
Hole transporting materials for perovskite solar cells: a chemical approach
Javier
Urieta-Mora
 a,
Inés
García-Benito
b,
Agustín
Molina-Ontoria
*b and
Nazario
Martín
*ab
a,
Inés
García-Benito
b,
Agustín
Molina-Ontoria
*b and
Nazario
Martín
*ab
aDepartamento Química Orgánica, Facultad C. C. Químicas, Universidad Complutense de Madrid, Av. Complutense s/n, 28040 Madrid, Spain. E-mail: nazmar@ucm.es; Web: http://www.nazariomartingroup.com/
bIMDEA-Nanociencia, C/ Faraday, 9, Ciudad Universitaria de Cantoblanco, 28049 Madrid, Spain. E-mail: agustin.molina@imdea.org
First published on 4th October 2018
Abstract
Photovoltaic solar cells based on perovskites have come to the forefront in science by achieving exceptional power conversion efficiencies (PCEs) in less than a decade of research. This “still young” generation of solar cells is currently rivalling, in PCEs, well-established technologies, such as cadmium telluride (CdTe) and silicon. Further improvements in device stability by means of innovative materials are yet to come, with technology becoming closer to meeting the market requirements. Emerging from this groundbreaking discovery, a great number of charge transporting materials have flourished, which is particularly true for hole transporting materials (HTMs). The huge number of molecules prepared stem from design and engineering of a wide variety of new and also chemically modified old molecules where organic synthesis has played a fundamental role. In this review, the contribution of chemistry through those synthetic protocols used for producing new and innovative HTMs from relatively simple organic molecules is presented in a rational and systematic manner. The variety and impact of synthetic strategies followed, the structure–property relationship and stability, conductivity and device performance are highlighted from a chemical viewpoint.
 Javier Urieta-Mora | Javier Urieta-Mora obtained his degree in Chemistry in 2014 from the Complutense University of Madrid and his master's degree in Organic Chemistry in 2015 from the same university. In September 2015, he started his PhD under the supervision of Dr Agustín Molina Ontoria and Prof. Nazario Martín. His research interests are focused on materials for energy. He is currently researching on the synthesis and characterization of new charge transporting materials for high-efficiency perovskite solar cells. |
 Inés García-Benito | Inés García Benito was born in Madrid (Spain) in 1988. She obtained her degree in Chemical Engineering in 2011 from Rey Juan Carlos University (Madrid, Spain). In 2017, she obtained her PhD in Organic Chemistry under the supervision of Prof. Nazario Martín León at the Universidad Complutense de Madrid. Her work was based on the design and synthesis of new hole-transporting materials to be used in photovoltaic applications. Currently, she is a postdoctoral researcher in Prof. Nazeeruddin's group at EPFL (Sion, Switzerland). Her research interests are focused on materials science, in particular, the development of new 2D perovskites. |
 Agustín Molina-Ontoria | Agustín Molina-Ontoria obtained his PhD from the Complutense University of Madrid under the supervision of Prof. Nazario Martín, where he focused on photoinduced electron transfer in covalently linked donor–acceptor systems through molecular wires of organic nature. From 2011 to 2014, he joined the lab of Prof. Luís Echegoyen as a post doctoral researcher, where he expanded his research interests to carbon nanostructures, such as carbon nano-onions and organic photovoltaic solar cells. In 2014, he moved to IMDEA Nanoscience, where he is currently undertaking a second postdoc, with a focus on the synthesis of easily attainable new organic hole-transporting materials for perovskite solar cells to establish the relationship between structure and photovoltaic performance. |
 Nazario Martín | Nazario Martín, FRSC, is full professor at UCM and vice-director of IMDEA-Nanoscience. Dr h.c. by La Havana (Cuba) and Castilla La Mancha (Spain) Universities, his research interests are focused on the molecular and supramolecular chemistry of carbon nanostructures in the context of chirality, molecular nanographenes, electron transfer, photovoltaic applications and nanoscience. He has published over 550 papers and supervised 39 theses. He is currently a member of the IAB of several international journals, including Chem. Soc. Rev., and he is the Editor-in-Chief of The Journal of Materials Chemistry (A, B and C). He is currently the President of the Confederation of Scientific Societies of Spain (COSCE). He has the “Advanced Grant” of the European Research Council (ERC) entitled “Chirallcarbon”. |
Introduction
The progress of humankind has been, and is still, intimately linked to energy. Since the Industrial Revolution (a major turning point), carbon-based fossil fuels have been by far our main energy source.1–3 However, an imminent and currently worrisome environmental contamination arose from their burning. As a result, this generation and future generations have to face new environmental challenges. In this regard, photovoltaics (PV) offers a feasible, clean and promising solution to overcome this problem, by harvesting and converting solar energy into electrical power. In the frame of a joint multidisciplinary study, recent results have shown not only a remarkable improvement in device efficiencies, but also a substantial reduction in fabrication costs. Notably, since the first silicon-based device prepared by Chapin in 19544 and the subsequent third generation PV technologies reported by Tang in 1986,5 no other PV technology has drawn so much attention than perovskite solar cells.6–8 Actually, one of the singular features of perovskites is their ambipolar behavior, n- and p-type conduction, which enables two different device architectures. Conventional n–i–p devices can be divided, in turn, into mesoscopic (in which the perovskite is infiltrated into a mesoporous scaffold of a metal oxide (TCO/c-TiO2/m-TiO2/perovskite/HTM/metal)) and planar heterojunction ((TCO/ETM/perovskite/HTM/metal), in which the perovskite is sandwiched between electron- and hole-selective contacts) structures. Both concepts have been transferred to the so-called inverted architectures (p–i–n), in which the aforementioned stack is inverted (TCO/HTM/perovskite/ETM/metal) (Fig. 1). | ||
| Fig. 1 Schematic illustration of the most common device architectures of PSCs: (a) (n–i–p) mesoporous and planar structures. (b) Energy diagram of the different components of a conventional PSC. (c) Inverted (p–i–n) mesoporous and planar structures. (d) Energy diagram of the different components of an inverted PSC. | ||
The scope of this review refers to the main classes of small organic molecules used as Hole Transporting Materials (HTMs) for perovskite solar cells. Furthermore, strategies and reaction types that chemists have used to construct a huge number of modified chemical structures with the ultimate goal of enhancing the device performance are specifically considered. Preparation of HTMs requires, however, rational chemical synthesis to obtain suitable materials for the fabrication of devices. In this regard, the imagination of chemists has produced a wide variety of chemical structures able to act as HTMs in both conventional n–i–p and inverted p–i–n perovskite solar cell (PSC) devices.
Most of the HTMs used in PSCs are organic semiconductors mainly due to their milder processing conditions compatible with perovskites. However, because of their organic nature, HTMs exhibit relatively low hole mobility and, therefore, they require the presence of a chemical dopant such as lithium-bis(trifluoromethanesulfonyl)imide (LiTFSI), thus increasing the device efficiency. Using the additive approach, first employed in solid-state dye sensitized solar cells (ss-DSSCs),9 allows perovskite solar cells to reach excellent power conversion efficiencies. However, the hygroscopic properties of additives promote device degradation.10 Currently, the vast majority of hole transporting materials reported for perovskite solar cells introduce additives to further improve the device performance. In this sense, 2,2′,7,7′-tetrakis[N,N-bis(p-methoxyphenyl)amino]-9,9′-spirobifluorene (spiro-OMeTAD) suffers from low hole mobility and conductivity in its pristine form, which leads to a decreased efficiency. Yet, this drawback can be circumvented by adding tert-butylpyridine (t-BP) and LiTFSI, both employed to modify the electrical properties of the HTM and the perovskite (Fig. 2).11–13 The presence of t-BP not only significantly improves the film-forming ability of the HTL (hole-transporting layer) by reducing the number of pinholes, but also avoids the aggregation of lithium salt by favouring its distribution.10,14 Recently, Snaith and coworkers showed that t-BP increases the photovoltaic performance and the steady-state efficiency of perovskite solar cells. The authors surmised that the main reason stems from the direct contact between the t-BP and the perovskite crystal, by inducing a p-doping of the surface of the perovskite film.15 On the other hand, LiTFSI is reported to boost conductivity in HTMs.16,17 This effect has extensively been studied in the case of spiro-OMeTAD and unveiled how it is affected by moisture and oxygen.18–20 Seok and coworkers introduced a cobalt(III) complex, tris[2-(1H-pyrazol-1-yl)-4-tert-butylpyridine]cobalt(III)tris[bis(trifluoromethylsulfonyl)imide] (FK209), as a p-type dopant for spiro-OMeTAD and studied the synergistic effect between LiTFSI and FK209 in combination with the spiro-OMeTAD (Fig. 2).21 Unlike FK209, LiTFSI does not chemically oxidize the HTM directly. Actually, FK209 produces an effective electron transfer to the electron donor (HTM), leaving the HTM partially oxidized (radical cation). As a consequence, higher conductivities are obtained due to the formation of additional holes, which leads to an increase in the charge carrier density. Co-doping the HTM with LiTFSI and FK209 also results in an increased open circuit voltage (Voc) due to the reduced carrier recombination at the interface between the perovskite and the HTM, which eventually leads to improved PCEs.22 Additionally, other p-type dopants, namely, phosphonic acid,23 tris(pentafluorophenyl)borane,24 F4-TCNQ,25 AgTFSI26 or Cu(II) complexes,27 have been employed as additives for photovoltaic applications.
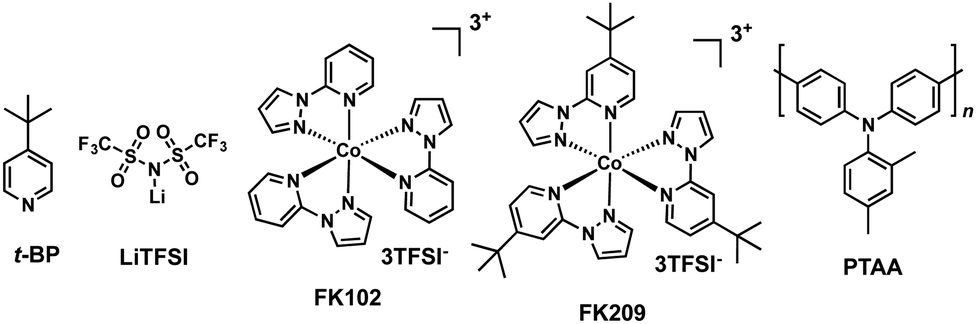 | ||
| Fig. 2 Molecular structures of t-BP, LiTFSI, FK102, FK209 and PTAA. | ||
This process is accompanied by a negative effect on device stability due to the complex oxidation process associated with undesired ion migration/interactions. Therefore, more recently, new and efficient dopant-free HTMs have been explored. Thus, the race to reach the efficiencies reported with doped HTMs in PSC devices using dopant-free HTMs has started and the values reached are becoming closer and almost comparable to the top record of 22.7%.28
Thus, in contrast to previous reviews on HTMs, we would like to emphasize different chemical structures used as the most significant HTMs together with the chemical strategies and reactions involved in their preparation29 – that is, focusing on the presentation of the main families of HTMs from a rational and systematic chemical approach.
In the following, we will present the different families of small organic HTMs synthesized in recent years organized according to the different chemical structures which constitute the core of the HTMs. Before starting, poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA), which is a polymeric structure of triphenylamines (TPAs), deserves a special mention as it currently holds the PCE world record for PSCs (Fig. 2).28 However, polymeric structures have certain drawbacks such as batch-to-batch reproducibility, difficult purification, and broad molar mass distribution. In this regard, small organic molecules offer potential advantages such as well-defined molecular weight, easy purification and good reproducibility. We will start with the 9,9′-spirobifluorene family since it is the core which is present in the most used and well-known HTM molecule, namely spiro-OMeTAD.
Hole transporting materials
Organic chemistry offers a wide variety of tools for designing and synthetizing an extensive assortment of organic molecules, “à la carte”, that can be employed as central scaffolds for HTMs. These molecules must meet general criteria. (i) They must be thermally and photochemically stable. (ii) The highest occupied molecular orbital (HOMO) must be slightly higher in energy than the valence band edge of the perovskite [−5.40 eV for MAPbI3 and −5.65 eV for (FAPbI3)0.85(MAPbBr3)0.15] to ensure an efficient extraction of the photogenerated holes. However, there is no consensus on what are the appropriate HOMO energy values.30–32 (iii) They must possess an optimal, or at least a reasonable, hole mobility for transporting the holes to the counter-electrode. (iv) High solubility is desirable for better film-forming properties. (v) A reduced tendency to crystallize (Tg > 100 °C) is required for avoiding phase transitions during device operation. (vi) For commercialization prospects must be easily attainable. (vii) Strong absorption is not an important requirement for obtaining highly efficient HTMs.The multiple and crucial tasks provided by the HTM include: (i) the aforementioned efficient extraction of the photogenerated holes, improving their transport to the counter-electrode and acting as an energetic barrier to block the electron transfer to the anode; (ii) improving the device stability by preventing the direct contact between the perovskite and the metal electrode (typically Au, Ag and Al) and protecting from diffusion of the electrode into the perovskite layer;33,34 (iii) suppressing the charge recombination losses at the interface between the perovskite and HTM by obtaining a fully and uniform coverage of the perovskite layer; and (iv) affecting the open circuit voltage (Voc), not only by the presence of dopants in the HTM layer,35 but also with a reduced energy disorder of the HTM layer (related to the chemical structure of the HTM),36 and by the different mobility/conductivity values of the HTM.
The spiro-OMeTAD molecule: background and synthesis
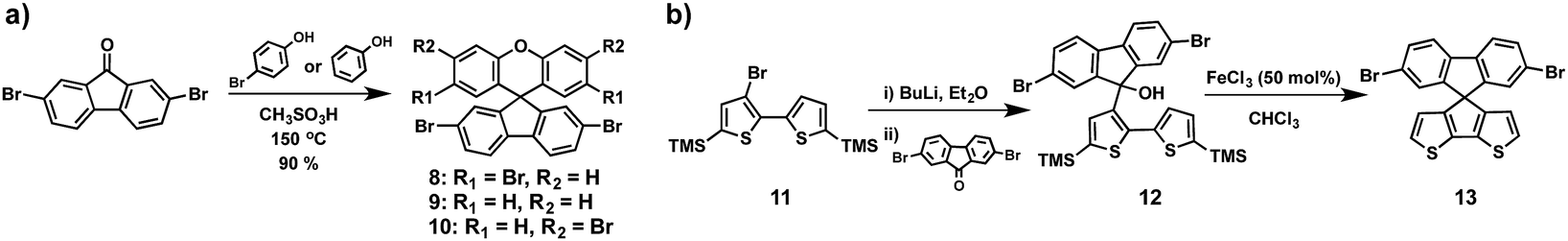
The “spiro concept” comprises two extended π-systems, which are connected to a tetrahedral sp3-hybridized atom, resulting in an orthogonal configuration between the aromatic rings.37,38 This spiro linkage improves the morphological stability, whereas the electronic properties of the electroactive moieties remain intact. Owing to its attractive properties, 9,9′-spirobifluorene has been exploited for organic electronics, mainly for photovoltaic applications.39,40 The synthesis of 9,9′-spirobifluorene was first published in 1930.41 Clarkson and Gomberg accomplished the preparation of the unsubstituted 9,9′-spirobifluorene (3) starting from 2-aminobiphenyl, which was transformed into the corresponding 2-iodobiphenyl (1) through Sandmeyer's reaction. Originally, 9-(biphenyl-2-yl)-9-fluorenol (2) is obtained in good yield, by first preparing the Grignard reagent of compound 1, followed by the addition of 9-fluorenone.42 Hitherto, this has been the most employed synthetic route for the preparation of spirobifluorene derivatives. In 1990, Tour and coworkers43 used an alternative route to synthesize 9-(biphenyl-2-yl)-9-fluorenol (2) by treating 2-iodobiphenyl (1) with 2 equivalents of t-BuLi at low temperatures (−78 °C) and quenching with 9-fluorenone. The fluorenol derivative undergoes a ring closure reaction and dehydration in refluxing acetic acid and catalytic amounts of hydrochloric acid, giving rise to 9,9′-spirobifluorene (3) in good yields. Finally, the four-fold bromination of the spirobifluorene requires an excess of bromine at room temperature in DMF or CH2Cl2 but few reports used stoichiometric amounts of bromine and a Lewis acid as catalyst (FeCl3) at 0 °C. The overall yield for the preparation of 2,2′,7,7′-tetrabromo-9,9′-spirobifluorene (4) spans a large range from 45% to 83%, as shown in Scheme 1.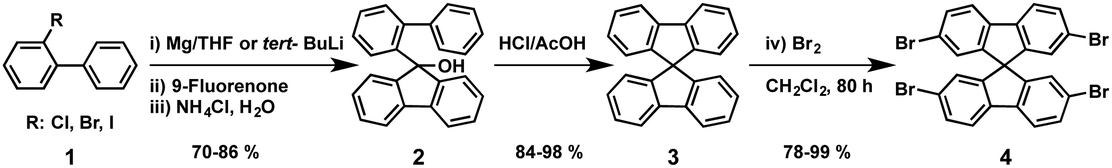 | ||
| Scheme 1 Synthesis of 2,2′,7,7′-tetrabromo-9,9′-spirobifluorene. | ||
The molecular structure of spiro-OMeTAD consists of a 9,9′-spirobifluorene central platform, which is functionalized with electron-donating p,p′-dimethoxydiphenylamine (DPA-OMe) at the 2, 2′, 7 and 7′ positions (Fig. 3). This structure emerged as an alternative material to TPD (N,N′-diphenyl-N,N′-bis(3-methylphenyl)4,4′-diamine) for organic electroluminescence devices, which suffered from low morphological stability of the amorphous film, owing to its low glass transition temperature (Tg = 62 °C).44 Introducing DPA-OMe units resulted not only in an improved morphological stability (Tg higher than 100 °C), but also in a better solubility and stronger electron donor properties compared to those without substituents on the periphery of the diphenylamines. The first ss-DSSCs incorporating spiro-OMeTAD as a solid charge-transport material were reported in 1998 by Grätzel and coworkers.9 Using dopants, they were able to obtain an overall efficiency of 0.74%, far from the 7–8% first reported using the liquid iodide/triiodide redox couple.45 More recently, PSCs experienced the same evolution, from liquid junction cells to spiro-OMeTAD-based solid-state heterojunctions, witnessing not only an astonishing improvement in their efficiency (9.7%–10.9%) but also an improvement in their stability.12,13 To date, spiro-OMeTAD has been the most highlighted and commonly used benchmark in PSCs. Using spiro-OMeTAD, high performances surpassing 20% have been recently achieved.46–48Table 1 summarizes the electronic properties and photovoltaic parameters of PSCs using spiro-derivatives discussed in this section.
 | ||
| Fig. 3 Molecular structures of spiro-OMeTAD and other related spiro-OMeTAD-type HTMs. | ||
| HTM | HOMOd (eV) | σ (S cm−1) | μ h (cm2 V−1 s−1) | V oc (V) | J sc (mA cm−2) | FF | PCE (%) | Architecturee | Perovskite | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a LiTFSI and t-BP. b LiTFSI, t-BP, FK209. c Dopant-free. d HOMO energy levels were converted from the original work using Fc/Fc+ at 5.1 eV vs. vacuum. e M = mesoporous, P = planar and IP = inverted planar configurations. | ||||||||||
| HTM-1 | −5.25 | — | — | 1.01 | 21.1 | 0.65 | 13.9 | M | MAPbI3 | 49 |
| HTM-2 | −5.16 | — | — | 1.02 | 21.2 | 0.78 | 16.7 | M | MAPbI3 | 49 |
| HTM-3 | −5.22 | — | 1.26 × 10−5 | 1.07 | 18.2 | 0.80 | 15.75 | IP | MAPbI3 | 50 |
| HTM-4 | −5.32 | — | 1.90 × 10−5 | 1.06 | 19.2 | 0.78 | 15.92 | IP | MAPbI3 | 50 |
| HTM-5 | −4.82 | — | 0.25 × 10−5 | 0.96 | 16.6 | 0.75 | 11.92 | IP | MAPbI3 | 50 |
| HTM-6 | −5.18 | 1.1 × 10−4 | 1.9 × 10−4 | 1.14 | 24.2 | 0.71 | 19.84 | M | MAPbI3 | 53 |
| HTM-7 | −5.18 | 1.9 × 10−4 | 5.5× 10−5 | 1.13 | 23.4 | 0.73 | 19.8 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 54 |
| HTM-8 | −5.41 | — | 2.2 × 10−5 | 1.01 | 18.0 | 0.73 | 13.25 | M | MAPbI3 | 55 |
| HTM-9 | −5.25 | — | 1.5 × 10−4 | 1.02 | 21.2 | 0.77 | 16.77 | M | MAPbI3 | 55 |
| HTM-10 | −5.41 | — | 3.3 × 10−5 | 1.00 | 18.9 | 0.74 | 14.06 | M | MAPbI3 | 55 |
| HTM-11 | −5.18 | — | — | 1.15 | 22.7 | 0.76 | 20.2 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 56 |
| HTM-12 | −5.24 | 3.26 × 10−4 | 8.25 × 10−5 | 0.95 | 21.3 | 0.67 | 13.6 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 59 |
| HTM-13 | −5.17 | 8.43 × 10−4 | 6.81 × 10−4 | 1.15 | 23.4 | 0.77 | 20.8 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 59 |
| HTM-14 | −5.22 | — | — | 1.05 | 19.5 | 0.69 | 14.19 | M | MAPbI3 | 60 |
| HTM-15 | −5.30 | — | 7.03 × 10−5 | 1.07 | 19.10 | 0.79 | 16.08 | P | MAPbI3 | 61 |
| HTM-16 | −5.29 | — | 9.14 × 10−5 | 1.10 | 20.60 | 0.80 | 18.06 | P | MAPbI3 | 61 |
| HTM-17 | −5.02 | — | 4.39 × 10−5 | 0.77 | 11.28 | 0.58 | 5.01 | P | MAPbI3 | 61 |
Pendant methoxy groups on the periphery of the arylamine units provide a higher solubility and tunability of the HOMO energy level of the HTMs. Furthermore, they also serve as binding sites to the underlying perovskites through the oxygen atom. The inductive electron σ-withdrawing nature of the –OMe group is balanced with its resonance electron-donating character. These electronic effects can be finely tuned by adjusting the positions of the substituents. In this regard, substitution at the para position results in an enhanced resonance electron-π-donating character but; in contrast, substitution at the meta position leads to a more pronounced σ-withdrawing behavior. Additionally, the steric effects at the ortho position can also affect the HOMO energy levels. Motivated by this strategy, Seok and co-workers accomplished the synthesis of spiro-OMeTAD derivatives attending to the position of the –OMe pendant groups on the diphenylamines, from para (spiro-OMeTAD) to meta (HTM-1) and ortho (HTM-2) substitutions (Fig. 3).49 To this end, tailored diarylamines were obtained from 4-bromoaniline and the corresponding anisidine in moderate yields (50%). The four-fold palladium-catalyzed Buchwald–Hartwig type amination reaction of the tailored diarylamines (DPA) with 2,2′,7,7′-tetrabromo-9,9′-spirobifluorene afforded the corresponding spiro-OMeTAD derivatives with different positions of the methoxy substituents. In this sense, the electronic effects, derived from each particular arrangement of the methoxy groups, are the main factor affecting the optical and electronic properties of these HTMs (Fig. 4). Compared with the pp-spiro-OMeTAD, HTM-1 and HTM-2 show significantly blue-shifted absorption onsets, implicating wider optical band-gaps with respect to the spiro-OMeTAD. The electrochemical behaviors of the new HTMs are indeed very similar. The ortho substitution has a similar electron donor strength to that observed for the para position (−5.16 eV). On the other hand, the meta substitution entails a deeper HOMO energy level owing to its σ-withdrawing behavior (−5.25 eV). CH3NH3PbI3-based mesoscopic devices incorporating the spiro-OMeTAD derivatives yielded efficiencies in the range of 14–16.7%, displaying similar open-circuit voltage (Voc) and short-circuit current (Jsc) values among them. The ortho-substituted spiro-OMeTAD (HTM-2) exhibits the highest efficiency with a markedly higher fill factor (FF) (0.77) than HTM-1, which is ascribed to its low series resistance (Rs) and high shunt resistance (Rsh). In contrast, as a consequence of the higher resistance, in devices comprising HTM-1, the performance is dramatically decreased due to its low FF of 0.65. Noteworthily, the PCE values of the commercial and synthesized pp-spiro-OMeTAD are indeed very similar.
