
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amide activation: an emerging tool for chemoselective synthesis
Daniel
Kaiser
 ,
Adriano
Bauer
,
Miran
Lemmerer
and
Nuno
Maulide
*
,
Adriano
Bauer
,
Miran
Lemmerer
and
Nuno
Maulide
*
Institute of Organic Chemistry, University of Vienna, Währinger Strasse 38, 1090 Vienna, Austria. E-mail: nuno.maulide@univie.ac.at
First published on 28th August 2018
Abstract
It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. However, a significant body of research on the selective activation of amides to achieve powerful transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product synthesis, highlighting the synthetic applications and the potential of this approach.
 Daniel Kaiser | Daniel Kaiser obtained his PhD in 2018 from the University of Vienna as an Austrian Academy of Sciences DOC-fellow under the supervision of Prof. N. Maulide, where his research was focussed on amide- and alkyne activation. He is currently working in the group of Prof. V. K. Aggarwal as an Erwin-Schrödinger postdoctoral fellow at the University of Bristol. |
 Adriano Bauer | Adriano Bauer obtained his MSc in 2015 from the University of Bologna, Italy, following a stay at Imperial College London under the guidance of Prof. A. C. Spivey, where he worked on the kinetic resolution of indolines with Prof. L. Gentilucci as a joint supervisor. Since 2016, he has been a PhD student in the group of Prof. N. Maulide at the University of Vienna. |
 Miran Lemmerer | Miran Lemmerer obtained his MSc from the University of Vienna in 2018, where he worked on Umpolung reactions of tertiary amides under the supervision of Prof. N. Maulide, following a five-month exchange in Lund, Sweden. He is currently planning to continue his education as a PhD student in Prof. N. Maulide's research group. |
 Nuno Maulide | Nuno Maulide was born in Lisbon, Portugal, in 1979. He obtained a PhD from the Université catholique de Louvain (I. Markó) in 2007. Following a postdoctoral stay at Stanford (B. M. Trost), he began his independent research career as a Max-Planck Group Leader at the MPI für Kohlenforschung (Germany) before assuming his current position as Full Professor for Organic Synthesis at the University of Vienna (Austria) and an ERC StG and CoG grantee. His research interests are broadly defined around the area of highly reactive intermediates and rearrangements. |
Introduction
In their classical representation, carboxamides comprise a central carbon atom possessing a double bond to oxygen and a single bond to nitrogen. This representation, however, is grossly incomplete given that nitrogen lone-pair delocalisation plays a crucial role in dictating the structure and reactivity of amides (Scheme 1). Indeed, this delocalisation imparts an increased stabilisation to the electrophilic carbonyl carbon, especially when compared to other carbonyl and carboxyl derivatives, resulting in considerably reduced reactivity towards nucleophiles.1–3 It has therefore long been textbook knowledge that, in contrast to acyl halides, anhydrides and esters, amides do not readily undergo addition of, for example, alcohols, amines or mild hydride sources, thereby precluding any chemoselective amide-transformations in the presence of other carbonyl functional groups. In addition, primary and secondary amides will not undergo addition of Grignard reagents or similar strong nucleophiles, but will rather be deprotonated at nitrogen. This difference in reactivity has led to a perception of amides as significantly less useful functional handles than their ester and acid chloride counterparts. To the same extent that amides are weak electrophiles at carbon, they have historically been shown to be powerful nucleophiles at oxygen, leading to the emergence of the field of “amide activation”.4–8 This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product synthesis, highlighting the synthetic applications and the potential of this approach. Owing to the recent development of alternative modes of amide activation, foremost relying on electronic effects, single electron reduction, and bulk or metal catalysis, an overview of these topics will be additionally presented.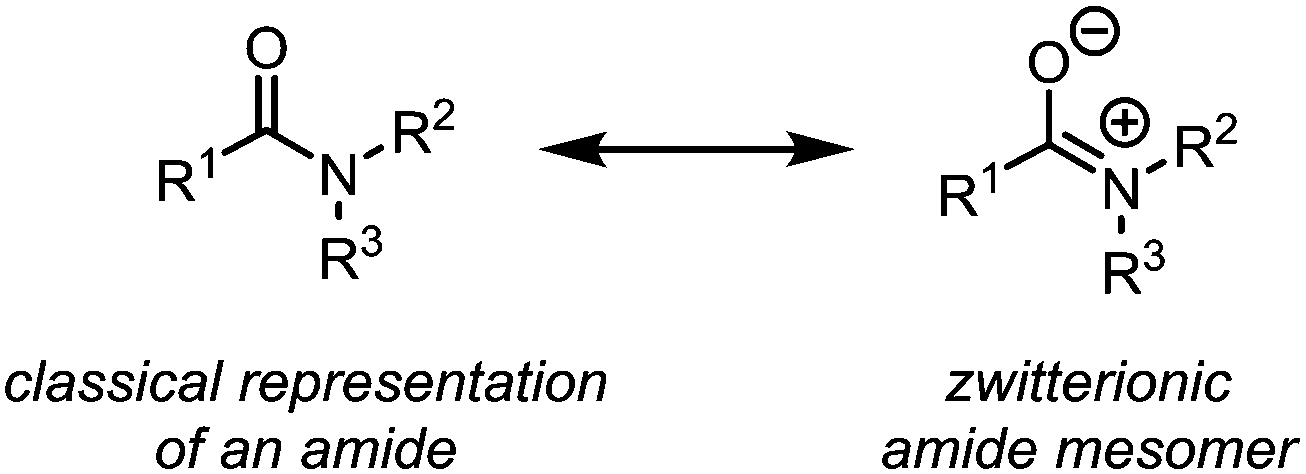 | ||
| Scheme 1 Dominating mesomeric structures of amides. | ||
The origins of electrophilic amide activation date back to the 19th century: as early as 1877 it was common knowledge that primary (NH2) amides can be converted to the corresponding nitriles through treatment with dehydration agents.9 However, all dehydration attempts of secondary and tertiary amides using phosphoric anhydride failed. In 1877, Wallach published a report on a successful reaction of secondary amides with phosphorus pentachloride (Scheme 2),9 in which several ground-breaking discoveries were detailed: first, the observed formation of α-dichloroamines, which decomposed smoothly to the corresponding α-chloroimines (imidoyl chlorides); second, the demonstration that the imidoyl chloride of N-phenyl acetamide dimerised under loss of hydrogen chloride to “base(s) of unknown nature” which contained chlorine. The latter was most likely the first instance of intermediate preparation of an α-chloroenamine (cf.Scheme 2). Moreover, Wallach recognised the highly electrophilic character of imidoyl chlorides and showed that they can be converted generally and easily to the corresponding amidines by treatment with amines.
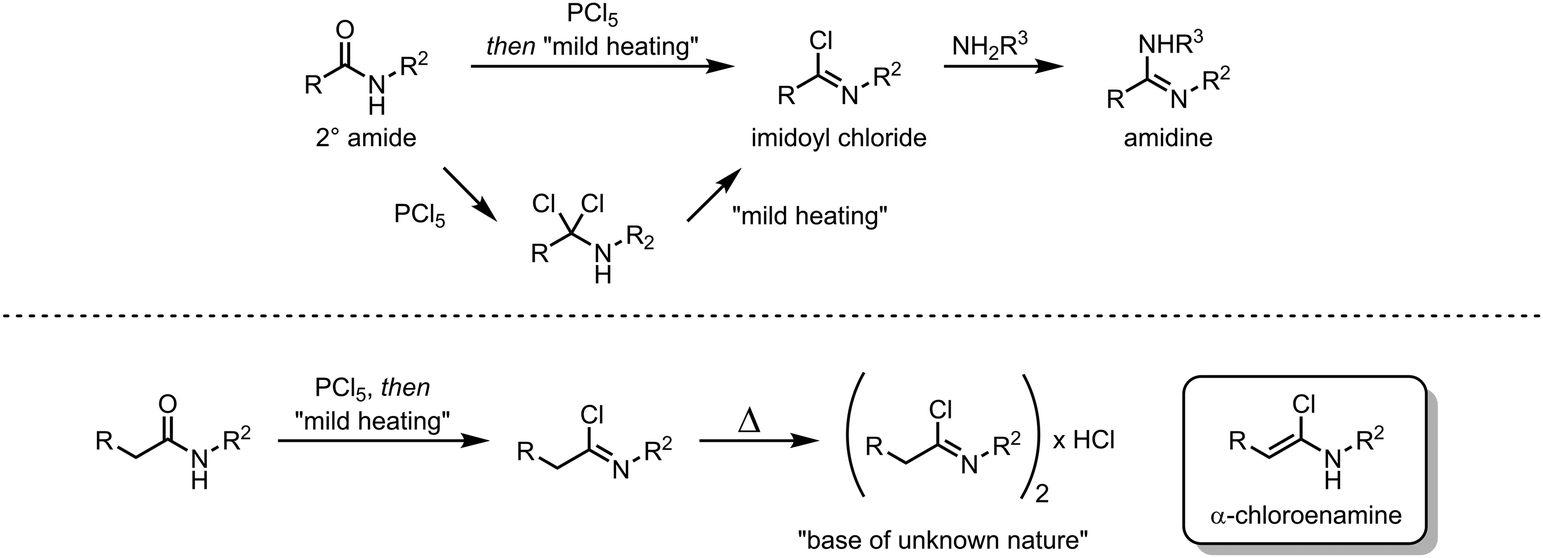 | ||
| Scheme 2 Early electrophilic activation of 2° amides, affording imidoyl chlorides (top) and α-chloroenamines (bottom). | ||
About 50 years later, for the first time, the formation of an α-chloroenamine was claimed: a tertiary amide was treated with phosphorus pentachloride to yield a compound, whose hydrolysis to the corresponding amide, unlike the related imidoyl chloride, was shown to be relatively difficult.10 Additionally, the substitution of the chloride and the formation of the corresponding amidine by treatment with aniline were observed. The structure, however, remained unconfirmed. Another 40 years passed before H. G. Viehe, L. Ghosez and co-workers published a new method for the preparation of alkyl and aryl α-chloroenamines from the corresponding 3° amides, using phosgene with subsequent deprotonation of the cationic imidoyl chloride salt.11 The chemical behaviour of these compounds was partly predictable based on knowledge gained from previous studies: the hydrolysis to the amide, the addition of elemental bromine or hydrogen chloride and subsequent elimination to the corresponding ynamines. However, the outstanding ability for nucleophilic substitution of the chloride seemed to surprise the authors: Grignard reagents, organolithium compounds, thiolates, alkoxides and lithium amides (deprotonated amines) yielded the substitution products in moderate to high yields (Scheme 3). Last but not least, Viehe et al. were able to show that α-chloroenamines can be readily employed in formal [2+2] cycloadditions with acetylenes, forming the corresponding cyclobutenone derivatives in good yields. The extraordinary reactivity was believed to be due to an equilibrium of the α-chloroenamine with the corresponding keteniminium chloride. This report marked the beginning of modern electrophilic amide activation in organic synthesis.
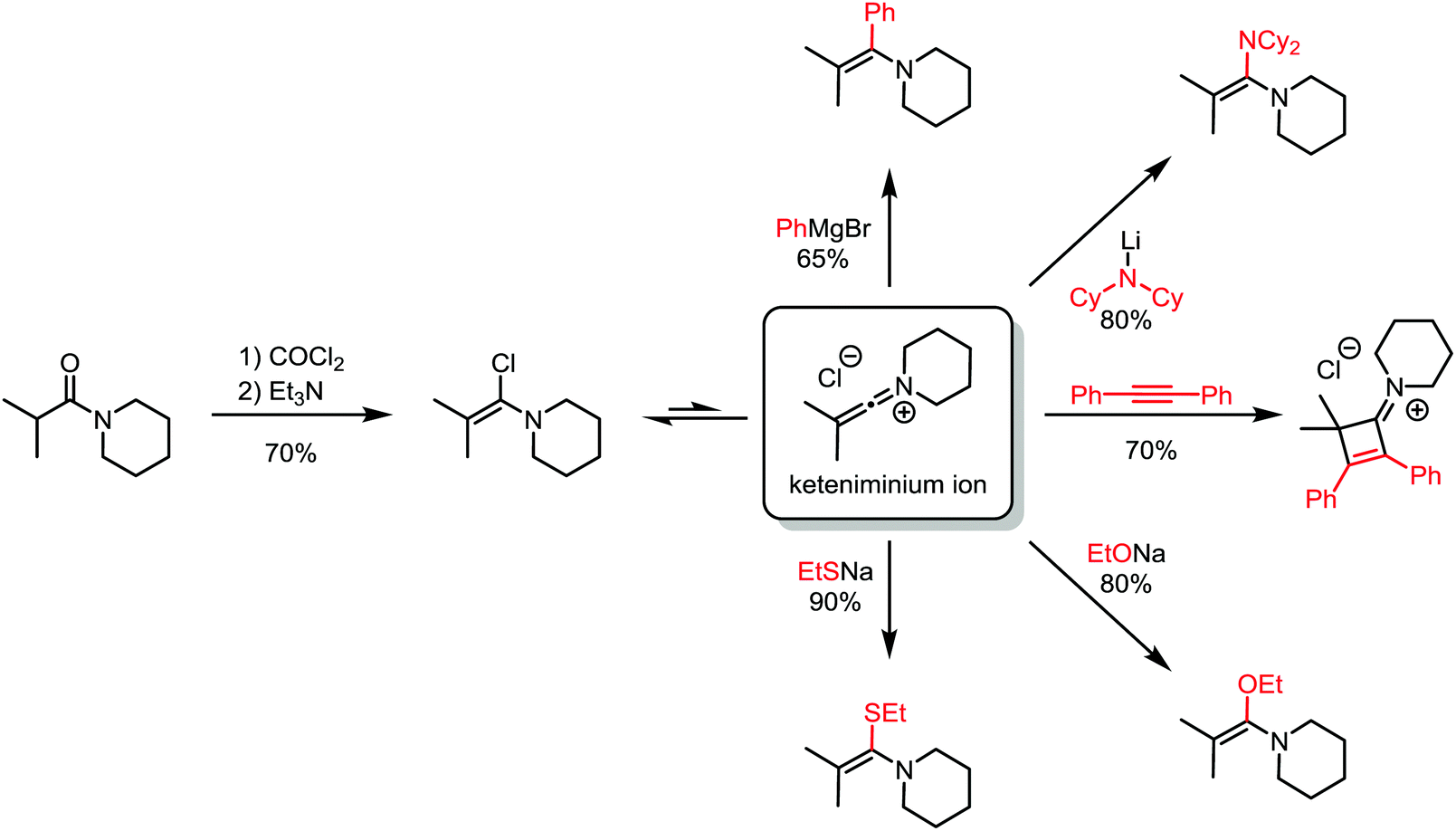 | ||
| Scheme 3 Products of addition of various nucleophiles to an α-chloroenamine-derived keteniminium ion. | ||
Keteniminium ions in formal [2+2] cycloadditions
Development of the methodology
In the 1970s, the Ghosez group initiated further investigations into the preparation of α-chloroenamines and their reactivity in cycloaddition reactions. They observed that, in contrast to ketenes, particularly electron-rich unsaturations are not necessary to perform cycloadditions with keteniminium ions: even the simplest alkene, ethylene, is a competent reaction partner in the [2+2]-cycloaddition of keteniminium ions at ambient temperature.12,13 Those reagents thus surpass ketenes in their reactivity, as ketenes require elevated temperatures to react with simple alkenes.14In the same year, [2+2]-cycloadditions of in situ formed keteniminium ions with conjugated dienes and alkenes were reported.15 The reactions were generally clean: the Diels–Alder product was absent, and the only observable side product was the parent amide. For non-symmetrical dienes, the least substituted double bond was favoured for the cycloaddition and both cis- and trans-alkenes formed the desired products with high stereospecificity.15 The scope was quickly extended to the synthesis of cyclobutenones using simple alkynes,16 and to the synthesis of 2-amino-1-azetines,17 and β-lactams using imines as cycloaddition partners.18
Hartman and Heine later showed that the cycloaddition of “keto”-keteniminium salt 2 (a keteniminium salt carrying two substituents at the α-position; the case where only one non-hydrogen substituent is present is usually denominated “aldo”, vide infra) to electron poor α,β-unsaturated esters (such as 3), ketones and amides was also possible.19 Interestingly, the major regioisomer did not correspond to the expected regiochemical outcome based on the polarity of the involved atoms (Scheme 4). This finding led the authors to propose a mechanism where the keteniminium ion reacts like a carbene in the first step, forming 4, followed by a preferred migration of the substituted alkyl group to lead to the observed cyclobutanone product 5. This mechanism was later found to be consistent with DFT computations in similar systems.20
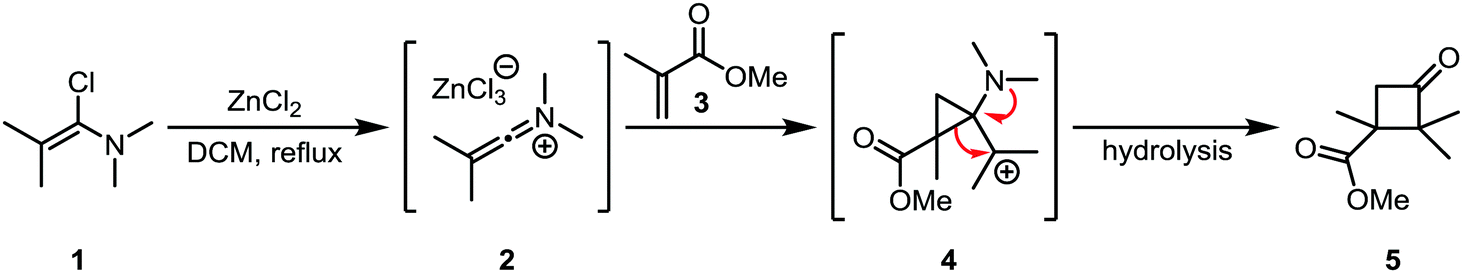 | ||
| Scheme 4 Formal carbene reactivity of a keteniminium ion, forming an aminocyclopropane intermediate. | ||
A common challenge in the formation of keteniminium salts from α-chloroenamines lies in the fact that the starting materials tend to exhibit considerable nucleophilicity (as enamines) and can react with the in situ formed keteniminium intermediates (Scheme 5, top). In particular, “aldo” keteniminium salts (7) were observed to readily react with their α-chloroenamine precursors (6).21 In the search for a viable alternative, Ghosez and co-workers turned to a combination of trifluoromethanesulfonic anhydride (triflic anhydride) and the non-nucleophilic base collidine for the activation of amides (Scheme 5, bottom). In doing so, the nucleophilicity of the precursors, 8 and 10, is considerably lowered, leading to suppression of the dimerisation reaction.21 Moreover, the amide can be used directly, obviating the need for the often-troublesome isolation of metastable α-chloroenamines. These two findings make this approach the method of choice for the generation of keteniminium salts from amides.
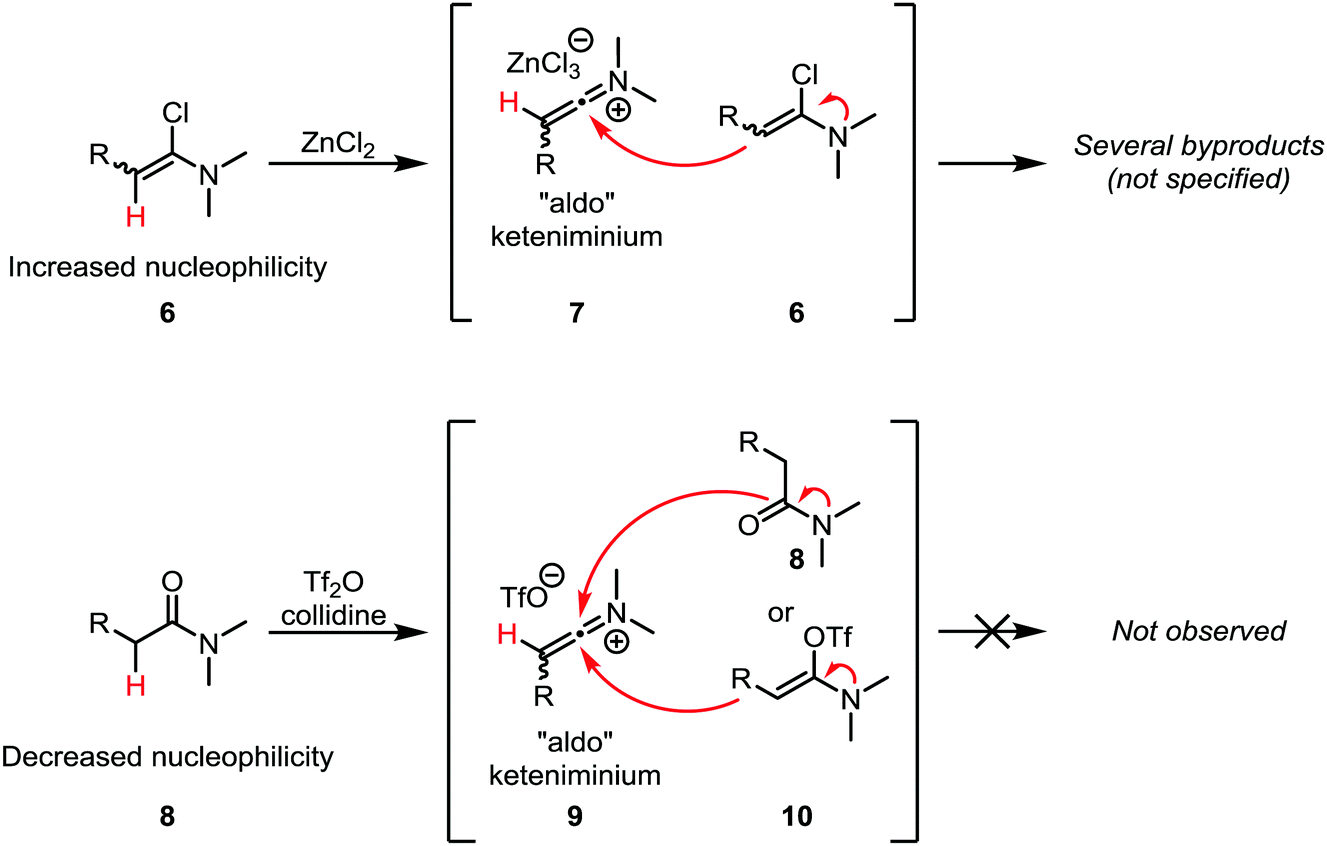 | ||
| Scheme 5 Discrepancy in the reactivities of α-chloroenamine- and amide-derived “aldo” keteniminium ions. | ||
Intramolecular [2+2]-cycloadditions of keteniminium salts have proven to be a viable alternative to cycloadditions of ketenes, since even large tether lengths are allowed, leading to a plethora of ring-fused cyclobutanone adducts. This is exemplified by the formation of structures such as medium-sized ring 13 (Scheme 6)22 and further downstream products,23,24 from amides such as 11.
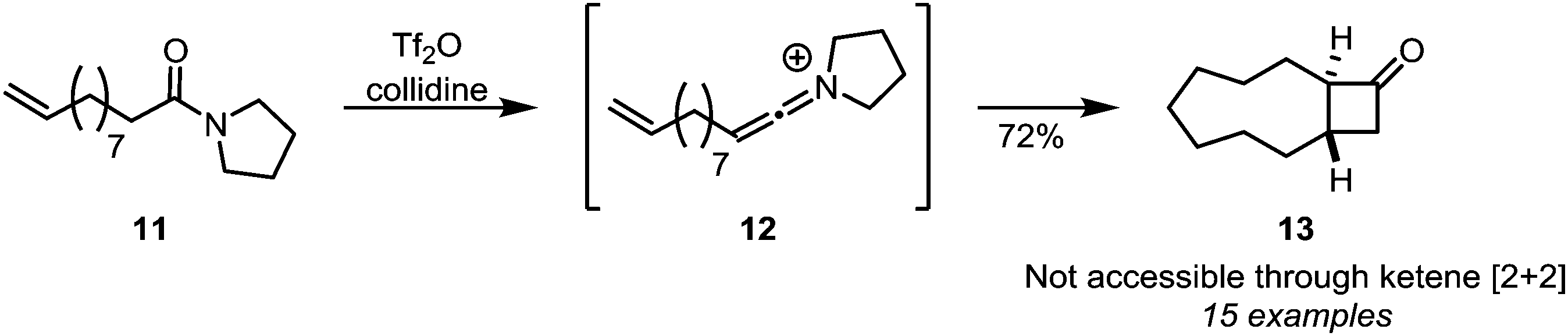 | ||
| Scheme 6 Intramolecular [2+2]-cycloaddition of keteniminium ions and alkenes. | ||
On the other hand, Snider and co-workers published a more critical view on keteniminium salts as [2+2]-cycloaddition reagents.25 Indeed, the authors showed that for a range of starting materials the ketene affords higher yields than its keteniminium congener, especially when a nucleophilic moiety is found within the framework. Even moieties with rather low nucleophilicity such as ethers can lead to considerable side reactions (vide infra),26 and the exploration of these shortcomings to achieve alternative reactivity will be discussed in more detail below. The group of Brady reconciled these data, concluding that “aldo” keteniminium salts are superior to “aldo” ketenes for the [2+2] cycloaddition but “keto” ketenes tend to outperform “keto” keteniminium salts.27
Unlike ketenes, keteniminium ions, inevitably bearing up to two substituents on nitrogen, can carry stereogenic information at the heteroatom (Scheme 7a). This has led to the development of traceless enantiopure auxiliaries (as represented by amide 14), enabling enantioselective syntheses of products of formal [2+2]-cycloaddition, such as 17,28 with C2-symmetry leading to a general solution.29 Enantiopure allylsilanes (18) can also be used efficiently for the diastereoselective assembly of cyclobutanones such as 20 by keteniminium [2+2]-cycloaddition chemistry (Scheme 7b).30
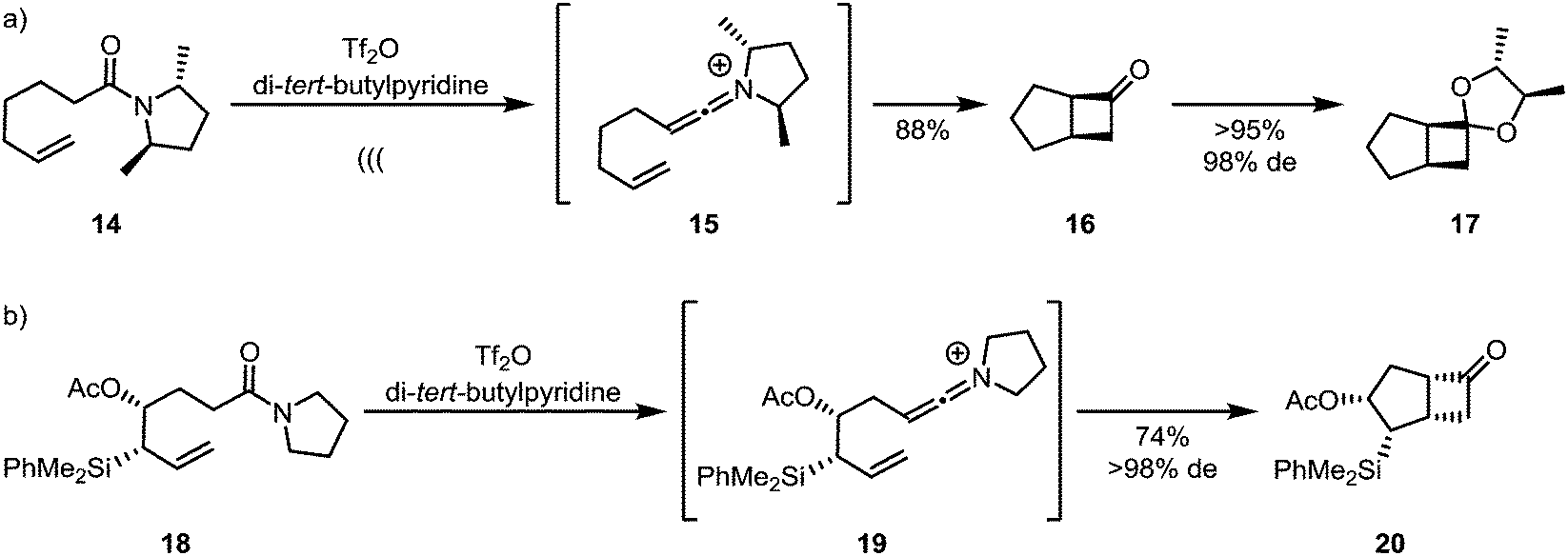 | ||
| Scheme 7 Diastereoselective keteniminium-[2+2]-cycloadditions, using (a) chiral auxiliaries and (b) chiral substrates. | ||
While the [2+2]-cycloaddition of ketenes and olefins is believed to be a concerted process, thermally allowed by a suprafacial–antarafacial approach of the two fragments, the cycloaddition of keteniminium salts to alkenes is very likely a stepwise process. The most convincing experimental evidence for a stepwise mechanism was reported in 1983, when the absence of stereospecificity was observed for the cycloaddition of 2-butene.31 Upon reaction of the keteniminium salt 22 with 2-cis-butene, the trans-cyclobutanone products 25b and ent-25b were also observed (Scheme 8). By carefully measuring the enantiomeric excesses of the different isomers, the authors proved that the absence of stereospecificity was not exclusively the result of isomerisation during hydrolytic workup, but rather also a consequence of rotation of the C–C bond of the (originally) olefinic fragment.
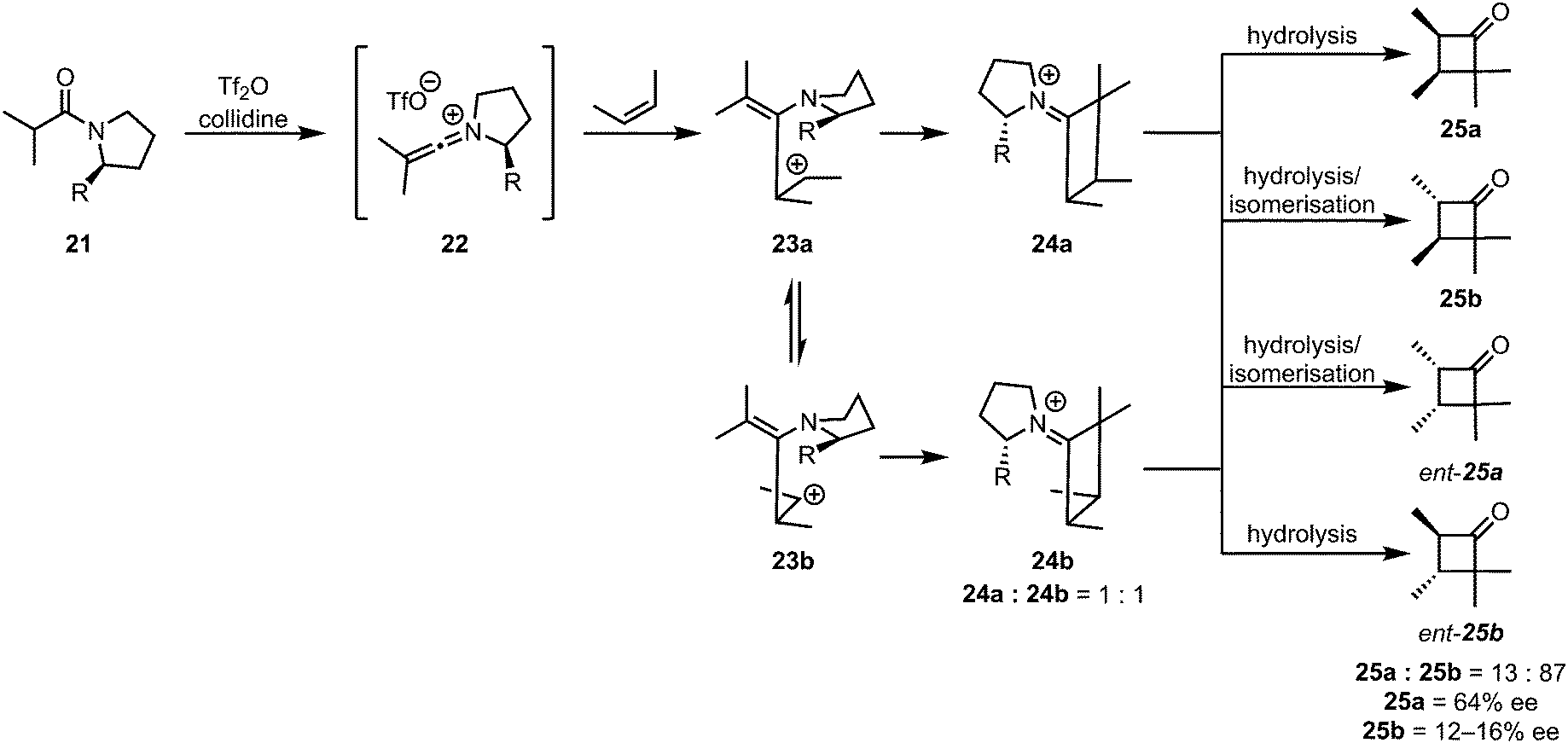 | ||
| Scheme 8 The lack of stereospecificity proves the stepwise nature of the addition of alkenes to keteniminium ions. | ||
Applications in total synthesis
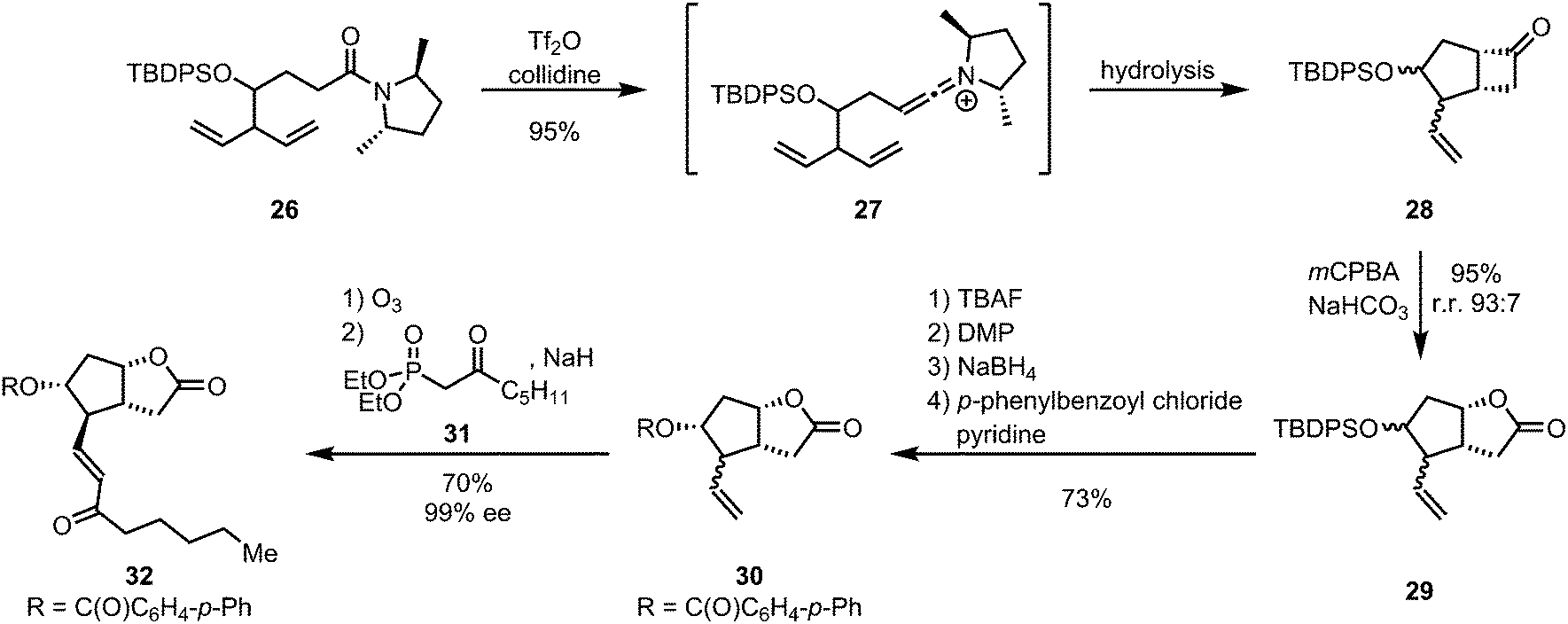 | ||
| Scheme 9 Ghosez's prostaglandin-core synthesis via keteniminium [2+2]-cycloaddition. | ||
De Mesmaeker and co-workers later showed that a similar strategy could be employed for the synthesis of GR-24, a synthetic analogue of the strigolactone plant hormones.35,36 In the same year, the methodology was extended to the synthesis of all four stereoisomers of 5-deoxystrigol (37) with high enantiomeric excesses (Scheme 10).37
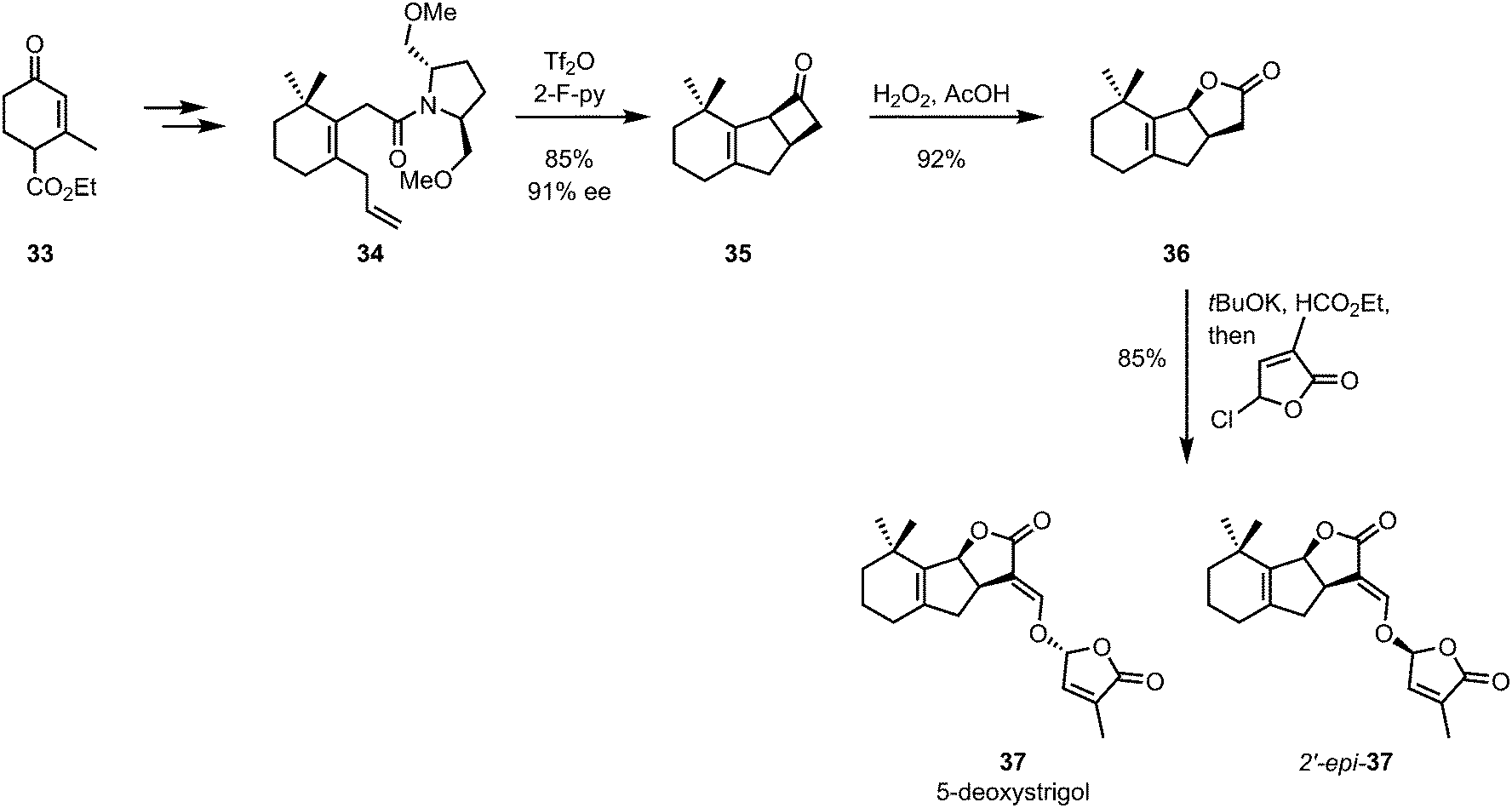 | ||
| Scheme 10 De Mesmaeker's 5-deoxystrigol synthesis employing a chiral auxiliary-mediated diastereoselective [2+2]-cycloaddition. | ||
The combination keteniminium ion [2+2]-cycloaddition/Baeyer–Villiger oxidation was also applied to the enantioselective formal syntheses of (−)-dihydroactinidiolide (44) and the related (−)-anastrephin (46) (Scheme 11). These insect pheromones were synthesised by a diastereoselective intramolecular [2+2]-cycloaddition of amide 40, affording intermediates 42 and epi-42. Stereogenic information was appended early on via sharpless asymmetric epoxidation of geraniol 38.38
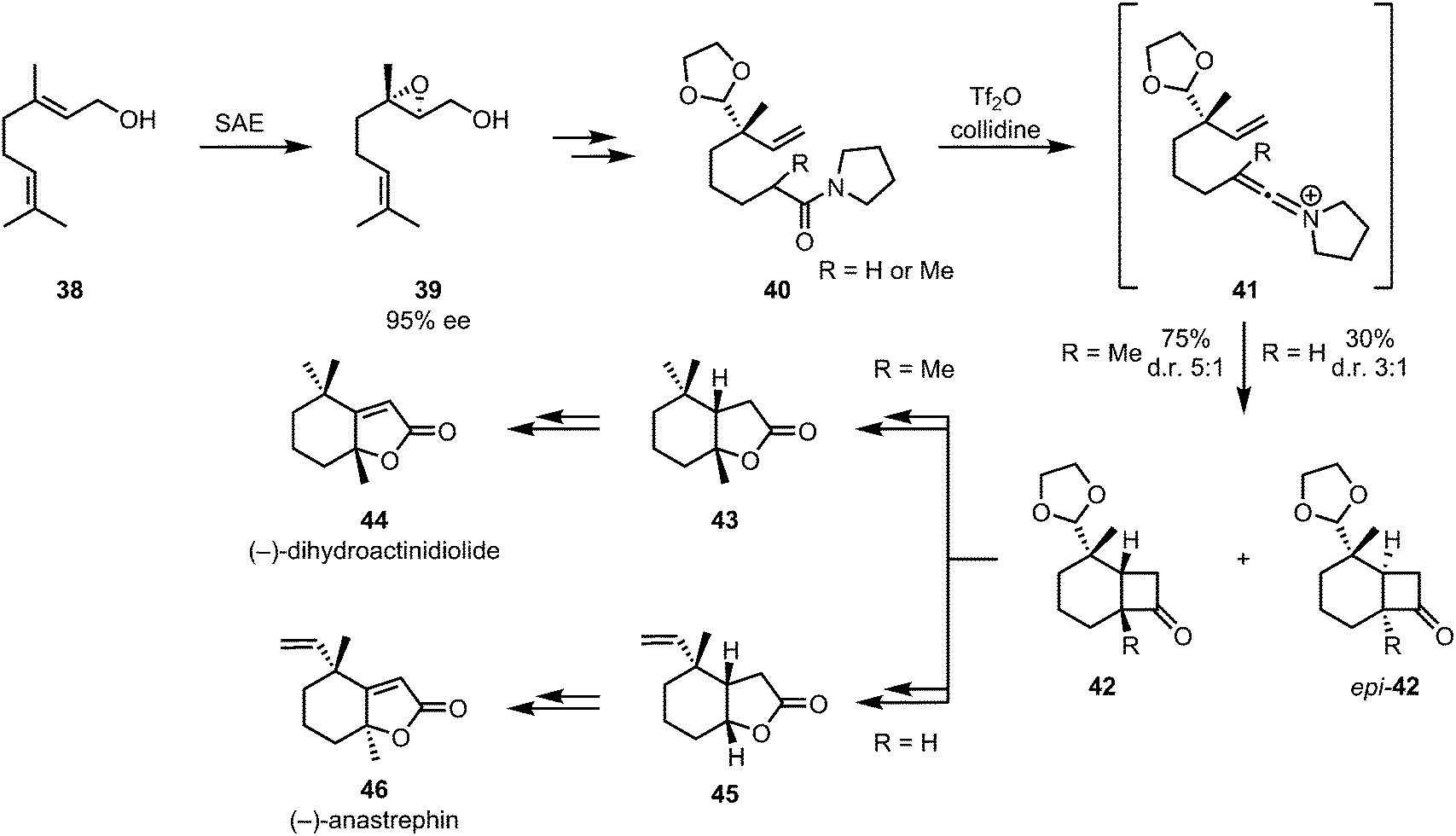 | ||
| Scheme 11 Diastereoselective keteniminium [2+2]-cycloaddition/Baeyer–Villiger sequence in Shishido's synthesis of insect pheromones. | ||
The ring expansion of cyclobutanones is obviously not limited to lactone formation by Baeyer–Villiger oxidation. As shown by Kim and Shim in their formal total synthesis of gibberellic acid (54), the cyclobutanone product (50) of an intramolecular keteniminium [2+2]-cycloaddition can be treated with diazomethane to yield the corresponding ring-expanded cyclopentanone compound 51, likely through a Tiffeneau–Demjanov-type rearrangement (Scheme 12). Unfortunately, low yields were observed for the cycloaddition and a low regioselectivity ratio for the ring expansion of approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was detected.39
:1 was detected.39
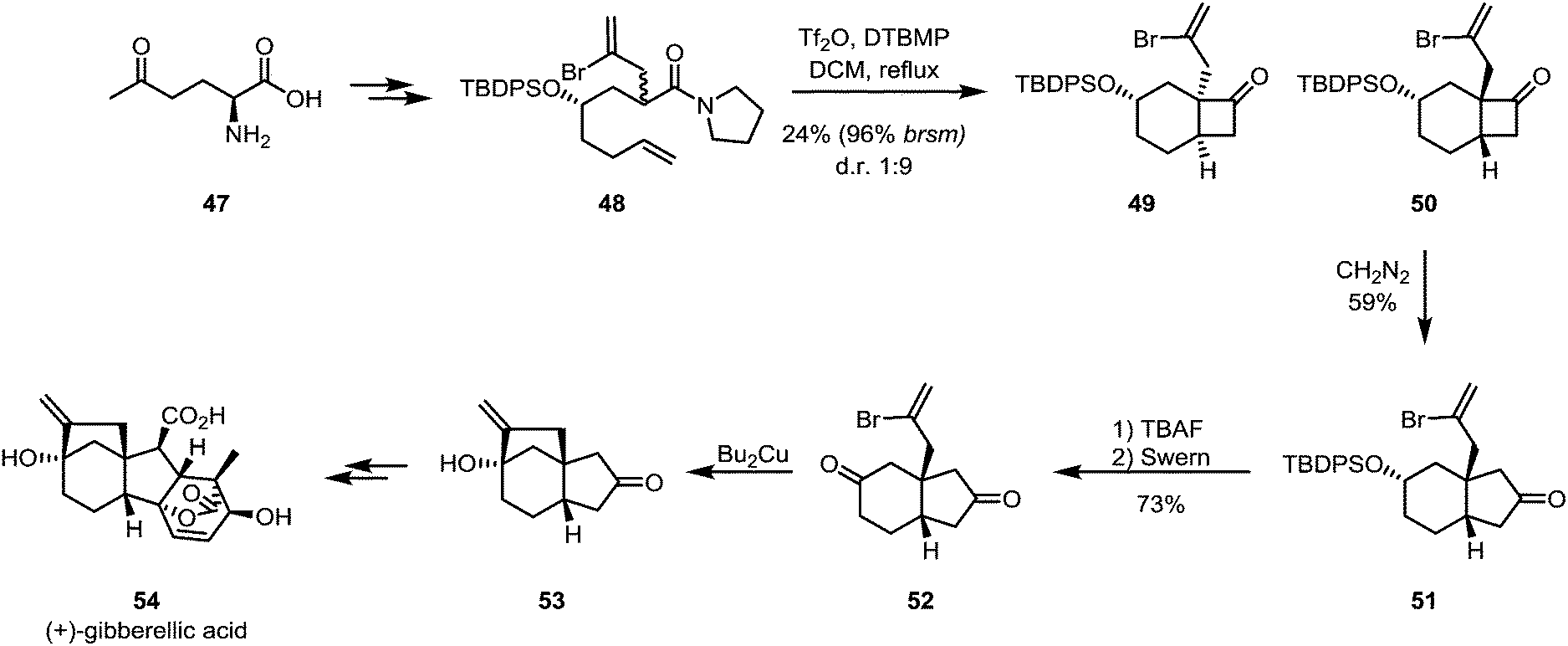 | ||
| Scheme 12 Kim's approach to cyclopentanone synthesis: [2+2]-cycloaddition, followed by ring-expansion. | ||
Addition of nucleophiles to keteniminium ions
While the addition of nucleophiles to most common carbonyl derivatives (ketones, aldehydes, esters) is a textbook approach to functional-group interconversion, such transformations are scarcely documented when it comes to amides. This lack in reactivity towards nucleophilic addition can be circumvented, however, by the activation of amides with electrophilic reagents such as oxalyl chloride, phosgene and foremost triflic anhydride (vide supra). This approach has been widely exploited in the addition of a variety of nucleophiles to the amide carbonyl, leading to the synthesis of a broad scope of functional groups and heterocyclic structures. Following the early reports by Ghosez described above,12 this general reactivity has experienced something of a renaissance in recent decades.Intermolecular addition of carbon nucleophiles
The intermolecular addition of carbon-based nucleophiles to keteniminium ions has given rise to a number of (orthogonal) procedures, allowing access to a wide range of functional groups and natural products.While the addition of π-unsaturated reactants such as alkenes to activated tertiary amides, as discussed above, leads to the ultimate formation of cyclobutanone or cyclobutenone structures, secondary amides can follow a different pathway. In a series of reports, Movassaghi et al. were able to establish a sequential amide activation/C-nucleophile addition sequence, affording pyridine and quinoline products (58) (Scheme 13).40,41 Therein, addition of an alkyne or enol ether to the intermittently formed nitrilium ion 56 leads to intermediates of type 57, prone to either metal-catalysed or spontaneous cyclisation. Besides the formation of highly substituted pyridine and quinoline cores, the use of N-aryl amides and cyclic silyl enol ethers notably allowed the synthesis of challenging mono- and diannulated quinolines.
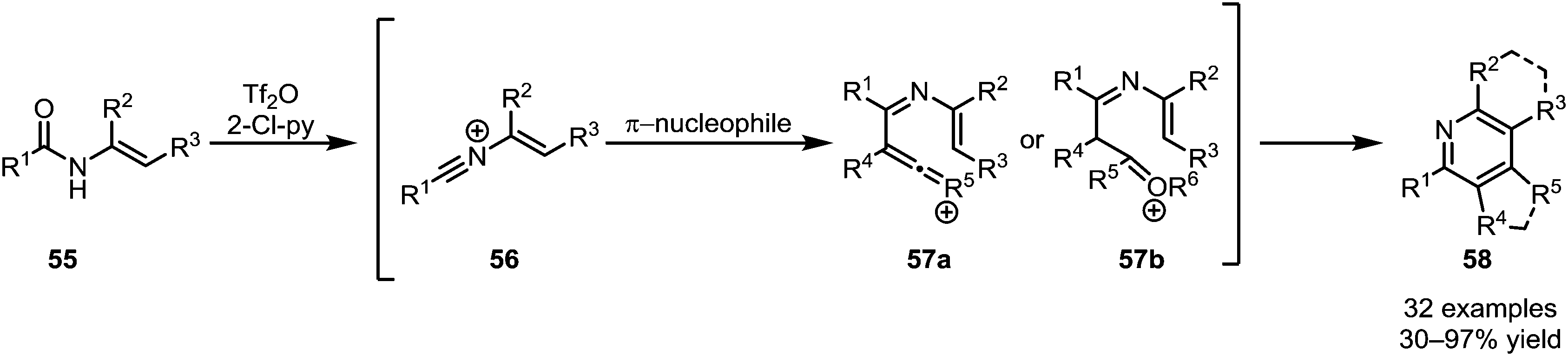 | ||
| Scheme 13 Movassaghi's synthesis of highly substituted pyridines and quinolines by addition of π-nucleophiles to activated secondary amides. | ||
Recent further work on this family of transformations has led to the development of highly modular processes, allowing access to a wide range of heterocyclic structures.42,43 While the aforementioned processes hinge on the addition of a two-carbon nucleophilic moiety, affording six-membered rings, Wang et al. were able to elegantly utilise ethyl diazoacetate (EDA) as a one-carbon synthon (Scheme 14).44 Forming substituted indole products (62), the authors exploited the inherent ambiphilic properties of EDA initially attacking the electrophilic nitrilium ion 60 and subsequently undergoing displacement of dinitrogen during cyclisation.
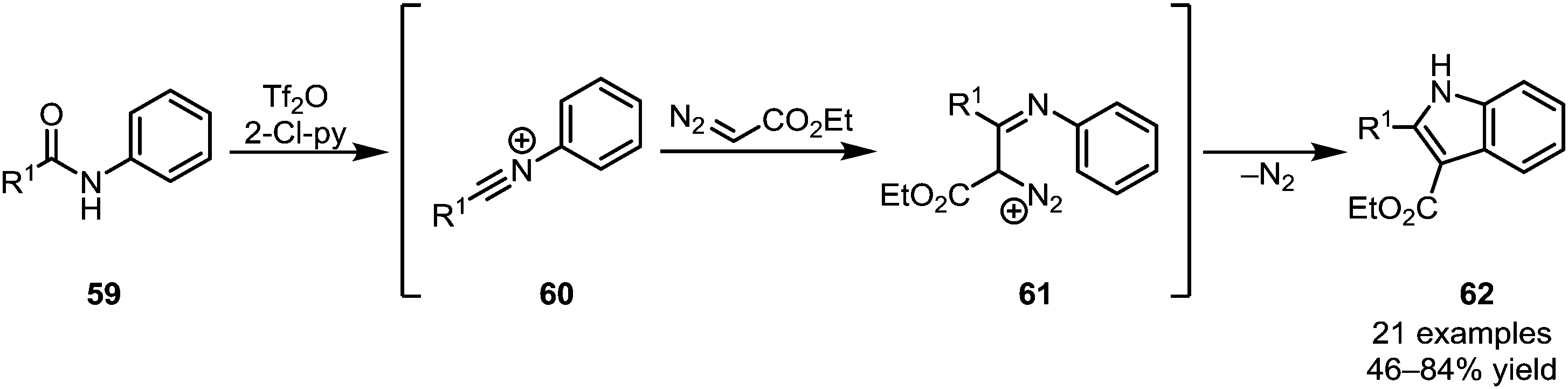 | ||
| Scheme 14 Wang's indole synthesis employing EDA. | ||
While the methods described above rely on the addition of π-nucleophiles to activated amides (thereby ultimately generating a new electrophilic center), the addition of (often sp3-hybridised) anionic carbon nucleophiles maintains the primary reactive site at the amide. The resulting imine or iminium ion can be functionalised in several ways downstream.
In 2012, the groups of Charette and Huang disclosed the selective formation of imines (64, and, after hydrolysis, ketones (65)) through monoaddition of organometallic reagents or enolates to activated secondary amides (63) (Scheme 15a).45,46 Despite displaying moderate electrophilic properties themselves, the imines can be obtained with high chemoselectivity, owing to the increased reactivity of the corresponding nitrilium ion precursors. Charette et al. further demonstrated the utility of electrophilic amide activation for chemoselective functionalisation through the synthesis of an unsymmetrical diketone 67 from diamide 66. Activation of the secondary amide moiety, followed by treatment with PhMgBr, effected initial imine formation at low temperatures. Subsequent selective addition of EtMgBr to the Weinreb amide function and ultimate acidic hydrolysis afforded 67 in a one-pot procedure and in excellent overall yield. In addition to ketones and imines, α-secondary amines, the corresponding products of in situ reduction, can be formed using mild reducing agents such as sodium borohydride or triethyl silane.47–49 This sequence has found widespread use in the synthesis of (α-chiral) amine-containing natural products (Scheme 15b). In this context, Huang et al. reported a wide range of total syntheses of cyclic alkaloids, employing various reducing agents. Iminium reduction with either LiAlH4 or Hantzsch ester hydride (HEH), for example, led to the synthesis of the alkaloid radicamine B (70), albeit with moderate diastereoselectivity, in 4 steps (three of which performed in one-pot) from a tartaric acid-derived enantiopure building block (Scheme 15b(1)).50 The same approach of electrophilic activation, followed by nucleophilic addition and hydride reduction, has further been used in the syntheses of the monocyclic alkaloids bgugaine, coniine, preussin and cassine (74)51 and a variety of quinolizidine-based alkaloids (76) (Scheme 15b(2)).52 Similarly, catalytic hydrogenation has also found application in amide-activation-based natural product synthesis, as shown by both Huang et al. and Oppolzer et al., in the 1977 synthesis of pumiliotoxin C (78) (Scheme 15b(3)).53–55
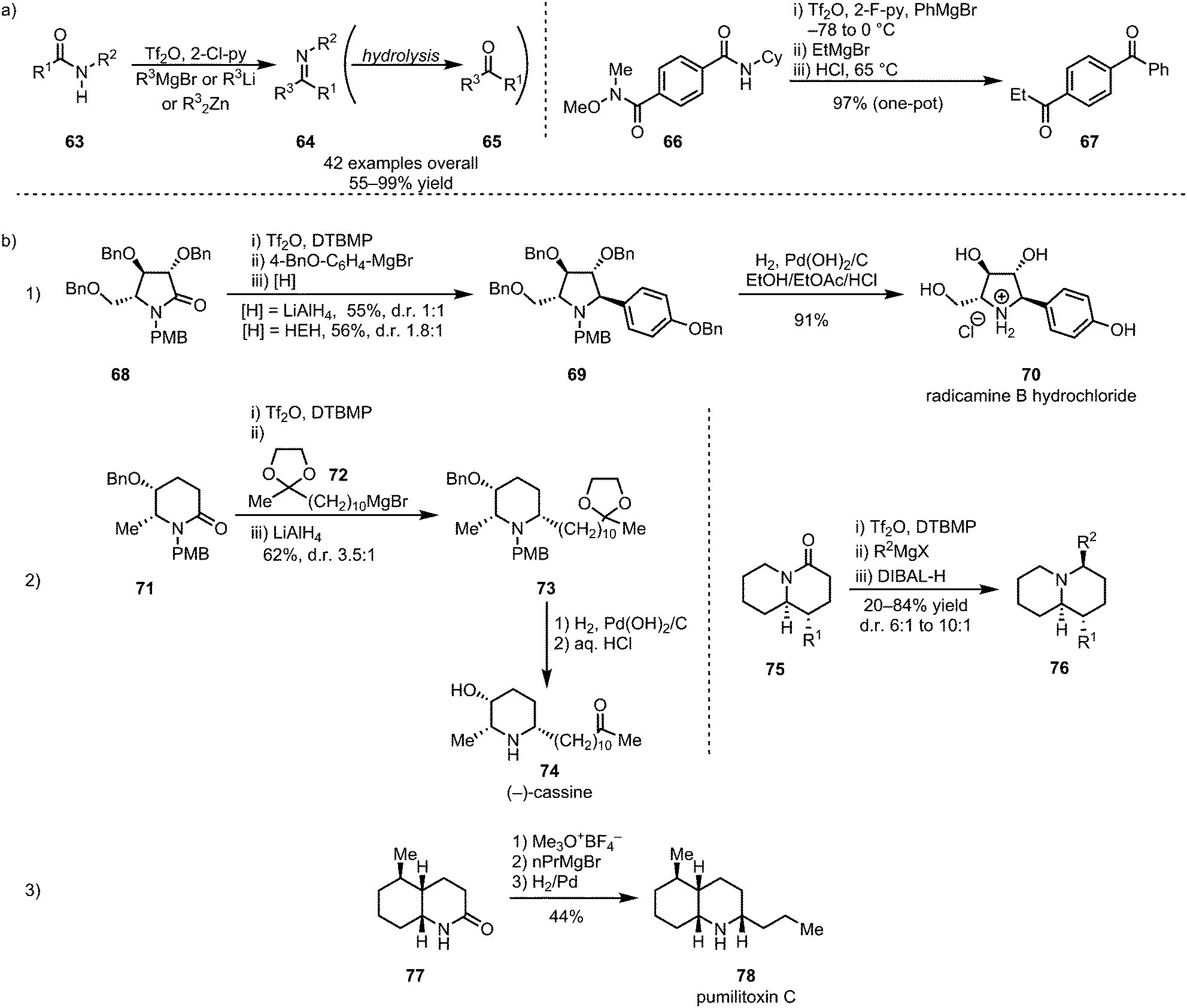 | ||
| Scheme 15 (a) Selective monoaddition of organometallic reagents for the synthesis of imines and ketones; and (b) application of a nucleophilic addition–reduction sequence to natural product total synthesis. | ||
The (sequential) intermolecular double addition of carbon nucleophiles is another viable process that enables the formation of valuable structures,56,57 such as biologically active sparteine derivatives (82) (Scheme 16a),58 and as such this approach has also found application in natural product synthesis.59–61 For example, this strategy has been applied to Huang's synthesis of FR901483 (86), using the added moieties for double cyclisation,60 and cephalotaxine (90), employing ring-closing metathesis for the formation of spirocyclic intermediate 89 (Scheme 16b).61
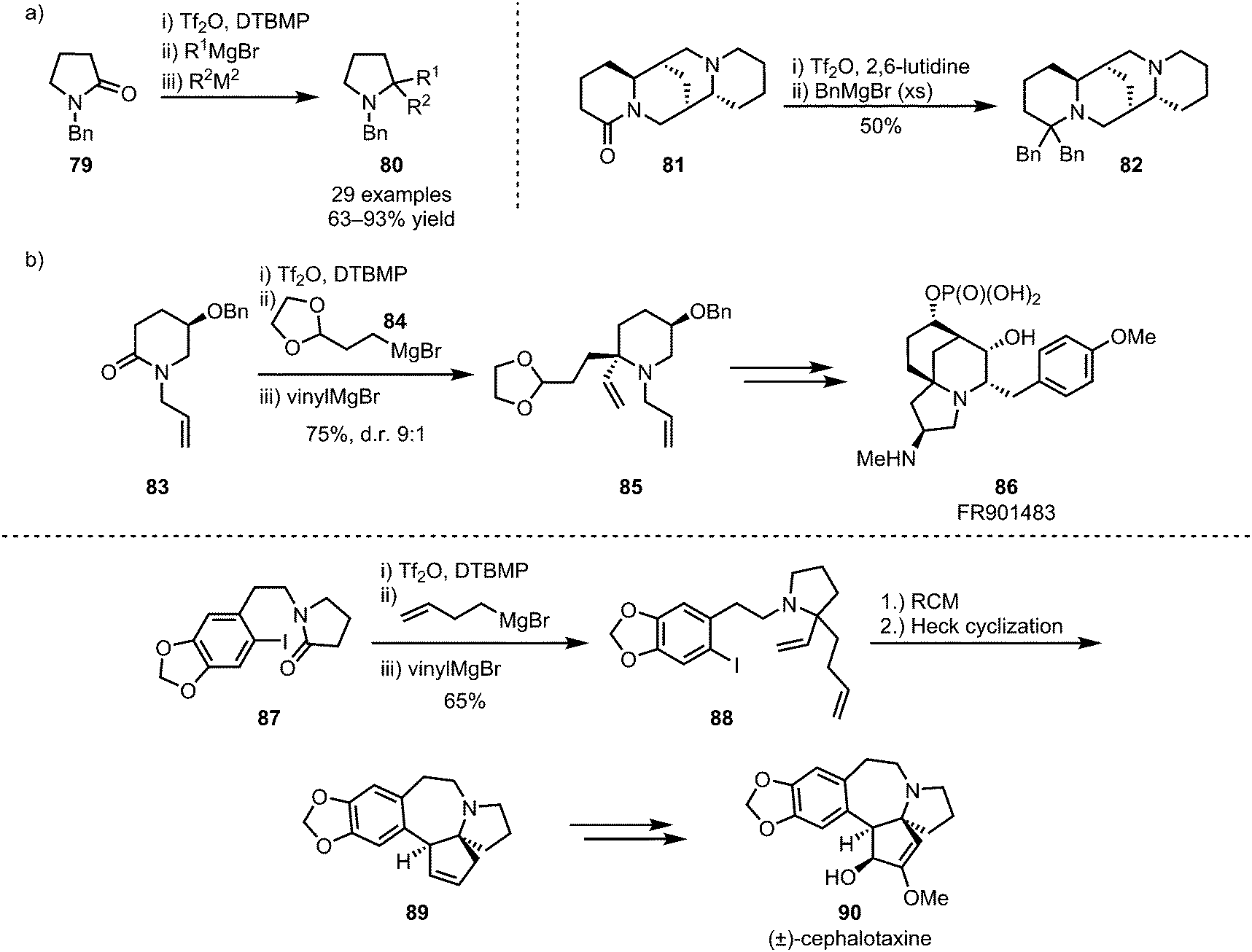 | ||
| Scheme 16 Sequential double addition of organometallic nucleophiles and its application in natural product synthesis. | ||
Intramolecular addition of carbon nucleophiles
Cyclic and polycyclic systems can be accessed by the intramolecular addition of tethered nucleophiles to activated amides. In this context, the activation of secondary amides of type 91, as disclosed by the group of Movassaghi, has been used to form substituted isoquinolines and β-carbolines (92) in a process reminiscent of the previously described intermolecular formation of heteroaromatics (Scheme 17a, cf.Scheme 13).62 Similarly, electron-rich aromatics have been used extensively as tethered nucleophiles in the development of new synthetic methodologies. In this regard, thiophenols,63 anisoles,64 and, most importantly, indoles65,66 have been widely employed (Scheme 17a). Indoles (such as 93, 95, 100 and 103), core structures of a large number of naturally occurring alkaloids, have moreover found extensive application in total synthesis (Scheme 17b–d).67–71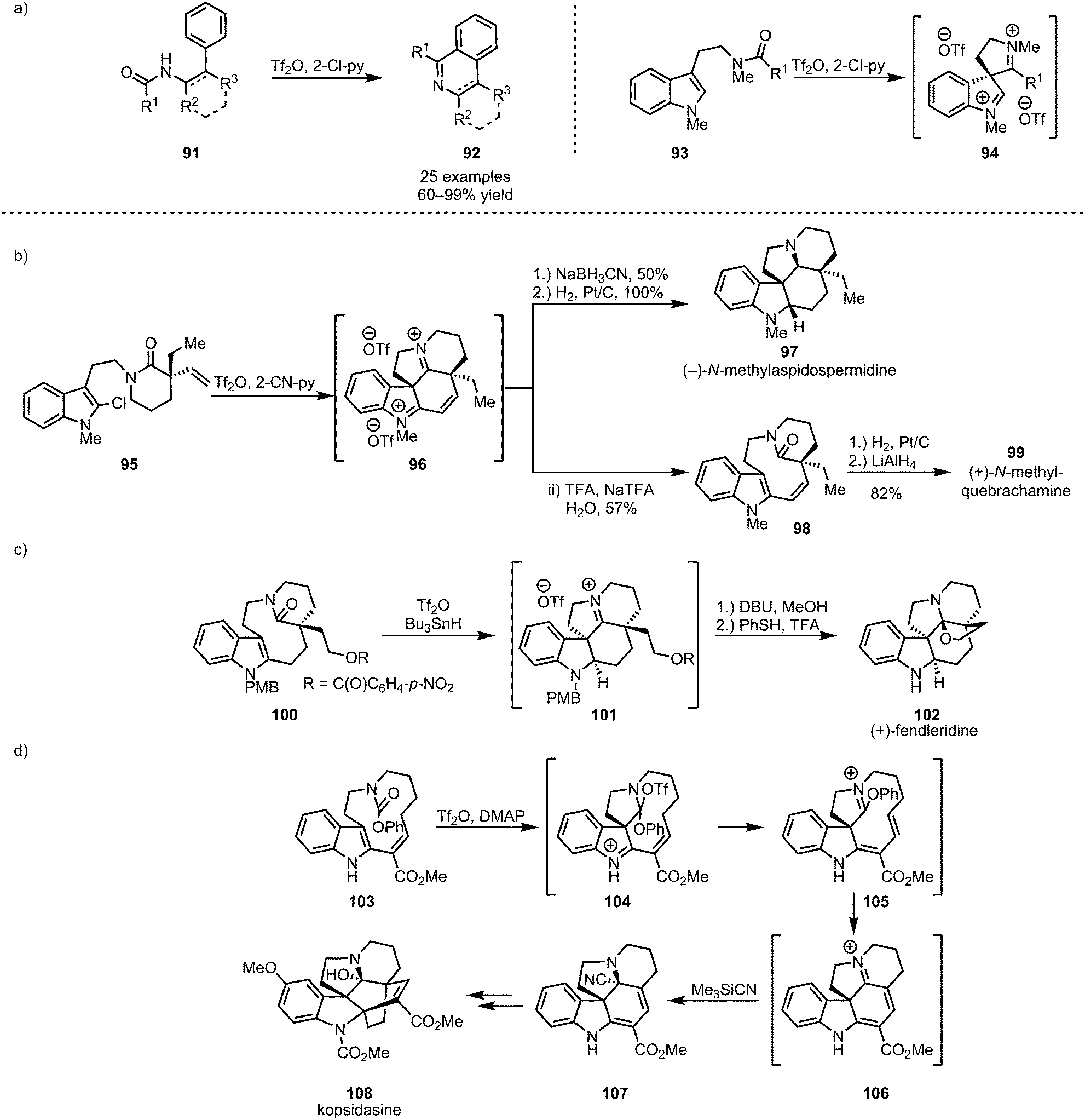 | ||
| Scheme 17 (a) Intramolecular trapping of activated amides with electron-rich aromatics and (b–d) the application of this approach to the synthesis of indoline-alkaloids. | ||
Nucleophilic alkenes and alkynes have also been shown to be suitable moieties for intramolecular trapping of activated amides, foremost in such cases where intramolecular [2+2]-cycloaddition is precluded for reasons of steric hindrance or ring-strain. Several efficient applications of silyl enol ethers (such as 109, 111 and 114)64,72 in synthetic methodologies and natural product synthesis73–75 have been reported in recent years, including the syntheses of tashiromine (113) and pervilleine B (116) (Scheme 18a). Noteworthy additions to this chemistry were made by the group of Bélanger, who employed the products of monoaddition of silyl enol ethers for 1,3-dipolar cycloadditions resulting in the synthesis of polycyclic natural product scaffolds (Scheme 18b).76,77 Huang and co-workers recently also disclosed the addition of enamines as alkene-carbanion equivalents for the reductive alkenylation of secondary amides, forming 1,3-diimino intermediates (121) and, as the products of reduction and elimination of an amino-moiety, allylic amines (121′) (Scheme 18c).78 An elegant use of a nucleophilic alkene (122) was additionally reported in 2002 by the group of Overman, triggering skeletal rearrangement to form [5,7]-bicyclic system 124 (Scheme 18d).79
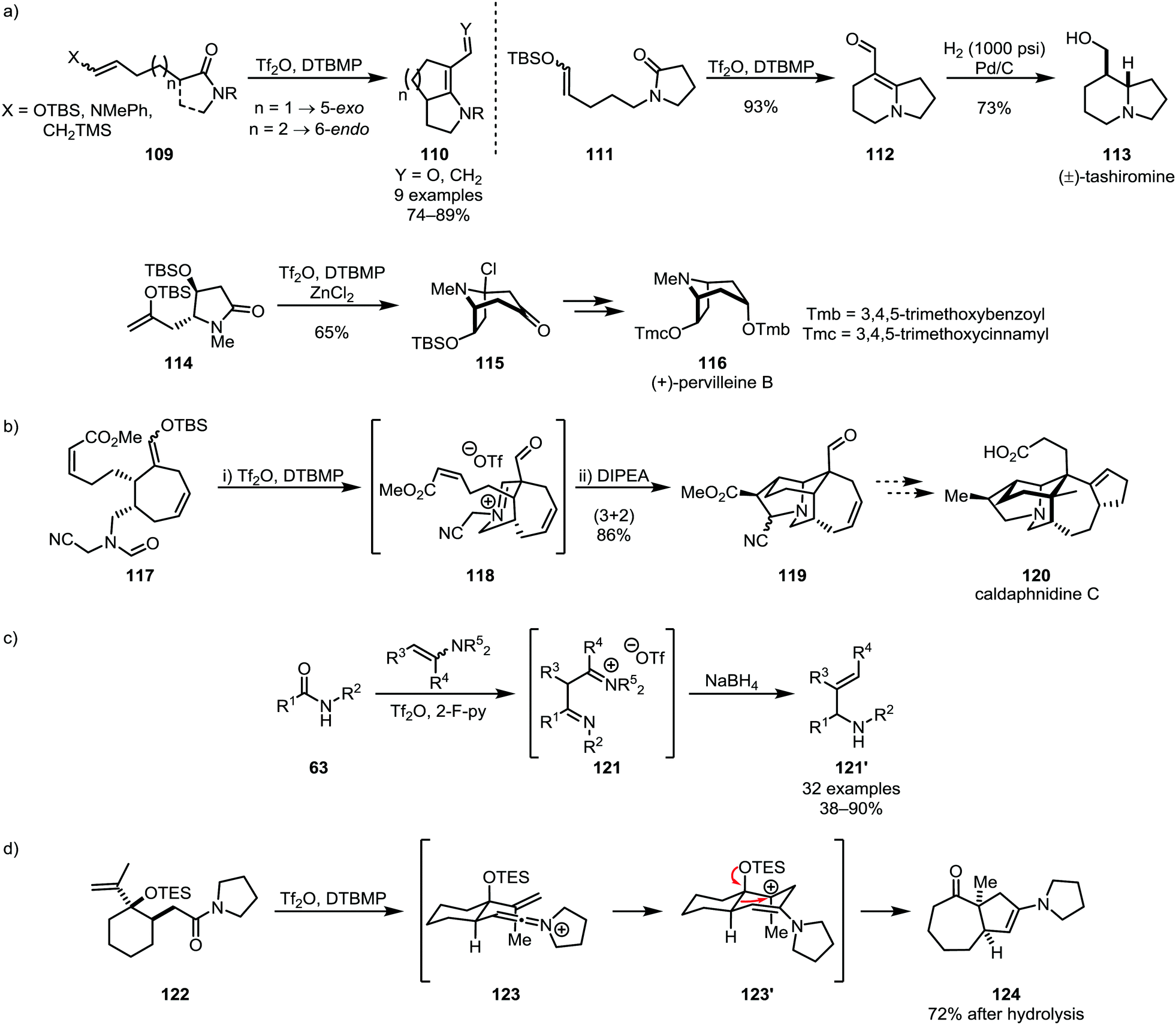 | ||
| Scheme 18 Silyl enol ethers, enamines and alkenes in the formation of natural product scaffolds and allylic amines through electrophilic amide activation. | ||
Reduction of activated amides
Although the reduction of activated thioamides is generally possible (vide infra), this approach has become virtually obsolete due to the advances in modern amide activation: in 2008 Charette and Barbe discovered that amides that were activated with triflic anhydride were smoothly converted into the corresponding amines (126) by the action of Hantzsch ester (Scheme 19, left).80 Later, the method was rendered more general, enabling the conversions of secondary amides (127) into imines (128), aldehydes (129) or amines (130), depending on the reaction conditions (Scheme 19, right).81 In the same year it was found that sodium borohydride can also be used as a reducing agent.82 This also constituted an improvement compared to previous reports, because enolisable amides were, for the first time, generally viable substrates. A modification of Charette's reduction was reported by the group of Huang,83 and the reduction of sterically hindered diisopropylamides—which cannot be successfully reduced with previously reported protocols—was recently further investigated by Song and co-workers.84 While these recent advances have not yet found applicability among the chemical industry,85 they feature in several total syntheses of natural products.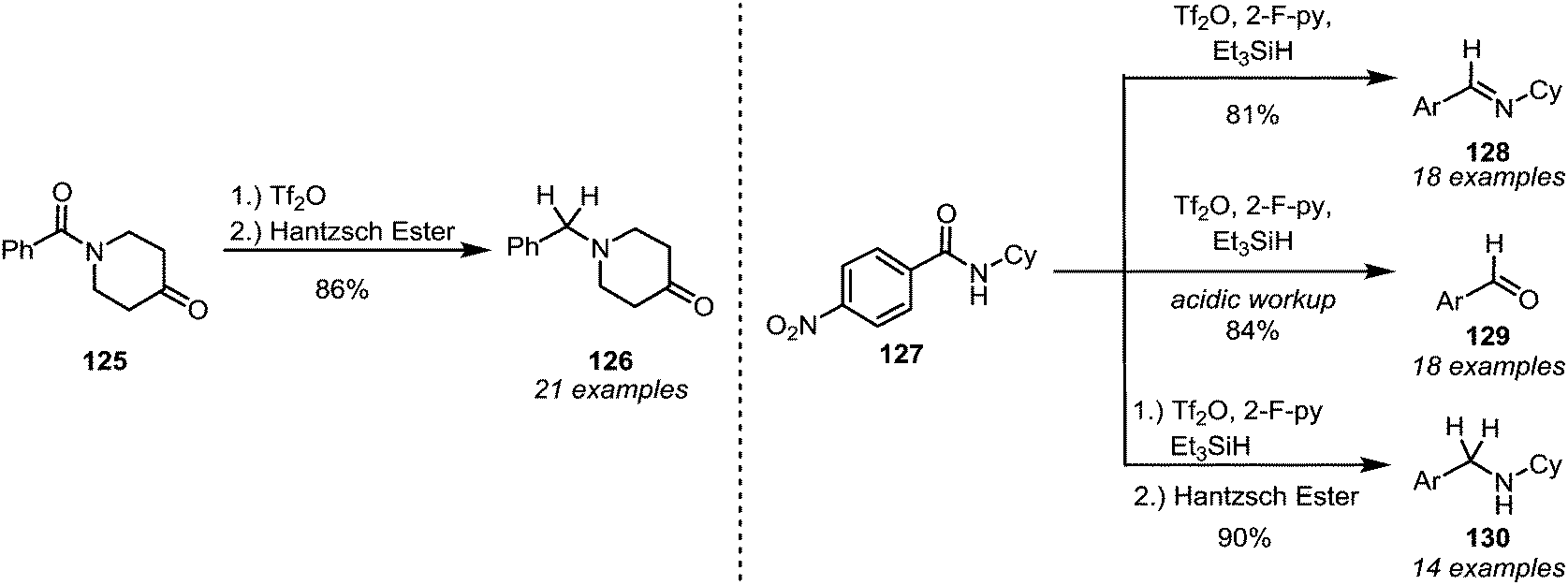 | ||
| Scheme 19 Mild and chemoselective reduction protocols for amides, based on prior electrophilic activation. | ||
In a late stage of the total synthesis of (−)-nakadomarin A (137) by Evans and co-workers, conventional methods employed for the reduction of amides (such as the use of DIBAL-H or LiAlH4) did not lead to a productive transformation of 137, as overreduction was observed for the five-membered ring lactam (Scheme 20).85–88
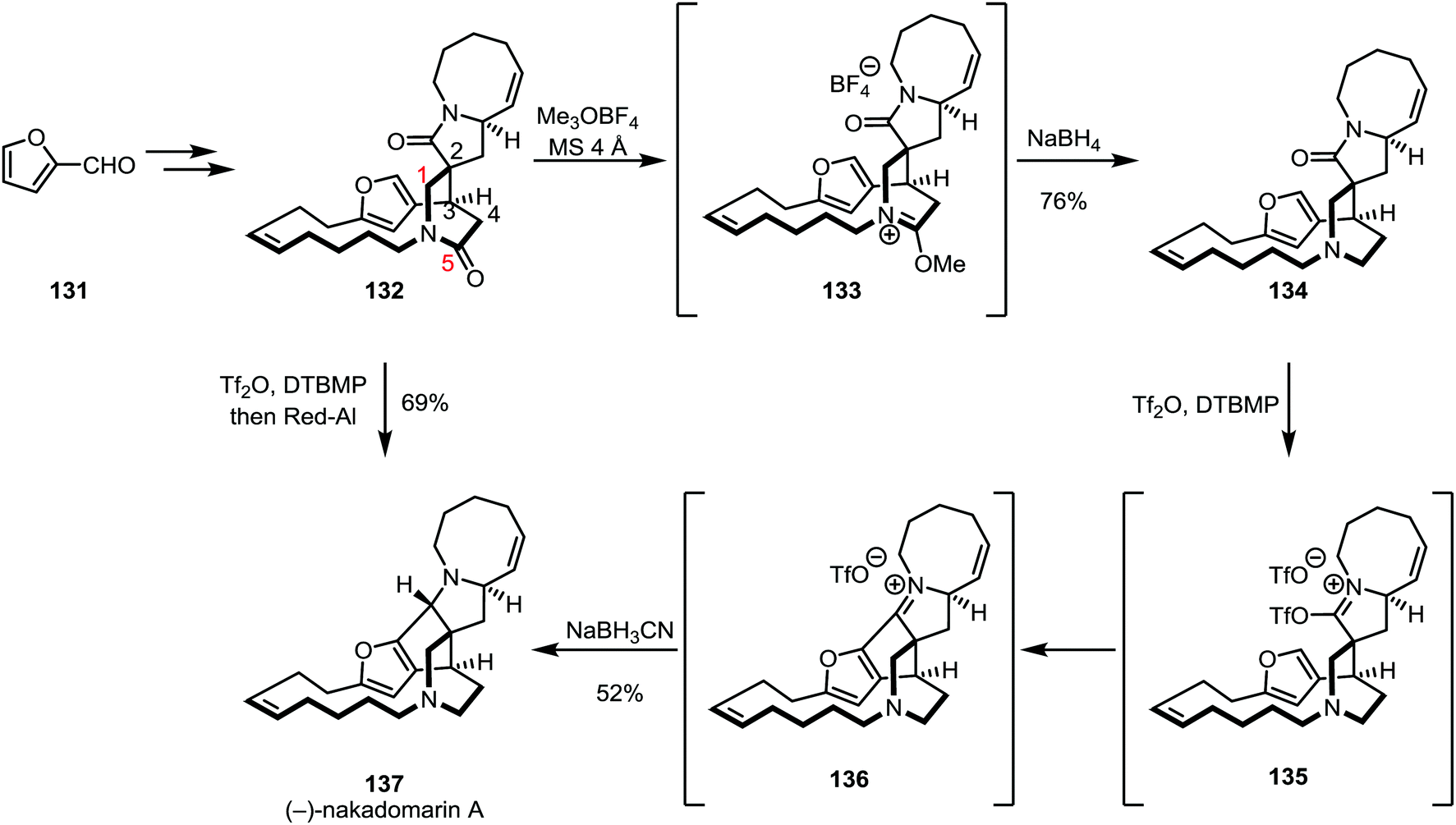 | ||
| Scheme 20 Evans’ triflic anhydride-based syntheses of (−)-nakadomarin A. | ||
Regioselective alkylation of the six-membered lactam oxygen was achieved using trimethyloxonium trifluoroborate and subsequent reduction with sodium borohydride afforded amine 134 in 76% yield. The authors observed a decrease in selectivity when using the slightly more hindered Meerwein's salt (Et3OBF4), suggesting that steric effects might play an important role in regioselectivity. The five-membered lactam was subsequently activated with triflic anhydride and a bulky pyridine base, triggering an intramolecular Friedel–Crafts cyclisation step. The resulting intermediate 136 was diastereoselectively reduced with sodium cyanoborohydride to yield (−)-nakadomarin in 52% yield. Later, this sequence was refined to a one pot procedure, using triflic anhydride followed by sodium(bis(2-methoxyethoxy))aluminium hydride (Red-Al) to afford the natural product in only one step from 132 and in 69% yield.
Zhang and co-workers encountered a similarly problematic amide reduction during their total synthesis of (±)-maistemonine (141) (Scheme 21).89 Several conditions were unsuccessfully tried to convert amide 139 to amine 140: BH3·Me2S, 9-BBN, Tf2O/Hantzsch ester, RhH(CO)(PPh3)3/Ph2SiH2, Et3OBF4/NaBH4/2,6-di-tert-butylpyridine and reduction of the corresponding thioamide. Eventually, the use of methyl triflate and sodium cyanoborohydride was found to give the desired product in 50% yield, with large amounts of the recovered starting material.
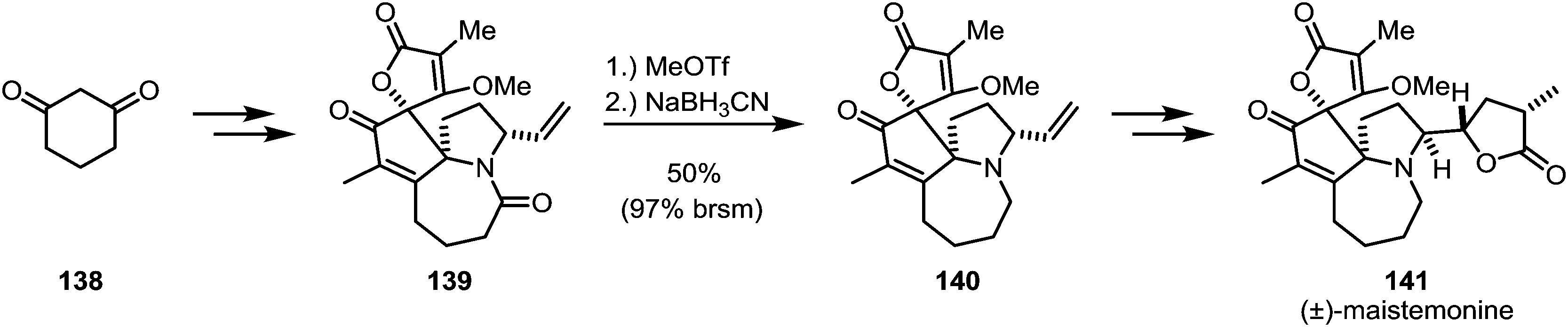 | ||
| Scheme 21 Zhang's synthesis of (±)-maistemonine, employing methyl triflate as the chemoselective activator. | ||
Intermolecular addition of heteroatom nucleophiles
Several heteroatom nucleophiles can also be used to intercept electrophilically activated amides. Apart from functional group interconversion and the formation of heterocycles, heteroatom nucleophiles have also been employed to mediate C–C- and (on exception) C–N-bond-forming rearrangements. In the context of secondary amides, early work dates back to 1969, when Abramovitch and Singer reported the N-pyridinylation of imidoyl chlorides with pyridine N-oxide90—a method that was optimised, generalised and mechanistically elucidated by Movassaghi et al. some 40 years later (Scheme 22).91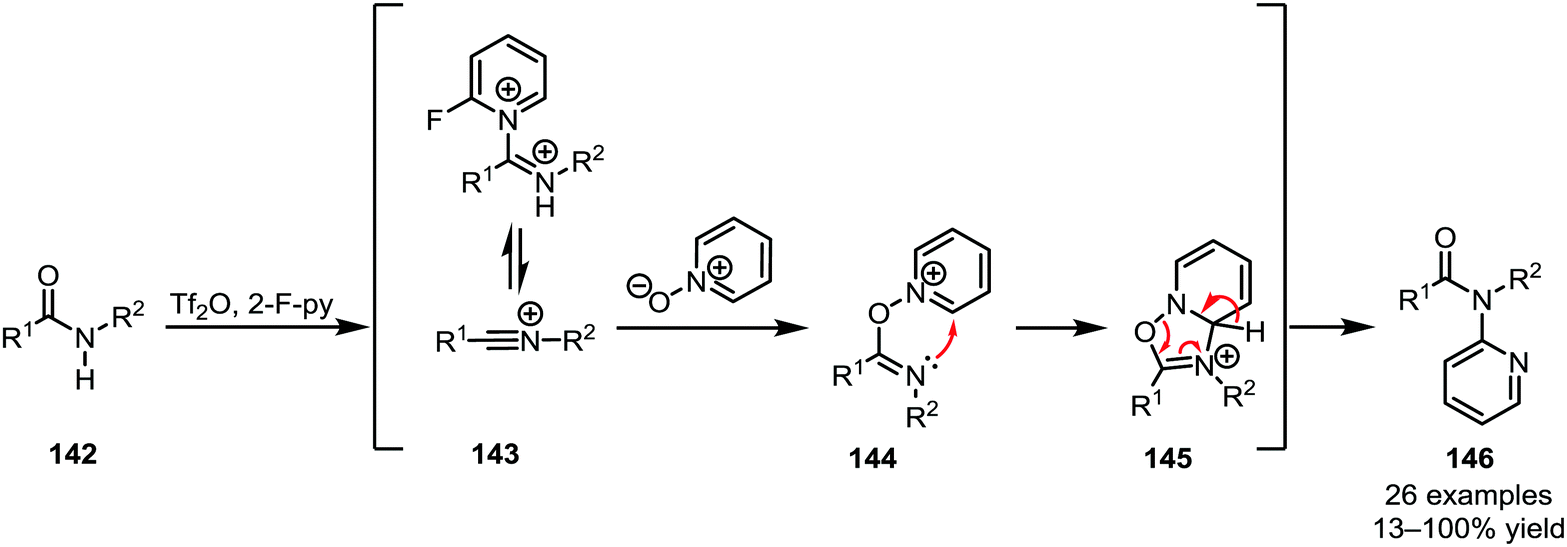 | ||
| Scheme 22 N-Pyridinylation of secondary amides with N-oxides. | ||
The products of heteroatom addition to keteniminium ions, taking the form of functionalised or substituted enol ethers, can be readily employed for [3,3]-sigmatropic rearrangements. An early example was reported by Welch in 1991, relying on methyl triflate for the activation of amide 147 and an alkoxide as the nucleophile (Scheme 23a).92 The allyl vinyl ether formed upon alkoxide addition enables the regeneration of the amide carbonyl and concomitant α-allylation via [3,3]-sigmatropic rearrangement to form 149. The chemoselectivity of triflic anhydride was exploited in a related approach reported by Maulide et al. in 2010 (Scheme 23b).26 Herein, a tethered allyl ether (150) served as the nucleophilic trapping agent, furnishing an intermediate allyl vinyl oxonium species (151) ideally poised for charge-accelerated [3,3]-sigmatropic shift. A series of ensuing studies was able to generalise this transformation, showing that a wide array of α-allylated (153) and -allenylated (155) carboxylate derivatives can be formed using this approach.93–95 The omission of a pyridine base in the reaction mixture prompted the discovery of a facile (macro)lactonisation procedure that was employed for the total synthesis of natural product 160 (Scheme 23c).96
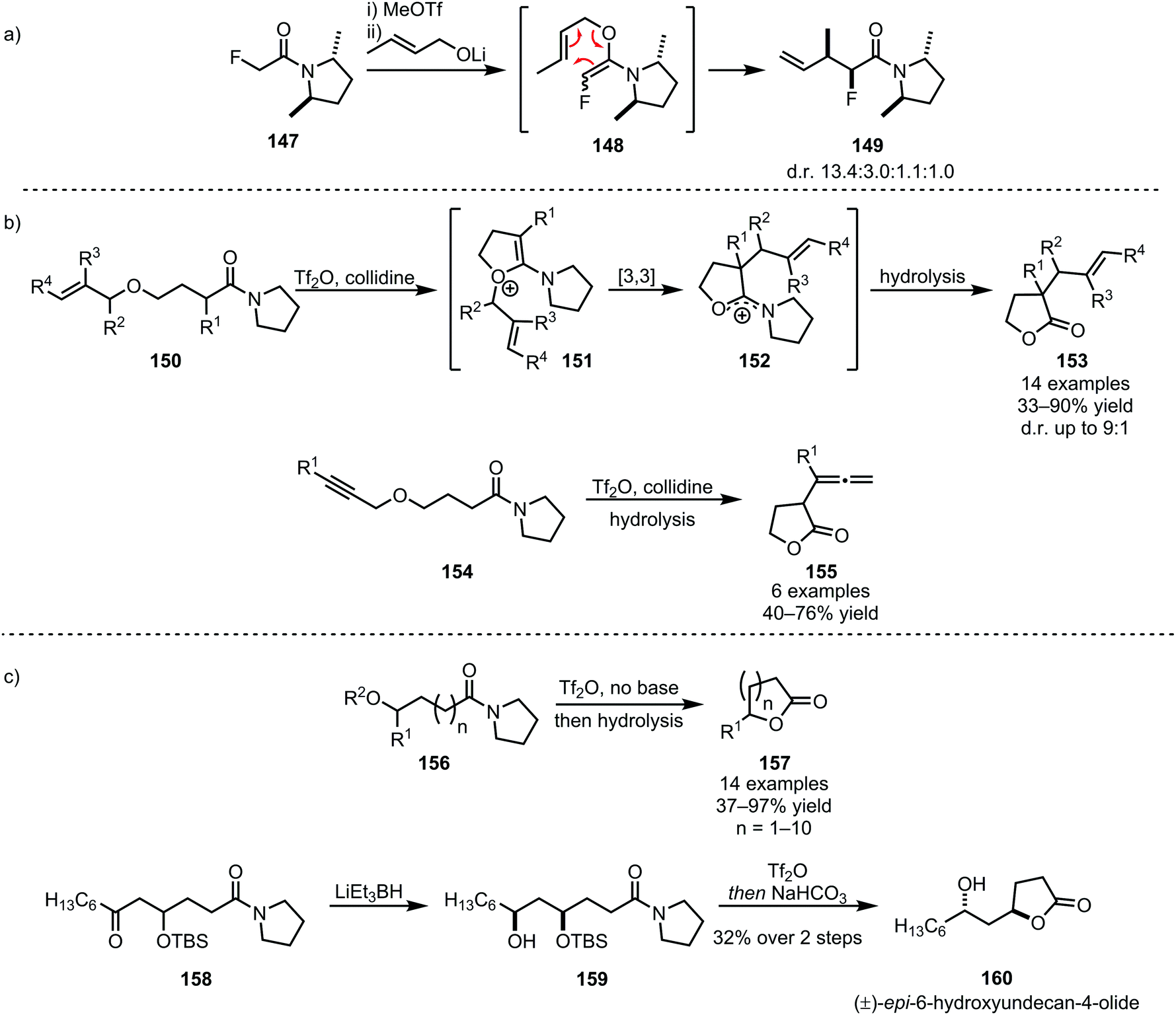 | ||
| Scheme 23 (a and b) [3,3]-Sigmatropic rearrangements in C–C-bond-forming reactions of amides; and (c) amide activation for chemoselective lactonisation of amides. | ||
A subsequent report detailing the enantioselective synthesis of α-allylated carboxylic acids (165) and aldehydes (166) using a chiral auxiliary is particularly noteworthy, as it allows the chemo- and stereoselective α-functionalisation of amide-substrates in the presence of a wide range of sensitive functional groups (Scheme 24a).97 Taking advantage of the same principle of nucleophilic addition to keteniminium ions followed by [3,3]-sigmatropic rearrangement, the Maulide group also reported the α-arylation of tertiary amides with both aryl sulfoxides and aryl hydroxamic acids, forming structures of types 170a and 170b, respectively (Scheme 24b).98,99
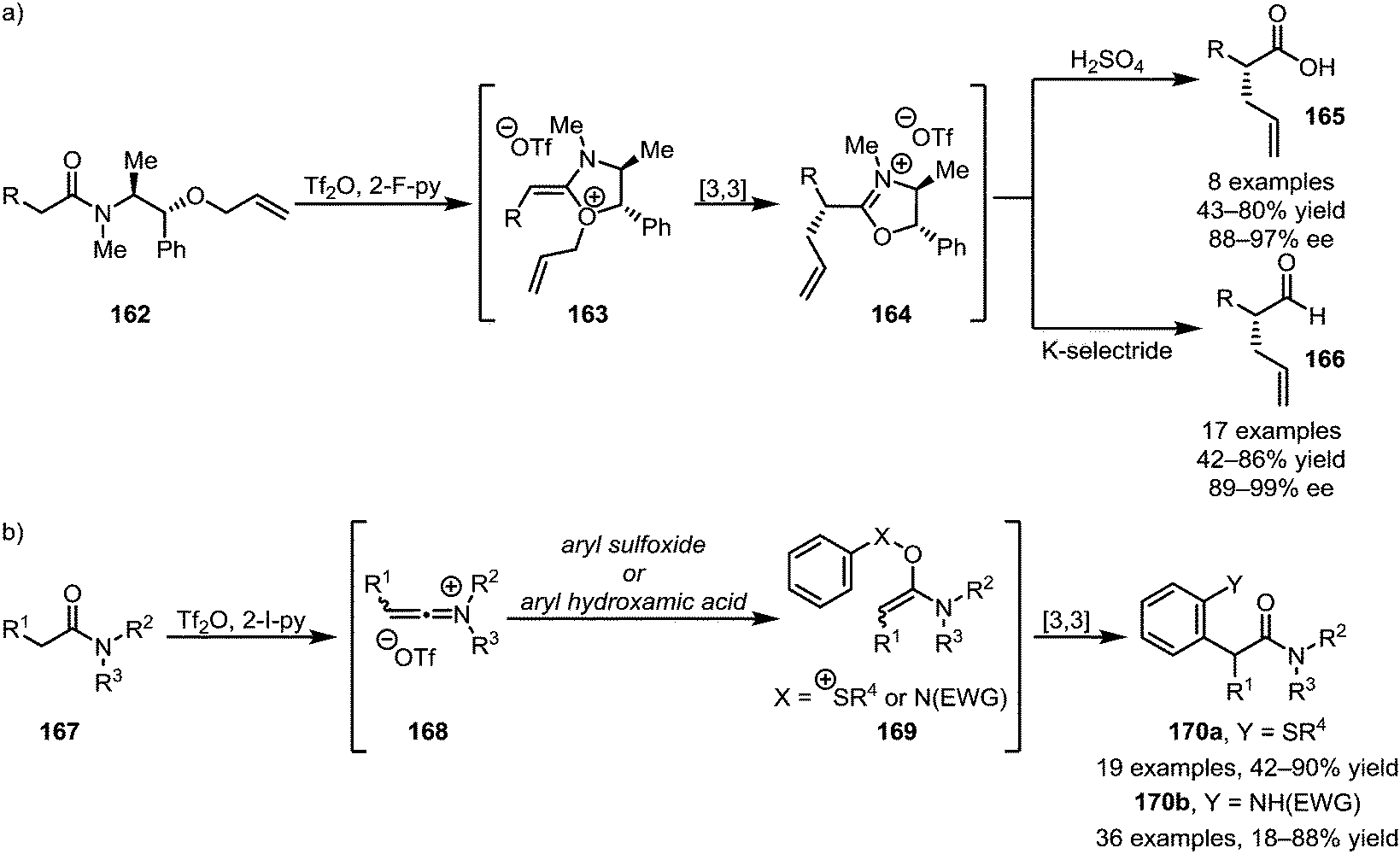 | ||
| Scheme 24 (a) Enantioselective α-allylation and (b) the use of aryl sulfoxides and aryl hydroxamic acids for the α-arylation of tertiary amides. | ||
In addition to C–C bond formation mediated by the initial heteroatom addition to activated amides, the direct functional group interconversion and heterocycle formation using heteroatom nucleophiles has also been widely explored (Scheme 25a). In this regard, the addition of ammonium sulfide to keteniminium ions readily affords the corresponding thioamides (172).100 Similarly, 1,2-aminothiols have been employed in the formation of thioxazolines (173)101 and triols enabled the formation of bridged orthoesters (174).102 In 2015, the group of Charette reported the use of hydrazides for the formation of 1,2,4-triazoles (175) and 3-aminoindazoles (176) from activated secondary and tertiary amides, respectively.103,104 In analogy to the previously mentioned pyridine and isoquinoline syntheses, Movassaghi et al. employed nitriles for the formation of pyrimidines (180) (Scheme 25b).105,106
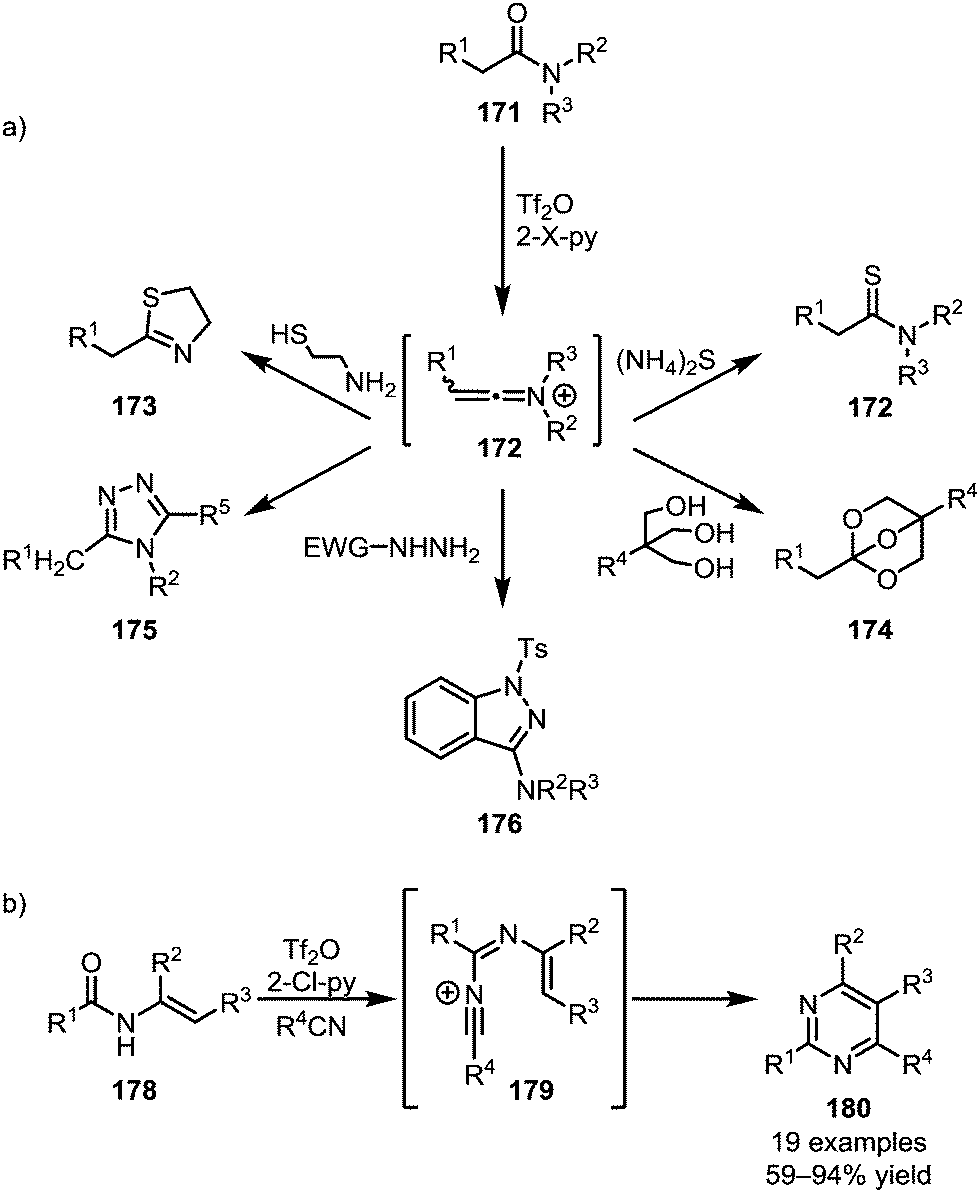 | ||
| Scheme 25 (a) Functionalisation of activated amides with heteroatom nucleophiles; and (b) Movassaghi's pyrimidine synthesis with nitriles. | ||
The addition of pyridine, known to form stable adducts with activated amides, has been utilised for the elegant synthesis of natural products (Scheme 26).107,108 Herein, the formation of a cationic pyridinium species (182) enables the nucleophilic addition of organometallic reagents to the pyridine's 2-position, forming dihydropyridines (183) (Scheme 26a). This approach has been applied to the syntheses of the polycyclic alkaloid tetraponerine T4 (186) (Scheme 26b) and, using a chiral auxiliary, (R)-(−)-coniine (189) in a stereoselective fashion (Scheme 26c).
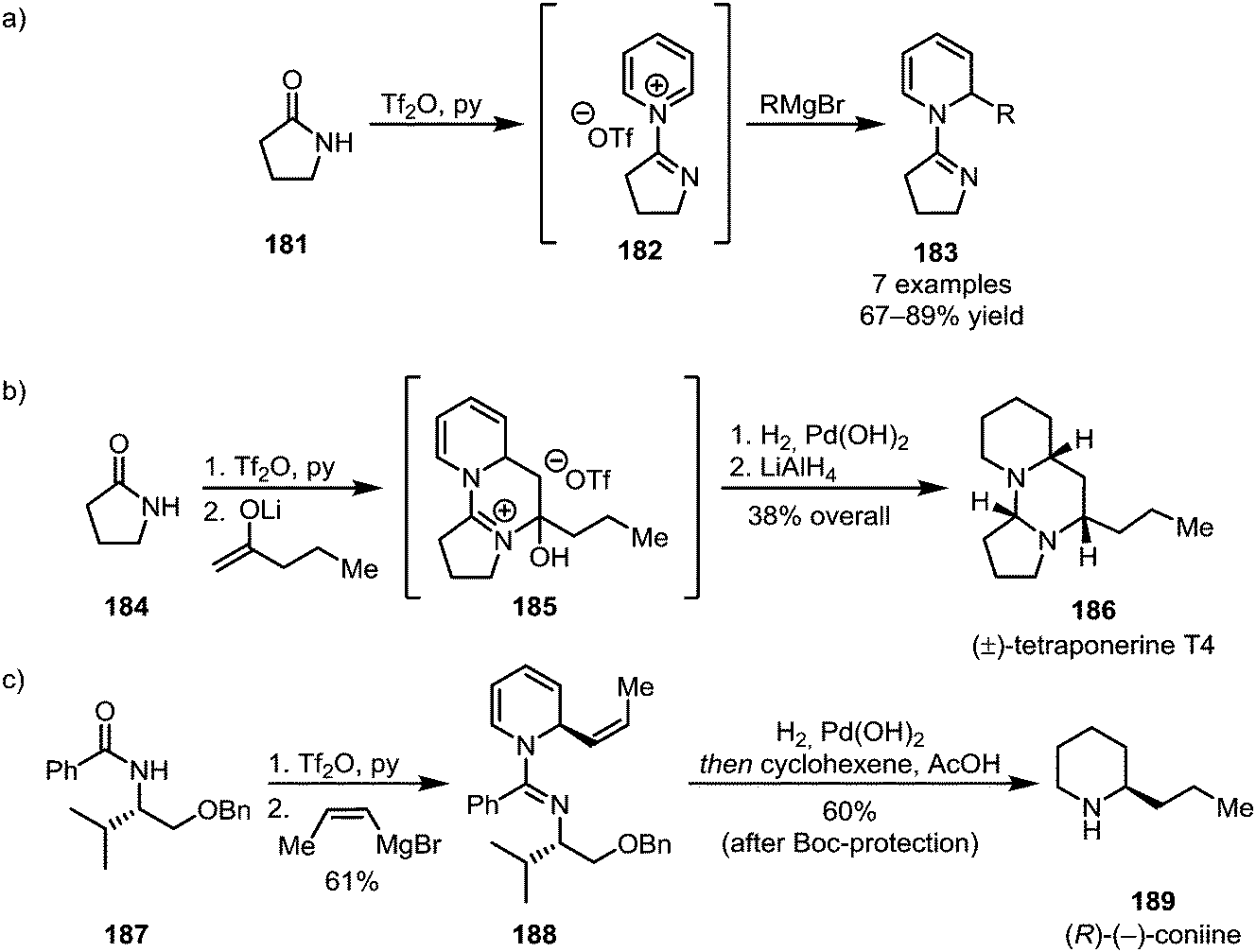 | ||
| Scheme 26 (a) Trapping of pyridinium adducts with organometallic reagents and (b and c) the use of this approach for natural product synthesis. | ||
Azides represent a special class of nucleophiles. Divergent reactivity of these reagents has been observed, depending on the nature of the activated amide employed. Secondary amides (190) have been engaged in the formation of both uncharged tetrazoles (191)109 and tetrazolium salts (192) (Scheme 27a).110 In contrast, an early report by Ghosez disclosed the formation of 2-amino-1-azirines (195) from tertiary amides (193) with sodium azide,111 and replacement of sodium azide with various alkyl azides was recently shown by the Maulide group to lead to a significant change in reactivity (Scheme 27b).112 In the event, a 2-amino-1-azirinium ion (197) is generated, the positive charge greatly facilitating aqueous hydrolysis and thereby leading to the formation of α-aminated amides (198).
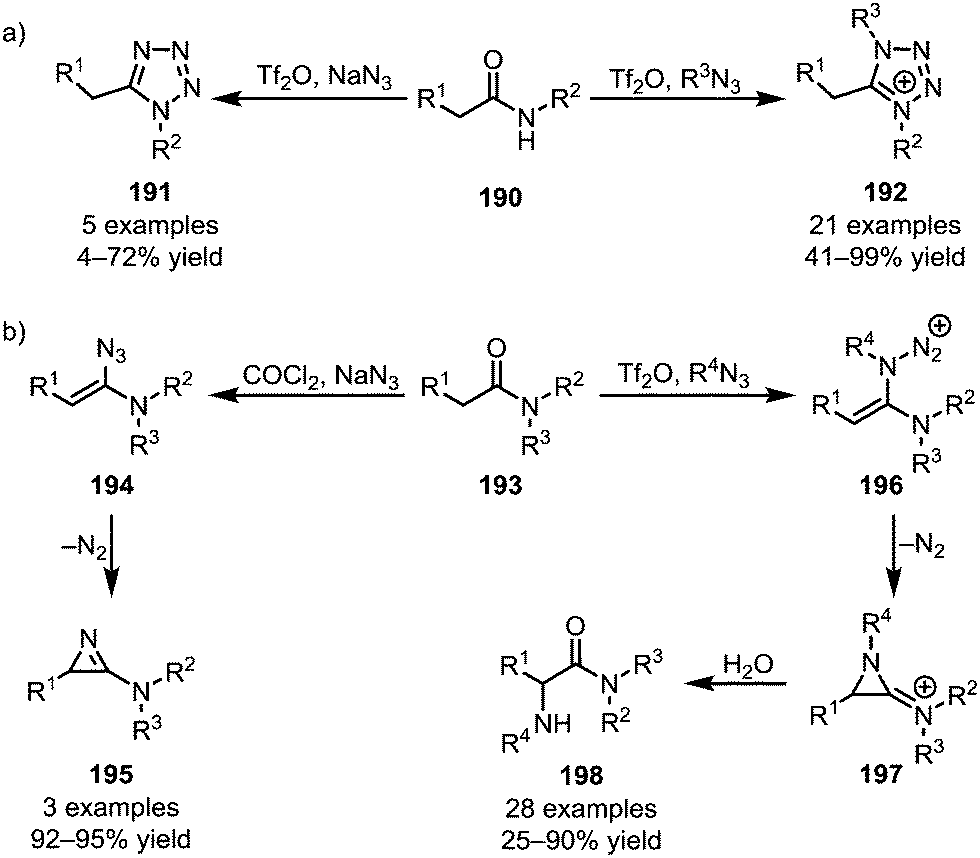 | ||
| Scheme 27 Applications of azides in the functionalisation of activated (a) secondary and (b) tertiary amides. | ||
Recently, the facile fragmentation of the N–O bond has led to a revival of Umpolung-chemistry in amide activation. In 1979, an isolated report on the formation of α-chlorinated amides (201) through Umpolung (via200), embedded in Ghosez's report on the α,β-desaturation of α-branched amides, was disclosed (Scheme 28a).113 Effectively reversing the natural polarity of the α-position to allow nucleophilic addition, this reactivity has gained increased interest in recent years, enabling novel disconnections for the α-functionalisation of amides. In this respect, and using triflic anhydride-mediated amide activation (thereby obviating the presence of nucleophilic counteranions), the Maulide group has reported intra- and intermolecular nucleophile addition for C–C bond formation at the α-position of amides, enabling the formation of lactams (204) and 1,4-dicarbonyls (205) (Scheme 28b).114,115 A related elegant example was recently disclosed by the Miyata group, employing isoxazolidine amides (206) in combination with organoaluminium nucleophiles for the α-arylation of amides (Scheme 28c).116 Moreover, chemoselective amide α-oxidation with N-oxides (employed for the transformation of linear amides) and TEMPO (preferred for branched and hindered amides) has been recently reported, allowing facile access to α-keto and α-hydroxy amides (210–213) (Scheme 28d).117
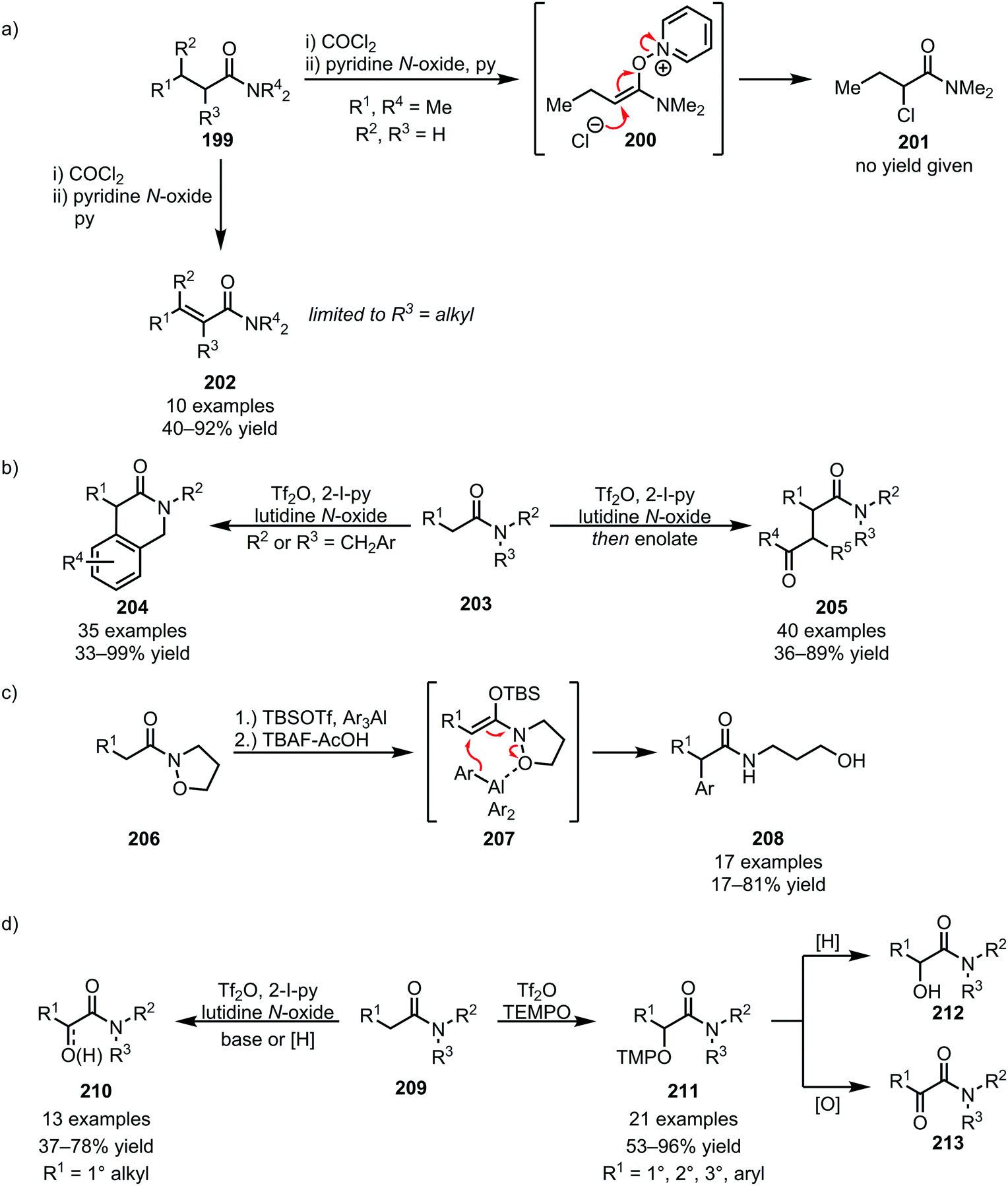 | ||
| Scheme 28 Umpolung-functionalisation of amides. | ||
Thioamide activation
The activation of thioamides (214) is less explored than the activation of amides. Nevertheless, interesting transformations have been observed for this chemistry (Scheme 29). Most commonly, thioamides are activated by electrophiles which are commonly perceived as “soft”, such as alkyl bromides or alkyl iodides.118 Although the validity of the HSAB principle has been recently questioned,119 such electrophiles are classically believed to have a high affinity towards the sulfur atom due to their similarities in HOMO/LUMO-properties and electrostatics.120–124 Alternatively, electrophiles classically perceived as “hard” (such as methyl triflate,125 or Meerwein's salt126,127) can also be employed, further supporting Mayr's criticism of the HSAB-principle.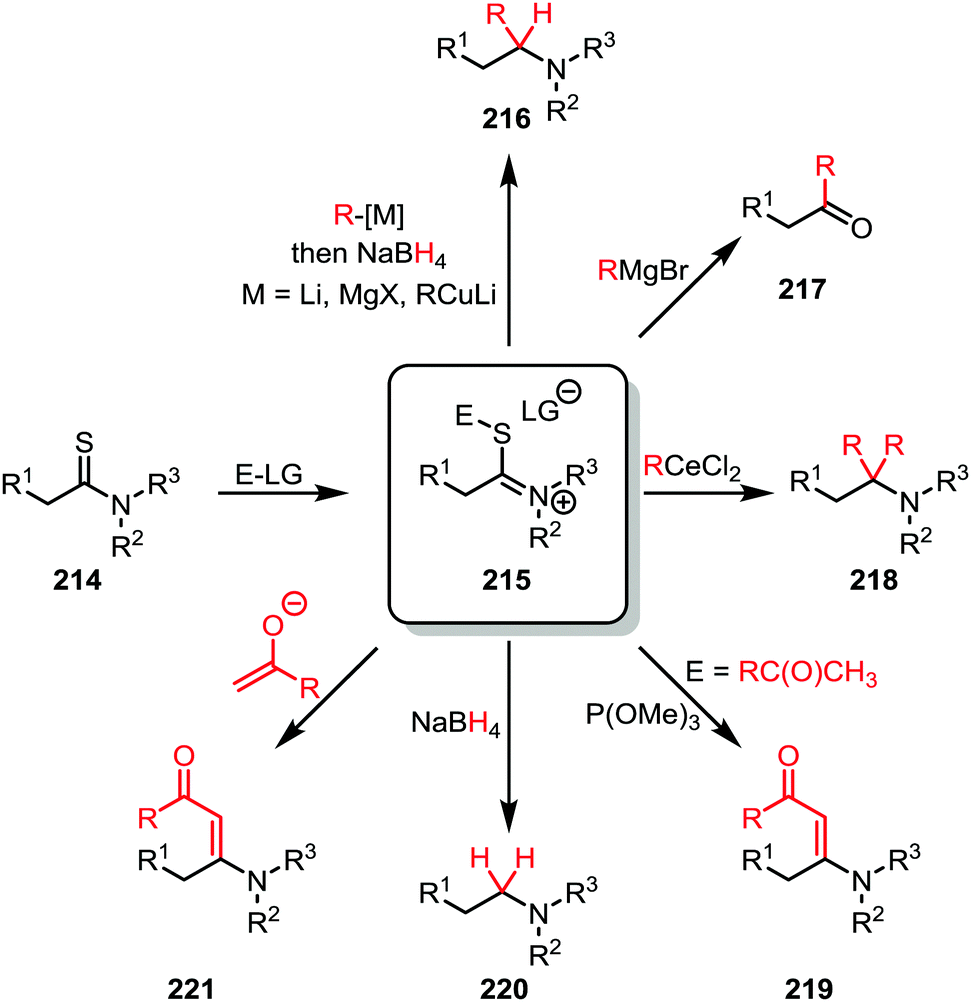 | ||
| Scheme 29 Functionalisation of activated thioamides. | ||
The iminium sulfide salt (215), generated upon alkylation of a thioamide (214), can be engaged by several carbon nucleophiles such as organocerium species,128 enolates,126 cuprates,120,121,123 or Grignard reagents121,122 (Scheme 29). Often a further reduction using a hydride source is employed following the first nucleophilic attack to provide an α-monosubstituted amine (216).
In 1989, the first enantioselective synthesis of peduncularine (225b) was reported by Speckamp and Hiemstra (Scheme 30).124 In the final stages of the synthesis, thioamide 222 was activated with methyl iodide to afford iminium sulfide 223. This species was isolated and subsequently treated with a Grignard reagent—quenching of the resulting intermediate with sodium borohydride provided the two epimeric amines 224a and 224b, which were readily converted to the natural product (and its epimer) by Fischer-indole synthesis. Incidentally, the authors were also able to show that epimer 225a, previously believed to correspond to isopeduncularine, does not in fact match the constitution of that natural product.
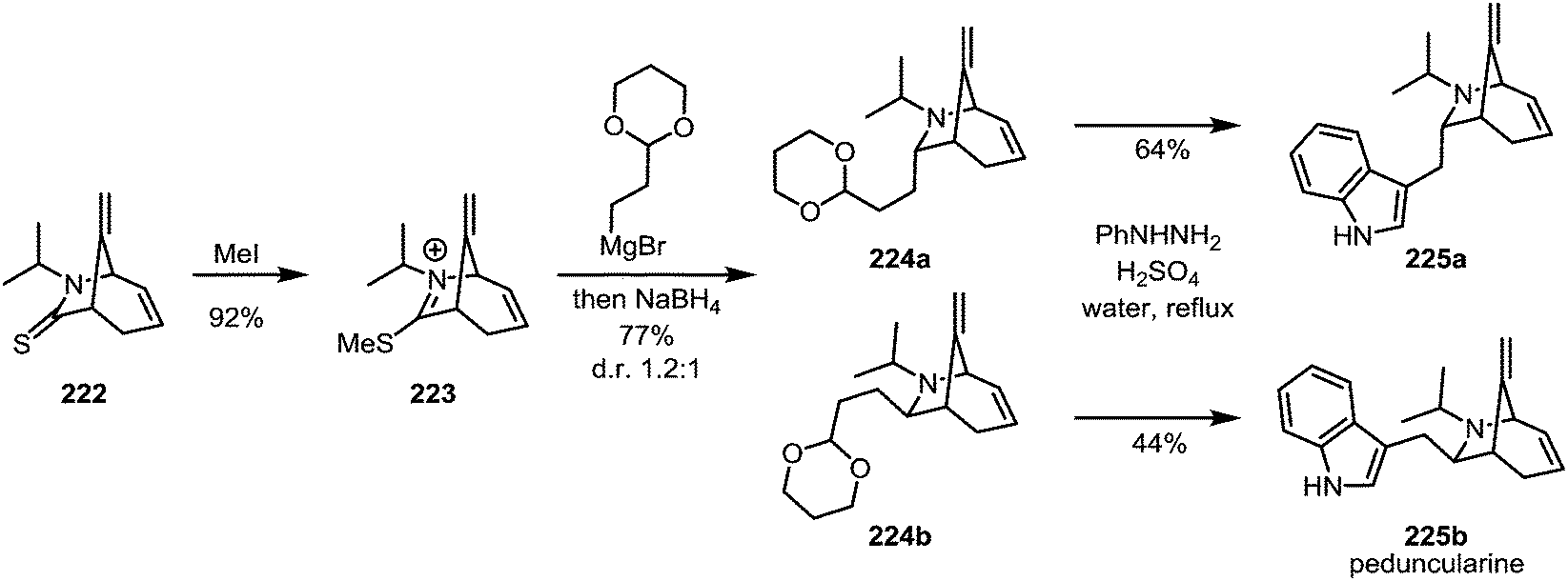 | ||
| Scheme 30 Speckamp and Hiemstra's synthesis of peduncularine, following the monoalkylation of an activated thioamide. | ||
While organocerium addition to activated thioamides usually leads to double alkylation, the addition of a Grignard reagent without further reductive workup can be used for the synthesis of ketones. For example, in the total synthesis of leucinostatin A (230) by Shibasaki and co-workers, methyl triflate was used for the activation of diallylthioamide 228, capture of which by ethylmagnesium bromide and subsequent hydrolysis afforded the desired ketone 229 (Scheme 31).125 Incorporation of the resulting building block (a protected amino acid) into a peptide framework led to the formation of the desired natural product.
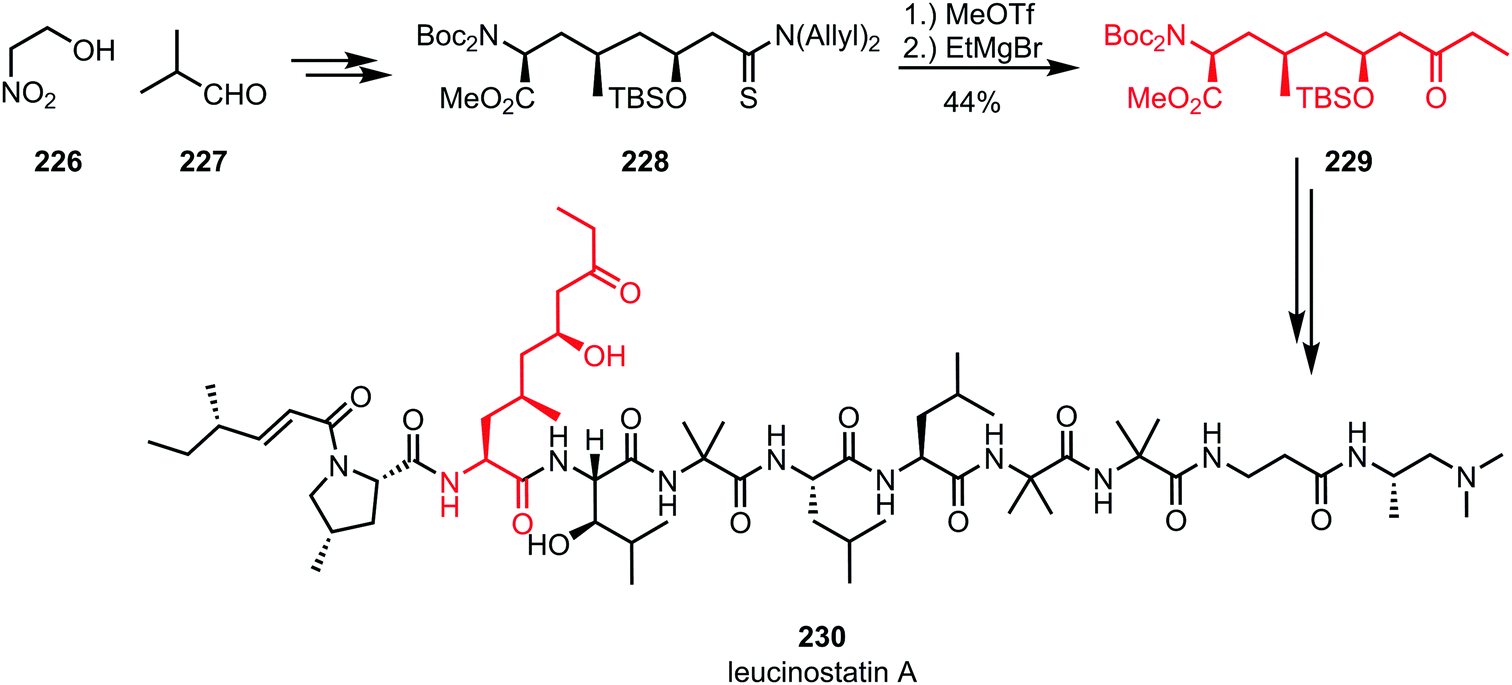 | ||
| Scheme 31 Abe's synthesis of the polypeptide leucinostatin A employs the chemoselective activation of a thioamide. | ||
In some cases the iminium sulfide salt surpasses the activated amide in its reactivity: in his total synthesis of (±)-methyl homodaphniphyllate (235), Heathcock faced ongoing difficulties to achieve cyclisation of 231 through an intramolecular enolate attack on the activated amide. Several approaches were unsuccessfully tried, until ultimately it was thioamide 233 which, upon electrophilic activation, allowed for the smooth formation of the cyclised product 234via enamine-addition in 80% yield (Scheme 32).126
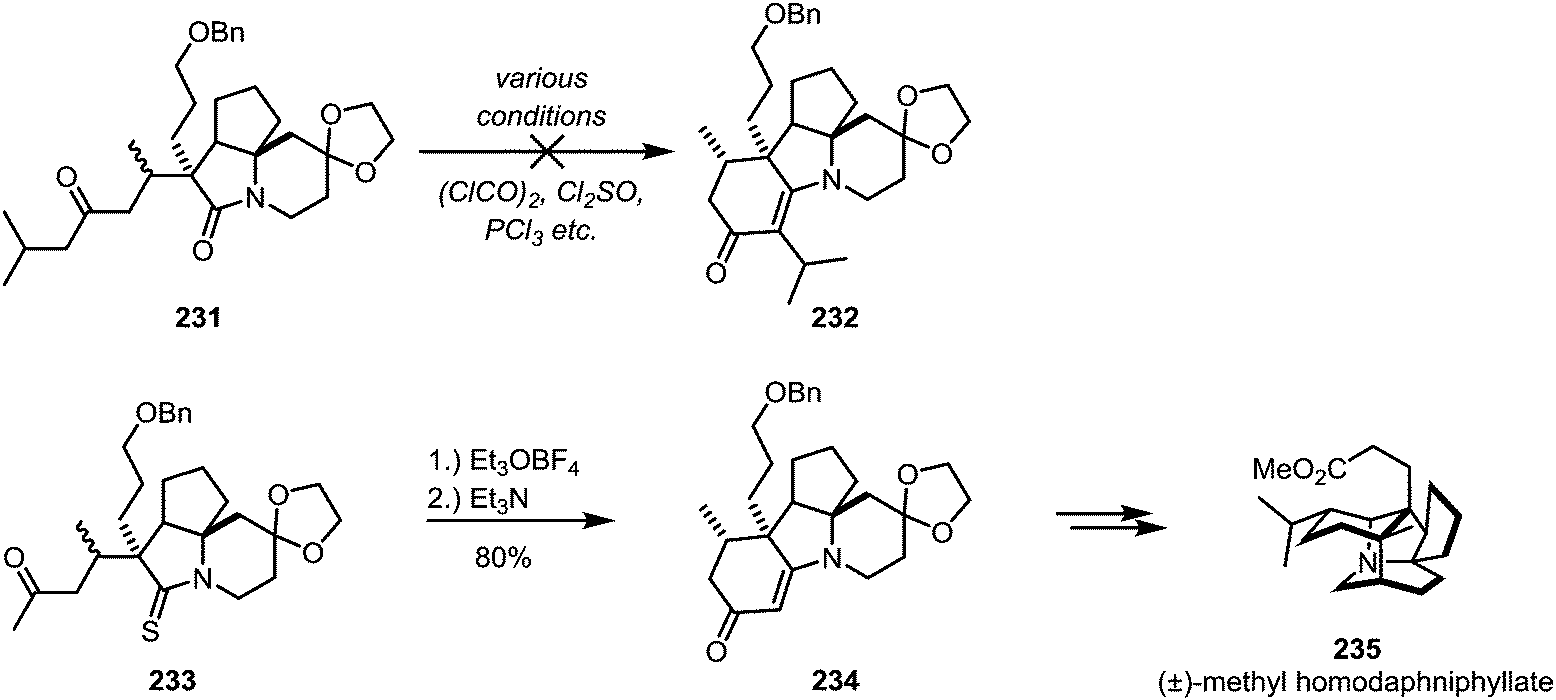 | ||
| Scheme 32 Activation of a thioamide with Meerwein's salt, as applied in Heathcock's synthesis of (±)-methyl homodaphniphyllate. | ||
A highly sophisticated variant of thioamide functionalisation is the so-called “sulfide contraction” developed by Eschenmoser and co-workers, where the activating agent also contains the eventual nucleophile (Scheme 33). Activation is accomplished using an α-bromoketone in combination with a base. The resulting thioimidate (237) is further treated with a phosphine under mild heating, during which deprotonation of the ketone α-position leads to the formation of a thiirane (239), which subsequently rearranges to form the imine-thiolate 240. Trapping of this intermediate with the phosphine then leads to the formation of an olefin (243) through a mechanism reminiscent of the Wittig olefination.118
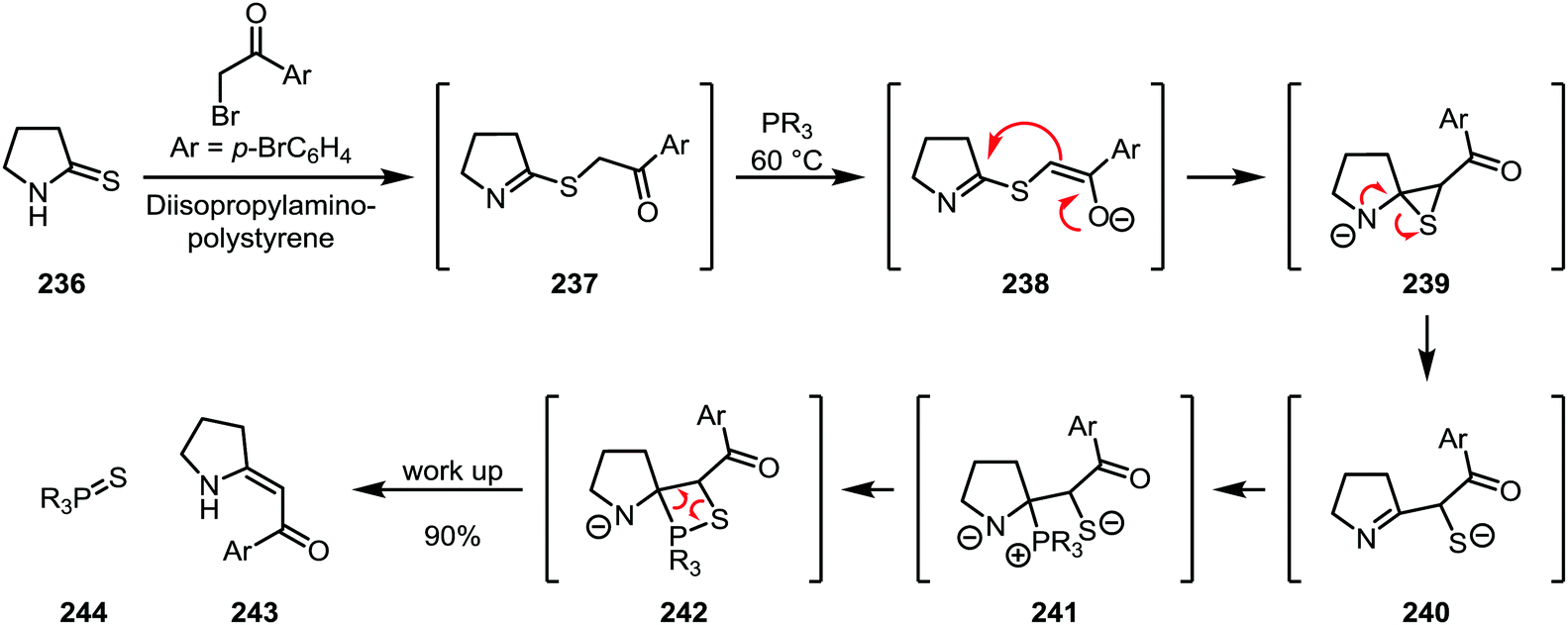 | ||
| Scheme 33 Eschenmoser sulfide contraction of thioamides. | ||
In addition to the aforementioned methods, activation of a thioamide can also be exploited for the clean reduction to the amine: in 1980, Raucher and Klein demonstrated that thioamide activation with triethyloxonium tetrafluoroborate (Meerwein's salt) can be followed by sodium borohydride-mediated reduction to readily afford the corresponding amine product (cf.Scheme 29, 220).127
Other activated amides and other activators
While the majority of approaches previously described capitalises on the nucleophilicity of the amide functionality, a further type of amide activation can be exploited when electron density is delocalised by incorporation of a second carbonyl moiety. Imides, possessing two carbonyls bound to nitrogen, are special cases of activated amides that show unique properties. In the following section, we aim to provide a brief introduction of selected imide-derivatisations against the background of functionalisation of suitable amides.SmI2-mediated functionalisation
The single-electron reductant samarium(II) iodide is known to readily afford ketyl radicals from various carbonyl and carboxyl derivatives. In 2005, Skrydstrup et al. published a method to transform N-acyl oxazolidinones into 1,4-dicarbonyls.129 In this report, SmI2 was used to selectively activate imide 245, forming radical anion 246. This intermediate can subsequently react with a range of Michael acceptors such as acrylic amides, esters or nitriles to form the desired 1,4-dicarbonyl compounds 247 (Scheme 34a). The reduction of amides with SmI2 has recently found a revival following Procter's and Szostak's developments of ring closing reactions. When treated with SmI2 and H2O, glutarimide 248 forms a radical intermediate which can then attack a tethered alkene to form indolizidines of type 250 (Scheme 34b).130 High regioselectivity is reported for this transformation through stabilisation of the reactive aminoketyl radical intermediate. A similar reaction with alkene-containing barbiturates has been shown to lead to the formation of highly complex ring systems.131–133 After single electron transfer from SmI2 to the amide, a sequence 5-exo-trig and 6-exo-trig cyclisations occurs, leading to a one-step construction of 253 (Scheme 34c).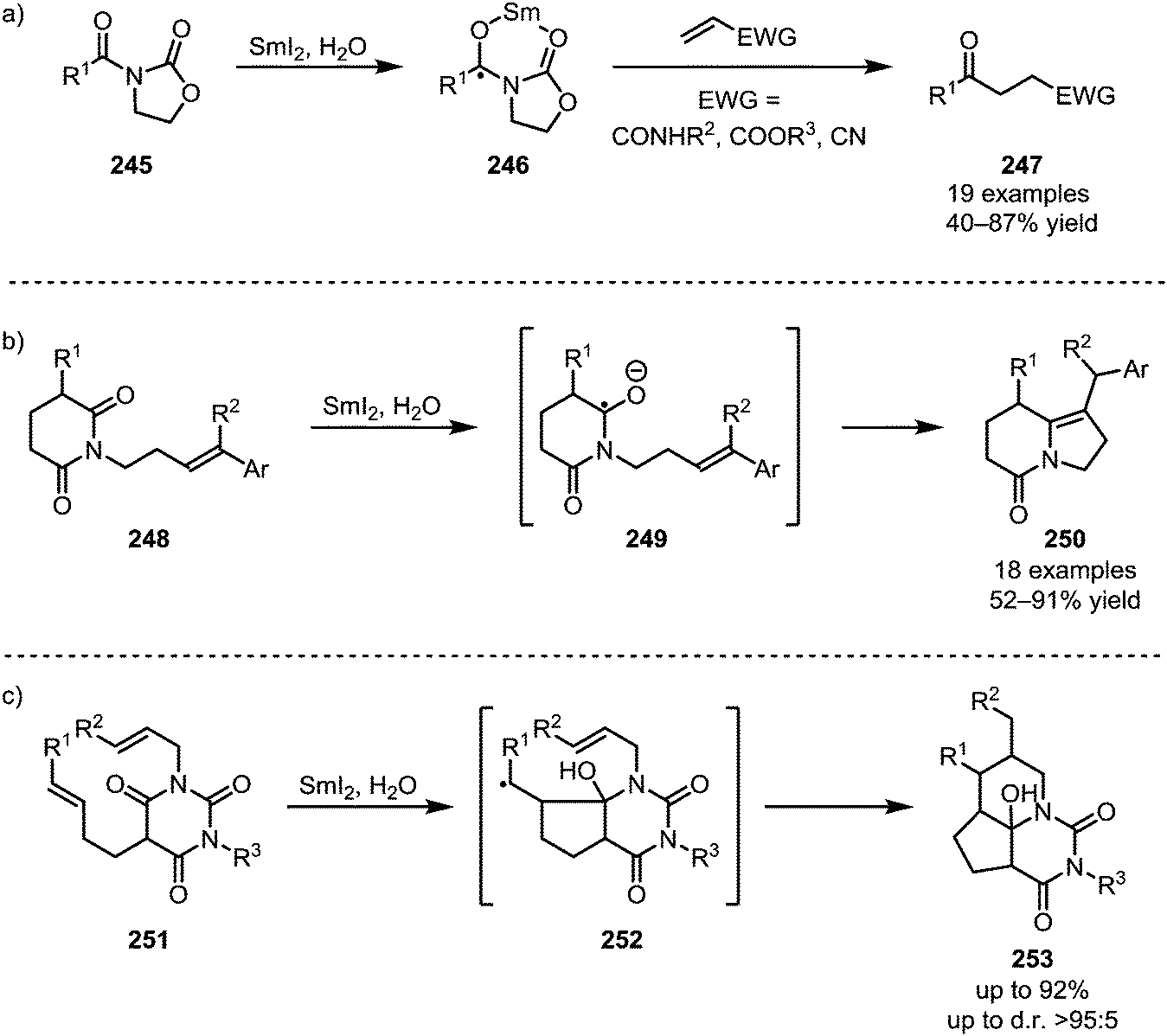 | ||
| Scheme 34 Activation of imide-derivatives with SmI2. | ||
Huang et al. were able to employ a similar procedure on a Boc-protected amide, applying this transformation to their total synthesis of (−)-securinine (259).134 A first use of SmI2 was reported for the reductive C–C coupling of amide 255, forming amine 256. At a later stage of the synthesis, SmI2 is used once again, effecting a second annulation through ketyl radical addition to a nitrile (Scheme 35).
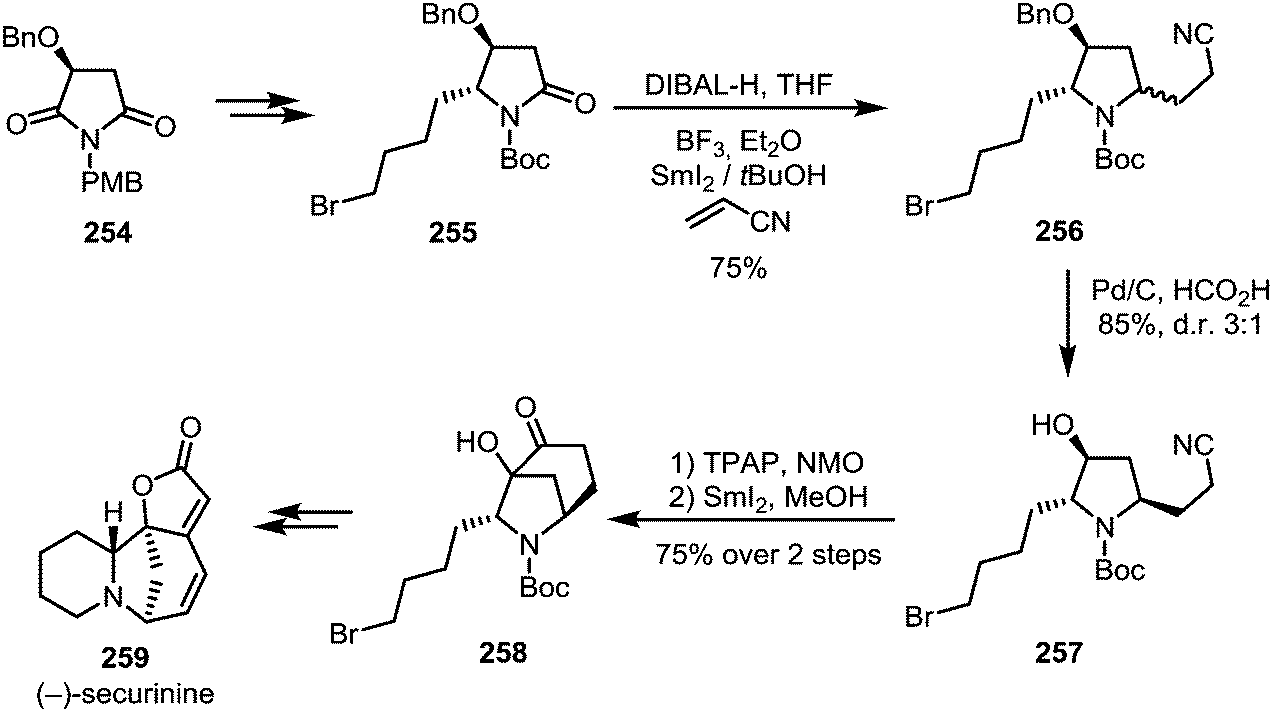 | ||
| Scheme 35 Total synthesis of (−)-securinine. | ||
The area of application of SmI2 in this context is, however, not limited to imides and imide-derivatives. In 2014, Procter et al. also reported that SmI2, in the presence of triethylamine, can be used to efficiently reduce simple, unactivated amides (260) to alcohols (261) with high chemoselectivity (Scheme 36).135 High yields are reported for 1°, 2° and 3° amides, such as compound 260a. Even amides connected to a chiral auxiliary (bearing a free alcohol) can be cleaved with retention of the configuration in the α-position (260b). Additionally, sterically demanding substituents (260c) give good yields of the target alcohol 261c. With the reduction of atenolol (260d) the authors were able to prove the additionally high functional group tolerance of this method and applicability to the formation of bioactive compounds.
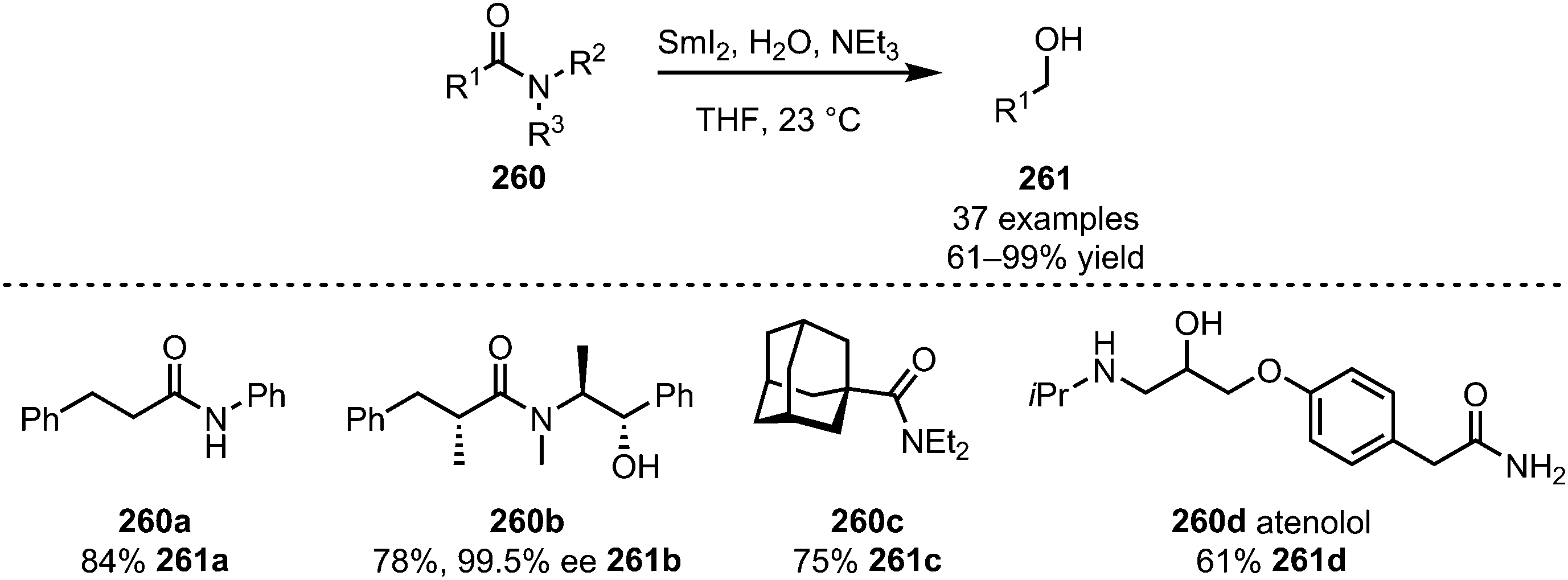 | ||
| Scheme 36 Chemoselective reduction of unactivated amides. | ||
Amide C–N bond activation using transition metal catalysts
While the samarium-mediated reactions described above rely on stoichiometric amounts of organometallic reagent, palladium- and nickel-complexes (most prominently) can be employed in catalytic amounts to activate the C–N bond central to the amide moiety. Thus, in recent years the introduction of transition metal catalysis to the field of amide C–N bond activation has ushered new reactivity pathways and enabled novel synthetic disconnections and transformations. The following subsection aims to provide an overview of the latest developments in this field—the information compiled hereafter has also been described in a series of excellent recent reviews.136–139In recent years, amides fitted with sterically demanding and electron-withdrawing groups have become the focus of intense investigation. These so-called “twisted amides” or “twistamides” display an enhanced tendency towards C–N bond cleavage, as the aforementioned steric bulk twists the C–N bond out of conjugation. Additionally, the siphoned electron density no longer stabilises the C–N bond, which is concomitantly elongated (Scheme 37).140,141 While a historic approach to ground state distortion was the use of bridged lactams (thereby employing actual amides),142 in recent years, the groups of Garg and Szostak have published an array of transformations using acyclic N-acylated amides, more properly to be termed imides (and derivatives thereof). In this regard, N-glutarimides, N-acyl-tert-butyl carbamates (Boc) and N-acyl-tosylamides (Ts) are also excellent activating groups for a wide range of reactions in which the C–N bond of the amide is activated.
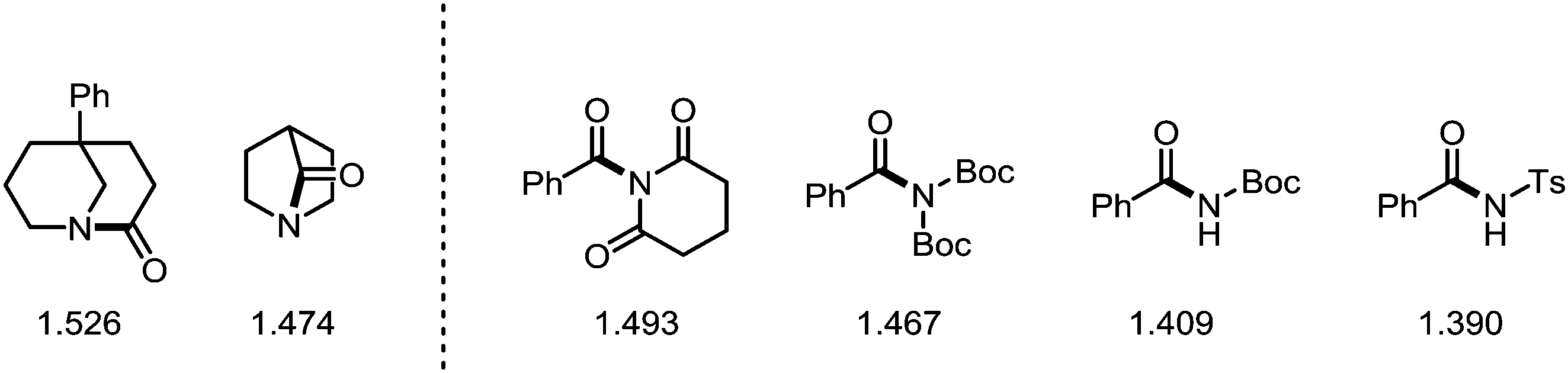 | ||
| Scheme 37 Several C–N bond lengths of strained or twisted amides and imides in Å. The C–N bond length is an indicator for its π-bond character. A longer bond is weaker and therefore intrinsically more activated towards cleavage. | ||
Electron-rich transition-metal complexes (such as palladium–NHC or nickel(COD)2) are able to effect oxidative addition into destabilised C–N bonds, leading to metallated intermediates (263) that can be engaged in typical cross-coupling catalytic cycles (Scheme 38).
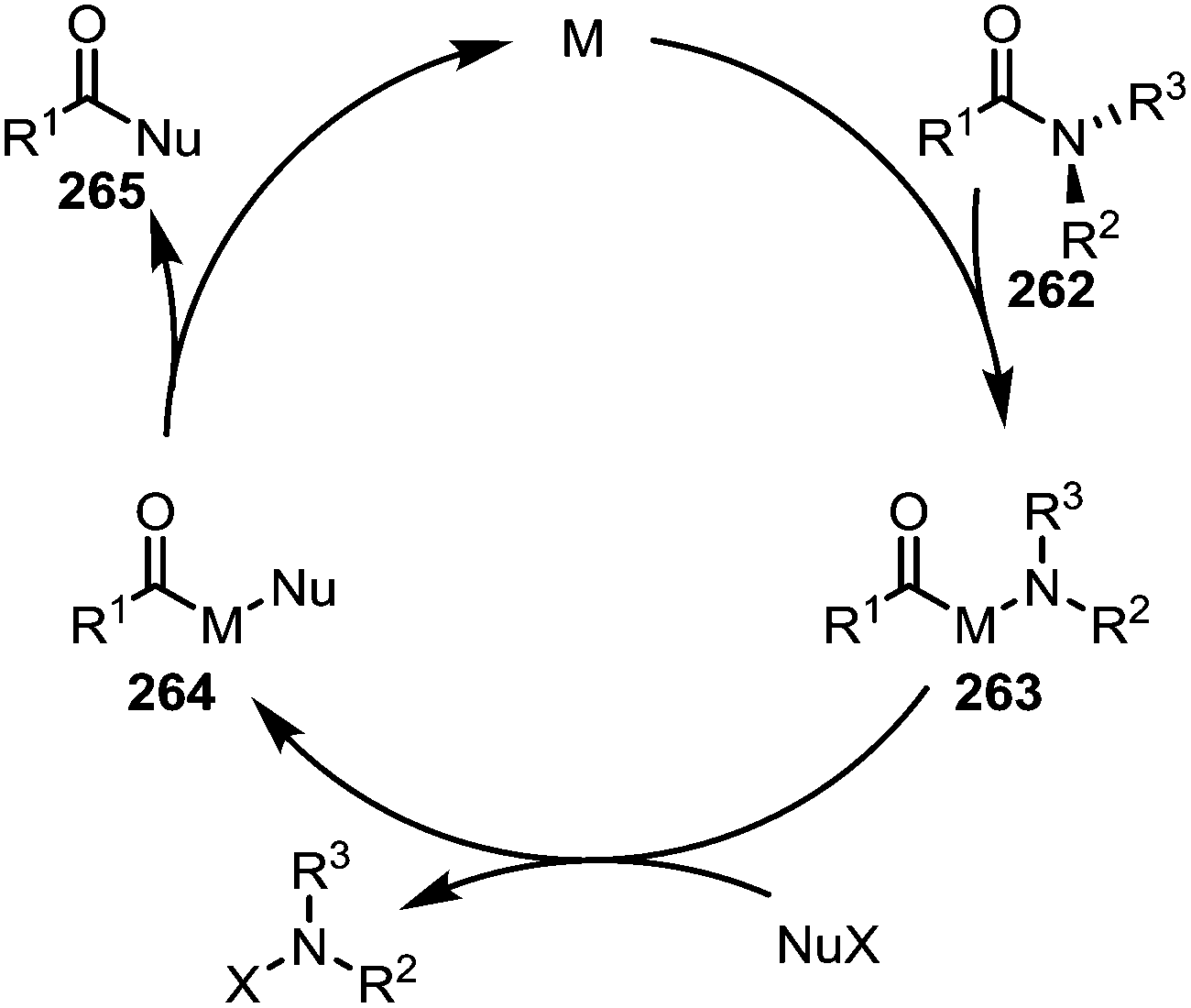 | ||
| Scheme 38 Mechanistic outline for the transition metal-catalysed activation and cleavage of amide C–N bonds for the formation of alternative carbonyl derivatives. | ||
Taking advantage of the insertion of such catalysts into the C–N bond, the groups of Garg143,144 and Hu145 in the case of nickel-catalysis, and Szostak in the case of palladium catalysis,146 have introduced a variety of transamidation protocols with primary and secondary amines (Scheme 39a). Other metals have also been employed in isolated cases: in this regard, lanthanide catalysts have been shown to activate primary amides for transamidation reactions with a range of amines (Scheme 39b, top),147 and—mimicking biological systems—especially tailored nicotinate amides can be alcoholised using zinc catalysis (Scheme 39b, bottom).148 Remarkably, in 2017, Szostak and co-workers reported a transamidation reaction that proceeds even in the absence of a transition metal catalyst, exploiting the increased electrophilicity of non-planar amides.149
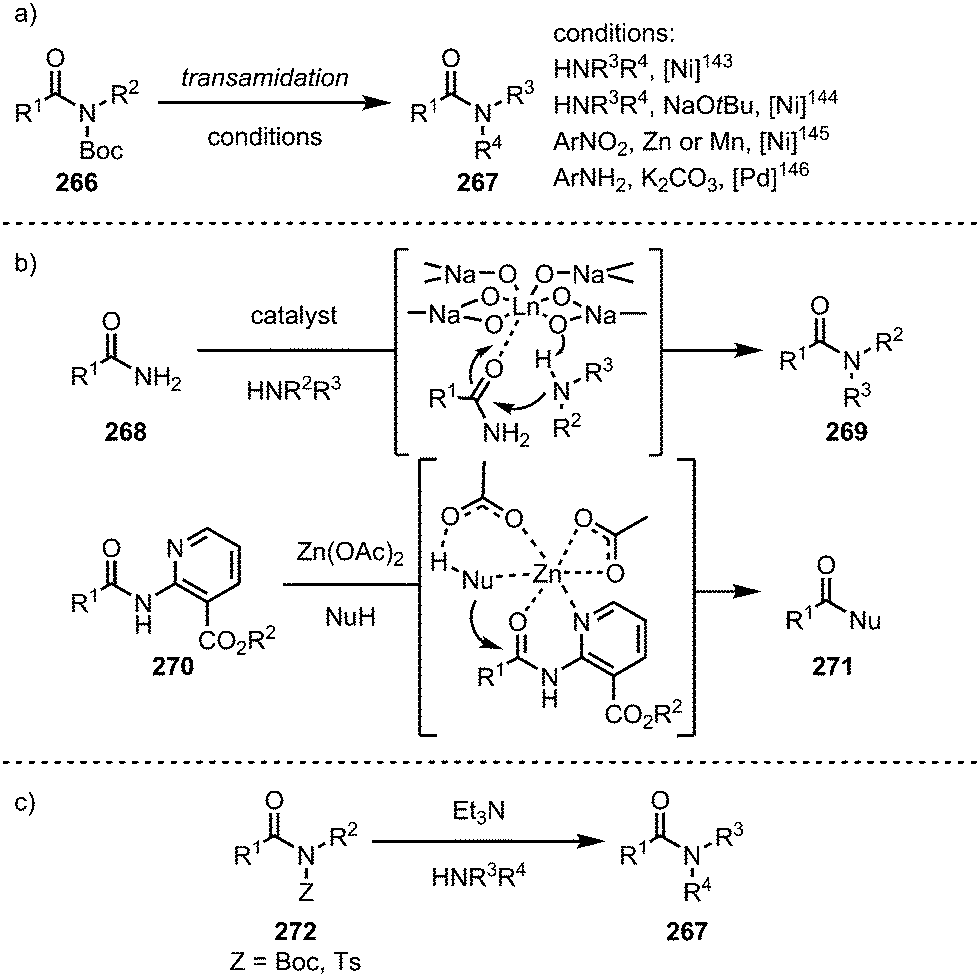 | ||
| Scheme 39 Various approaches to transition metal-catalysed and metal-free transamidation. | ||
Similar to the metal-catalysed transamidations mentioned above, treatment of the intermediates formed through nickel- or palladium-insertion (cf.263, Scheme 38) into the C–N bond with a range of alcohols leads to facile ester formation (Scheme 40).150,151
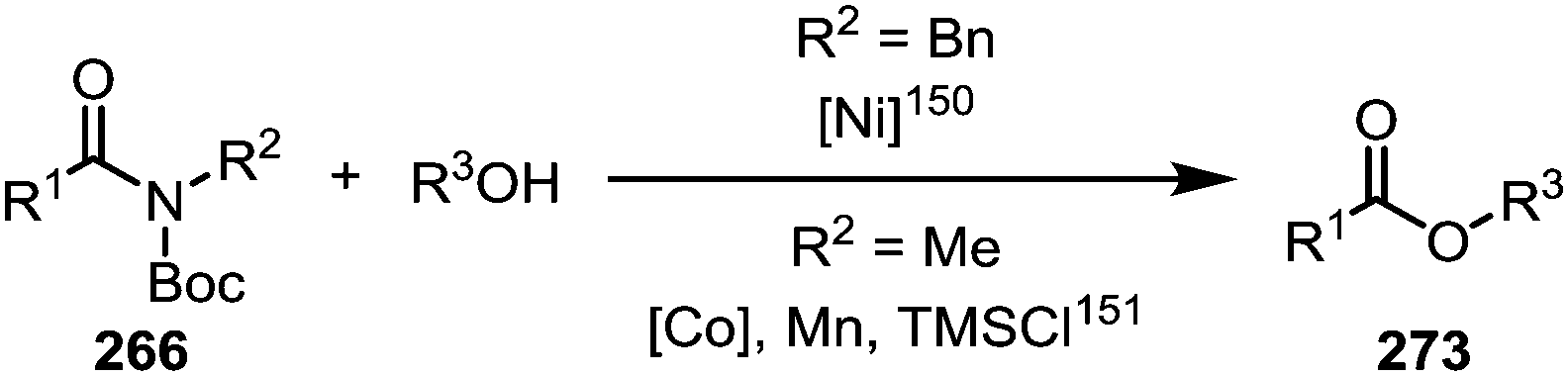 | ||
| Scheme 40 Amide-to-ester conversion of activated amides using transition metal catalysts. | ||
A vast array of procedures enabling the selective formation of ketones from twisted amides has been disclosed in recent years (Scheme 41). Once more, nickel and palladium complexes have to be proven prime catalysts for these transformations, hinging on C–N bond activation, transmetalation and final reductive elimination of the organometallic intermediate. In the context of palladium-catalysed ketone syntheses, N-acylsaccharins (274) have been shown to be privileged species, finding application in Suzuki–Miyaura cross-couplings,152 Sonogashira couplings,153—however, if a glutarimide is used instead of an N-acylsaccharin, the decarbonylated (vide infra) Sonogashira-product is obtained154—and direct couplings with aromatic heterocycles (Scheme 41a).155 Additional examples of Suzuki–Miyaura couplings have been reported for N-glutarimides,156,157 and Boc,158 as well as sulfonyl substituted amides (Scheme 41b).159,160
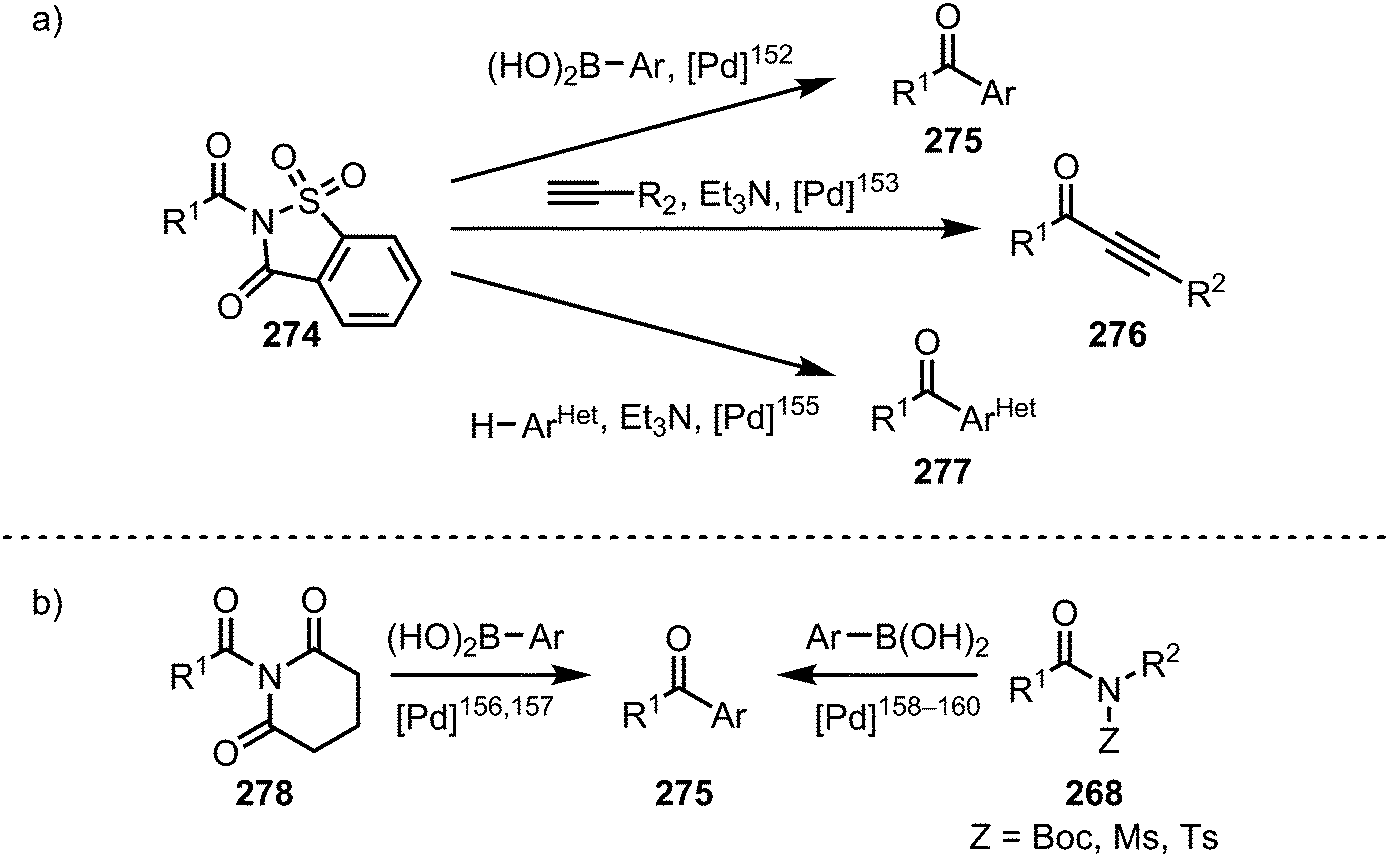 | ||
| Scheme 41 Palladium-catalysed ketone syntheses through C–N bond cleavage of acyl-substituted amides. | ||
Literature reports of the conversion of amides to ketones under nickel catalysis have mainly relied on protocols of Negishi–161–164 and Suzuki–Miyaura coupling (Scheme 42).165 The application of this type of transformation to the synthesis of bioactive compounds is exemplified by Scheme 42b, which shows the formation of ketone 283, a nanomolar antiproliferative agent, via a Suzuki coupling in good yield on a gram scale.166
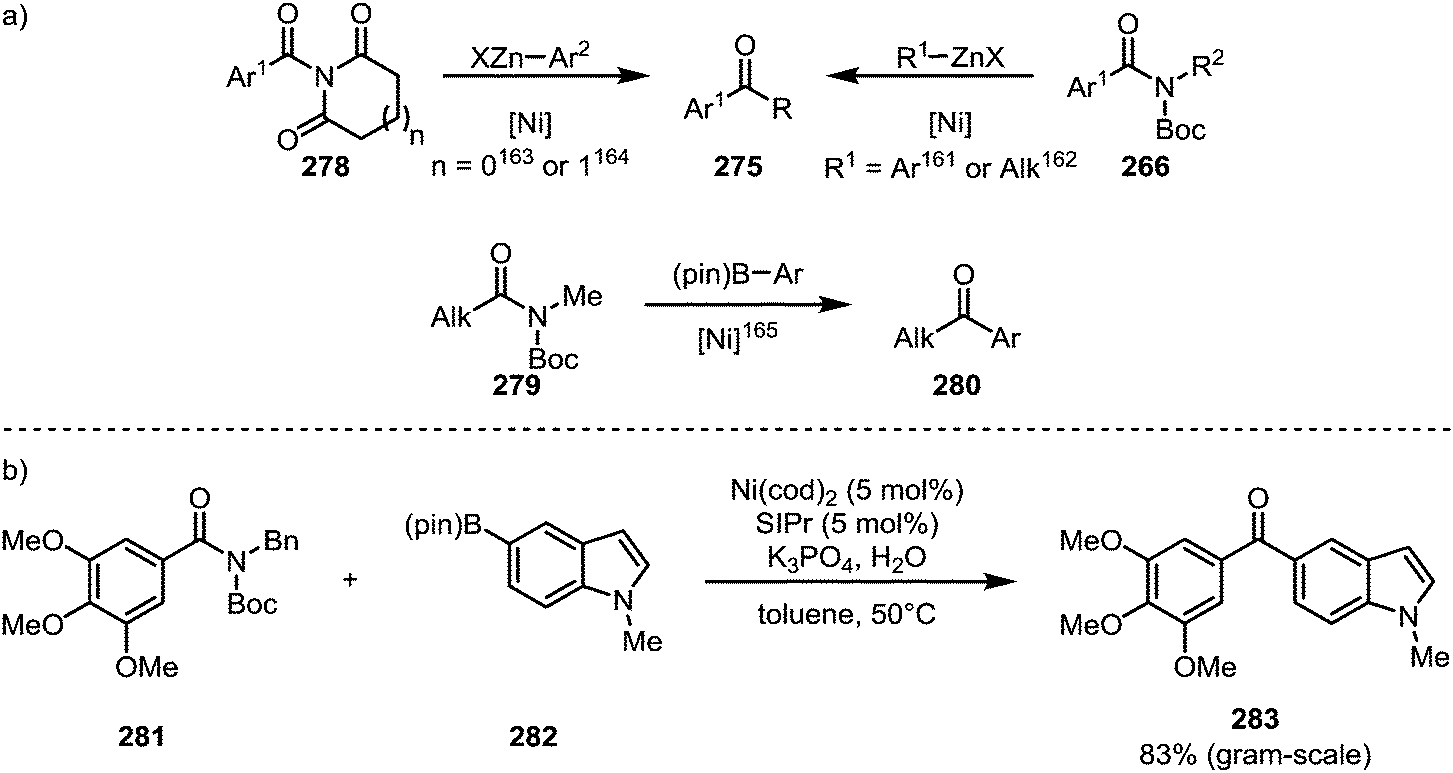 | ||
| Scheme 42 Nickel-catalysed ketone syntheses through C–N bond cleavage of acyl-substituted amides. | ||
Garg and co-workers furthermore brought forth an elegant example of a nickel-catalysed Mizoroki–Heck cyclisation, leading to the formation of products (285) containing α-quaternary ketones (Scheme 43a).167 A similar approach was recently also adopted by Stanley and co-workers, who were able to suppress the terminating β-hydride elimination through addition of arylboronic esters, leading to the products of carboacylation (287) through transmetalation and subsequent reductive elimination (Scheme 43b).168 Szostak and co-workers have, in turn, reported the intermolecular decarbonylative Heck-reaction of distorted amides (288, 289)—their work, however, showed that it proceeded with loss of the carbonyl functionality (Scheme 43c).169,170
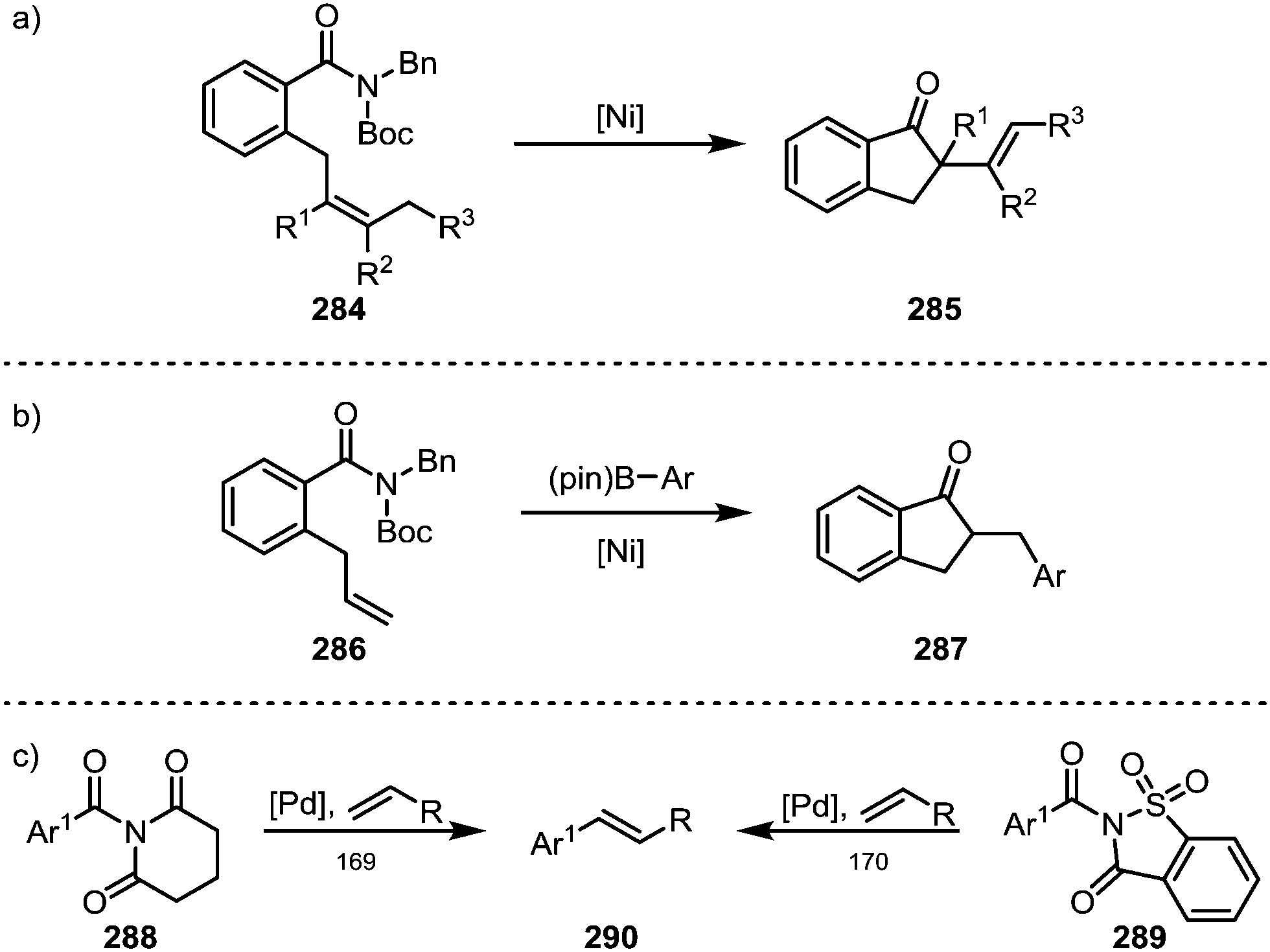 | ||
| Scheme 43 Nickel-catalysed Mizoroki–Heck-type cyclisations onto twisted amides. | ||
Decarbonylative transition metal catalysed transformations
As hinted at in Scheme 43c, while the reductive elimination of intermediates such as 263 (cf.Scheme 38) can afford new carboxyl and carbonyl derivatives, the loss of carbon monoxide (CO) leads to alternative decarbonylative pathways (Scheme 44).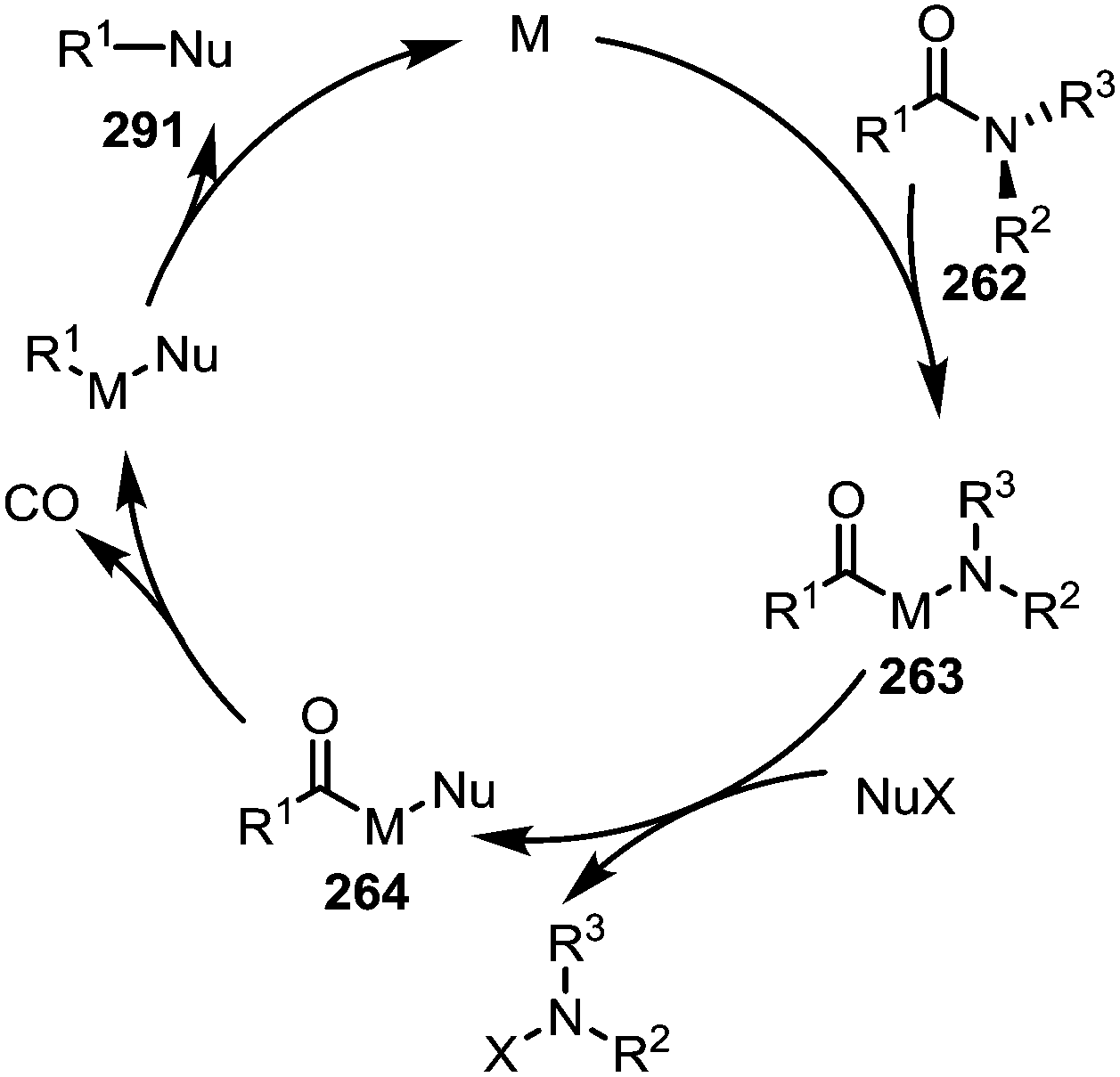 | ||
| Scheme 44 Mechanistic outline for the transition metal-catalysed activation and decarbonylative cleavage of amide C–N bonds. | ||
Such processes involving the loss of CO can, for example, be enforced by the use of elevated temperatures (∼150 °C).139,171 For example, a Suzuki-type biaryl coupling, affording 292, can take place if the decarbonylation step is induced prior to reductive elimination (Scheme 45a, left).171,172 Alkynes can be decarbonylatively added to arenes and alkenes through a nickel-catalysed Sonogashira-type coupling reaction, forming 293 (Scheme 45a, right).173 In addition, aromatic compounds can be functionalised with sulfides (295),174 phosphonates (296),175 cyanides (297),176 boronic esters (298),177 or amines (299)178 (Scheme 45b). Recently, it has been shown that twistamides can also be employed in the directed cross-coupling to C–H activated aryl moieties (300), affording biaryl compounds (301) (Scheme 45c).179,180
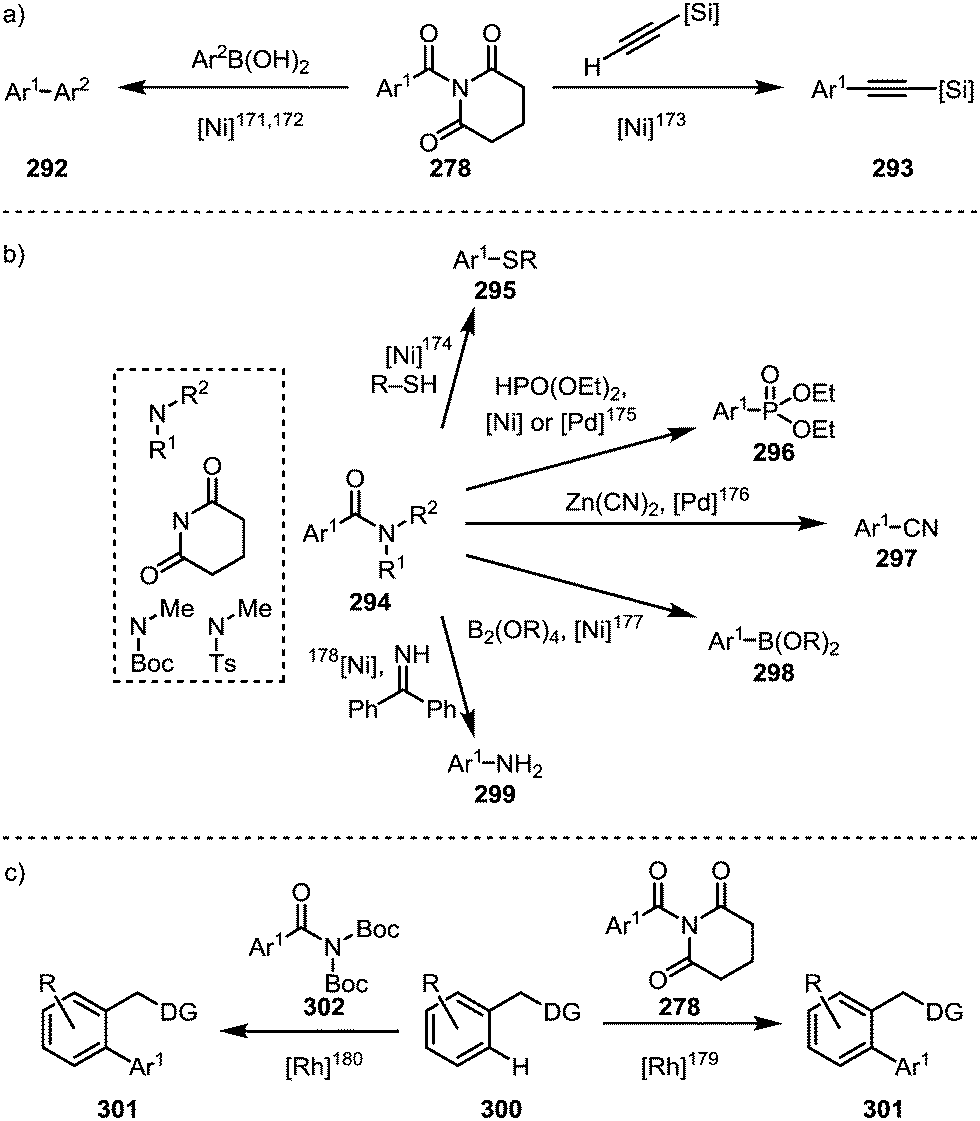 | ||
| Scheme 45 Transition metal-catalysed decarbonylative couplings mediated through initial amide C–N bond activation. | ||
A report by Rueping and co-workers has additionally shown the possibility of conducting a complete reductive removal of amides (and esters) using nickel acetate and silanes (Scheme 46a).181 A 2017 report by Shi and co-workers features the description of a mechanistically intriguing retro-hydroamidocarbonylation of Boc-activated amides (304), leading to the facile formation of a wide array of olefinic products (306) through β-hydride elimination of 305 (Scheme 46b).182
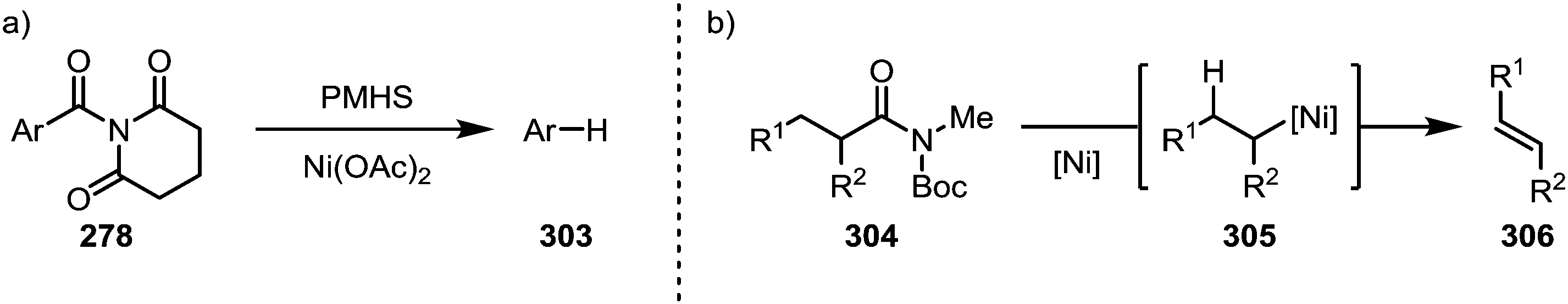 | ||
| Scheme 46 (a) Reductive and (b) alkene-forming amide-cleavage/decarbonylation reactions. | ||
Further privileged amides in selective transformations
While the above transformations focus on N-acyl amide derivatives, other types of amides have also been shown to exhibit unique properties for (selective) transition metal-catalysed or even metal-free transformations.Azetidine amides (307) represent such a privileged class of amides, as Szostak and co-workers were able to show that their treatment with organolithium and Grignard reagents in the absence of any catalyst selectively affords the corresponding ketones (Scheme 47a).183 Remarkably, the suppression of double addition is possible through the formation of highly stable tetrahedral intermediates, thereby providing a viable alternative to the venerable Weinreb amides (308).184 Apart from N-acyl amides and the aforementioned azetidine amides, anilides (309) also present a special class of activated amides (Scheme 47b), as shown by detailed mechanistic analyses in reports by Garg and co-workers,185 as well as Szostak and co-workers,186 who highlighted the influence of the aryl moiety on resonance energies and dihedral angles. Planar amides derived from aromatic azacycles (310) are activated in an electronic way, ultimately resulting in similar reactivity. In 2017, Maiti demonstrated a decarbonylative reduction of pyrazole-amides,187 while Szostak reported the synthesis of ketones (311) from pyrrole-amides (Scheme 47c).188
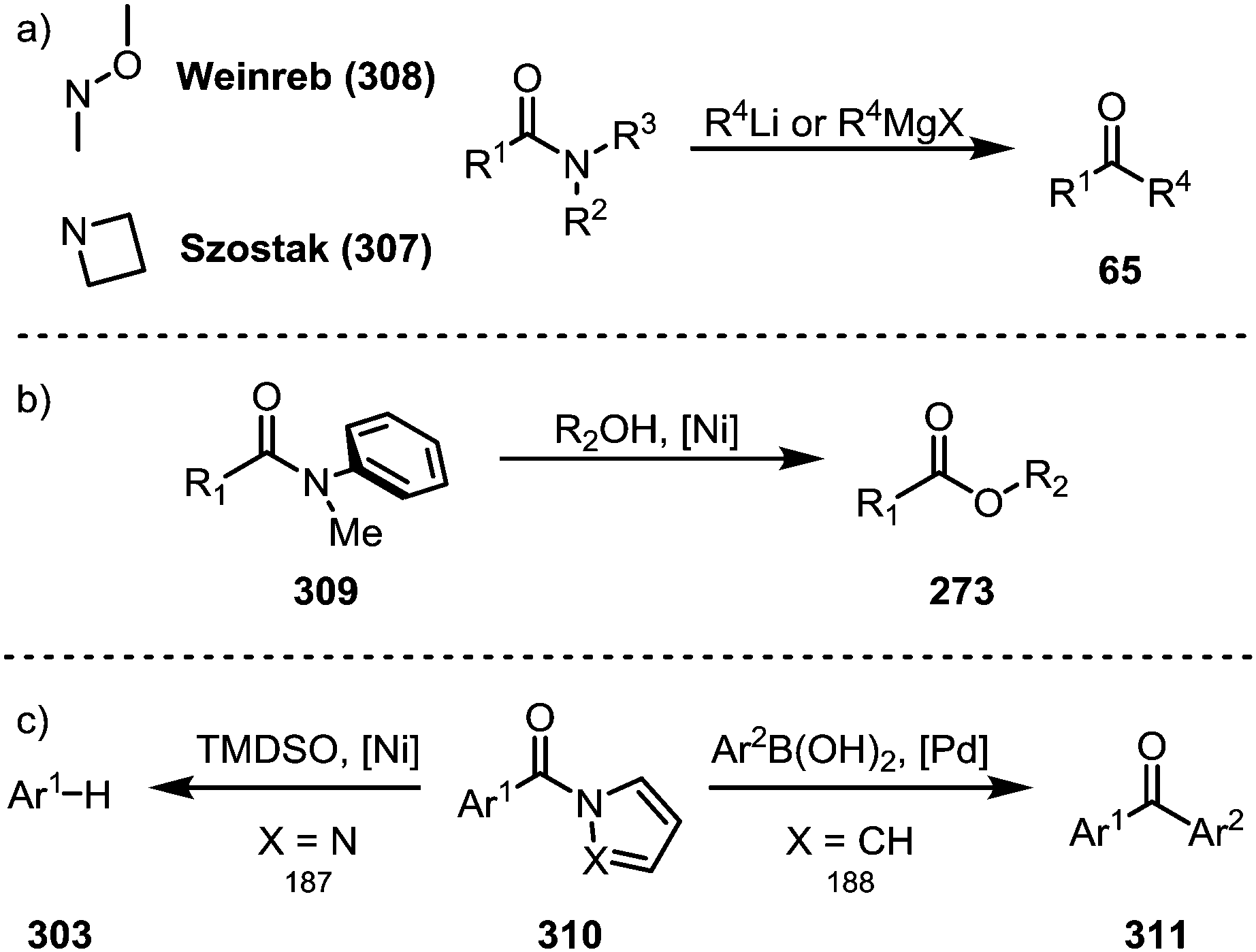 | ||
| Scheme 47 (a) Azetidine amides as privileged electrophiles; and (b and c) transition metal-catalysed C–N bond activation of amides not fitted with an electron-withdrawing group. | ||
Conclusion
Within this review article, we have aimed at providing an overview of means to exploit the intrinsic electronic and geometric properties of amides for their selective functionalisation. Owing to the pronounced nucleophilicity of the amide carbonyl functionality, electrophilic amide activation and transition metal-catalysed amide C–N bond activation have received a great amount of attention in the last few decades, especially in the context of methodology development. Examples for the application of these reactions in total syntheses were provided for most of the classes of transformations. The secondary aim of this review is to inspire chemists to adopt these new techniques, whenever conventional transformations fail to provide satisfactory synthetic results. Very much an actively evolving subfield of organic synthesis, amide activation holds considerable potential for further elaboration and application in the synthesis of bioactive unnatural and natural products alike.Conflicts of interest
The authors have no conflicts of interest to report.Acknowledgements
We are grateful to the University of Vienna for continued support of our research programs. Funding by the FWF (P30226), the Austrian Academy of Sciences (DOC fellowship to D. K.) and the ERC (CoG 682002) is gratefully acknowledged.Notes and references
- A. Greenberg, C. M. Breneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science, Wiley, New York, 2003 Search PubMed.
- A. J. Bennet, Q.-P. Wang, H. Slebocka-Tilk, V. Somayaji and R. S. Brown, J. Am. Chem. Soc., 1990, 112, 6383–6385 CrossRef CAS.
- H. Slebocka-Tilk and R. S. Brown, J. Org. Chem., 1987, 52, 805–808 CrossRef CAS.
- D. Kaiser and N. Maulide, J. Org. Chem., 2016, 81, 4421–4428 CrossRef CAS PubMed.
- B. B. Snider, Chem. Rev., 1988, 88, 793–811 CrossRef CAS.
- I. L. Baraznenok, V. G. Nenajdenko and E. S. Balenkova, Tetrahedron, 2000, 56, 3077–3119 CrossRef CAS.
- C. Madelaine, V. Valerio and N. Maulide, Chem. – Asian J., 2011, 6, 2224–2239 CrossRef CAS PubMed.
- N. Kumagai and M. Shibasaki, Chem. – Eur. J., 2016, 22, 15192–15200 CrossRef CAS PubMed.
- O. Wallach, Liebigs Ann. Chem., 1877, 184, 1–127 CrossRef.
- J. v. Braun and A. Heymons, Chem. Ber., 1929, 62, 409–413 CrossRef.
- L. Ghosez, B. Haveaux and H. G. Viehe, Angew. Chem., Int. Ed. Engl., 1969, 8, 454–455 CrossRef CAS.
- L. Ghosez, Angew. Chem., Int. Ed. Engl., 1972, 11, 852–853 CrossRef CAS.
- A. Sidani, J. Marchand-Brynaert and L. Ghosez, Angew. Chem., Int. Ed. Engl., 1974, 13, 267–268 CrossRef.
- R. Bisceglia and C. J. Cheer, J. Chem. Soc., Chem. Commun., 1973, 59, 165–166 RSC.
- J. Marchand-Brynaert and L. Ghosez, J. Am. Chem. Soc., 1972, 94, 2870–2871 CrossRef CAS.
- C. Hoornaert, A. M. Hesbain-Frisque and L. Ghosez, Angew. Chem., Int. Ed. Engl., 1975, 14, 569–570 CrossRef.
- J. Marchand-Brynaert, M. Moya-Portuguez, D. Lesuisse and L. Ghosez, J. Chem. Soc., Chem. Commun., 1980, 173–174 RSC.
- M. De Poortere, J. Marchand-Brynaert and L. Ghosez, Angew. Chem., Int. Ed. Engl., 1974, 13, 267–268 CrossRef.
- H. G. Heine and W. Hartmann, Angew. Chem., Int. Ed. Engl., 1981, 20, 782–783 CrossRef.
- W. J. Ding and D. C. Fang, J. Org. Chem., 2001, 66, 6673–6678 CrossRef CAS PubMed.
- J. B. Falmagne, J. Escudero, S. Taleb-Sahraoui and L. Ghosez, Angew. Chem., Int. Ed. Engl., 1981, 20, 879–880 CrossRef.
- I. Marko, B. Ronsmans, A. M. Hesbain-Frisque, S. Dumas and L. Ghosez, J. Am. Chem. Soc., 1985, 107, 2192–2194 CrossRef CAS.
- C. Genicot, B. Gobeaux and L. Ghosez, Tetrahedron Lett., 1991, 32, 3827–3830 CrossRef CAS.
- C. Schmit, S. Sahraoui-Taleb, E. Differding, C. G. Dehasse-De Lombaert and L. Ghosez, Tetrahedron Lett., 1984, 25, 5043–5046 CrossRef CAS.
- B. B. Snider and R. A. H. F. Hui, J. Org. Chem., 1985, 50, 5167–5176 CrossRef CAS.
- C. Madelaine, V. Valerio and N. Maulide, Angew. Chem., Int. Ed., 2010, 49, 1583–1586 CrossRef CAS PubMed.
- W. T. Brady, Y. S. F. Giang, L. Weng and M. M. Dad, J. Org. Chem., 1986, 52, 2216–2220 CrossRef.
- L. Y. Chen and L. Ghosez, Tetrahedron Lett., 1990, 31, 4467–4470 CrossRef CAS.
- C. Houge, A. M. Frisque-Hesbain, A. Mockel and L. Ghosez, J. Am. Chem. Soc., 1982, 104, 2920–2921 CrossRef CAS.
- J. M. Adam, L. Ghosez and K. N. Houk, Angew. Chem., Int. Ed. Engl., 1999, 38, 2728–2730 CrossRef CAS PubMed.
- H. Saimoto, C. Houge, A. M. Hesbain-Frisque, A. Mockel and L. Ghosez, Tetrahedron Lett., 1983, 24, 2251–2254 CrossRef CAS.
- L. Ghosez, I. Marko and A. M. Hesbain-Frisque, Tetrahedron Lett., 1986, 27, 5211–5214 CrossRef CAS.
- L. Y. Chen and L. Ghosez, Tetrahedron: Asymmetry, 1991, 2, 1181–1184 CrossRef CAS.
- D. Depré, L. Y. Chen and L. Ghosez, Tetrahedron, 2003, 59, 6797–6812 CrossRef.
- M. Lachia, P. M. J. Jung and A. De Mesmaeker, Tetrahedron Lett., 2012, 53, 4514–4517 CrossRef CAS.
- M. Lachia, H. C. Wolf and A. De Mesmaeker, Bioorg. Med. Chem. Lett., 2014, 24, 2123–2128 CrossRef CAS PubMed.
- M. Lachia, P. Y. Dakas and A. De Mesmaeker, Tetrahedron Lett., 2014, 55, 6577–6581 CrossRef CAS.
- O. Irie and K. Shishido, Chem. Lett., 1995, 53–54 CrossRef CAS.
- P. J. Shim and H. D. Kim, Tetrahedron Lett., 1998, 39, 9517–9520 CrossRef CAS.
- M. Movassaghi, M. D. Hill and O. K. Ahmad, J. Am. Chem. Soc., 2007, 129, 10096–10097 CrossRef CAS PubMed.
- M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 4592–4593 CrossRef CAS PubMed.
- T. Wezeman, S. Zhong, M. Nieger and S. Bräse, Angew. Chem., Int. Ed., 2016, 55, 3823–3827 CrossRef CAS PubMed.
- L. H. Li, Z. J. Niu and Y. M. Liang, Chem. – Eur. J., 2017, 23, 15300–15304 CrossRef CAS PubMed.
- S. L. Cui, J. Wang and Y. G. Wang, J. Am. Chem. Soc., 2008, 130, 13526–13527 CrossRef CAS PubMed.
- W. S. Bechara, G. Pelletier and A. B. Charette, Nat. Chem., 2012, 4, 228–234 CrossRef CAS PubMed.
- K. J. Xiao, A. E. Wang, Y. H. Huang and P. Q. Huang, Asian J. Org. Chem., 2012, 1, 130–132 CrossRef CAS.
- K. J. Xiao, A. E. Wang and P. Q. Huang, Angew. Chem., Int. Ed., 2012, 51, 8314–8317 CrossRef CAS PubMed.
- P. Q. Huang, Y. H. Huang, K. J. Xiao, Y. Wang and X. E. Xia, J. Org. Chem., 2015, 80, 2861–2868 CrossRef CAS PubMed.
- A. Romanens and G. Bélanger, Org. Lett., 2015, 17, 322–325 CrossRef CAS PubMed.
- J. C. Liao, K. J. Xiao, X. Zheng and P. Q. Huang, Tetrahedron, 2012, 68, 5297–5302 CrossRef CAS.
- K. J. Xiao, Y. Wang, K. Y. Ye and P. Q. Huang, Chem. – Eur. J., 2010, 16, 12792–12796 CrossRef CAS PubMed.
- C. Lindemann and C. Schneider, Synthesis, 2016, 828–844 CAS.
- H. Q. Deng, X. Y. Qian, Y. X. Li, J. F. Zheng, L. Xie and P. Q. Huang, Org. Chem. Front., 2014, 1, 258–266 RSC.
- W. Oppolzer, C. Fehr and J. Warneke, Helv. Chim. Acta, 1977, 60, 48–58 CrossRef CAS PubMed.
- K. J. Xiao, Y. Wang, Y. H. Huang, X. G. Wang and P. Q. Huang, J. Org. Chem., 2013, 78, 8305–8311 CrossRef CAS PubMed.
- K. J. Xiao, J. M. Luo, K. Y. Ye, Y. Wang and P. Q. Huang, Angew. Chem., Int. Ed., 2010, 49, 3037–3040 CrossRef CAS PubMed.
- P. Q. Huang, W. Ou, K. J. Xiaoa and A. E. Wang, Chem. Commun., 2014, 50, 8761–8763 RSC.
- V. S. Gawali, S. Simeonov, M. Drescher, T. Knott, O. Scheel, J. Kudolo, H. Kählig, U. Hochenegg, A. Roller, H. Todt and N. Maulide, ChemMedChem, 2017, 12, 1819–1822 CrossRef CAS PubMed.
- H. H. Huo, X. E. Xia, H. K. Zhang and P. Q. Huang, J. Org. Chem., 2013, 78, 455–465 CrossRef CAS PubMed.
- H. H. Huo, H. K. Zhang, X. E. Xia and P. Q. Huang, Org. Lett., 2012, 14, 4834–4837 CrossRef CAS PubMed.
- K. J. Xiao, J. M. Luo, X. E. Xia, Y. Wang and P. Q. Huang, Chem. – Eur. J., 2013, 19, 13075–13086 CrossRef CAS PubMed.
- M. Movassaghi and M. D. Hill, Org. Lett., 2008, 10, 3485–3488 CrossRef CAS PubMed.
- A. Lumbroso, J. Behra, A. Kolleth, P. Y. Dakas, U. Karadeniz, S. Catak, S. Sulzer-Mossé and A. De Mesmaeker, Tetrahedron Lett., 2015, 56, 6541–6545 CrossRef CAS.
- G. Bélanger, V. Darsigny, M. Doré and F. Lévesque, Org. Lett., 2010, 12, 1396–1399 CrossRef PubMed.
- J. W. Medley and M. Movassaghi, Org. Lett., 2013, 15, 3614–3617 CrossRef CAS PubMed.
- K. L. White, M. Mewald and M. Movassaghi, J. Org. Chem., 2015, 80, 7403–7411 CrossRef CAS PubMed.
- K. L. White and M. Movassaghi, J. Am. Chem. Soc., 2016, 138, 11383–11389 CrossRef CAS PubMed.
- T. Kang, K. L. White, T. J. Mann, A. H. Hoveyda and M. Movassaghi, Angew. Chem., Int. Ed., 2017, 56, 13857–13860 CrossRef CAS PubMed.
- M. Mewald, J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2014, 53, 11634–11639 CrossRef CAS PubMed.
- J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2012, 51, 4572–4576 CrossRef CAS PubMed.
- P. Magnus, L. Gazzard, L. Hobson, A. H. Payne, T. J. Rainey, N. Westlund and V. Lynch, Tetrahedron, 2002, 58, 3423–3443 CrossRef CAS.
- G. Bélanger, R. Larouche-Gauthier, F. Ménard, M. Nantel and F. Barabé, Org. Lett., 2005, 7, 4431–4434 CrossRef PubMed.
- G. Bélanger, R. Larouche-Gauthier, F. Ménard, M. Nantel and F. Barabé, J. Org. Chem., 2006, 71, 704–712 CrossRef PubMed.
- F. Lévesque and G. Bélanger, Org. Lett., 2008, 10, 4939–4942 CrossRef PubMed.
- S. Y. Huang, Z. Chang, S. C. Tuo, L. H. Gao, A. E. Wang and P. Q. Huang, Chem. Commun., 2013, 49, 7088–7090 RSC.
- G. Bélanger, J. Boudreault and F. Lévesque, Org. Lett., 2011, 13, 6204–6207 CrossRef PubMed.
- J. Boudreault, F. Lévesque and G. Bélanger, J. Org. Chem., 2016, 81, 9247–9268 CrossRef CAS PubMed.
- A.-E. Wang, C.-C. Yu, T.-T. Chen, Y.-P. Liu and P.-Q. Huang, Org. Lett., 2018, 20, 999–1002 CrossRef CAS PubMed.
- L. E. Overman and J. P. Wolfe, J. Org. Chem., 2002, 67, 6421–6429 CrossRef CAS PubMed.
- G. Barbe and A. B. Charette, J. Am. Chem. Soc., 2008, 130, 18–19 CrossRef CAS PubMed.
- G. Pelletier, W. S. Bechara and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 12817–12819 CrossRef CAS PubMed.
- S. H. Xiang, J. Xu, H. Q. Yuan and P. Q. Huang, Synlett, 2010, 1829–1832 CAS.
- P. Q. Huang, Q. W. Lang and Y. R. Wang, J. Org. Chem., 2016, 81, 4235–4243 CrossRef CAS PubMed.
- P. Xiao, Z. Tang, K. Wang, H. Chen, Q. Guo, Y. Chu, L. Gao and Z. Song, J. Org. Chem., 2018, 83, 1687–1700 CrossRef CAS PubMed.
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS.
- S. Bonazzi, B. Cheng, J. S. Wzorek and D. A. Evans, J. Am. Chem. Soc., 2013, 135, 9338–9341 CrossRef CAS PubMed.
- Interestingly, an alternative synthesis showed that the placement of the amide carbonyl at carbon 1, rather than carbon 5 (cf.132, Scheme 20), allowed the regioselective reduction with DIBAL-H.
- P. Jakubec, A. F. Kyle, J. Calleja and D. J. Dixon, Tetrahedron Lett., 2011, 52, 6094–6097 CrossRef CAS.
- Z. H. Chen, Z. M. Chen, Y. Q. Zhang, Y. Q. Tu and F. M. Zhang, J. Org. Chem., 2011, 76, 10173–10186 CrossRef CAS PubMed.
- R. A. Abramovitch and G. M. Singer, J. Am. Chem. Soc., 1969, 91, 5672–5673 CrossRef CAS.
- J. W. Medley and M. Movassaghi, J. Org. Chem., 2009, 74, 1341–1344 CrossRef CAS PubMed.
- T. Yamazaki, J. T. Welch, J. S. Plummer and R. H. Gimi, Tetrahedron Lett., 1991, 32, 4267–4270 CrossRef CAS.
- M. Padmanaban, L. C. R. Carvalho, D. Petkova, J. W. Lee, A. S. Santos, M. M. B. Marques and N. Maulide, Tetrahedron, 2015, 71, 5994–6005 CrossRef CAS.
- V. Valerio, C. Madelaine and N. Maulide, Chem. – Eur. J., 2011, 17, 4742–4745 CrossRef CAS PubMed.
- B. Peng, D. O’Donovan, I. D. Jurberg and N. Maulide, Chem. – Eur. J., 2012, 18, 16292–16296 CrossRef CAS PubMed.
- V. Valerio, D. Petkova, C. Madelaine and N. Maulide, Chem. – Eur. J., 2013, 19, 2606–2610 CrossRef CAS PubMed.
- B. Peng, D. Geerdink and N. Maulide, J. Am. Chem. Soc., 2013, 135, 14968–14971 CrossRef CAS PubMed.
- B. Peng, D. Geerdink, C. Farès and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 5462–5466 CrossRef CAS PubMed.
- S. Shaaban, V. Tona, B. Peng and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 10938–10941 CrossRef CAS PubMed.
- A. B. Charette and M. Grenon, J. Org. Chem., 2003, 68, 5792–5794 CrossRef CAS PubMed.
- A. B. Charette and P. Chua, J. Org. Chem., 1998, 63, 908–909 CrossRef CAS.
- A. B. Charette and P. Chua, Tetrahedron Lett., 1997, 38, 8499–8502 CrossRef CAS.
- W. S. Bechara, I. S. Khazhieva, E. Rodriguez and A. B. Charette, Org. Lett., 2015, 17, 1184–1187 CrossRef CAS PubMed.
- P. Cyr, S. Régnier, W. S. Bechara and A. B. Charette, Org. Lett., 2015, 17, 3386–3389 CrossRef CAS PubMed.
- M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 14254–14255 CrossRef CAS PubMed.
- O. K. Ahmad, J. W. Medley, A. Coste and M. Movassaghi, Org. Synth., 2012, 89, 549–561 CrossRef CAS.
- A. B. Charette, S. Mathieu and J. Martel, Org. Lett., 2005, 7, 5401–5404 CrossRef CAS PubMed.
- A. B. Charette, M. Grenon, A. Lemire, M. Pourashraf and J. Martel, J. Am. Chem. Soc., 2001, 123, 11829–11830 CrossRef CAS PubMed.
- E. W. Thomas, Synthesis, 1993, 767–768 CrossRef CAS.
- V. Tona, B. Maryasin, A. de la Torre, J. Sprachmann, L. González and N. Maulide, Org. Lett., 2017, 19, 2662–2665 CrossRef CAS PubMed.
- M. Rens and L. Ghosez, Tetrahedron Lett., 1970, 11, 3765–3768 CrossRef.
- V. Tona, A. de la Torre, M. Padmanaban, S. Ruider, L. González and N. Maulide, J. Am. Chem. Soc., 2016, 138, 8348–8351 CrossRef CAS PubMed.
- R. Da Costa, M. Gillard, J. B. Falmagne and L. Ghosez, J. Am. Chem. Soc., 1979, 101, 4381–4383 CrossRef CAS.
- D. Kaiser, A. de la Torre, S. Shaaban and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 5921–5925 CrossRef CAS PubMed.
- D. Kaiser, C. J. Teskey, P. Adler and N. Maulide, J. Am. Chem. Soc., 2017, 139, 16040–16043 CrossRef CAS PubMed.
- N. Takeda, E. Futaki, Y. Kobori, M. Ueda and O. Miyata, Angew. Chem., Int. Ed., 2017, 56, 16342–16346 CrossRef CAS PubMed.
- A. de la Torre, D. Kaiser and N. Maulide, J. Am. Chem. Soc., 2017, 139, 6578–6581 CrossRef CAS PubMed.
- M. Roth, P. Dubs, E. Gotschi and A. Eschenmoser, Helv. Chim. Acta, 1971, 54, 710–734 CrossRef CAS.
- H. Mayr, M. Breugst and A. R. Ofial, Angew. Chem., Int. Ed., 2011, 50, 6470–6505 CrossRef CAS PubMed.
- P. Mateo, J. E. Cinqualbre, M. Meyer Mojzes, K. Schenk and P. Renaud, J. Org. Chem., 2017, 82, 12318–12327 CrossRef CAS PubMed.
- M. Amat, J. Hidalgo, N. Llor and J. Bosch, Tetrahedron: Asymmetry, 1998, 9, 2419–2422 CrossRef CAS.
- M. Amat, N. Llor, J. Hidalgo, C. Escolano and J. Bosch, J. Org. Chem., 2003, 68, 1919–1928 CrossRef CAS PubMed.
- S. Lee, M. Bae, J. In, J. H. Kim and S. Kim, Org. Lett., 2017, 19, 254–257 CrossRef CAS PubMed.
- W. J. Klaver, H. Hiemstra and W. N. Speckamp, J. Am. Chem. Soc., 1989, 111, 2588–2595 CrossRef CAS.
- H. Abe, H. Ouchi, C. Sakashita, M. Kawada, T. Watanabe and M. Shibasaki, Chem. – Eur. J., 2017, 23, 11792–11796 CrossRef CAS PubMed.
- C. H. Heathcock, S. K. Davidsen, S. G. Mills and M. A. Sanner, J. Org. Chem., 1992, 57, 2531–2544 CrossRef CAS.
- S. Raucher and P. Klein, Tetrahedron Lett., 1980, 21, 4061–4064 CrossRef CAS.
- S. Britto, P. Renaud and M. Nallu, Spectrochim. Acta, Part A, 2014, 120, 489–493 CrossRef CAS PubMed.
- C. M. Jensen, K. B. Lindsay, R. H. Taaning, J. Karaffa, A. Mette Hansen and T. Skrydstrup, J. Am. Chem. Soc., 2005, 127, 6544–6545 CrossRef CAS PubMed.
- S. Shi, R. Lalancette, R. Szostak and M. Szostak, Chem. – Eur. J., 2016, 22, 11949–11953 CrossRef CAS PubMed.
- H. M. Huang and D. J. Procter, J. Am. Chem. Soc., 2016, 138, 7770–7775 CrossRef CAS PubMed.
- H. M. Huang and D. J. Procter, J. Am. Chem. Soc., 2017, 139, 1661–1667 CrossRef CAS PubMed.
- H. M. Huang, P. Bonilla and D. J. Procter, Org. Biomol. Chem., 2017, 15, 4159–4164 RSC.
- X. Zheng, J. Liu, C. X. Ye, A. Wang, A. E. Wang and P. Q. Huang, J. Org. Chem., 2015, 80, 1034–1041 CrossRef CAS PubMed.
- M. Szostak, M. Spain, A. J. Eberhart and D. J. Procter, J. Am. Chem. Soc., 2014, 136, 2268–2271 CrossRef CAS PubMed.
- C. Liu and M. Szostak, Chem. – Eur. J., 2017, 23, 7157–7173 CrossRef CAS PubMed.
- J. E. Dander and N. K. Garg, ACS Catal., 2017, 7, 1413–1423 CrossRef CAS PubMed.
- R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888 RSC.
- G. Meng, S. Shi and M. Szostak, Synlett, 2016, 2530–2540 CAS.
- C. Liu and M. Szostak, Chem. – Eur. J., 2017, 23, 7157–7173 CrossRef CAS PubMed.
- G. Meng, S. Shi, R. Lalancette, R. Szostak and M. Szostak, J. Am. Chem. Soc., 2018, 140, 727–734 CrossRef CAS PubMed.
- R. Szostak, J. Aube and M. Szostak, J. Org. Chem., 2015, 80, 7905–7927 CrossRef CAS PubMed.
- E. L. Baker, M. M. Yamano, Y. Zhou, S. M. Anthony and N. K. Garg, Nat. Commun., 2016, 7, 11554–11558 CrossRef PubMed.
- J. E. Dander, E. L. Baker and N. K. Garg, Chem. Sci., 2017, 8, 6433–6438 RSC.
- C. W. Cheung, M. L. Ploeger and X. Hu, ACS Catal., 2017, 7, 7092–7096 CrossRef CAS.
- G. Meng, P. Lei and M. Szostak, Org. Lett., 2017, 19, 2158–2161 CrossRef CAS PubMed.
- H. Sheng, R. Zeng, W. Wang, S. Luo, Y. Feng, J. Liu, W. Chen, M. Zhu and Q. Guo, Adv. Synth. Catal., 2017, 359, 302–313 CrossRef CAS.
- C. C. D. Wybon, C. Mensch, K. Hollanders, C. Gadais, W. A. Herrebout, S. Ballet and B. U. W. Maes, ACS Catal., 2018, 8, 203–218 CrossRef CAS.
- Y. Liu, S. Shi, M. Achtenhagen, R. Liu and M. Szostak, Org. Lett., 2017, 19, 1614–1617 CrossRef CAS PubMed.
- L. Hie, E. L. Baker, S. M. Anthony, J.-N. Desrosiers, C. Senanayake and N. K. Garg, Angew. Chem., Int. Ed., 2016, 55, 15129–15132 CrossRef CAS PubMed.
- Y. Bourne-Branchu, C. Gosmini and G. Danoun, Chem. – Eur. J., 2017, 23, 10043–10047 CrossRef CAS PubMed.
- C. Liu, G. Meng, Y. Liu, R. Liu, R. Lalancette, R. Szostak and M. Szostak, Org. Lett., 2016, 18, 4194–4197 CrossRef CAS PubMed.
- M. Cui, H. Wu, J. Jian, H. Wang, C. Liu, D. Stelck and Z. Zeng, Chem. Commun., 2016, 52, 12076–12079 RSC.
- L. Liu, D. Zhou, M. Liu, Y. Zhou and T. Chen, Org. Lett., 2018, 20, 2741–2744 CrossRef CAS PubMed.
- S. Karthik and T. Gandhi, Org. Lett., 2017, 19, 5486–5489 CrossRef CAS PubMed.
- P. Lei, G. Meng and M. Szostak, ACS Catal., 2017, 7, 1960–1965 CrossRef CAS.
- G. Meng and M. Szostak, Org. Lett., 2015, 17, 4364–4367 CrossRef CAS PubMed.
- G. Meng, S. Shi and M. Szostak, ACS Catal., 2016, 6, 7335–7339 CrossRef CAS.
- C. Liu, Y. Liu, R. Liu, R. Lalancette, R. Szostak and M. Szostak, Org. Lett., 2017, 19, 1434–1437 CrossRef CAS PubMed.
- X. Li and G. Zou, Chem. Commun., 2015, 51, 5089–5092 RSC.
- S. Shi and M. Szostak, Org. Lett., 2016, 18, 5872–5875 CrossRef CAS PubMed.
- B. J. Simmons, N. A. Weires, J. E. Dander and N. K. Garg, ACS Catal., 2016, 6, 3176–3179 CrossRef CAS.
- S. Shi and M. Szostak, Synthesis, 2017, 49, 3602–3608 CrossRef CAS.
- S. Ni, W. Zhang, H. Mei, J. Han and Y. Pan, Org. Lett., 2017, 19, 2536–2539 CrossRef CAS PubMed.
- T. B. Boit, N. A. Weires, J. Kim and N. K. Garg, ACS Catal., 2018, 8, 1003–1008 CrossRef CAS PubMed.
- N. A. Weires, E. L. Baker and N. Garg, Nat. Chem., 2016, 8, 75–79 CrossRef CAS PubMed.
- J. M. Medina, J. Moreno, S. Racine, S. Du and N. K. Garg, Angew. Chem., Int. Ed., 2017, 56, 6567–6571 CrossRef CAS PubMed.
- J. A. Walker Jr., K. L. Vickerman, J. N. Humke and L. M. Stanley, J. Am. Chem. Soc., 2017, 139, 10228–10231 CrossRef PubMed.
- G. Meng and M. Szostak, Angew. Chem., Int. Ed., 2015, 54, 14518–14522 CrossRef CAS PubMed.
- C. Liu, G. Meng and M. Szostak, J. Org. Chem., 2016, 81, 12023–12030 CrossRef CAS PubMed.
- C.-L. Ji and X. Hong, J. Am. Chem. Soc., 2017, 139, 15522–15529 CrossRef CAS PubMed.
- S. Shi, G. Meng and M. Szostak, Angew. Chem., Int. Ed., 2016, 55, 6959–6963 CrossRef CAS PubMed.
- W. Srimontree, A. Chatupheeraphat, H.-H. Liao and M. Rueping, Org. Lett., 2017, 19, 3091–3094 CrossRef CAS PubMed.
- S.-C. Lee, H.-H. Liao, A. Chatupheeraphat and M. Rueping, Chem. – Eur. J., 2018, 24, 3608–3612 CrossRef CAS PubMed.
- C. Liu and M. Szostak, Angew. Chem., Int. Ed., 2017, 56, 12718–12722 CrossRef CAS PubMed.
- S. Shi and M. Szostak, Org. Lett., 2017, 19, 3095–3098 CrossRef CAS PubMed.
- J. Hu, Y. Zhao, J. Liu, Y. Zhang and Z. Shi, Angew. Chem., Int. Ed., 2016, 55, 8718–8722 CrossRef CAS PubMed.
- H. Yue, L. Guo, H.-H. Liao, Y. Cai, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 4282–4285 CrossRef CAS PubMed.
- G. Meng and M. Szostak, ACS Catal., 2017, 7, 7251–7256 CrossRef CAS.
- G. Meng and M. Szostak, Org. Lett., 2016, 18, 796–799 CrossRef CAS PubMed.
- H. Yue, L. Guo, S.-C. Lee, X. Liu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 3972–3976 CrossRef CAS PubMed.
- J. Hu, M. Wang, X. Pu and Z. Shi, Nat. Commun., 2017, 8, 14993 CrossRef PubMed.
- C. Liu, M. Achtenhagen and M. Szostak, Org. Lett., 2016, 18, 2375–2378 CrossRef CAS PubMed.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS.
- L. Hie, N. F. Fine Nathel, T. K. Shah, E. L. Baker, X. Hong, Y.-F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 524, 79–83 CrossRef CAS PubMed.
- R. Szostak, G. Meng and M. Szostak, J. Org. Chem., 2017, 82, 6373–6378 CrossRef CAS PubMed.
- A. Dey, S. Sasmal, K. Seth, G. K. Lahiri and D. Maiti, ACS Catal., 2017, 7, 433–437 CrossRef CAS.
- G. Meng, R. Szostak and M. Szostak, Org. Lett., 2017, 19, 3596–3599 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |