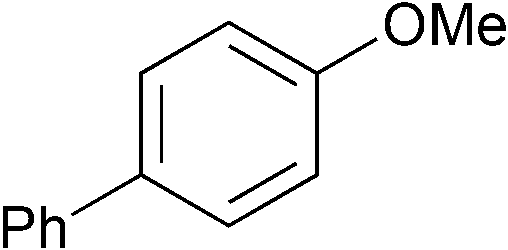
|
p-Tol | 1 (5) | 100 | 16 | 59 | 34 |
| 14 | p-Tol | 5 (5) | 100 | 16 | 84 | 70 [65] | |
| 15 | As above | o-Tol (2.0) | 5 (5) | 100 | 16 | 51 | 49 [43] |
| 16d |
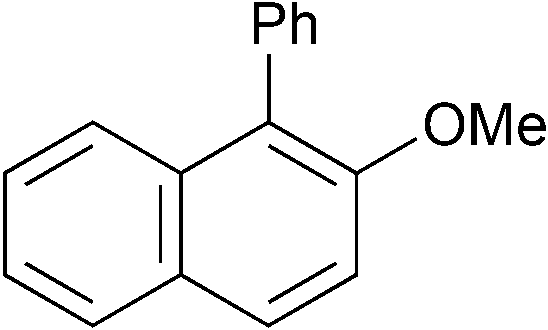
|
p-Tol | 1 (1) | 80 | 16 | 82 | 70 |
| 17 | p-Tol | 1 (1) | 100 | 16 | 70 | 60 | |
| 18d | p-Tol | 5 (1) | 80 | 16 | 54 | 41 | |
| 19 | p-Tol | 5 (1) | 100 | 16 | 91 | 85 [83] | |
| 20 | o-Tol (2.0) | 1 (1) | 100 | 16 | 59 | 40 | |
| 21 | o-Tol (2.0) | 5 (1) | 100 | 16 | 76 | 61 [59] | |
| 22d |
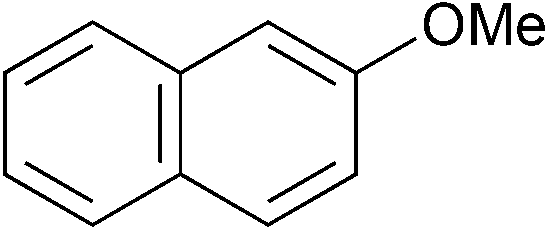
|
o-Tol | 1 (1) | 80 | 16 | >99 | 95 |
| 23d | o-Tol | 5 (1) | 80 | 16 | >99 | 97 [82] | |
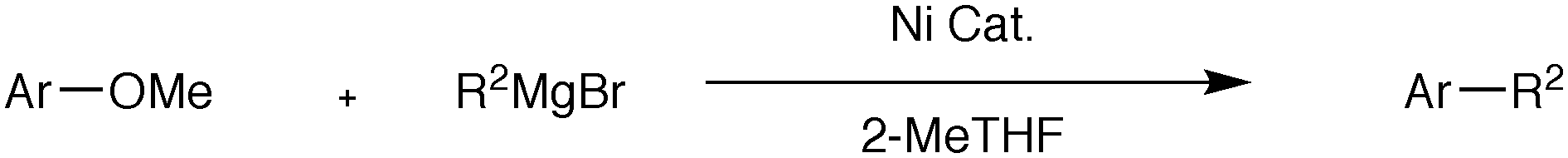
A final ortho-substituted substrate of particular interest was 2-(2-methoxyphenyl)-pyridine, since there is potential for an ortho-directing effect to have an impact on the nickel catalysis. At room temperature, a significant amount of the desired product is formed in the presence of the nickel catalysts (Scheme 3). However this is let down by low chemoselectivity with unknown side products, and the demethylated phenol being present. The ortho-directing group therefore has a profound and positive effect on the reactivity of the substrate with Ni catalysts, although this can be difficult to control. Even lower chemoselectivity was observed at higher temperatures. In fact, while there is no sign of a background (uncatalysed) reaction at room temperature, more chemoselective coupling can be observed without any catalyst at 80 °C (71% isolated yield). This appears to be the first Meyers ether cleavage4 that has been reported using a pyridyl directing group and highlights that this chemistry is potentially general whenever there is a nearby mesomerically electron-withdrawing group that also can coordinate to magnesium.
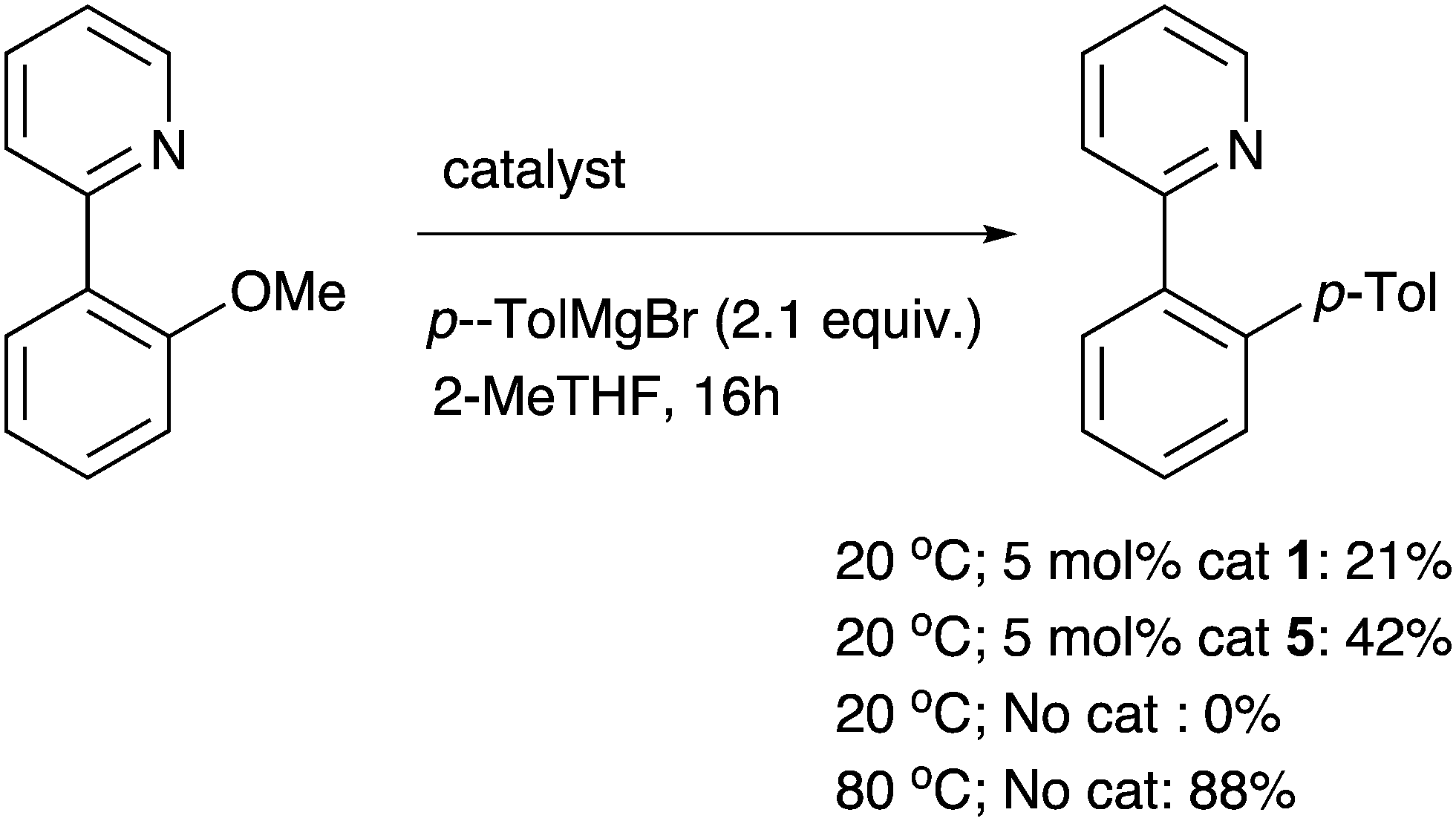 | ||
| Scheme 3 A potentially chelating substituent enables Ni-catalysed ether cleavage at room temperature, and also an uncatalysed reaction at higher temperature (% product vs. internal standard is shown, with conversions for Ni-catalysed reactions both 85%; 71% isolated yield from uncatalysed rct., see ESI†). | ||
We also examined some further examples of Ni/NHC catalyst systems with varying steric properties and found a similar trend regarding ligand sterics; the NHC ligands, IDM and ICy with the smallest% buried volume13 gave improved results relative to generally more favoured NHC ligands such as IPr, IPr* and SiPr (see ESI†). Ni/ICy has been used in ether cleavages using alkynyl and alkyl Grignard reagents recently,2q,r The findings with the carbenes are consistent with a direct and strong ligand steric effect on these reactions.
For the reactions described here, we suggest the use of cheap, air stable, commercially available [NiCl2(PnBu3)2] as the preferred catalyst.
There is possibly an argument that high catalyst loadings are perfectly acceptable with some cheap iron-based cross-coupling catalysts (used for more activated electrophiles),10 but the ligands often have a significant cost, so large scale cross-couplings are likely to need substrate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :catalyst ratios less than 100:1. In the case of nickel, the requirement to remove all the nickel due to its toxicity creates further costs.11 Couplings at lower catalyst loadings than the loadings of 1 to 5 mol% used in the literature were studied next using 2-methoxynaphthalene as substrate.
:catalyst ratios less than 100:1. In the case of nickel, the requirement to remove all the nickel due to its toxicity creates further costs.11 Couplings at lower catalyst loadings than the loadings of 1 to 5 mol% used in the literature were studied next using 2-methoxynaphthalene as substrate.
In the reaction of p-TolMgBr with 2-methoxynaphthalene at 80 °C using [NiCl2(PCy3)2], there is no advantage gained when using [NiCl2(PnBu3)2] (Table 2, entries 1 and 3). However, at 100 °C, we were able to deliver high yields of product using 0.25 mol% of [NiCl2(PnBu3)2]; better than can be achieved using [NiCl2(PCy3)2] as catalyst (Table 2, entries 5 and 6). From a mechanistic viewpoint, there is clearly a ligand steric effect on the productivity of these reactions at higher temperatures, with smaller cone angle, electron-donating phosphine-based catalysts giving better results. This could be a direct effect in that a less crowded Ni(PR3)n species of some form undergoes C–O bond cleavage more effectively, or an indirect effect relating to catalyst stability. Initial data from sampling experiments throughout this work suggest both catalysts 1 and 5 remain active for extended periods of time, suggesting the former explanation of a ligand effect on the catalytic cycle. Time sampling experiments for reactions carried out at 0.1 mol% of catalyst showed quite unusual behaviour (Fig. 1).
|
|
||||
|---|---|---|---|---|
| Entrya | Catalyst (mol%) | Temp (°C) | Conversionb (%) | Productb (%) |
| a Reactions were carried out on the scale of aryl ether (0.50 mmol), Grignard reagent (0.75 mmol in Et2O, 0.5 M, 1.5 equiv.), 2-MeTHF (2.25 mL) in sealed vessels for 16 hours unless otherwise noted. b Conversions and yields were determined by 1H NMR using 1-methylnaphthalene as an internal standard [yield of isolated product in square brackets]. c 69 hours reaction time. | ||||
| 1 | 1 (0.5) | 80 | 39 | 38 |
| 2 | 1 (0.5) | 100 | 75 | 59 |
| 3 | 5 (0.5) | 80 | 17 | 16 |
| 4 | 5 (0.5) | 100 | 93 | 89 |
| 5 | 5 (0.25) | 100 | 83 | 71 |
| 6c | 5 (0.1) | 100 | 62 | 56 |

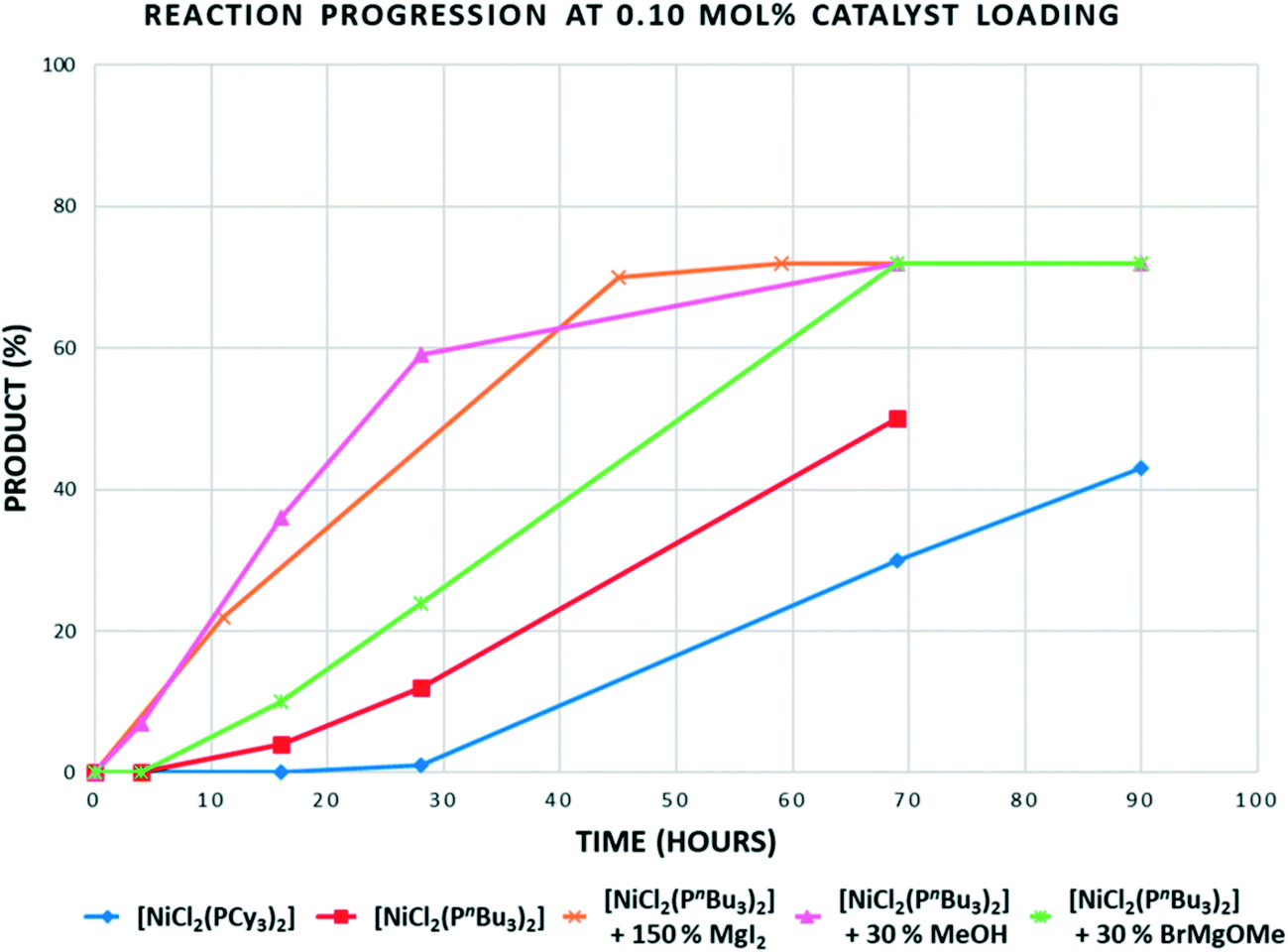 | ||
| Fig. 1 Time profile for the reaction of 2-methoxynaphthalene with p-tolyl magnesium bromide using 0.1 mol% of the Ni catalysts shown at 100 °C. “30% MeOH” = 0.3 equiv. of MeOH added to solution containing catalyst and substrate, prior to Grignard addition, “30% BrMgOMe” = 0.3 equiv. of MeOH added to Grignard prior to addition of solution containing catalyst and substrate, (see ESI† for further details). | ||
Most catalysts would give highest rates at the highest substrate concentration (i.e. most catalysts being first order in substrate), but also when catalyst is freshest: it is normal for some degradation to further limit rates after days have passed for the majority of cross-coupling catalysts. The results obtained were therefore quite unexpected. There is almost no conversion in the first 16 hours for either catalyst. After around a day, the reaction begins to accelerate with, as we now expected, the smaller cone angle tri-n-butylphosphine catalysts proceeding faster. A time profile where the degree of conversion increases in this way can either imply an induction period where a pre-catalyst is converted into the active species, or autocatalysis from a product of the reaction. Since the reduction of NiL2Cl2 to Ni(0) using a Grignard is a fast reaction, this is not likely to be the origin of the induction period. When considering the mechanism for this type of reaction, the formation of some higher order Ni nanoparticle or multinuclear species is not outside the realms of possibility and could exhibit a time profile like this. However, an added possibility in these reactions is that various magnesium salts that could be formed during these reactions might promote the reaction. Using this experimental set-up, without in situ monitoring, it is not possible to study the kinetics of these reactions and fully study autocatalytic behaviour and other promotion effects. However, the preliminary data does seem to indicate a possible role of Mg salts other than the often-postulated role for the Grignard as promoter. First of all an excess of anhydrous magnesium iodide was added to the reaction in order to see if there was any indication that just a simple Mg salt2a could remove the induction period: which it clearly does (Fig. 1, brown line). A more realistic concentration and identity of Mg salt were studied next. Some of the likely Mg salts formed during the reactions were added by quenching a slightly larger excess of Grignard with methanol to make ‘Mg(OMe)Br’. This was done in two different ways, just in case either methanol had a negative effect on the catalyst. While this study does not delve into the details of the promotion effect (or even if there is significance in the improved performance when methanol is added to the catalyst first), it is clear the magnesium salts reduce the induction period. A full kinetic study using in situ techniques on a range of Ni catalysed Ni–O bond cleavages using various promoters would be welcome.
The role of the magnesium salts produced during the coupling reactions could therefore be important in the mechanism for the couplings of naphthalenes at least (a preliminary attempt to use these additives to improve yields in one of the more problematic couplings in Table 1 did not significantly improve the result). The data are consistent with the induction period being a result of autocatalysis from the Mg salts produced during the reaction, presumably assisting in the C–O bond cleavage. There have been previous proposals for a nickelate complex undergoing oxidative addition after transmetalation from the Grignard,12a or an alternative in which the Grignard or an organoaluminium reagent acts as an activator for the otherwise conventional oxidative addition process.2y,3a Our working hypothesis is that for these activated substrates, oxidative addition, or conceivably transmetalation, is promoted by the Mg salts, formally [MgBr(OMe)], that are formed during the reaction.
Conclusions
Of course, streamlined ligands have been used in other examples of cross-coupling catalysis,2r,14 but they are more often overlooked. Meanwhile, examples of a unambiguous ligand steric effect favouring smaller ligands are unusual. There is a tendency for methodology chemists to initially minimise reaction temperatures, aiming at convenience for lab-scale research, which might lead to streamlined ligands giving poor performance under those conditions. However, both less activated reactions and reactions needing to be run using low catalyst loadings will often be operated at higher temperatures. We suggest there is a case for re-examining streamlined ligands in other reactions at higher temperatures. The ligand n-Bu3P is perhaps especially worthwhile since it is already produced at large scale, with a good range of metal salts commercially available or made in a trivial manner. The exact nature of the active catalyst in these reactions is unclear, but the fact that elevated temperatures seem to be needed to reach the working temperature of the catalyst, along with the very low activity of bidentate systems, may suggest the active species is formed after a quite challenging ligand dissociation. Once this has occurred, it releases a highly active and unhindered catalyst. The less-hindered catalyst has been validated to be more effective than Cy3P/nickel in over 10 challenging cross-couplings, but also to have an effect on activated substrates when carried out at higher temperature. This aspect also includes what seem to be the lowest catalysts loadings used in this type of Ni catalysis. A full kinetic and mechanistic study covering a full range of ligands and substrates at high and low temperatures would be a useful topic for further study.12 Target-focussed applications of Ni complexes of streamlined ligands at higher temperatures should also be considered following on from this work.Experimental
General information
Full experimental details and characterisation data are available in ESI.†All manipulations were carried out under an inert atmosphere of nitrogen or argon using standard Schlenk techniques. Solvents were dried and degassed before use.
Research data that underpins this article (NMR f.i.d.'s) can be accessed at doi 10.17630/f1b1bb53-eb4e-4ae4-ada3-1168bfa5df9a.
Example procedure of Ni-catalysed Grignard cross-coupling of aryl ethers, Table 1, entry 10: to an oven dried microwave vial equipped with a stirrer bar was added [NiCl2(PnBu3)2] (13.4 mg, 0.025 mmol). The vial was sealed with a crimp cap and flushed with argon for 30 minutes. 2-Methoxybiphenyl (92.1 mg, 0.50 mmol) and 1-methylnaphthalene (60 μL, 0.42 mmol, internal standard) were added to a flame dried Schlenk flask under an inert atmosphere. 2-MeTHF (2.25 mL) was then added to the Schlenk flask to make a solution. A t0 sample (approximately 10 μL) was taken and analysed by 1H NMR (to calibrate the ratio of internal standard to starting material). The solution containing the electrophile and internal standard was added to the nickel catalyst via syringe. PhMgBr (439 μ, 0.75 mmol, 1.7 M in Et2O) was added dropwise over 15 minutes. The reaction mixture was stirred vigorously in an oil bath at 100 °C for 16 hours. Upon cooling to room temperature, approximately 20 μL of the crude reaction mixture was added to a vial and quenched with CDCl3. The resulting mixture was then filtered through a small cotton wool plug into an NMR tube and a 1H NMR was run to assess the ratio between SM and desired product. Saturated aqueous ammonium chloride (5 mL) was added to the reaction mixture and the aqueous phase was extracted three times with ethyl acetate (3 × 5 mL), dried over sodium sulfate and concentrated under reduced pressure. Purification via column chromatography on silica gel (eluent petroleum ether) gave 1,1′:2′,1′′-terphenyl (98.6 mg, 86%) as a colourless oil with data in accordance with the literature.15δH (500 MHz, CDCl3); 7.49–7.42 (4H, m, CArH), 7.26–7.20 (6H, m, CArH), 7.19–7.14 (4H, m, CArH); δC (125 MHz, CDCl3) 141.6 (2C, CAr), 140.7 (2C, CAr), 130.7 (2C, CArH), 130.0 (4C, CArH), 128.0 (4C, CArH), 127.6 (2C, CArH), 126.6 (2C, CArH); m/z HRMS (EI+) C18H14+ ([M]+) requires 230.1096; found 230.1092 (−1.7 ppm).
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thanks the EPSRC for funding in the initial stages of this work from EP/J018139/1 and from the DTG.Notes and references
- (a) H. Zeng, Z. Qiu, A. Domínguez-Huerta, Z. Hearne, Z. Chen and C.-J. Li, ACS Catal., 2017, 7, 510 CrossRef CAS; (b) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081 RSC; (c) J. F. Cívicos, D. A. Alonso and C. Nájera, Eur. J. Org. Chem., 2012, 3670 CrossRef; (d) B.-J. Lie, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 1728 CrossRef PubMed; (e) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346 CrossRef CAS PubMed; (f) J. Albaneze-Walker, R. Raju, J. A. Vance, A. J. Goodman, M. R. Reeder, J. Liao, M. T. Maust, P. A. Irish, P. Espino and D. R. Andrews, Org. Lett., 2009, 11, 1463 CrossRef CAS PubMed; (g) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717 CrossRef CAS PubMed.
- (a) M. Tobisu, T. Takahira, T. Morioka and N. Chatani, J. Am. Chem. Soc., 2016, 138, 6711 CrossRef CAS PubMed; (b) L. Guo, X. Liu, C. Baumann and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 15415 CrossRef CAS PubMed; (c) Z.-C. Cao, Q.-Y. Luo and Z.-J. Shi, Org. Lett., 2016, 18, 5978 CrossRef CAS PubMed; (d) Z.-K. Yang, D.-Y. Wang, H. Minami, H. Ogawa, T. Ozaki, T. Saito, K. Miyamoto, C. Wang and M. Uchiyama, Chem. – Eur. J., 2016, 22, 15693 CrossRef CAS PubMed; (e) C. Wang, T. Ozaki, R. Takita and M. Uchiyama, Chem. – Eur. J., 2012, 18, 3482 CrossRef CAS PubMed; (f) P. Leowanawat, N. Zhang and V. Percec, J. Org. Chem., 2012, 77, 1018 CrossRef CAS PubMed; (g) M. Tobisu, T. Shimasaki and N. Chatani, Chem. Lett., 2009, 38, 710 CrossRef CAS; (h) M. Tobisu, T. Shimasaki and N. Chatani, Angew. Chem., Int. Ed., 2008, 47, 4866 CrossRef CAS PubMed; (i) E. Wenkert, E. L. Michelotti and C. S. Swindell, J. Am. Chem. Soc., 1979, 101, 2246 CrossRef CAS; (j) D. Heijnen, J.-B. Gualtierotti, V. Hornillos and B. L. Feringa, Chem. – Eur. J., 2016, 22, 3991 CrossRef CAS PubMed; (k) J. Zhang, J. Xu, Y. Xu, H. Sun, Q. Shen and Y. Zhang, Organometallics, 2015, 34, 5792 CrossRef CAS; (l) M. J. Iglesias, A. Prieto and M. C. Nicasio, Org. Lett., 2012, 14, 4318 CrossRef CAS PubMed; (m) L.-G. Xie and Z.-X. Wang, Chem. – Eur. J., 2011, 17, 4972 CrossRef CAS PubMed; (n) F. Zhao, Y.-F. Zhang, J. Wen, D.-G. Yu, J.-B. Wei, Z. Xi and Z.-J. Shi, Org. Lett., 2013, 15, 3230 CrossRef CAS PubMed; (o) C. Zarate, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2017, 139, 1191 CrossRef CAS PubMed; (p) X. Liu, C.-C. Hsiao, I. Kalvet, M. Leiendecker, L. Guo, F. Schoenebeck and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6093 CrossRef CAS PubMed; (q) M. Tobisu, T. Takahira, A. Ohtsuki and N. Chatani, Org. Lett., 2015, 17, 680 CrossRef CAS PubMed; (r) M. Tobisu, T. Takahira and N. Chatani, Org. Lett., 2015, 17, 4352 CrossRef CAS PubMed; (s) T. Morioka, A. Nishizawa, K. Nakamura, M. Tobisu and N. Chatani, Chem. Lett., 2015, 44, 1729 CrossRef CAS; (t) C. Zarate, R. Manzano and R. Martin, J. Am. Chem. Soc., 2015, 137, 6754 CrossRef CAS PubMed; (u) M. Tobisu, A. Yasutome, H. Kinuta, K. Nakamura and N. Chatani, Org. Lett., 2014, 16, 5572 CrossRef CAS PubMed; (v) M. Leiendecker, C.-C. Hsiao, L. Guo, N. Alandini and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 12912 CrossRef CAS PubMed; (w) B.-T. Guan, S.-K. Xiang, T. Wu, Z.-P. Sun, B.-Q. Wang, K.-Q. Zhao and Z.-J. Shi, Chem. Commun., 2008, 1437 RSC; (x) J. W. Dankwardt, Angew. Chem., Int. Ed., 2004, 43, 2428 CrossRef CAS PubMed; (y) J. Cornella and R. Martin, Org. Lett., 2013, 15, 6298 CrossRef CAS PubMed.
- Similar naphthyl effect is apparent in reductive cleavage reactions, see (a) M. Tobisu, T. Morioka, A. Ohtsuki and N. Chatani, Chem. Sci., 2015, 6, 3410 RSC and ref's therein; (b) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439 CrossRef CAS PubMed; (c) P. Álvarez-Bercedo and R. Martin, J. Am. Chem. Soc., 2010, 132, 17352 CrossRef PubMed.
- (a) M. Jacques, Curr. Org. Chem., 2011, 15, 2413 CrossRef; (b) T. G. Gant and A. I. Meyers, Tetrahedron, 1994, 50, 2297 CrossRef CAS; (c) A. I. Meyers, R. Gabel and E. Mihelich, J. Org. Chem., 1978, 43, 1372 CrossRef CAS.
- S. M. Leckie, G. J. Harkness and M. L. Clarke, Chem. Commun., 2014, 50, 11511 RSC.
- (a) C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559 CrossRef CAS PubMed; (b) M. Beller, G. Centi and L. Sun, ChemSusChem, 2017, 10, 6 CrossRef CAS PubMed.
- (a) J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602 CrossRef CAS PubMed; (b) P. Varanasi, P. Singh, M. Auer, P. D. Adams, B. A. Simmons and S. Singh, Biotechnol. Biofuels, 2013, 6, 14 CrossRef CAS PubMed; (c) M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29 CrossRef CAS; (d) C. Amen-Chen, H. Pakdel and C. Roy, Biomass Bioenergy, 1997, 13, 25 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS.
- Catalysts 1, 5 and 6 are commercially available. See ESI† for further details and syntheses of catalysts 2-4.
- (a) O. M. Kuzmina, A. K. Steib, A. Moyeux, G. Cahiez and P. Knochel, Synthesis, 2015, 47, 1696 CrossRef CAS; (b) R. B. Bedford, Acc. Chem. Res., 2015, 48, 1485 CrossRef CAS PubMed; (c) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (d) W. M. Czaplik, M. Mayer, J. Cvengroš and A. J. von Wangelin, ChemSusChem, 2009, 2, 396 CrossRef CAS PubMed; (e) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317 CrossRef CAS PubMed.
- K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150 CrossRef CAS PubMed.
- (a) H. Ogawa, H. Minami, T. Ozaki, S. Komagawa, C. Wang and M. Uchiyama, Chem. – Eur. J., 2015, 21, 13904 CrossRef CAS PubMed; (b) P. Kelley, G. A. Edouard, S. Lin and T. Agapie, Chem. – Eur. J., 2016, 22, 17173 CrossRef CAS PubMed; (c) T. Wititsuwannakul, Y. Tantirungrotechai and P. Surawatanawong, ACS Catal., 2016, 6, 1477 CrossRef CAS; (d) L. Xu, L. W. Chung and Y.-D. Wu, ACS Catal., 2016, 6, 483 CrossRef CAS; (e) J. Cornella, E. Gómez-Bengoa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1997 CrossRef CAS PubMed; (f) J. Cornella and R. Martin, Org. Lett., 2013, 15, 6298 CrossRef CAS PubMed.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841 RSC.
- A. D. Sun, K. Leung, A. D. Restinvo, N. A. LaBerge, H. Takasaki and J. A. Love, Chem. – Eur. J., 2014, 20, 3162 CrossRef CAS PubMed.
- B. J. Groombridge, S. M. Goldup and I. Larrosa, Chem. Commun., 2015, 51, 3832 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of experimental procedures, analytical data and NMR spectra are available in the ESI. See DOI: 10.1039/c7cy01205e |
| This journal is © The Royal Society of Chemistry 2018 |