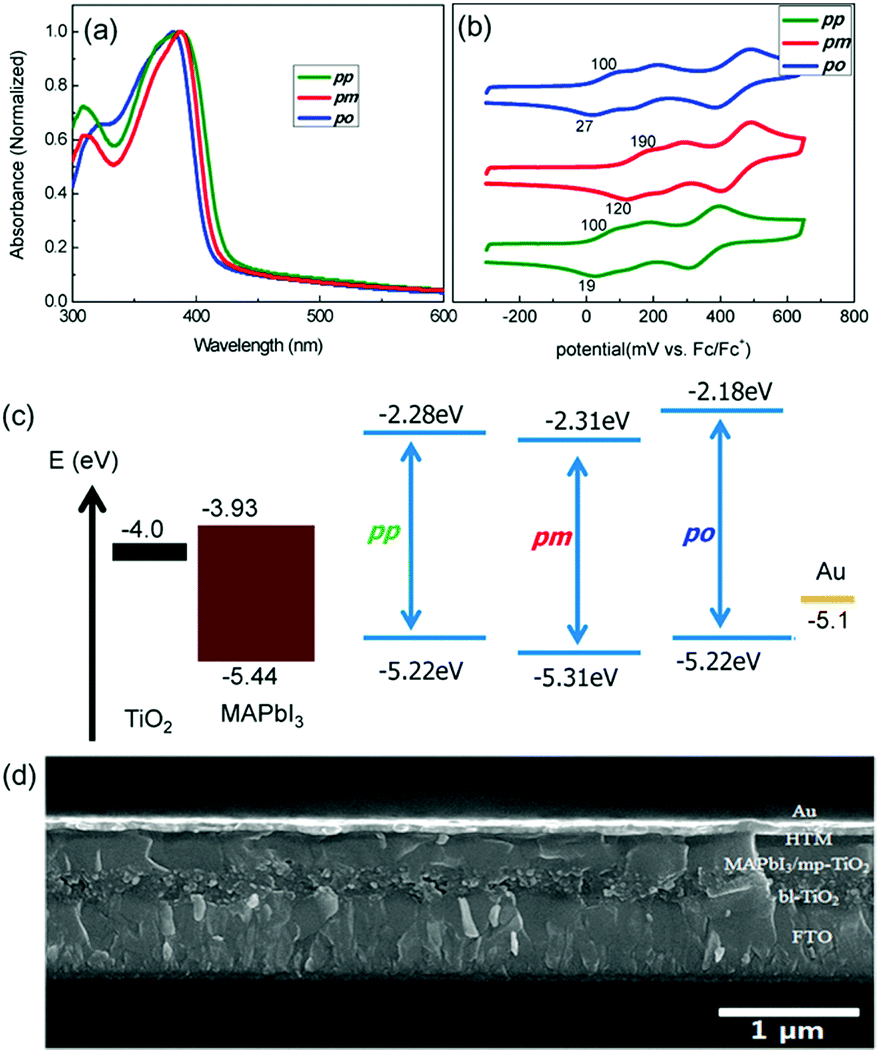 | ||
| Fig. 4 (a) UV-vis absorption spectra of spiro-OMeTAD (pp), HTM-1 (pm) and HTM-2 (po) in chlorobenzene. (b) Cyclic voltammograms of spiro-OMeTAD, HTM-1 and HTM-2. (c) Energy diagram of the different components of the PSC. (d) Cross-sectional SEM image showing the different layers of a PSC device. Reproduced from ref. 49 with permission from American Chemical Society, copyright 2014. | ||
The Hammett strategy for the modulation of the HOMO energy level is not only applicable to the position of the substituents, but also to the modification of the donor or acceptor strength of the pendant group. Huang and co-workers reported three new spiro-OMeTAD derivatives and explored how introducing different pendant groups (ethyl, methylsulfanyl and N,N-dimethylamino) at the para position of the diphenylamines, in order to determine how these substituents affect their electronic properties and photovoltaic performance (Fig. 3).50 These derivatives were synthesized using a stepwise protocol, following the spiro-OMeTAD procedure. The main challenge of these syntheses lies in the preparation of custom-made diphenylamines (Scheme 2). 4-Ethyl-N-(4-ethylphenyl)aniline (7) and N1-(4-(dimethylamino)phenyl)-N4,N4-dimethylbenzene-1,4-diamine (6) derivatives were obtained in reasonable yields using the standard Buchwald–Hartwig reaction. On the other hand, bis(4-methylthiophenyl)amine (5) is obtained in a higher yield (65%). Attending to the “inductive” and “resonance” parameters, nitrogen substituents are stronger electron donors than the oxygen ones owing to their stronger π-donating ability. On the other hand, increasing the electronegativity of the substituent does not necessarily affect the donor ability, but the inductive parameter does. Given these general trends, the stronger π-donation ability of the N,N-dimethylamino group leads to a higher HOMO energy (−4.82 eV) in comparison with spiro-OMeTAD. The –OMe substituent in spiro-OMeTAD exhibits similar π-donating character to –SMe, but at the same time inductively electron σ-withdrawing character, which results in a deeper HOMO energy level for HTM-4 with –SMe (−5.32 eV) than that of spiro-OMeTAD (−5.16 eV). Finally, the ethyl groups in the para position of the diphenylamine (HTM-3) do not have a negligible effect on its HOMO energy. The thermal behavior of the new spiro-derivatives unveiled both their amorphous and crystalline nature. The spiro-based HTMs were evaluated in inverted PSCs with an ITO/HTM/MAPbI3/PCBM/ZnO-NP/Al architecture. The best device had a PCE of 15.92%, which was obtained by incorporating HTM-4, followed by HTM-3 (15.75%), HTM-5 (11.92%) and spiro-OMeTAD (11.55%). The deeper HOMO energies of HTM-4 and HTM-3 are surmised to be the reason for their higher Voc values as compared with HTM-5 and spiro-OMeTAD. Scanning electron microscopy (SEM) of the as-prepared perovskite on top of the HTMs revealed a strong impact on the grain size formation of MAPbI3, which is affected by the different hydrophobic surfaces of the HTMs. Thus, the smaller perovskite grain sizes stem from the lower hydrophobic surface of HTM-5, in comparison with the larger grain size of the perovskites attained when using spiro-OMeTAD, HTM-4 and HTM-3. Time-resolved photoluminescence (TRPL) revealed faster dynamics along with lower average decay times for HTM-3 (67 ns), HTM-5 (8 ns), and HTM-4 (53 ns) than spiro-OMeTAD (131 ns), which is attributed to an improved hole extraction and charge dissociation.
 | ||
| Scheme 2 Synthetic route for the preparation of molecularly modified arylamines. | ||
Spiro-like-based cores for HTMs
Many examples of the preparation of spiro-like structures have been reported in the literature (Fig. 5). Most of them require multi-step synthetic procedures, which is in contrast to the simplicity of one-pot approaches to obtain spiro derivatives.51Table 1 gathers the electronic properties and photovoltaic parameters of PSCs using spiro-like HTMs discussed in the section.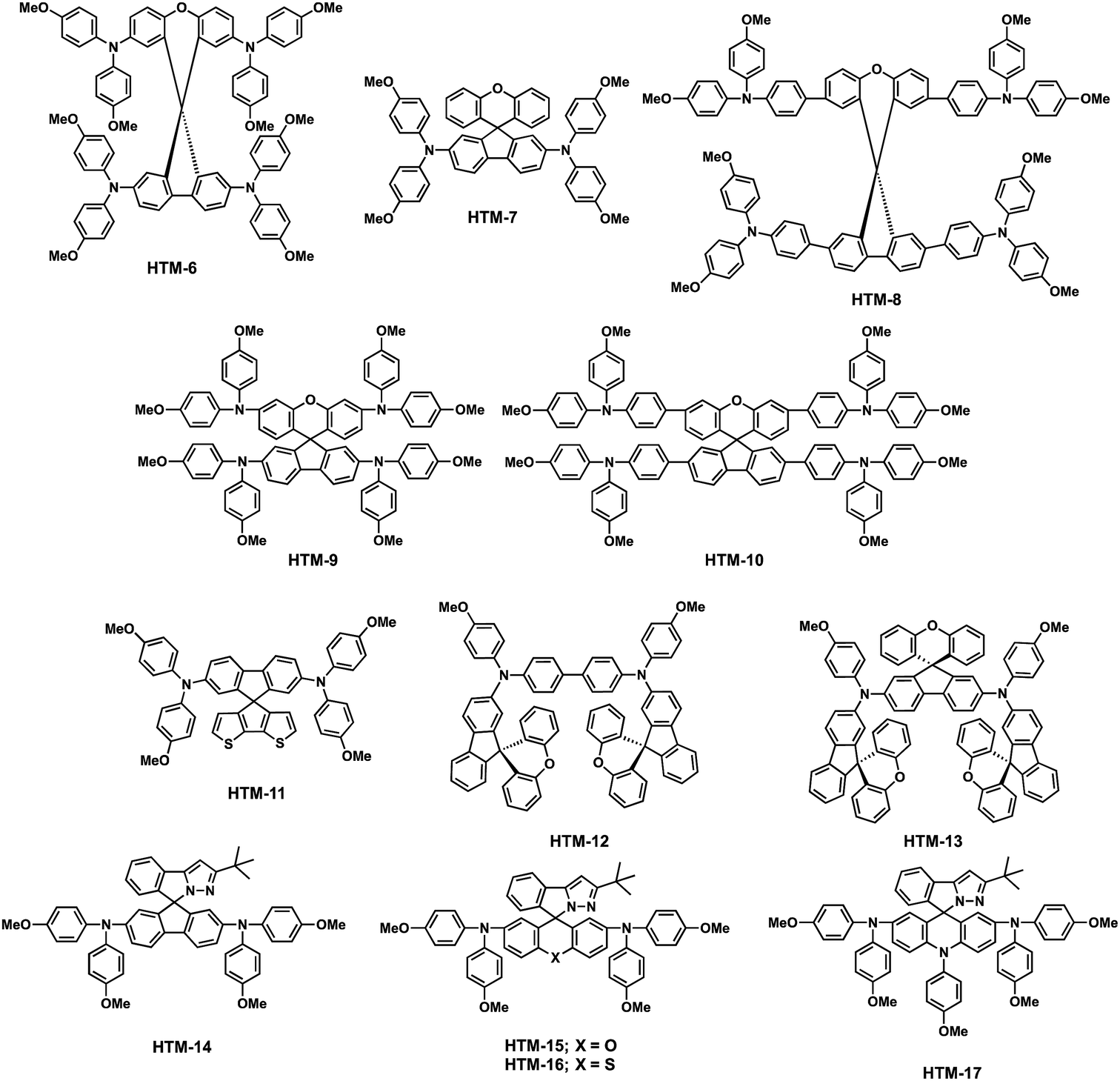 | ||
| Fig. 5 Molecular structures of spiro-type HTMs. | ||
The group of Huang serendipitously accomplished the preparation of the spiro(fluorene-9,9′-xanthene) (SFX) core based on a one-pot route (Scheme 3a).52 Starting from commercially available materials, 2,7-dibromo-9-fluorenone reacts with phenol or 4-bromophenol in the presence of methanesulfonic acid to give the corresponding SFX (8, 9 and 10) in excellent yields (>90%) and, importantly, no column chromatography is necessary for the following synthetic steps (Scheme 3). Adopting this unexpected methodology, Hagfeldt, Sun and co-workers reported in 2016 the synthesis and photovoltaic application of highly efficient SFX based HTMs for solid-state DSSCs and for PSCs (Fig. 5).53 The new four-armed diphenylamine (DPA)-substituted SFX was prepared in just two steps with an overall yield of 74%. Owing to the structural similarity with spiro-OMeTAD, HTM-6 exhibits not only similar HOMO energy, but also similar absorption and emission properties. Additionally, according to theoretical calculations, HTM-6 shows a higher hole transport due to the smaller reorganization energy when compared with spiro-OMeTAD. This was further confirmed by hole mobility measurements, 1.9 × 10−4 cm2 V−1 s−1 for HTM-6 and 8.1 × 10−5 cm2 V−1 s−1 for spiro-OMeTAD. Doped HTM-6 was incorporated in mesoscopic devices with the normal configuration of FTO/c-TiO2/m-TiO2/perovskite/HTM-6/Au, which demonstrated a record PCE value of 19.84%, outperforming spiro-OMeTAD. Afterwards, the same group reported the synthesis of two-armed SFX substituted with two DPA groups on the fluorene unit, HTM-7 (Fig. 5).54 The molecular structure was unambiguously confirmed by X-ray diffraction of a single crystal. HTM-7 exhibits similar electrochemical, optical and mobility values to those previously reported four-armed HTM-6. Devices based on HTM-7 afforded a PCE value of 19.8%. A striking finding is that after 5 weeks the sealed device showed only 2.5% PCE loss.
 | ||
| Scheme 3 Synthetic strategies for the preparation of other related spiro-type central scaffolds. | ||
DPAs (HTM-6 and HTM-9) and TPAs (HTM-8 and HTM-10) were covalently linked to the SFX central scaffold through inexpensive copper-catalyzed Ullmann N-aryl coupling and Suzuki cross-coupling reactions, respectively (Fig. 5).55 The increased conjugation of the TPA-functionalized SFX derivatives results in a red-shifted absorption of around 50 nm with respect to those substituted with DPAs. In addition, no difference in the absorption was observed regarding the position of the substitution. A similar trend was observed from the CV measurements. The stronger electron-donating character of the DPA results in a higher HOMO energy level (−5.18 and −5.25 eV) in comparison with the TPA (−5.41 eV). The hole mobilities of doped HTMs were determined using SCLC. The 3′,6′-position of the DPA in the xanthene ring on HTM-9 improves by one order of magnitude the hole mobility (1.5 × 10−4 cm2 V−1 s−1) of its analogues (3.3–9.9 × 10−5 cm2 V−1 s−1). Due to this benefit, the PCEs of mesoscopic devices using CH3NH3PbI3 reach values up to 16.77%, which are not only superior to those of the other SFX-derivatives but also to spiro-OMeTAD. Additionally, devices containing HTM-9 using mixed cation and mixed anion perovskites lead to higher efficiencies, up to 17.7%.
An original HTM was reported by Nazeeruddin and coworkers (HTM-11) (Fig. 5). This derivative comprises a dissymmetrical spiro-like structure with half fluorene and the other half cyclopenta[2,1-b:3,4-b′]dithiophene.56 The synthesis of TMS-capped bithiophene 11 was conveniently performed in three synthetic steps, starting from commercially available 2,3-dibromothiophene and following previous reports.57 By treating 11 with n-BuLi and further quenching it by adding 2,7-dibromofluorenone successfully afforded fluorenol 12. Finally, the latter is subjected to ring closure reaction and dehydration, using FeCl3 in CHCl3 (Scheme 3b). HTM-11 was prepared in a 33% overall yield.
Theoretical calculations by Rego and coworkers predicted that the interaction of spiro-OMeTAD/perovskite occurs through the methoxy groups on the periphery of the arylamines and the surface of the perovskite terminated on PbI6 octahedra. Attractive electrostatic interactions with Pb(II) are responsible for this connection, while those with iodides are respulsive.58 On the other hand, HTM-11 exhibited not only the aforementioned interactions with the surface of the perovskite but also possibly additional thiophene–iodide interactions (Fig. 6). This may result in an enhanced interfacial coupling between HTM-11 and the perovskite, thus facilitating the injection of the holes from the latter to the HTM. Both, spiro-OMeTAD and HTM-11, showed similar optical and electrochemical properties (−5.16 eV vs. −5.18 eV). The authors compared the photovoltaic behaviors of HTM-11 and spiro-OMeTAD in combination with different perovskite-based (MAPbBr3, MAPbI3 and mixed perovskite) mesoscopic devices. Excellent results were obtained using (FAPbI3)0.85(MAPbBr3)0.15, with efficiencies up to 20.2%. Under the same conditions, spiro-OMeTAD performed worse than HTM-11. In addition, no device degradation was observed after two months’ storage under dry conditions in the dark.
 | ||
| Fig. 6 Docking of the spiro-OMeTAD and HTM-11 (FDT) on the MAI-terminated (110) perovskite surface. Attractive electrostatic interactions between the HTM and the perovskite surface are highlighted. Reproduced from ref. 56 with permission from Springer Nature, copyright 2016. | ||
Recently, the groups of Sun, Jen and Hagfeldt expanded this approach by further tailoring two new SFX-based HTMs (Fig. 5).59 These novel 3D HTMs display two (HTM-12) or three (HTM-13) SFX moieties in their structure. Increasing the number of SFX units results in an improved film-forming ability and, therefore, a reduced tendency to create pinholes that can compromise the device performance. The synthesis of these derivatives was accomplished in a one-pot procedure by assembling the 4,4′-dibromo-1,1′-biphenyl or dibromo-SFX (14) with 4-methoxyaniline first and, in a second step, with the monobromo-SFX (15) to obtain HTM-12 and HTM-13 in excellent yields, 81% and 84%, respectively (Scheme 4). Both HTMs exhibit not only higher Td (441 °C for HTM-12 and 446 °C for HTM-13) than that of spiro-OMeTAD (Td = 422 °C) but also higher glass transition temperatures (Tg). HTM-12 and HTM-13 absorb light only in the UV region, whereas, after doping, chemically oxidized HTMs exhibit moderate absorbances at 493 and 525 nm, respectively, which correspond to the formation of the radical cation of the HTMs.59 Slightly deeper HOMO energy levels (−5.24 eV for HTM-12 and −5.17 eV for HTM-13) than spiro-OMeTAD (−5.16 eV) were determined from CV, indicating an excellent band alignment with the valence band edge of the perovskites, which can lead to an effective extraction of the charges. This was further confirmed by measuring the steady-state photoluminescence (PL). When deposited on top of the perovskites, HTM-13 shows a stronger quenching of the PL than HTM-12 and spiro-OMeTAD, implying a more efficient hole extraction between HTM-13 and the perovskite. The photo-induced current decay further corroborates the faster dynamics of HTM-13, owing to its higher hole mobility. Conventional mesoporous (FAPbI3)0.85(MAPbBr3)0.15-based devices with HTM-12, using LiTFSI, t-BP and FK209 as dopants, exhibit a maximum PCE of 13.6%, while the champion device employing HTM-13 yields a PCE of 20.8%, which is higher than that reported for spiro-OMeTAD under the same conditions. HTM-12 exhibits much lower efficiency due to its lower solubility and, therefore, poor film formation during the deposition process. On the other hand, introducing an additional bulky xanthene unit (HTM-13) results in a more distorted and extended 3D structure, providing a more soluble (up to 70 mg mL−1) material. The high solubility of HTM-13 eventually leads to high quality film formation and coverage, which is of paramount importance to reduce the charge recombination losses at the interface between the HTM and the perovskite.59 Furthermore, HTM-13 has longer-lasting device stability (after 6 months the PCE decreased 7%) than spiro-OMeTAD (10% of PCE losses after 6 months) not only in aging control experiments but also under ambient air conditions with a humidity of 60%.
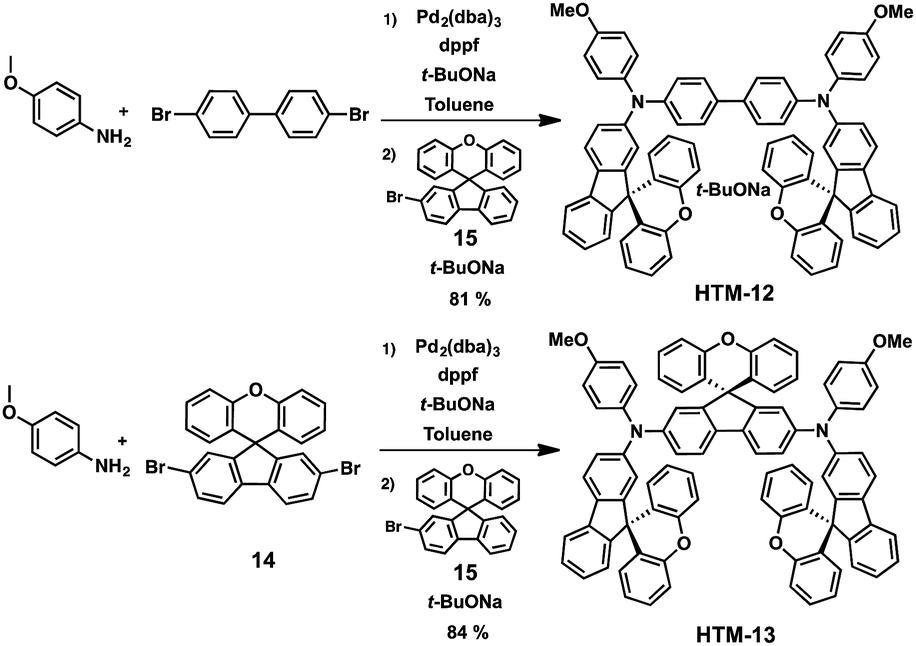 | ||
| Scheme 4 Synthetic routes for the preparation of HTM-12 and HTM-13. | ||
Other modifications of the spiro system incorporate a C–N linkage, such as phenylpyrazole/fluorene (HTM-14), xanthene (HTM-15), thioxanthene (HTM-16) and acridine (HTM-17), resulting in asymmetric and electronically diverse spiro-based HTMs (Fig. 5).60,61 The synthetic strategy previously presented to obtain spiro-OMeTAD can also be applied for the preparation on these C–N derivatives. By treating 2-bromophenyl pyrazole derivatives with n-BuLi and quenching with the corresponding ketone, followed by hydrolysis in a mixture of acetic acid and concentrated hydrochloric acid at room temperature, all derivatives can be obtained in four to five steps in overall yields higher than 20%. Furthermore, HTM-15, HTM-16 and HTM-17 exhibit 70 nm blue-shifted absorption values when compared to spiro-OMeTAD due to the disrupted conjugation over the xanthene, thioxanthene and acridine, while HTM-14 displays an identical optical band gap. These HTMs are electronically diverse, as can be seen from their HOMO energy levels, which are strongly affected by the nature of their substituents. Interestingly, HTM-15 (−5.30 eV), HTM-16 (−5.29 eV) and HTM-14 (−5.22 eV) display deeper HOMO levels than spiro-OMeTAD (−5.16 eV). Conversely, acridine is a strong electron donating unit whereby HTM-17 exhibits minor stabilization of the HOMO level (−5.02 eV). The thioxanthene derivative exhibits the highest hole mobility due to the larger polarizable radius of S than O. The lower hole mobility of HTM-17 probably stems from the steric hindrance of the N-methoxyphenyl substitution, which hampers the intermolecular interactions.61 The new HTMs were tested using a planar configuration (ITO/SnO2/C60/MAPbI3/HTM/MoO3/Ag) with and without dopants (LiTFSI and t-BP). Owing to HTM-16's deeper HOMO energy and higher hole mobility, HTM-16-based planar PSCs display PCEs of 18.06% with dopants and 11.7% in dopant-free devices, which are higher than those of spiro-OMeTAD-based devices (16.15% and 8.51%) and those of HTM-15 (16.08% and 9.13%). Moreover, the lower performance of HTM-17 (5.01% and 0.48%) is ascribed to the unmatched HOMO level.
Carbocyclic cores
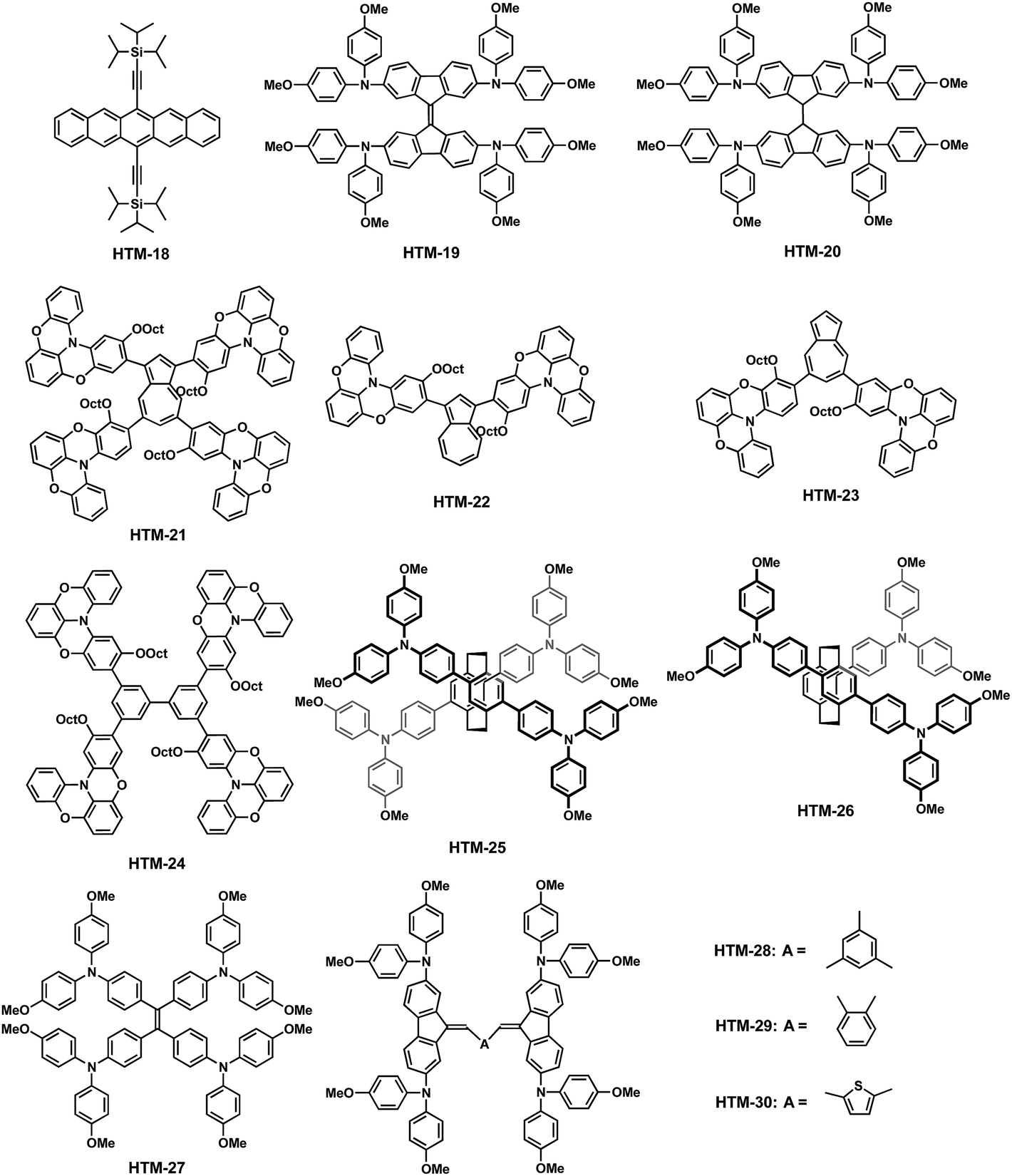
In addition to the aforementioned spiro-HTMs, a wide variety of small molecules having a carbocyclic structure as the central core have also been used as efficient HTMs. The electronic properties and the photovoltaic parameters of the following carbocyclic-based HTMs including their device architectures are shown in Table 2. Ahmad and coworkers reported the use of 6,13-bis(triisopropylsilylethynyl)pentacene (HTM-18) as a HTM for dopant-free PSCs (Fig. 7).62 This was one of the first examples of dopant-free HTMs. The TIPS groups at the C-6 and C-13 positions of the pentacene central scaffold render not only solubility to the acene, which simplify its processing and purification, but also promote self-assembly into 2D columnar π-stacking structures.63 The distance between planes is significantly smaller in the TIPS-pentacene (3.33 Å) than in the unsubstituted pentacene (6.27 Å), indicating rather high hole mobilities.64–66 From a synthetic viewpoint, the preparation of TIPS-pentacene requires only two synthetic steps. The first step is the condensation of phthalaldehyde and 1,4-cyclohexanedione under basic conditions, followed by the addition to the carbonyl group and subsequent reduction, in nearly quantitative yield. HTM-18 absorption is characterized by three long-wavelength bands at 643, 592 and 549 nm, with a deeper HOMO (−5.43 eV) energy level than spiro-OMeTAD. Upon doping (LiTFSI and t-BP), the oxidation potential is cathodically-shifted and chemically irreversible. MAPbI3-based PSCs employing undoped HTM-18 gave a remarkable 11.5% efficiency under AM 1.5G irradiation. In turn, upon doping, the efficiency of the devices decreases to 8.20% due to the generation of trap sites and disorder in the packing of the HTM.62 Although the efficiency was not high enough compared to other doped-HTMs, the use of undoped-HTMs is envisaged to improve the long-term stability of PSCs.| HTM | HOMOd (eV) | σ (S cm−1) | μ h (cm2 V−1 s−1) | V oc (V) | J sc (mA cm−2) | FF | PCE (%) | Architecturee | Perovskite | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a LiTFSI and t-BP. b LiTFSI, t-BP, FK209. c Dopant-free. d HOMO energy levels were converted from the original work using Fc/Fc+ at 5.1 eV vs. vacuum. e M = mesoporous, P = planar and IP = inverted planar configurations. | ||||||||||
| HTM-18 | −5.43 | — | — | 0.91 | 20.84 | 0.60 | 11.51 | M | MAPbI3 | 62 |
| HTM-19 | −5.21 | 3.40 × 10−5 | 7.0 × 10−4 | 1.02 | 22.3 | 0.77 | 17.8 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 67 |
| HTM-20 | −5.18 | 8.65 × 10−5 | 1.58 × 10−4 | 1.15 | 24.2 | 0.71 | 19.80 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 71 |
| HTM-21 | −5.12 | — | 2.1 × 10−4 | 1.08 | 21.7 | 0.71 | 16.5 | M | MAPbI3 | 75 |
| HTM-22 | −5.20 | — | 7.6 × 10−5 | 0.91 | 15.9 | 0.67 | 9.7 | M | MAPbI3 | 75 |
| HTM-23 | −5.50 | — | 6.3 × 10−6 | 0.91 | 17.3 | 0.54 | 8.5 | M | MAPbI3 | 75 |
| HTM-24 | −5.35 | — | 2.4 × 10−5 | 0.70 | 2.31 | 0.28 | 0.45 | M | MAPbI3 | 75 |
| HTM-25 | −5.27 | — | 6.32 × 10−4 | 1.05 | 21.8 | 0.78 | 17.9 | P | MAPbI3 | 77 |
| HTM-26 | −5.28 | — | 5.1 × 10−4 | 1.03 | 21.4 | 0.74 | 16.3 | P | MAPbI3 | 78 |
| HTM-27 | −5.00 | — | 5.92 × 10−5 | 0.89 | 17.32 | 0.72 | 11.02 | M | MAPbI3 | 82 |
| HTM-28 | −5.18 | 1.6 × 10−5 | 7.0 × 10−6 | 1.14 | 21.7 | 0.71 | 17.77 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 86 |
| HTM-29 | −5.14 | 4.2 × 10−5 | 2.3 × 10−5 | 1.12 | 23.2 | 0.75 | 19.47 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 86 |
| HTM-30 | −5.16 | 4.9 × 10−5 | 8.0 × 10−5 | 1.14 | 22.5 | 0.77 | 19.96 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 86 |
 | ||
| Fig. 7 Molecular structures of carbocyclic core-based HTMs. | ||
In 2016, Nazeeruddin and coworkers reported a simple and easy method to obtain a HTM based on 9,9′-bifluorenylidene as the central moiety (HTM-19) (Fig. 7).67 9,9′-Bifluorenylidene is considered to be a rare case of a homomerous bistricyclic aromatic ene, owing to its central 5-member rings, which induce planarity in the final structure.68 The synthesis of a 9,9′-bifluorenylidene central scaffold was first reported in 1875 by de la Harpe and van Dorp.69 To date, there have been several approaches to obtain 9,9′-bifluorenylidene through facile and convenient methodologies, namely, from fluorenones or by Pd-catalyzed double cross-coupling. The authors employed a straightforward two-step synthetic procedure. Firstly, commercially available 2,7-dibromo-9H-fluoren-9-one was treated with Lawesson's reagent in refluxing toluene, to afford 2,2′,7,7′-tetrabromo-9,9′-bifluorenylidene. This reductive dimerization occurs via the formation of the corresponding thione followed by a carbine dimerization.70 The subsequent four-fold Buchwald–Hartwig C–N coupling reaction furnishes HTM-19 in 33% yield for the two-step synthesis. HTM-19 exhibits good thermal stability and a narrower band gap due to red-shifted absorption in the visible region relative to spiro-OMeTAD, which is not only attributed to its larger degree of conjugation but also to an intramolecular charge transfer from the DPA to the electron acceptor 9,9′-bifluorenylidene. The electron-donating diphenylamine moieties adjust the HOMO level to −5.21 eV. The lateral conductivities of HTM-19 and spiro-OMeTAD are rather similar when prepared in the lab, in sharp contrast to the higher conductivity of the commercial spiro-OMeTAD. Mesoscopic mixed perovskite (FAPbI3)0.85(MAPbBr3)0.15-based devices containing doped-HTM-19 were compared to lab-made spiro-OMeTAD. Interestingly, HTM-19 reaches a PCE of 17.8% in comparison to the 17.4% reached by the lab-made spiro-OMeTAD. Both show noticeable hysteresis. In this case, HTM-19 loses 70 mV in Voc, which is, however, countervailed by a gain of almost 2 mA cm−2 in Jsc. In turn, mesoscopic mixed perovskite (FAPbI3)0.85(MAPbBr3)0.15-based devices incorporating commercially available spiro-OMeTAD yield a PCE of 18.4% with improved Voc, Jsc and FF values and no hysteresis.
Later, in 2016, Sun, Hagfeldt and coworkers studied the influence of 3D molecular structures on the PV performance.71 Toward this end, they designed a HTM by replacing the twisted spiranic bond between the two fluorene halves in spiro-OMeTAD by a covalent “C–C” bond (HTM-20) and compared to the more rigid “C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C” linkage in HTM-19 (Fig. 8). The synthesis of HTM-20 relied on a facile two-step synthesis, firstly, a C–N amination reaction between 2,7-dibromofluorene and bis(4-methoxyphenyl)amine. Subsequently, the latter was treated with n-BuLi in the presence of cobalt(II) chloride to yield HTM-20 in excellent overall yield (71%). Compared with HTM-19, a change in the 3D geometry of the HTMs was observed, such as the rigid central connection for spiro-OMeTAD and HTM-19 compared to a rotatable linkage in HTM-20.
C” linkage in HTM-19 (Fig. 8). The synthesis of HTM-20 relied on a facile two-step synthesis, firstly, a C–N amination reaction between 2,7-dibromofluorene and bis(4-methoxyphenyl)amine. Subsequently, the latter was treated with n-BuLi in the presence of cobalt(II) chloride to yield HTM-20 in excellent overall yield (71%). Compared with HTM-19, a change in the 3D geometry of the HTMs was observed, such as the rigid central connection for spiro-OMeTAD and HTM-19 compared to a rotatable linkage in HTM-20.
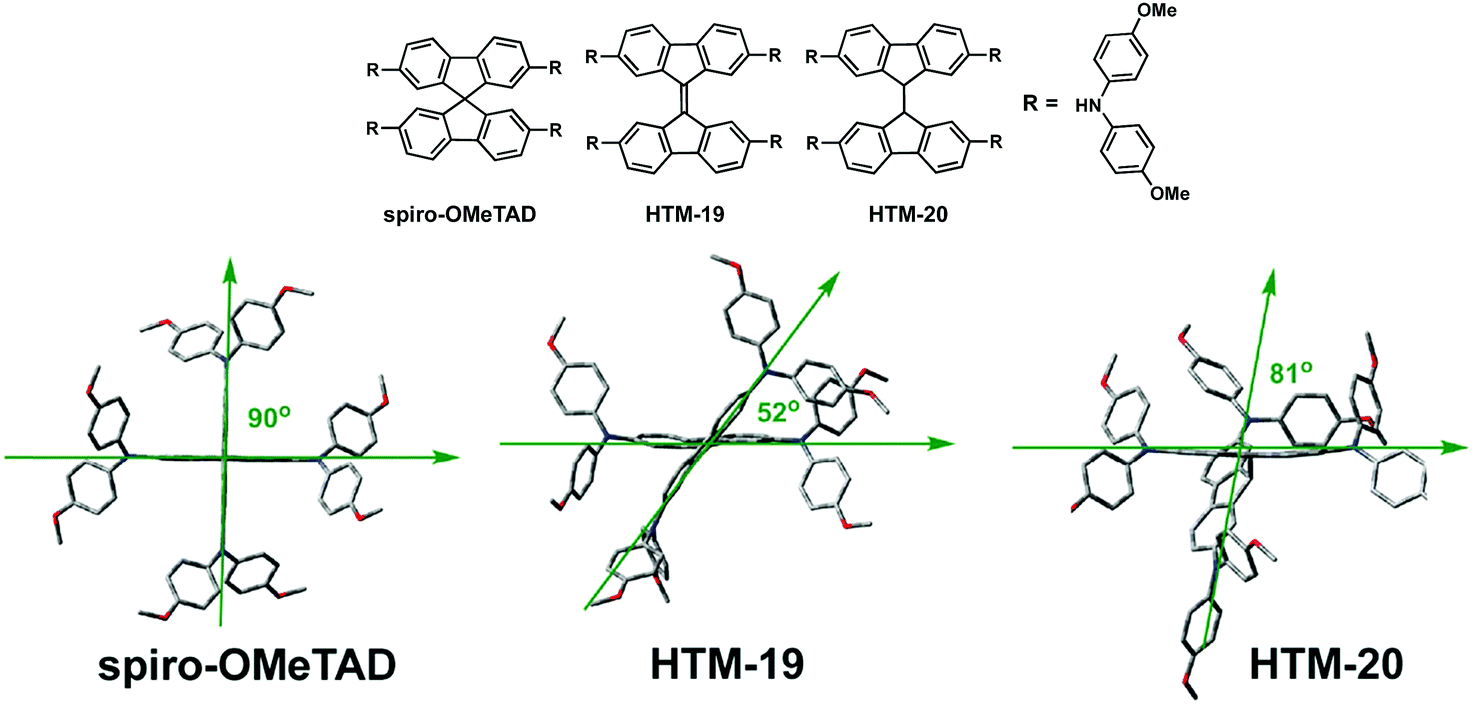 | ||
| Fig. 8 Optimized geometries calculated at the DFT level for spiro-OMeTAD, HTM-19 and HTM-20. Reproduced from ref. 71 with permission from Wiley-VCH, copyright 2016. | ||
Conventional mesoscopic (FAPbI3)0.85(MAPbBr3)0.15-based devices employing HTM-19 and HTM-20 record maximum PCEs of 16.6% and 19.8%, respectively, with the latter outperforming the benchmark spiro-OMeTAD (18.9%). The superior performance of HTM-20-based devices is attributed to a combination of efficient hole extraction at the HTM/perovskite interface and an efficient hole transport, in turn owing to the good film forming ability of HTM-20.
Scott, Murata and coworkers disclosed four molecules using two different central scaffolds, an azulene72–74 derivative and biphenyl, which are covalently linked to an atypical oxygen-bridged triphenylamine (HTM-21, HTM-22, HTM-23 and HTM-24) (Fig. 7).75 Standard Ir-catalyzed C–H activation successfully furnished the boronic ester-substituted azulene and biphenyl. On the other hand, the custom-made electron-donating counterpart, oxygen-bridged TPA, can be prepared in an additional six-step sequence in relatively good overall yield (53%) (Scheme 5).76 The Buchwald–Hartwig amination reaction between 2,6-difluoroaniline (16) and 4-iodoanisole affords the DPA derivative 17, which reacts by an Ullman C–N coupling in the presence of copper for the formation of the TPA moiety 18. Then, cleavage of methoxy units with BBr3 generates the trihydroxy intermediate, which, in one-pot reaction, undergoes a double-cyclization under basic conditions by substitution of the fluorine atoms, and then, the remaining hydroxy group is alkylated using 1-bromooctane, to form derivative 19. Finally, bromination reaction using NBS under mild conditions led to the formation of the selective monobrominated oxygen-bridged triphenylamine 20. This TPA was covalently attached to the corresponding azulene or biphenyl central scaffolds. The synthesis of different azulene-based or biphenyl-based HTMs is achieved by means of the palladium-catalyzed Suzuki–Miyaura cross-coupling reaction with the corresponding donor moiety, affording HTM-21, HTM-22, HTM-23 and HTM-24 in yields ranging from 46 to 76%. All HTMs exhibit strong absorption in the UV region (<400 nm) associated with π–π* transitions. Furthermore, azulene-based HTMs displayed a weak and broad peak in the visible region due to the intramolecular charge transfer transition of the azulene moiety. Mesoscopic MAPbI3-based devices incorporating doped-HTM-21, HTM-22, HTM-23 and HTM-24 yield a wide range of PCEs: from a poor 0.45% for the biphenyl derivative to a remarkable 16.5% for the tetra-substituted azulene HTM-21, which displayed a conductivity (2.1 × 10−4 cm2 V−1 s−1) comparable to that of spiro-OMeTAD. In addition, a high hole-collecting ability at the HTM/perovskite interface was observed for HTM-21 using time-resolved microwave conductivity (TRMC) in comparison to the other HTMs. These results suggest that the horizontal face-on orientation of this derivative on top of the perovskite layer could facilitate charge collection, enhanced by the two-dimensionally expanded molecular shape of the π-conjugated scaffold in tetra-substituted azulene HTM-21.
 | ||
| Scheme 5 Synthesis of molecularly engineered oxygen-bridged triphenylamine (20). | ||
The use of [2,2]paracyclophane-based HTMs has been reported in two different works by Son and co-workers (HTM-25 and HTM-26) (Fig. 7).77,78 [2,2′]-Paracyclophane (PcP) has received a lot of interest in molecular electronics due to its fascinating structure and physical properties.79–81 PcP consists of two parallel benzene rings linked through ethylene bridges, with a ring–ring spacing similar to that of van der Waals (∼3.09 Å), thus conferring a strong electronic coupling through the space between the rings. The interesting properties of PcP as a HTM arise from their dense packing in the thin-film, owing to the rigid shape of the central backbone, which, at the same time, enhances the charge transport ability in the three-dimensional structure. The authors described a tetra-substituted paracyclophane (PcP) mimicking the spiro-OMeTAD structure using p-methoxytriphenylamines as donor units for fine tuning the HOMO energy level (HTM-25). This work was followed by the study of multi-armed PcP-based HTMs. For the synthesis of HTM-25 and HTM-26, tetra-brominated or tri-brominated compounds were prepared from commercially available [2,2′]-paracyclophane in very low yields (∼17%). Next, four-fold palladium-catalyzed cross-coupling reaction with the triphenylamine boronic ester successfully furnished HTM-25 in a low 5% overall yield. The optoelectronic properties of HTM-25 are similar to those of spiro-OMeTAD; the maximum absorption of the latter (365 nm) is slightly blue-shifted, while the emission maximum (470 nm) is strongly red-shifted. The energy band gap estimated from the absorption and emission spectra is 2.96 eV, which is 0.08 eV lower than the value obtained for spiro-OMeTAD. The HOMO energy levels (−5.27 eV for HTM-25 and −5.28 eV for HTM-26) were determined for thin-films using photoelectron spectroscopy in air (PESA), showing slightly lower values than spiro-OMeTAD (−5.16 eV). PcP-based HTMs were evaluated in planar PSCs with a FTO/TiO2/MAPbI3/HTM/Au architecture. The best performing HTM-25, using LiTFSI and t-BP as dopants, afforded a PCE of 18.0% with no hysteresis, while HTM-26 achieved a remarkable 16.4%.
In 2015, Palomares, Martín and coworkers developed, for the first time, a tetraarylethene-based (TAE) HTM, substituted with p,p′-dimethoxydiphenylamine (HTM-27) (Fig. 7).82 This central scaffold presents a simple molecular structure based on four phenyl rings, which are linked to a central ethene rod through single bonds. Its propeller-like conformation shows π–π stacking interactions, and also multiple C–H⋯π intermolecular interactions take place between the hydrogen of the phenyl rings of one TAE and the π electrons of an adjacent molecule. These interactions lock the molecular conformation, providing highly emissive properties in the aggregated state. Furthermore, the TAE structure can be envisaged as an evolution of the spiro-concept in going from the “open” to “closed” form of the fluorene rings and from the “C–C” configuration to a “CC” configuration between the two halves. HTM-27 requires only two synthetic steps from commercially available 4,4′-diaminobenzophenone, which involves a two-fold copper-catalysed Ullmann reaction with 4-iodoanisole, followed by the McMurry reductive coupling reaction of ketones using a low-valent titanium reagent. The final compound was obtained in 72% yield. Conventional mesoporous MAPbI3-based devices with undoped HTM-27 reached a PCE of 11.02%, slightly lower than the undoped spiro-OMeTAD (13.53%). Shortly after, Ko and coworkers reported the same molecule, but, in this case, using dopants (LiTFSI and t-BP). Mesoporous MAPbI3-based devices using HTM-27 exhibited a PCE of 13.09%, while spiro-OMeTAD, under the same conditions, reached a similar photovoltaic performance (13.87%).83 The chemical versatility of this central scaffold has been used by the scientific community for preparing HTMs, for instance, by introducing different electron-donating units, which resulted in a PCE of 16.74%.84 Very recently, a TAE-based HTM end-capped with p-methoxytriphenylamine (TPA-OMe) was employed as dopant-free in tin-based PSCs.85 {en}FASnI3-based devices using this HTM displayed a PCE of 7.02%, which was higher than those obtained for the doped spiro-OMeTAD (5.20%) and the doped PTAA (6.67%).
Nazeeruddin, Getautis and coworkers reported in 2016 a series of simple molecules based on small central units, benzene (HTM-28 and HTM-29) or thiophene (HTM-30) (Fig. 7).86 We decided not to split these derivatives for comparison purposes in the discussion. The new HTMs showed a simple structure and required a two-step synthesis and were obtained in moderate overall yields, from 28 to 35%. Initially, a condensation reaction under basic conditions (NaOH) between the corresponding di- or trialdehydes and 2,7-dibromofluorene, followed by the subsequent standard Buchwald–Hartwig reaction led to HTMs 28–30. Interestingly, the charge mobilities measured, using xerographic time-of-flight (XTOF), for HTM-29 and HTM30 were higher than that of spiro-OMeTAD, with values ranging from 4 to 8 × 10−5 cm2 V−1 s−1. The incorporation of an extra addend in HTM-28 results in much lower charge mobility, presumably due to its bulkier size, which prevents a better organization and tighter packing and, consequently, the charge transport.86 To demonstrate the applicability of these HTMs, (FAPbI3)0.85(MAPbBr3)0.15-based devices were prepared. Impressive power conversion efficiencies of 19.5% and 20.0% were achieved using HTM-29 and HTM-30, respectively, which confirms HTM-30 as one of the most effective low-cost HTMs reported so far in the literature.
Nitrogen-containing central scaffolds
Triazatruxene (TAT) is one of the most frequently used building blocks for organic photovoltaic applications due to its chemical versatility, which allows an easy modification of its optical and electronic properties.87 TAT consists of three indole units fused to a benzene central ring in a C3 symmetric planar π-extended conjugated structure, which provides high hole mobility owing to its strong π–π intermolecular interactions and electron donating properties. Nazeeruddin and co-workers reported in 2015 a straightforward synthesis of a series of new TAT-based HTMs endowed with different electron-donating side arms (HTM-31, HTM-32, HTM-33 and HTM-34) (Fig. 9).88 The preparation of the symmetrically functionalized tribrominated TAT comprises three synthetic steps with an overall yield of 39% (Scheme 6a). Firstly, trimerization of 2-indolinone in the presence of phosphoryl chloride leads to the formation of tris-N-unsubstituted-TAT (21), which represents the most convenient synthetic approach for the preparation of unsubstituted TAT in terms of yield and reaction time. Another approach to obtain unsubstituted TAT is based on bromine-catalyzed cyclotrimerization of indoles followed by the subsequent reductive dehalogenation. Alkyl chains are introduced under basic conditions with an excess of alkyl halides to improve the solubility of the TAT-core (22). The regioselective bromination using NBS gives rise to 3,8,13-tribromotriazatruxene (23). Finally, the different electron donor units, methoxy, p-methoxybenzene, p,p′-dimethoxydiphenylamine (OMe-DPA) and (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-N,N-bis(4-methoxyphenyl)aniline, are covalently linked by three-fold Ullmann, Suzuki and Buchwald–Hartwig coupling reactions. The photovoltaic performance of the doped-triazatruxene HTMs is evaluated using mesoscopic PSC devices made of the mixed anion and mixed cation perovskite (FAPbI3)0.85(MAPbBr3)0.15. The average PCE of the new derivatives span 8.9% to 17.7% with little hysteresis. | ||
| Fig. 9 Molecular structures of triazatruxene-based HTMs. | ||
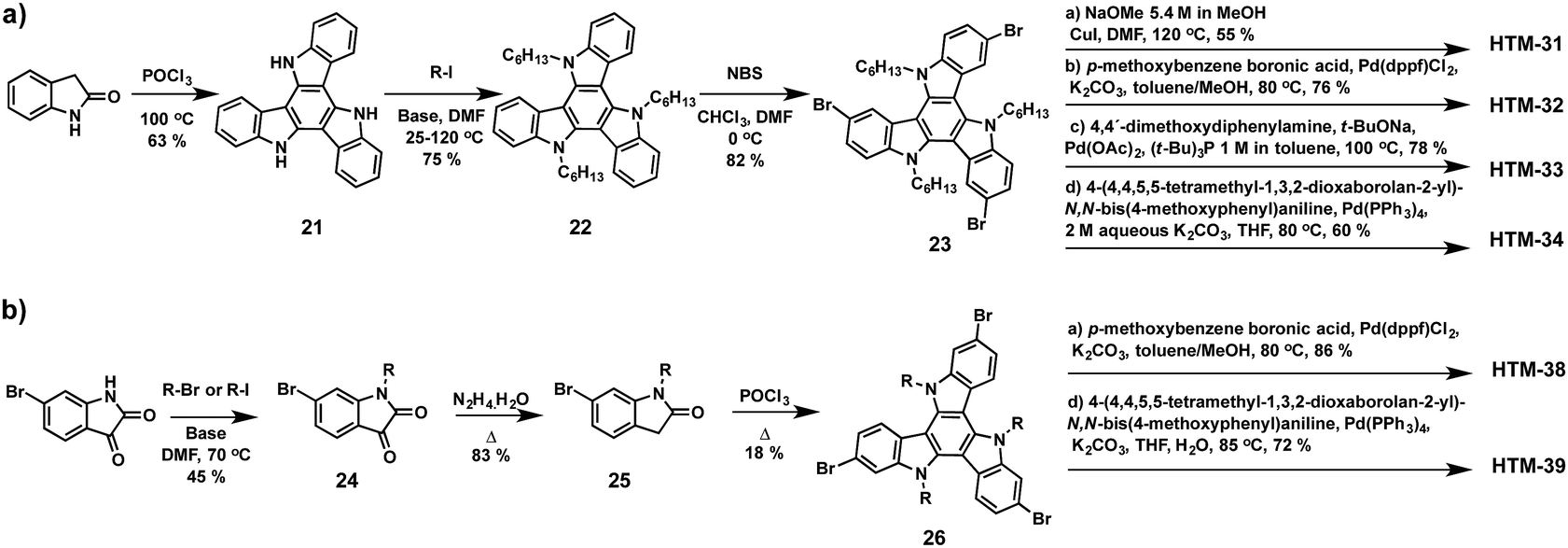 | ||
| Scheme 6 Synthetic strategies for the preparation of 2,7,12- and 3,8,13-isomers of the triazatruxene as a central scaffold and representative examples of the corresponding HTMs. | ||
HTM-32 and HTM-34 exhibit excellent PCEs of 18.3% and 16.8%, compared with that obtained for the spiro-OMeTAD.
The device performances of the nitrogen-containing HTMs are summarized in Table 3.
| HTM | HOMOd (eV) | σ (S cm−1) | μ h (cm2 V−1 s−1) | V oc (V) | J sc (mA cm−2) | FF | PCE (%) | Architecturee | Perovskite | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a LiTFSI and t-BP. b LiTFSI, t-BP, FK209. c Dopant-free. d HOMO energy levels were converted from the original work using Fc/Fc+ at 5.1 eV vs. vacuum. e M = mesoporous, P = planar and IP = inverted planar configurations. | ||||||||||
| HTM-31 | −5.12 | — | 5.0 × 10−4 | 0.99 | 16.0 | 0.57 | 9.0 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 88 |
| HTM-32 | −5.24 | — | 2.8 × 10−5 | 1.14 | 20.7 | 0.77 | 18.3 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 88 |
| HTM-33 | −5.00 | — | 6.0 × 10−5 | 0.96 | 18.4 | 0.67 | 11.9 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 88 |
| HTM-34 | −5.20 | — | 2.0 × 10−4 | 1.13 | 20.4 | 0.72 | 16.8 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 88 |
| HTM-35 | −5.51 | — | 5.0 × 10−7 | 1.05 | 16.01 | 0.53 | 8.88 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 89 |
| HTM-36 | −5.26 | — | 2.6 × 10−4 | 1.13 | 21.70 | 0.78 | 19.03 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 89 |
| HTM-37 | −5.34 | — | 1.1 × 10−5 | 1.11 | 19.31 | 0.69 | 14.87 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 89 |
| HTM-38 | −5.28 | — | 2.41 × 10−4 | 1.08 | 22.8 | 0.77 | 18.8 | P | MAPbI3 | 90 |
| HTM-39 | −5.35 | — | 9.45 × 10−5 | 1.03 | 22.3 | 0.74 | 16.9 | P | MAPbI3 | 90 |
| HTM-40 | −5.17 | — | — | 0.94 | 14.43 | 0.64 | 8.62 | M | MAPbI3 | 91 |
| HTM-41 | −5.41 | — | — | 0.90 | 16.51 | 0.66 | 9.83 | M | MAPbI3 | 91 |
| HTM-42 | −5.36 | — | 1.69 × 10−3 | 1.05 | 23.55 | 0.77 | 19.05 | M | [FAPbI3]0.92[MAPbBr3]0.08 | 92 |
| HTM-43 | −5.17 | — | 5.2 × 10−4 | 1.07 | 22.5 | 0.74 | 17.8 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 93 |
| HTM-44 | −4.91 | — | — | 0.82 | 4.2 | 0.61 | 2.1 | M | Csx[MA0.17FA0.83](100−x) Pb[I0.83Br0.17]3 | 94 |
| HTM-45 | −5.29 | — | — | 1.11 | 21.1 | 0.75 | 17.6 | M | Csx[MA0.17FA0.83](100−x) Pb[I0.83Br0.17]3 | 94 |
| HTM-46 | −5.29 | — | 1.06 × 10−4 | 0.99 | 17.33 | 0.63 | 10.79 | M | MAPbI3 | 96 |
| HTM-47 | −5.15 | — | 4.92 × 10−4 | 0.92 | 18.39 | 0.70 | 11.86 | M | MAPbI3 | 96 |
| HTM-48 | −5.38 | — | 1.1 × 10−4 | 1.09 | 20.85 | 0.77 | 17.5 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 97 |
| HTM-49 | −5.30 | — | 1.2 × 10−4 | 1.13 | 21.71 | 0.77 | 18.9 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 97 |
More recently, the same group has developed a series of molecularly engineered TAT derivatives with a D–π–A structure (HTM-35, HTM-36 and HTM-37) (Fig. 9).89 The new HTMs comprise different π-conjugated spacers bridging the TAT-core and dicyanovinyl (DCV) acceptor groups. TAT-derivatives are synthetized by a Pd-catalyzed Suzuki–Miyaura cross-coupling reaction between the tribromoTAT and the π-bridges in good yields, followed by a Vilsmeier–Haack formylation. Finally, the dicyanovinyl groups are covalently connected to the aldehyde-terminated derivatives by a three-fold Knoevenagel condensation, affording new HTMs in overall yields ranging from 18 to 30% (without taking into account the synthesis of the spacers). Compared to their star-shaped analogues, previously described, HTM-35, HTM-36 and HTM-37 exhibit significantly red-shifted absorption to the visible range due the charge-transfer bands, with maxima centered at 487, 515 and 569 nm. In particular, HTM-36 shows a highly-ordered face-on organization, determined by GIWAXS, which is beneficial for charge transport through the vertical direction along a stacked structure (Fig. 10).89 This effect results in a one and two-orders of magnitude enhanced hole mobility in comparison with less ordered equivalent HTM-35 and HTM-37, respectively. (FAPbI3)0.85(MAPbBr3)0.15-based PSCs employing undoped HTM-36 afforded an impressive 19%. On the other hand, dopant-free HTM-35 and HTM-36 exhibit significantly decreased Jsc and FF values owing to their greater charge recombination, which is surmised to be the reason for the lower LUMO energy level.
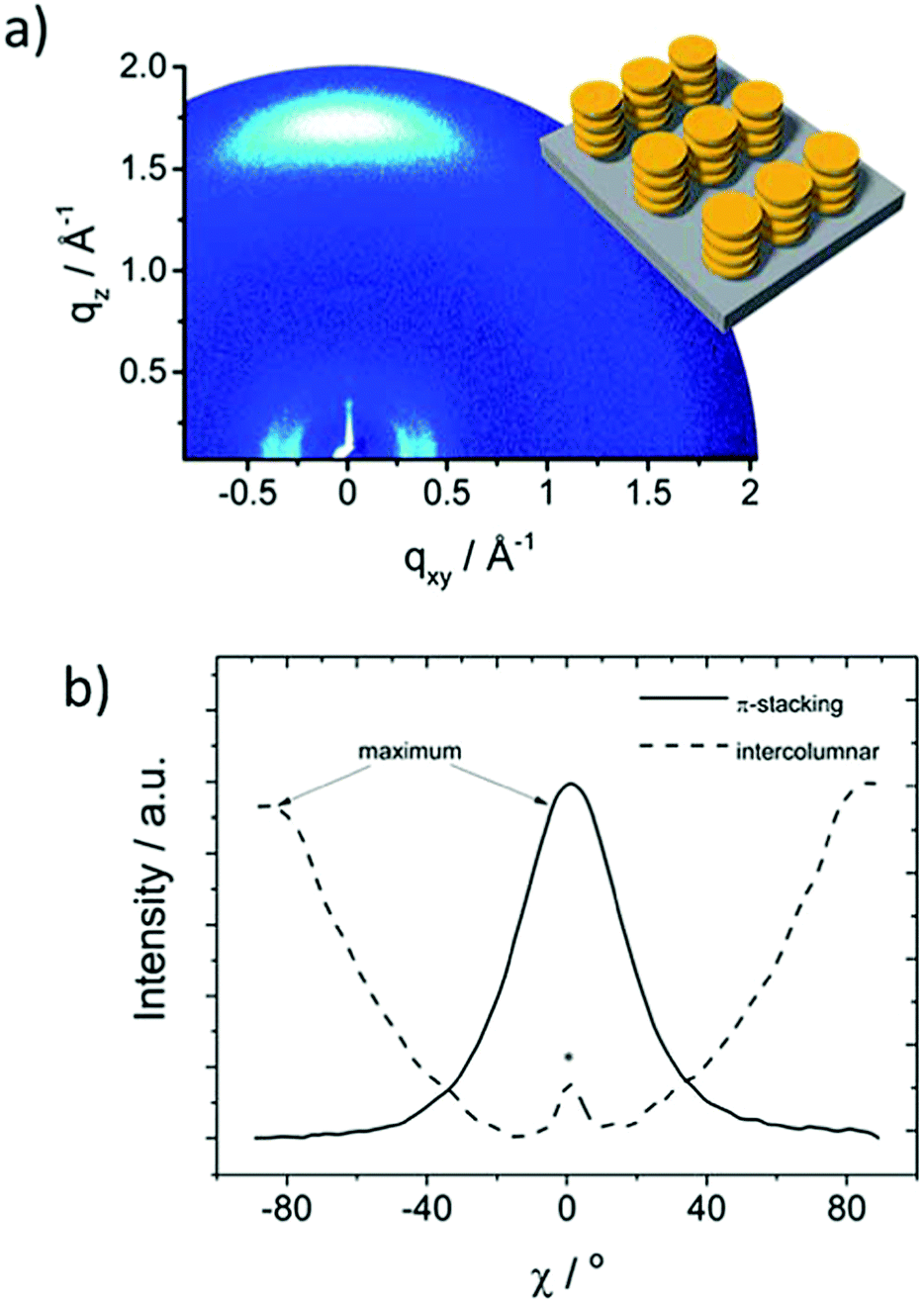 | ||
| Fig. 10 Morphology characterization: (a) two-dimensional GIWAXS pattern of HTM-36 and (b) azimuthally integrated intensity of the π-stacking and intercolumnar reflections. Reproduced from ref. 89 with permission from the Royal Society of Chemistry, copyright 2017. | ||
Shielding the perovskite from moisture with a thin interlayer of poly-N-vinylcarbazole (PVK), sandwiched in between the perovskite and the HTM, not only suppresses charge recombination but also facilitates an efficient charge transport, which results in enhanced Voc, Jsc and FF and, therefore, higher PCE. To this end, Mayor, Su and co-workers synthesized two triazatruxene derivatives with different electron donor units, namely p-methoxybenzene HTM-38 and OMe-TPA HTM-39 (Fig. 9).90 The preparation of the 2,7,12-isomer (26) takes place in a lower yield (18%) than its 3,8,13-isomer (63%) (Scheme 6b). Planar CH3NH3PbI3-based PSCs with a thin interlayer of PVK and using doped-HTM-38 and HTM-39 exhibit PCE values of 18.8% and 16.9%, respectively. In contrast, devices without this interfacial layer result in inferior performance – 14.1% for spiro-OMeTAD, 13.9% for HTM-39 and 15.5% for HTM-38. Additionally, the PVK-protection results in longer-lasting device stability in aging control experiments. In a 35% humid atmosphere at room temperature PSCs incorporating PVK and HTM-38 or spiro-OMeTAD maintain 90% and 87% of their initial PCEs after 700 hours, which is attributed to a slower moisture penetration into the perovskite layer. On the other hand, the same devices without the protection of the PVK interlayer retain only 68% and 73%, respectively.
The chemical versatility of the TAT core allows the modulation of their properties through a rational design. Ahmad and co-coworkers designed and synthesized a much simpler TAT-based HTM-40 and HTM-41 substituted in the N-position with different alkyl or aryl groups (Fig. 9).91 The new derivatives are obtained in good overall yields of 21% for HTM-40 (three synthetic steps) and 34% for HTM-41 (two synthetic steps). Both compounds show a narrow optical range owing to their limited absorption in the visible region. The HOMO energy level for HTM-40 shifts from −5.17 to −5.41 eV for HTM-41, as a result of the weaker electron-donating properties of the latter. Mesoscopic CH3NH3PbI3-based PSCs using these dopant-free HTMs exhibit efficiencies as high as 8.62% for HTM-40 and 9.83% for HTM-41, which were further improved by adding LiTFSI to efficiencies higher than 10%.
Seok and co-workers have very recently designed and synthesized linear fluorinated indolo[3,2-b]indole (HTM-42) as a core of a new family of HTMs (Fig. 11).92 The introduction of fluorine substituents into the structure promotes intramolecular interactions (C–F⋯H), thereby providing tighter molecular packing, which leads to a higher hole mobility (1.69 × 10−3 cm2 V−1 s−1) compared to spiro-OMeTAD (2.17 × 10−4 cm2 V−1 s−1). The synthetic pathway toward the preparation of the core indolo[3,2-b]indole requires a challenging nine-step synthesis and results in an overall yield of less than 5% (Scheme 7). Regioselective bromination of 5-fluoro-2-nitroaniline with NBS in acetic acid yields derivative 27, which is treated with NaNO2 under acidic conditions, giving rise to the diazonium salt intermediate, which is transformed into the corresponding aryl halide using KI to afford compound 28. The latter undergoes a Sonogashira cross-coupling (Pd/Cu catalytic system) reaction with (trimethylsilyl)acetylene (TMSA), followed by deprotection of the TMS group using non-conventional conditions. A further Sonogashira reaction between compounds 30 and 28 successfully furnishes compound 31. It is worth mentioning that strict exclusion of oxygen is necessary to avoid Glaser homocoupling side reactions. Compound 31 undergoes an oxidation of the acetylene group in a high yield (81%). Subsequent reduction of the nitro groups employing standard conditions (SnCl2/HCl), followed by an intramolecular cyclization reaction, leads to 2,7-dibromo-3,8-difluoro-5,10-dihydroindolo[3,2-b]indole (33) as the central scaffold. The latter is sequentially N-alkylated, using 1-bromohexane and a base in refluxing anhydrous THF, and cross-coupled by the two-fold Suzuki–Miyaura reaction with 5′-hexyl-2,2′-bithiophene-5-boronic acid pinacol ester to successfully obtain HTM-42. Mesoscopic [FAPbI3]0.92[MAPbBr3]0.08-based PSCs using HTM-42 shows an average PCE of 19.05%, with noticeable hysteresis (19.8% for reverse scan and 18.3% for forward scan), which is significantly higher than that measured for spiro-OMeTAD under the same conditions (16.8%).
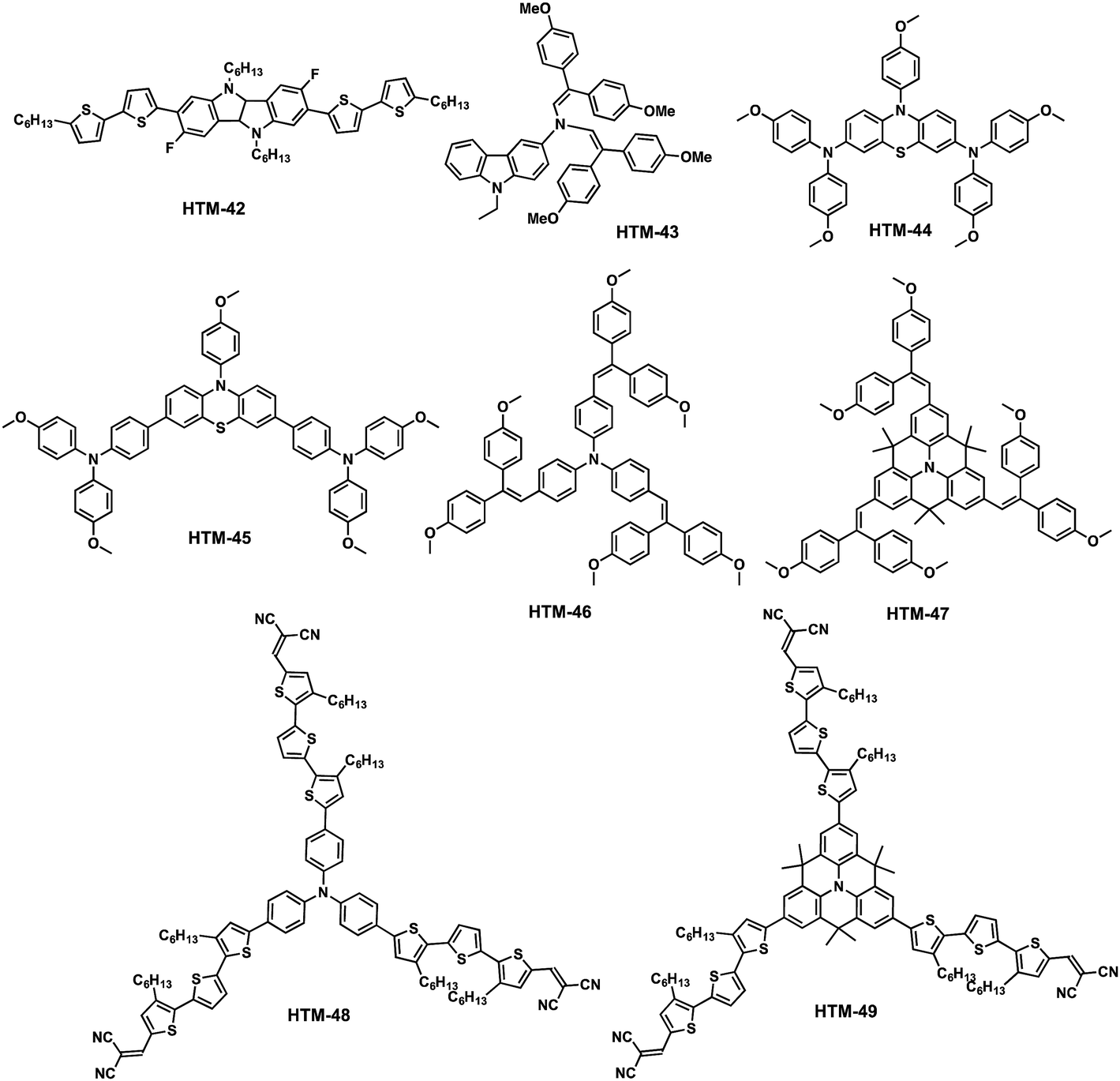 | ||
| Fig. 11 Molecular structures of arylamine-based central core HTMs. | ||
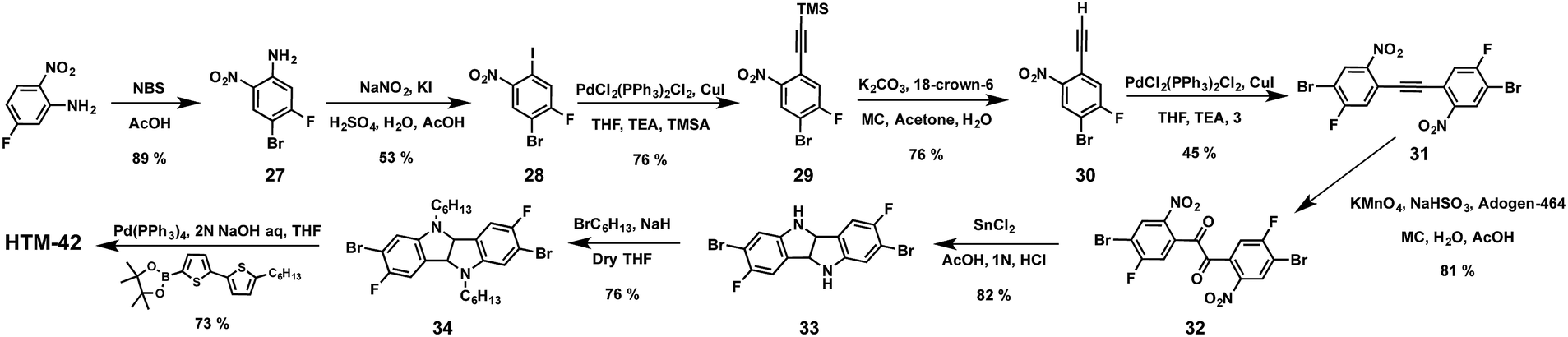 | ||
| Scheme 7 Synthetic strategies for the preparation of indolo[3,2-b]indole-based HTM-42. | ||
Nazeeruddin and coworkers designed, synthesized and incorporated an extraordinarily simple molecule, based on a carbazole conjugated enamine, as HTM-43 for PSCs (Fig. 11).93 The one-step acid-catalyzed condensation of 3-amino-9-ethylcarbazole, which is commercially available, and 2,2-bis(4-methoxyphenyl)acetaldehyde successfully affords HTM-43 in 64%. Noteworthily, no column chromatography is needed for purifying this compound. HTM-43 exhibits good thermal stability until 380 °C and a glass transition temperature during the second heating cycle (Tg) of 111 °C, which is high enough for photovoltaic devices. The optical properties of HTM-43 reveal a limited absorbance in the visible range, with an absorption peak at 440 nm but with a relatively low absorption coefficient (≈1.0 × 104 M−1 cm−1). After chemical oxidation with TFSI, HTM-43 shows an absorption maximum centered at 1020 nm. Its HOMO energy was determined in the solid state by photoelectron spectroscopy in air (PESA) to be −5.01 eV and in solution, using electrochemistry, to be −5.17 eV. HTM-43 was incorporated in mesoscopic CH3NH3PbI3-based PSCs, yielding an efficiency of 17.8%. Additionally, HTM-43 was employed in planar CH3NH3PbIxCl3−x-based PSCs, displaying remarkable PCE values of 16.9%, slightly lower than that of spiro-OMeTAD.
Abate and co-workers presented two relatively simple molecules using phenothiazine as the central scaffold endowed with different arylamines (HTM-44 and HTM-45) (Fig. 11).94 Phenothiazine is a useful moiety for photovoltaic applications due to its low price and its flexible electron-rich properties, which can be easily tuned for generating new interesting derivatives not only for photovoltaic applications but also for medicinal chemistry.95 In this work, the authors prepared the phenothiazine-based core in a straightforward two-step synthetic sequence, starting from commercial phenothiazine; first, a C–N palladium-catalyzed Buchwald–Hartwig reaction to introduce p-anisole followed by a selective bromination at the 3- and 6-positions to obtain the dibromo derivative. The aforementioned HTM-44 could be prepared via the Buchwald–Hartwig C–N cross-coupling reaction with DPA-OMe catalyzed by Pd(AcO)2/dppf under basic conditions. Alternatively, HTM-45 was prepared by a Suzuki–Miyaura C–C cross-coupling reaction between dibromophenothiazine and the boronic ester of triphenylamine. The two new HTMs are obtained in relatively low overall yields: 22% for HTM-44 and 25% for HTM-45.
Cyclic voltammograms of the new HTMs show a strong electron-donating ability for HTM-44, with a HOMO energy of −4.91 eV, while HTM-45 has a HOMO energy of −5.29 eV. These values are slightly lower than that of spiro-OMeTAD, measured under the same conditions. The values observed are consistent with DFT calculations that predict the HOMO of HTM-44 mainly located in the phenothiazine scaffold, whereas in HTM-45 it is distributed along the TPA moiety. The new HTMs were tested in mesoscopic devices using the triple cation perovskite (generic form: Csx(MA0.17FA0.83)(100−x)Pb(I0.83Br0.17)3), displaying efficiencies of 2.1% and 17.6% for HTM-44 and HTM-45, respectively. The poor performance of HTM-44 is attributed to an oxidative degradation of the HTM due to its lower HOMO energy. In contrast, HTM-45 shows a similar performance to spiro-OMeTAD (17.5%), probably due to its higher conductivity, which has a positive influence on the FF.
Star-shaped arylamine moieties have been used by Ko, Nazeeruddin and coworkers for synthesizing a TPA-based derivative (HTM-46) and a rigid quinolizino acridine (HTM-47), which were functionalized with the incorporation of bis(O-methoxyphenyl)ethene as peripheral groups (Fig. 11).96 Both HTMs were prepared in a single step from their corresponding brominated scaffolds via the Suzuki cross-coupling reaction in moderate yields (around 50%) with 2-[2,2-bis(4-methoxyphenyl)ethenyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane. HTM-46 and HTM-47 present strong absorptions at 396 and 414 nm, respectively, and HOMO energies of −5.29 and −5.15 eV. The rigid and planar structure of HTM-47 explains the red-shifted maximum and its stronger donor ability. Both doped-HTMs were tested in mesoscopic devices in combination with the standard CH3NH3PbI3, achieving power conversion efficiencies of 11.86% for HTM-47 and 10.79% for HTM-46, which are slightly higher than those values achieved for dopant-free-based devices (9.18 and 7.45%, respectively).
Adopting the same approach as that for previously described HTM-36, the same group reported two new star-shaped dopant-free HTMs (HTM-48 and HTM-49) (Fig. 11).97 To this end, they combined the latter central scaffolds (TPA and the quinolizino acridine) with a π-conjugated alkyl-substituted terthiophene spacer bridging the core and dicyanovinyl (DCV) electron accepting-groups. The presence of the triarylamine and the quinolizino acridine guarantees optimal electron-donating properties and, thus, excellent band alignment with the valence band edge of the perovskite. The undoped HTM-48 and HTM-49 were incorporated in mesoscopic devices with the normal configuration of FTO/c-TiO2/m-TiO2/(FAPbI3)0.85(MAPbBr3)0.15/HTM/Au, which demonstrated a record PCE value of 18.9% for HTM-49 and 17.5% for HTM-48, outperforming undoped spiro-OMeTAD (7.5%). Furthermore, the authors studied the maximum power output (MPO) decay of the devices. HTM-49-based devices resulted in long-lasting device stability under continuous light soaking experiments. Actually, after 1300 hours the device efficiency decreased to 65% of its initial performance. In contrast, HTM-48 and spiro-OMeTAD decreased to 25% and 15%, respectively. This result further confirmed that the stability of the HTMs is an extrinsic degradation factor of the PSCs and by improving their stability it is possible to improve the long-term stability of the devices.
Sulfur-containing HTMs
Thiophene-based compounds are appealing building blocks with interesting optoelectronic properties for application in a variety of fields, namely organic photovoltaics,98,99 organic field-effect transistors100 and organic light-emitting diodes (OLEDs),101 to name a few. The photovoltaic parameters of sulfur-containing HTMs including their device architectures are listed in Table 4.| HTM | HOMOc (eV) | σ (S cm−1) | μ h (cm2 V−1 s−1) | V oc (V) | J sc (mA cm−2) | FF | PCE (%) | Architectured | Perovskite | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a LiTFSI, t-BP, FK209. b LiTFSI, t-BP, FK102. c HOMO energy levels were converted from the original work using Fc/Fc+ at 5.1 eV vs. vacuum. d M = mesoporous, P = planar and IP = inverted planar configurations. | ||||||||||
| HTM-50 | −5.11 | — | — | 1.05 | 19.1 | 0.65 | 13.2 | M | MAPbI3 | 102 |
| HTM-51 | −5.28 | — | — | 0.95 | 18.9 | 0.61 | 11.0 | M | MAPbI3 | 105 |
| HTM-52 | −5.16 | 6.0 × 10−6 | — | 1.04 | 20.4 | 0.72 | 16.0 | M | MAPbI3 | 106 |
| HTM-53 | −5.20 | 1.3 × 10−5 | — | 1.09 | 20.6 | 0.77 | 17.0 | M | MAPbI3 | 106 |
| HTM-54 | −5.40 | 2.80 × 10−5 | — | 1.07 | 21.9 | 0.77 | 18.2 | M | MAPbI3 | 106 |
| HTM-55 | −5.33 | 5.3 × 10−4 | — | 1.09 | 23.04 | 0.75 | 18.97 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 110 |
| HTM-56 | −5.18 | 1.3 × 10−4 | — | 1.10 | 22.50 | 0.73 | 18.17 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 110 |
| HTM-57 | −5.32 | 5.8 × 10−4 | — | 1.07 | 21.75 | 0.78 | 18.13 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 113 |
| HTM-58 | −5.32 | 1.5 × 10−4 | — | 1.05 | 20.94 | 0.78 | 17.28 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 113 |
| HTM-59 | −5.31 | 4.5 × 10−5 | — | 1.03 | 19.60 | 0.78 | 15.66 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 113 |
| HTM-60 | −5.32 | 4.2 × 10−6 | — | 1.03 | 14.92 | 0.63 | 9.67 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 113 |
| HTM-61 | −5.23 | 2.1 × 10−4 | 7.66 × 10−5 | 1.15 | 23.12 | 0.64 | 16.9 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 115 |
| HTM-62 | −5.21 | 3.5 × 10−4 | 1.34 × 10−4 | 1.13 | 23.59 | 0.75 | 20.1 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 115 |
Grätzel, Grimsdale and co-workers reported in 2014 a simple HTM based on 3,4-ethylenedioxythiophene (EDOT) bearing two TPAs (HTM-50) (Fig. 12).102 EDOT represents a versatile building block for the preparation of functional organic materials.103,104HTM-50 was prepared in 82% yield using a one-pot reaction, involving the bromination of pristine EDOT, which was used without further purification in a subsequent Suzuki cross-coupling reaction with the molecularly engineered TPA. Conventional mesoscopic devices using CH3NH3PbI3 displayed efficiencies ranging from 10.6% in dopant-free to 13.8% using LiTFSI, t-BP and tris[2-(1H-pyrazol-1-yl)pyridine]cobalt(III) tris(hexafluorophosphate) (FK102). These values were comparable to those obtained using doped-spiro-OMeTAD (13.7%), making HTM-50 an excellent candidate since it presents a much simpler structure.
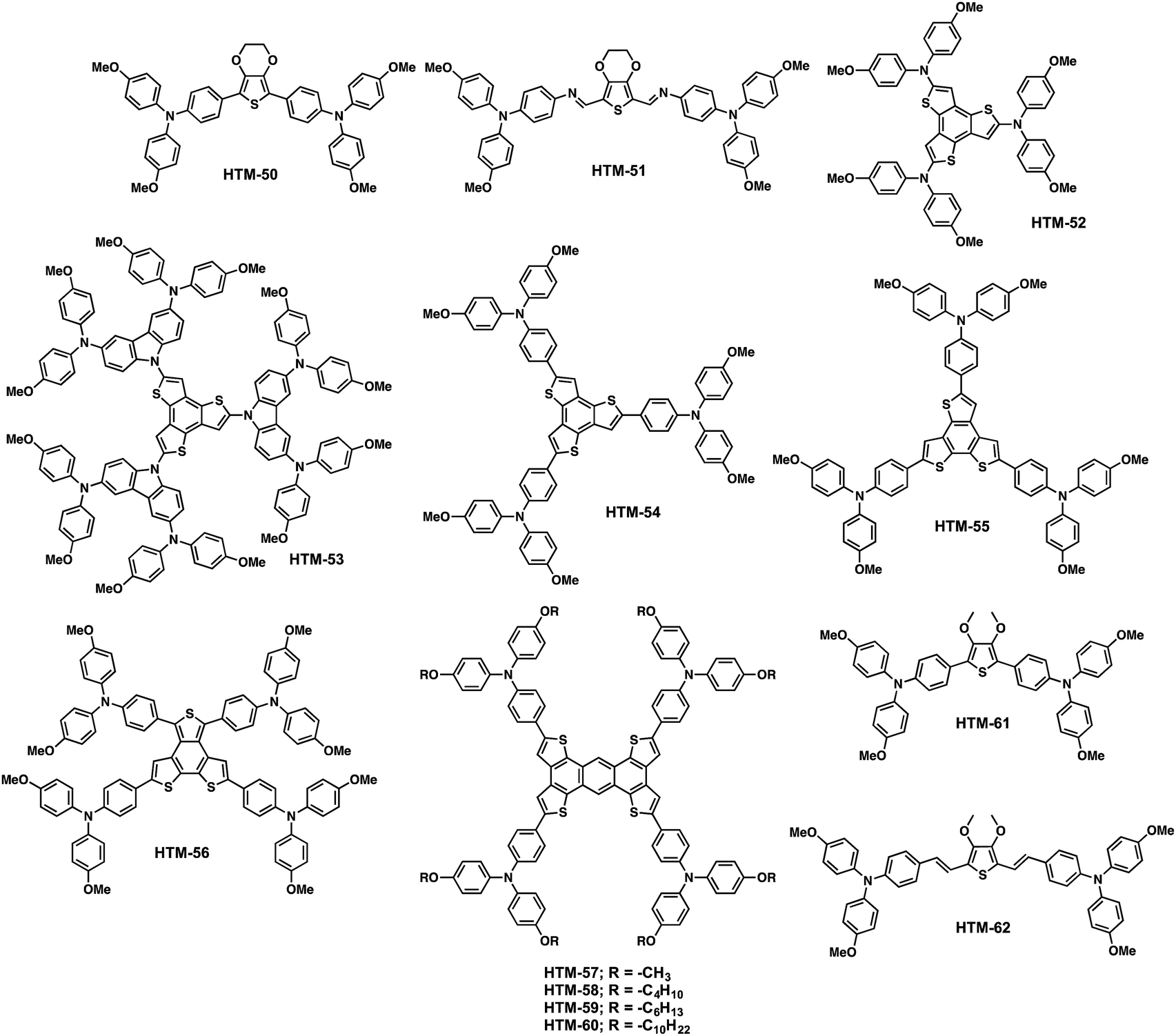 | ||
| Fig. 12 Molecular structures of thiophene-based HTMs. | ||
This result was followed by the work of Docampo and co-workers, who designed an analogous structure based on an azomethine bond that allows using a less complicated synthetic route based on Schiff-base condensation chemistry instead of palladium catalysed cross-coupling reactions (HTM-51) (Fig. 12).105 Furthermore, this alternative path provides a cost-effective and more environmentally friendly route, with water being the only by-product. Incorporation of the azomethine bond results in a redshift and broadening of the absorbance as a consequence of the growing π-conjugation and its electron-withdrawing properties. HTM-51 exhibits a lower HOMO energy level (−5.28 eV compared to −5.11 eV for HTM-50), which results in a better alignment with the valence band of the perovskite material. However, the PCE value using planar (CH3NH3PbI3) perovskite-based photovoltaics with doped-HTM-51 (LiTFSI, t-BP and FK102) exceeded 11%, which is relatively lower compared to its palladium-catalysed homologous HTM-50.
In 2016, Martín and coworkers reported three new benzotrithiophene (BTT) based HTMs, bearing DPA-OMe (HTM-52), DPA-OMe-substituted carbazole (HTM-53) and TPA-OMe (HTM-54) (Fig. 12).106 The BTT structure consists of three thiophene rings fused to a central benzene ring. Benzo[1,2-b:3,4-b′:5,6-b′′]trithiophene is a potential π-core for a new class of organic semiconductors, due to its planar and rigid π-framework that can promote strong intermolecular interactions. The first synthesis of the BTT compound dates back to the early work of Proetzsch et al. in 1972107 and Jayasuriya et al. in 1989.108 However, its tedious synthesis hindered its wide use. Thanks to the more recently established straightforward procedure for the synthesis of this central scaffold reported by Kuwabara and co-workers,109 it is now available in a multigram scale in the laboratory. Following this protocol, the authors were able to prepare the BTT central scaffold followed by bromination reaction at the α position. Cross-coupling reaction with the corresponding arylamine moiety provided HTM-52, HTM-53 and HTM-54 in overall yields ranging from 14 to 17%. The stronger electron-donating character of p-methoxydiphenylamine and p-methoxydiphenylamine-substituted carbazole raised the HOMO energy levels of HTM-52 (−5.2 eV) and HTM-53 (−5.2 eV) compared to the weaker p-methoxytriphenylamine-based HTM-54 (−5.4 eV). The PV response of these new HTMs was studied with two different perovskites, the standard MAPbI3 and the (FAPbI3)0.85(MAPbBr3)0.15. Using LiTFSI, t-BP and FK209, in conventional MAPbI3-based devices, HTM-54 gave an excellent performance of 18.2%, with almost no hysteresis. On the other hand, devices with HTM-53 present a high Voc (1.09 V), resulting in PCEs up to 17%. HTM-52 shows the lowest performance (16%, 13.9 ± 1.5%), which they claimed to be related to its low solubility in chlorobenzene compared to HTM-53 and HTM-54. A similar behavior was observed in (FAPbI3)0.85(MAPbBr3)0.15-based devices, with HTM-52 being the least effective HTM, leading to a lower fill factor and Jsc. In contrast, compounds HTM-53 and HTM-54 behave similarly, and remarkable PCE values of up to 17.5% were obtained. As a general trend, the previously mentioned chemical structures seem to favour the formation of homogeneous thin films along with a good conductivity, which lead to excellent FF values (>70%) comparable to those obtained for spiro-OMeTAD. The conductivity values of HTM-52 (6.0 × 10−6 S cm−1), HTM-53 (1.3 × 10−5 S cm−1) and HTM-54 (2.79 × 10−5 S cm−1) were in good agreement with the device performance.
The same group reported the isomerism effect on the photovoltaic performance of different isomers of BTT (HTM-55 and HTM-56) having bbb-BTT-2 and bbc-BTT-3 as the molecular cores, respectively (Fig. 12 and 13).110 For this purpose, the authors selected the asymmetric three b-fused thiophene, namely benzo[1,2-b:3,4-b′:6,5-b′′]trithiophene (bbb-BTT-2) (Fig. 13), which was first synthesized in 1989108 and has been employed in a wide number of applications. However, this synthetic approach could not be applied to bbc-BTT (two b- and one c-fused thiophene, C2v symmetry) as described by Perrine and coworkers. bbc-BTT-3 was eventually synthesized by Müllen and coworkers in 2011.111,112 The protocol described for building the third thiophene ring requires the use of the Wilkinson catalyst in combination with sulfur powder in refluxing benzene. The scope of this reaction is severely limited due to its low yield. After the three-fold and four-fold bromination at the α position of the thiophene rings, TPA-OMe units were introduced by a Suzuki cross-coupling reaction, which successfully furnished the new HTM-55 and HTM-56. Changing the arrangement of the sulfur atoms in the case of HTM-55 and HTM-56 resulted in a wider absorbance in the visible range. The absorption band of HTM-56 was significantly red-shifted in comparison to the other two three-armed compounds (HTM-54 and HTM-55) due to the enhanced π-electron delocalization of its core (Fig. 14a). It is reported that the broad absorption presented by HTM-55 could be attributed to the direct conjugation between the two cis thiophene rings, in contrast to the meta arrangement with the central benzene ring in HTM-54. Theoretical calculations show that the C2v-symmetric core of HTM-56 exhibits a single/double C–C bond pattern characteristic of a cis-bithiophene where an extra thiophene ring is linked to the central β positions, favoring an efficient π-conjugation path. On the other hand, the central core of HTM-55 is considered to be an intermediate case where the aromatic character of the central benzene ring is still maintained although it presents larger differences between the CC bond lengths compared with the central core of its homologous three-armed HTM-54 (Fig. 14b). The HOMO energy levels were also affected by the different arrangements of the sulfur atoms; while HTM-55 (−5.3 eV) exhibited a low-lying HOMO energy similar to HTM-54 (−5.4 eV), HTM-56 (−5.2 eV) showed a raised HOMO energy level similar to spiro-OMeTAD. The photovoltaic performances of the isomers of BTT were evaluated using mesoscopic PSC devices made of compositionally engineered perovskite (FAPbI3)0.85(MAPbBr3)0.15. The doped-HTM-55 (LiTFSI, t-BP, FK209) demonstrated a PCE of 19%, virtually hysteresis-free. On the other hand, HTM-56-based devices achieved a PCE of 18.2%. Both HTMs outperformed their brother isomer HTM-54.
 | ||
| Fig. 13 Molecular structures of the seven possible benzotrithiophene isomers. | ||
 | ||
| Fig. 14 (a) UV-vis absorption spectra (solid lines) of BTT-3, BTT-4 and BTT-5 in CH2Cl2. (b) Optimized bond lengths (in Å) calculated at the B3LYP/6-31G** level, in CH2Cl2 solution, for the isomeric BTTs cores bbb-BTT-1, bbb-BTT-2 and bbc-BTT-3. | ||
π-Extended, sulfur-rich compounds have also been used as promising candidates for hole-transporting purposes. Martín and co-workers reported a flat and sulfur-rich polycyclic aromatic central scaffold, anthra[1,2-b:4,3-b′:5,6-b′′:8,7-b′′′]tetrathiophene (ATT), which is endowed with p-alkoxytriphenylamines of different alkyl chain lengths (HTM-57, HTM-58, HTM-59 and HTM-60) (Fig. 12).113 The synthesis of these HTMs proceeds smoothly under standard conditions and can be achieved in 4 synthetic steps in good overall yields, ranging from 37 to 58% (Scheme 8). Different TPAs were attached to the central scaffold in order to study the influence of the length of the alkyl chain on the photovoltaic performance. The brominated-central unit can be synthesized in an overall yield of 80%, and no further purification (column chromatography) was needed for any of the synthetic steps.
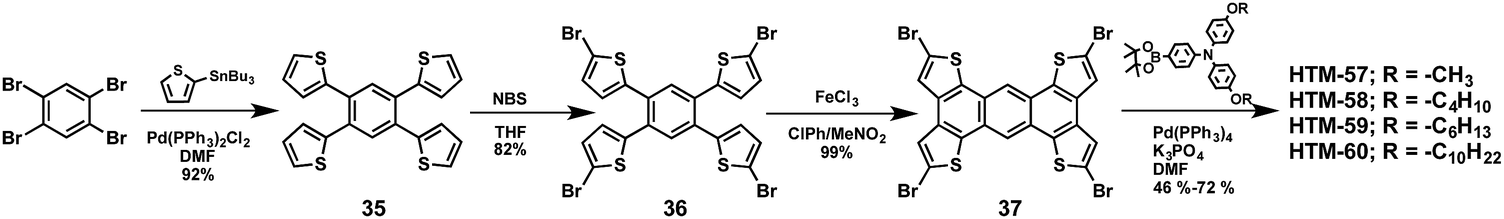 | ||
| Scheme 8 Synthetic strategies for the preparation of anthratetrathiophene as the central scaffold and the respective HTMs. | ||
Firstly, commercially available 1,2,4,5-tetrabromobenzene reacts with 2-(tributyltin)thiophene through a Pd-catalyzed Stille coupling reaction. Then, four-fold α-selective bromination reaction employing NBS in THF bears the “open” core (36). Finally, the Scholl reaction mediated by a Lewis acid such as FeCl3 in combination with nitromethane afforded, in a quantitative yield, the π-extended structure (37). This methodology represents a facile approach for building polycyclic aromatic π-conjugated materials.114 TPAs with different alkyl chains (from methoxy to decyloxy) were linked to the brominated scaffold through a four-fold Suzuki coupling in DMF. ATT derivatives show a strong absorption band in the 300–500 nm range, with a maximum centered at 400 nm (ε = 1.5 × 10−5 M−1 cm−1), and HOMO energies of −5.3 eV were estimated in solution using cyclic voltammetry.
Conventional mesoporous (FAPbI3)0.85(MAPbBr3)0.15-based devices incorporating sulfur-rich HTMs, using LiTFSI, t-BP and FK209 as dopants, exhibit a maximum PCE of 18.13% for HTM-57. Upon increasing the length of the peripheral alkyl chains, the device efficiency decreases to 17.28% for HTM-58, 15.66% for HTM-59, and 9.67% for HTM-60. Photoluminescence (PL) studies show a strong quenching of the perovskite signal when using ATT as well as spiro-OMeTAD molecules. A stronger quenching is observed for HTM-57, which reveals a more efficient hole extraction at the interface with the perovskite. Furthermore, HTM-57 and HTM-58 show remarkable values of conductivity (5.8 × 10−4 and 1.5 × 10−4 S cm−1, respectively), which are attributed to a relatively highly ordered structure in the device due to two main driving forces, π–π stacking and S⋯S interactions.
Recently, Grätzel and coworkers reported two simple molecules based on a thiophene motif to be used as low-cost efficient HTMs (HTM-61 and HTM-62) (Fig. 12).115 3,4-Methoxy functionalized-thiophene is employed as a scaffold bearing TPAs, which are connected by a single and double bond. HTM-61 is synthesized using a Suzuki cross-coupling reaction with a standard boronic ester, while HTM-62 is prepared by a Horner–Wadsworth–Emmons reaction in 53% yields in both cases. In comparison with HTM-61, the light-harvesting properties of HTM-62 are strongly improved, owing to the better conjugation and the enhancement of the planarity, which eventually leads to a higher hole mobility.115 Both compounds have a suitable HOMO energy of −5.23 eV for HTM-61 and −5.21 eV for HTM-62, which are similar to the HOMO energy of spiro-OMeTAD and had a good alignment with the valance band edge of the perovskite. The hole-transport properties of pristine HTMs were evaluated for hole-only devices using time-of-flight (TOF) measurements, obtaining values of 7.66 × 10−5 and 1.34 × 10−4 cm2 V−1 s−1 for HTM-61 and HTM-62, respectively. The new HTMs were tested using a mesoscopic configuration (FTO/c-TiO2/m-TiO2/MAPbI3/HTM/Au) with dopants (LiTFSI, t-BP and FK209). Noteworthily, HTM-62 achieves a PCE of 20.1%, with respect to the 20.6% reached by spiro-OMeTAD. In contrast, HTM-61-based devices gave a power conversion efficiency of 16.9%. As a consequence of the lower Jsc and FF, the authors conjectured that the lower efficiency displayed by the latter is according to its lower hole mobility and conductivity. In addition, devices based on HTM-62 showed higher stability than that of HTM-61 and spiro-OMeTAD under a relative humidity of 30% or 85% at room temperature, aged without encapsulation in the dark and under continuous illumination.
Oxygen and silicon-containing HTMs
The presence of oxygen in HTMs has attracted the attention of different research groups. The device performances of oxygen- and silicon-containing HTMs including their device architectures are shown in Table 5. Grimsdale and coworkers reported a simple and inexpensive furan-based HTM, which comprises an electron-rich furan ring covalently linked to TPA-OMe units (HTM-63) (Fig. 15).116 The one-step Suzuki cross-coupling reaction between commercially available 2,5-dibromofuran and a TPA boronic acid derivative furnished the desired HTM in 65% yield. The UV-vis absorption spectrum of HTM-63 shows a red-shifted maximum, in comparison to spiro-OMeTAD, which is ascribed to the better conjugation in HTM-63. The estimated HOMO energy level from CV measurements of HTM-63 is similar to that of spiro-OMeTAD. Furthermore, the hole reorganization energy of HTM-63 determined from theoretical calculations is 79 meV larger than that of spiro-OMeTAD, in sharp contrast to the hole mobility, which is higher for HTM-63 due to its large electronic coupling. Mesoscopic MAPbI3-based devices incorporating doped-HTM-63 yield PCEs as high as 13.1%. The steady-state photoluminescence of the perovskites is strongly quenched after the deposition of HTM-63 (ca. 97%) and spiro-OMeTAD (ca. 93%), indicating a more efficient hole extraction at the perovskite MAPbI3/HTM-63 interface when compared to MAPbI3/spiro-OMeTAD.| HTM | HOMOd (eV) | σ (S cm−1) | μ h (cm2 V−1 s−1) | V oc (V) | J sc (mA cm−2) | FF | PCE (%) | Architecturee | Perovskite | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a LiTFSI, t-BP, FK209. b LiTFSI, t-BP, FK102. c F4-TCNQ. d HOMO energy levels were converted from the original work using Fc/Fc+ at 5.1 eV vs. vacuum. e M = mesoporous, P = planar and IP = inverted planar configurations. | ||||||||||
| HTM-63 | −5.15 | — | — | 1.10 | 19.63 | 0.61 | 13.1 | M | MAPbI3 | 116 |
| HTM-64 | −5.01 | — | — | 1.01 | 21.56 | 0.65 | 14.20 | M | MAPbI3 | 117 |
| HTM-65 | −5.14 | — | — | 1.03 | 21.22 | 0.64 | 14.07 | M | MAPbI3 | 117 |
| HTM-66 | −5.33 | — | 7.2 × 10−5 | 0.92 | 18.9 | 0.65 | 11.0 | M | MAPbI3 | 118 |
| HTM-67 | −5.16 | — | — | 0.96 | 16.8 | 0.72 | 11.7 | M | MAPbI3 | 121 |
| HTM-68 | −5.17 | — | — | 0.97 | 19.4 | 0.72 | 13.5 | M | MAPbI3 | 121 |
| HTM-69 | −5.39 | — | 2.96 × 10−4 | 1.07 | 23.08 | 0.72 | 19.06 | IP | MAPbI3 | 122 |
| HTM-70 | −5.34 | — | 6.3 × 10−5 | 0.99 | 19.85 | 0.77 | 14.15 | IP | MAPbI3 | 122 |
 | ||
| Fig. 15 Molecular structures of oxygen- and silicon-based HTMs. | ||
Shi and coworkers designed and synthetized two new extended π-conjugated HTMs based on a dibenzofuran unit, substituted in positions 3 and 6 with different methoxyaniline moieties (HTM-64 and HTM-65) (Fig. 15).117HTM-64 and HTM-65 exhibit good thermal properties and limited absorption in the visible region. The HOMO energy levels differ from each other due to the different electronic natures of the substituents (−5.01 eV for HTM-64 and −5.14 eV for HTM-65). Mesoscopic devices were prepared employing the following architecture: FTO/c-TiO2/m-TiO2/MAPbI3/HTM/Ag. The new HTMs were deposited using LiTFSI, t-BP and FK209 as dopants. The champion HTM-64-based device has a PCE of 14.20%. On the other hand, HTM-65-based devices showed similar photovoltaic performances with a maximum PCE of 14.07%. These values are slightly lower than those obtained for spiro-OMeTAD-based devices (15.04%) under the same conditions.
Mhaisalkar, Grimsdale and coworkers successfully introduced a silafluorene central core endowed with TPAs (HTM-66) (Fig. 15).118
Typically, silole-containing derivatives show enhanced charge transport properties compared to their carbon counterparts, which is attributed to an effectively induced crystallinity due to the longer C–Si bond that reduces the steric hindrance.119,120HTM-66 shows a simple structure, which was obtained in one-step synthesis in moderate yields. The introduction of the silafluorene results in a π-extended conjugation, causing a red-shift in the absorption onset in comparison with spiro-OMeTAD. Mesoscopic HTM-66-based devices display a PCE of 11%, which is slightly lower than that measured for spiro-OMeTAD (12.3%) under standard conditions. Although both devices show similar performances, the HTM-66-based device shows a substantially lower Voc than spiro-OMeTAD (0.92 vs. 0.975 V), despite the deeper HOMO energy of HTM-66 (−5.33 vs. −5.16 eV). Thus, the inferior charge extraction ability and lower conductivity are assumed to be the reasons for that difference in Voc.
Nazeeruddin, Ko and coworkers previously reported two different two-armed sililothiophene-based central scaffolds HTM-67 and HTM-68, using TPAs as the donor units (Fig. 15).121 The synthesis of these new derivatives followed a well-established procedure, which includes the preparation of the molecularly engineered TPA and the bromination of the sililothiophenes (Scheme 9). The synthesis of the latter derivatives comprises four additional synthetic steps. Bromination of bithiophene leads to tetrabromo-derivative 38, which is then selectively debrominated with Zn to afford dibromo-derivative 39. Compound 39 is treated with n-BuLi and quenched with di-n-hexyldichlorosilane or diphenyldichlorosilane, yielding derivatives 40 and 41, respectively, which are subsequently brominated with NBS. Finally, the two-fold Stille cross-coupling reaction between both counterparts leads to compounds HTM-67 and HTM-68. The different substitution on the sp3 silicon atom (HTM-67: alkyl chains and HTM-68: phenyl) does not have a strong impact on the optical and electrochemical properties. In turn, both doped-HTMs exhibit different photovoltaic behaviors in mesoscopic MAPbI3-based devices. HTM-68 displays a PCE of 13.5%, which is higher than that obtained for HTM-67 (11.7%). In contrast, the PCEs of these new HTMs were lower than the PCE achieved for spiro-OMeTAD-based devices (15.2%).
 | ||
| Scheme 9 Synthetic strategies for the preparation of a dithienosilole central scaffold. | ||
Very recently, Li and coworkers introduced two new X-shaped silicon-based HTMs for p–i–n PSCs,122 which is a less explored approach of HTMs (HTM-69 and HTM-70) (Fig. 15). As mentioned earlier, the introduction of silicon atoms into the structure of π-conjugated systems influences not only the optical but also the hole transport properties of the materials. The new four-armed substituted HTMs were prepared following a straightforward two-step synthesis by assembling the tetra(4-bromophenyl)silane with the corresponding electron-donating units. HTM-based devices were prepared employing the planar p–i–n architecture ITO/HTMs/MAPbI3/PCBM/Al. The doped/annealed based devices showed a PCE of 14.15% for HTM-70 and 18.23% for HTM-69, in sharp contrast to the undoped-devices, whose efficiency decreased to 6%. Furthermore, by depositing PCBM/C60/BCP as a triple cathode buffer layer, the PCE boosted up to 19.06%.
Metallomacrocycle-based HTMs
Porphyrins and phthalocyanines (Pc) have been extensively exploited as sensitizers for DSSCs123–125 and as scaffolds for the preparation of electron-donating materials for small-molecule bulk-heterojunction solar cells (SMBHJs).126–129 These two-dimensional macrocycles exhibit very appealing properties, such as intense absorption in the near-infrared region, high chemical flexibility and thermal stability, which make them good candidates as hole-transporting materials for PSCs.130,131 The photovoltaic parameters of the metallomacrocycle-based HTMs are summarized in Table 6.| HTM | HOMOd (eV) | σ (S cm−1) | μ h (cm2 V−1 s−1) | V oc (V) | J sc (mA cm−2) | FF | PCE (%) | Architecuree | Perovskite | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a LiTFSI and t-BP. b LiTFSI, t-BP, FK209. c Dopant-free. d HOMO energy levels were converted from the original work using Fc/Fc+ at 5.1 eV vs. vacuum. e M = mesoporous, P = planar and IP = inverted planar configurations. | ||||||||||
| HTM-71 | −5.16 | — | 2.04 × 10−4 | 0.99 | 22.82 | 0.73 | 16.60 | M | MAPbI3 | 132 |
| HTM-72 | −5.15 | — | 1.53 × 10−5 | 1.01 | 17.80 | 0.59 | 10.55 | M | MAPbI3 | 132 |
| HTM-73 | −5.31 | — | 2.89 × 10−4 | 1.07 | 21.61 | 0.66 | 15.36 | M | MAPbI3 | 134 |
| HTM-74 | −5.23 | — | 3.06 × 10−4 | 1.10 | 22.69 | 0.71 | 17.78 | M | MAPbI3 | 134 |
| HTM-75 | −5.20 | 4.0 × 10−5 | — | 1.05 | 22.6 | 0.73 | 17.5 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 136 |
| HTM-76 | −5.26 | 5.0 × 10−7 | — | 1.03 | 20.19 | 0.66 | 13.3 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 138 |
| HTM-77 | −5.13 | 8.0 × 10−5 | — | 1.05 | 20.28 | 0.80 | 17.1 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 138 |
| HTM-78 | −5.09 | 6.0 × 10−7 | — | 1.10 | 20.16 | 0.69 | 15.5 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 138 |
| HTM-79 | −5.06 | 1.97 × 10−4 | 1.90 × 10−4 | 1.08 | 23.1 | 0.73 | 18.3 | M/V2O5 | [FAPbI3]0.85[MAPbBr3]0.15 | 140 |
In 2016, Chen and co-workers devised a series of trans-substituted porphyrins with bilateral alkylamines of different lengths (HTM-71 and HTM-72) (Fig. 16).132 The new materials are synthesized in 5 synthetic steps in 7% overall yield, involving the condensation reaction of dipyrromethane with the corresponding aldehyde, which is the simplest route for obtaining trans-A2B2-porphyrins,133 bromination reaction and the Sonogashira cross-coupling reaction. The UV-vis spectra clearly reveal the minor contribution to light absorption by the porphyrin-based HTMs, with an absorption onset at 800 nm, due to the strong absorption of the perovskite film. Additionally, HTM-71 displays both Soret and Q bands significantly red-shifted and broadened, which suggest more effective aggregation of the porphyrins bearing shorter alkyl chains. On the other hand, increasing the length of the alkyl chain has no significant influence on the electrochemical properties and, therefore, both derivatives exhibit similar HOMO energy levels, suitable for hole scavenging from the perovskite film. Doped-HTM-71 and -HTM-72 based PSCs using CH3NH3PbI3 yielded PCE values of 16.6% and 10.5%, respectively, with noticeable hysteresis. These values were slightly lower than that obtained for spiro-OMeTAD under the same conditions (18.0%). The hole mobility (measured by space charge limited current) of HTM-71 (2.04 × 10−4 cm2 V−1 s−1) was one order of magnitude higher than that of HTM-72 (1.53 × 10−5 cm2 V−1 s−1). Thus, the longer bilateral alkyl chains hamper the hole transport to the Au electrode. More recently, Zhu and co-workers developed HTMs based on a highly symmetric Zn- and-Cu-porphyrin containing TPAs at each of the four meso positions (HTM-73 and HTM-74) (Fig. 16).134HTM-73 exhibits blue-shifted Q bands and split Soret bands in comparison with HTM-74. In addition, both compounds are highly stable and start decomposing above 430 °C. Mesoscopic compositionally engineered perovskite (FAPbI3)0.85(MAPbBr3)0.15-based devices containing doped-HTM-73 and HTM-74 reached 15.36% and 17.78%, respectively, with noticeable hysteresis. HTM-73-based devices exhibit lower Voc, Jsc and FF values stemming from its lower solubility and the formation of pinholes within the HTM film, which causes fast charge recombination and a lower FF. Additionally, HTM-74-based devices stored after 30 days (40–45% humidity at rt) retained 85% of their initial PCE, in sharp contrast to the 45% of devices incorporating spiro-OMeTAD.
 | ||
| Fig. 16 Molecular structures of metallomacrocycle-based HTMs. | ||
Seok and co-workers combined different doping ratios of commercially available tert-butyl-CuPc (HTM-75) with po-spiro-OMeTAD (HTM-2) in mesoporous PSCs, which has a positive effect on the PCE (Fig. 16).135 Transient photovoltage decay (TPD) experiments revealed that the addition of HTM-75 as a dopant retards the charge recombination. The best-performing device based on HTM-2 delivers an excellent performance of 17.5%. A remarkable improvement in the photovoltaic performance was achieved as a consequence of doping the HTM layer with increasing amount of HTM-75. Devices doped with 4.8% of HTMs show an improvement in the Voc and FF, reaching average efficiencies of 19.4%, with only little hysteresis. Retarding the charge recombination by doping HTM-2 with HTM-75 resulted in an enhanced FF, which clearly points to a reduction of the electron leakage current.
Furthermore, the same group investigated the impact of interface engineering not only on the PCE but also on the thermal stability of the device using HTM-75.136 Evidence of the strong interaction at the HTM/perovskite interface was experimentally determined by means of Raman spectroscopy.
An intense peak due to the deformation of the pyrrole ring appeared at 1600 cm−1 for the HTM/perovskite composite, while this peak for the pristine HTM-75 is typically very weak.137
Theoretical calculations predicted that the face-on HTM-75/perovskite interaction is preferred over the edge-on (2.22 eV vs. 0.51 eV) (Fig. 17). The authors proposed that the highly-ordered face-on organization of HTM-75 on top of the perovskite, along with an intimate contact between them, leads to improved hole transfer without contact resistance. Mesoscopic (FAPbI3)0.85(MAPbBr3)0.15-based devices employing doped-HTM-75 yielded a remarkable PCE of 18.8%. Noteworthily, these devices were stable for 200 hours without encapsulation under ambient conditions.
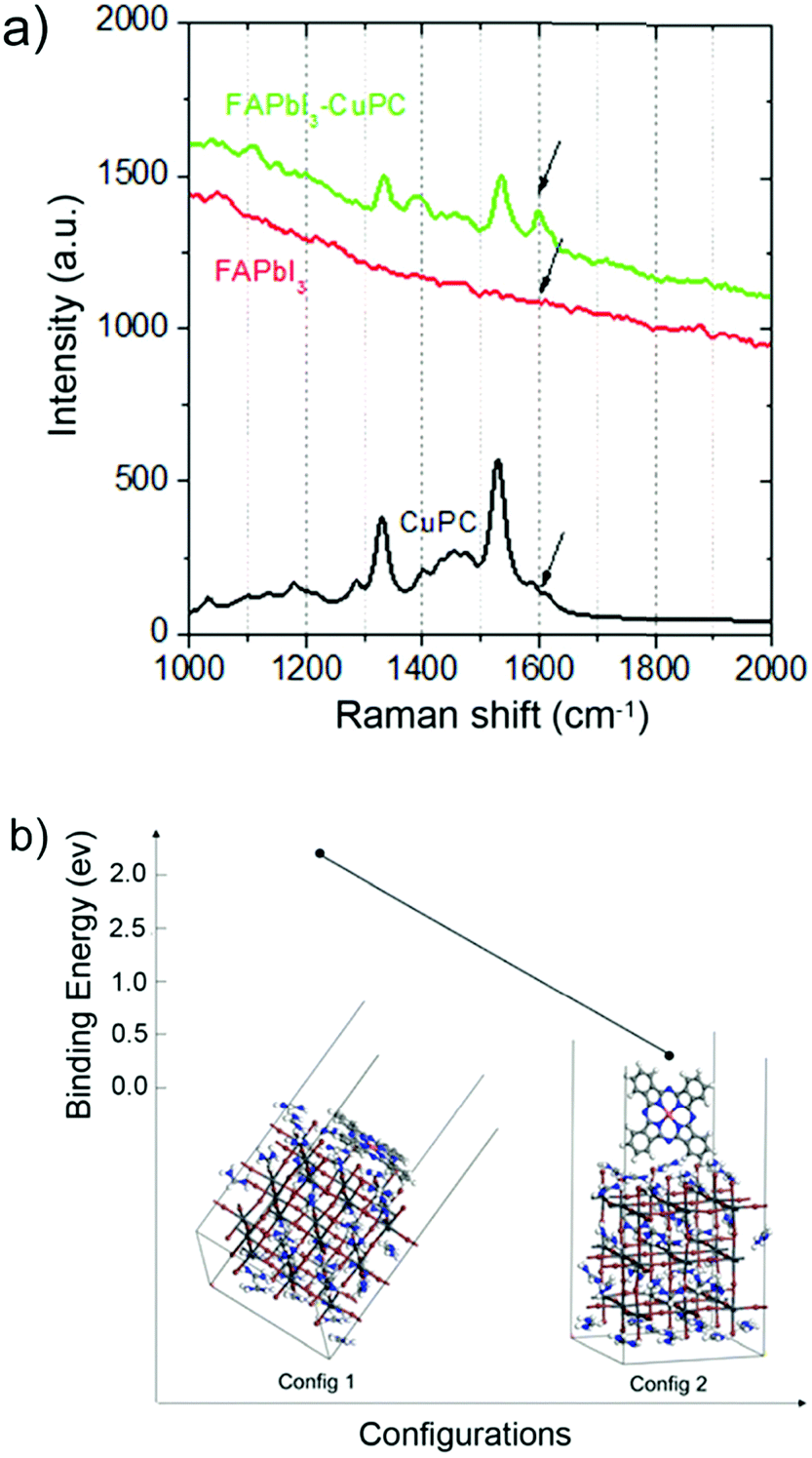 | ||
| Fig. 17 (a) Raman spectra of powder HTM-75, FAPbI3 powder and a composite powder of FAPbI3:HTM-75. (b) Theoretical calculation of face-on and edge-on orientations between the perovskite and HTM-75. Reproduced from ref. 136 with permission from the Royal Society of Chemistry, copyright 2017. | ||
Torres, Nazeeruddin and coworkers studied three Pc derivatives, endowed with different peripheral substituents, tert-butyl (HTM-76), hexylthiophene (HTM-77) and hexylbisthiophene (HTM-78), as HTMs for PSCs (Fig. 16).138 Extending the aromatic π-conjugation of the Pc is an effective method to tune their absorption properties. As a consequence, the typical Q-band absorption maxima are bathochromically-shifted to 665 nm for HTM-76, to 690 nm for HTM-77 and to 702 nm for HTM-78, along with a marked tendency toward aggregation. The HOMO energy level can be tuned by increasing the conjugation of the peripheral substituents. Thus, the lowest HOMO energy is for the commercial HTM-76 (−5.26 eV). Introducing four thiophenes results in an HOMO energy of −5.13 eV and the addition of four bisthiophenes leads to a raised HOMO energy (−5.09 eV). Mesoscopic devices based on doped HTM-76, HTM-77 and HTM-78 using the mixed-ion perovskite (FAPbI3)0.85(MAPbBr3)0.15 yield an overall efficiency of 13.3%, 17.1 and 15.5%, respectively, with noteworthy hysteresis. The lateral conductivity of the thin film supported the PCE trend. Thus, the lowest conductivity was found for HTM-76 (6.0 × 10−7 S cm−1) and for HTM-78 (6.0 × 10−7 S cm−1), whereas HTM-77 (8.0 × 10−5 S cm−1) exhibited comparable conductivity to that of spiro-OMeTAD (8.7 × 10−5 S cm−1). These findings boost the PCE for previously described ZnPc-based HTM-77.139
Sun and co-workers introduced an interface modification on perovskite-based devices, incorporating a buffer layer of V2O5 on top of the nickel(II)-based Pc acting as a HTM (HTM-79) (Fig. 16).140 The performance of the new HTM/V2O5 was investigated in mesoscopic devices. Initially, HTM-79 was incorporated without dopants, resulting in average PCE values of 9.9%. Adding dopants (LiTFSI), efficiencies of 17% are obtained, as a result of the increasing conductivity. Furthermore, incorporating an extra buffer layer (V2O5) on top of HTM-79 yields not only an enhanced overall efficiency until 17.6% but also lengthens the lifetime after the aging period by retaining 75% of their initial PCE after 30 days. On the other hand, spiro-OMeTAD decays more than 50% in the same period of time.
π-Conjugated oligomeric-based HTMs
In addition to the aforementioned HTMs, novel low band gap small organic molecules, constructed using building blocks derived from organic photovoltaics, have been exploited in PSCs. In this section, we include the most recent π-conjugated oligomeric structures, which exhibit strong absorbance in the visible region and are based on D–A or A–D–A-type derivatives (Fig. 18). The photovoltaic properties of the π-conjugated oligomeric-based HTMs including their optoelectrical properties are listed in Table 7.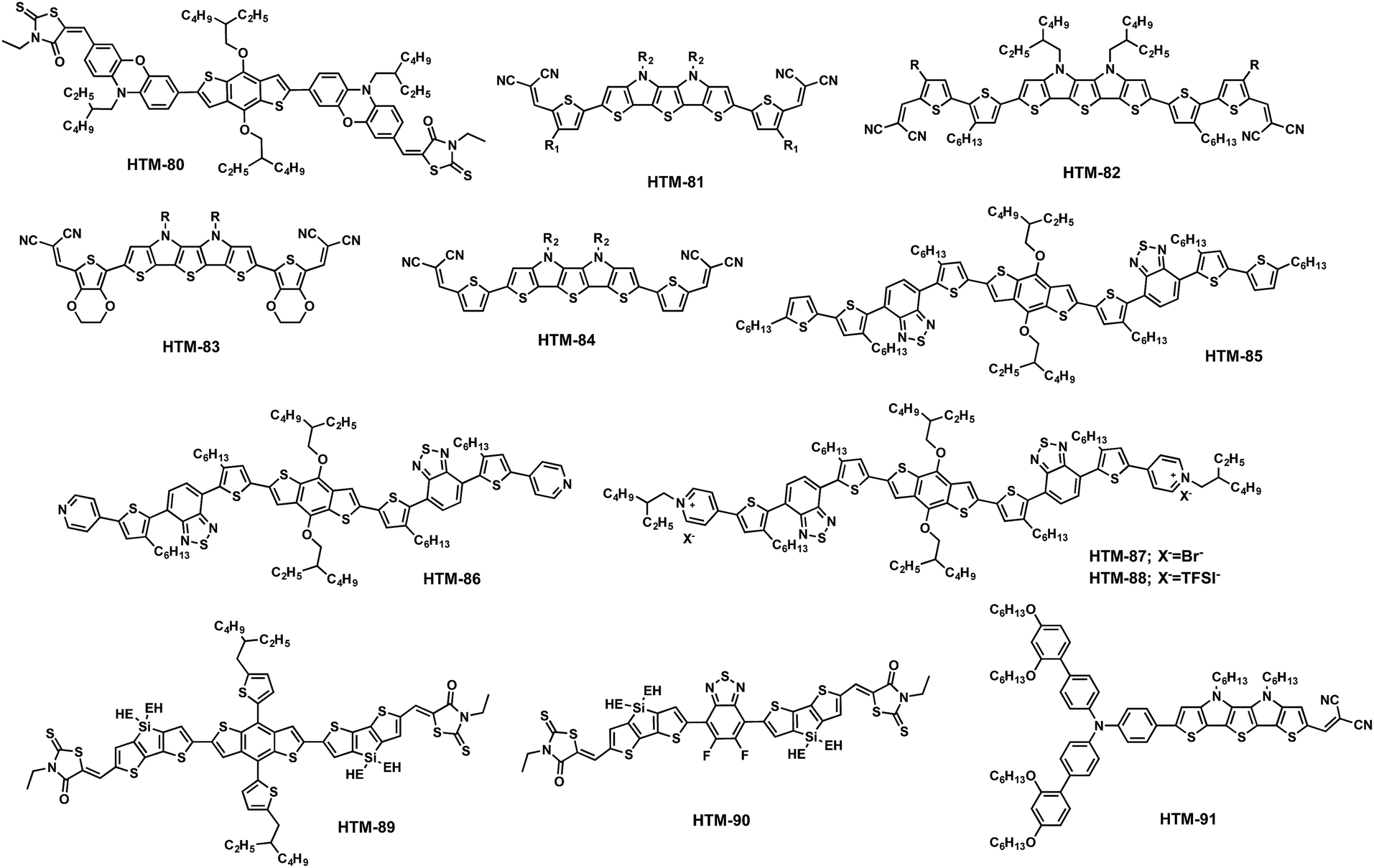 | ||
| Fig. 18 Molecular structures of π-conjugated oligomers as HTMs. | ||
| HTM | HOMOc (eV) | σ (S cm−1) | μ h (cm2 V−1 s−1) | V oc (V) | J sc (mA cm−2) | FF | PCE (%) | Architectured | Perovskite | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a LiTFSI and t-BP. b Dopant-free. c HOMO energy levels were converted from the original work using Fc/Fc+ at 5.1 eV vs. vacuum. d M = mesoporous, P = planar. | ||||||||||
| HTM-80 | −5.29 | 1.16 × 10−3 | 2.71 × 10−4 | 1.03 | 18.8 | 0.68 | 13.1 | P | MAPbI3 | 141 |
| HTM-81 | −5.26 | — | 7.3 × 10−5 | 0.99 | 16.4 | 0.65 | 10.5 | M | MAPbI3 | 144 |
| HTM-82 | −5.10 | — | 6.6 × 10−5 | 0.90 | 15.2 | 0.68 | 9.5 | M | MAPbI3 | 144 |
| HTM-83 | −5.20 | — | 4.2 × 10−5 | 0.95 | 16.50 | 0.72 | 11.24 | M | MAPbI3 | 145 |
| HTM-84 | −5.28 | — | 3.5 × 10−5 | 0.90 | 15.84 | 0.70 | 10.04 | M | MAPbI3 | 145 |
| HTM-85 | −5.26 | 1.16 × 10−4 | 3.18 × 10−4 | 1.00 | 18.4 | 0.76 | 13.9 | M | MAPbI3 | 146 |
| HTM-86 | −5.33 | — | 1.1 × 10−4 | 1.04 | 19.83 | 0.63 | 13.0 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 147 |
| HTM-87 | −5.28 | — | 3.03 × 10−4 | 1.05 | 21.94 | 0.66 | 15.1 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 147 |
| HTM-88 | −5.28 | — | 3.24 × 10−4 | 1.10 | 22.84 | 0.69 | 17.4 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 147 |
| HTM-89 | −5.09 | — | 1.0 × 10−4 | 1.05 | 21.2 | 0.73 | 16.2 | P | MAPbI3−xClx | 148 |
| HTM-90 | −5.15 | — | 2.4 × 10−6 | 0.98 | 17.1 | 0.37 | 6.2 | P | MAPbI3−xClx | 148 |
| HTM-91 | −5.16 | — | 9.5 × 10−5 | 1.05 | 22.2 | 0.73 | 16.9 | M | [FAPbI3]0.85[MAPbBr3]0.15 | 149 |
Sun and coworkers developed a benzodithiophene-based HTM endowed with phenoxazine-bridges and end-capped with 3-ethylrhodanine as an electron-accepting group (HTM-80) (Fig. 18).141 The new HTM can be obtained in a five-step sequence, without considering the synthesis of molecularly engineered distannylated benzodithiophene (BDT), which requires an extra four- to five-step synthetic protocol, starting from thiophene-3-carboxylic acid (Scheme 10a). HTM-80 absorbs over a wide spectral range (350–650 nm) with a maximum at 521 nm. In the cast-film, the absorption maximum is bathochromically shifted by 36 nm, relative to that in solution. Based on the SCLC method, dopant-free HTMs display hole mobilities of 2.71 × 10−4 cm2 V−1 s−1 for HTM-80 and 1.32 × 10−4 cm2 V−1 s−1 for spiro-OMeTAD. The undoped HTM-80 was incorporated in devices with a planar configuration of ITO/ZnO/PC70BM/CH3NH3PbI3/HTM-80/Ag, reaching a PCE of 13.2%, which was higher to those obtained for the doped-spiro-OMeTAD.
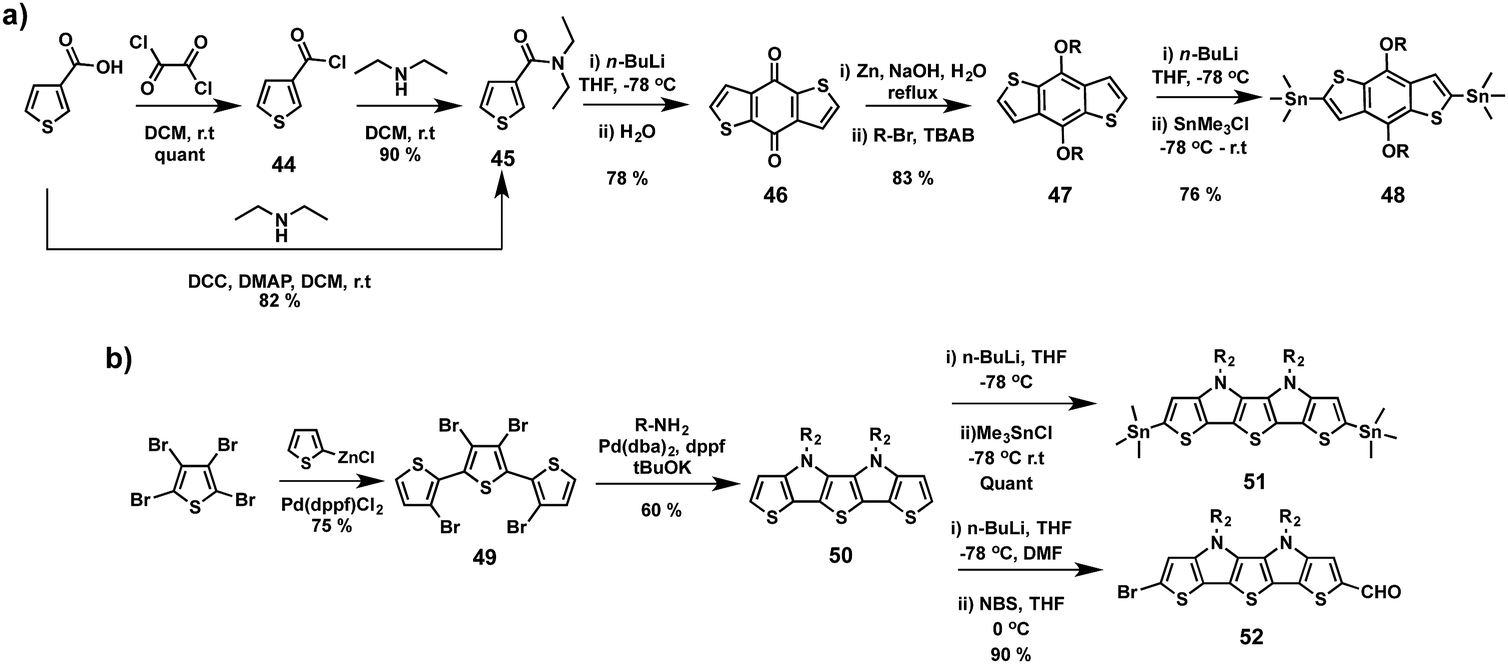 | ||
| Scheme 10 (a) Synthesis of molecularly engineered distannylated benzodithiophene (BDT). (b) Synthetic strategies for the preparation of S,N-heteropentacenes. | ||
Heteroacenes containing both thiophenes and pyrroles have been successfully employed in organic photovoltaics,142,143 as well as in PSCs. These ladder-type π-conjugated systems are envisaged to exhibit improved OFET performance owing to their additional intermolecular interactions (hydrogen bonding, π-stacking and sulfur–sulfur). The introduction of the pyrrole ring allows the anchoring of solubilizing chains on the N for solution-processing of these HTMs. In 2014, Bäuerle, Gräztel and coworkers reported a symmetrical thiophene–pyrrole-based HTM endowed with different thiophene spacers and end-capped with dicyanovinylene acceptor units (HTM-81 and HTM-82) (Fig. 18).144 The synthesis of a bis-stannylated S,N-heteropentacene central scaffold (51) was accomplished in three synthetic steps in 45% overall yield (Scheme 10b). Initially, a Pd-catalyzed Negishi coupling reaction between tetrabromothiophene and (3-bromothien-2-yl)zinc(II)chloride to yield the corresponding tetrabromo-terthiophene derivative (49). The latter was converted into the S,N-heteropentacene (50) by a Pd-catalyzed Buchwald–Hartwig amination reaction. Finally, the molecularly engineered central core (51) was quantitatively obtained by treating the latter with n-BuLi and quenching it with trimethyltin chloride. HTM-81 and HTM-82 were prepared by a two-fold Pd-catalyzed Stille cross-coupling reaction in 36% overall yield, without considering the synthesis of different thiophene linkers. HTMs exhibit strong absorption in the visible range, at 655 nm for HTM-81 and at 630 nm HTM-82, due to the presence of a charge-transfer-band. Incorporating an additional thiophene ring in HTM-82 (−5.10 eV), the HOMO energy level is increased in comparison with HTM-81 (−5.26 eV). Conventional mesoporous MAPbI3-based devices with both undoped-HTMs reached 10.5% and 9.5% for HTM-81 and HTM-82, respectively. Noteworthily, incident-photon-to-current conversion efficiency spectra reveal a noticeable improvement of the photocurrent from 400 to 800 nm, which was attributed to the contribution of HTM-81.
Later, in 2015, the same groups reported on two new S,N-heteropentacene HTMs following a similar concept. The central scaffold in HTM-83 and HTM-84 is flanked by EDOT or the thiophene ring, and end-capped with dicyanovinylene acceptor units (Fig. 18).145 The synthesis of these two novel derivatives proceeds smoothly following the synthetic methodologies followed for the aforementioned HTMs. In comparison with HTM-81 and HTM-82, the light-harvesting properties of HTM-83 and HTM-84 were significantly enhanced owing to their improved packing within the film as a result of the absence of long alkyl chains in the thiophene ring. Undoped HTM-83 and HTM-84 were incorporated in mesoscopic CH3NH3PbI3-based PSCs, yielding a maximum overall efficiency of 11.24% and 10.04%, respectively.
Sun, Kloo and coworkers disclosed another benzodithiophene-based derivative as a HTM (HTM-85) (Fig. 18). BDT was flanked with alkylated thiophene–benzothiadiazole π-conjugated spacers, and was successfully employed in bulk-heterojunction small-molecule organic solar cells (BHJ-SMOSCs).146 These 2D co-planar structures benefit the charge carrier transport due to the strong π–π intermolecular interaction when assembled into a thin film. Different alkyl chains were attached to the backbone to ensure its sufficient solubility in common organic solvents. The synthetic route of the new HTM took four steps without considering the synthesis of the distannylated BDT (Scheme 10a). Hole-only devices were prepared and measured by SCLC; a hole-mobility of 3.18 × 10−4 cm2 V−1 s−1 was obtained, which is two times higher than that for spiro-OMeTAD. A similar trend was observed in the conductivity without using any additives. Mesoscopic HTM-85-based devices, using CH3NH3PbI3, displayed a PCE of 13.4%.
Later and following the previous design, the same authors presented three new HTMs (HTM-86, HTM-87 and HTM-88) (Fig. 18).147 Appending pyridine moieties as end-capping to the BDT central scaffold, the authors evaluated the ionization effect of the pyridine/pyridinium unit and studied the effect of introducing different counter-ions (Br− and TFSI−) on the photovoltaic performance. In comparison with pristine pyridine HTM-86, the ionization of the pyridine end-capping units in HTM-87 and HTM-88 reflected a broader and red-shifted absorbance of about 50 nm. In addition, the ionic materials displayed lower reorganization energies, estimated by DFT calculations (163 and 157 meV for HTM-87 and HTM-88vs. 271 meV for HTM-86), which could imply faster charge transfer rates for the ionic HTMs. A similar trend was observed from hole mobility measurements using hole-only devices. The new HTMs were applied as additive-free HTMs in mesoscopic (FAPbI3)0.85(MAPbBr3)0.15-based PSCs. Through device optimization, PSCs containing HTM-88 yielded a remarkable average PCE of 17.4%, which is slightly lower than the performance of the doped spiro-OMeTAD (17.9%). On the other hand, HTM-86 and HTM-87 presented lower efficiency values (15.1 and 13.0%). Additionally, a stability test was performed under ambient conditions. Within 30 days, the PCEs of spiro-OMeTAD decreased from 17.4% to 6.4%, while HTM-86 and HTM-88 preserved 64% and 76% of their initial PCEs. The authors surmised that the higher hydrophobicity of the new HTMs prevents the water molecules from penetrating into the perovskite layer.
Yang and co-workers have also explored A–D–A systems as HTMs for PSCs.148 Dithienosilole was employed as a π-bridge for covalently linking electron-accepting rhodanines to the central scaffold (Fig. 18). Electron-donating alkylthienyl-substituted benzo[1,2-b:4,5-b′]dithiophene (HTM-89) and electron-withdrawing 5,6-difluoro-2,1,3-benzothiadiazole (HTM-90) were employed as central cores, which effectively tuned their HOMO/LUMO energy levels. HTM-89 can be synthesized by a Stille cross-coupling reaction between bis(trimethylstannyl)benzodithiophene and the corresponding brominated derivative of the dithienosililocarbaldehyde, which was previously prepared by a Vilsmeier–Haack reaction on bis(2-ethylhexyl)dithienosilole, followed by bromination reaction. Then, Knoevenagel condensation with dicarbaldehyde in the presence of catalytic base afforded HTM-89 in moderate overall yield (34%) starting from the expensive starting material bis(trimethylstannyl)benzodithiophene.
The synthetic preparation of HTM-90 involves a Stille cross-coupling reaction of the 4,7-dibromo-5,6-difluorobenzothiadiazole and the 2-(trimethyltin)dithienosilole, followed by a double Vilsmeier–Haack reaction using POCl3 and a final Knoevenagel condensation catalysed by a base, with an overall yield down to 30%. The hole mobilities, measured by SCLC, of HTM-89 and HTM-90 were 2.4 × 10−6 and 1.0 × 10−4 cm2 V−1 S−1, respectively. Both HTMs were tested in non-doped devices in a planar conventional device structure of ITO/TiO2/CH3NH3PbI3−xClx/HTM/MoO3/Ag. PSCs based on dopant-free HTM-89 showed a remarkable PCE of 16.2%. In sharp contrast, HTM-90 exhibited a poor PCE of 6.2%, which was attributed to its low-lying HOMO level (−5.5 eV) that does not guarantee an efficient hole injection and due to its low carrier mobility.
In 2016, Bäuerle, Gräztel and coworkers studied in detail the effect of D–B–A (donor–bridge–acceptor) systems as HTMs for PSCs, in which D is a molecularly engineered TPA, A is a dicyanovinylene group, and the B group is the previously described S,N-heteropentacene (HTM-91) (Fig. 18).149 To this aim, asymmetric derivative 52 was prepared by reacting derivative 50 with n-BuLi and quenching it with DMF (Scheme 10b). A subsequent bromination reaction yielded the asymmetric bridge 52. Finally, the donor moiety was introduced through the Suzuki cross-coupling reaction and the acceptor counterpart through a Knoevenagel condensation in the presence of malonitrile to successfully afford HTM-91. This showed not only strong and broad absorbance in the visible region with an optical band gap 1.71 eV in the thin-film, but also excellent band alignment with the valence band edge of the perovskite. Conventional mesoporous (FAPbI3)0.85(MAPbBr3)0.15-based devices with HTM-91, using LiTFSI and t-BP as additives and chlorobenzene as processing solvent, exhibit a maximum PCE of 13.9%. Significant improvements in all photovoltaic parameters were achieved by solvent modification. Therefore, using 1,1′,2,2′-tetrachloroethane as solvent yielded a maximum PCE of 16.9%. More recently, the same group successfully improved the photovoltaic performance of these D–B–A systems by tailoring the donor moiety to a PCE up to 17.7% using the triple-cation perovskite.150 In turn, the PCE of the device significantly decreased by increasing the length of the π-conjugated S,N-heteroacene bridge.
Conclusions and further perspectives
Since the advent of perovskites in 2009, tremendous advances have been made in every aspect of PSCs, from fundamental knowledge and material properties to device configurations and stability, among others. PCEs have also rapidly increased, exceeding 22%, revolutionizing the PV field and delivering one of the hottest topics in science.The leading role of the perovskite in the device needs a good supporting actor (charge selective contacts) for achieving highly efficient PSCs. This crucial role is attributed to spiro-OMeTAD, which is typically the HTM of choice and represents the benchmark for PSC devices. Alternatively, a wide variety of chemical structures have been investigated as p-type materials, opening new avenues to boost the PCEs.
In this review, molecular design and the organic synthesis of new small HTMs and their impact on the photovoltaic performance have been presented in a systematic way, paying special attention to the chemistry behind them.
From a structural viewpoint, basic designs of new HTMs rely on a variety of different central scaffolds mainly decorated with arylamine derivatives. Their three-dimensional propeller structures are beneficial for improving solution processability. In addition, strong electron-donating properties and excellent hole-injection render an appropriate band alignment with the valence band edge of the perovskite. It seems clear that spiro-like HTMs to date have achieved superior performances and it is presumed that they will continue to do so in doped-PSCs. However, as stated in this review, the vast majority of such HTMs need dopants to reach efficiencies higher than 17%. Of course, this is not a general rule and exceptions are currently rather limited.151–154
In contrast to non-planar spiro-like compounds, a variety of planar and aromatic molecules have been studied as central scaffolds for innovative HTMs. Furthermore, the presence of heteroatoms namely O, S, N, and Si, and their combinations has allowed determining their influence in the search for optimized and more stable HTMs at lower costs and with the ultimate goal of producing PSC devices at a competitive level. Needless to say, there are important drawbacks to solve. Metallomacrocycle-based compounds such as porphyrins and phthalocyanines are robust and well-known materials that together with the aforementioned carbo- and heterocycle-containing central scaffolds are potential candidates for use as highly efficient HTMs in PSCs.
An alternative means to successfully prepare highly efficient HTMs is by employing a D–π–A approach, in which the π-extended conjugation allows face-on organization through strong π–π interactions, affording remarkable charge-transport properties. “Self-doping” HTMs of ionic organic molecules is an effective method to reach high efficiencies by avoiding the use of dopants. This approach helps in enhancing charge extraction and significantly improving conductivity.155
Another chemical approach that could attract a lot of attention is the exploration of new crosslinkable HTMs. There are just a couple of examples with efficiencies of 18.3%, in which a HTM bearing a vinyl group reacts with pentaerythritol tetrakis(3-mercaptopropionate) (PETMP) through a well-known (first reported in 1905) thiol–ene “click chemistry”.156 In this sense, the crosslinkable-HTM not only exhibits a higher hole mobility and morphological stability, but also enhances hydrophobicity, which protects the perovskite layer from decomposition.
Currently, the future commercialization of PSCs is driven by the development of HTMs away from the elemental structure–property relationship understanding. Yet, there is no doubt that organic compounds will continue being an important part of the PSC architecture. It is our hope that this review gathering the most significant progress carried out on HTMs – systematically organized according to the chemical families forming the central scaffold – over the past years be a stimulus for the chemical community working in this fascinating area of organic compounds for energy. Considering the power of synthetic organic chemistry, no doubt that the preparation of innovative and more efficient molecules will be only limited by the imagination of chemists.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Research Council (ERC-320441-Chirallcarbon), the Spanish Ministry of Economy and Competitiveness (MINECO) of Spain (Projects CTQ2014-52045-R, CTQ2015-71936-REDT, CTQ2016-81911-REDT and CTQ2017-83531-R), the CAM (PHOTOCARBON project S2013/MIT-2841), and the Unidad de Excelencia Severo Ochoa SEV-2016-0686 is acknowledged.Notes and references
- N. Martín, Adv. Energy Mater., 2017, 7, 1601102 CrossRef.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- D. M. Chapin, C. S. Fuller and G. L. Pearson, J. Appl. Phys., 1954, 25, 676–677 CrossRef CAS.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- Z. Song, C. L. McElvany, A. B. Phillips, I. Celik, P. W. Krantz, S. C. Watthage, G. K. Liyanage, D. Apul and M. J. Heben, Energy Environ. Sci., 2017, 10, 1297–1305 RSC.
- M. Cai, Y. Wu, H. Chen, X. Yang, Y. Qiang and L. Han, Adv. Sci., 2017, 4, 1600269 CrossRef PubMed.
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissortel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS.
- E. J. Juarez-Perez, M. R. Leyden, S. Wang, L. K. Ono, Z. Hawash and Y. Qi, Chem. Mater., 2016, 28, 5702–5709 CrossRef CAS.
- J. Krüger, R. Plass, L. Cevey, M. Piccirelli, M. Grätzel and U. Bach, Appl. Phys. Lett., 2001, 79, 2085–2087 CrossRef.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- H. S. Kim, C. R. Lee, J. H. Im, K. B. Lee, T. Moehl, A. Marchioro, S. J. Moon, R. Humphry-Baker, J. H. Yum, J. E. Moser, M. Grätzel and N. G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- S. Wang, M. Sina, P. Parikh, T. Uekert, B. Shahbazian, A. Devaraj and Y. S. Meng, Nano Lett., 2016, 16, 5594–5600 CrossRef CAS PubMed.
- S. N. Habisreutinger, N. K. Noel, H. J. Snaith and R. J. Nicholas, Adv. Energy Mater., 2017, 7, 1601079 CrossRef.
- J. Xia, N. Masaki, M. Lira-Cantu, Y. Kim, K. Jiang and S. Yanagida, J. Am. Chem. Soc., 2008, 130, 1258–1263 CrossRef CAS PubMed.
- B. Fang, H. Zhou and I. Honma, Appl. Phys. Lett., 2005, 86, 261909 CrossRef.
- A. Abate, T. Leijtens, S. Pathak, J. Teuscher, R. Avolio, M. E. Errico, J. Kirkpatrik, J. M. Ball, P. Docampo, I. McPherson and H. J. Snaith, Phys. Chem. Chem. Phys., 2013, 15, 2572–2579 RSC.
- Z. Hawash, L. K. Ono and Y. Qi, Adv. Mater. Interfaces, 2016, 3, 1600117 CrossRef.
- L. K. Ono, P. Schulz, J. J. Endres, G. O. Nikiforov, Y. Kato, A. Kahn and Y. Qi, J. Phys. Chem. Lett., 2014, 5, 1374–1379 CrossRef CAS PubMed.
- J. H. Noh, N. J. Jeon, Y. C. Choi, M. K. Nazeeruddin, M. Grätzel and S. I. Seok, J. Mater. Chem. A, 2013, 1, 11842–11847 RSC.
- H. Xi, S. Tang, X. Ma, J. Chang, D. Chen, Z. Lin, P. Zhong, H. Wang and C. Zhang, ACS Omega, 2017, 2, 326–336 CrossRef CAS.
- C.-C. Chung, T.-W. Tsai, H.-P. Wu and E. W.-G. Diau, J. Phys. Chem. C, 2017, 121, 18472–18479 CrossRef CAS.
- T. Ye, J. Wang, W. Chen, Y. Yang and D. He, ACS Appl. Mater. Interfaces, 2017, 9, 17923–17931 CrossRef CAS PubMed.
- D.-Y. Chen, W.-H. Tseng, S.-P. Liang, C.-I. Wu, C.-W. Hsu, Y. Chi, W.-Y. Hung and P.-T. Chou, Phys. Chem. Chem. Phys., 2012, 14, 11689–11694 RSC.
- B. Xu, J. Huang, H. Ågren, L. Kloo, A. Hagfeldt and L. Sun, ChemSusChem, 2014, 7, 3252–3256 CrossRef CAS PubMed.
- C. Chen, W. Zhang, J. Cong, M. Cheng, B. Zhang, H. Chen, P. Liu, R. Li, M. Safdari, L. Kloo and L. Sun, ACS Energy Lett., 2017, 2, 497–503 CrossRef CAS.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376 CrossRef CAS PubMed.
- L. Calió, S. Kazim, M. Grätzel and S. Ahmad, Angew. Chem., Int. Ed., 2016, 55, 14522–14545 CrossRef PubMed.
- S. M. Park, S. M. Mazza, Z. Liang, A. Abtahi, A. M. Boehm, S. R. Parkin, J. E. Anthony and K. R. Graham, ACS Appl. Mater. Interfaces, 2018, 10, 15548–15557 CrossRef CAS PubMed.
- R. A. Belisle, P. Jain, R. Prasanna, T. Leijtens and M. D. McGehee, ACS Energy Lett., 2016, 1, 556–560 CrossRef CAS.
- L. E. Polander, P. Pahner, M. Schwarze, M. Saalfrank, C. Koerner and K. Leo, APL Mater., 2014, 2, 081503 CrossRef.
- L. Zhao, R. A. Kerner, Z. Xiao, Y. L. Lin, K. M. Lee, J. Schwartz and B. P. Rand, ACS Energy Lett., 2016, 1, 595–602 CrossRef CAS.
- E. M. Sanehira, B. J. Tremolet de Villers, P. Schulz, M. O. Reese, S. Ferrere, K. Zhu, L. Y. Lin, J. J. Berry and J. M. Luther, ACS Energy Lett., 2016, 1, 38–45 CrossRef CAS.
- J.-P. Correa-Baena, W. Tress, K. Domanski, E. H. Anaraki, S.-H. Turren-Cruz, B. Roose, P. P. Boix, M. Grätzel, M. Saliba, A. Abate and A. Hagfeldt, Energy Environ. Sci., 2017, 10, 1207–1212 RSC.
- Y. Shao, Y. Yuan and J. Huang, Nat. Energy, 2016, 1, 15001 CrossRef CAS.
- L.-H. Xie, J. Liang, J. Song, C.-R. Yin and W. Huang, Curr. Org. Chem., 2010, 14, 2169–2195 CrossRef CAS.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS PubMed.
- J. Yi, Y. Wang, Q. Luo, Y. Lin, H. Tan, H. Wang and C.-Q. Ma, Chem. Commun., 2016, 52, 1649–1652 RSC.
- X.-F. Wu, W.-F. Fu, Z. Xu, M. Shi, F. Liu, H.-Z. Chen, J.-H. Wan and T. P. Russell, Adv. Funct. Mater., 2015, 25, 5954–5966 CrossRef CAS.
- R. G. Clarkson and M. Gomberg, J. Am. Chem. Soc., 1930, 52, 2881–2891 CrossRef CAS.
- C. Poriel, Y. Ferrand, S. Juillard, P. Le Maux and G. Simonneaux, Tetrahedron, 2004, 60, 145–158 CrossRef CAS.
- J. M. Tour, R. Wu and J. S. Schumm, J. Am. Chem. Soc., 1990, 112, 5662–5663 CrossRef CAS.
- J. Salbeck, N. Yu, J. Bauer, F. Weissörtel and H. Bestgen, Synth. Met., 1997, 91, 209–215 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- X. Li, D. Bi, C. Yi, J.-D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, 353, 52–62 CrossRef PubMed.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano, J.-P. Correa Baena, J.-D. Decoppet, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Grätzel and A. Hagfeldt, Sci. Adv., 2016, 2, e1501170 Search PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- N. J. Jeon, H. G. Lee, Y. C. Kim, J. Seo, J. H. Noh, J. Lee and S. I. Seok, J. Am. Chem. Soc., 2014, 136, 7837–7840 CrossRef CAS PubMed.
- Z. Hu, W. Fu, L. Yan, J. Miao, H. Yu, Y. He, O. Goto, H. Meng, H. Chen and W. Huang, Chem. Sci., 2016, 7, 5007–5012 RSC.
- F. Bischoff and H. Adkins, J. Am. Chem. Soc., 1923, 45, 1030–1033 CrossRef CAS.
- L.-H. Xie, F. Liu, C. Tang, X.-Y. Hou, Y.-R. Hua, Q.-L. Fan and W. Huang, Org. Lett., 2006, 8, 2787–2790 CrossRef CAS PubMed.
- B. Xu, D. Bi, Y. Hua, P. Liu, M. Cheng, M. Grätzel, L. Kloo, A. Hagfeldt and L. Sun, Energy Environ. Sci., 2016, 9, 873–877 RSC.
- D. Bi, B. Xu, P. Gao, L. Sun, M. Grätzel and A. Hagfeldt, Nano Energy, 2016, 23, 138–144 CrossRef CAS.
- K. Liu, Y. Yao, J. Wang, L. Zhu, M. Sun, B. Ren, L. Xie, Y. Luo, Q. Meng and X. Zhan, Mater. Chem. Front., 2017, 1, 100–110 RSC.
- M. Saliba, S. Orlandi, T. Matsui, S. Aghazada, M. Cavazzini, J.-P. Correa-Baena, P. Gao, R. Scopelliti, E. Mosconi, K.-H. Dahmen, F. De Angelis, A. Abate, A. Hagfeldt, G. Pozzi, M. Grätzel and M. K. Nazeeruddin, Nat. Energy, 2016, 1, 15017 CrossRef CAS.
- G. Pozzi, S. Orlandi, M. Cavazzini, D. Minudri, L. Macor, L. Otero and F. Fungo, Org. Lett., 2013, 15, 4642–4645 CrossRef CAS PubMed.
- A. Torres and L. G. C. Rego, J. Phys. Chem. C, 2014, 118, 26947–26954 CrossRef CAS.
- B. Xu, J. Zhang, Y. Hua, P. Liu, L. Wang, C. Ruan, Y. Li, G. Boschloo, E. M. J. Johansson, L. Kloo, A. Hagfeldt, A. K. Y. Jen and L. Sun, Chem, 2017, 2, 676–687 CAS.
- Y. Wang, T.-S. Su, H.-Y. Tsai, T.-C. Wei and Y. Chi, Sci. Rep., 2017, 7, 7859 CrossRef PubMed.
- Y. Wang, Z. Zhu, C.-C. Chueh, A. K. Y. Jen and Y. Chi, Adv. Energy Mater., 2017, 7, 1700823 CrossRef.
- S. Kazim, F. J. Ramos, P. Gao, M. K. Nazeeruddin, M. Grätzel and S. Ahmad, Energy Environ. Sci., 2015, 8, 1816–1823 RSC.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed.
- G. Giri, E. Verploegen, S. C. B. Mannsfeld, S. Atahan-Evrenk, D. H. Kim, S. Y. Lee, H. A. Becerril, A. Aspuru-Guzik, M. F. Toney and Z. Bao, Nature, 2011, 480, 504 CrossRef CAS PubMed.
- C. D. Sheraw, T. N. Jackson, D. L. Eaton and J. E. Anthony, Adv. Mater., 2003, 15, 2009–2011 CrossRef CAS.
- J. S. Brooks, D. L. Eaton, J. E. Anthony, S. R. Parkin, J. W. Brill and Y. Sushko, Curr. Appl. Phys., 2001, 1, 301–306 CrossRef.
- K. Rakstys, M. Saliba, P. Gao, P. Gratia, E. Kamarauskas, S. Paek, V. Jankauskas and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2016, 55, 7464–7468 CrossRef CAS PubMed.
- P. U. Biedermann, John J. Stezowski and I. Agranat, Eur. J. Org. Chem., 2001, 15–34 CrossRef CAS.
- C. de la Harpe and W. A. van Dorp, Ber. Dtsch. Chem. Ges., 1875, 8, 1048–1050 CrossRef.
- A. Levy, M. Goldschmidt and I. Agranat, Lett. Org. Chem., 2006, 3, 579–584 CrossRef CAS.
- J. Zhang, Y. Hua, B. Xu, L. Yang, P. Liu, M. B. Johansson, N. Vlachopoulos, L. Kloo, G. Boschloo, E. M. J. Johansson, L. Sun and A. Hagfeldt, Adv. Energy Mater., 2016, 6, 1601062 CrossRef.
- A. St. Pfau and P. Plattner, Helv. Chim. Acta, 1936, 19, 858–879 CrossRef CAS.
- P. A. Plattner and A. St. Pfau, Helv. Chim. Acta, 1937, 20, 224–232 CrossRef CAS.
- D. Copland, D. Leaver and W. B. Menzies, Tetrahedron Lett., 1977, 18, 639–640 CrossRef.
- H. Nishimura, N. Ishida, A. Shimazaki, A. Wakamiya, A. Saeki, L. T. Scott and Y. Murata, J. Am. Chem. Soc., 2015, 137, 15656–15659 CrossRef CAS PubMed.
- A. Wakamiya, H. Nishimura, T. Fukushima, F. Suzuki, A. Saeki, S. Seki, I. Osaka, T. Sasamori, M. Murata, Y. Murata and H. Kaji, Angew. Chem., Int. Ed., 2014, 53, 5800–5804 CrossRef CAS PubMed.
- S. Park, J. H. Heo, J. H. Yun, T. S. Jung, K. Kwak, M. J. Ko, C. H. Cheon, J. Y. Kim, S. H. Im and H. J. Son, Chem. Sci., 2016, 7, 5517–5522 RSC.
- S. Park, J. H. Heo, C. H. Cheon, H. Kim, S. H. Im and H. J. Son, J. Mater. Chem. A, 2015, 3, 24215–24220 RSC.
- P. Nirmalraj, D. Thompson, A. Molina-Ontoria, M. Sousa, N. Martín, B. Gotsmann and H. Riel, Nat. Mater., 2014, 13, 947 CrossRef CAS PubMed.
- M. P. Eng, S. Shoaee, A. Molina-Ontoria, A. Gouloumis, N. Martín and J. R. Durrant, Phys. Chem. Chem. Phys., 2011, 13, 3721–3729 RSC.
- S. Wolfrum, J. R. Pinzon, A. Molina-Ontoria, A. Gouloumis, N. Martín, L. Echegoyen and D. M. Guldi, Chem. Commun., 2011, 47, 2270–2272 RSC.
- L. Cabau, I. García-Benito, A. Molina-Ontoria, N. F. Montcada, N. Martín, A. Vidal-Ferran and E. Palomares, Chem. Commun., 2015, 51, 13980–13982 RSC.
- H. Choi, K. Do, S. Park, J.-S. Yu and J. Ko, Chem. – Eur. J., 2015, 21, 15919–15923 CrossRef CAS PubMed.
- L. Zhu, Y. Shan, R. Wang, D. Liu, C. Zhong, Q. Song and F. Wu, Chem. – Eur. J., 2017, 23, 4373–4379 CrossRef CAS PubMed.
- W. Ke, P. Priyanka, S. Vegiraju, C. C. Stoumpos, I. Spanopoulos, C. M. M. Soe, T. J. Marks, M.-C. Chen and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 388–393 CrossRef CAS PubMed.
- T. Malinauskas, M. Saliba, T. Matsui, M. Daskeviciene, S. Urnikaite, P. Gratia, R. Send, H. Wonneberger, I. Bruder, M. Grätzel, V. Getautis and M. K. Nazeeruddin, Energy Environ. Sci., 2016, 9, 1681–1686 RSC.
- X.-C. Li, C.-Y. Wang, W.-Y. Lai and W. Huang, J. Mater. Chem. C, 2016, 4, 10574–10587 RSC.
- K. Rakstys, A. Abate, M. I. Dar, P. Gao, V. Jankauskas, G. Jacopin, E. Kamarauskas, S. Kazim, S. Ahmad, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2015, 137, 16172–16178 CrossRef CAS PubMed.
- K. Rakstys, S. Paek, P. Gao, P. Gratia, T. Marszalek, G. Grancini, K. T. Cho, K. Genevicius, V. Jankauskas, W. Pisula and M. K. Nazeeruddin, J. Mater. Chem. A, 2017, 5, 7811–7815 RSC.
- P.-Y. Su, L.-B. Huang, J.-M. Liu, Y.-F. Chen, L.-M. Xiao, D.-B. Kuang, M. Mayor and C.-Y. Su, J. Mater. Chem. A, 2017, 5, 1913–1918 RSC.
- F. J. Ramos, K. Rakstys, S. Kazim, M. Grätzel, M. K. Nazeeruddin and S. Ahmad, RSC Adv., 2015, 5, 53426–53432 RSC.
- I. Cho, N. J. Jeon, O. K. Kwon, D. W. Kim, E. H. Jung, J. H. Noh, J. Seo, S. I. Seok and S. Y. Park, Chem. Sci., 2017, 8, 734–741 RSC.
- M. Daskeviciene, S. Paek, Z. Wang, T. Malinauskas, G. Jokubauskaite, K. Rakstys, K. T. Cho, A. Magomedov, V. Jankauskas, S. Ahmad, H. J. Snaith, V. Getautis and M. K. Nazeeruddin, Nano Energy, 2017, 32, 551–557 CrossRef CAS.
- R. Grisorio, B. Roose, S. Colella, A. Listorti, G. P. Suranna and A. Abate, ACS Energy Lett., 2017, 2, 1029–1034 CrossRef.
- S. P. Massie, Chem. Rev., 1954, 54, 797–833 CrossRef CAS.
- H. Choi, S. Park, S. Paek, P. Ekanayake, M. K. Nazeeruddin and J. Ko, J. Mater. Chem. A, 2014, 2, 19136–19140 RSC.
- S. Paek, P. Qin, Y. Lee, K. T. Cho, P. Gao, G. Grancini, E. Oveisi, P. Gratia, K. Rakstys, S. A. Al-Muhtaseb, C. Ludwig, J. Ko and M. K. Nazeeruddin, Adv. Mater., 2017, 29, 1606555 CrossRef PubMed.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. Shakya Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. J. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2011, 133, 3272–3275 CrossRef CAS PubMed.
- P. Liu, Y. Wu, H. Pan, Y. Li, S. Gardner, B. S. Ong and S. Zhu, Chem. Mater., 2009, 21, 2727–2732 CrossRef CAS.
- J. H. Seo, E. B. Namdas, A. Gutacker, A. J. Heeger and G. C. Bazan, Adv. Funct. Mater., 2011, 21, 3667–3672 CrossRef CAS.
- H. Li, K. Fu, A. Hagfeldt, M. Grätzel, S. G. Mhaisalkar and A. C. Grimsdale, Angew. Chem., Int. Ed., 2014, 53, 4085–4088 CrossRef CAS PubMed.
- J. Roncali, P. Blanchard and P. Frere, J. Mater. Chem., 2005, 15, 1589–1610 RSC.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- M. L. Petrus, T. Bein, T. J. Dingemans and P. Docampo, J. Mater. Chem. A, 2015, 3, 12159–12162 RSC.
- A. Molina-Ontoria, I. Zimmermann, I. García-Benito, P. Gratia, C. Roldán-Carmona, S. Aghazada, M. Grätzel, M. K. Nazeeruddin and N. Martín, Angew. Chem., Int. Ed., 2016, 55, 6270–6274 CrossRef CAS PubMed.
- R. Proetzsch, D. Bieniek and F. Korte, Tetrahedron Lett., 1972, 13, 543–544 CrossRef.
- N. Jayasuriya, J. Kagan, J. E. Owens, E. P. Kornak and D. M. Perrine, J. Org. Chem., 1989, 54, 4203–4205 CrossRef CAS.
- T. Kashiki, S. Shinamura, M. Kohara, E. Miyazaki, K. Takimiya, M. Ikeda and H. Kuwabara, Org. Lett., 2009, 11, 2473–2475 CrossRef CAS PubMed.
- I. García-Benito, I. Zimmermann, J. Urieta-Mora, J. Arago, A. Molina-Ontoria, E. Orti, N. Martín and M. K. Nazeeruddin, J. Mater. Chem. A, 2017, 5, 8317–8324 RSC.
- X. Guo, S. R. Puniredd, M. Baumgarten, W. Pisula and K. Müllen, J. Am. Chem. Soc., 2012, 134, 8404–8407 CrossRef CAS PubMed.
- X. Guo, S. Wang, V. Enkelmann, M. Baumgarten and K. Müllen, Org. Lett., 2011, 13, 6062–6065 CrossRef CAS PubMed.
- I. Zimmermann, J. Urieta-Mora, P. Gratia, J. Aragó, G. Grancini, A. Molina-Ontoria, E. Ortí, N. Martín and M. K. Nazeeruddin, Adv. Energy Mater., 2017, 7, 1601674 CrossRef.
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed.
- F. Zhang, Z. Wang, H. Zhu, N. Pellet, J. Luo, C. Yi, X. Liu, H. Liu, S. Wang, X. Li, Y. Xiao, S. M. Zakeeruddin, D. Bi and M. Grätzel, Nano Energy, 2017, 41, 469–475 CrossRef CAS.
- A. Krishna, D. Sabba, J. Yin, A. Bruno, P. P. Boix, Y. Gao, H. A. Dewi, G. G. Gurzadyan, C. Soci, S. G. Mhaisalkar and A. C. Grimsdale, Chem. – Eur. J., 2015, 21, 15113–15117 CrossRef CAS PubMed.
- Y. Shi, K. Hou, Y. Wang, K. Wang, H. Ren, M. Pang, F. Chen and S. Zhang, J. Mater. Chem. A, 2016, 4, 5415–5422 RSC.
- A. Krishna, D. Sabba, J. Yin, A. Bruno, L. J. Antila, C. Soci, S. Mhaisalkar and A. C. Grimsdale, J. Mater. Chem. A, 2016, 4, 8750–8754 RSC.
- H.-Y. Chen, J. Hou, A. E. Hayden, H. Yang, K. N. Houk and Y. Yang, Adv. Mater., 2010, 22, 371–375 CrossRef CAS PubMed.
- M. C. Scharber, M. Koppe, J. Gao, F. Cordella, M. A. Loi, P. Denk, M. Morana, H.-J. Egelhaaf, K. Forberich, G. Dennler, R. Gaudiana, D. Waller, Z. Zhu, X. Shi and C. J. Brabec, Adv. Mater., 2010, 22, 367–370 CrossRef CAS PubMed.
- A. Abate, S. Paek, F. Giordano, J.-P. Correa-Baena, M. Saliba, P. Gao, T. Matsui, J. Ko, S. M. Zakeeruddin, K. H. Dahmen, A. Hagfeldt, M. Grätzel and M. K. Nazeeruddin, Energy Environ. Sci., 2015, 8, 2946–2953 RSC.
- R. Xue, M. Zhang, G. Xu, J. Zhang, W. Chen, H. Chen, M. Yang, C. Cui, Y. Li and Y. Li, J. Mater. Chem. A, 2018, 6, 404–413 RSC.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- A. Yella, C.-L. Mai, S. M. Zakeeruddin, S.-N. Chang, C.-H. Hsieh, C.-Y. Yeh and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 2973–2977 CrossRef CAS PubMed.
- M.-E. Ragoussi, M. Ince and T. Torres, Eur. J. Org. Chem., 2013, 6475–6489 CrossRef CAS.
- C.-L. Wang, J.-Y. Hu, C.-H. Wu, H.-H. Kuo, Y.-C. Chang, Z.-J. Lan, H.-P. Wu, E. Wei-Guang Diau and C.-Y. Lin, Energy Environ. Sci., 2014, 7, 1392–1396 RSC.
- K. Gao, L. Li, T. Lai, L. Xiao, Y. Huang, F. Huang, J. Peng, Y. Cao, F. Liu, T. P. Russell, R. A. J. Janssen and X. Peng, J. Am. Chem. Soc., 2015, 137, 7282–7285 CrossRef CAS PubMed.
- N. F. Montcada, S. Arrechea, A. Molina-Ontoria, A. I. Aljarilla, P. de la Cruz, L. Echegoyen, E. Palomares and F. Langa, Org. Electron., 2016, 38, 330–336 CrossRef CAS.
- A. Varotto, C.-Y. Nam, I. Radivojevic, J. P. C. Tomé, J. A. S. Cavaleiro, C. T. Black and C. M. Drain, J. Am. Chem. Soc., 2010, 132, 2552–2554 CrossRef CAS PubMed.
- W. Ke, D. Zhao, C. R. Grice, A. J. Cimaroli, G. Fang and Y. Yan, J. Mater. Chem. A, 2015, 3, 23888–23894 RSC.
- F. J. Ramos, M. Ince, M. Urbani, A. Abate, M. Grätzel, S. Ahmad, T. Torres and M. K. Nazeeruddin, Dalton Trans., 2015, 44, 10847–10851 RSC.
- H.-H. Chou, Y.-H. Chiang, M.-H. Li, P.-S. Shen, H.-J. Wei, C.-L. Mai, P. Chen and C.-Y. Yeh, ACS Energy Lett., 2016, 1, 956–962 CrossRef CAS.
- J. S. Lindsey, Acc. Chem. Res., 2010, 43, 300–311 CrossRef CAS PubMed.
- S. Chen, P. Liu, Y. Hua, Y. Li, L. Kloo, X. Wang, B. Ong, W.-K. Wong and X. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 13231–13239 CrossRef CAS PubMed.
- J. Seo, N. J. Jeon, W. S. Yang, H.-W. Shin, T. K. Ahn, J. Lee, J. H. Noh and S. I. Seok, Adv. Energy Mater., 2015, 5, 1501320 CrossRef.
- Y. C. Kim, T. Y. Yang, N. J. Jeon, J. Im, S. Jang, T. J. Shin, H. W. Shin, S. Kim, E. Lee, S. Kim, J. H. Noh, S. I. Seok and J. Seo, Energy Environ. Sci., 2017, 10, 2109–2116 RSC.
- T. V. Basova and B. A. Kolesov, J. Struct. Chem., 2000, 41, 770–777 CrossRef CAS.
- K. T. Cho, O. Trukhina, C. Roldán-Carmona, M. Ince, P. Gratia, G. Grancini, P. Gao, T. Marszalek, W. Pisula, P. Y. Reddy, T. Torres and M. K. Nazeeruddin, Adv. Energy Mater., 2017, 7, 1601733 CrossRef.
- P. Gao, K. T. Cho, A. Abate, G. Grancini, P. Y. Reddy, M. Srivasu, M. Adachi, A. Suzuki, K. Tsuchimoto, M. Grätzel and M. K. Nazeeruddin, Phys. Chem. Chem. Phys., 2016, 18, 27083–27089 RSC.
- M. Cheng, Y. Li, M. Safdari, C. Chen, P. Liu, L. Kloo and L. Sun, Adv. Energy Mater., 2017, 7, 1602556 CrossRef.
- M. Cheng, B. Xu, C. Chen, X. Yang, F. Zhang, Q. Tan, Y. Hua, L. Kloo and L. Sun, Adv. Energy Mater., 2015, 5, 1401720 CrossRef.
- A. Mishra, D. Popovic, A. Vogt, H. Kast, T. Leitner, K. Walzer, M. Pfeiffer, E. Mena-Osteritz and P. Bäuerle, Adv. Mater., 2014, 26, 7217–7223 CrossRef CAS PubMed.
- H. Kast, A. Mishra, G. L. Schulz, M. Urdanpilleta, E. Mena-Osteritz and P. Bäuerle, Adv. Funct. Mater., 2015, 25, 3414–3424 CrossRef CAS.
- P. Qin, H. Kast, M. K. Nazeeruddin, S. M. Zakeeruddin, A. Mishra, P. Bäuerle and M. Grätzel, Energy Environ. Sci., 2014, 7, 2981–2985 RSC.
- C. Steck, M. Franckevicius, S. M. Zakeeruddin, A. Mishra, P. Bäuerle and M. Grätzel, J. Mater. Chem. A, 2015, 3, 17738–17746 RSC.
- C. Chen, M. Cheng, P. Liu, J. Gao, L. Kloo and L. Sun, Nano Energy, 2016, 23, 40–49 CrossRef CAS.
- M. Cheng, K. Aitola, C. Chen, F. Zhang, P. Liu, K. Sveinbjörnsson, Y. Hua, L. Kloo, G. Boschloo and L. Sun, Nano Energy, 2016, 30, 387–397 CrossRef CAS.
- Y. Liu, Z. Hong, Q. Chen, H. Chen, W.-H. Chang, Y. Yang, T.-B. Song and Y. Yang, Adv. Mater., 2016, 28, 440–446 CrossRef CAS PubMed.
- D. Bi, A. Mishra, P. Gao, M. Franckevičius, C. Steck, S. M. Zakeeruddin, M. K. Nazeeruddin, P. Bäuerle, M. Grätzel and A. Hagfeldt, ChemSusChem, 2016, 9, 433–438 CrossRef CAS PubMed.
- N. Arora, C. Wetzel, M. I. Dar, A. Mishra, P. Yadav, C. Steck, S. M. Zakeeruddin, P. Bäuerle and M. Grätzel, ACS Appl. Mater. Interfaces, 2017, 9, 44423–44428 CrossRef CAS PubMed.
- R. Azmi, S. Y. Nam, S. Sinaga, Z. A. Akbar, C.-L. Lee, S. C. Yoon, I. H. Jung and S.-Y. Jang, Nano Energy, 2018, 44, 191–198 CrossRef CAS.
- H. Chen, W. Fu, C. Huang, Z. Zhang, S. Li, F. Ding, M. Shi, C.-Z. Li, A. K. Y. Jen and H. Chen, Adv. Energy Mater., 2017, 7, 1700012 CrossRef.
- F. Zhang, X. Liu, C. Yi, D. Bi, J. Luo, S. Wang, X. Li, Y. Xiao, S. M. Zakeeruddin and M. Grätzel, ChemSusChem, 2016, 9, 2578–2585 CrossRef CAS PubMed.
- Z. Li, Z. Zhu, C.-C. Chueh, S. B. Jo, J. Luo, S.-H. Jang and A. K. Y. Jen, J. Am. Chem. Soc., 2016, 138, 11833–11839 CrossRef CAS PubMed.
- J. Zhang, B. Xu, L. Yang, A. Mingorance, C. Ruan, Y. Hua, L. Wang, N. Vlachopoulos, M. Lira-Cantú, G. Boschloo, A. Hagfeldt, L. Sun and E. M. J. Johansson, Adv. Energy Mater., 2017, 7, 1602736 CrossRef.
- Z. Li, Z. Zhu, C. Chueh, J. Luo and A. K. Y. Jen, Adv. Energy Mater., 2016, 6, 1601165 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |