
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Liposomes and polymersomes: a comparative review towards cell mimicking
Emeline
Rideau
 a,
Rumiana
Dimova
b,
Petra
Schwille
c,
Frederik R.
Wurm
*a and
Katharina
Landfester
*a
a,
Rumiana
Dimova
b,
Petra
Schwille
c,
Frederik R.
Wurm
*a and
Katharina
Landfester
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de; landfest@mpip-mainz.mpg.de
bMax Planck Institute for Colloids and Interfaces, Wissenschaftspark Potsdam-Golm, Am Mühlenberg, 14476 Potsdam, Germany
cMax Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany
First published on 4th September 2018
Abstract
Cells are integral to all forms of life due to their compartmentalization by the plasma membrane. However, living organisms are immensely complex. Thus there is a need for simplified and controllable models of life for a deeper understanding of fundamental biological processes and man-made applications. This is where the bottom-up approach of synthetic biology comes from: a stepwise assembly of biomimetic functionalities ultimately into a protocell. A fundamental feature of such an endeavor is the generation and control of model membranes such as liposomes and polymersomes. We compare and contrast liposomes and polymersomes for a better a priori choice and design of vesicles and try to understand the advantages and shortcomings associated with using one or the other in many different aspects (properties, synthesis, self-assembly, applications) and which aspects have been studied and developed with each type and update the current development in the field.
 Emeline Rideau | Emeline Rideau is currently a postdoctoral researcher under the supervision of Prof. Dr Katharina Landfester at the Max Planck Institute for Polymer Research, Germany. She obtained her BSc in biomolecular sciences from the University of St Andrews (UK) and her DPhil from the University of Oxford (UK) in organic chemistry in 2016 with Prof. Stephen Fletcher. Her current research interests include synthesis and self-assembly of block copolymers into polymersomes and hybrid vesicles as cellular mimics. |
 Rumiana Dimova | Rumiana Dimova obtained her PhD from Bordeaux University (France) in 1999. Afterwards she joined the Max Planck Institute of Colloids and Interfaces as a postdoctoral fellow, where she became a group leader in 2000. Since then she has been leading the Biophysics Lab in the Department of Theory and BioSystems. Her main research interests are in the field of membrane biophysics. In 2014, she was awarded the EPS Emmy Noether distinction for women in physics. |
 Petra Schwille | Petra Schwille obtained her PhD in 1996 with Manfred Eigen at the MPI for Biophysical Chemistry in Göttingen. After a postdoctoral stay at Cornell University (Ithaca, NY) she established a research group at the MPI Göttingen in 1999 and accepted a call as professor and chair of biophysics at the BIOTEC of the TU Dresden in 2002. In 2011, she was appointed as a scientific member of the Max Planck Society and director at the MPI of Biochemistry, Martinsried. Her research interests range from single molecule biophysics to bottom-up synthetic biology of artificial cells. |
 Frederik R. Wurm | Frederik R. Wurm is currently heading the research group “Functional Polymers” at the Max Planck Institute for Polymer Research (MPIP). In his interdisciplinary research, Frederik designs materials with molecular-defined functions for degradable polymers, nanocarriers with controlled blood interactions, adhesives, and phosphorus flame-retardants. He has published more than 150 research articles to date. He received his PhD in 2009 (JGU Mainz, D). After a two-year stay at EPFL (CH) as a Humboldt fellow, he joined the department “Physical Chemistry of Polymers” at MPIP and finished his habilitation in Macromolecular Chemistry in 2016. |
 Katharina Landfester | Katharina Landfester received her doctoral degree in Physical Chemistry after working in 1995 at the MPI for Polymer Research (MPIP). After a postdoctoral stay at the Lehigh University (Bethlehem, PA), she worked at the MPI of Colloids and Interfaces in Golm leading the mini-emulsion group. From 2003 to 2008, she was a professor at the University of Ulm. She joined the Max Planck Society in 2008 as one of the directors of the MPIP. Her research focuses on creating functional colloids for new material and biomaterial applications. |
I. Introduction
Cells, prokaryotic and eukaryotic, are undeniably integral to all forms of life and even essential for life apparition due to their compartmentalization.1–4 Within the compartments, sensitive entities like the genetic material, are protected, processes are regulated and can statistically occur due to concentration effects, as is the case in protein synthesis. The driving force of reproduction and growth is made possible via cell division and energy is generated through the use of proton gradients between cell membranes using adenosine triphosphate (ATP)-synthase or between specialized organelles like the chloroplasts. Moreover, cells only exist because of the barrier that separates the intracellular components from the extracellular media: its membrane.The fundamental importance of cell membranes has incited scientists not only to study them, but also to create synthetic analogs and models, such as bilayer structures based on natural or synthetic molecules ranging from lipids to surfactants and amphiphilic block copolymers.
But why are synthetic cells necessary, when natural cells are so efficient, well-designed and a product of billions of years of evolution?
From the biological point of view, there has been a lot of effort to elucidate the inherent complexity of life, and biotechnology and bioengineering try to use this knowledge to improve man-made technology. However as life is so immensely complex, the usage and control of organisms in biotechnology are highly difficult.1 Thus there is a need for simplified and controllable models of life both for a deeper understanding of fundamental biological processes and further man-made applications.
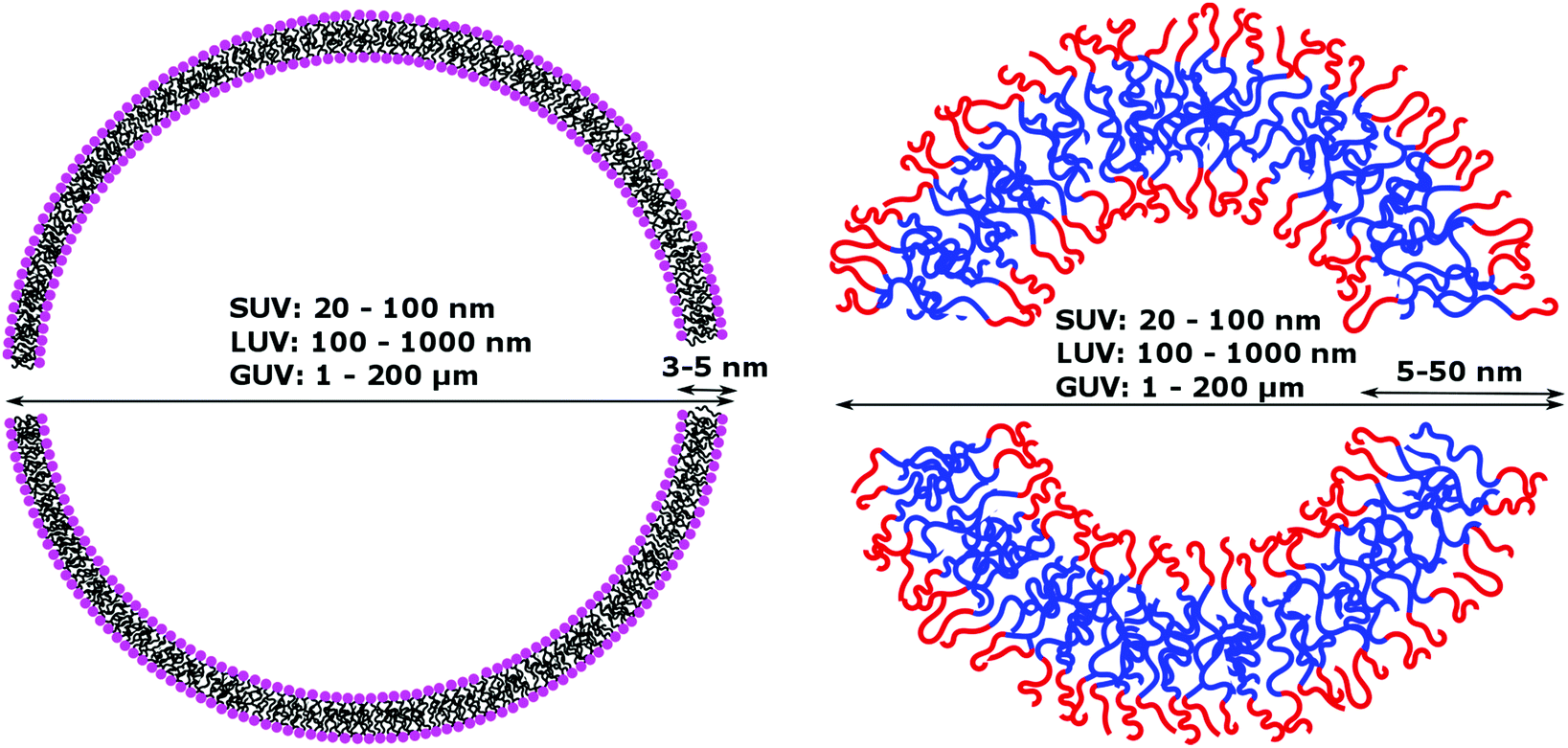
Natural cell membranes consist of a phospholipid bilayer, to which a cornucopia of proteins and small molecules (cholesterol, carbohydrates etc.) are bound.5 Hence it is not surprising that natural lipids (mostly phospholipids)6,7 or their synthetic alternatives can self-assemble into vesicles (spherical bilayer) on their own, termed liposomes (liposomes – from the Greek some = “body of”). Liposomes are a simplified model of a cell membrane without its ornaments. Liposomes have been extremely well studied since the 1960s, but they are unstable and sensitive and have poor modular chemical functionality as we discuss in Section II of this review.8 Thus, despite lipids being highly biocompatible, there has been a great motivation in recent years to use versatile, easily obtainable and tunable entities, which behave in many aspects like lipids: amphiphilic polymers.1,9–11 The general physical properties of lipids and amphiphilic polymers are similar: they both are composed of a polar end (hydrophilic or charged head in lipids typically a phosphate moiety and 1 or 2 hydrophilic block(s) in polymers) covalently bound to a hydrophobic tail (1–2 aliphatic chains in lipids and 1 hydrophobic bock in polymers).12 Thus based on these facts amphiphilic polymers can also self-assemble into polymeric vesicles, or polymersomes (Fig. 1).
 | ||
| Fig. 1 Subunits, 2D and 3D structures of liposomes and polymersomes. | ||
In this review, we will first explore the differences and similarities in physical properties between liposomes and polymersomes to try to understand their advantages and limitations (Section II). We will also briefly explain the different methods to obtain those vesicles (Section III) and discuss stimuli-responsive vesicles as primordial functionalization of those assemblies towards cell mimicking (Section IV). Finally, we summarize recent progress in creating artificial cells (Section V).
II. Comparison of physical properties
Despite their similar amphiphilic nature, liposomes and polymersomes exhibit important physical differences summarized in Table 1. Those properties have significant repercussion in the application of the vesicles notably from the perspective of generating synthetic protocells.|
|
|||
|---|---|---|---|
| Liposomes | Polymersomes | Eukaryotic cells | |
| Molecular weight of building compounds | 102–103 g mol−1 | 103–104 g mol−1 | 102–105 g mol−1 |
| Membrane thickness | 3–5 nm | 5–50 nm | 8–10 nm (including membrane proteins) |
| Diameter | SUV: 20–100 nm | Animal cells: 10–30 μm | |
| LUV: 100–1000 nm | Plant cells: 10–100 μm | ||
| GUV: 1–200 μm | |||
| Encapsulating volume (calculated) | SUV: 10−15–10−13 μL | SUV: 0–10−13 μL | Animal cells: 10−6–10−5 μL |
| LUV: 10−13–10−9 μL | LUV: 0–10−9 μL | Plant cells: 10−6–10−3 μL | |
| GUV: 10−9–10−3 μL | GUV: 10−9–10−3 μL | ||
(a) Size
Phospholipids have a molecular weight of 100–1000 g mol−1 whereas commonly used amphiphilic block copolymers typically have molecular weights Mn of >1000 g mol−1 as polymers are easier to synthesize controllably and in a low dispersity fashion than oligomers.12 The molecular weight characteristics of the vesicular subunits result in the bilayer thickness of the self-assembled vesicles to be 3–5 nm for liposomes.2,12,13 For polymersomes, reported values are variable, but typically thicker, on average 5–50 nm.2,8,14 The membrane thickness of polymersomes was found to be dependent on the polymer Mn or degree of polymerization, especially for the hydrophobic block,2,11,12,15,16 hence explaining its much greater range. In comparison the cell membrane is 8–10 nm thick, including the lipid bilayer, membrane proteins and small molecules (cholesterol and carbohydrates).17 In this respect, the appropriately designed polymersomes compare better to natural cells than liposomes, despite the latter being chemically more similar to a cell. Liposome membranes are thinner than cell membranes due to the absence of membrane proteins.In terms of diameter, liposomes and polymersomes can both form small vesicles (nm range) or giant vesicles (>1 μm) depending on the method used for preparation.15 The obtained vesicles are usually characterized and classified based on their diameter and lamellar properties (Fig. 2). These classifications are not well defined and vary between authors. The term small unilamellar vesicles (SUVs) is often used for vesicles of 20–100 nm diameter, large unilamellar vesicles (LUVs) for 100 nm–1 μm vesicles and giant unilamellar vesicles (GUVs) for vesicles of >1 μm diameter.9 In this review we commonly refer to SUVs for the smaller vesicles typically with diameters around 100 nm and GUVs for the macroscopic vesicles. In comparison, animal cells are typically 10–30 μm large and plant cells 10–100 μm also depending on their type and thus most model cells will focus on the formation and properties of GUVs. Even cell organelles like mitochondria are macroscopic (typically 1 × 2 μm)18 and though typically represented by SUVs for simplification, proper models should account for their large size. Sometimes vesicles are not unilamellar and can be referred to as multilamellar (MLV) (several bilayers) or multivesicular (MVV) (vesicles in vesicles also called vesosomes) and are usually larger in order to accommodate the volume of internal structures.9,19 MLV and UV, even of similar size, have very different physicochemical properties like permeability, stability, elasticity, toughness etc. which lead to different applications. MLV are particularly common in the development of drug delivery systems.9 Moreover, despite block copolymer membranes being thicker than lipid membranes, it has been shown that small-sized ion-channels can span a triblock copolymer1 and grafted polymer membranes.13 These interesting results prove that polymeric membranes possess a high degree of adaptability to form thinner membrane rafts to incorporate the peptidic channel and thus are also biocompatible. We noticed that even though lipid-GUVs have been relatively well studied, reports on polymeric GUVs have only become increasingly more popular since 2012.
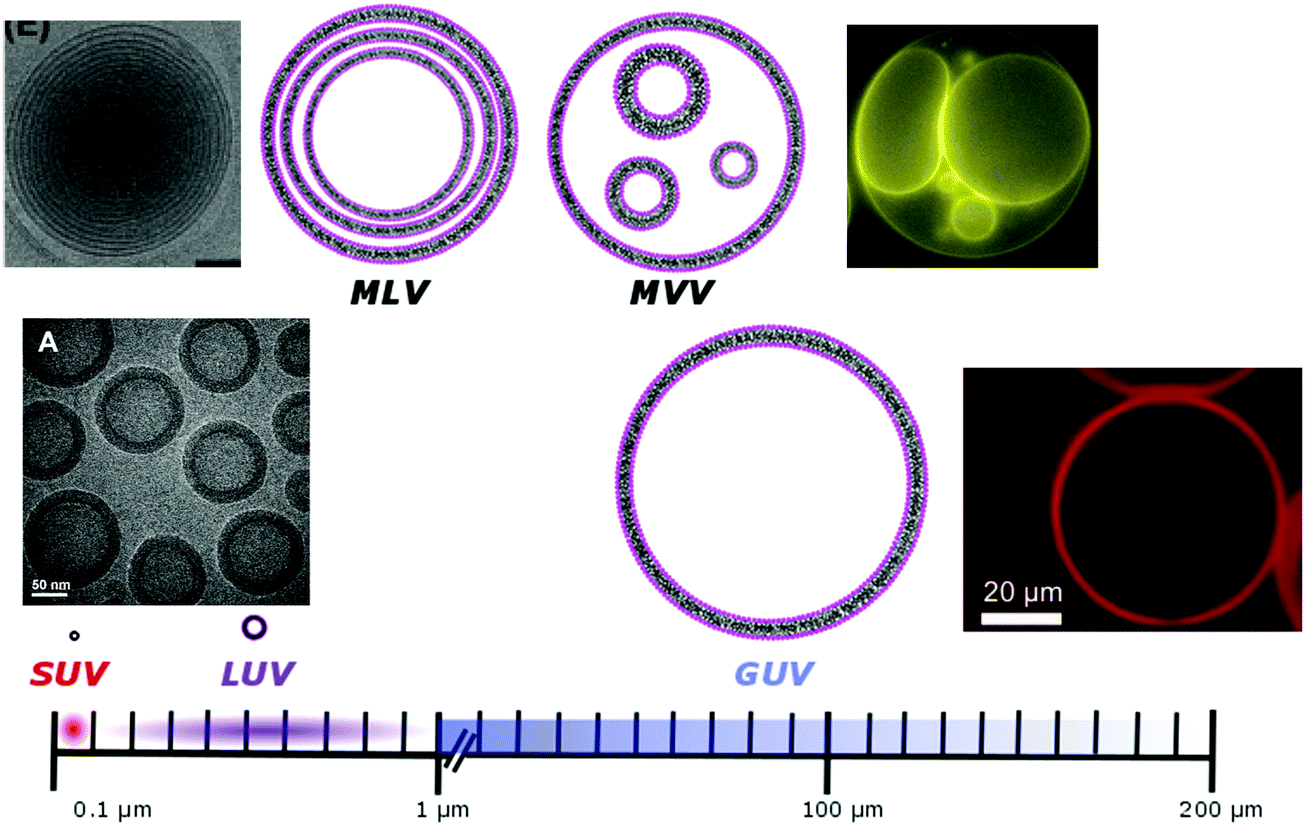 | ||
| Fig. 2 Different classifications of possible vesicles. SUV/LUV/GUV: small/large/giant unilamellar vesicles; MLV: multilamellar vesicles; MVV: multivesicular vesicles. Microscopy images: SUV/LUV: Adapted from ref. 20 with permission from The Royal Society of Chemistry. GUV: Reprinted from J. Petit, I. Polenz, J. C. Baret, S. Herminghaus and O. Baumchen, Eur. Phys. J. E: Soft Matter Biol. Phys., 2016, 39, 59. https://doi.org/10.1140/epje/i2016-16059-8.21. MLV: reprinted from S. Allen, O. Osorio, Y.-G. Liu and E. Scott, Facile assembly and loading of theranostic polymersomes via multi-impingement flash nanoprecipitation, J. Controlled Release, 262, 91–103. Copyright (2017), with permission from Elsevier.22 MVV: adapted from K. Oglęcka, J. Sanborn, A. N. Parikh and R. S. Kraut, Front. Physiol., 2012, 3, 120 (https://www.frontiersin.org/articles/10.3389/fphys.2012.00120/full).23 | ||
Because membranes are essentially just a compartmentalization, the encapsulating volume of the vesicles is an important property to consider. Based on the membrane thickness and vesicular size, we calculated the estimated internal volume to determine how the differences in membrane thickness affect the encapsulation efficiency and how these relate to eukaryotic cells (Table 1). For SUVs and LUVs, the encapsulated volume is small (>10−9 μL) and the variation in internal volume for polymersomes is high and greatly dependent on the membrane thickness, whereas liposomes are much less affected as the membrane thickness is insignificant compared to the vesicular size. For GUVs, in the case of both polymersomes and liposomes, the membrane thickness is negligible compared to the vesicle size and a well-designed synthetic vesicle could fit the volume of a eukaryotic cell (∼10−4 μL) for protocell synthesis purposes.
(b) Permeability and stability
Vesicles, independently of their amphiphilic building blocks, have the significant advantage over other carriers to be able to encapsulate both hydrophilic and hydrophobic cargos within their lumen and membrane respectively (Fig. 3a). However, liposomes are always described as problematically leaky (Fig. 3b), resulting in poor retention efficiency of cargos.24 Their chemical properties, size, saturation levels and method of preparation can modulate their permeability.6,9,25 The high permeability of liposomes is deemed a consequence of their high lateral fluidity linked to their low molecular weight and is discussed in more detail in Section Ic (Fig. 3c).12,26 The high dynamic mobility of membranes is also found in cells, making liposomes a good choice for artificial cell generation in this respect. In contrast, polymersomes retain their cargos with greater efficiency. Polymersomes are thermodynamically more inert as their lateral diffusivity is very low.11,12 This property often depends on entanglement and varies with the overall Mn of the polymer and the nature of its hydrophobic block and is also determined by the glass-transition temperature (Tg) of the hydrophobic block.16,27,28 The polymersomes’ low membrane permeability can also be problematic in certain applications (e.g. nanoreactors) as passive diffusion of small molecules through the membrane is low and does not accurately mimic cell fluidity.8,29 However, because of the versatility of polymer chemistry, advanced polymersomes can be functionalized for tunable permeability and lateral mobility and for example the grafted polymer PDMS-g-PEO generates vesicles of similar thickness (5 nm) and high fluidity to those of liposomes and cells.13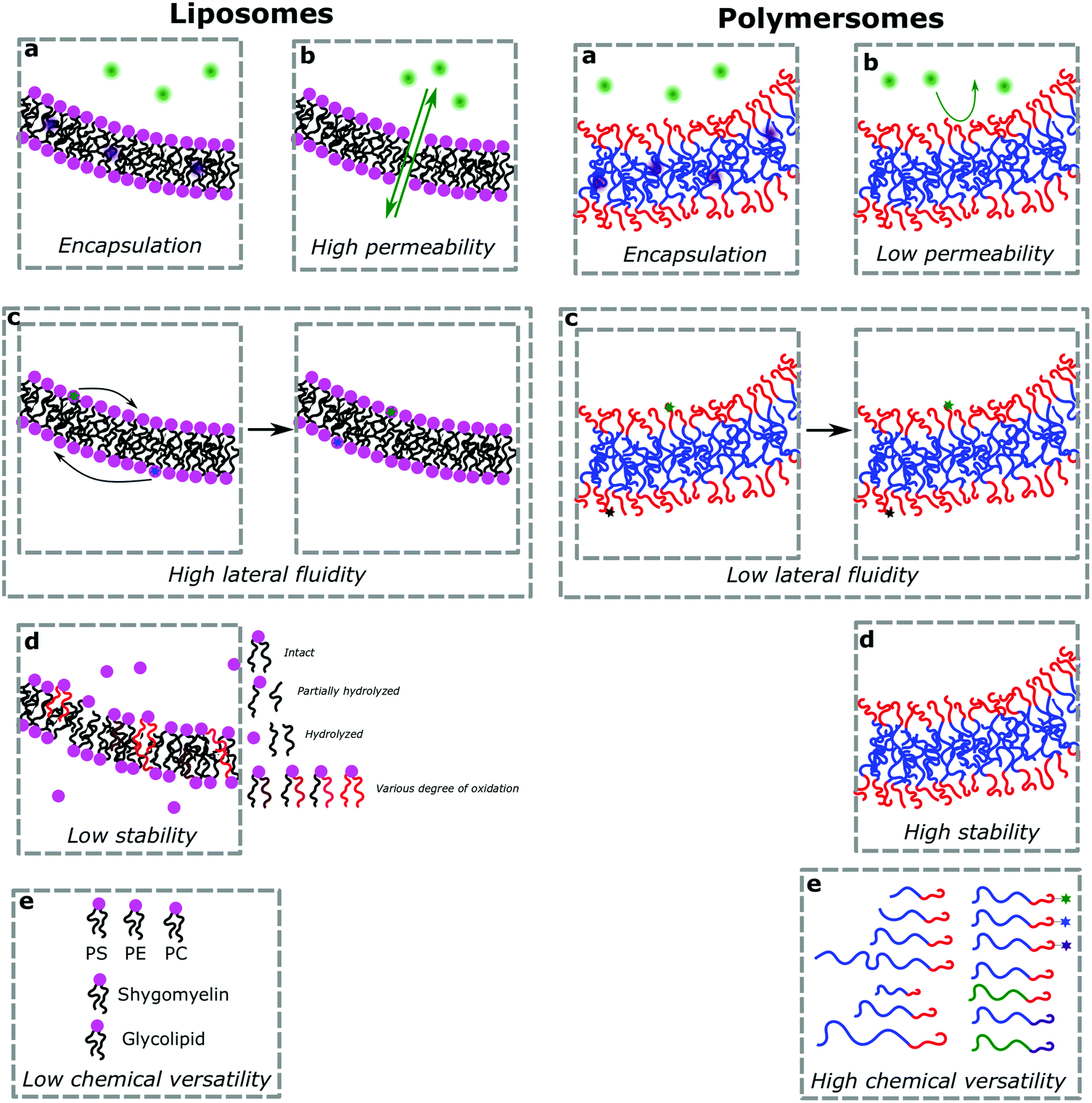 | ||
| Fig. 3 Comparison of properties between liposomes (left) and polymersomes (right). | ||
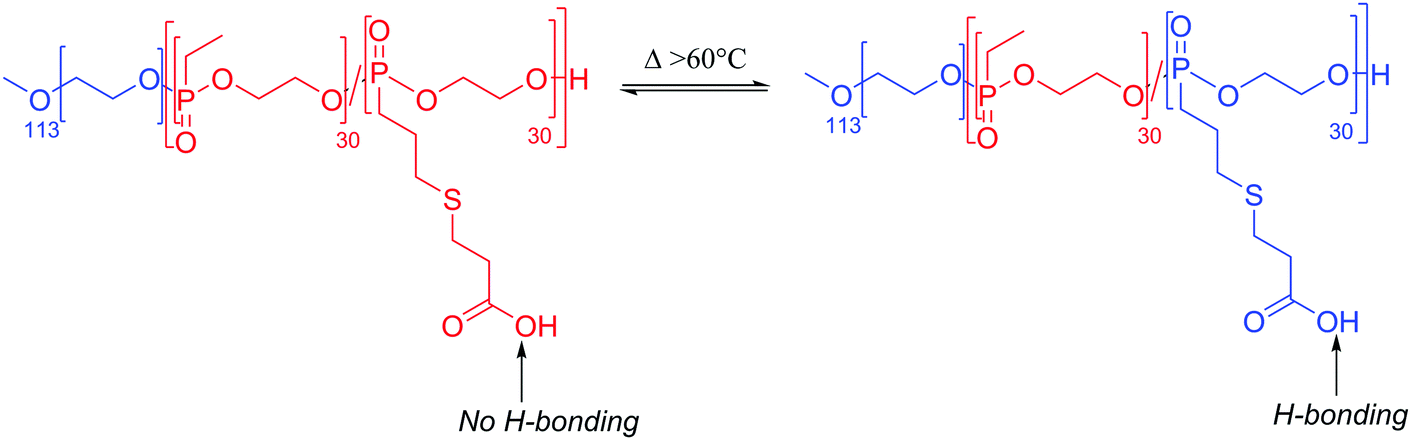
As a further consequence of the amphiphiles’ chemical nature and lateral fluidity, liposomes are short-lived while polymersomes have a greater life-span (Fig. 3d). Stability can mean the chemical stability of the lipids or the physical stability of the vesicles (number, size, structure) and also is dependent on the storing conditions. Liposomes can be stable for months when handled appropriately and stored under an inert gas at low temperature.9 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) SUVs were reported to be stable for 48 h, phosphatidylethanolamine (PE) SUVs for 12 days, and LUVs for weeks.6 The issue of liposome stability arises from the lipids’ chemical instability rather than entropically favored disassembly.6,9,30 This is why the overall stability of liposomes is dependent on the nature of the lipids used. Their unsaturated fatty acid chains are prone to oxidation by reactive oxygen species (ROS) which causes changes in the overall liposome properties such as its permeability. In addition, the ester moieties can undergo hydrolysis, the covalent bond between the hydrophilic and hydrophobic fractions is cleaved, degrading the liposomes (Scheme 1). Oxidative degradation can be minimized by protecting lipids from light, keeping them under an inert atmosphere, at low temperature (−20 °C), in a highly pure state (no heavy metals, free fatty acids or lyso-phosphatidylcholine (lyso-PC)) and sometimes stabilized by antioxidants while storing them.6,30 However, such measures are harder to maintain once they are self-assembled into liposomes in water or injected in vivo.
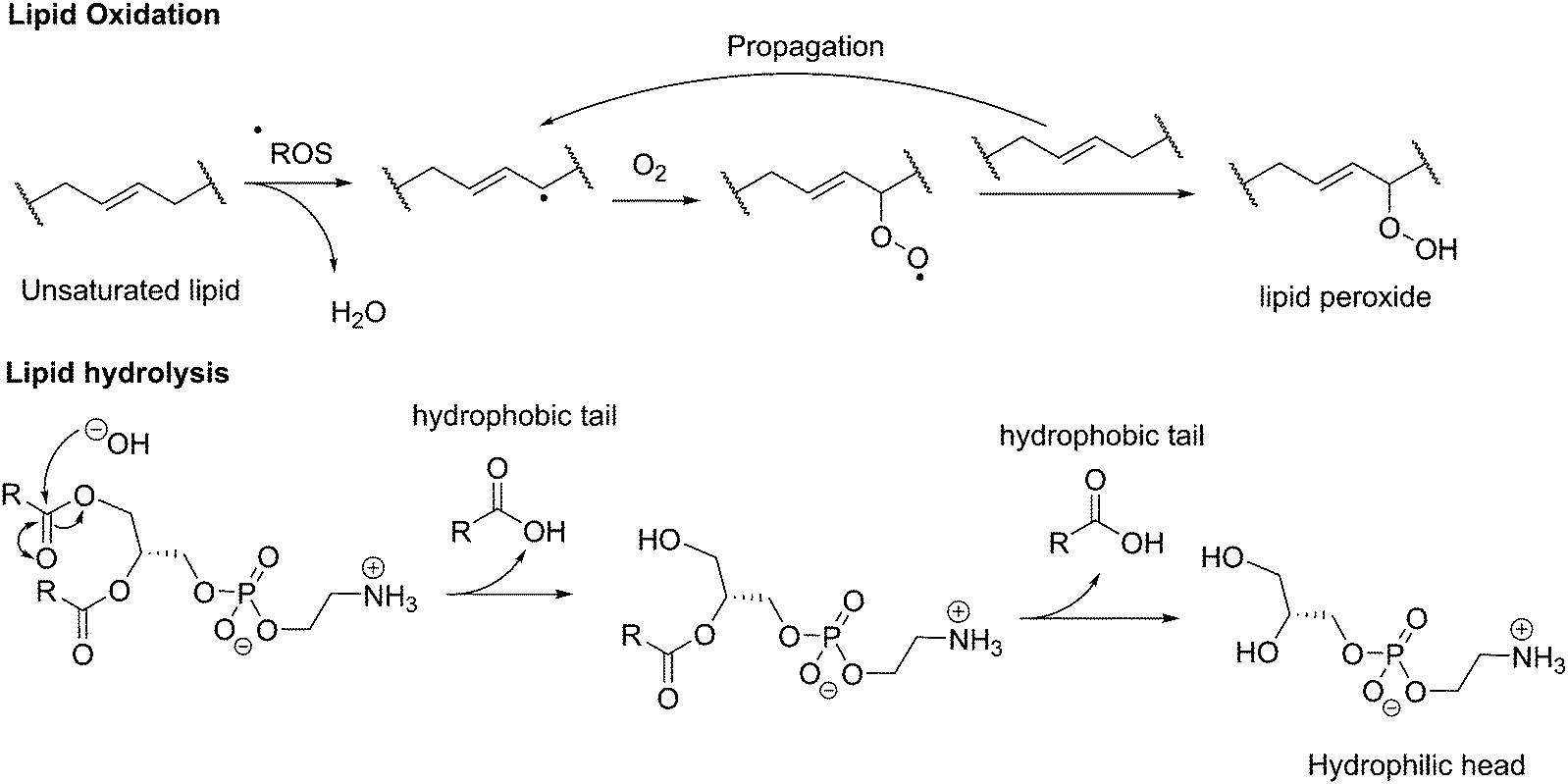 | ||
| Scheme 1 Chemical instability of lipids due to oxidation or hydrolysis. ROS: reactive oxygen species. | ||
These chemical instability challenges are significantly improved when using block copolymers.1,24,28,31 Even though they are still prone to oxidative addition and potential cross-linking of the olefins (if present), the covalent bonds between the blocks are more difficult to break than the ester linkage used in phospholipids (e.g. ether bond in poly(butadiene)-block-poly(ethylene oxide) (PB-b-PEO)). Moreover, degradation of some of the repeating units of the polymers has a lower impact than on the low molecular weight lipids. Hence polymersomes have a greatly increased chemical stability lifetime compared to liposomes and were shown to be stable after at least one month.32,33 Even up to 6 months of stability at room temperature has been reported which is probably not the limit.34,35 Polymersomes are seemingly more stable than liposomes due to their membrane thickness, entanglement, and lateral diffusivity.2 Despite the general trend that polymersomes are more stable and less permeable than liposomes, such properties are entirely dependent on many different aspects most importantly the size of the polymeric vesicles, the nature of the amphiphiles, the method of preparation, and storage conditions. The stability and permeability of polymersomes are consequently highly variable and unfortunately actual values are rarely mentioned and assumptions on these properties should be used cautiously.
(c) Mechanical and rheological properties
Presumably, the most essential mechanical properties of polymer and lipid membranes are bending and stretching elasticity as they are related to the vesicle resistance to deformation and stretching experienced during drug delivery (resistance in the high shear rate of blood circulation and deformation through tiny vessels such as capillaries) or during cellular processes (e.g. division, fusion, osmotic shock). The membrane response to bending and stretching is characterized by the bending rigidity and the area compressibility or stretching elasticity modulus (the shear elastic modulus is zero for fluid membranes). Table 2 shows data about the mechanical, rheological and electrical properties of polymer and lipid membranes, which are easy to assess from measurements on GUVs.26,36–38 Depending on the membrane thickness, polymersome membranes can exhibit significant stiffness as demonstrated by the values of the bending rigidity modulus. For comparison, we have included data for lipid membranes in the gel phase as some of their properties appear to be close to those of polymersomes. Even though the bending rigidity of polymersomes might approach that of gel-phase lipid membranes, the response of the former to a deformation will differ significantly from that of a gel-phase vesicle because the latter is not fluid: gel-phase vesicles relax much faster after deformation induced e.g. by electric pulses.39| Membrane material property | Polymer membranesa | Lipid membranes in the fluid phase | Lipid membranes in the gel phase |
|---|---|---|---|
| a The values reported for polymersomes depend on the diblock copolymer used (and the respective membrane thickness) and can vary strongly. b Data from ref. 38 and 43–49; the value for gel-phase lipid membranes corresponds to the bending rigidity of DMPC measured at around 5 degrees below the main phase transition temperature of the lipid. c Data from ref. 43, 46, 47 and 50. d Data from ref. 43, 44 and 51. e Data from ref. 39 and 42. f Data from ref. 41–43 and 52. g Data from ref. 53 and 54. | |||
| Bending rigidityb | 35–400kBT | 20kBT | 350kBT |
| Stretching elasticityc | 120 mN m−1; 470 mN m−1 | 200 mN m−1 | 850 mN m−1 |
| Lysis tensionf | 20–30 mN m−1 | 5–10 mN m−1 | >15 mN m−1 |
| Critical poration potentiale | 4–9 V | 1 V | 10 V |
| Pore edge tensiong | 10–50 pN | 10–50 pN | — |
| Shear surface viscosityd | 2 × 10−6 N s m−1 | 3–7 × 10−9 N s m−1 | Diverges |
Upon stretching, membrane tension builds up. When it reaches the so-called lysis tension, which is about 5–10 mN m−1 for phosphatidylcholine membranes, the membrane ruptures.40,41 The lysis tension of polymersomes is high, and exceeds 20 mN m−1,42 which is why they are referred to as tough vesicles. The membrane lysis tension is directly related to the critical transmembrane potential leading to poration of vesicles. The critical poration potential of gel-phase vesicles and polymersomes is several times higher compared to that of fluid membranes39,42 (one of the reasons being the larger membrane thickness). In fluid membranes, pores will reseal to avoid the contact of water with the hydrophobic part of the bilayer and the energy penalty for this is contained in the so-called pore edge tension. The resealing of pores in polymersomes is slowed down, because of the higher membrane viscosity,43 and is arrested in gel-phase membranes39 (upon poration, gel-phase GUVs develop cracks which do not reseal even within minutes as the membrane viscosity diverges44). One study estimated the shear viscosity of polymersome membrane to be 500 times higher than that of fluid lipid membranes.43 Consequently, molecular mobility in the polymersome membrane is much less compared to liposomes where diffusion of membrane probes is fast. The latter also leads to faster domain coarsening in phase separated fluid vesicles.
(d) Chemical versatility
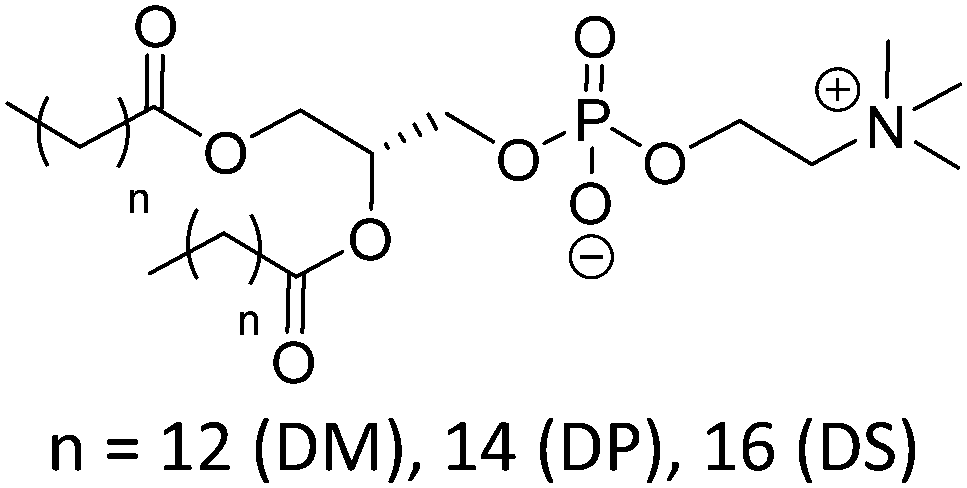
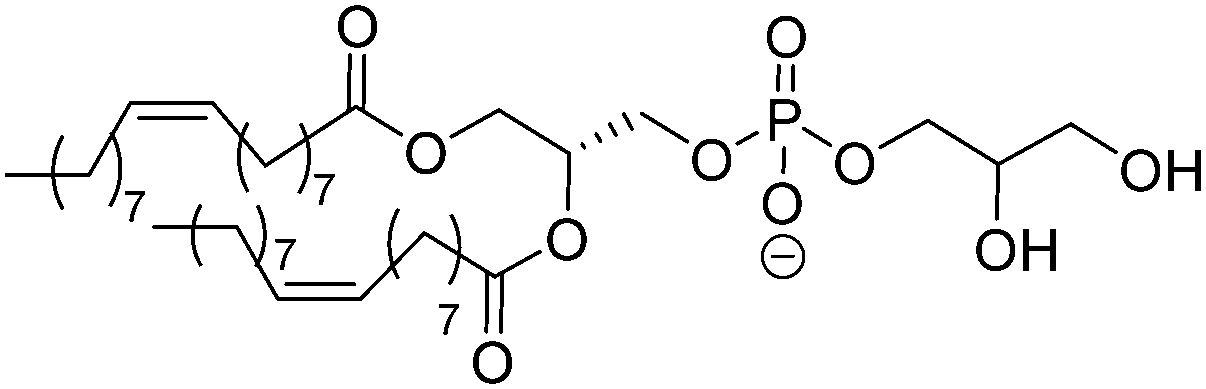
Natural and synthetic lipids55,56 are already highly specialized molecules that suffer from a large limitation of the available chemical functionalization space (Fig. 3e). Their physical and chemical properties are relatively restricted as small modifications can lead to large repercussions in the properties of their small molecules and thus their self-assembly. The modification of the head of lipids is achievable but is restricted to hydrophilic moieties so that lipids still self-assemble into vesicles. The modification of the lipid tail(s) requires often several synthetic steps as shown for example for lipid 1 in Scheme 8 which is synthesized in 9 steps.57From Table 3, we observe that commonly used lipids forming liposomes are exclusively natural phospholipids and even sometimes used as a naturally occurring mixture such as soy-PC or hydro soy-PC. Their structures and sizes are fairly similar (molecular weight ∼ 800 g mol−1) and only vary in aliphatic chain lengths (13–17Cs long; 0–2 degree of saturation and always cis) and end groups (primary amine, tertiary amine, diol, or carboxylic acid) which determine the overall charge of the lipids as the phosphate diester moiety is always deprotonated at pH 7 (pKa < 7). Despite their relative low chemical diversity, these lipids have widespread gel-liquid crystal transition temperatures (Tm) even among the same type of phospholipids (−22 to 60 °C). Most importantly, Tm can be higher than room temperature, a property that is significant for liposome self-assembly (see Section III). Tm depends on the nature of the amphiphiles, notably their saturation level, and represents the temperature at which the amphiphile's physical state changes from an ordered gel phase to a disordered liquid crystalline phase.25 A few groups synthesized non-natural lipids for their self-assembly into vesicles. For example, the group of Zumbühl synthesized modified diamidophospholipids, which self-assembled into interesting d-form vesicles or cuboid liposomes by using hydrogen bonding networks from well-designed lipids.58–61
| Lipid | Structure | T m (°C) | Charge at pH 7 pKa62 |
|---|---|---|---|
| T m: gel–liquid crystal transition temperature. For lipid abbreviations see the table of abbreviations. | |||
| Phosphatidylcholine (PC) | |||
| DMPC |
|
23 | Zwitterionic |
| DPPC | 41 | ≤1 (P(O)(OR)2OH) | |
| DSPC | 55 | ||
| DOPC |
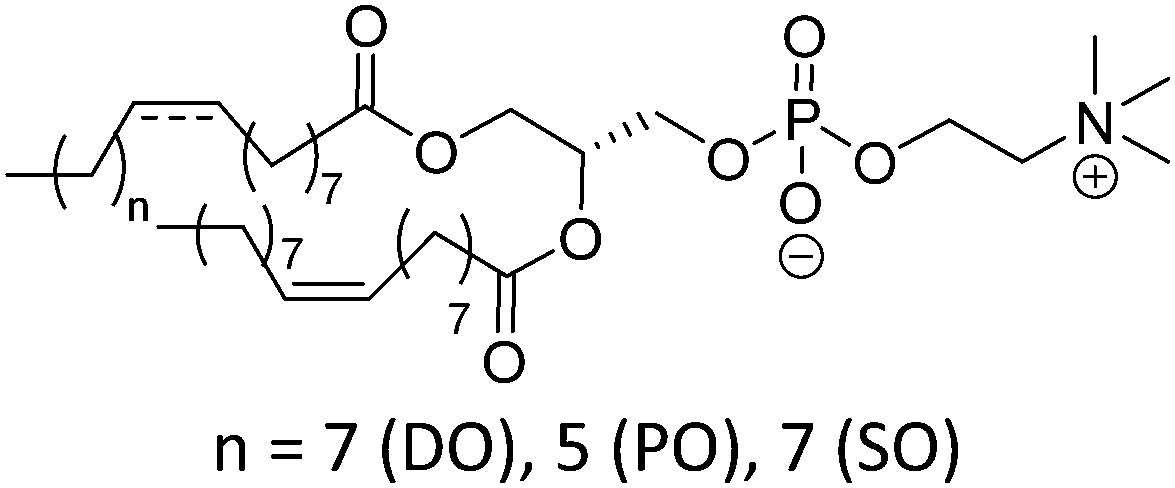
|
−22 | |
| POPC | −2 | ||
| SOPC | 6 | ||
| Soy PC (mixture) |
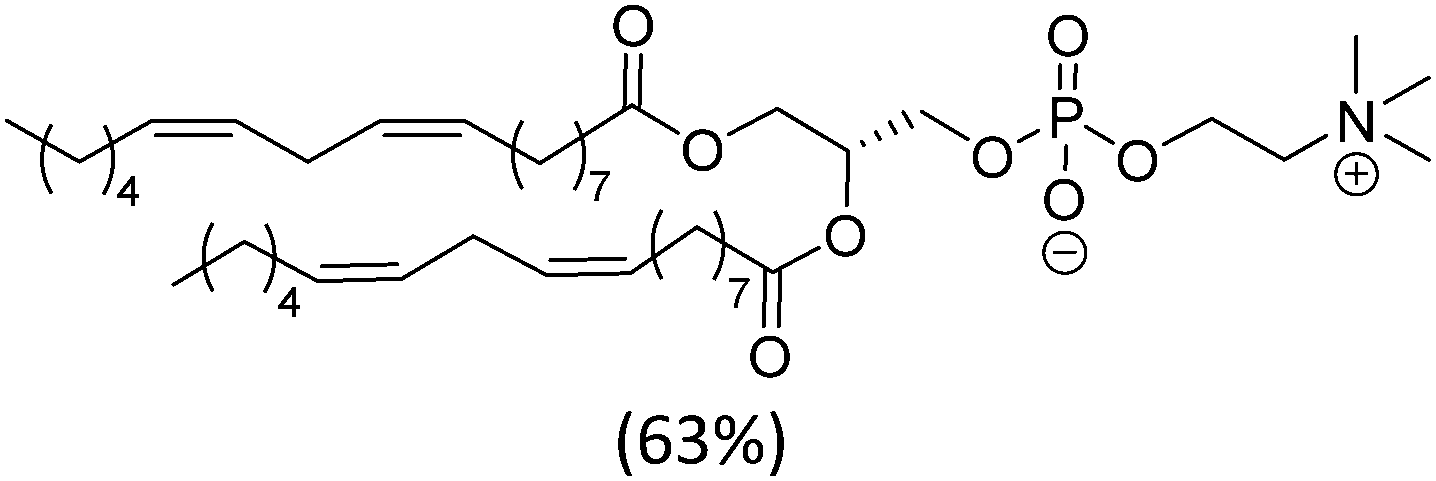
|
−20 | |
| Hydro Soy PC (mixture) |
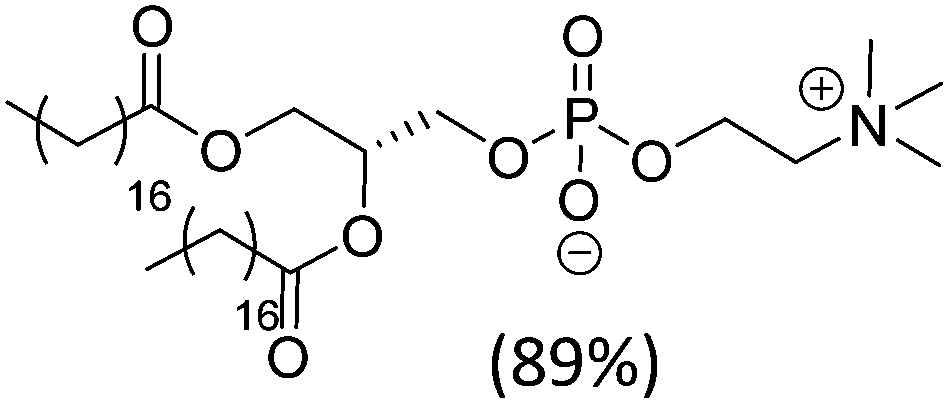
|
52 | |
| Phosphatidylglycerol (PG) | |||
| DMPG |
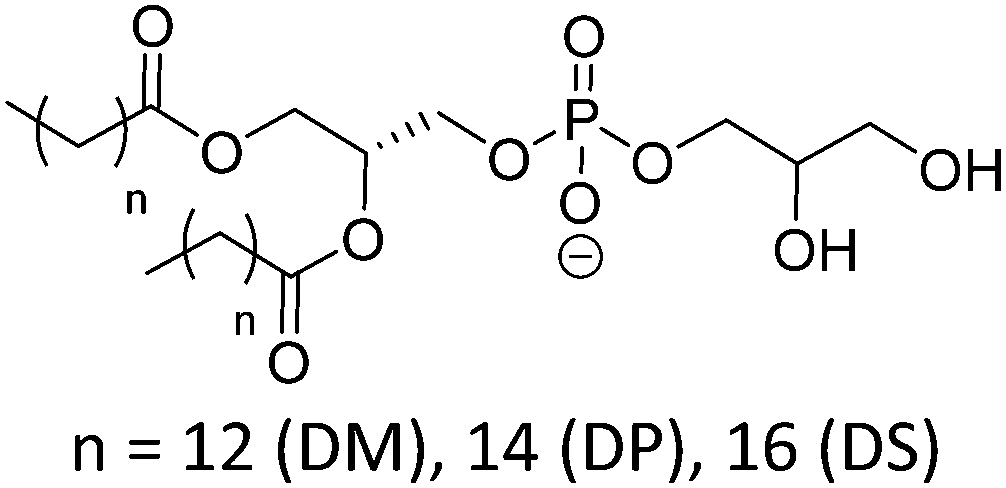
|
23 | Negative |
| DPPG | 41 | ∼3.0 (P(O)(OR)2OH) | |
| DSPG | 55 | ||
| DOPG |
|
–18 | |
| Phosphatidylethanolamine (PE) | |||
| DMPE |
|
50 | Zwitterionic |
| DPPE | 60 | ≤1.7 (P(O)(OR)2OH) | |
| ≥9.6 (NH3+) | |||
| Phosphatidylserine (PSe) | |||
| DMPS |
|
38 | Negative |
| DPPS | 51 | ∼3 (P(O)(OR)2OH) | |
| ∼4 (COOH) | |||
| ≥9.7 (NH3+) | |||
| Sphingomyelin (SM) | |||
| Palmitoyl-SM |
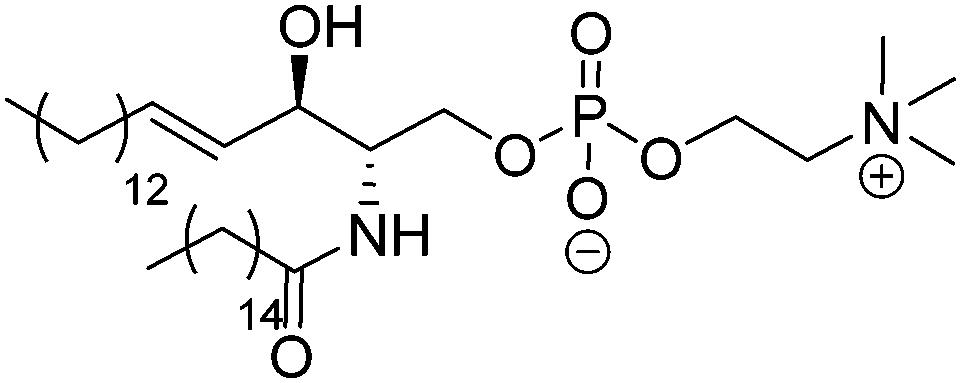
|
41 | Zwitterionic |
| ≤1 (P(O)(OR)2OH) | |||
| ∼17 (OH) | |||
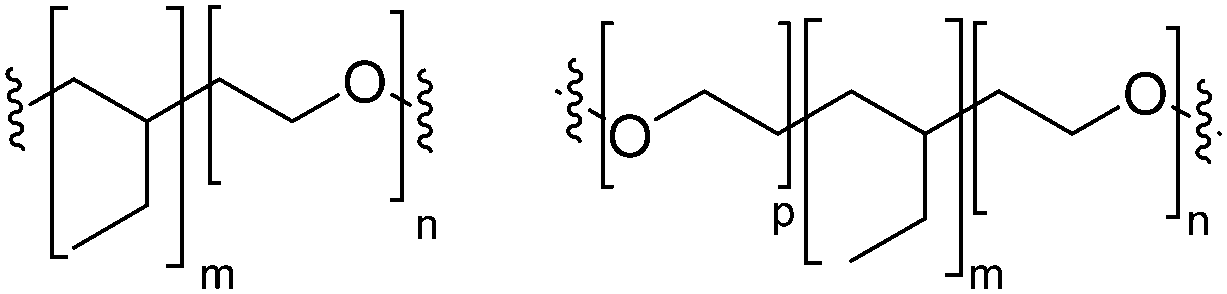
Block copolymers have the advantage of being adjustable and easily tunable to desired characteristics of the vesicles (Fig. 3e).2,28 Primarily, a large pool of blocks (Table 4 lists some typical examples) is available and their overall molecular weight Mn and block ratio can be controlled via classical ionic or controlled radical polymerization techniques.1,3,24 These designable properties allow for the formation of a variety of polymersomes with adjustable shape, fluidity, entanglement, permeability, stability, and responsiveness,11,12 and the polymers can be tailored to appropriate physical properties or biocompatibility.1 Contrary to lipids, block copolymers are however polydispersed. Even if their distribution is most often low, thanks to modern polymerization, polymers’ dispersity can affect their self-assembly and reproducibility can be challenging when using different batches of the same polymers.15 The most common polymer architecture used in forming polymersomes are di-(AB) or tri-block copolymers (ABA or rarely ABC) (where A (and C) is the hydrophilic block and B the hydrophobic block) (Table 4);16 however, polymersomes formed from branched, grafted comb or star polymers have also been reported,63–65 emphasizing the superior synthetic versatility of polymers over lipids (Fig. 4).
| Copolymer | Structure | Charge at pH 7 pKa |
|---|---|---|
| PB: poly(butadiene); PEO: poly(ethylene oxide); PEE: poly(ethyl ethylene); PS: polystyrene; P2VP: poly(2-vinylpyridine); PDMS: polydimethylsiloxane; PCL: polycaprolactone; PIB: polyisobutylene; PAA: polyacrylic acid; PMOXA: poly-2-methyl-2-oxazoline; b: block; g: grafted. | ||
| PB-b-PEO |
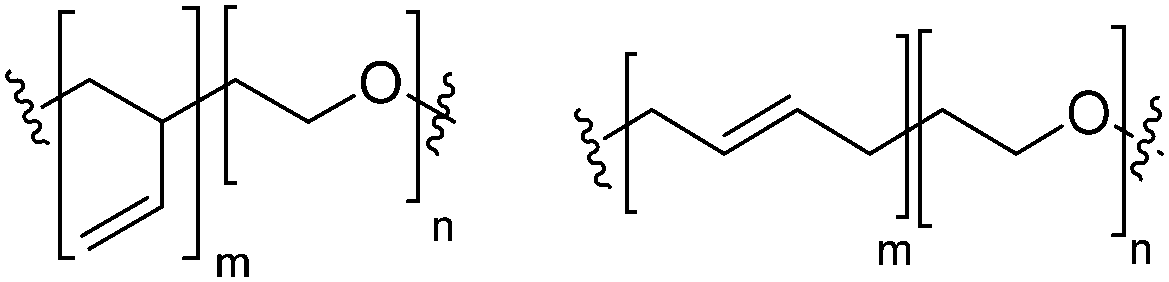
|
Neutral |
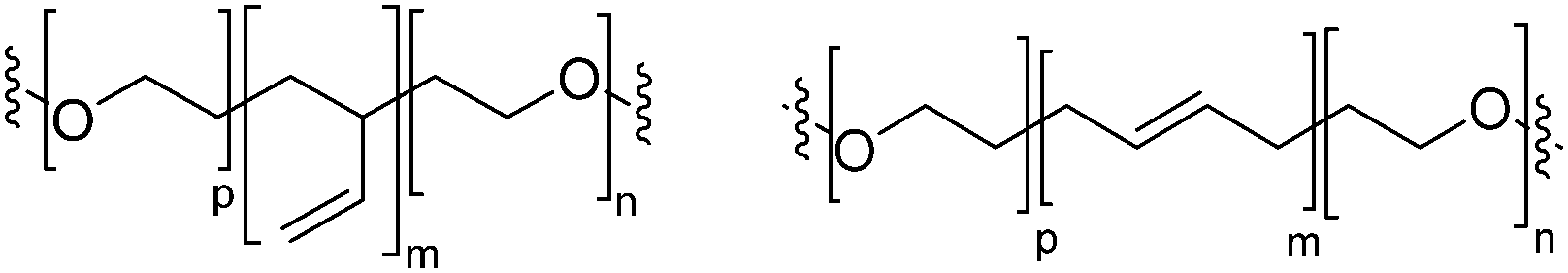
|
||
| PEE-b-PEO |
|
|
| PS-b-PEO |
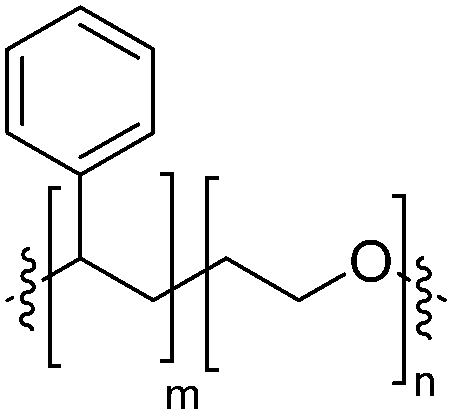
|
|
| P2VP-b-PEO |
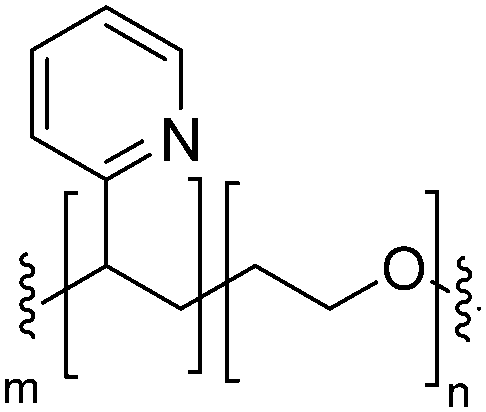
|
|
| PDMS-b-PEO |
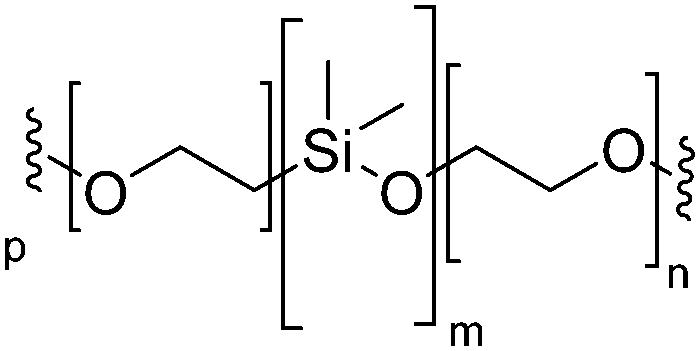
|
|
| PDMS-g-PEO |
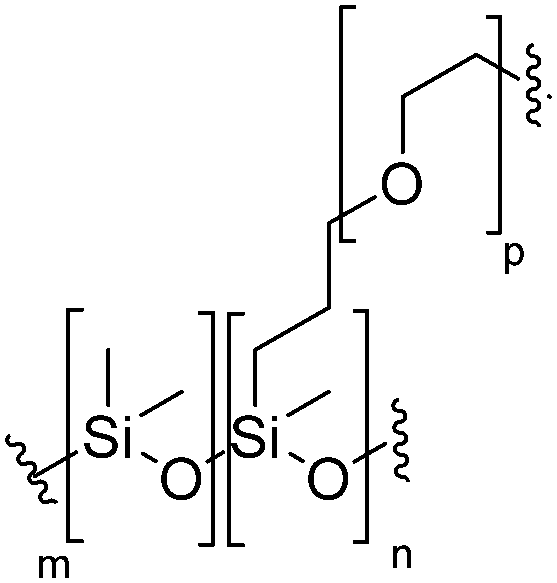
|
|
| PCL-b-PEO |
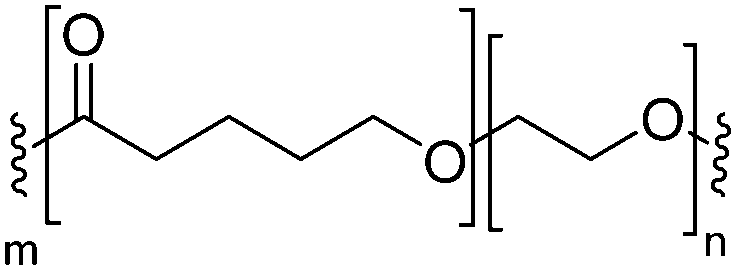
|
|
| PIB-b-PEO |

|
|
| PS-b-PAA |
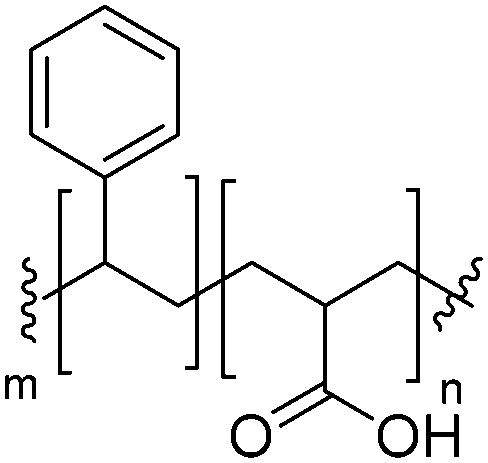
|
Negative |
| pKa PAA: 4.25 | ||
| PDMS-b-PMOXA |
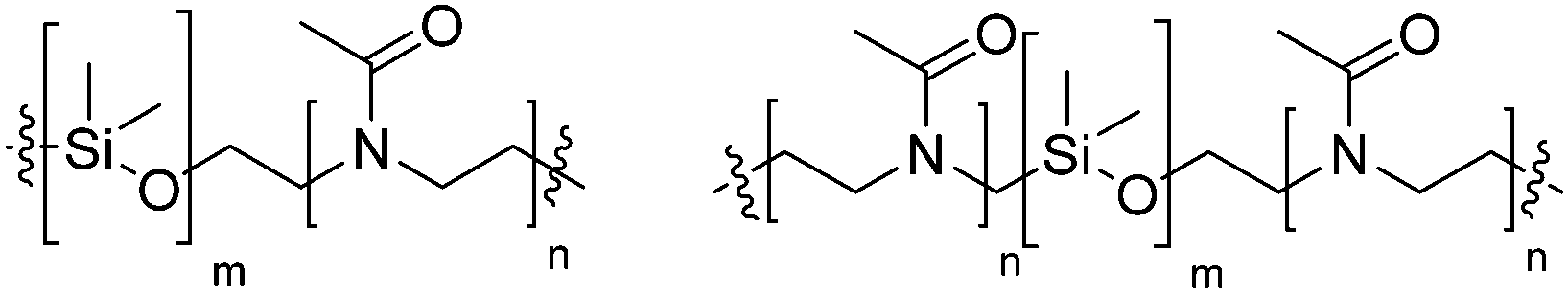
|
Neutral |
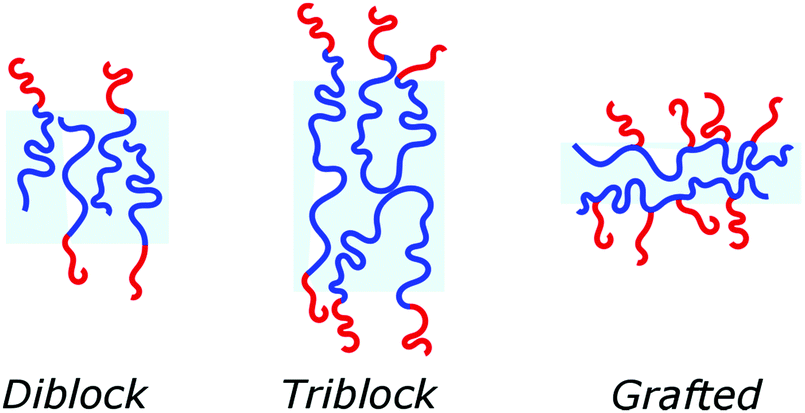 | ||
| Fig. 4 Different types of block copolymer, which can self-assemble into polymersomes. | ||
The amphiphilic composition is the driving force for the self-assembly of lipids and block copolymers into various structures.66 As the hydrophilic and hydrophobic moieties are chemically incompatible, but covalently coupled, no microphase separation takes place.1,15 Their rearrangement into vesicles is entropically favored, as it reduces non-favored interactions in aqueous media by aggregating the hydrophobic groups together away from polar solvent. The hydrophobic and hydrophilic groups are said to be microphase-separated. However, while natural lipids have been designed to self-assemble only into vesicles, as block copolymers can be synthesized with a wide variety of volume ratios of the blocks, they can self-assemble into different morphologies (entropically favored microphase separations).15 The nature of the polymer and the hydrophilic/hydrophobic ratio are the major parameters which determine whether polymersomes (bilayer), spherical micelles (monolayer), cylindrical micelles, rods, lamellae (bilayer), hexagonal cylinders etc. are formed.24
The microphase separation depends on 3 parameters: the volume fractions of the blocks (f), the degree of polymerization (N) and the Flory–Huggins parameter χ (degree of block incompatibility).66 Theoretical phase diagrams between f and the segregation parameter χN using self-consistent mean-field theory are used to determine which block ratios are more likely to yield the desired morphology. The critical packing parameter Pc = v/(a·l) is also used to predict the most likely morphology of the self-assembled aggregate (where v is the volume of the hydrophobic block, a is the area of the hydrophilic block and l is the length of the block copolymer).16,24 For Pc < 1/3, spherical micelles are formed, for 1/3 < Pc < 1/2, cylindrical micelles, 1/2 < Pc < 1 vesicles, for Pc = 1, planar bilayers, and for Pc > 1, inverted structures. Pc was first established for surfactants and lipids; however, it is generally also accepted for amphiphilic block copolymers. However, determining these parameters is not trivial, especially as a block copolymer head group is more indefinite than those in surfactants or lipids. A less accurate but more straightforward prediction of microphase separation of the block copolymer pre-synthesis is the hydrophilic weight fraction fw = Mn(hydrophilic block)/Mn(polymer).16,24,35 Which aggregates are formed greatly depends on the nature of the blocks (hydrophilicity or hydrophobicity, sterics, degrees of freedom, polymer polydispersity, rigidity, etc.) but as a general rule 0.25 < fw < 0.40 yields polymersomes.12 However, predicting self-assembly morphologies is highly complex as thermodynamics and kinetics properties can lead to the same polymer generating different morphologies depending on the method used. For example, PEO45-b-PMMA51 (PMMA: poly(methyl methacrylate)) was shown to form micelles (29 nm in diameter) when using acetone injection in water and polymersomes (550 nm) by the addition of water/benzyl alcohol to the polymeric solution in THF/MeOH.67 The determination of self-assembly morphologies by the block copolymer packing parameter has been well exemplified by polymerization-induced self-assembly (PISA) (see Section III Vesicle preparation).68
(e) Mixed vesicles and phase separation
Both liposomes and polymersomes have their own advantages and shortcomings. However, the careful design of functional polymersomes or liposomes for customizing the vesicle properties can be synthetically demanding and challenging.One alternative to obtain vesicles with the advantage of both polymersomes and liposomes is to form capsosomes (liposomes in a polymer shell) (Fig. 5).2,3,69,70 This multi-compartmentalized scaffold allows for the desired permeable membrane of the liposomes and the extra stability of the polymeric nanocarriers. These systems are interesting for cell mimicry as the cell uses organelles to further compartmentalize different functions. However, such systems are not trivial to generate and diverge from the simple modelling of a membrane (multiple bilayers rather than a single bilayer).
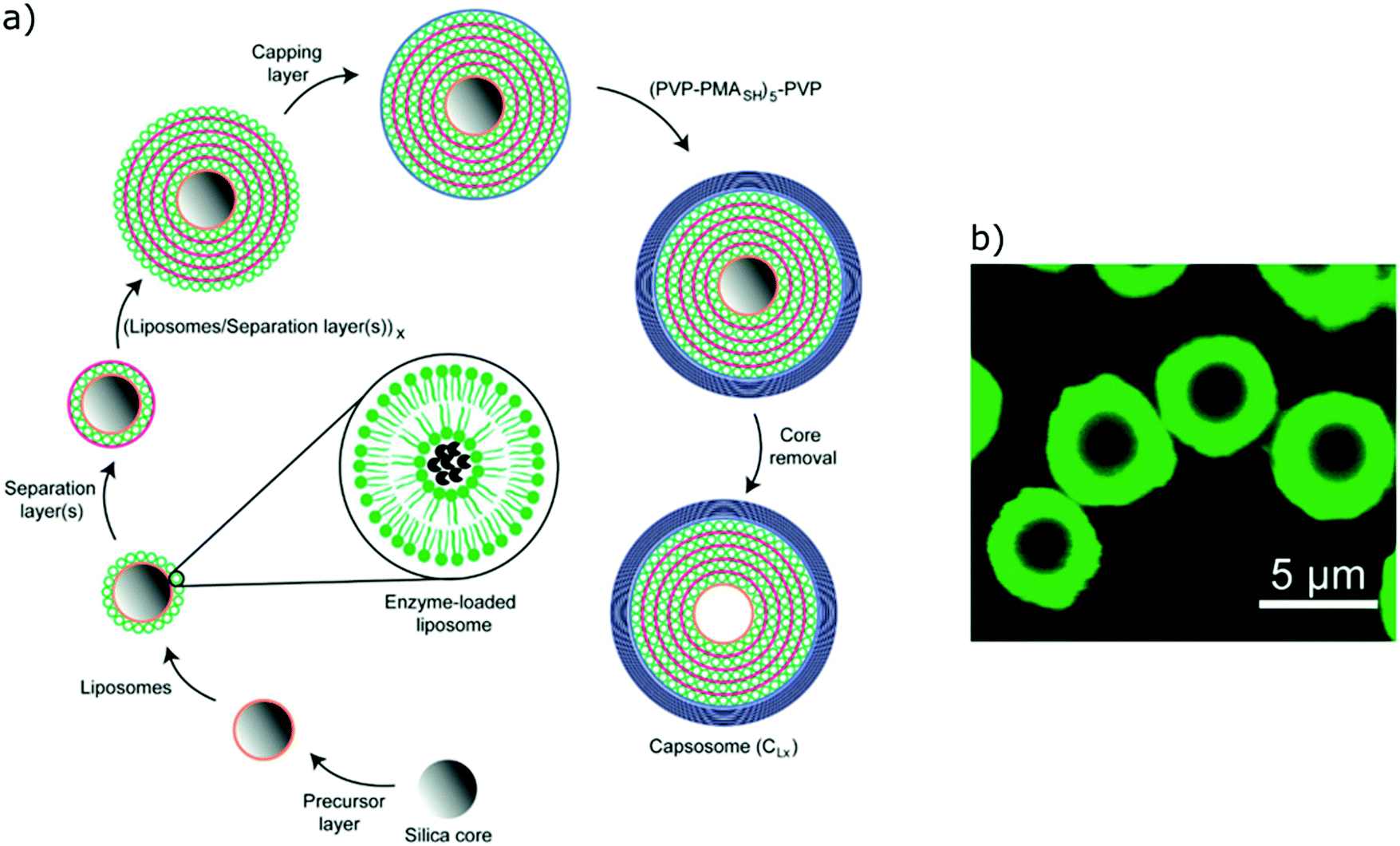 | ||
| Fig. 5 (a) Schematic representation of capsosomes. (b) Confocal laser scanning microscopy of a capsosome with eight liposome deposition steps. Adapted with permission from R. Chandrawati, L. Hosta-Rigau, D. Vanderstraaten, S. A. Lokuliyana, B. Städler, F. Albericio and F. Caruso, ACS Nano, 2010, 4(3), 1351–1361 (https://pubs.acs.org/doi/abs/10.1021/nn901843j).69 | ||
An increasingly promising alternative for membrane studies and for the pharmaceutical industry is generating lipid–polymer hybrid vesicles, also called lipopolymersomes, HLP (hybrid liposome–polymersome) or HUV (hybrid unilamellar vesicles).13,14,71,72 Here the membranes are formed from mixing both lipids and polymers. Hybrid vesicles can offer an easier platform to finely control the physical properties of vesicles with the best characteristic of both types (biocompatibility and biofunctionality and synthetic control of robustness, permeability and responsiveness).8 Indeed, a great variety of vesicles with little additional synthetic effort can be efficiently obtained. This variability can be achieved by playing with the nature and functionalization of lipids and polymers and their respective ratio resulting in a library of vesicles with many different properties. Despite the advantages and interests related to hybrid vesicles, only a few systems have been described and almost exclusively GUVs presumably due to the difficulty in distinguishing between phase separation on smaller vesicles, which is normally achieved using selective fluorescent tagging.11,73 A key parameter for the formation of stable hybrid vesicles is the compatibility between the block copolymer and the lipid (chemical structure, size, thermodynamic parameters etc.) and their ratio.8 At first glance, it seems surprising that such different amphiphiles can form hybrid vesicles as lipids form 3–5 nm thick membranes while block copolymers form on average thicker membranes (5–50 nm). Thus, hybrid vesicle self-assembly can suffer from geometrical incompatibility. Nonetheless, hybrid structures have been described for polymers of relatively high molecular weights, for example with PC/PE/Lissamine rhodamine-PE (Liss Rhod-PE) and PMOXA-b-PDMS-b-PMOXA (PMOXA: poly(2-methyl-2-oxazoline); PDMS: polydimethylsiloxane) (Mn = 9000 g mol−1).74
The main property to take into account when dealing with hybrid vesicles is their phase separation.8,11,14 Homogeneous vesicles (statistically distributed lipid and polymer), vesicles with domains (phase separation within the membrane) or complete phase separation of the lipids and the polymer (coexistence of liposomes and polymersomes) can form (Fig. 6). The nature, size difference, ratio of lipids and polymers and the method of formation determine which type of phase separation is obtained. If the size difference is large, the formation of domains would be entropically favored in order to minimize the exposure of hydrophobic moieties to water or to hydrophilic moieties.8 The demixing of polymer and lipids is particularly advantageous for studying the concept of lateral heterogeneity found in plasma membranes and further discussion can be found in Section IV.
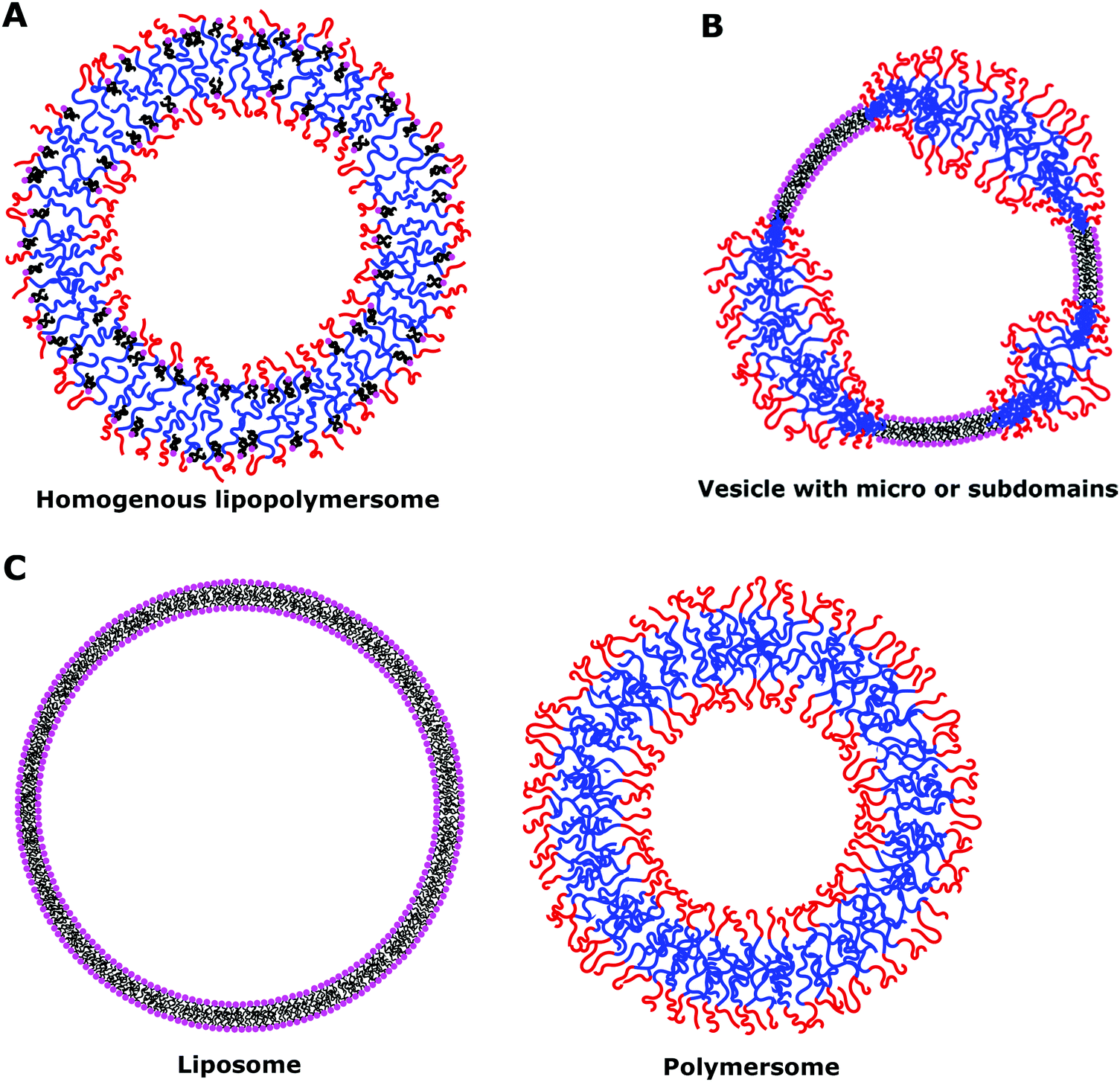 | ||
| Fig. 6 Possible scenarios of phase separation of lipids and block copolymer vesicles. | ||
III. Vesicle preparation
Over the years, many different preparation methods for liposomes have been developed, most of which have also been applied to the formation of polymersomes.2,15 Those methods dictate the size, yield, polydispersity, and type (unilamellar, multilamellar, vesosomes) of the resulting vesicles. In 2018, some of the mechanisms behind the formation of polymersomes were elucidated.75 Conditions to self-assembly liposomes and polymersomes may vary due to the difference in stability, solubility, and stiffness of the lipid and polymer membranes. The nature of the amphiphile (chemical properties, molecular weight, polydispersity), additives (ions, small molecules, surfactants) or organic solvent used, concentration of the amphiphile, temperature, aqueous medium etc. can all affect the size, yield and polydispersity of the vesicles.15,76Another important aspect of liposome formation is that vesicles will only be obtained at temperatures above the Tm of the lipid used.6,25,77 Having to form liposomes above their Tm can be problematic when encapsulating temperature-sensitive entities (e.g. protein, DNA etc.).6,25,77 For block copolymers, the Tg and Tm are dependent on the degree of polymerization and polymeric architecture and thus much less defined than for lipids. In fact, the Tg of polystyrene (PS) can vary from −10 °C at a Mn = 500 g mol−1 up to 100 °C at a Mn > 104 g mol−1.78 In terms of blocks, PB and PDMS have Tgs well below room temperature (PB(1,2) Tg ≈ −21 °C (Mn = 105k); PB(1,4) Tg ≈ −77 °C (Mn = 50k); PDMS Tg ≈ −127 °C),78 while polycaprolactone (PCL) and PEO have a low Tg but a Tm above room temperature (PEO, Tg ≈ −54 °C and Tm ≈ 66 °C (Mn = 90k); PCL Tg ≈ −72 °C and Tm ≈ 65 °C (Mn = 60k))78 and others have only transition temperatures above room temperature (PS ≈ 100 °C (Mn = 1k); polyacrylic acid (PAA) ≈ 70 °C (Mn = 1.8k);78 PMOXA ≈ 55 °C (Mn = 4k)79). Nonetheless, block copolymer self-assembly is usually carried out at room temperature regardless of the Tg and of the block used. It remains unclear why formation of polymersomes at room temperature using high Tg or Tm blocks is possible in opposition to lipids.
Despite the variety of methods developed, they can be classified into two types: solvent free methods and solvent displacement ones (Table 5).6,15,76,80 In solvent free techniques, only the amphiphiles are hydrated in an aqueous medium and thus no organic solvents are present in the vesicle solution. In solvent displacement techniques, the amphiphiles are dissolved in an organic solvent and then placed in aqueous medium followed by removal of the organic solvent.
| Methods | Type | Size | Additives | Polydispersity | Ref. |
|---|---|---|---|---|---|
| Film rehydration | Solvent free | SUV, MLV | — | Large | 6, 15, 76 and 80 |
| Solid rehydration | Solvent free | SUV, MLV | — | Large | 81 and 82 |
| Electroformation | Solvent free | GUV | — | Low | 46, 67, 77 and 83 |
| Gel-assisted hydration | Solvent free | GUV | Agarose or PVA | Low | 83–85 |
| Solvent injection (ethanol, ether) | Solvent displacement | SUV | Solvent | Large | 15 and 25 |
| Emulsion phase transfer | Solvent displacement | GUV | Solvent, surfactant | Low | 86–88 |
| Microfluidics | Solvent displacement | SUV or GUV | Solvents, surfactant, sugars, polyelectrolytes, polymers etc. | Low | 21 and 89–92 |
(a) Solvent free methods
Solvent free methods are all variations of film rehydration (Fig. 7).6,15,76,80 Before the formation of the vesicles, the amphiphiles are dissolved in the organic solvent and placed on a solid surface (e.g. glass, Teflon, metal wires) before evaporating the solvent entirely to form a thin layer on that surface (Fig. 10a). Then addition of the aqueous medium causes swelling of the film and ultimately forms vesicles once the bilayer leaves the surface. The swelling and budding stage can be facilitated by mechanical stirring or sonication. Film rehydration generates MLVs and SUVs with broad size distributions, but is nonetheless the most commonly used method to form vesicles as the procedure is straightforward, no special equipment is required and it reproducibly yields a high number of vesicles completely free of organic solvents. Solid rehydration is conceptually similar but uses a bulk powder of the amphiphiles rather than a cast solution. Due to the problematic high polydispersity obtained with film hydration, commonly sequential extrusion is used to achieve better distribution control. This technique consists of repeatedly passing a crude heterogeneous mixture of vesicles (usually MLVs) through a polycarbonate membrane with small pores (typically 100 nm diameter but can be any desired size) to obtain homogeneously distributed SUVs of diameter matching the pore size.30,80,93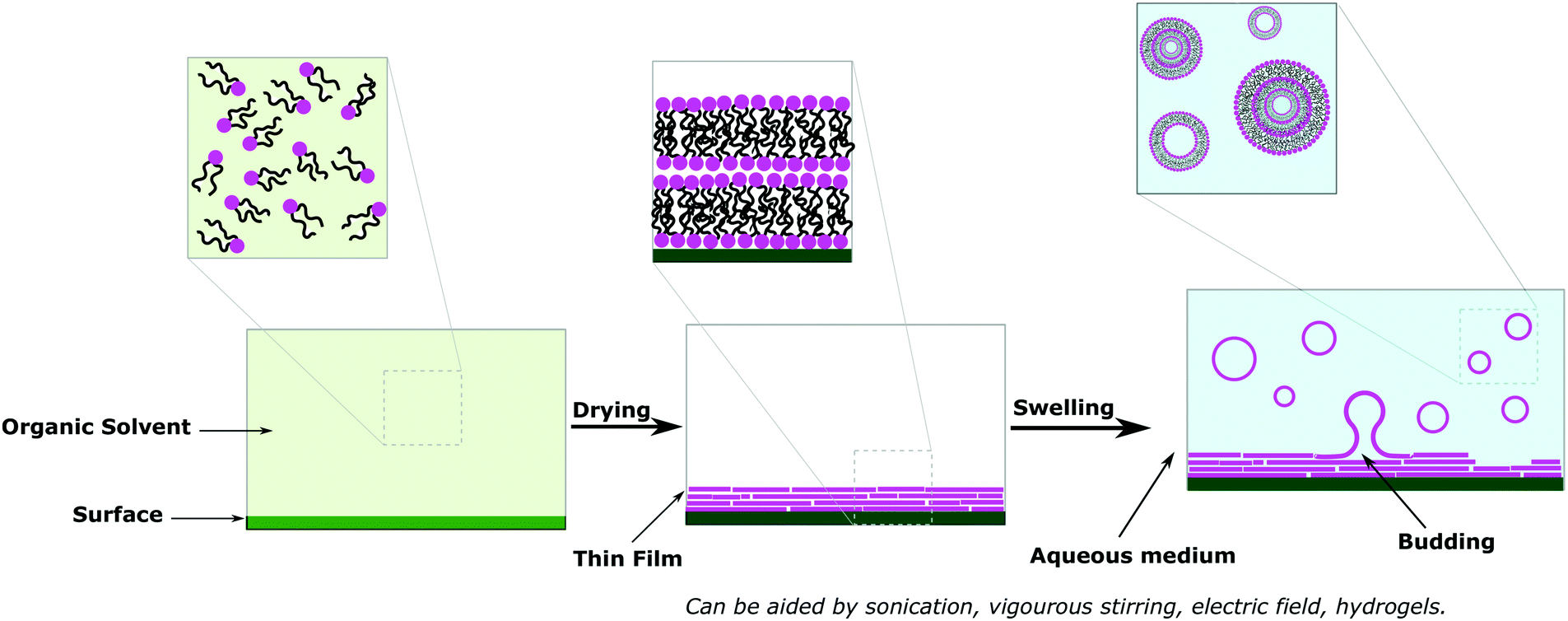 | ||
| Fig. 7 Self-assembly of lipids and block copolymer vesicles using solvent-free methods. | ||
Aided film rehydration methods such as electroformation or gel-assisted hydration are also quite popular as they allow the formation of GUVs with much lower polydispersity than simple film hydration.15 These methods are also desirable as they allow the formation of vesicles for challenging amphiphiles.9 Electroformation was pioneered in 1986 on liposomes, and consists of depositing a thin film of amphiphiles on electrodes (Pt or indium titanium oxide (ITO) glass).77,83,94 After addition of the aqueous medium, an alternating current is applied facilitating the swelling and release of the vesicles. The electric current is thought to cause fluctuation in the film and interlayer repulsion resulting in detachment from the electrodes.9 Electroformation is the method of choice to obtain liposome GUVs and was reproduced for polymersomes in 1999 on Pt wires.46 And has been repeated for a larger pool of block copolymers and extended to ITO glass.43,67,95–97 Remarkably, despite the fact that electroformation is well-established for liposomes, special in-house devices are almost exclusively used and poorly detailed. A recent study by Dionzou et al. clearly pointed out that the nature of the block copolymer and conditions needed to reproducibly obtain polymersome GUVs are challenging to determine.67
In gel-assisted hydration,83–85,98,99 the surface is pre-coated with dissolved agarose or poly(vinyl alcohol) (PVA) in warm water, and dried to form a thin gel layer before forming the amphiphile layer. The driving force is the swelling of the hydrogel when the coated surface is placed in aqueous medium. This method has only been described more recently (2009)98 and has been gaining in popularity. It also requires more readily available equipment (coverslip rather than an electroformation chamber). However, PVA or agarose could also get embedded in the vesicular membrane modifying its properties and behavior. One major drawback of all film hydration methods is the difficulty in encapsulating water-soluble molecules.80 Indeed, any desired entities need to somehow intercalate between the amphiphilic film layers which can be difficult for macromolecules and charged molecules but had nonetheless been reported.77
Another approach that has become very popular recently is polymerization-induced self-assembly (PISA), which has been used as a solvent-free polymer-specific methodology to form vesicles.68,100–103 This method relies on the solubility differences commonly in water between the monomer and polymer: as the hydrophilic monomer polymerized on a hydrophilic macroinitiator, also called the stabilizer-block; the polymer becomes increasingly more hydrophobic, entropically driving the amphiphile self-assembly. After reaching an initial critical degree of polymerization, micellar nucleation occurs. Further polymerization leads to spherical nanoparticle assemblies (first spheres then worms, other intermediate structures, and finally vesicles) (Fig. 8). Targeted assemblies can be achieved by adjusting the amphiphile concentration or the degree of polymerization of the stabilizer-block. PISA has been achieved by using diverse polymerizations but by far the most commonly used one is reversible addition fragmentation transfer (RAFT) polymerization to form diblock copolymers but also triblock copolymers and uses organosulfur based chain-transfer agents (CTAs). Most commonly, PISA is limited to self-assembled structures in the nanometer regime (nanoparticles, worms, or SUVs of 10–100 nm). Recently, Albertsen et al. described the photo-initiated PISA of 2-hydroxypropyl methacrylate (HPMA) onto PEO, forming GUVs (up to 10 μm).101 The Ru-photosensitizer generates radicals upon exposure to 450 nm radiation, which induce the polymerization of HPMA. These GUVs underwent what the author describes as phoenix behaviors where the change in osmotic pressure during self-assembly leads to the cyclic growth and collapse of the vesicles, catalyzed by exposure to blue light. Over time the GUVs thus gradually shrank, and increased in number demonstrating that SUVs might be more thermodynamically stable than GUVs. PISA perfectly exemplifies the versatility of block copolymers compared to lipids and allows the shortcutting of the different stages of assembly. However, because the method relies on hydrophilicity modulation, PISA is limited to a certain pool of specific blocks (typically HPMA) exhibiting the correct properties but has nonetheless shown incredible diversity.
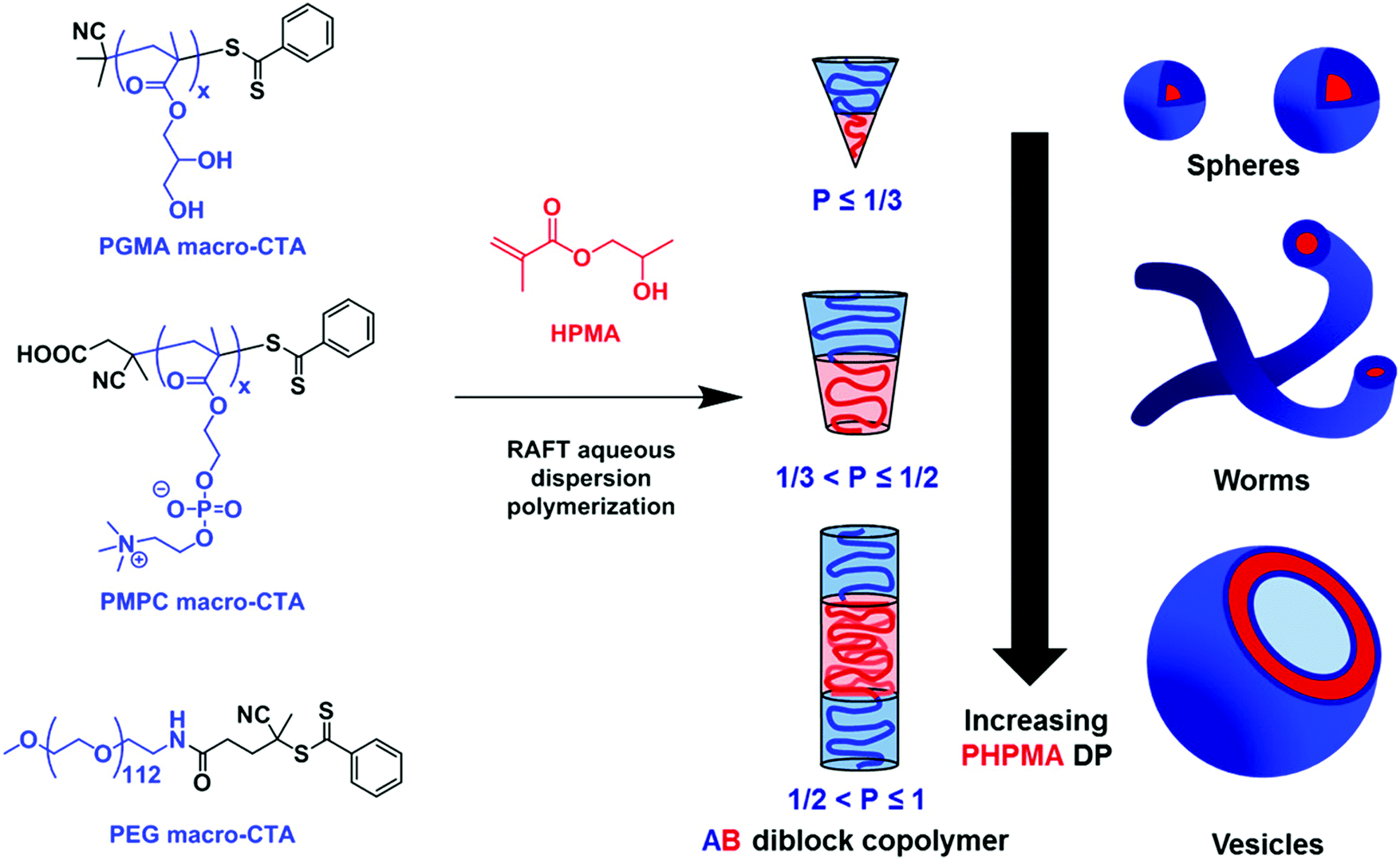 | ||
| Fig. 8 Polymerization-induced self-assembly (PISA) of 2-hydroxypropyl methacrylate (HPMA). A variety of macromolecular chain transfer agents (CTAs) were used to produce various assemblies depending on the degree of polymerization of HPMA. Reprinted with permission from N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136(29), 10174–10185. Copyright (2014) American Chemical Society68 (https://pubs.acs.org/doi/abs/10.1021/ja502843f). | ||
(b) Solvent displacement methods
Solvent displacement methods define any techniques where organic solvents and the aqueous phase coexist to promote the self-assembly of the amphiphiles before removal of the organic phase. These techniques have the great advantage of facilitating the efficient encapsulation of water-soluble molecules but have the difficulty of organic solvent removal. Organic solvent molecules trapped in the hydrophobic bilayer or dissolved in the aqueous phase would cause exposure of sensitive materials (protein, DNA, small molecule) to an organic solvent, which might sometimes limit their application since the organic solvent can either inactivate the biological macromolecule or be toxic.25 Those methods also often require the use of additives such as surfactants (typically small molecules or block copolymers containing PEG), which get embedded in the membrane as also amphiphilic.104–106Solvent injection consists of slowly adding a solution of amphiphiles in organic solvent (most of the time ether or ethanol) to an aqueous medium under vigorous stirring (Fig. 9).15,25 Such vesicles are SUVs, often MLVs and have a broad size distribution but it is an easy and efficient method to obtain vesicles like film hydration. Vesicle polydispersity and lamellarity can be homogenized by extrusion. Similar methods use reduced pressure, dialysis and/or centrifugation as means to remove organic solvents after mixing of the aqueous and organic phases to obtain similar vesicle populations.
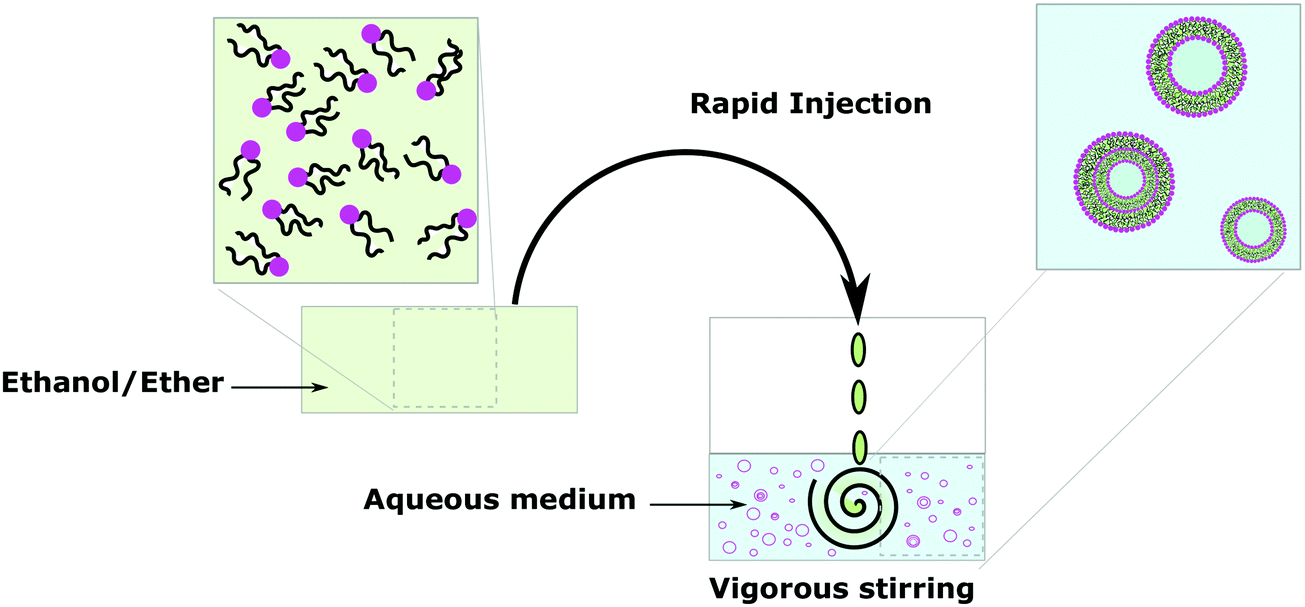 | ||
| Fig. 9 Self-assembly of lipids and block copolymer vesicles by using solvent injection. | ||
A more controlled manner to form vesicles in defined size distribution is by using emulsion phase transfer (Fig. 10).80,86–88 An initial w/o (water-in-oil) emulsion is prepared by vigorously stirring a small quantity of aqueous media to a solution of amphiphiles in an organic solvent, effectively forming water droplets coated with the amphiphile. Then the emulsion is poured on top of an oil-in-water biphasic system where amphiphiles dissolved in the organic solvent should have assembled into a monolayer at the interface. Due to their density difference, the monocoated water droplets from the emulsion sink to the aqueous phase through the amphiphilic saturated interface generating w/o/w double emulsions. Water droplet migration can be facilitated by using sucrose in the inner water droplet and equiosmotic aqueous glucose in the biphasic system due to their density difference80 and/or centrifugation (emulsion–centrifugation),91–93 which can also (partially) remove the trapped organic solvent, thus generating low polydispersed vesicles. As the outer and inner layers of the vesicles are formed at separate stages, complete control of the vesicular structure is possible and as such asymmetric vesicles can be obtained (see Section Va Asymmetric membranes for more details).86,87 Moreover, encapsulation can be achieved by doping the inner water droplet with desired hydrophilic cargos contrasting with the solvent-free methods.
 | ||
| Fig. 10 Self-assembly of lipids and block copolymer vesicles by using emulsion phase transfer. | ||
An improvement to the emulsion phase transfer methods is microfluidics. Microfluidics is probably the most advanced solvent displacement method and has allowed exquisite control and progress in constructing artificial cells.21,89,90,107 This water-in-oil-in-water (w/o/w) double emulsion technique selectively forms vesicles homogeneous in size from SUVs to GUVs depending on the requirement.80,108 Amphiphile-stabilized water-in-oil (w/o) emulsions are first prepared using organic solvents immiscible with water at the first junction. Then these droplets flow to a second junction through an aqueous phase coated with an amphiphile to obtain w/o/w double emulsions (Fig. 11). A monolayer of amphiphile is obtained at both the w/o interphases. Solvent extraction from the double emulsion to form vesicles is a process called dewetting.105,109 Dewetting of the double emulsion can be achieved by using ethanol in the outer aqueous phase21,110 surfactants,105 octanol as the oil-phase rather than the traditional hexane or chloroform,111 or solvent evaporation.109,112 Absolute dewetting is a fundamentally challenging process and most vesicles formed by solvent-displacement methods still have, in the best cases, trace amounts of solvents embedded within their membrane. Moreover, testing for remaining solvent is rarely done in advanced systems.90,113 Highly complex mixtures of components are required for solvent removal, stabilization and formation (surfactant, sugars, polyelectrolytes, non-amphiphilic polymers etc.) which are imbedded in the membrane or in the vesicular lumen.21,89,105,114,115 These impurities will affect the vesicle properties and impair potential encapsulated bio-components and are often overlooked. In addition, as the inner amphiphilic monolayer is much smaller than the outer monolayer, considerable rearrangement needs to occur which sometimes leads to undesired aggregation within the membrane if the number of amphiphiles is in excess.80,105,109,112 The final size of the vesicle is controlled by the initial inner water droplet, achieving very low size distributions. This method also shows a high encapsulation efficiency of a variety of proteins and small molecules and it was also proven that these entities can be co-encapsulated, an important aspect to generate advanced protocells.
 | ||
| Fig. 11 Production of water-in-oil-in-water (w/o/w) double emulsion by using a microfluidic device. Amphiphile-stabilized water-in-oil (w/o) emulsions are first prepared using organic solvents (middle fluid, MF) immiscible with water (inner fluid, IF) at the first junction. The droplets flow to a second junction through an aqueous phase (outer fluid, OF) coated with an amphiphile to obtain w/o/w double emulsions. Adapted from J. Petit, I. Polenz, J. C. Baret, S. Herminghaus, O. Baumchen, Eur. Phys. J. E: Soft Matter Biol. Phys., 2016, 39, 59. https://doi.org/10.1140/epje/i2016-16059-8.21. | ||
Pico-injection microfluidics has been developed as a novel means for post self-assembly encapsulation in droplet-supported vesicles.107,113,116 This active encapsulating technique is used for the manipulation of compartments after their formation and the sequential addition of multiple components which would not be otherwise incompatibly added in bulk. By means of electroporation, single drop injection is carried out, precisely controlling the cargo concentration in each vesicle. Recently, Weiss et al. described the sequential loading of transmembrane F0F1-ATP synthase and integrin embedded into the lipid bilayer and the encapsulation of cytoskeletal proteins resulting in actin filaments and microtubule reconstitution by high-throughput microfluidics pico-injection (Fig. 12).113 Pico-injection has proven to be an excellent technique to achieve the controlled encapsulation of complex bio-relevant macromolecules in the membrane or inside droplet-stabilized GUVs. The technique is however still rarely used as it is so far limited to lipid-droplets and requires highly specialized equipments.
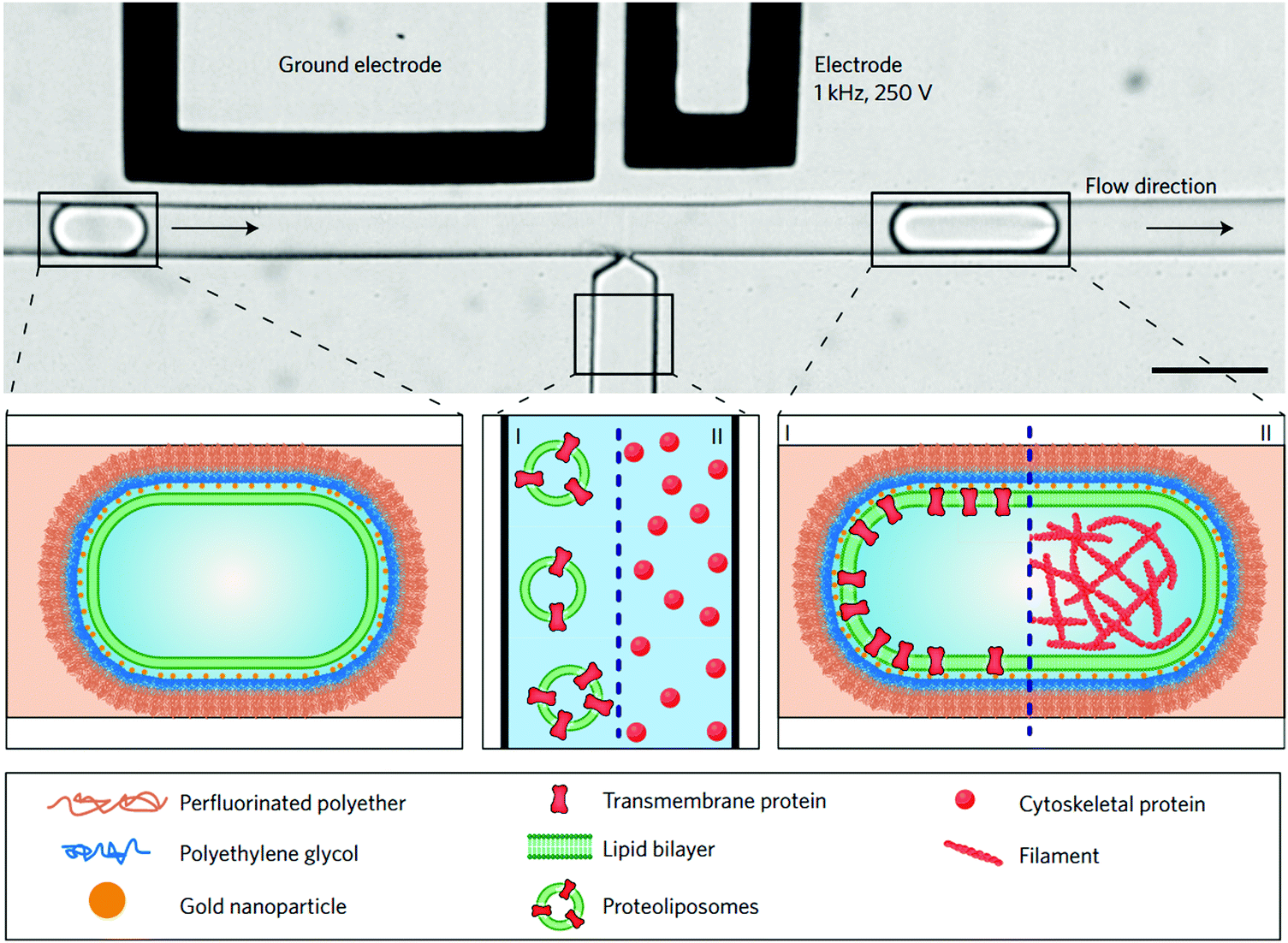 | ||
| Fig. 12 Sequential bottom-up assembly of transmembrane and cytoskeletal proteins into droplet-stabilized GUVs by high throughput pico-injection. Scale bar 50 μm. Reproduced with permission from M. Weiss, J. P. Frohnmayer, L.T. Benk, B. Haller, J. W. Janiesch, T. Heitkamp, M. Börsch, R. B. Lira, R. Dimova, R. Lipowsky, E. Bodenschatz, J. C. Baret, T. Vidakovic-Koch, K. Sundmacher, I. Platzman and J. P. Spatz, Nat. Mater., 2018, 17, 89–96.103 | ||
(c) Microscopy observation
Depending on the size of the vesicles obtained, different microscopy techniques can be used for the observation and characterization of the vesicles (Fig. 13).15 For GUVs, optical microscopy is most frequently used, as it is non-destructive and allows for direct visualization under physiological conditions.1 Alternatives such as fluorescence microscopy are also popular as the enclosed compartment with encapsulated soluble fluorescent dyes and/or the membrane using lipophilic fluorophores either by covalent or non-covalent linkage can be simultaneously labelled specifically with fluorochromes with high sensitivity (typically 50 fluorescent molecules per mL or 1 fluorescent molecule per 1000 lipids).1 However, due to the limited resolution of conventional optical microscopy only GUVs can be easily observed.1 For SUVs and LUVs, transmission electron microscopy (TEM) is favored as it has a resolution 105 higher than optical microscopy.15 Cryo-TEM is preferred as it allows for the observation of a sample in its hydrated state. TEM can also be used to determine the membrane thickness of SUVs and GUVs.9,12,15 However, the electrons may affect the state of the specimen and it also has to be considered that most of the experiments are not conducted under wet conditions, but require drying or freezing of the sample. State-of-the-art techniques for super-resolution such as scanning near-field optical microscopy (SNOM), photo-activated localization microscopy (PALM), stimulated emission depletion microscopy (STED), or structured illumination microscopy (SIM) would allow the wet imaging of smaller vesicles but are still very rarely used.117–119 | ||
| Fig. 13 Microscopy images obtained for different techniques. TEM: transmission electron microscopy. Film hydration: Reprinted from K. Kita-Tokarczyk, J. Grumelard, T. Haefele, W. Meier, Block copolymer vesicles—using concepts from polymer chemistry to mimic biomembranes, Polymer, 46(11), 3540–3563. Copyright (2005), with permission from Elsevier.15 Electroformation: in-house image. Solvent injection: adapted from ref. 20 with permission from The Royal Society of Chemistry. Microfluidics: Reprinted with permission from J. Petit, I. Polenz, J. C. Baret, S. Herminghaus and O. Baumchen, Eur. Phys. J. E: Soft Matter Biol. Phys., 2016, 39, 59. https://doi.org/10.1140/epje/i2016-16059-8.21. | ||
IV. Stimuli-responsive vesicles
Liposomes and polymersomes can be chemically modified (functionalized) to tune their properties, which is particularly important for certain biomedical applications.15,76,120–122An important application of small vesicles is drug delivery.24 To be efficient, such vesicles must achieve two characteristics: delivery of the drugs at the desired site and release of the vesicle content only at that location. Frequently, a responsive stimulus is necessary for specific and efficient drug delivery. Most stimuli-responsive or ‘smart’ vesicles release their cargo upon activation either due to (a) full disruption (disassembly) usually caused by making the hydrophobic moiety of the amphiphile more hydrophilic or (b) tuning the vesicles’ permeability for a slow controlled release of encapsulated molecules (‘breathing vesicles’) or (c) changing their supramolecular morphology.28 These changes can be reversible or irreversible and arise from changes to the supramolecular assemblies (e.g. H-bonding network) or chemical changes in the molecular unit.123 Functionalization can also be used in nanoreactors to enhance chemical processes.24 Moreover, stimuli-responsive behaviors are intrinsically the basis of biological processes (cell signaling, immunity, energy and small molecule production etc.) and is an important step in biomimetic studies.24 Stimuli can be of biological or chemical nature such as pH changes, response to ROS, enzyme, glucose etc. or physical stimuli such as light, temperature, ultra-sound, magnetic fields, pressure etc.24,124,125 and can be combined for multiple responsivities.
Liposomes are usually functionalized to overcome their low stability, leakiness, and low blood circulation times. PEGylated lipids (PEG: poly(ethylene glycol)) have been shown to be successful in forming liposomes as PEG is hydrophilic and its covalent attachment to the hydrophilic head of the lipid does not modify its amphiphilicity.9,30,126 PEG-stabilized liposomes are thus the most prominent class of functional liposomes and are most commonly formed by covalently grafting a PEG chain to lipids and doping the liposomes with these specialized lipids. The synthesis of PEGylated lipids is achieved by adding a terminal activated PEG chain (for example with imidazole) to an amine terminated lipid (PE) (Scheme 2), which forms MLV liposomes (112–136 nm).126 PEGylated liposomes are also called “stealth” liposomes. The addition of biocompatible PEG chains increasingly helped the bioavailability of liposomes in vivo by reducing potential interactions with immune cells (e.g. macrophages) by coating the liposome surface and reducing liposome aggregation.19,127 These “stealth” liposomes have greatly increased blood circulation time from 1 h to 1–2 days and thus improved drug distribution.1,12,19
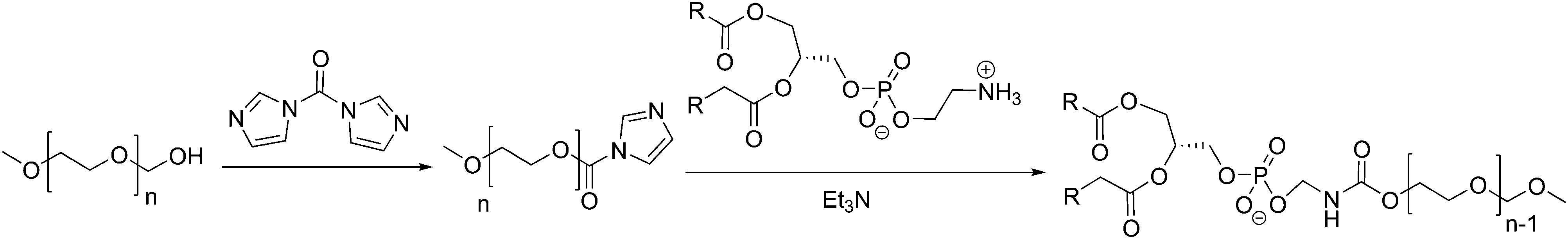 | ||
| Scheme 2 Example synthesis of PEGylated-lipids by Allen et al.126 | ||
(a) Biological or chemical stimuli-responsive vesicles
pH-Sensitive LUVs and SUVs are the most studied release systems as one can make use of physiological pH-gradients.122,128,129 Extracellularly, inflammatory tissues and tumors are more acidic than healthy tissues, or intracellularly endosomes and lysosomes also show acidic conditions (below pH 7), making pH change an ideal trigger for drug delivery.125pH-Sensitive liposomes are commonly generated using a small amount of anionic lipid derivatives such as N-succinyldioleoyl-phosphatidylethanolamine (COPE),130 cholesteryl hemicussinate (CHEMS),131–133 a cholesterol derivative, or α-tocopherol acid succinate131 implemented in neutral lipid vesicles. In response to an acidic environment, for example below 5.8 (pKa of CHEMS),133 the negative moiety of these lipids gets protonated switching from vesicles to inverted micelle structures. pH-Sensitive colorimetric reversible LUVs (≤800 nm) were described using the small amphiphilic molecule polydiacetylene (PDA) and could be used for bio-imaging.134 By its nature, PDA's structure is analogous to a lipid as it is composed of a hydrophilic head (acetohydrazide), rather than the much longer repeated hydrophilic moiety found in block copolymers. Upon addition of HCl, the hydrogen bonding network of the amide moiety is disrupted, replaced by an ionic stabilization of the terminal amine by chloride anion and presumably some charge repulsion causing a rearrangement of the macromolecular assembly without vesicular disassembly (Scheme 3). This supramolecular assembly of the vesicles allows them to undergo photo-polymerization visualized as a shift from a red to a blue color. In the neutral form, polymerization of the diacetylene groups is not possible due to geometrical constraints determined by the hydrogen bonding network of the head group. Later Yuan and Hanks also added an amphiphilic fluorophore BO558 in these PDA vesicles (≤450 nm) which resulted in a reversible switching of the fluorescence (on when neutral and off when acidic) which could also be used in bio-imaging.135
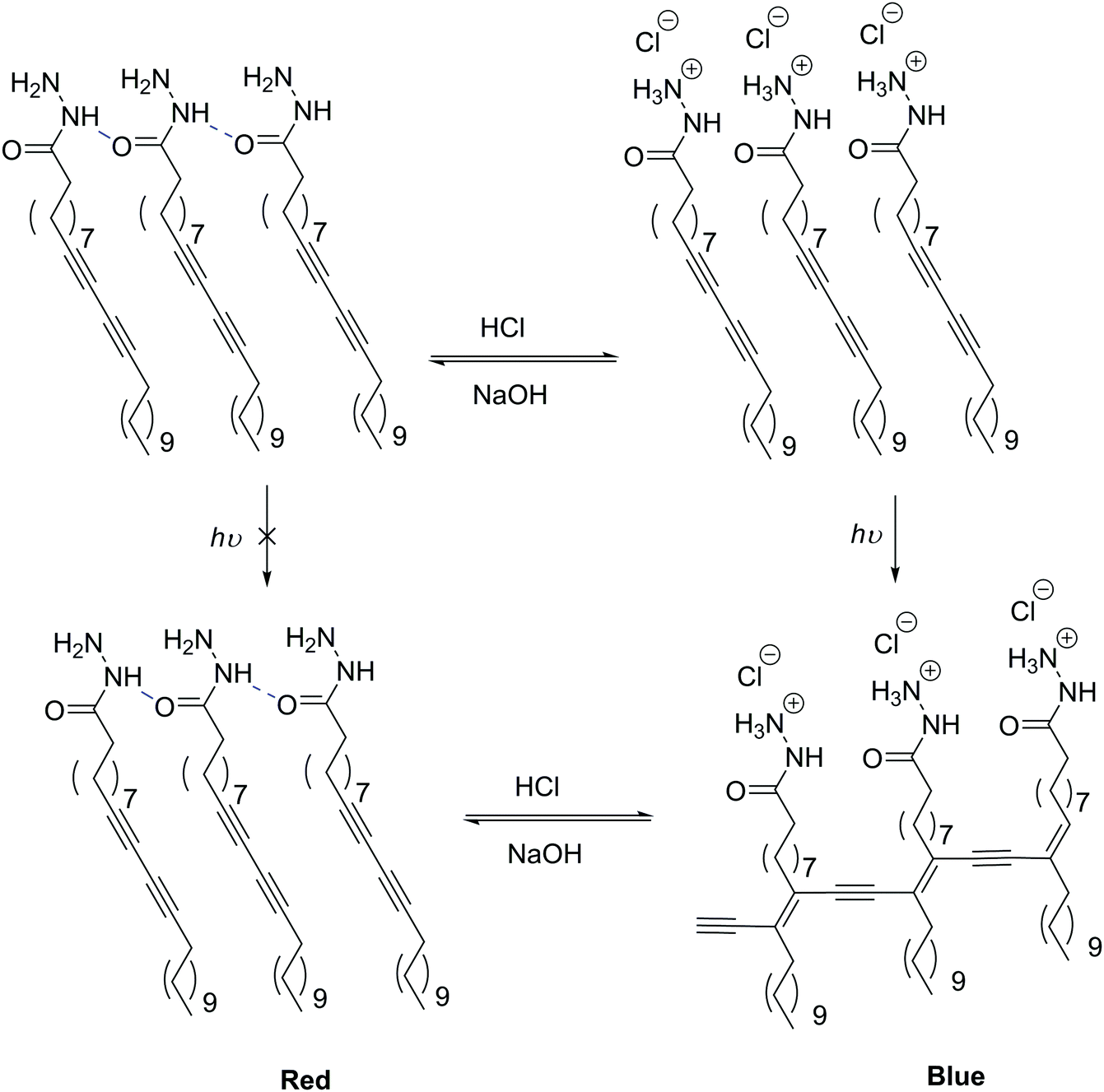 | ||
| Scheme 3 pH-Responsive SUVs due to a supramolecular change in the head group hydrogen bonding network.134 | ||
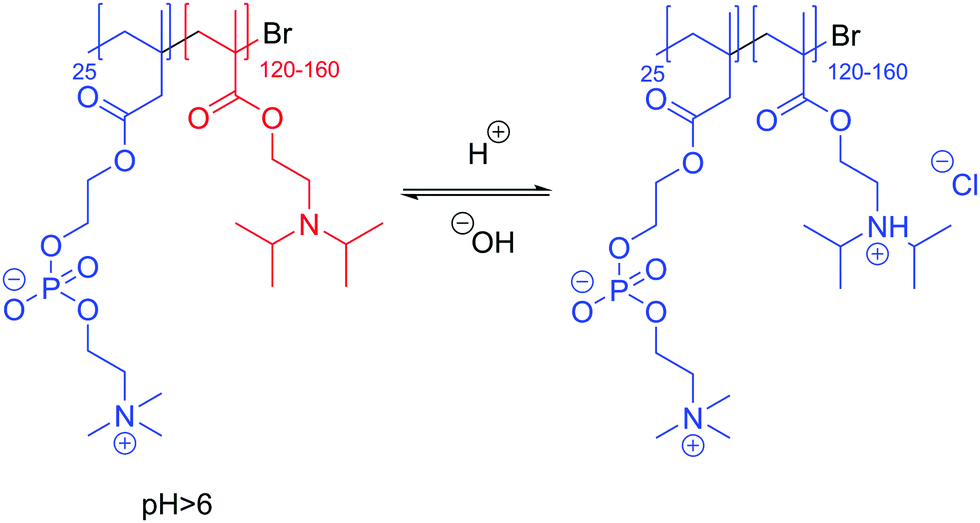
pH-Responsive polymersomes (exclusively LUVs or smaller) can also be formed from hydrolytic, acid cleavable polymers, or are most commonly composed of ionizable blocks such as a polyacid (e.g. carboxylic or sulfonic acid) or a polybase (e.g. amines).15,24,124,125 In the presence of different pH values, their hydrophobic fraction becomes more water-soluble by ionization modification and thus the polymersomes disassemble. pH-Responsiveness has the advantage of being virtually instantaneous compared to other chemical modifications such as hydrolysis. Armes and coworkers136 described a poly(2-methacryloyloxyethyl phosphorylcholine)-b-poly(2-(diisopropylamino)ethyl methacrylate) (PMPC-b-PDPA) block copolymer where PMPC is a phospholipid-like hydrophilic block and PDPA a polybase (Scheme 4). At pH > 6, PDPA (pKa 6.3) acts as a hydrophobic block and stable polymersomes (100 nm) can be formed. Upon lowering the pH, the tertiary amine is protonated, becoming hydrophilic and causing vesicles’ disassembly. The authors further proved that the anti-cancerous drug doxorubicin can be efficiently encapsulated and retained when mixed in the presence of PMPC-b-PDPA at pH 2 and then adjusted to pH 7.4. These polymersomes were subsequently used to provide efficient targeted intracellular delivery of drugs, antibodies, DNA, etc.125 In a more advanced system, Rodriguez-Hernandez and Lecommandoux reported reversible poly(glycolic acid)-b-poly-L-lysine (PGA-b-PLys) polymersomes (100–200 nm) where the PGA block is hydrophobic and PLys hydrophilic at pH < 4, and the hydrophilicities of the blocks revert at pH > 10 where the PGA block is hydrophilic and PLys hydrophobic.137
 | ||
| Scheme 4 Example of pH-responsive polymers bearing the polybase PDPA. Blue: hydrophilic. Red: hydrophobic.136 | ||
Eisenberg and coworkers detailed pH-responsive PEO45-b-PS130-b-PDEA120 (PDEA: poly(2-diethylaminoethyl methacrylate)) LUVs clearly exemplifying the ‘breathing’ effect of the membrane.138 The pH-responsive block PDEA causes a change in vesicle sizes (from 250 nm to 480 nm), wall thickness (25 nm to 80 nm), structure and appearance (apparent swelling of the membrane at lower pH) (Fig. 14). These transformations undoubtedly affect the overall polymersome permeability creating a notably more porous membrane for protons.
 | ||
| Fig. 14 Cryo-TEM image of a pH-responsive PEO-b-PS-b-PDEA polymersome at different pH values. Reprinted with permission from S. Yu, T. Azzam, I. Rouiller and A. Eisenberg, J. Am. Chem. Soc., 2009, 131(30), 10557–10566. Copyright (2009) American Chemical Society.138 | ||
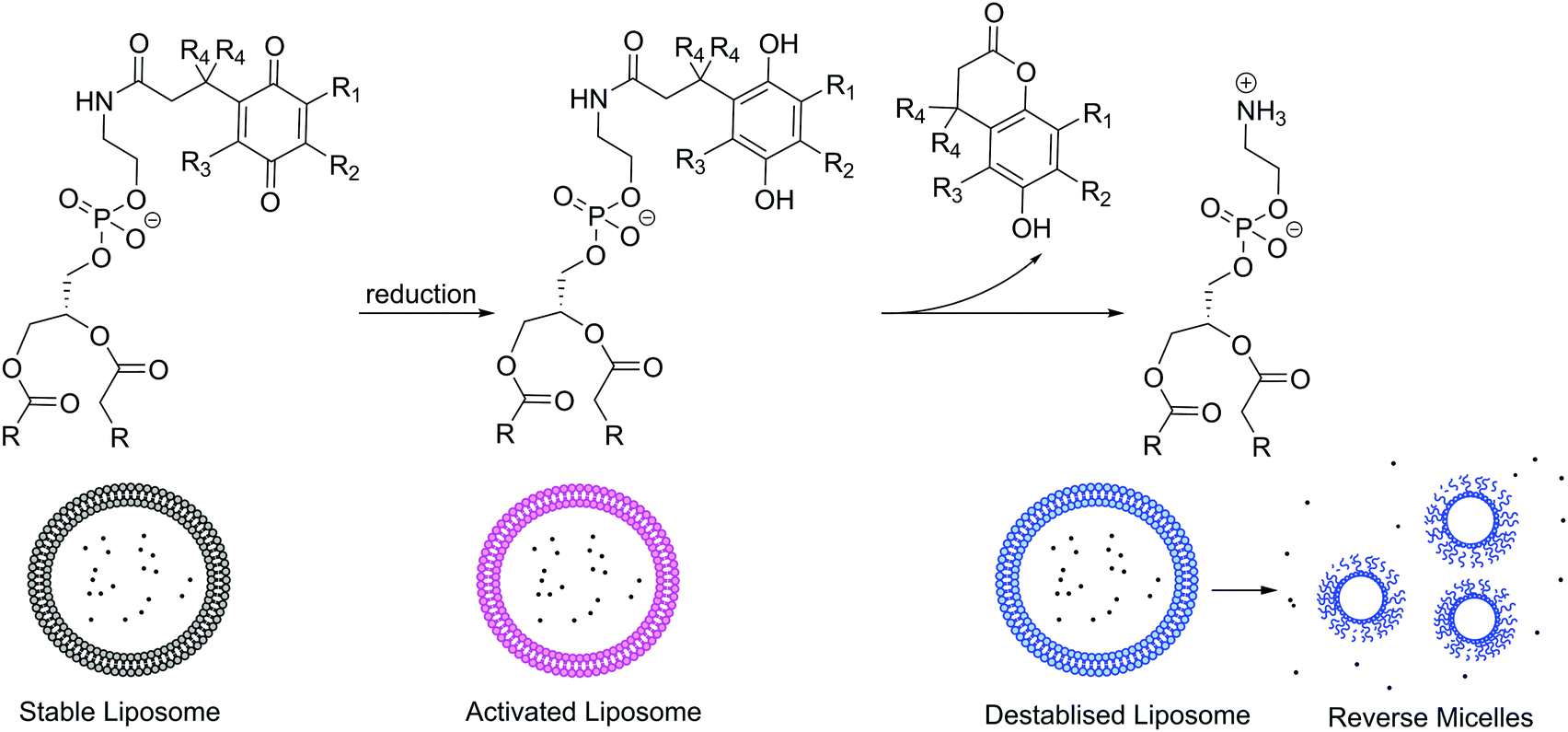
Many biological processes are triggered by changes of redox potential. Thus, similarly to pH gradients, reduction or oxidation sensitive moieties can conveniently be triggered for controlled release or imaging particularly for intracellular delivery due to the redox potential difference between the different organelles and the cytosol.125 In healthy cells, ROS such as peroxides, hydroxyl radicals, singlet oxygen etc. are produced as a byproduct of aerobic metabolism and are kept under control by antioxidants, while any damage to cells is repaired. Infection, inflammation, cancer, and many other diseases cause oxidative stress, an overproduction of ROS in cells, which cannot be controlled by the body's natural defenses. Thus, redox-responsive vesicles are attractive for pharmaceutical purposes. Reduction or oxidation sensitive moieties such as ferrocene, disulfide bonds (cleavage under reductive conditions), quinoline etc. can all be implemented in the lipid's head or tail or polymer blocks.139 For example, quinoline functionalized DOPE forms stable liposomes (100 nm); however, under reducing conditions, quinoline switches to hydroquinone that can rearrange as a lactone, releasing the free PE lipid.140 This lipid is not capable of maintaining a bilayer and rearranges into reverse micelles, releasing its cargo (Scheme 5).
 | ||
| Scheme 5 Example of redox responsive lipids bearing quinoline groups.140 | ||
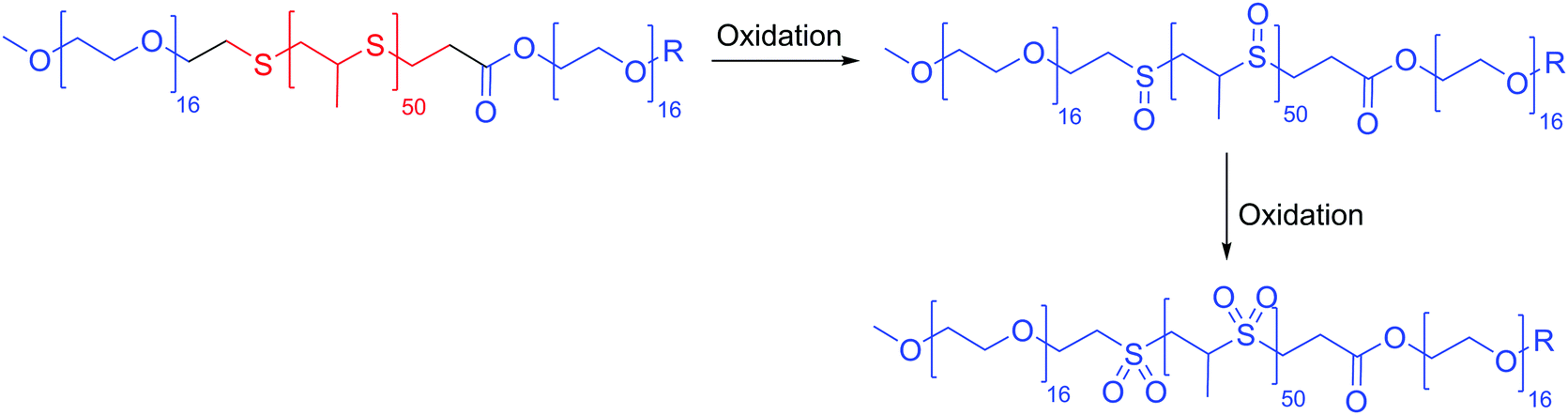
Many redox responsive polymersomes have also been described. The first oxidative responsive polymersomes (100 nm) were described in detail by Napoli et al. in 2004 using PEO-b-PPS-b-PEO (PPS: polyphenylene sulfide) (Scheme 6).27 The thioether in the hydrophobic block becomes oxidized to a sulfoxide initially and ultimately sulfone becoming hydrophilic and thus changing the morphology of the polymersomes to micelles. The ferrocenylsilane block or in general ferrocene containing polymers have been used to form redox active organometallic vesicles.15 These polymersomes are disrupted in the presence of high concentration of ROS released by the diseased cells causing the oxidation of ferrocene to ferrocenium which destroys the vesicles.24
 | ||
| Scheme 6 Oxidative responsive polymersomes by Napoli et al.27 | ||
Enzymes, being highly specialized biological entities, have superior efficiency for selective and sensitive delivery of vesicular cargo. Enzyme-responsive polymersomes were used for drug delivery as certain enzymes are overexpressed in pathological regions.24 For example, Cathepsin B is found in abundance in tumor tissues and specifically degrades Gly–Phe–Leu–Gly (GFLG) sequences.141 Lee et al. used a PEO-b-PDLLA (PDLLA: poly(D,L-lactide)) block copolymer with a GFLG linker which self-assembles into SUVs (124 nm). The block copolymer is cleaved in the presence of Cathepsin B (at tumor site) causing disassembly and the release of any drugs for example the anti-epidermal growth factor receptor (anti-EGFR), a chemotherapy agent.142
(b) Physical stimuli-responsive vesicles
Temperature is another popular trigger for release of vesicular cargo as temperature modulates cell activity. Thermoresponsive liposomes rely on the transition temperature of natural phospholipids rather than synthetically modified lipids. Typically DPPC and MSPC (1-myristoyl-2-stearoyl-sn-glycero-3-phosphocholine) are used as these lipids have a Tm between 41 and 45 °C, a physiologically relevant temperature range especially for targeted drug delivery.121,143–146 The thermo-responsiveness of the liposomes can be modulated by using a mixture of lipids. Nonetheless, lipid-only temperature sensitive liposomes remain rare as lipid modification is difficult and the simple doping of liposomes with thermosensitive polymers represents a much more achievable modulative platform for thermoresponsive vesicles. For example Pippa et al. formed C12H25-PNIPAM-COOH/DPPC (PNIPAM: poly(N-isopropylacrylamide)) hybrid vesicles (100–200 nm) behaving similarly to pure polymeric vesicles.147Multiple thermoresponsive polymersomes have been described most frequently also relying on PNIPAM as the thermoresponsive block.124,125,128 PNIPAM can undergo a phase transition from hydrophilic to hydrophobic upon heating at its lower critical solution temperature (LCST) of ∼32 °C. Thus, at the physiological temperature of ∼37 °C, the PNIPAM block is hydrophobic by displacing the hydrogen bond network responsible for its solvation (hydrophilicity) below its LCST. The block copolymer is thus amphiphilic and PEO-b-PNIPAM can self-assemble into polymersomes (>1 μm) (Fig. 15). Below the LCST, the thermoresponsive block is solvated in water by favored hydrogen bonding and causes the polymersome to dissociate releasing any encapsulated substances. LCST polymers such as PNIPAM can thus be used to release drugs locally when using hypothermic patches or ice packs, local cryosurgery probes or tissue freezing.148
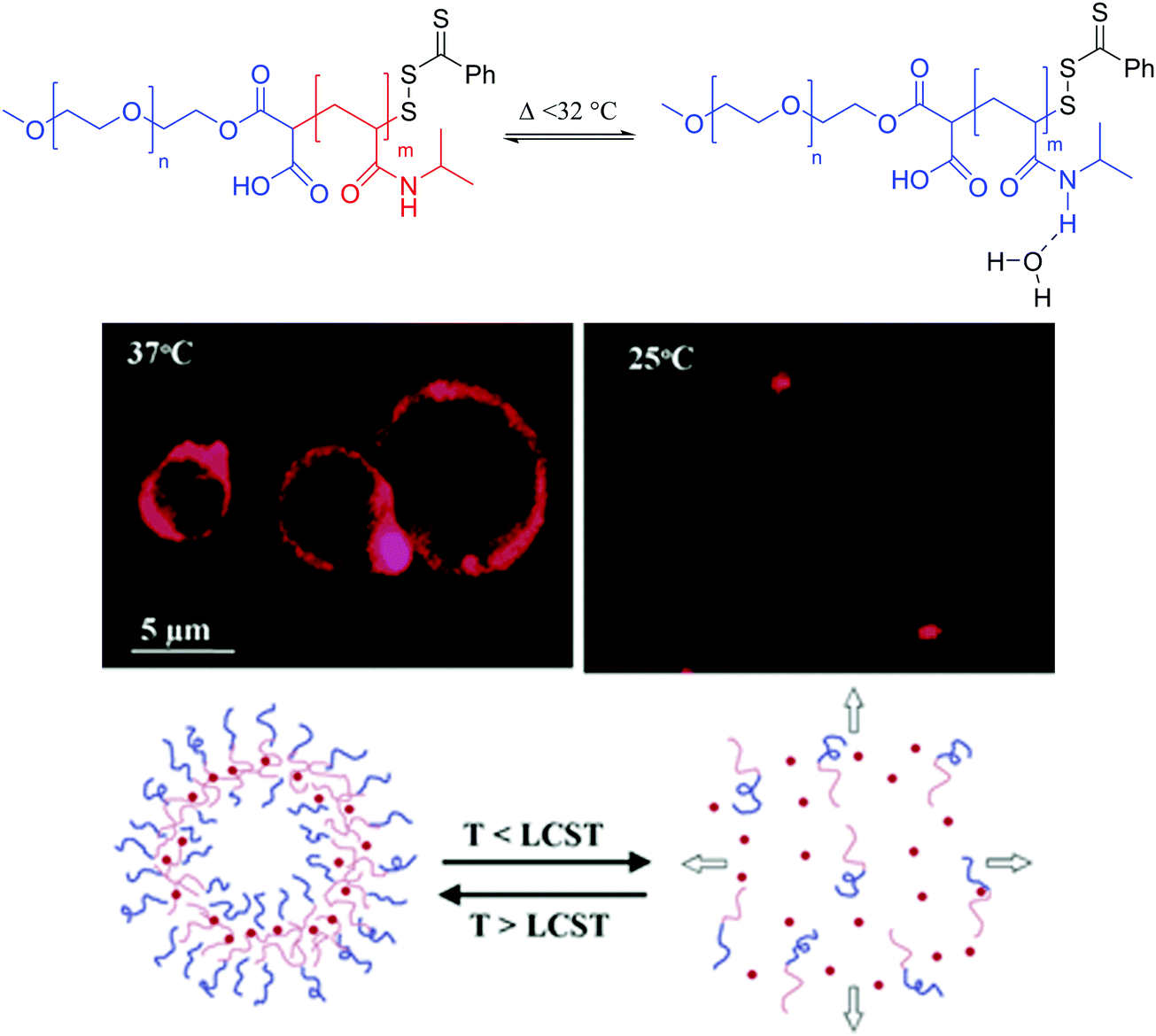 | ||
Fig. 15 Thermoresponsive block copolymer bearing the thermosensitive PNIPAM block with LCST behavior. Reprinted with permission from S. Qin, Y. Geng, D.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) E. Discher and S. Yang, Adv. Mater., 2006, 18, 2905–2909. Copyright (2006) Wiley.148 E. Discher and S. Yang, Adv. Mater., 2006, 18, 2905–2909. Copyright (2006) Wiley.148 | ||
Interestingly, almost all thermoresponsive polymersomes use LCST responsive moieties, which intuitively seem more troublesome to use than an upper critical solution temperature (UCST) block since the polymersomes have to be stored at elevated temperatures and release their cargo at lower temperatures. Tumor tissues or infection sites tend to have more elevated temperature (+2–5 °C) than healthy tissues.149 Therefore, polymers with an UCST in water would be much more interesting and easier to use than LCST polymers. Only very recently was a poly(phosphonate) block (PPE) shown to have a UCST.123 PPE-b-PEO was subsequently proven to self-assemble into polymersomes (100–200 nm) at room temperature and to fully disassemble upon heating up to 60 °C as the PPE block became hydrophilic and to reversibly re-assemble when cooling (Scheme 7). Unfortunately, full disintegration of the polymersomes did not occur at the physiological temperature; however, these block copolymers are a compelling initial proof of concept that polymersomes with UCST behavior are achievable and further work should be done to tune these PPE blocks to a UCST close to 40 °C.
 | ||
| Scheme 7 Thermosensitive PPE block with UCST behavior.123 | ||
Light can initiate a simple, non-invasive, efficient, and on-demand trigger since time, exposure, wavelength, and intensity can be well controlled, and it is easier to handle than chemical responses.24,125 However, most light-responsive moieties used in vesicles are triggered by UV or visible light irradiation which limits the vesicle application to very close to the surface area of the body,125 and near-infrared sensitive assemblies would be more appropriate for deep tissue drug-release. Bayer et al. developed photo-responsive liposomes (<200 nm) using a synthetic derivative of PC with a 2-nitrobenzyl moiety and showed the release of an hydrophobic cargo (Nile red) upon irradiating the sample at 350 nm (Scheme 8).57
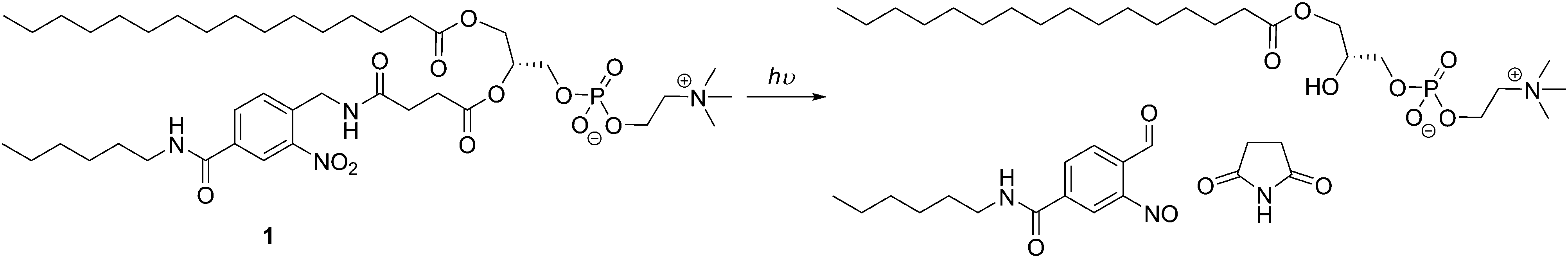 | ||
| Scheme 8 Photo-responsive modified PC which can assemble into photo-responsive liposomes.57 | ||
Other examples of photo-labile phospholipids were described by Suzuki et al., who used glycerophosphocholines presenting two terminal 2-nitrobenzyl groups.150 By UV irradiation at 350 nm the vesosomes release their double-stranded DNA (dsDNA) cargo into the outer GUV (10 μm diameter). Ces and coworkers described UV–C responsive PC derivatives composed of diacetylene.151 The UV irradiation catalyzes the polymerization of diacetylene effectively cross-linking the phospholipid aliphatic chains creating pores in the membrane, a process already described in Scheme 3.
Photo-responsive polymersomes have been described using azobenzene (reversible cis/trans isomerization), spiropyran (ring opening), O-nitrobenzyl (photocleavage of block junction), coumarin, 2-diazo-1,2-naphthoquinone etc. moieties which also change hydrophobicity (conformational changes) or cause cleavage leading to the dissociation of the vesicles.16,125 Not all light-triggered polymeric systems result in irreversible disassembly of polymersomes. For example, Su et al. described light-responsive PAA-b-PAzoM (PAzoM: poly{6-[4-(4-methylphenylazo)phenoxy]hexyl acrylate)} polymersomes (2–4 μm) which can switch between spherical polymersomes and earlike shaped ones depending on the isomerization of the azobenzene moiety.152
Vesicles, whether liposomes or polymersomes, can be modified in a variety of ways. Their primary purpose is the encapsulation and release of drugs. By encapsulating drugs, they are protected against enzymatic degradation and the encapsulation can improve the biodistribution and reduce side-effects.19,25,76 SUVs make efficient carriers of small molecules in contrast to most other nanomedicine agents,28 and have been shown to deliver both hydrophilic (encapsulated) and hydrophobic compounds (embedded in the membrane) to target tissues.127 We noticed that by far almost all functionalized or stimuli responsive vesicles are small (LUVs or SUVs) and the few micron-sized vesicles which have been reported are only a few μm (<5 μm). For biomedical purposes, LUVs are more adapted, as they are more malleable, can easily circulate in the body and can even be internalized into cells. Nonetheless there is a clear scarcity of functional and/or stimuli-responsive GUVs (ideally >10 μm) which are essential for characterization of the membrane mechanical properties and for the generation of protocells and biomimetic compartments.
V. Mimicking cell functions in vesicles
Vesicles have been extensively studied and developed from a biomedical perspective because they are biocompatible. Nonetheless, because of their compartmentalization effect, vesicles can also be advantageous to use for enhancing chemical reactions and biosynthesis as reactors or to generate protocells.153–155 For example, incompatible reactions could be achieved by simply compartmentalizing them.4 Ultimately a cell can also be described as a highly evolved microreactor which encapsulates many nanoreactors (organelles). Constructing simplified synthetic cells is an increasingly expanding area because it contributes to an understanding of fundamental biological processes and provides new and unique prospects for future therapeutic applications, from self-regulating bioreactors to new biomedical devices.90,107,156–159 Ultimately, the goal would be to understand cell processes sufficiently using models so that we could replace natural living cells with functional synthetic cells.(a) Membrane functionality
We have already shown that synthetic compartmentalization can be readily achieved with liposomes and polymersomes. However, cell membranes are very complex: they are asymmetric, have lipid rafts, use a variety of means to transport ions and small molecules through the membrane in a controlled manner, communicate between cells, act together to form tissues etc. | ||
| Fig. 16 Distribution of phospholipids in human red blood cells. Reproduced with permission from D. Marquardt, B. Geier and G. Pabst, Membranes, 2015, 5(2), 180–196 (http://www.mdpi.com/2077-0375/5/2/180).161 | ||
Lipid-only asymmetric liposomes have been described as they naturally entropically favor specific lateral and transversal lipid phase separation due to the intrinsic properties of the lipid such as the angle and charge of hydrocarbon chains, as depicted in Fig. 16.161,163–165 However, controlling asymmetry in liposomes to understand the principles behind their distribution or achieve unnatural distributions can mostly only be achieved by tuning the chemical composition of their chemical environment for example through pH modulation.164 Phosphatidylglycerol (PG) could selectively (80–90%) rearrange to equilibrate on the inner layer of the SUVs (100 nm) when the intravesicular environment was basic and on the outer leaflet when the intravesicular environment was acidic while PE remained equally distributed. The basis behind such preferential tuning is that PGs are acidic and thus rearrange on the proton depleted side of the membrane as a means to equilibrate the transmembrane proton gradient.
Inducing asymmetry within the vesicle membrane can also be achieved due to membrane curvature. Tuning the chemical composition of the amphiphiles can dictate a preferred orientation of the amphiphile within the membrane.166–169 This is particularly advantageous for polymersomes as the block copolymer length is easily adjustable synthetically in contrast to liposomes as discussed in Section IId. Mixing two diblock copolymers with differently sized hydrophilic blocks could result in a hydrophilic block with the smaller volume in the inner membrane and the larger one on the outer membrane for example with PS295-b-PAAn (n = 12 or 74) polymersomes (100 nm).167 More frequently ABC triblock copolymers are described to form asymmetric membranes presumably as they are easier to design and control than diblock copolymers. Meier and his group investigated two similar ABC polymers PEO-b-PDMS-b-PMOXA170 and PEO-b-PCL-b-PMOXA35 which formed asymmetric SUVs (∼100 nm) and GUVs (∼5 μm) respectively. Asymmetry is achieved by tuning the block length. PEO45-b-PDMS65-b-PMOXA346 was determined to form vesicles with the PMOXA block oriented on the outside and PEO40-b-PDMS45-b-PMOXA97 with the reverse arrangement. In analogy to liposomes, Liu and Eisenberg described asymmetric vesicles (120 nm) which can switch orientation depending on pH changes, impeccably exemplifying the influence of the chemical composition on asymmetry.171 PAA26-b-PS890-b-P4VP40 (P4VP: poly(4-vinyl pyridine)) has two relatively short hydrophilic blocks and thus their orientation in polymersomes cannot be dictated by membrane curvature alone. Both PAA and P4VP are however pH sensitive. At low pH values, the blocks are protonated, thus PAA is neutrally charged, whereas P4VP is positively charged, while at high pH they are deprotonated, PAA is negatively charged and P4VP is neutral. When the blocks carry charges, intra- and intermolecular repulsive interactions are exhibited. Hence, the charged block swells and effectively has a larger volume than the neutral one. Following the membrane curvature restriction, the charged block would be favored on the outer layer of the polymersome membrane (Fig. 17).
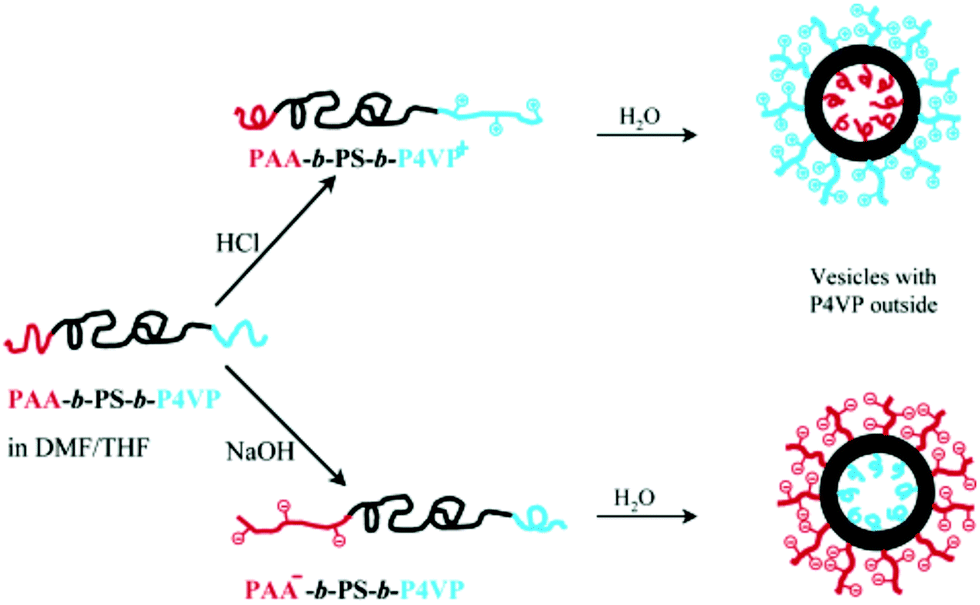 | ||
| Fig. 17 Reversible asymmetric polymersome from PAA26-b-PS890-b-P4VP40. Reprinted with permission from (F. Liu and A. Eisenberg, J. Am. Chem. Soc., 2003, 125(49), 15059–15064).171 Copyright (2003) American Chemical Society. https://pubs.acs.org/doi/abs/10.1021/ja038142r. | ||
Generating asymmetry in lipid and polymer membranes has recently been largely simplified by solvent double emulsion methodologies such as microfluidics or emulsion phase transfer which allows the highly controlled construction of each layer in the vesicular bilayer separately regardless of the nature of the amphiphiles.86,87,90,162,172,173 Weitz and coworkers described the self-assembly of asymmetric liposomes (0.5–2 μm) by emulsion phase transfer with POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and POPS (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine).86 The aliphatic chains of the lipids are the same but POPS has a small anionic head group and POPC a large zwitterionic head group. Both asymmetric vesicles (with POPC or POPS on the inner layer) were generated despite the unfavoured curvature of POPC in the inner layer. In an interesting recent example, Peyret et al. developed hybrid POPC/PB-b-PEO asymmetric GUVs (∼10 μm) using emulsion–centrifugation (Fig. 18).87 A polymer-stabilized sucrose(aq)/toluene droplet is initially formed and poured into a lipid-stabilized glucose(aq)/toluene interface, leading to the formation of GUVs with an outer lipid monolayer and an inner polymer monolayer. The reverse hybrid asymmetric vesicles were more difficult to achieve due to the low stability of the POPC-droplets. Interestingly, polymer only asymmetric membranes have yet to be synthesized by these methods.
 | ||
| Fig. 18 Preparation of asymmetric hybrid GUVs by emulsion–centrifugation. (a) Preparation of sucrose filled polymer-stabilized droplets in toluene. (b) Crossing the amphiphile-stabilized glucose/toluene interface helped by centrifugation, forming (c) asymmetric hybrid vesicles. Adapted from A. Peyret, E. Ibarboure, J.-F. Le Meins and S. Lecommandoux, Adv. Sci., 2017, 5(1) with permission from Wiley. (https://onlinelibrary.wiley.com/doi/abs/10.1002/advs.201700453).87 | ||
As previously discussed, a major drawback of microfluidics and droplet emulsion transfer methodologies is the unassessed contamination of the membrane with organic solvents which alter the asymmetric membrane's properties such as in this case the transversal diffusion of the amphiphile.89,115,161 With an emphasis on reducing solvent impurities, Takeuchi and coworkers developed lipidic GUVs (∼4 μm) formed from the non-uniform cleavage of an asymmetric DOPC/DOPS (1,2-dioleoyl-sn-glycero-3-phosphocholine/1,2-dioleoyl-sn-glycero-3-phosphoserine) planar lipid bilayer by a pulsed microfluidic jet flow.115 Sinusoidal undulations cause deformation in the planar membrane generating two types of GUVs. The larger ones (150 μm) contained a large amount of solvent while the smaller ones (3–20 μm) did not. The authors showed that transversal diffusion is inhibited by the presence of n-decane in the vesicular bilayer of the larger GUVs while the smaller ones efficiently mimic the transversal diffusion found in apoptotic cells.
Mixtures of lipids are frequently used to generate liposomes more physiologically relevant to model membranes.114,179–182 These mixed lipid membranes can phase separate, due to differences in the melting temperature of the lipids, their aliphatic chains (>2 carbons) or headgroups.8,127 It is not well understood what dictates transversal versus lateral heterogeneity in lipid membranes. Moreover, beyond the nature of the lipids themselves, mixing of lipids is also greatly influenced by temperatures, impurities (even in trace amounts), lipid ratios and lipid compositions in each vesicle. An increasing number of model membranes using liposomes have shown that lipid domains can be formed when using the appropriate lipids and ratios.176,183–186 Because of its role in lipid rafts, cholesterol phase separation has also been studied extensively. For example, DOPC/DPPC (50/50) and DOPC/DSPC (50/50) (DSPC 1,2-distearoyl-sn-glycero-3-phosphocholine) GUVs (30 μm) both exhibited gel-fluid phase separation but by adding 10 mol% of cholesterol, the vesicles became homogeneous (domains disappeared) and with 20 mol% cholesterol, new fluid domains (1–20 μm) were formed (liquid ordered) which differ from the original domains (liquid disordered). For cholesterol ≥50 mol%, domains were no longer visualized by optical microscopy. The authors also showed that cholesterol affects lipid fluidity in a similar way for DPPC and DSPC and more strongly between cholesterol and SM. The driving force for lipid raft formation in the plasma membrane might be due to hydrogen bonding between SM and cholesterol, which was not observed with PC-cholesterol. The phase diagrams of ternary (and even quaternary) mixtures are now extensively studied on GUVs.187–189
Hybrid polymer-only vesicles are rarer.190–192 In the last decade, the group of Battaglia has specialized in studying the surface topology of LUVs using mixed polymers. For example, diverse ratios of PMPC25-b-PDPA70 mixed with PEO23-b-PDPA15 were analyzed regarding their domain formation in polymersomes (200 nm) and their internalization in cells.191 The authors showed that by mixing 25 mol% PEO-b-PDPA and 75 mol% PMPC-b-PDPA, PEO-rich domains become visible. As a result, the PMPC block shrinks which affects the overall cell uptake kinetics of the polymersomes. Fewer hybrid polymersomes are internalized than the pure PMPC polymersomes suggesting that PMPC chains have a higher affinity for cell membranes than the PEO chains. When using the opposite ratio (75 mol% PEO-b-PDPA and 25 mol% PMPC-b-PDPA), PMPC domains are formed within a PEO matrix and surprisingly its uptake kinetics is very similar to that of the pure PMPC polymersomes and faster uptake kinetics for larger polymersomes. Thus, the presence of domains and their size affect the cellular uptake of these polymersomes. Discher and coworkers reported that the PAA block of PAA-b-PBO can be cross-linked by calcium or copper chelators in hybrid polymersome GUVs (10–20 μm) (Fig. 19).190 When PAA-b-PBO was mixed in different ratios with fluorescently labelled PB-b-PEO in the presence of Ca2+, demixing from the neutral amphiphiles was observed in accordance with the PAA-b-PBO and PB-b-PEO ratios. The presence of the calcium chelator even allowed the thermodynamic stability of the domains.
 | ||
| Fig. 19 Lateral phase separation of PAA-b-PBO and PB-b-PEO with 25% (a), 50% (b) and 75% (c) of PAA-b-PBO. Scale bar 2 μm. Reprinted with permission from Springer Customer Service Centre GmbH: Springer Nature, D. A. Christian, A. Tian, W. G. Ellenbroek, I. Levental, K. Rajagopal, P. A. Janmey, A. J. Liu, T. Baumgart and D. E. Discher, Spotted vesicles, striped micelles and Janus assemblies induced by ligand binding, Nat. Mater., Copyright (2009).190 | ||
In terms of polymer–lipid hybrid vesicles, domain formation is frequent as the chemical incompatibilities between lipids and polymer are greater than those for their respective identical amphiphiles (compare Section IIe). Most hybrid vesicles are made from diblock, triblock, or grafted polymers with PDMS, PB, or PIB (polyisobutylene) as the hydrophobic block and PEO or PMOXA as the hydrophilic blocks. In terms of lipids, PE or PC is almost exclusively used.8 Whether the membrane is homogeneous, heterogeneous or completely separated is difficult to determine pre-emptively and is highly sensitive to conditions. Chemin et al. described PIB-b-PEO and DPPC to form a heterogeneous membrane at a narrow ratio of 20–28% of block copolymer when using PIB87-b-PEO17 and in contrast only homogeneous vesicles when using PIB37-b-PEO48 regardless of the lipid/polymer molar composition.127 Nam et al. also showed that when using POPC (>30 mol%) and PB46-b-PEO30 (>70 mol%) HLPs are obtained; POPC (35–65 mol%) and PB46-b-PEO30 (35–65 mol%) give no vesicles; and POPC (70–100 mol%) and PB46-b-PEO30 (0–30 mol%) give a mixture of liposomes and HLPs.71 Similar to lipid-only and polymer-only hybrid vesicles, domain formation in polymer-hybrid vesicles can also be controlled by external stimuli or membrane composition. Beales and coworkers could control domain formation and properties of PB-b-PEO and DLPC (1,2-dilauroyl-sn-glycero-3-phosphocholine), DCPC (1,2-dicetyl-sn-glycero-3-phosphocholine) or DPPC GUVs (20 μm) via thermally driven phase separation, inclusion of cholesterol or macromolecular additives (surfactants, enzymes, cyclodextrins).174 When cooled slowly, only a few large phospholipid-rich domains are formed, while fast cooling generates numerous small domains.
Domain instability in heterogeneous membranes may result in equilibration by fission into separate vesicles.72,127 However, this challenge is only observed for polymer–lipid hybrid vesicles presumably due to the much greater chemical differences between lipids and polymers than lipid–lipid or the few polymer–polymer vesicles that have been tested. The formation of stable domains in GUVs is however essential for protocells. Using PDMS-g-(PEO)2 and DPPC, Dao et al. described the formation of stable nanodomains in hybrid polymer/lipid SUVs (100 nm).177 Lipid rich or polymer rich domains co-existed in the same vesicles. Domain formation in lipid/polymer hybrid membranes was also obtained in GUVs. Unfortunately, in the case of GUVs fission of the vesicles was observed after a few hours resulting in pure liposomes and polymersomes as the most entropically stable phase of the amphiphile self-assembly.127 The different outcome between SUVs and GUVs suggests that the curvature of the membrane greatly affects domain stabilization and stable micro-domain formation in polymer–lipid GUVs remains a challenge.
Bassereau and her group worked extensively on the incorporation of transmembrane proteins such as Ca2+-ATPsynthase, the photoactivable proton pump bacteriorhodopsin (BR) or the voltage-dependent K+ channel, KvAP, into liposome GUVs.199–201 These advanced liposome GUVs (10–100 μm) were generated by the initial detergent-mediated reconstitution of solubilized membrane proteins into proteoliposome SUVs (100–200 nm). The SUVs were then fused by electroformation or gel-assisted swelling. The proteins could be oriented into the membrane by a transfer mechanism from the mixed micelles to the lipid membrane (more details of this method are given below). Thus the proteins retained their biological activity for example when using BR and valinomycin, another K+ selective carrier, large transmembrane pH gradients can be generated upon light stimulus (Fig. 20a and b).200 Internal pH variations could be measured based on the fluorescence of pyranine, a photosensitive dye. In the presence of BR alone, a positive transmembrane electrical potential is generated upon light stimulus, retro-inhibiting the proton pumping. The GUVs’ permeability to protons was low (Fig. 20c). Valinomycin compensates for the electrical potential by releasing K+ externally, allowing for the acidification of the vesicles.
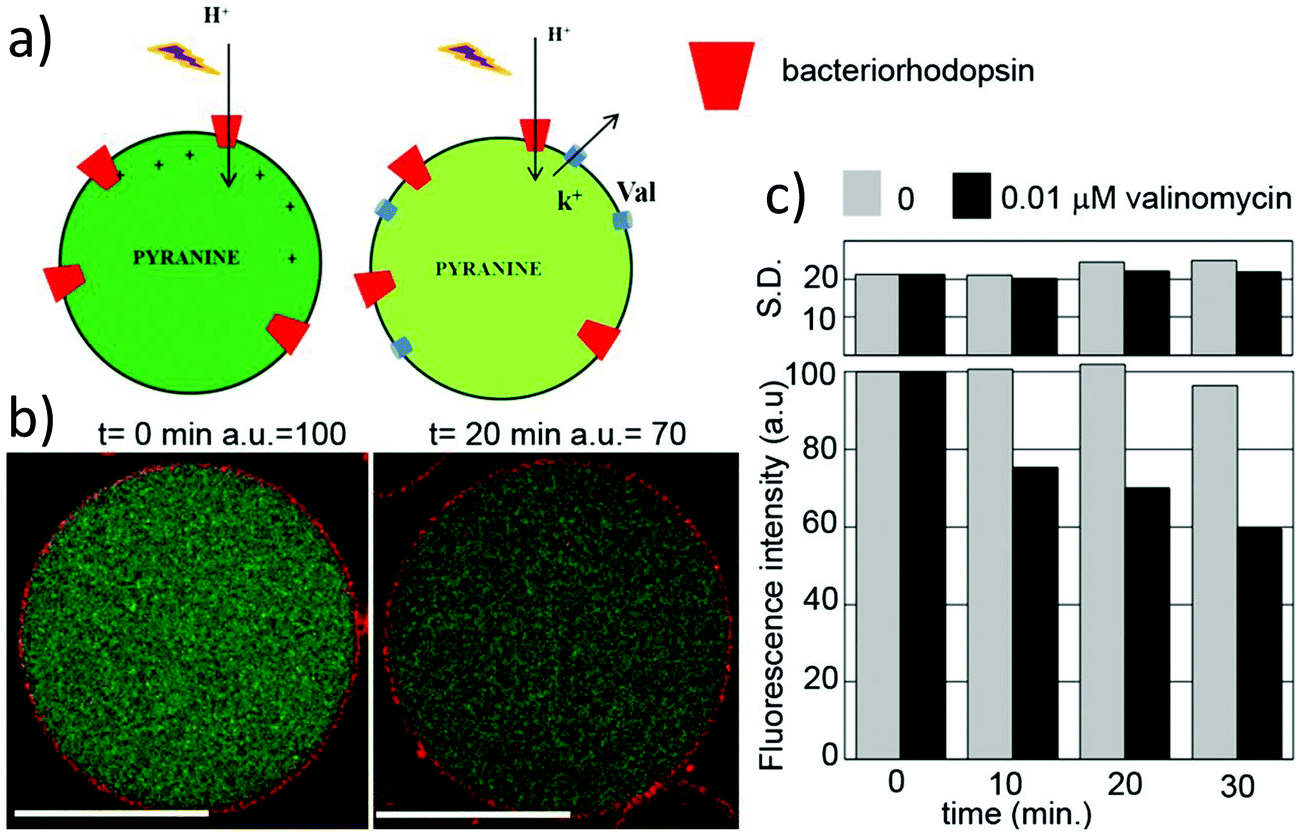 | ||
| Fig. 20 Incorporation of transmembrane proteins into liposome GUVs. (A and B) Light-induced proton pumping induces an internal acidification of GUVs. (C) Normalized pyranine fluorescence intensity in the presence or absence of valinomycin. Reprinted with permission from M. Dezi, A. Di Cicco, P. Bassereau and D. Lévy, Detergent-mediated incorporation of transmembrane proteins in giant unilamellar vesicles with controlled physiological contents. Proc. Natl. Acad. Sci. U. S. A., 2013, 110(18), 7276–7281.200 | ||
A major challenge of reconstitution of most transmembrane proteins into vesicles is their orientation.202,203 In 2017, Mavelli and coworkers reconstituted a photosynthetic reaction center (RC), an integral membrane protein, into liposome GUVs (20 μm) by droplet emulsion transfer.202 RC was initially extracted from purple bacteria using lauryldimethylamine N-oxide (LDAO) in aqueous solution generating micellar-stabilized RC. The RC-micelles were then emulsified in a lipid-rich oil phase yielding w/o droplets, which were subsequently transferred into an oil/water interphase forming the GUVs by centrifugation. The RC-micelles are thought to deliver their cargo during the w/o droplet phase driven by hydrophobic interactions. The major strength of this study comes from the asymmetry of the RC-micelles in terms of hydrophilicity/hydrophobicity, which guides the orientation of the membrane yielding 90% physiological orientation of the transmembrane proteins into the GUVs. The RC@lipid-GUVs were then used to generate a light-induced pH gradient across the membrane.
In terms of polymersomes, in 2016, Messager et al. described the use of membrane-spanning DNA nanopores NP-3C in PMPC25-b-PDPA72 polymersomes (100–200 nm).197 The NP-3C were fabricated through the self-assembly of oligonucleotides and the pore diameters could be customized. The polymersomes exhibited size-dependent permeability: substrates can be freely transported in and out of the polymersomes while larger enzymes were retained. As a proof of principle, the hydrolytic enzyme trypsin was encapsulated and the substrate peptide B-NAR-AMC (Boc-Gln-Ala-Arg-7-amido-4-methylcoumarin) was added outside the vesicle. Trypsin cleaved the substrate B-NAR-AMC to release the fluorescent product AMC that could then be quantified. In 2017, Castiglione and coworkers described the use of mutant Outer Membrane Protein F (OmpF G119D) channels in a PMOXA-b-PDMS-b-PMOXA nanoreactor (110 nm) used for an incompatible multi-enzyme cascade (Fig. 21).29 One enzyme (N-acyl-D-glucosamine2-epimerase (AGE)) and its allosteric activator ATP were encapsulated inside the polymersome while the other enzymes of the cascade (N-acetylneuraminate lyase (NAL) and CMP-sialic acid synthetase (CSS)) were immobilized on the outer surface of the vesicle as well as their respective cofactors (pyruvate and cytidine triphosphate (CTP)). The cascade is normally incompatible as AGE is strongly inhibited by CTP. The OmpF G119D had the advantage over the wildtype OmpF that the exchange of the glycine at position 119 to aspartate introduces a negative charge in the channel, increasing cation selectivity. As a result, selective massive transport of the neutrally charged substrates (N-acetylglucosamine (GlcNAc) and N-acetylmannosamine (ManNAc)) remained possible while the problematic negatively charged allosteric cofactors (CTP and ATP) were segregated outside and inside the polymersome respectively proving the value of compartmentalization. In 2018, from the same perspective Palivan and coworkers used extensively modified OmpF notably by adding cysteine residues to make the membrane channel redox-responsive.204 Impressively the author used these modified OmpF polymersome SUVs (100 nm) to generate artificial organelles that could be integrated in vitro in HeLa cells and in vivo in zebrafish embryos, exemplifying the potential of cell mimicking in medicine and materials science.
 | ||
| Fig. 21 Multi-enzyme cascade carried out in a polymersome nanoreactor. Reprinted with permission from L. Klermund, S. T. Poschenrieder and K. Castiglione, ACS Catal., 2017, 7(6), 3900–3904.29 Copyright (2017) American Chemical Society. https://pubs.acs.org/doi/abs/10.1021/acscatal.7b00776. | ||
In 2018, van Hest and coworkers described breathing polymersomes (100 nm) that could also be used to switch the membrane permeability on and off dependent on the pH.205 Upon intravesicular addition of acidic fuels (HCl and urea) and a substrate, the polymersomes expanded and horseradish peroxidase (HRP) could react with its substrate. Simultaneously, modification of urea into ammonia by urease effectively lowered the system's pH-value and thus switched off the permeability of the nanoreactor once a critical pH-value was reached. The systems lack transport selectivity compared to transmembrane proteins, but clearly demonstrated the feedback induced temporal control of the HRP's activity based on controllable permeability of the polymersome membranes.
In cells, small molecules are not exclusively transported via channels spanning the membrane. Many ions, molecules and even viruses or bacteria are selectively transported through the membrane by endocytosis. Endocytosis is an important mechanism of cells but due to its complexity it is not well understood. Landfester and coworkers described the uptake of PS (16 nm) or SiO2 nanoparticles (NPs) (14 to 57 nm) into PDMS-b-PMOXA polymersomes (∼100 nm) (Fig. 22).206,207 These systems allow us to understand transmembrane transport in general as a physical process in the absence of any protein or supplementary energy and also how the uptake of engineered nanoparticles can be optimized for biomedical applications. The NPs are encapsulated in vesicles in four steps (Fig. 22d): (1) recognition of the NPs at the vesicle surface, (2) engulfing initiation, (3) entire coverage of the NPs by the membrane, and (4) complete internalization. The internalization of these particles was proven to be faster for SiO2 particles (8 min) compared to PS nanoparticles (15 min), and to be strongly concentration and size dependent. Similarly, Meinel et al. studied the induced phagocytic uptake of PS particles (1 μm) in egg-PC liposome GUVs (20 μm).208 In order to engulf particles, the GUV needs to be significantly deformed and this was induced by optical tweezers. The optical tweezers has the advantage of applying controlled 3D force (femto-Newton) without mechanical contact.
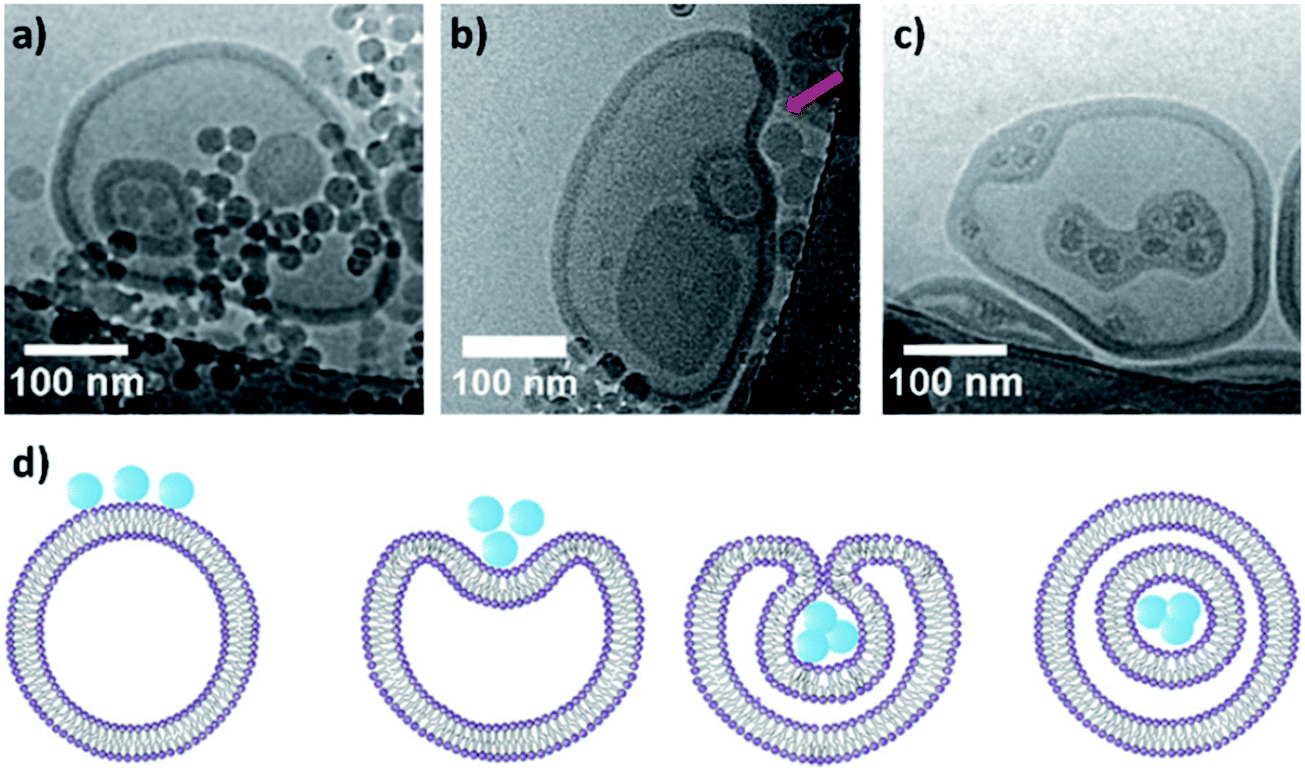 | ||
| Fig. 22 Cryo-TEM micrographs of the encapsulation of PS and SiO2 nanoparticles into polymersomes. Reprinted with permission from Jaskiewicz, A. Larsen, I. Lieberwirth, K. Koynov, W. Meier, G. Fytas, A. Kroeger and K. Landfester, Angew. Chem., Int. Ed., 2012, 51, 4613–4617. Copyright (2012) Wiley.206 | ||
Terminally-modified liposomes for recognition or docking are rare as difficult to modify. Thus liposomes can be decorated by incorporating surface proteins, like MHC, in a similar fashion by which transmembrane transporters are added to the membrane or by PEGylation as discussed in Section IV but has always been done from a medical perspective. For block copolymers, the addition of small moieties at their terminal end has a small impact on their self-assembly properties compared to liposomes. Commonly, compatible click moieties (azide/alkyne or maleimide/thiol), or Michael addition partners (carboxyl and amines) are often implemented on the terminal position of the block copolymers to add commercially available entities pre or post self-assembly to the polymersomes. Biotin is a commonly used ligand for biological functionalization due to its high binding affinity for the proteins streptavidin and avidin. In 2018, Landfester and coworkers used acrylate functionalized PB-b-PEO GUVs (20 μm) to conjugate amine-functionalized biotin post-assembly.106 These biotinylated GUVs were used to mimic the adhesive properties of cells by characterizing the interaction of polymersomes with neutravidin-coated glass surfaces. Similarly, Broz et al. showed that biotin-functionalized PMOXA-b-PDMS-b-PMOXA polymersomes (100–250 nm) would bind to streptavidin and oligonucleotide polyguanylic acid (polyG), which specifically targets the macrophage A1 scavenger receptor and thus could be used for diagnostic or therapeutic use.227 Kubilis et al. described glycopolymer (p(NβGluEAM5-b-BA50)) protocells (20 μm) capable of interacting with lectin-functionalized particles.97 Carbohydrate–lectin interaction is a common means of recognition between extracellular ligands and cells and notably initiates processes such as inflammatory cascade, fertilization, cancer cell metastasis and virus docking. The glycol-GUVs were shown to bind to glucose-specific lectin Concanavalin A (Con A)-functionalized polystyrene beads and not carboxylate-modified PS beads. The authors made the parallel of this specific membrane recognition to the binding of virus to mammalian cells for subsequent uptake and infection.
(b) Compartmentalized processes
We have just discussed how the membranes of vesicles themselves have been modified to mimic the cell plasma membrane. The simplest membrane can also be used to encapsulate processes with increasing complexity just like living cells. Compartmentalization of modules allows protection from the extra-vesicular environment, especially when dealing with incompatible processes, concentration of diverse functionality in a single entity rather than in bulk or diluted in solution, and is the basis of all cellular processes (metabolism, division, growth, communication, motility etc.).90,107,154In 2014, Peters et al. developed a fully synthetic polymersomes-in-polymersome multi-compartmentalized system mimicking cell hierarchical construct (organelles in cells).229 The authors chose a cascade reaction involving the initial oxidation of non-fluorescent substrate 1 by the phenylacetone monooxygenase (PAMO) and its NADPH cofactor followed by ester hydrolysis by Candida antarctica lipase B (CalB) or alcalase yielding an alcohol (Fig. 23). The alcohol is then oxidized to an aldehyde using alcohol dehydrogenase (ADH) and NAD+ and undergoes spontaneous β-elimination to yield fluorescent resorufin 5. Alcalase is sometimes used as a substitute for CalB because this protease is incompatible with other enzymes and a successful reaction cascade proves the importance of compartmentalization. In order to use this cascade to mimic a cell, CaIB/alcalase and ADH were encapsulated independently in PS40-b-PIAT50 (PIAT: poly(3-(isocyano-L-alanyl-aminoethyl)thiophene)) polymersomes (187 nm and 318 nm respectively). The CalB/alcalase nanoreactors and ADH nanoreactors were then encapsulated in a large PB-b-PEO polymersome (20–60 μm) along with substrate 1, PAMO and NAD+. The cascade conversion was observed by confocal fluorescence microscopy by following the quantitative increase of fluorescence over time. The fluorescence was confined to inner compartments as ADH interacts electrostatically with 5 preventing its diffusion out of the nanoreactors. Similarly, Voit and co-workers also studied enzymatic cascade reactions in pH-switchable adamantyl-functionalized polymersomes as artificial organelles into large polymersomes (1 μm) possessing temperature- and pH-responsiveness and PEG surface functionalization.231
 | ||
| Fig. 23 Reaction cascade performed in a complex multi-hierarchical protocell. Reprinted with permission from R. J. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150. Copyright (2013) Wiley.229 | ||
Another example of multi-compartmentalization was demonstrated in 2017 by generating hybrid liposomes-in-polymersomes.146 POPC, DMPC, diC15-PC (1,2-dipentadecanoyl-sn-glycero-3-phosphocholine), or DPPC SUVs (<400 nm) were first prepared by film hydration and subsequently used as the inner emulsion droplet for the emulsion–centrifugation method to form PB-b-PEO GUVs (20 μm). The loading and co-loading of the different liposomes in polymersome GUVs was established. Moreover because lipids have a different Tm, hydrophilic cargo can selectively be released from SUVs when the liposomes are subjected to temperature above their Tm. The temperature-controlled release of first methylene blue from diC15-PC SUVs at 37 °C and then fluorescein from DPPC SUVs at 45 °C into the polymersome GUV lumen was demonstrated, exemplifying the significant progress made in recent years to build ever-more complex systems with different ‘organelles’ within a single GUV.
As a matter of fact, artificial organelles have even been implemented in living cells, going beyond the generation of protocells for medical purposes.233 In this study for example, the authors generate liposomes/fluorescent gold nanoclusters in a polymer shell (capsosomes, 2–3 μm) which are readily taken up by macrophages. The capsosomes are composed of both glucose oxidase (GOx) liposomes and HRP liposomes effectively converting glucose into the fluorescent resorufin, retaining its activity within the macrophages. In contrast, Elani et al. attempted to bridge the gap between artificial protocells and biological cells by building living-cells-in-liposomes.235 By microfluidics, a variety of human cells as well as E. coli could be encapsulated into POPC GUVs (70 μm). A three step cascade converting lactose into fluorescent resorufin was subsequently demonstrated by coupling the cellular pathways with non-cellular free enzymes. Furthermore, the liposomes were also demonstrated to provide an effective protective barrier for the living cells against toxins such as Cu2+ thus demonstrating the advancement artificial systems can bring to living cells.
In 2005, Choi and Montemagno designed one of the first biomimetic polymersomes: PEtOz-b-PDMS-b-PEtOz (PEtOz: poly(2-ethyl-2-oxazo-line)) SUVs (<200 nm) capable of generating ATP by coupling the activity of photoactive BR and F0F1-ATP synthase (Fig. 24a).238 BR is a light-driven proton pump, which creates a proton gradient across the polymersome. This gradient is then used by the ATP-synthase to synthesize ATP. Montemagno's model proved that polymersomes can be used for biosynthesis through the coupled effect of two transmembrane proteins in a single vesicle and thus that a fundamental cellular module can be recreated in a complete synthetic approach towards the generation of functional synthetic organelles.
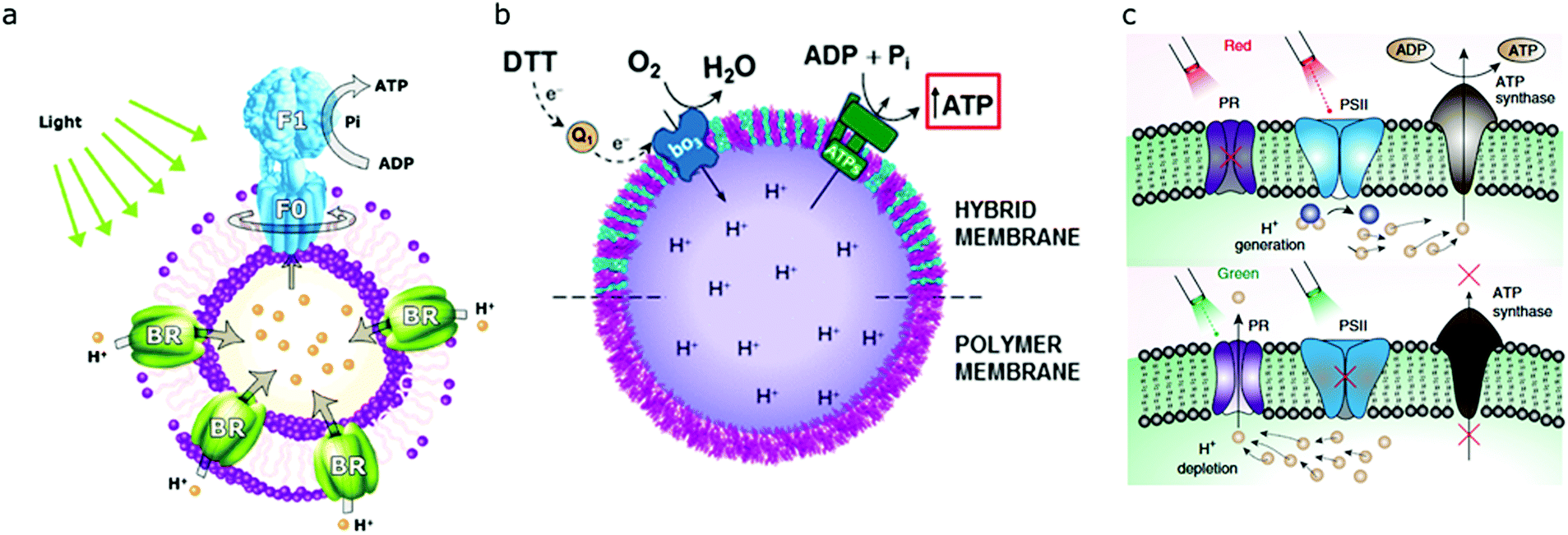 | ||
| Fig. 24 ATP-synthase based proteovesicles as cellular mimics for the generation of ATP using an electrochemical proton gradient. (a) Bacteriorhodopsin (BR) and ATP-synthase reconstituted in PEtOz-b-PDMS-b-PEtOz polymersomes. Adapted with permission from H. J. Choi and C. D. Montemagno, Nano Lett., 2005, 5, 2538–2542. Copyright (2005) American Chemical Society.238 (b) Cytochrome bo3 quinol oxidase (Bo3) and ATP synthase reconstituted in PDMS-g-PEO polymersomes and PDMS-g-PEO/lipid hybrid vesicles. Reprinted (adapted) with permission from L. Otrin, N. Marusic, C. Bednarz, T. Vidakovic-Koch, I. Lieberwirt, K. Landfester and K. Sundmacher, Nano Lett., 2017, 17, 6816–6821. Copyright (2017) American Chemical Society.13 (c) Complementary photoconverters (PSII and PR) and an ATP-synthase reconstituted in lipid SUVs inside lipid GUVs. Reprinted by permission from Springer Customer Service Centre GmbH: Springer Nature, K. Y. Lee, S. J. Park, K. A. Lee, S. H. Kim, H. Kim, Y. Meroz, L. Mahadevan, K. H. Jung, T. K. Ahn, K. K. Parker and K. Shin, Photosynthetic artificial organelles sustain and control ATP-dependent reactions in a protocellular system, Nat. Biotechnol., Copyright (2018).236 | ||
In 2017, from the same perspective, Sundmacher and coworkers detailed the generation of artificial mitochondria by coupling ATP-synthase with cytochrome bo3 quinol oxidase into PDMS-g-PEO and hybrid PDMS-g-PEO/POPC or soy PC SUVs (100 nm) and GUVs (50 μm) (Fig. 24b).13 Like BR, bo3 acts as a proton pump by oxidizing ubiquinol and generating water from O2, driving the production of ATP into the outer media. PDMS-g-PEO was described as particularly advantageous over other polymers as generating only 5 nm wide membranes and also having a fluidity comparable to liposomes, contrary to other commonly used polymers, accounting for the retained bo3's activity in the membrane.
In 2018, Lee et al. designed a switchable photosynthetic liposome SUVs (100 nm) in liposome GUVs (10–100 μm) as a superb advancement towards artificial cells (multi-compartmentalization, ATP-regeneration and cytoskeleton formation).236 The authors used two photoconverters, a plant-derived photosystem II (PSII) and a bacteria-derived proteorhodopsin (PR), to control the transmembrane proton gradients in the SUVs and thus ATP generation from ADP by ATP-synthase as artificial mitochondria (Fig. 24c). Red light activates PSII's electron-transports chains in the SUVs facilitating ATP synthesis in the GUV lumen while green light activates direct proton pumping by PR, impeding ATP synthesis.
The cytoskeleton has many functions, amongst which the most important ones are cell shape and support, endocytosis, mobility and division, and thus has been gathering extensive focus in cell mimicking.113,236,253–261 In the study by Lee et al. described above, the photo-switchable generation of ATP was coupled to ATP-dependent actin polymerization leading to morphological changes in the lipid GUV membrane.236 This process was achieved by embedding magnesium ionophores in the GUV membrane and G-actin in the lumen. Upon ATP synthesis, G-actin nucleation was triggered in the presence of Mg2+ and formed actin filaments. The growth of these filaments led to the formation of F-actin spheres which deformed the membrane eventually rupturing the vesicles. The membrane deformation by the polymerization of actin could also be controlled by forming liposomes with liquid-disordered (Ld) domains composed of polyunsaturated phospholipids which established favorable interactions with actin and liquid-ordered (Lo) domains composed of sphingomyelin, cholesterol and polysaturated-PEG2000-PE weakly interacting with actin. The Ld domain's strong attraction between Ld and actin filaments locally deformed the membrane into teardrops (Fig. 25). Weiss et al. used lipid/polymer droplet-stabilized GUVs (dsGUVs) as a protocellular platform.113 The reconstitution of actin filaments and microtubules was achieved by sequential pico-injection of G-actin or tubulin. The authors showed the importance of pico-injection over pre-mixing in the vesicles as the encapsulation of microtubules inhibited the formation of the dsGUVs.
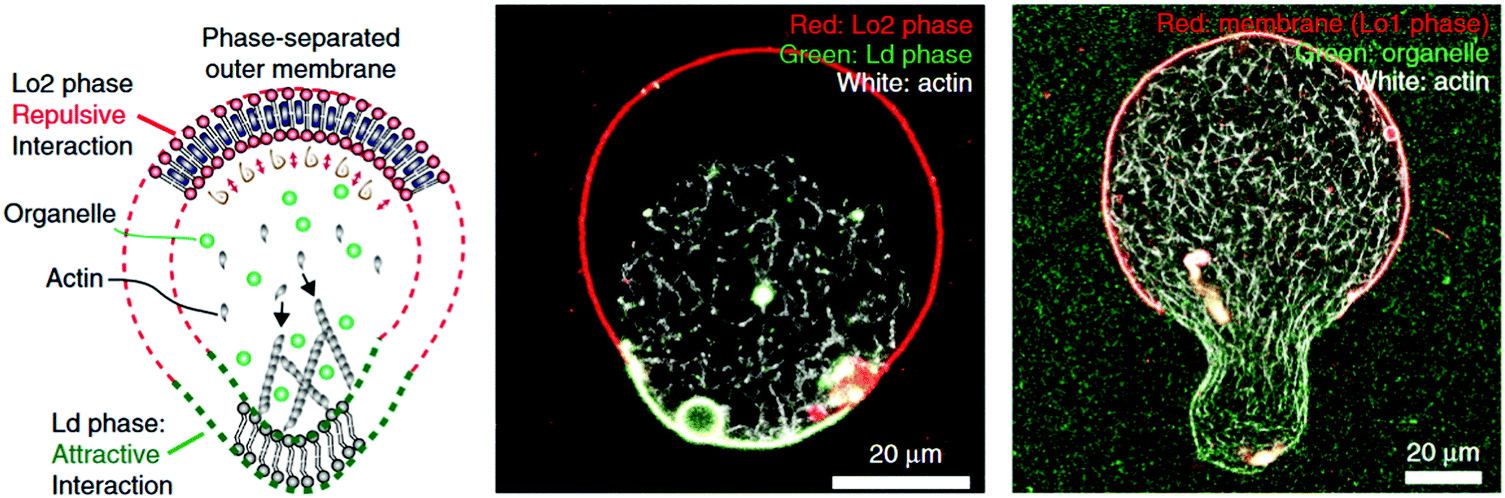 | ||
| Fig. 25 Control of actin polymerization and membrane shape of the protocellular systems. Liquid-disordered domain (Ld); liquid-ordered domains (Lo1 or Lo2). Reprinted with permission from Springer Customer Service Centre GmbH: Springer Nature, K. Y. Lee, S. J. Park, K. A. Lee, S. H. Kim, H. Kim, Y. Meroz, L. Mahadevan, K. H. Jung, T. K. Ahn, K. K. Parker and K. Shin, Photosynthetic artificial organelles sustain and control ATP-dependent reactions in a protocellular system, Nat. Biotechnol., Copyright (2018).236 | ||
As a very elegant mimic of a cell, Martino et al. reproduced the cell-free expression of bacterial proteins within polymersomes.243 The GUVs (126 μm) were synthesized from PEO-b-PLA (PLA: poly(lactic acid)) and a PLA homopolymer which was used to strengthen the membrane using a microfluidic capillary device (Fig. 26a). The polymersome encapsulated an E. coli ribosomal expression kit with the DNA plasmid of a MreB fusion protein with red fluorescent protein (MreB-RFP). Assessing protein expression was achieved by monitoring the resulting increasing fluorescence. The production of protein is a pillar in cell mimicry but cells also deliver their expressed protein to the extracellular environment. Thus, there is also interest in triggered-release. Using negative osmotic shock, pores were formed in the membrane and thus the polymersomes were rendered semi-permeable allowing the triggered release of the newly expressed proteins while retaining the polymersomes by subsequent self-sealing (Fig. 26b). The light-induced osmotic shock release of SUV liposomes and polymersomes (100 nm) from PB-b-PEO GUVs (10 μm) has also been described.251
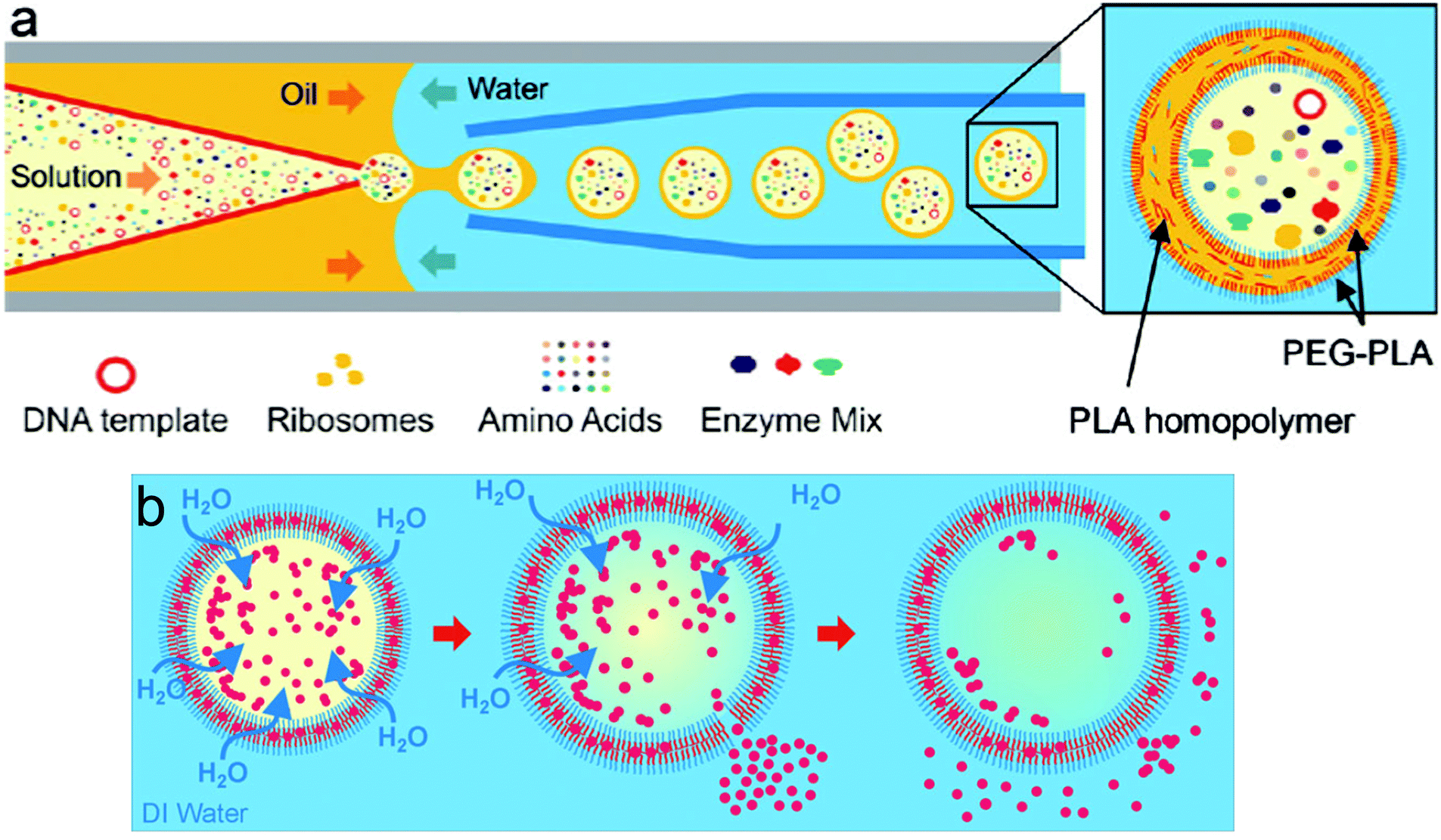 | ||
| Fig. 26 Protein expressing polymersome GUVs generated by microfluidics. Schematic representation of (a) the methodology used to generate these artificial cells and (b) protein release under osmotic shock and subsequent self-sealing. PEG: poly(ethylene glycol). PLA: poly(lactic acid). Reprinted with permission from C. Martino, S. H. Kim, L. Horsfall, A. Abbaspourrad, S. J. Rosser, J. Cooper and D. A. Weitz, Angew. Chem., Int. Ed., 2012, 51, 6416–6420. Copyright (2012) Wiley.243 | ||
Most artificial cell studies focus on the basic processes common to all cells. However, because cells are specialized (blood cells, neurons, skin cells etc.), certain functionalities are specific to a type of cell.262 Red blood cells are of particular interest because they can also be used in medicine as specialized oxygen carriers. Multiple studies using vesicles mimicking red blood cells by encapsulating hemoglobin (Hb) have been described.263–266 Encapsulating hemoglobin does not affect its oxygen binding properties and O2 off-loading can be regulated by membrane thickness of appropriately selected block copolymers to obtain a moderate O2 release and avoid side effects. Arifin and Palmer demonstrated that PB-b-PEO (250 nm) encapsulating bovine Hb exhibited oxygen affinities comparable to human erythrocytes consistent with values required for efficient oxygen delivery in the systemic circulation and unlike liposomes composed of natural lipids, these polymersomes did not induce Hb oxidation.263 However, this example and many others exclusively involved SUVs. Najer et al. recently developed red blood cell (RBC) membrane mimics as a potential cure for malaria.266 During infection, malaria parasites enter RBCs by binding to heparan sulfate in order to reproduce. By using polymersomes expressing RBC receptor-like molecules on their outer layer, the parasites would bind to the polymersome and hence their reproduction cycle would be inhibited. PMOXA5-b-PDMS58-b-PMOXA5/PDMS65-b-heparin12 hybrid polymersomes (7 μm) were shown to specifically bind to the parasite ligand and even interact with the viable parasite.
VI. Conclusions
Vesicles are interesting carriers as offering a great variety in terms of size, permeability, fluidity, methods of preparation etc. and thus have been used in an array of applications ranging from nanoreactors to cell mimicry via FDA-approved medicines. Liposomes and polymersomes present many different properties. Liposomes are closer mimics to eukaryotic cell membranes but are difficult to handle and use whereas polymersomes are tougher, malleable, stable vesicles. Nonetheless stability does not always rhyme with cell mimicry as fluidity is a key parameter of cell membranes. Thus the silver-lining to design accomplished artificial cells lies in controllability. However, despite the arsenal of polymer chemistry, polymersome studies, especially polymeric GUVs, are so far mostly restricted to commercially available amphiphilic polymers in a similar way that liposomes are only self-assembled from natural phospholipids. Regardless of their properties, liposomes and polymersomes have both been extensively used to build artificial cells. Recent years have seen a wide range of cell processes and functionalities implemented in these synthetic membranes, which are just starting to be combined into state-of-the-art vesicles slowly building up to generating protocells. Lipid/polymer hybrid vesicles have gained interest as a good compromise between liposomes and polymersomes for a greater control and adaptability of physicochemical properties to any desired functionality and applications, optimal for cell mimicry. As polymersomes have shown great potential, we predict that the era of polymersomes, in parallel with hybrid vesicles, has yet to come when more interdisciplinary works are established to allow the full versatility of polymers’ elaborate chemistries to be used in the field of cell mimicry.Abbreviations
| ADH | Alcohol dehydrogenase |
| ADP | Adenosine diphosphate |
| AGE | N-Acyl-D-glucosamine2-epimerase |
| AMC | 7-Amido-4-methylcoumarin |
| anti-EGFR | Anti-epidermal growth factor receptor |
| ATP | Adenosine triphosphate |
| B | Block |
| B-NAR-AMC | Boc-Gln-Ala-Arg-7-amido-4-methylcoumarin |
| bo3 | Cytochrome bo3 quinol oxidase |
| BR | Bacteriorhodopsin |
| CalB | Candida antarctica lipase B |
| Con A | Concanavalin A |
| CSS | CMP-sialic acid synthetase |
| CTAs | Chain-transfer agents |
| CTP | Cytidine triphosphate |
| DC-FCCS | Dual-color fluorescence cross-correlation spectroscopy |
| DCPC | 1,2-Dicetyl-sn-glycero-3-phosphocholine (lipid) |
| DEX | Dextran (hydrophilic block) |
| diC15-PC | 1,2-Dipentadecanoyl-sn-glycero-3-phosphocholine (lipid) |
| DLPC | 1,2-Dilauroyl-sn-glycero-3-phosphocholine (lipid) |
| DMPC | 1,2-Dimyristoyl-sn-glycero-3-phosphocholine (lipid) |
| DMPE | 1,2-Dimyristoyl-sn-glycero-3-phosphoethanolamine (lipid) |
| DMPG | 1,2-Dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (lipid) |
| DMPS | 1,2-Dimyristoyl-sn-glycero-3-phospho-L-serine (lipid) |
| DOPC | 1,2-Dioleoyl-sn-glycero-3-phosphocholine (lipid) |
| DOPE | 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (lipid) |
| DOPG | 1,2-Di-(9Z-octadecenoyl)-sn-glycero-3-phospho-(1′-rac-glycerol) (lipid) |
| DOPS | 1,2-Dioleoyl-sn-glycero-3-phosphoserine (lipid) |
| DP | Degree of polymerization |
| DPPC | 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (lipid) |
| DPPE | 1,2-Dipalmitoyl-sn-glycero-3-phosphoethanolamine (lipid) |
| DPPG | 1,2-Dipalmitoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (lipid) |
| DPPS | 1,2-Dipalmitoyl-sn-glycero-3-phospho-L-serine (lipid) |
| dsDNA | Double-stranded DNA |
| dsGUVs | Droplet-stabilized GUVs |
| DSPC | 1,2-Distearoyl-sn-glycero-3-phosphocholine (lipid) |
| DSPG | 1,2-Distearoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (lipid) |
| eGFP | Enhanced green fluorescent protein |
| Egg PC | L-α-Phosphatidylcholine (egg, chicken) (lipid) |
| Egg SM | Sphingomyelin (egg, chicken) (lipid) |
| F | Volume fractions |
| F-Actin | Filamentous actin |
| FDA | Food and drug administration |
| FRET | Förster resonance energy transfer |
| f w | Hydrophilic weight fraction |
| G | Grafted |
| G-actin | Globular actin |
| GlcNAc | N-Acetylglucosamine |
| GOx | Glucose oxidase |
| GUV | Giant unilamellar vesicle |
| Hb | Hemoglobin |
| HLP | Hybrid liposome–polymersome |
| HPMA | 2-Hydroxypropyl methacrylate |
| HRP | Horseradish peroxidase |
| HUV | Unilamellar hybrid vesicles |
| Hydro Soy PC | L-α-Phosphatidylcholine, hydrogenated (Soy) (lipid) |
| ITO | Indium titanium oxide |
| KvAP | Voltage-dependent K+ channel from Aeropyrum Pernix |
| LCST | Lower critical solution temperature |
| Ld | Liquid-disordered |
| LDAO | Lauryldimethylamine N-oxide |
| Liss Rhod | 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine-N-Lissamine rhodamine B sulfonyl (lipid) |
| Lo | Liquid-ordered |
| LUV | Large unilamellar vesicle |
| lyso-PC | Lysophosphatidylcholines (lipid) |
| ManNAc | N-Acetylmannosamine |
| MHC | Major histocompatibility complex |
| MLV | Multilamellar vesicles |
| M n | Number average molecular weight |
| MSPC | (1-Myristoyl-2-stearoyl-sn-glycero-3-phosphocholine) (lipid) |
| MVV | Multivesicular vesicles |
| MW | Molecular weight |
| N | Degree of polymerization |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADPH | Reduced nicotinamide adenine dinucleotide phosphate |
| NAL | N-Acetylneuraminate lyase |
| NP-3C | DNA nanopore |
| NPs | Nanoparticles |
| o/w | Oil-in-water |
| OmpF | Outer membrane protein F |
| P2VP | Poly(2-vinylpyridine) (hydrophilic block) |
| P4VP | Poly(4-vinyl pyridine) (hydrophilic block) |
| PAA | Polyacrylic acid (hydrophilic block) |
| PALM | Photo-activated localization microscopy |
| Palmitoyl-SM | N-Palmitoyl-D-erythro-sphingosylphosphorylcholine (lipid) |
| PAMO | Phenylacetone monooxygenase |
| PAzoM | Poly{6-[4-(4-methylphenylazo)-phenoxy]hexyl acrylate} (responsive block) |
| PBA | Poly(butyl acrylate) (hydrophobic block) |
| PBD or PB | Polybutadiene (hydrophobic block) |
| PC | Phosphatidylcholine (lipid) |
| P c | Critical packing parameter |
| PCL | Polycaprolactone (hydrophobic block) |
| PDA | Polydiacetylenes (amphiphilic block) |
| PDEA | Poly(2-diethylaminoethyl methacrylate) (responsive block) |
| PDLLA | Poly(D,L-lactide) (hydrophobic block) |
| PDMS | Polydimethylsiloxane (hydrophobic block) |
| PDPA | Poly(2-(diisopropylamino)ethyl methacrylate) (responsive block) |
| PE | Phosphatidylethanolamine (lipid) |
| PEE | Poly(ethyl ethylene) (hydrophobic block) |
| PEO or PEG | Poly(ethylene oxide) or Poly(ethylene glycol) (hydrophilic block) |
| PEtOz | Poly(2-ethyl-2-oxazo-line) (hydrophilic block) |
| PG | Phosphatidylglycerol (lipid) |
| PGA | Poly(glycolic acid) (hydrophobic block) |
| PI | Phosphatidylinositol (lipid) |
| PIAT | Poly(3-(isocyano-L-alanyl-aminoethyl)thiophene) (hydrophilic block) |
| PIB | Polyisobutylene (hydrophobic block) |
| PISA | Polymerization-induced self-assembly |
| PLA | Poly(lactic acid) (hydrophobic block) |
| PLys | Poly-L-lysine (responsive block) |
| PMASH | Thiol-functionalized poly(methacrylic acid) |
| PMMA | Poly(methyl methacrylate) (hydrophobic block) |
| PMOXA | Poly-2-methyl-2-oxazoline (hydrophilic block) |
| PMPC | Poly(2-(methacryloyloxy)ethyl phosphorylcholine) (hydrophilic block) |
| PNIPAM | Poly(N-isopropylacrylamide) (responsive block) |
| polyG | Polyguanylic acid (hydrophilic block) |
| POPC | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (lipid) |
| POPS | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (lipid) |
| PPE | Poly(phosphonate) (responsive block) |
| PPS | Polyphenylene sulfide (responsive block) |
| PR | Proteorhodopsin |
| PS | Polystyrene (hydrophobic block) |
| PSe | Phosphatidylserine (lipid) |
| PSII | Plant-derived photosystem II |
| PVA | Poly(vinyl alcohol) |
| PVP | Poly(N-vinylpyrrolidone) |
| RAFT | Reversible addition fragmentation transfer |
| RBC | Red blood cell |
| RC | Reaction center |
| ROS | Reactive oxygen species |
| SIM | Structured illumination microscopy |
| SM | Sphingomyelin/Sphingosylphosphorylcholine (lipid) |
| SNOM | Scanning near-field optical microscopy |
| SOPC | 1-Stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (lipid) |
| Soy PC | L-α-Phosphatidylcholine (Soy) (lipid) |
| STED | Stimulated emission depletion microscopy |
| SUV | Small unilamellar vesicle |
| TEM | Transmission electron microscopy |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| T g | Glass-transition temperature |
| T m | Gel-liquid crystal transition temperature/Melting temperature |
| UCST | Upper critical solution temperature |
| UV | Ultraviolet |
| w/o | Water-in-oil |
| w/o/w | Water-in-oil-in-water |
| X | Flory–Huggins parameter |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the MaxSynBio consortium which is jointly funded by the Federal Ministry of Education and Research of Germany and the Max Planck Society. Open Access funding provided by the Max Planck Society.References
- X. Y. Zhang, P. Tanner, A. Graff, C. G. Palivan and W. Meier, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2293–2318 CrossRef.
- R. Chandrawati and F. Caruso, Langmuir, 2012, 28, 13798–13807 CrossRef PubMed.
- M. Li, X. Huang, T. Y. D. Tang and S. Mann, Curr. Opin. Chem. Biol., 2014, 22, 1–11 CrossRef PubMed.
- M. C. M. van Oers, F. P. J. T. Rutjes and J. C. M. van Hest, Curr. Opin. Biotechnol., 2014, 28, 10–16 CrossRef PubMed.
- A. I. Lamond, Nature, 2002, 417, 383 CrossRef.
- F. Szoka and D. Papahadjopoulos, Annu. Rev. Biophys. Bioeng., 1980, 9, 467–508 CrossRef PubMed.
- J. Li, X. L. Wang, T. Zhang, C. L. Wang, Z. J. Huang, X. Luo and Y. H. Deng, Asian J. Pharm. Sci., 2015, 10, 81–98 CrossRef.
- J. F. Le Meins, C. Schatz, S. Lecommandoux and O. Sandre, Mater. Today, 2013, 16, 397–402 CrossRef.
- A. Jesorka and O. Orwar, Annu. Rev. Anal. Chem., 2008, 1, 801–832 CrossRef PubMed.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576–3590 RSC.
- S. Winzen, M. Bernhardt, D. Schaeffel, A. Koch, M. Kappl, K. Koynov, K. Landfester and A. Kroeger, Soft Matter, 2013, 9, 5883–5890 RSC.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef PubMed.
- L. Otrin, N. Marusic, C. Bednarz, T. Vidakovic-Koch, I. Lieberwirt, K. Landfester and K. Sundmacher, Nano Lett., 2017, 17, 6816–6821 CrossRef PubMed.
- M. Schulz and W. H. Binder, Macromol. Rapid Commun., 2015, 36, 2031–2041 CrossRef PubMed.
- K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540–3563 CrossRef.
- V. Malinova, S. Belegrinou, D. D. Ouboter and W. P. Meier, Adv. Polym. Sci., 2010, 224, 113–165 Search PubMed.
- M. H. Ross and W. Pawlina, Histology, Lippincott Williams & Wilkins, 2006 Search PubMed.
- R. Milo and R. Phillips, Cell Biology by the Numbers, Taylor & Francis Group, 2015 Search PubMed.
- M. L. Immordino, F. Dosio and L. Cattel, Int. J. Nanomed., 2006, 1, 297–315 CrossRef PubMed.
- S. A. Meeuwissen, S. M. C. Bruekers, Y. C. Chen, D. J. Pochan and J. C. M. van Hest, Polym. Chem., 2014, 5, 489–501 RSC.
- J. Petit, I. Polenz, J. C. Baret, S. Herminghaus and O. Baumchen, Eur. Phys. J. E: Soft Matter Biol. Phys., 2016, 39, 59 CrossRef PubMed.
- S. Allen, O. Osorio, Y.-G. Liu and E. Scott, J. Controlled Release, 2017, 262, 91–103 CrossRef PubMed.
- K. Oglęcka, J. Sanborn, A. N. Parikh and R. S. Kraut, Front. Physiol., 2012, 3, 120 Search PubMed.
- H. L. Che and J. C. M. van Hest, J. Mater. Chem. B, 2016, 4, 4632–4647 RSC.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102 CrossRef PubMed.
- J. F. Le Meins, O. Sandre and S. Lecommandoux, Eur. Phys. J. E: Soft Matter Biol. Phys., 2011, 34, 14 CrossRef PubMed.
- A. Napoli, M. Valentini, N. Tirelli, M. Muller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef PubMed.
- L. Messager, J. Gaitzsch, L. Chierico and G. Battaglia, Curr. Opin. Pharmacol., 2014, 18, 104–111 CrossRef PubMed.
- L. Klermund, S. T. Poschenrieder and K. Castiglione, ACS Catal., 2017, 7, 3900–3904 CrossRef.
- S. Vemuri and C. T. Rhodes, Pharm. Acta Helv., 1995, 70, 95–111 CrossRef PubMed.
- S. T. Poschenrieder, S. K. Schiebel and K. Castiglione, Eng. Life Sci., 2018, 18, 101–113 CrossRef.
- F. H. Meng, C. Hiemstra, G. H. M. Engbers and J. Feijen, Macromolecules, 2003, 36, 3004–3006 CrossRef.
- N. P. Kamat, M. H. Lee, D. Lee and D. A. Hammer, Soft Matter, 2011, 7, 9863–9866 RSC.
- C. Sanson, C. Schatz, J. F. Le Meins, A. Soum, J. Thevenot, E. Garanger and S. Lecommandoux, J. Controlled Release, 2010, 147, 428–435 CrossRef PubMed.
- E. V. Konishcheva, U. E. Zhumaev and W. P. Meier, Macromolecules, 2017, 50, 1512–1520 CrossRef.
- R. Dimova, S. Aranda, N. Bezlyepkina, V. Nikolov, K. A. Riske and R. Lipowsky, J. Phys.: Condens. Matter, 2006, 18, S1151–S1176 CrossRef PubMed.
- R. Dimova, in Advances in Planar Lipid Bilayers and Liposomes, ed. A. Iglič, Academic Press, 2012, vol. 16, pp. 1–50 Search PubMed.
- R. Dimova, Adv. Colloid Interface Sci., 2014, 208, 225–234 CrossRef PubMed.
- R. L. Knorr, M. Staykova, R. S. Gracia and R. Dimova, Soft Matter, 2010, 6, 1990–1996 RSC.
- D. Needham and R. M. Hochmuth, Biophys. J., 1989, 55, 1001–1009 CrossRef PubMed.
- K. Olbrich, W. Rawicz, D. Needham and E. Evans, Biophys. J., 2000, 79, 321–327 CrossRef PubMed.
- H. Aranda-Espinoza, H. Bermudez, F. S. Bates and D. E. Discher, Phys. Rev. Lett., 2001, 8720, 4 Search PubMed.
- R. Dimova, U. Seifert, B. Pouligny, S. Forster and H. G. Dobereiner, Eur. Phys. J. E: Soft Matter Biol. Phys., 2002, 7, 241–250 Search PubMed.
- R. Dimova, B. Pouligny and C. Dietrich, Biophys. J., 2000, 79, 340–356 CrossRef PubMed.
- U. Seifert and R. Lipowsky, in Structure and Dynamics of Membranes (Handbook of Biological Physics), ed. R. Lipowsky and E. Sackmann, Elsevier, Amsterdam, 1995, vol. 1a, pp. 403–463 Search PubMed.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef PubMed.
- W. Rawicz, K. C. Olbrich, T. McIntosh, D. Needham and E. Evans, Biophys. J., 2000, 79, 328–339 CrossRef PubMed.
- K. R. Mecke, T. Charitat and F. Graner, Langmuir, 2003, 19, 2080–2087 CrossRef.
- H. Bermudez, D. A. Hammer and D. E. Discher, Langmuir, 2004, 20, 540–543 CrossRef PubMed.
- D. Needham and E. Evans, Biochemistry, 1988, 27, 8261–8269 CrossRef PubMed.
- R. Dimova, C. Dietrich, A. Hadjiisky, K. Danov and B. Pouligny, Eur. Phys. J. B, 1999, 12, 589 CrossRef.
- E. Evans and D. Needham, J. Phys. Chem., 1987, 91, 4219–4228 CrossRef.
- H. Bermúdez, H. Aranda-Espinoza, D. A. Hammer and D. E. Discher, EPL, 2003, 64, 550 CrossRef.
- T. Portet and R. Dimova, Biophys. J., 2010, 99, 3264–3273 CrossRef PubMed.
- M. Watanabe, K. Kawano, K. Toma, Y. Hattori and Y. Maitani, J. Controlled Release, 2008, 127, 231–238 CrossRef PubMed.
- A. G. Kohli, P. H. Kierstead, V. J. Venditto, C. L. Walsh and F. C. Szoka, J. Controlled Release, 2014, 190, 274–287 CrossRef PubMed.
- A. M. Bayer, S. Alam, S. I. Mattern-Schain and M. D. Best, Chem. – Eur. J., 2014, 20, 3350–3357 CrossRef PubMed.
- M. N. Holme, I. A. Fedotenko, D. Abegg, J. Althaus, L. Babel, F. Favarger, R. Reiter, R. Tanasescu, P. L. Zaffalon, A. Ziegler, B. Muller, T. Saxer and A. Zumbuehl, Nat. Nanotechnol., 2012, 7, 536–543 CrossRef PubMed.
- R. Tanasescu, M. A. Lanz, D. Mueller, S. Tassler, T. Ishikawa, R. Reiter, G. Brezesinski and A. Zumbuehl, Langmuir, 2016, 32, 4896–4903 CrossRef PubMed.
- S. Matviykiv, M. Buscema, T. Meszaros, G. Gerganova, T. Pfohl, A. Zumbuhl, J. Szebeni and B. Muller, Proc. SPIE, 2017, 10162 Search PubMed.
- F. Neuhaus, D. Mueller, R. Tanasescu, S. Balog, T. Ishikawa, G. Brezesinski and A. Zumbuehl, Angew. Chem., Int. Ed., 2017, 56, 6515–6518 CrossRef PubMed.
- D. Marsh, CRC handbook of lipid bilayers, CRC Press, 1990 Search PubMed.
- H. Q. Yin, S. W. Kang and Y. H. Bae, Macromolecules, 2009, 42, 7456–7464 CrossRef.
- W. Y. Ayen, B. Chintankumar, J. P. Jain and N. Kumar, Polym. Adv. Technol., 2011, 22, 158–165 CrossRef.
- H. Y. Chang, Y. L. Lin, Y. J. Sheng and H. K. Tsao, Macromolecules, 2013, 46, 5644–5656 CrossRef.
- Y. Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- M. Dionzou, A. Morere, C. Roux, B. Lonetti, J. D. Marty, C. Mingotaud, P. Joseph, D. Goudouneche, B. Payre, M. Leonetti and A. F. Mingotaud, Soft Matter, 2016, 12, 2166–2176 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef PubMed.
- R. Chandrawati, L. Hosta-Rigau, D. Vanderstraaten, S. A. Lokuliyana, B. Stadler, F. Albericio and F. Caruso, ACS Nano, 2010, 4, 1351–1361 CrossRef PubMed.
- R. Chandrawati, M. P. van Koeverden, H. Lomas and F. Caruso, J. Phys. Chem. Lett., 2011, 2, 2639–2649 CrossRef.
- J. Nam, P. A. Beales and T. K. Vanderlick, Langmuir, 2011, 27, 1–6 CrossRef PubMed.
- T. P. T. Dao, F. Fernandes, E. Ibarboure, K. Ferji, M. Prieto, O. Sandre and J. F. Le Meins, Soft Matter, 2017, 13, 627–637 RSC.
- T. P. T. Dao, A. Brulet, F. Fernandes, M. Er-Rafik, K. Ferji, R. Schweins, J. P. Chapel, F. M. Schmutz, M. Prieto, O. Sandre and J. F. Le Meins, Langmuir, 2017, 33, 1705–1715 CrossRef PubMed.
- T. Ruysschaert, A. F. P. Sonnen, T. Haefele, W. Meier, M. Winterhaltert and D. Fournier, J. Am. Chem. Soc., 2005, 127, 6242–6247 CrossRef PubMed.
- S. T. Poschenrieder, M. Hanzlik and K. Castiglione, J. Appl. Polym. Sci., 2017, 135, 46077 CrossRef.
- P. V. Pawar, S. V. Gohil, J. P. Jain and N. Kumar, Polym. Chem., 2013, 4, 3160–3176 RSC.
- M. I. Angelova and D. S. Dimitrov, Faraday Discuss., 1986, 81, 303–311 RSC.
- H. D. Chen, S. Werner and H. Brenner, Clin. Gastroenterol. Hepatol., 2017, 15, 1483–1484 CrossRef.
- H. M. L. Lambermont-Thijs, F. S. van der Woerdt, A. Baumgaertel, L. Bonami, F. E. Du Prez, U. S. Schubert and R. Hoogenboom, Macromolecules, 2010, 43, 927–933 CrossRef.
- P. Walde, K. Cosentino, H. Engel and P. Stano, ChemBioChem, 2010, 11, 848–865 CrossRef PubMed.
- J. C. M. Lee, H. Bermudez, B. M. Discher, M. A. Sheehan, Y. Y. Won, F. S. Bates and D. E. Discher, Biotechnol. Bioeng., 2001, 73, 135–145 CrossRef PubMed.
- H. Kukula, H. Schlaad, M. Antonietti and S. Forster, J. Am. Chem. Soc., 2002, 124, 1658–1663 CrossRef PubMed.
- T. P. T. Dao, M. Fauquignon, F. Fernandes, E. Ibarboure, A. Vax, M. Prieto and J. F. Le Meins, Colloids Surf., A, 2017, 533, 347–353 CrossRef.
- A. Weinberger, F. C. Tsai, G. H. Koenderink, T. F. Schmidt, R. Itri, W. Meier, T. Schmatko, A. Schroder and C. Marques, Biophys. J., 2013, 105, 154–164 CrossRef PubMed.
- A. C. Greene, D. Y. Sasaki and G. D. Bachand, J. Visualized Exp., 2016, e54051 Search PubMed.
- S. Pautot, B. J. Frisken and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10718–10721.
- A. Peyret, E. Ibarboure, J.-F. Le Meins and S. Lecommandoux, Adv. Sci., 2018, 5, 1700453 CrossRef PubMed.
- K. Tsumoto, Y. Hayashi, J. Tabata and M. Tomita, Colloids Surf., A, 2018, 546, 74–82 CrossRef.
- D. van Swaay and A. deMello, Lab Chip, 2013, 13, 752–767 RSC.
- C. Martino and A. deMello, Interface Focus, 2016, 6, 20160011 CrossRef PubMed.
- Y. Elani, Biochem. Soc. Trans., 2016, 44, 723–730 CrossRef PubMed.
- K. Kamiya and S. Takeuchi, J. Mater. Chem. B, 2017, 5, 5911–5923 RSC.
- L. D. Mayer, M. J. Hope and P. R. Cullis, Biochim. Biophys. Acta, 1986, 858, 161–168 CrossRef.
- M. Fidorra, L. Duelund, C. Leidy, A. C. Simonsen and L. A. Bagatolli, Biophys. J., 2006, 90, 4437–4451 CrossRef PubMed.
- L. Theogarajan, S. Desai, M. Baldo and C. Scholz, Polym. Int., 2008, 57, 660–667 CrossRef.
- A. M. Eissa, M. J. P. Smith, A. Kubilis, J. A. Mosely and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5184–5193 CrossRef.
- A. Kubilis, A. Abdulkarim, A. M. Eissa and N. R. Cameron, Sci. Rep., 2016, 6, 32414 CrossRef PubMed.
- K. S. Horger, D. J. Estes, R. Capone and M. Mayer, J. Am. Chem. Soc., 2009, 131, 1810–1819 CrossRef PubMed.
- A. C. Greene, I. M. Henderson, A. Gomez, W. F. Paxton, V. VanDelinder and G. D. Bachand, PLoS One, 2016, 11, e0158729 CrossRef PubMed.
- C. Gonzato, M. Semsarilar, E. R. Jones, F. Li, G. J. P. Krooshof, P. Wyman, O. O. Mykhaylyk, R. Tuinier and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 11100–11106 CrossRef PubMed.
- A. N. Albertsen, J. K. Szymanski and J. Perez-Mercader, Sci. Rep., 2017, 7, 41534 CrossRef PubMed.
- K. Wang, Y. X. Wang and W. Q. Zhang, Polym. Chem., 2017, 8, 6407–6415 RSC.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- J. C. Baret, Lab Chip, 2012, 12, 422–433 RSC.
- N. N. Deng, M. Yelleswarapu and W. T. S. Huck, J. Am. Chem. Soc., 2016, 138, 7584–7591 CrossRef PubMed.
- J. Petit, L. Thomi, J. Schultze, M. Makowski, I. Negwer, K. Koynov, S. Herminghaus, F. R. Wurm, O. Baumchen and K. Landfester, Soft Matter, 2018, 14, 894–900 RSC.
- K. Göpfrich, I. Platzman and J. P. Spatz, Trends Biotechnol., 2018, 36, 938–951 CrossRef PubMed.
- B. Yu, R. J. Lee and L. J. Lee, Methods Enzymol., 2009, 465, 129–141 Search PubMed.
- R. C. Hayward, A. S. Utada, N. Dan and D. A. Weitz, Langmuir, 2006, 22, 4457–4461 CrossRef PubMed.
- S. Y. Teh, R. Khnouf, H. Fan and A. P. Lee, Biomicrofluidics, 2011, 5, 044113 CrossRef PubMed.
- S. Deshpande, Y. Caspi, A. E. C. Meijering and C. Dekker, Nat. Commun., 2016, 7, 10447 CrossRef PubMed.
- H. C. Shum, D. Lee, I. Yoon, T. Kodger and D. A. Weitz, Langmuir, 2008, 24, 7651–7653 CrossRef PubMed.
- M. Weiss, J. P. Frohnmayer, L. T. Benk, B. Haller, J. W. Janiesch, T. Heitkamp, M. Borsch, R. B. Lira, R. Dimova, R. Lipowsky, E. Bodenschatz, J. C. Baret, T. Vidakovic-Koch, K. Sundmacher, I. Platzman and J. P. Spatz, Nat. Mater., 2018, 17, 89–96 CrossRef PubMed.
- L. R. Arriaga, S. S. Datta, S. H. Kim, E. Amstad, T. E. Kodger, F. Monroy and D. A. Weitz, Small, 2014, 10, 950–956 CrossRef PubMed.
- K. Kamiya, R. Kawano, T. Osaki, K. Akiyoshi and S. Takeuchi, Nat. Chem., 2016, 8, 881–889 CrossRef PubMed.
- A. R. Abate, T. Hung, P. Mary, J. J. Agresti and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19163–19166 CrossRef PubMed.
- S. W. Hell, Science, 2007, 316, 1153–1158 CrossRef PubMed.
- G. Battaglia, C. LoPresti, M. Massignani, N. J. Warren, J. Madsen, S. Forster, C. Vasilev, J. K. Hobbs, S. P. Armes, S. Chirasatitsin and A. J. Engler, Small, 2011, 7, 2010–2015 CrossRef PubMed.
- S. W. Hell, S. J. Sahl, M. Bates, X. W. Zhuang, R. Heintzmann, M. J. Booth, J. Bewersdorf, G. Shtengel, H. Hess, P. Tinnefeld, A. Honigmann, S. Jakobs, I. Testa, L. Cognet, B. Lounis, H. Ewers, S. J. Davis, C. Eggeling, D. Klenerman, K. I. Willig, G. Vicidomini, M. Castello, A. Diaspro and T. Cordes, J. Phys. D: Appl. Phys., 2015, 48, 443001 CrossRef.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef PubMed.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Pharmaceutics, 2017, 9, 12 CrossRef PubMed.
- P. S. Zangabad, S. Mirkiani, S. Shahsavari, B. Masoudi, M. Masroor, H. Hamed, Z. Jafari, Y. D. Taghipour, H. Hashemi, M. Karimi and M. R. Hamblin, Nanotechnol. Rev., 2018, 7, 95–122 Search PubMed.
- T. Wolf, T. Rheinberger, J. Simon and F. R. Wurm, J. Am. Chem. Soc., 2017, 139, 11064–11072 CrossRef PubMed.
- M. H. Li, Fund. Biomed. Technol., 2011, 5, 291–331 Search PubMed.
- X. L. Hu, Y. G. Zhang, Z. G. Xie, X. B. Jing, A. Bellotti and Z. Gu, Biomacromolecules, 2017, 18, 649–673 CrossRef PubMed.
- T. M. Allen, C. Hansen, F. Martin, C. Redemann and A. Yau-Young, Biochim. Biophys. Acta, 1991, 1066, 29–36 CrossRef.
- M. Chemin, P. M. Brun, S. Lecommandoux, O. Sandre and J. F. Le Meins, Soft Matter, 2012, 8, 2867–2874 RSC.
- O. Onaca, R. Enea, D. W. Hughes and W. Meier, Macromol. Biosci., 2009, 9, 129–139 CrossRef PubMed.
- A. Blanazs, M. Massignani, G. Battaglia, S. P. Armes and A. J. Ryan, Adv. Funct. Mater., 2009, 19, 2906–2914 CrossRef.
- R. Nayar and A. J. Schroit, Biochemistry, 1985, 24, 5967–5971 CrossRef PubMed.
- M. Z. Lai, N. Duzgunes and F. C. Szoka, Biochemistry, 1985, 24, 1646–1653 CrossRef PubMed.
- I. M. Hafez and P. R. Cullis, Biochim. Biophys. Acta, 2000, 1463, 107–114 CrossRef.
- W. Kulig, P. Jurkiewicz, A. Olzynska, J. Tynkkynen, M. Javanainen, M. Manna, T. Rog, M. Hof, I. Vattulainen and P. Jungwirth, Biochim. Biophys. Acta, 2015, 1848, 422–432 CrossRef PubMed.
- U. Jonas, K. Shah, S. Norvez and D. H. Charych, J. Am. Chem. Soc., 1999, 121, 4580–4588 CrossRef.
- Z. Z. Yuan and T. W. Hanks, Polymer, 2008, 49, 5023–5026 CrossRef.
- J. Z. Du, Y. Q. Tang, A. L. Lewis and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 17982–17983 CrossRef PubMed.
- J. Rodriguez-Hernandez and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef PubMed.
- S. Y. Yu, T. Azzam, I. Rouiller and A. Eisenberg, J. Am. Chem. Soc., 2009, 131, 10557–10566 CrossRef PubMed.
- R. L. McCarley, Annu. Rev. Anal. Chem., 2012, 5, 391–411 CrossRef PubMed.
- W. Ong, Y. M. Yang, A. C. Cruciano and R. L. McCarley, J. Am. Chem. Soc., 2008, 130, 14739–14744 CrossRef PubMed.
- T. Thambi, J. H. Park and D. S. Lee, Biomater. Sci., 2016, 4, 55–69 RSC.
- J. S. Lee, T. Groothuis, C. Cusan, D. Mink and J. Feijen, Biomaterials, 2011, 32, 9144–9153 CrossRef PubMed.
- M. B. Yatvin, J. N. Weinstein, W. H. Dennis and R. Blumenthal, Science, 1978, 202, 1290–1293 CrossRef PubMed.
- D. C. Turner, D. Moshkelani, C. S. Shemesh, D. Luc and H. L. Zhang, Pharm. Res., 2012, 29, 2092–2103 CrossRef PubMed.
- I. Levacheva, O. Samsonova, E. Tazina, M. Beck-Broichsitter, S. Levachev, B. Strehlow, M. Baryshnikova, N. Oborotova, A. Baryshnikov and U. Bakowsky, Colloids Surf., B, 2014, 121, 248–256 CrossRef PubMed.
- A. Peyret, E. Ibarboure, N. Pippa and S. Lecommandoux, Langmuir, 2017, 33, 7079–7085 CrossRef PubMed.
- N. Pippa, A. Meristoudi, S. Pispas and C. Demetzos, Int. J. Pharm., 2015, 485, 374–382 CrossRef PubMed.
- S. H. Qin, Y. Geng, D. E. Discher and S. Yang, Adv. Mater., 2006, 18, 2905–2909 CrossRef.
- F. H. Meng, Z. Y. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef PubMed.
- K. Suzuki, K. Machida, K. Yamaguchi and T. Sugawara, Chem. Phys. Lipids, 2018, 210, 70–75 CrossRef PubMed.
- J. W. Hindley, Y. Elani, C. M. McGilvery, S. Ali, C. L. Bevan, R. V. Law and O. Ces, Nat. Commun., 2018, 9, 1093 CrossRef PubMed.
- W. Su, K. Han, Y. H. Luo, Z. Wang, Y. M. Li and Q. J. Zhang, Macromol. Chem. Phys., 2007, 208, 955–963 CrossRef.
- L. Schoonen and J. C. M. van Hest, Adv. Mater., 2016, 28, 1109–1128 CrossRef PubMed.
- B. C. Buddingh and J. C. M. van Hest, Acc. Chem. Res., 2017, 50, 769–777 CrossRef PubMed.
- W. K. Spoelstra, S. Deshpande and C. Dekker, Curr. Opin. Biotechnol., 2018, 51, 47–56 CrossRef PubMed.
- P. Schwille, Science, 2011, 333, 1252–1254 CrossRef PubMed.
- M. Marguet, C. Bonduelle and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 512–529 RSC.
- Y. Tu, F. Peng, A. Adawy, Y. Men, L. K. Abdelmohsen and D. A. Wilson, Chem. Rev., 2016, 116, 2023–2078 CrossRef PubMed.
- V. Balasubramanian, B. Herranz-Blanco, P. V. Almeida, J. Hirvonen and H. A. Santos, Prog. Polym. Sci., 2016, 60, 51–85 CrossRef.
- B. Fadeel and D. Xue, Crit. Rev. Biochem. Mol. Biol., 2009, 44, 264–277 CrossRef PubMed.
- D. Marquardt, B. Geier and G. Pabst, Membranes, 2015, 5, 180–196 CrossRef PubMed.
- L. Lu, J. W. Schertzer and P. R. Chiarot, Lab Chip, 2015, 15, 3591–3599 RSC.
- A. Peyret, H. Zhao and S. Lecommandoux, Langmuir, 2018, 34, 3376–3385 CrossRef PubMed.
- M. J. Hope, T. E. Redelmeier, K. F. Wong, W. Rodrigueza and P. R. Cullis, Biochemistry, 1989, 28, 4181–4187 CrossRef PubMed.
- A. Callan-Jones, B. Sorre and P. Bassereau, Cold Spring Harbor Perspect. Biol., 2011, 3, a004648 Search PubMed.
- L. B. Luo and A. Eisenberg, Langmuir, 2001, 17, 6804–6811 CrossRef.
- L. B. Luo and A. Eisenberg, J. Am. Chem. Soc., 2001, 123, 1012–1013 CrossRef PubMed.
- Y. L. Zhang, F. Wu, W. E. Yuan and T. Jin, J. Controlled Release, 2010, 147, 413–419 CrossRef PubMed.
- A. F. Mason and P. Thordarson, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3817–3825 CrossRef.
- R. Stoenescu and W. Meier, Chem. Commun., 2002, 3016–3017, 10.1039/b209352a,.
- F. T. Liu and A. Eisenberg, J. Am. Chem. Soc., 2003, 125, 15059–15064 CrossRef PubMed.
- P. C. C. Hu, S. Li and N. Malmstadt, ACS Appl. Mater. Interfaces, 2011, 3, 1434–1440 CrossRef PubMed.
- K. Karamdad, R. V. Law, J. M. Seddon, N. J. Brooks and O. Ces, Chem. Commun., 2016, 52, 5277–5280 RSC.
- J. Nam, T. K. Vanderlick and P. A. Beales, Soft Matter, 2012, 8, 7982–7988 RSC.
- K. Simons and E. Ikonen, Nature, 1997, 387, 569–572 CrossRef PubMed.
- L. Bagatolli and P. B. Sunil Kumar, Soft Matter, 2009, 5, 3234–3248 RSC.
- T. P. T. Dao, F. Fernandes, M. Er-Rafik, R. Salva, M. Schmutz, A. Brulet, M. Prieto, O. Sandre and J. F. Le Meins, ACS Macro Lett., 2015, 4, 182–186 CrossRef.
- P. A. Beales, S. Khan, S. P. Muench and L. J. Jeuken, Biochem. Soc. Trans., 2017, 45, 15–26 CrossRef PubMed.
- W. H. Binder, V. Barragan and F. M. Menger, Angew. Chem., Int. Ed., 2003, 42, 5802–5827 CrossRef PubMed.
- F. A. Heberle and G. W. Feigenson, Cold Spring Harbor Perspect. Biol., 2011, 3, a004630 Search PubMed.
- R. J. Brea, A. K. Rudd and N. K. Devaraj, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8589–8594 CrossRef PubMed.
- C. M. Rosetti, A. Mangiarotti and N. Wilke, Biochim. Biophys. Acta, 2017, 1859, 789–802 CrossRef PubMed.
- C. Dietrich, L. A. Bagatolli, Z. N. Volovyk, N. L. Thompson, M. Levi, K. Jacobson and E. Gratton, Biophys. J., 2001, 80, 1417–1428 CrossRef PubMed.
- S. L. Veatch and S. L. Keller, Biochim. Biophys. Acta, Mol. Cell Res., 2005, 1746, 172–185 CrossRef PubMed.
- P. F. F. Almeida, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 72–85 CrossRef PubMed.
- A. J. Garcia-Saez and P. Schwille, FEBS Lett., 2010, 584, 1653–1658 CrossRef PubMed.
- A. R. Honerkamp-Smith, S. L. Veatch and S. L. Keller, Biochim. Biophys. Acta, 2009, 1788, 53–63 CrossRef PubMed.
- T. M. Konyakhina, J. Wu, J. D. Mastroianni, F. A. Heberle and G. W. Feigenson, Biochim. Biophys. Acta, 2013, 1828, 2204–2214 CrossRef PubMed.
- N. Bezlyepkina, R. S. Gracia, P. Shchelokovskyy, R. Lipowsky and R. Dimova, Biophys. J., 2013, 104, 1456–1464 CrossRef PubMed.
- D. A. Christian, A. Tian, W. G. Ellenbroek, I. Levental, K. Rajagopal, P. A. Janmey, A. J. Liu, T. Baumgart and D. E. Discher, Nat. Mater., 2009, 8, 843–849 CrossRef PubMed.
- C. LoPresti, M. Massignani, C. Fernyhough, A. Blanazs, A. J. Ryan, J. Madsen, N. J. Warren, S. P. Armes, A. L. Lewis, S. Chirasatitsin, A. J. Engler and G. Battaglia, ACS Nano, 2011, 5, 1775–1784 CrossRef PubMed.
- L. Ruiz-Perez, L. Messager, J. Gaitzsch, A. Joseph, L. Sutto, F. L. Gervasio and G. Battaglia, Sci. Adv., 2016, 2, e1500948 Search PubMed.
- R. Stoenescu, A. Graff and W. Meier, Macromol. Biosci., 2004, 4, 930–935 CrossRef PubMed.
- D. Wong, T. J. Jeon and J. Schmidt, Nanotechnology, 2006, 17, 3710–3717 CrossRef.
- M. Lomora, M. Garni, F. Itel, P. Tanner, M. Spulber and C. G. Palivan, Biomaterials, 2015, 53, 406–414 CrossRef PubMed.
- T. Einfalt, R. Goers, I. A. Dinu, A. Najer, M. Spulber, O. Onaca-Fischer and C. G. Palivan, Nano Lett., 2015, 15, 7596–7603 CrossRef PubMed.
- L. Messager, J. R. Burns, J. Kim, D. Cecchin, J. Hindley, A. L. B. Pyne, J. Gaitzsch, G. Battaglia and S. Howorka, Angew. Chem., Int. Ed., 2016, 55, 11106–11109 CrossRef PubMed.
- I. L. Jorgensen, G. C. Kemmer and T. G. Pomorski, Eur. Biophys. J., 2017, 46, 103–119 CrossRef PubMed.
- P. Girard, J. Pecreaux, G. Lenoir, P. Falson, J. L. Rigaud and P. Bassereau, Biophys. J., 2004, 87, 419–429 CrossRef PubMed.
- M. Dezi, A. Di Cicco, P. Bassereau and D. Levy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7276–7281 CrossRef PubMed.
- M. Garten, S. Aimon, P. Bassereau and G. E. S. Toombes, J. Visualized Exp., 2015, 52281 Search PubMed.
- E. Altamura, F. Milano, R. R. Tangorra, M. Trotta, O. H. Omar, P. Stano and F. Mavelli, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3837–3842 CrossRef PubMed.
- L. Dai, L. M. Tan, Y. L. Jiang, Y. Shi, P. Wang, J. P. Zhang and Z. Y. Otomo, Chem. Phys. Lett., 2018, 705, 78–84 CrossRef.
- T. Einfalt, D. Witzigmann, C. Edlinger, S. Sieber, R. Goers, A. Najer, M. Spulber, O. Onaca-Fischer, J. Huwyler and C. G. Palivan, Nat. Commun., 2018, 9, 1127 CrossRef PubMed.
- H. L. Che, S. P. Cao and J. C. M. van Hest, J. Am. Chem. Soc., 2018, 140, 5356–5359 CrossRef PubMed.
- K. Jaskiewicz, A. Larsen, I. Lieberwirth, K. Koynov, W. Meier, G. Fytas, A. Kroeger and K. Landfester, Angew. Chem., Int. Ed., 2012, 51, 4613–4617 CrossRef PubMed.
- K. Jaskiewicz, A. Larsen, D. Schaeffel, K. Koynov, I. Lieberwirth, G. Fytas, K. Landfester and A. Kroeger, ACS Nano, 2012, 6, 7254–7262 CrossRef PubMed.
- A. Meinel, B. Trankle, W. Romer and A. Rohrbach, Soft Matter, 2014, 10, 3667–3678 RSC.
- A. J. van Rensen, M. H. Wauben, M. C. Grosfeld-Stulemeyer, W. van Eden and D. J. Crommelin, Pharm. Res., 1999, 16, 198–204 CrossRef.
- T. Nakamura, R. Moriguchi, K. Kogure, N. Shastri and H. Harashima, Mol. Ther., 2008, 16, 1507–1514 CrossRef PubMed.
- M. Maji, S. Mazumder, S. Bhattacharya, S. T. Choudhury, A. Sabur, M. Shadab, P. Bhattacharya and N. Ali, Sci. Rep., 2016, 6, 27206 CrossRef PubMed.
- B. J. Lestini, S. M. Sagnella, Z. Xu, M. S. Shive, N. J. Richter, J. Jayaseharan, A. J. Case, K. Kottke-Marchant, J. M. Anderson and R. E. Marchant, J. Controlled Release, 2002, 78, 235–247 CrossRef PubMed.
- J. A. Opsteen, R. P. Brinkhuis, R. L. M. Teeuwen, D. W. P. M. Lowik and J. C. M. van Hest, Chem. Commun., 2007, 3136–3138, 10.1039/b704568a,.
- W. M. Pardridge, Drug Discovery Today, 2007, 12, 54–61 CrossRef PubMed.
- G. P. Robbins, R. L. Saunders, J. B. Haun, J. Rawson, M. J. Therien and D. A. Hammer, Langmuir, 2010, 26, 14089–14096 CrossRef PubMed.
- S. Egli, H. Schlaad, N. Bruns and W. Meier, Polymers, 2011, 3, 252–280 CrossRef.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef PubMed.
- J. Ingale, A. Stano, J. Guenaga, S. K. Sharma, D. Nemazee, M. B. Zwick and R. T. Wyatt, Cell Rep., 2016, 15, 1986–1999 CrossRef PubMed.
- Z. Q. Pang, W. Lu, H. L. Gao, K. L. Hu, J. Chen, C. L. Zhang, X. L. Gao, X. G. Jiang and C. Q. Zhu, J. Controlled Release, 2008, 128, 120–127 CrossRef PubMed.
- S. F. M. van Dongen, M. Nallani, S. Schoffelen, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Macromol. Rapid Commun., 2008, 29, 321–325 CrossRef.
- R. Nehring, C. G. Palivan, S. Moreno-Flores, A. Mantion, P. Tanner, J. L. Toca-Herrera, A. Thunemann and W. Meier, Soft Matter, 2010, 6, 2815–2824 RSC.
- S. Egli, M. G. Nussbaumer, V. Balasubramanian, M. Chami, N. Bruns, C. Palivan and W. Meier, J. Am. Chem. Soc., 2011, 133, 4476–4483 CrossRef PubMed.
- B. Iyisan, A. C. Siedel, H. Gumz, M. Yassin, J. Kluge, J. Gaitzsch, P. Formanek, S. Moreno, B. Voit and D. Appelhans, Macromol. Rapid Commun., 2017, 38, 1700486 CrossRef PubMed.
- J. J. Lin, P. Ghoroghchian, Y. Zhang and D. A. Hammer, Langmuir, 2006, 22, 3975–3979 CrossRef PubMed.
- M. Felici, M. Marza-Perez, N. S. Hatzakis, R. J. M. Nolte and M. C. Feiters, Chem. – Eur. J., 2008, 14, 9914–9920 CrossRef PubMed.
- S. Domes, V. Filiz, J. Nitsche, A. Fromsdorf and S. Forster, Langmuir, 2010, 26, 6927–6931 CrossRef PubMed.
- P. Broz, S. M. Benito, C. Saw, P. Burger, H. Heider, M. Pfisterer, S. Marsch, W. Meier and P. Hunziker, J. Controlled Release, 2005, 102, 475–488 CrossRef PubMed.
- Z. Fu, M. A. Ochsner, H.-P. M. de Hoog, N. Tomczak and M. Nallani, Chem. Commun., 2011, 47, 2862–2864 RSC.
- R. J. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef PubMed.
- Y. Elani, R. V. Law and O. Ces, Nat. Commun., 2014, 5, 5305 CrossRef PubMed.
- X. L. Liu, P. Formanek, B. Voit and D. Appelhans, Angew. Chem., Int. Ed., 2017, 56, 16233–16238 CrossRef PubMed.
- N.-N. Deng, M. Yelleswarapu, L. Zheng and W. T. S. Huck, J. Am. Chem. Soc., 2017, 139, 587–590 CrossRef PubMed.
- M. Godoy-Gallardo, C. Labay, V. D. Trikalitis, P. J. Kempen, J. B. Larsen, T. L. Andresen and L. Hosta-Rigau, ACS Appl. Mater. Interfaces, 2017, 9, 15907–15921 CrossRef PubMed.
- T. Trantidou, M. Friddin, Y. Elani, N. J. Brooks, R. V. Law, J. M. Seddon and O. Ces, ACS Nano, 2017, 11, 6549–6565 CrossRef PubMed.
- Y. Elani, T. Trantidou, D. Wylie, L. Dekker, K. Polizzi, R. V. Law and O. Ces, Sci. Rep., 2018, 8, 4564 CrossRef PubMed.
- K. Y. Lee, S. J. Park, K. A. Lee, S. H. Kim, H. Kim, Y. Meroz, L. Mahadevan, K. H. Jung, T. K. Ahn, K. K. Parker and K. Shin, Nat. Biotechnol., 2018, 36, 530–535 CrossRef PubMed.
- B. Pitard, P. Richard, M. Duñarach, G. Girault and J.-L. Rigaiud, Eur. J. Biochem., 1996, 235, 769–778 CrossRef PubMed.
- H. J. Choi and C. D. Montemagno, Nano Lett., 2005, 5, 2538–2542 CrossRef PubMed.
- X. Feng, Y. Jia, P. Cai, J. Fei and J. Li, ACS Nano, 2016, 10, 556–561 CrossRef PubMed.
- T. Nilsson, C. R. Lundin, G. Nordlund, P. Adelroth, C. von Ballmoos and P. Brzezinski, Sci. Rep., 2016, 6, 24113 CrossRef PubMed.
- O. Biner, T. Schick, Y. Müller and C. Ballmoos, FEBS Lett., 2016, 590, 2051–2062 CrossRef PubMed.
- T. F. Zhu and J. W. Szostak, J. Am. Chem. Soc., 2009, 131, 5705–5713 CrossRef PubMed.
- C. Martino, S. H. Kim, L. Horsfall, A. Abbaspourrad, S. J. Rosser, J. Cooper and D. A. Weitz, Angew. Chem., Int. Ed., 2012, 51, 6416–6420 CrossRef PubMed.
- J. M. Hernandez, A. J. Kreutzberger, V. Kiessling, L. K. Tamm and R. Jahn, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12037–12042 CrossRef PubMed.
- M. D. Hardy, J. Yang, J. Selimkhanov, C. M. Cole, L. S. Tsimring and N. K. Devaraj, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8187–8192 CrossRef PubMed.
- R. J. R. W. Peters, M. Nijemeisland and J. C. M. Van Hest, Angew. Chem., Int. Ed., 2015, 54, 9614–9617 CrossRef PubMed.
- K. P. Adamala, D. A. Martin-Alarcon, K. R. Guthrie-Honea and E. S. Boyden, Nat. Chem., 2016, 9, 431 CrossRef PubMed.
- S. Sun, M. Li, F. Dong, S. Wang, L. Tian and S. Mann, Small, 2016, 12, 1920–1927 CrossRef PubMed.
- M. Nijemeisland, L. K. E. A. Abdelmohsen, W. T. S. Huck, D. A. Wilson and J. C. M. van Hest, ACS Cent. Sci., 2016, 2, 843–849 CrossRef PubMed.
- F. Caschera and V. Noireaux, Artif. Life, 2016, 22, 185–195 CrossRef PubMed.
- A. Peyret, E. Ibarboure, A. Tron, L. Beaute, R. Rust, O. Sandre, N. D. McClenaghan and S. Lecommandoux, Angew. Chem., Int. Ed., 2017, 56, 1566–1570 CrossRef PubMed.
- S. Koike and R. Jahn, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9883–E9892 CrossRef PubMed.
- A. P. Liu, D. L. Richmond, L. Maibaum, S. Pronk, P. L. Geissler and D. A. Fletcher, Nat. Phys., 2008, 4, 789 Search PubMed.
- D. Merkle, N. Kahya and P. Schwille, ChemBioChem, 2008, 9, 2673–2681 CrossRef PubMed.
- L.-L. Pontani, J. van der Gucht, G. Salbreux, J. Heuvingh, J.-F. Joanny and C. Sykes, Biophys. J., 2009, 96, 192–198 CrossRef PubMed.
- F.-C. Tsai, B. Stuhrmann and G. H. Koenderink, Langmuir, 2011, 27, 10061–10071 CrossRef PubMed.
- Y. T. Maeda, T. Nakadai, J. Shin, K. Uryu, V. Noireaux and A. Libchaber, ACS Synth. Biol., 2012, 1, 53–59 CrossRef PubMed.
- K. Carvalho, F.-C. Tsai, E. Lees, R. Voituriez, G. H. Koenderink and C. Sykes, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16456–16461 CrossRef PubMed.
- G. Rivas, S. K. Vogel and P. Schwille, Curr. Opin. Chem. Biol., 2014, 22, 18–26 CrossRef PubMed.
- C. Kurokawa, K. Fujiwara, M. Morita, I. Kawamata, Y. Kawagishi, A. Sakai, Y. Murayama, S. I. M. Nomura, S. Murata, M. Takinoue and M. Yanagisawa, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7228–7233 CrossRef PubMed.
- S. Dhir, S. Salahub, A. S. Mathews, S. K. Kumaran, C. D. Montemagno and S. Abraham, Chem. Commun., 2018, 54, 5346–5349 RSC.
- S. Majumder and A. P. Liu, Phys. Biol., 2018, 15, 013001 CrossRef PubMed.
- D. R. Arifin and A. F. Palmer, Biomacromolecules, 2005, 6, 2172–2181 CrossRef PubMed.
- S. Rameez, H. Alosta and A. F. Palmer, Bioconjugate Chem., 2008, 19, 1025–1032 CrossRef PubMed.
- S. Rameez, U. Banerjee, J. Fontes, A. Roth and A. F. Palmer, Macromolecules, 2012, 45, 2385–2389 CrossRef PubMed.
- A. Najer, S. Thamboo, C. G. Palivan, H. P. Beck and W. Meier, Chimia, 2016, 70, 288–291 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |