
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Environmentally responsive plasmonic nanoassemblies for biosensing
Xiaoling
Wu
ab,
Changlong
Hao
ab,
Jatish
Kumar
 c,
Hua
Kuang
*ab,
Nicholas A.
Kotov
de,
Luis M.
Liz-Marzán
cf and
Chuanlai
Xu
*ab
c,
Hua
Kuang
*ab,
Nicholas A.
Kotov
de,
Luis M.
Liz-Marzán
cf and
Chuanlai
Xu
*ab
aState Key Lab of Food Science and Technology, Jiangnan University, Wuxi, Jiangsu 214122, People's Republic of China. E-mail: kuangh@jiangnan.edu.cn; xcl@jiangnan.edu.cn
bInternational Joint Research Laboratory for Biointerface and Biodetection, Collaborative Innovationcenter of Food Safety and Quality Control in Jiangsu Province, School of Food Science and Technology, Jiangnan University, Wuxi, Jiangsu 214122, People's Republic of China
cCIC biomaGUNE and CIBER-BBN, Paseo de Miramón 182, 20014 Donostia-San Sebastian, Spain
dDepartment of Chemical Engineering, University of Michigan, Ann Arbor, MI 48109, USA
eBiointerfaces Institute, University of Michigan, Ann Arbor, MI 48109, USA
fIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
First published on 8th May 2018
Abstract
Assemblies of plasmonic nanoparticles enable new modalities for biosensing. Engineered superstructures from metal nanoparticles can enhance the plasmon resonances and chiroptical activity of nanoscale dispersions. Such phenomena are keys to the fabrication of highly sensitive, selective and fast-responding detection platforms, making them promising candidates for clinical applications. This tutorial review summarizes and discusses recent advances in this area. The topics covered in the review include the basic strategies adopted for assembly and engineering of plasmonic nanoparticles, optical properties of the assembled nanostructures and their applications to both in vitro and in vivo detection of biological compounds. We also offer our vision of the future prospects of this field of research. Among emerging applications in this area are novel nanosensors and platforms, for food safety, environmental monitoring, health safeguarding, as well as biodefense.
 Xiaoling Wu | Xiaoling Wu received her PhD degree in 2014 under the direction of Prof. Chuanlai Xu. Following an initial period as a lecturer, she was promoted to associate professor at Jiangnan University in 2016. Her research is focused on plasmonic NP assemblies and bioapplications. |
 Changlong Hao | Changlong Hao is currently a postdoctoral researcher in Professor Chuanlai Xu's group in Jiangnan University. He received his PhD degree from Jiangnan University in June 2015 under the supervision of Prof. Chuanlai Xu. He was a postdoctoral researcher at the National Center for Nanoscience and Technology, Beijing, from July 2015 to August 2017 under the supervision of Prof. Zhiyong Tang. His current research focuses on the design and preparation of chiral inorganic assemblies and their applications in catalysis and biosensing. |
 Jatish Kumar | Jatish Kumar is currently a Marie Curie postdoctoral researcher in Prof. Luis M. Liz-Marzán's group in Spain. He obtained his PhD in Chemistry from CSIR-NIIST, India, in 2012 under the supervision of Prof. K. George Thomas, and subsequently worked as a JSPS postdoctoral fellow in Prof. Tsuyoshi Kawai's group at NAIST, Nara, Japan. His research interests include synthesis and assembly of chiral nanomaterials for biodetection. |
 Hua Kuang | Hua Kuang received her PhD degree at China Agricultural University. She is a full professor with research focused on the analytical aspects of nanoassemblies and instrumental characterizations. |
 Nicholas A. Kotov | Nicholas A. Kotov obtained his PhD in chemistry from Moscow State University in 1990. He is currently Cejka Professor at the Department of Chemical Engineering of the University of Michigan in Ann Arbor, MI. His research interests encompass biomimetic inorganic nano-structures, represented in this review by self-organization phenomena, chirality of nanoparticles and selected biosensing effects of plasmonic nanomaterials. |
 Luis M. Liz-Marzán | Luis Liz-Marzán has been Ikerbasque Professor and Scientific Director of CIC biomaGUNE, in San Sebastián (Spain), since September 2012. He graduated in chemistry from the University of Santiago de Compostela, was a postdoc at Utrecht University and Professor at the University of Vigo (1995–2012). He has been Invited Professor at several universities and research centers worldwide and has received numerous research awards. His major research activity is devoted to understanding the growth mechanisms of metal nanocrystals, to tailor their surface chemistry and direct their self-assembly. He also works on the design of biomedical applications based on the plasmonic properties of well-defined metal nanoparticles and nanostructures. |
 Chuanlai Xu | Chuanlai Xu received his BS, MS and PhD degrees from Jiangnan University. He is a full professor and his research is focused on NPs synthesis, NP-based biodetection, controllable nanoassemblies, and optical and theoretical properties of nanoassemblies. |
Key learning points(1) Basic strategies for the fabrication of plasmonic superstructures(2) Composition-, morphology- and configuration-dependent optical properties (3) Methods for signal amplification (4) Ultrasensitive and highly selective optical responses in nanoparticle assemblies due to collective effects (5) Multi-modal optical responses in vivo and in vitro. |
1. Introduction
When excited by electromagnetic radiation, plasmonic nanomaterials, such as gold and silver, can confine and manipulate photons at the nanoscale by the collective oscillation of conduction electrons, which is known as localized surface plasmon resonance (LSPR). The resonant wavelength (significantly exceeding their dimensions) is dependent on the local dielectric environment and the geometry (size, shape, composition, and orientation) of the nanostructures.1 When nanoparticles (NPs) are located in close proximity to each other, their individual surface plasmons couple, resulting in high electric fields localized in the gaps between the NPs. These regions with enhanced electric fields are often referred to as “hot spots”.2 The enhanced electromagnetic field near the surface of NPs or in the hot spots can result in a variety of unique emerging phenomena, such as plasmon-enhanced absorption, surface enhanced Raman scattering (SERS), and plasmon-enhanced luminescence. Due to these unique effects, plasmonic nanoassemblies can find numerous applications in biosensing, photocatalysis, metamaterials, light harvesting, photothermal transduction, photovoltaics, and non-linear optics.3,4 Owing to their vast potential, the assembly of plasmonic NPs into desired geometries is currently one of the most dynamic fields of research in nanotechnology, drawing the attention of researchers across scientific disciplines, from materials science to chemistry, physics, biology and medicine.5,6 These intrinsically attractive features can be attributed to the following advantages of these materials: (1) intricate or hierarchical superstructures; (2) synergistic amplification of the signals; (3) rapid sensing of multiple components.Controllable and tunable optical properties, such as LSPR, SERS, circular dichroism (CD) and photoluminescence (PL), can be engineered for plasmonic nanoassemblies, based on the judicious selection of building blocks and assembly strategies, which result in tailored morphologies. Both bottom-up and top-down manufacturing processes have been utilized for the construction of nanoassemblies. Among the different strategies, self-assembly driven by non-covalent interactions has been one of the most successful approaches. As a result, a great variety of plasmonic nanoassemblies with diverse geometries and varying numbers of components have been reported. These include plasmonic dimers,7,8 trimers,9 tetramers,10 core–satellite,11 propeller-like,12 pyramids,13,14 toroids,15 side-by-side (SBS),16 end-to-end (ETE), single NP/nanorod (NR) helices,5,17 double NP helices,18 stair-like NR helices,19 coil-like NR helices, twisted chains, layer-by-layer (LBL) assembled20,26 and other films,22,23 ordered arrays,24 supraparticles21 and superlattices.25 The particular configuration of each of these assemblies determines their respective optical response. Of specific interest is the chiral activity in asymmetric assemblies, whereas symmetric assemblies remain, in the first approximation, achiral.8,26 Engineered nanoassemblies are mainly composed of plasmonic NPs, represented by AuNPs,3 AgNPs,13 Au nanostars,27 AuNRs,16 Au@Ag NPs,28 Au@SiO2 NPs,6 as well as non-plasmonic NPs, such as magnetic NPs (Fe3O4 NPs),25 up-converting NPs (UCNPs),11 CdTe NPs,21 and silver sulfide (Ag2S) NPs,14 as building blocks. In addition, small organic molecules, supramolecular polymers29 and biomolecules such as peptides,17 proteins,23 oligonucleotides,13 DNA origami,15 and cellulose fibers,22 have been used to fabricate plasmonic assemblies with targeted geometrical features. Importantly, small environmental modifications can precisely regulate the self-assembly behavior, leading to enhanced properties in some selected assemblies, which can be exploited for the development of ultrasensitive sensing devices.16,30 It is however challenging to design rational nanostructures with augmented plasmonic features to achieve synergistic amplification effects in nanoassemblies. Another key task involves triggering such amplified effects rapidly and efficiently, as well as integrating them into sensing devices. Even though many sensors based on plasmonic materials have been reported, fabrication of robust, highly selective and fast-responding sensors which can satisfy daily life requirements are still in the development stage. Therefore, it is essential to address the recent progress of plasmonic nanoassemblies for detection applications.
In this tutorial review, we provide basic concepts of plasmonic nanoassembly engineering and some strategies that have been proposed for their use as sensors. We summarize and discuss recent significant advances and future directions of this field. We particularly focus on non-covalent chemistry-driven self-assembly of plasmonic nanomaterials, and resulting optical responses for the design of novel biosensors.
2. Strategies for the assembly of plasmonic nanostructures
2.1 DNA-mediated assembly
A variety of biomolecules have been used as linkers or templates to direct plasmonic self-assembly. These include DNA, peptides, proteins, lipids, viruses and other biopolymers. DNA, owing to its precise and predictable programmability by Watson–Crick base pairing, excellent biocompatibility, feasible integration of functional nucleic acids, and convenient synthetic routes, is one of the most versatile and robust building blocks for the synthesis of complex three-dimensional NP assemblies. Of particular interest is DNA origami, with unique addressability and custom design structures which enable a wide variety of plasmonic engineering designs with collective physicochemical properties and enhanced optical response. NP dimers constitute the most basic structures among the various assemblies. For example, Lohmüller et al. demonstrated the synthesis of AuNP dimers with controlled interparticle spacing using a DNA origami template.7 The DNA origami block comprised an 8064-nucleotide-long scaffolding DNA strand, which was folded into shape by 200 short DNA strands (∼40 nucleotides long). Thiolated single-stranded DNA-modified AuNPs were then hybridized onto the origami structure by three 15-nucleotide-long linking strands. Two AuNPs were closely placed on two sides of the three-layered DNA origami block, by linking DNA molecules (Fig. 1a). The distance between the NPs could be controlled by the thickness of the origami sheet and the linking DNA molecules. Molecules placed at the resulting hot spots exhibited intense SERS response, which was highly dependent on the direction of polarization with respect to the dimer axis. On the other hand, Liu and coworkers adopted an attractive strategy toward the synthesis of reconfigurable AuNR dimers exhibiting distinct chiral properties.8 A switchable DNA origami template was used, which was made of two 14-helix bundles folded from a long single-stranded DNA scaffold with the help of hundreds of staple strands. The two DNA bundles were linked together by the scaffold strand passing twice between them at one point, which regulated the angle between the two DNA bundles. Eight unpaired bases were introduced to each DNA connector to avoid Holliday junction formation. Two complementary DNA-functionalized AuNRs were robustly hosted on each DNA bundle through twelve binding sites with reconfigurable conformation (Fig. 1b). The synthesis of DNA origami-templated heterogeneous trimers with nanometer precision and high assembly yield was demonstrated by Liedl and co-workers.9 A cylindrical DNA bundle was constructed with three equally spaced attachment sites, and by specific complementary sequence modification and hybridization, two AuNPs and one AgNP were specifically bound on the designed outer sites and middle site, respectively (Fig. 1c). The resulting heterotrimers showed an interparticle gap of 40 nm between the two AuNPs, which was pivotal to ultrafast energy transfer by plasmon coupling. DNA-origami technology has also been employed for the fabrication of more attractive and complex superstructures. As shown in Fig. 1d, AuNP helices with a designated configuration (right- or left-handed), and designed helical parameters with 34 nm diameter and 57 nm helical pitch, have been successfully synthesized by the attachment of nine AuNPs on the surface of 24-helix DNA origami bundles.5 Each attachment site was composed of three-staple extensions, each of them being a 15-nucleotide-long single-stranded oligonucleotide. The AuNPs were functionalized with multiple thiol-modified DNA strands, complementary to such staple extensions. Interestingly, the geometry of the plasmonic nanohelices, including helical pitch and diameter, was tailored by varying the NP size. In another example, a toroidal plasmonic superstructure was assembled on a hierarchical DNA origami.15 Four curved DNA origami monomers were linked via head–head and tail–tail connectors, resulting in an intact origami ring formation. Specifically, each origami monomer was functionalized with 24 AuNPs, displaying a toroidal diameter of 120 nm (Fig. 1e). The approaches described herein can in principle result in a plethora of assembled nanostructures possessing tunable optical responses. | ||
| Fig. 1 DNA-mediated plasmonic nanoassemblies (a–e). AuNP dimer (a),7 AuNR dimers (b),8 Au–Ag–Au NP trimers (c),9 AuNP helices (d),5 and toroidal structures (e)15 assembled with DNA origami as the template. (f–j) AgNP pyramids (f),13 propeller-like AuNR–UCNP tetramers (g),12 AuNR dimer–UCNP core–satellite assemblies (h),11 AgNP superlattices (i)31 and AuNR ETE structures (j)16 assembled utilizing DNA-programmable methods. Reproduced with permission: ref. 7, 11 and 15 Copyright 2014, 2016, 2017 American Chemical Society (ACS); ref. 5, 8, 9 and 16 Copyright 2012, 2013, 2014, 2017 Macmillan Publishers Ltd; ref. 12, 13 and 31 Copyright 2013, 2015, 2016 Wiley-VCH Verlag GmbH & Co. | ||
DNA-bridged plasmonic superstructures can also be constructed without resorting to DNA origami templates. Xu et al. demonstrated the assembly of Ag pyramids with high yield through a simple and well-designed DNA frame.13 Four AgNPs were modified with four different single-stranded DNA fragments, which could be complementarily hybridized with each other. In particular, each single-stranded DNA was segmented into three parts, which constituted the six sides of the pyramids. AgNPs were located at the apexes of the pyramids, acting as the connecting vertices (Fig. 1f). The diameter of the pyramids was calculated to be 48 ± 2.4 nm, and the gap between adjacent NPs was estimated to be 8 ± 0.3 nm. Importantly, by embedding cancer biomarker-specific aptamers in the single-stranded DNA sequences, pyramids with tunable gaps were obtained. These nanostructures exhibited strong SERS response, capable of attomolar detection of the selected disease biomarkers. Moreover, propeller-like NR–UCNP assemblies and AuNR dimer–UCNP core–satellite superstructures were also reported, based on conventional DNA hybridization, without the fabrication of DNA templates (Fig. 1g and h).11,12 By design of DNA linkers with well-defined interparticle spacing, Mirkin and coworkers demonstrated the DNA-mediated assembly of plasmonic superlattices.31 Face-centered cubic or body-centered cubic AgNP superlattices and binary Ag–AuNP superlattices were fabricated, wherein NPs were located at the end sites of DNA oligonucleotides with three terminal cyclic disulfide groups as robust anchors (Fig. 1i). In a different approach, polymerase chain reaction (PCR)-driven NR assemblies were reported by Xu, Kotov and coworkers.16 AuNRs preferentially bound with thiol-terminated primers at their ends or side facets could act as “monomers” for the PCR replication system in ETE or SBS assemblies, respectively. Specifically, the length and complexity of the assemblies were strongly dependent on the number of PCR thermal cycles (Fig. 1j). The dihedral angle between the adjacent NRs in SBS assemblies acquired negative values of −9.0 for dimers, −7.2 for trimers, −8.0 for tetramers, and −7.0 for pentamers. The consistently negative values corresponded to the right-rotating enantiomers of the SBS assemblies. Importantly, the PCR-driven assembly approach opens a new avenue for the synthesis of large-scale nanoassemblies.
2.2 Peptide/protein-mediated assembly
Similar to DNA hybridization, peptides and proteins can also be used as building blocks to direct plasmonic NP assemblies. This can be achieved through the localized and precise deposition of NPs onto designed supramolecular templates or through antibody–antigen specific recognition systems. Specifically, peptides can be self-assembled into hierarchical superstructures with varying morphologies. The wide range of chemical functional groups present on the template can be used for the oriented nucleation and stabilization of metal NPs on its surface. Rosi and co-workers demonstrated the peptide-based double-helical assembly of AuNPs.18 Peptide nanofiber assemblies comprising L-amino acids (C12-L-PEPAu conjugates, C12-L-PEPAu = [C11H23CO]-AYSS-GAPPMAPPF) were used to achieve left-handed AuNP double helices, by mixing the C12-L-PEPAu conjugates with Au precursor solution and HEPES buffer. The resulting AuNP double helices displayed quantifiable metrics, including regular pitch (∼82 nm), interhelical distance (∼7.5 nm) and interparticle distance (∼1.7 nm), for spherical AuNPs with an average diameter of 5.7 nm (Fig. 2a). The left-handedness of the double helices was derived from the left-handed twist in the nanofiber templates formed from the C12-L-PEPAu conjugates. Similarly, right-handed AuNP double helices were achieved by the use of templates possessing the opposite (D) isomer, namely C12-D-PEPAu conjugates. Likewise, controllable and well-defined AuNP single helices were obtained based on other peptide conjugates, C18-(PEPM-oxAu)2, where PEPM-oxAu was AYSSGAPPMoxPPF.17 Importantly, the C18-(PEPM-oxAu)2 conjugates were arranged perpendicular to the faces in cross-β architecture and assembled into helical ribbons instead of typical twisted nanofibers. As the inner surface of the helical ribbons was sterically hindered, the AuNPs bound only on the outer surfaces via the exposed polyproline α-helices (Fig. 2b). Therefore, the resulting structures were single helices and not typical double helices, exhibiting exceptionally strong chiroptical activity. Stupp and coworkers reported ordered arrays of AgNPs on peptide nanofibers.24 Novel peptide-amphiphiles were self-assembled into supramolecular nanofibers with aldehyde groups on their surface. The aldehyde moieties were employed to reduce two Ag+ ions into Ag2 clusters, being oxidized into carboxylic acid groups (via the Tollens reaction) without an external reducing agent or additives for nucleation control. Reduction of Ag+ ions and subsequent nucleation in the medium led to the formation of uniform and spatially ordered AgNPs over the nanofibers (Fig. 2c). In general, plasmonic nanostructures can be controllably assembled by carefully designing the peptide conjugate templates.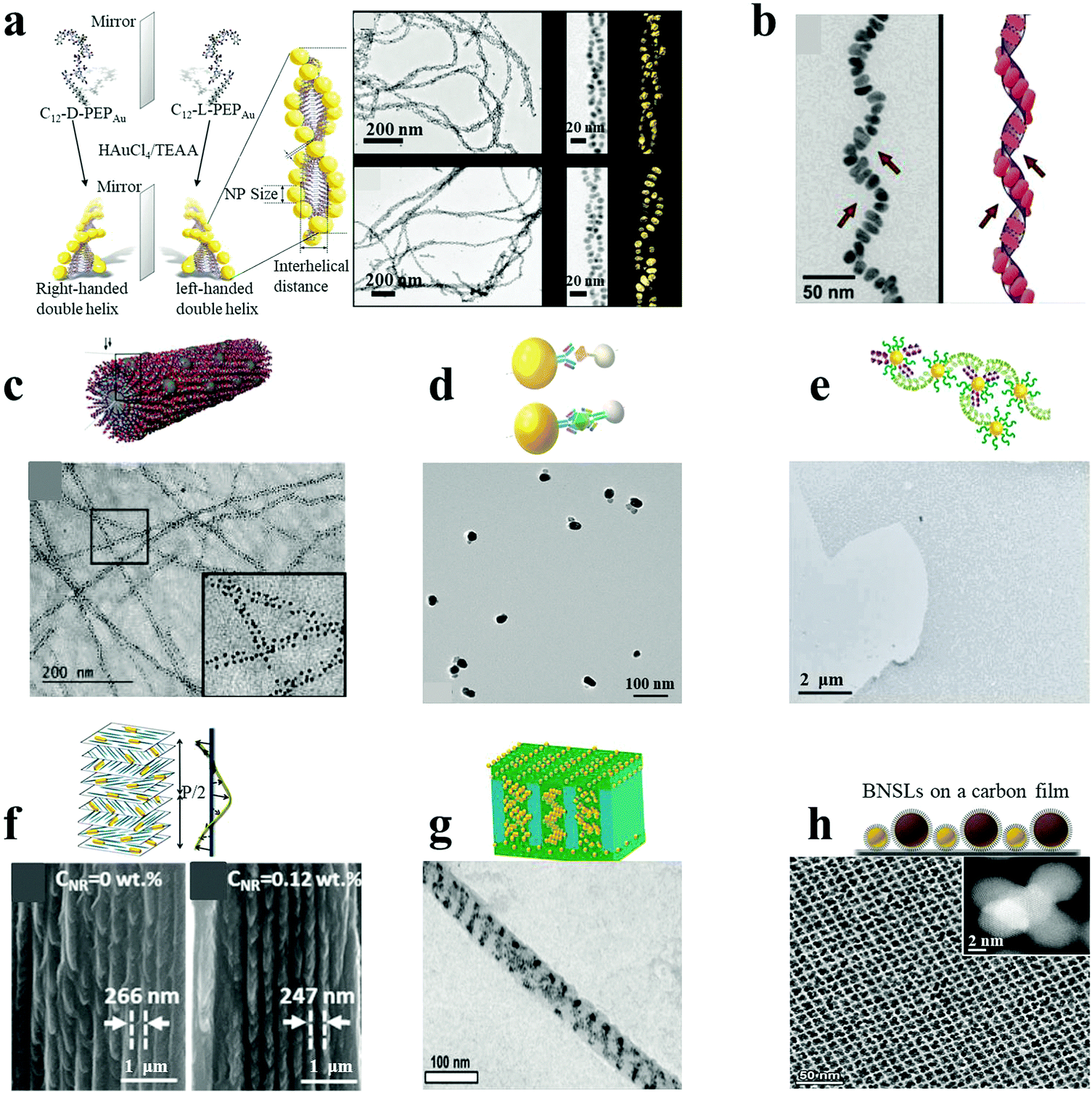 | ||
| Fig. 2 Peptide/protein as well as other supramolecular/polymeric template-mediated plasmonic nanoassemblies (a–c). AuNP double helices (a),18 AuNP single helices (b)17 and AgNP ordered arrays (c)24 were assembled with peptides as the template. (d and e) Au–Ag NP heterodimers (d)4 and massive AuNP film (e)23 were assembled under the direction of proteins. (f and g) Composite AuNR–CNCs film (f)22 and diverse AuNP helices via cylindrical confinement (g)29 were assembled with supramolecular or polymer assemblies as templates. (h) Non-close-packed AuNP array superlattices prepared by post-assembly etching method.25 Reproduced with permission: ref. 4, 17, 18, 22–24 and 29 Copyright 2013, 2014, 2016, 2017 ACS; ref. 25 Copyright 2017 American Association for the Advancement of Science (AAAS). | ||
Similar strategies to those used for peptide-directed NP assemblies can be employed in the case of proteins because they share the same basic functional groups in the amino acid building blocks. However, additional approaches have been reported for protein-mediated NP assembly, based on unique and specific antibody–antigen affinity pairs. As shown in Fig. 2d, Au–Ag NP heterodimers were assembled with high yield through the typical sandwich immunology mode or competitive recognition format.4 In the case of the competitive recognition format, antigen-modified AgNPs were directly recognized by antibody-coated AuNPs, leading to the formation of heterodimers after immunology incubation. In the case of the sandwich format, AuNPs and AgNPs were respectively functionalized with monoclonal antibodies to protein analytes and complementary secondary antibodies, which were linked together by the protein analytes. The NP pairs acquired scissor-like geometry, with the long axes of the NPs forming an angle of ∼9°, corresponding to right-handed rotating enantiomers. In a similar but slightly different approach, Dujardin and coworkers reported the fabrication of massive assembled AuNP films based on repeat protein pair formation.23 The artificial protein pairs, named α-repeat protein pairs, possessed high mutual affinity due to their highly stable periodic architecture and nanomolar dissociation constants. Peptide-primed AuNPs were functionalized with selected interacting proteins and targets by ligand exchange, resulting in high affinity pairs, which drove the self-assembly of AuNPs into a massive film (Fig. 2e).
2.3 Other templates
In addition to biological systems (DNA, peptides and proteins), a variety of other templates have also been employed for the controlled assembly of plasmonic NPs. These include organic assemblies, polymeric substrates and silica nanostructures, to name a few. Some of the earlier work in this area was carried out by using LBL assembly by Kotov and Liz-Marzan.20 This approach was extended by Kotov and coworkers to chiroplasmonic assemblies whose nanoscale architecture was controlled by the macroscale strain.26 Similar approaches can be applied to other plasmonic materials, such as metallic carbon nanotubes.26Plasmonic NPs dispersed or incorporated into supramolecular polymers can self-organize into nanoscale or even microscale superstructures with ordered geometries and three-dimensional (3D) patterns. Kumacheva and co-workers demonstrated the formation of chiral plasmonic films based on cellulose nanocrystal (CNC) assemblies as templates.22 CNCs can assemble into chiral, nematic-ordered, cholesteric liquid crystalline phase layers, forming a left-handed helical structure by the anticlockwise rotation of adjacent CNC layers. Oppositely charged AuNRs can co-assemble with CNCs by mixing within an aqueous suspension and slow water evaporation, so that the NRs are oriented parallel to the CNCs’ long axes and perpendicular to the direction of the left-handed helix (Fig. 2f). The resulting composite plasmonic films with tunable geometries (such as helical pitch and NR dimensions) show tailorable chiroptical activities ranging from the visible to the near-IR spectral regions. A lamellar composite film was assembled by Kramer's group, based on a symmetric block copolymer of polystyrene-b-poly(2-vinylpyridine) (PS-b-P2VP) and PS-coated AuNPs via a selective solvent casting method.32 A two-phase macrostructure was achieved, with a low NPs concentration in the lamellar surface layers and high NPs concentration in the hexagonal substrate layers, presenting NP-induced phase transitions in the self-assembled diblock-copolymer films. Pablo and coworkers assembled block copolymer–NP composite films on chemically nano-patterned substrates.33 They focused on the experimental and theoretical investigation of the location and distribution of NPs within the ordered film, which depended on the thermodynamic state of the composite in equilibrium with the surface. By subjecting supramolecular block-copolymer assemblies to cylindrical confinement, Xu and coworkers succeeded in fabricating complex 3D NP assemblies.29 Block-copolymers formed hexagonally packed cylindrical assemblies, induced by drop-casting the macromolecules onto an anodic aluminum oxide membrane and thermal annealing at 110 °C. By incorporating various amounts of AuNPs, complex 3D nanocomposite nanowires including double rows of spherical NP clusters, single NP arrays, stacked NP rings, single or double helical ribbons could be fabricated (Fig. 2g). In a recent report by Klajn and co-workers, a series of non-close-packed NP superlattices were prepared by a post-assembly etching method.25 Briefly, binary NP superlattices were self-assembled at the diethylene glycol–air interface via evaporation from monodisperse colloids of AuNPs and Fe3O4 NPs, which were subsequently transferred onto a carbon film. The ligands on the NPs were then removed by thermally induced desorption. The resulting tunable porous NP arrays were observed after the selective etching of one component from the binary NP superlattices and the resulting reorganization, exemplified by AuNP tetrahedral quintets formed by dissolving the supporting Fe3O4 NPs (Fig. 2h). Template-less assemblies can also be formed with a variety of plasmonic materials, taking advantage of the intrinsic ability of NPs to self-assemble.
3. Properties of plasmonic nanoassemblies
3.1 SERS effects
SERS is based on the large amplification of the Raman scattering signal from analytes in close proximity to a plasmonic substrate. Typically observed enhancement factors (EFs) range from 102 to 106.28 The huge enhancement mainly originates from two mechanisms: (i) electromagnetic enhancement due to magnification of the electric field and (ii) chemical enhancement by charge-transfer processes between the metal and the analyte.6 Conventional SERS methods are restricted by low or unreliable signals arising from rough noble metal surfaces (Au, Ag or Cu) or uncontrollable hot-spots in random assemblies. Recently, discrete plasmonic nanoassemblies with strong electromagnetic fields and modulated hot-spots have been widely used as SERS-substrates. In a representative example, Suh et al. reported nanogap-tailorable SERS active nanodumbbells, with a single Cy3 dye molecule located between two DNA-tethered AuNPs, followed by Ag shell deposition on the surface of AuNP heterodimers.28 No Raman signals were detected for monomeric Au–Ag core–shell NPs (5 nm or 10 nm Ag shell), AuNP heterodimers without an Ag shell, or Au–Ag core–shell nanodumbbells with thin Ag shell thickness (<3 nm), due to insufficient electromagnetic enhancement (Fig. 3a). Raman signals with an EF of 1012 could be observed from Au–Ag nanodumbbells with ∼5 nm thick Ag shells. However, a larger shell thickness of 10 nm yielded less reproducible SERS signals due to the misplacement of Cy3 dye or formation of improper junctions. In general, an effective electromagnetic enhancement is critical for SERS techniques. In this respect, nanostars have attracted much attention, as they possess multiple tip branches which produce strong intrinsic hot-spots. Uniform 3D Au nanostar dimers with sub-10 nm gaps were fabricated by Toma and co-workers on silicon pillars, via a top-down method.27 The SERS performance of the Au nanostar homodimers was greatly dependent on the polarization of the incident light with respect to the dimer, pillar height, and interparticle spacing. A maximum enhancement factor of 107 was achieved for an optimum geometry of such Au nanostar dimers (Fig. 3b). Sen et al. recently demonstrated much higher EF values originating from the plasmon coupling between the sharp tips and cores of Au nanostar dimers, even up to 2 × 1010 at 7 nm interparticle gaps with 633 nm laser excitation, which were strong enough for single analyte detection.2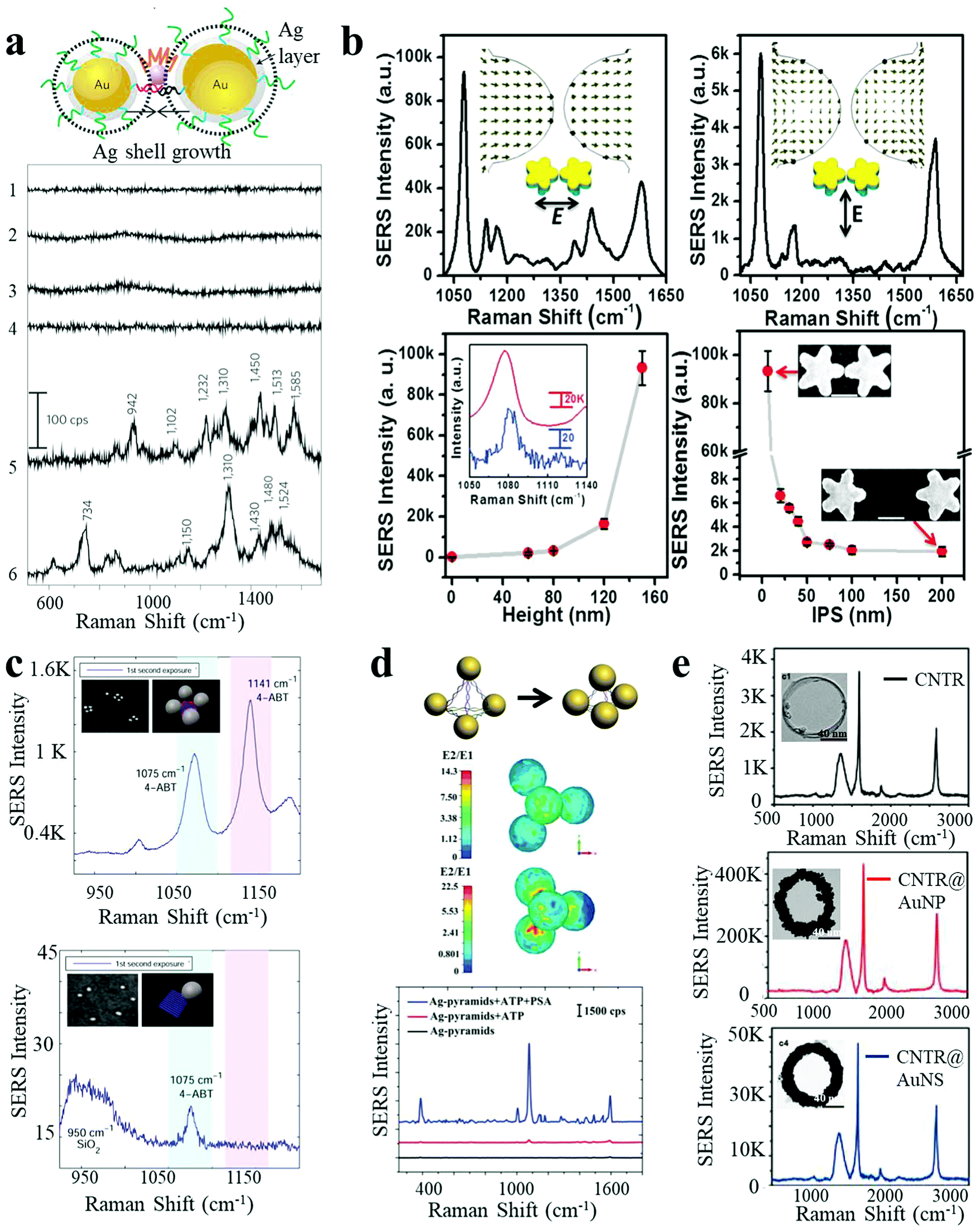 | ||
| Fig. 3 SERS response of plasmonic nanoassemblies. (a) Au–Ag core–shell nanodumbbells with tailorable SERS by Ag-shell growth-based gap engineering.28 (b) 3D Au nanostar dimers with sub-10 nm gap and dependence of SERS intensity on the incident laser polarization direction, pillar height and the interparticle spacing.27 (c) SERS spectra of AuNP tetramers with interparticle hot spots and their corresponding monomer control.10 (d) Ag-pyramids with DNA-frame-driven shorter gap-based SERS response and corresponding electric field simulations.13 (e) SERS spectra of carbon nanotube ring, carbon nanotube ring coated with AuNPs and carbon nanotube ring coated with a complete Au shell.35 Reproduced with permission: ref. 28 Copyright 2010 Macmillan Publishers Ltd; ref. 13 and 27 Copyright 2014, 2015 Wiley-VCH Verlag GmbH & Co.; ref. 10 and 35 Copyright 2014, 2016 ACS. | ||
Besides dimeric structures, more complex assemblies have also been constructed as efficient SERS substrates. Finkelstein et al. built AuNP tetramers using DNA-origami as templates.10 The hot spots resulting from the small gaps between closely spaced NPs endowed the assembled tetramers with significant Raman signal enhancement (larger than a hundred times) per NP, as compared to isolated NPs (Fig. 3c). Based on the fourth-power law for SERS enhancement, the value of the real component of permittivity for the metal is the main factor determining the efficiency. Hence, much larger SERS enhancements can be predicted for Ag as compared to Au at their corresponding LSPR maxima. Therefore, Ag nanomaterials constitute a better choice of SERS substrates in terms of signal intensity.34
In a report by Xu et al., nanoscale pyramids of Ag NPs were assembled using DNA bridges with specific geometry determined by the DNA frame.13 The efficiency of the plasmonic resonances between the NPs in the assembly was relatively small in the expanded state but could be enhanced by the collapse of the nanopyramids, leading to intense plasmon coupling (Fig. 3d). Importantly, multiplexing Raman reporters were simultaneously embedded into the “DNA frame”, making it suitable for subsequent multiplexed biosensing. In addition to materials based on noble metals, carbon nanomaterials have also found application as Raman substrates. Chen et al. reported a SERS-active AuNP–plasmonic hybrid nanostructure comprising carbon nanotube rings coated with AuNPs.35 As shown in Fig. 3e, pure carbon nanotube rings presented intrinsic but low Raman activity, which showed a 110-fold increase after AuNP coating. However, the SERS performance dropped significantly upon complete coating of an Au nanoshell, presumably due to the removal of the plasmon coupling effect.
3.2 Chiroptical properties
Chirality is observed in a wide variety of objects in nature. Many biological molecules, such as nucleic acids, proteins, and sugars, are chiral and have preferred handedness in living organisms. However, these chiral bio- or organic molecules typically display modest chiroptical activity and limited spectral range. Nanoscale engineering is beneficial to the development of new types of chiral materials with the capacity (i) to rotate the polarization of light and (ii) for differential absorption of right- and left-circularly polarized light to a larger extent. Plasmonic NPs and their assemblies exhibit strong optical activity in a wide electromagnetic spectral range, and therefore have attracted much attention, leading to the rise of the field of chiral nanomaterials. Due to the enhanced chiroptical activity, chiral plasmonic nanomaterials can find applications in biosensing and photonic devices.Homo- and heterodimers made of presumably spherical NPs are not expected to display chirality, according to basic symmetry considerations. However, Xu and Kotov reported chiral Au–Ag NP heterodimers, exhibiting a characteristic bisignate line shape in the visible wavelength range (350–700 nm). The structures exhibited a high chiral anisotropy factor (g) of 2.05 × 10−2.4 The strong optical activity was attributed to the prolate shape of the NPs and a consistent dihedral angle, with scissor-like geometry, of the NP dimer. Pronounced CD spectral changes could be observed with the variation in conformation of the dimer. By manipulating the dihedral angles of AuNR dimers, the CD “wave” could be altered through periodic alterations of sign and amplitude in the plasmon region (500–900 nm, Fig. 4a).8 Such spatial and temporal reconfiguration may enable in situ optical monitoring of biological processes or dynamic transitions, as well as optical super fluid which could be switched between positive and negative refractive indices.
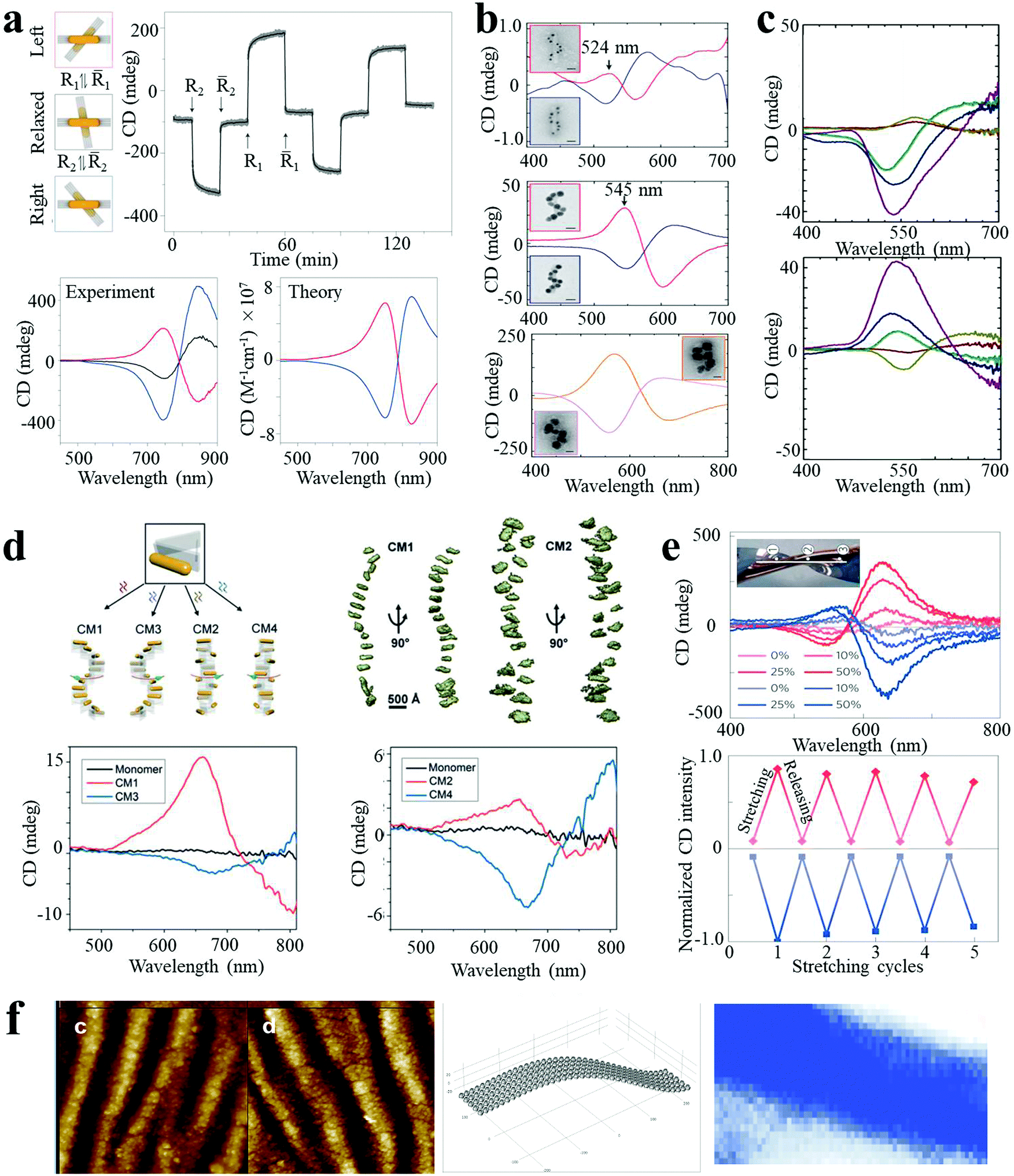 | ||
| Fig. 4 Chiroptical activities of plasmonic nanoassemblies. (a) In situ monitoring of CD signals over time at 725 nm for the different states of AuNR dimer.8 (b and c) Ag deposition induced CD enhancement of AuNP single nanohelices (b)5 and double nanohelices (c).18 (d) Tunable CD of stair-like NR helices and coil-like NR helices.19 (e) CD spectra, peak values for reversible stretching cycles, (f) geometry of NP assemblies and CD surface map of AuNP multilayers.26 Reproduced with permission: ref. 5, 8 and 26 Copyright 2012, 2014, 2016 Macmillan Publishers Ltd; ref. 18 Copyright 2013 ACS; ref. 19 Copyright 2017 Wiley-VCH Verlag GmbH & Co. | ||
Plasmon enhanced optical activity has not only been observed in these simple assemblies, but also in twisted or helical NP or NR superstructures. Liedl and co-workers fabricated chiral AuNP single nanohelices on DNA origami templates, with characteristic bisignate signatures in the visible range.5 The ellipticity value showed a giant increase (a factor of 400 fold) when AuNPs of larger size were used (Fig. 4b). Moreover, the CD intensity was further enhanced (up to 108 M−1 cm−1) by deposition of an Ag shell on the AuNP helices. CD enhancement induced by Ag deposition was also observed in AuNP double nanohelices. As shown in Fig. 4c, the amplitude of the CD peaks was dramatically increased when increasing the Ag shell thickness, with a significant concomitant blue shift.18 Recently, Liu and co-workers reported programmable AuNR superstructures with tailorable plasmonic chiroptical activity.19 Using well-designed DNA aptamers, the NR superstructures were driven to form distinct chiral conformations (stair- or coil-like) and handedness (right- and left-handed), with characteristic peak–dip and dip–peak spectral line shapes (Fig. 4d).
Besides the “rigid” system modifications (templates, changes of interparticle gap or sizes of building blocks, and Ag deposition) described above, the optical activity can also be modulated by the macroscale stretching of “flexible” solid-state substrates. Kotov et al. reported strain-modulated chiroptical nanocomposites made from LBL assembly of AuNP multilayer films on twisted elastic substrates.26 As shown in Fig. 4e, the AuNP multilayer films displayed strong CD bands at the plasmon wavelength, attributed to linear effects of circular extinction and the S-like shape of the NP chains. The ellipticity values increased considerably with increasing strain (by at least ten fold for stretching to 50%) and reverted back as the stress was released, depicting cyclically modulated optical activity by mechanical stretching. These experiments constitute a demonstration of chirality transfer from the macro- to the nanoscale. Similar behavior was obtained for nanocomposites made of carbon nanotubes, expanding the chiroptical activity to the near-IR part of the spectrum.
3.3 Photoluminescence response
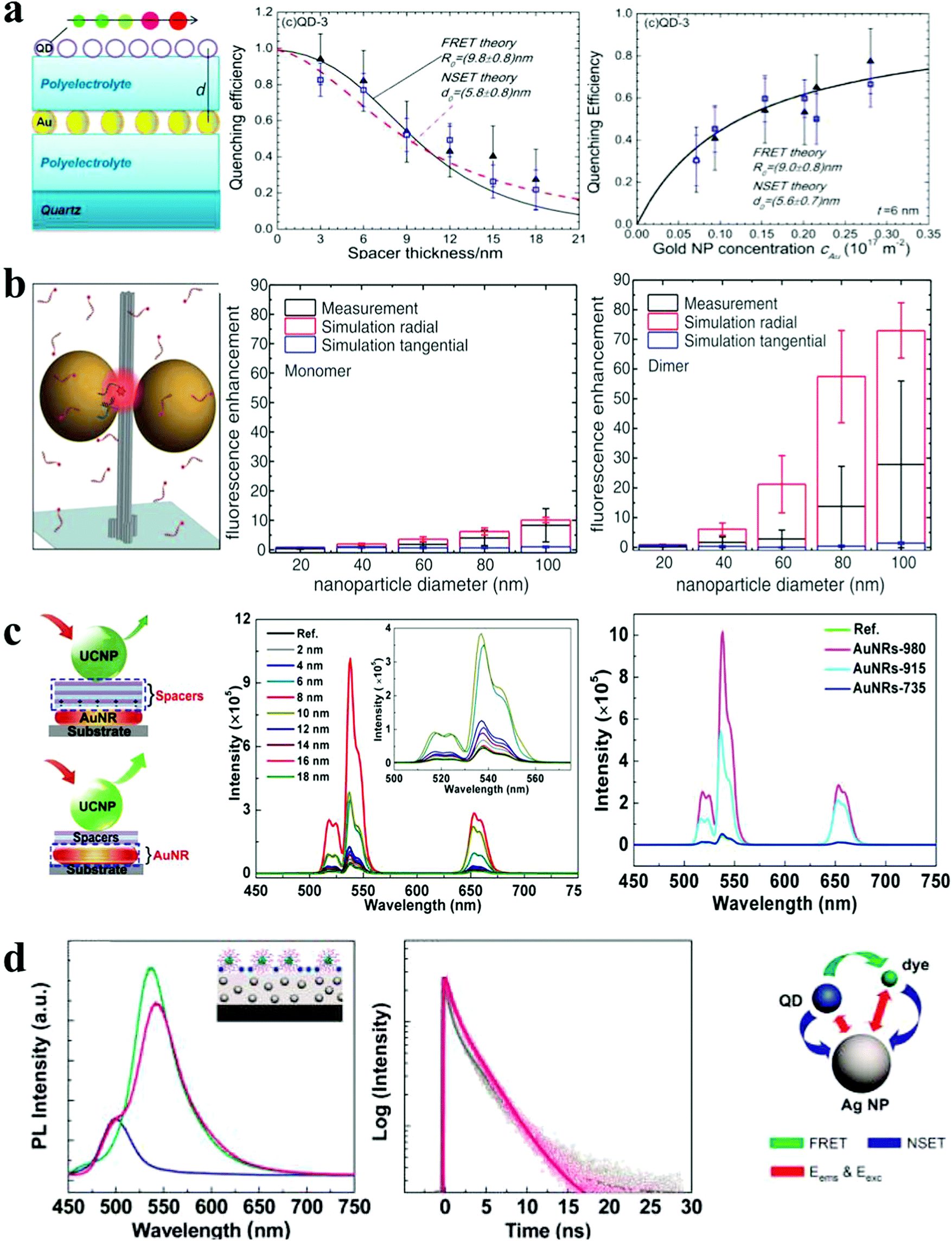 | ||
| Fig. 5 Photoluminescence response of plasmonic nanoassemblies. (a) Distance and concentration dependence of PL quenching efficiency of QD monolayers deposited on top of a AuNP monolayer.21 (b) The mean fluorescence enhancement of dye at docking sites of AuNP nanoantennas with varied NP sizes.3 (c) Distance- and spacer-dependent plasmon-enhanced fluorescence in UCNP.20 (d) Steady-state and time-resolved fluorescence monitoring of hybrid assemblies of AuNP film, QDs and dyes.36 Reproduced with permission: ref. 21 and 36 Copyright 2012 ACS; ref. 3 Copyright 2012 AAAS; ref. 20 Copyright 2015 Macmillan Publishers Ltd. | ||
4. Emerging sensor applications
4.1 LSPR shift-based sensors
A shift in LSPR wavelength can be observed upon refractive index changes in the environment around NPs. In general, LSPR shift-based biosensing primarily relies on target-mediated NP aggregation. However, Jiang et al. reported a dispersion-dominated LSPR method for glutathione detection by target-directed prevention of arginine-modified AuNP aggregation via mercury–thiol interaction.37 A nanomolar limit of detection (LOD) was achieved which enabled the determination of intracellular glutathione levels. In a recent report by Stevens et al., a similar reversed LSPR strategy was employed which was further improved by polymerization-based signal amplification for better sensitivity.38 Polymerization may occur in the presence of free radicals generated from glucose oxidase, which can be inhibited by the presence of catalase. Even a very low monomer-to-polymer conversion can trigger the NP aggregation, resulting in high sensitivity to the presence of radical-generating enzyme targets (catalase sensitivity down to 1 ng mL−1) (Fig. 6a). Tan et al. established a colorimetric sensor platform for detection of exosomal surface proteins, based on a panel of aptamers-complexed AuNPs.39 The AuNPs were non-specifically bound to aptamers by noncovalent complexation, which protected the NPs from aggregation. In the presence of exosome surface proteins, aptamers strongly bound to the proteins, leading to aggregation, which resulted in an obvious color change. The measurement of the exosomal level of CD63 was achieved. This platform also enabled the differentiation of various exosomal proteins within minutes by capturing and profiling exosomal protein information. Large and long-range NP aggregates are unpredictable, uncontrollable and unstable, and show limited sensitivity improvement and a narrow dynamic concentration range of the target analytes. However, Kim et al. demonstrated an ultrasensitive LSPR biosensor based on oriented NP aggregation.40 As shown in Fig. 6b, oriented AuNP dimers were selectively assembled from asymmetrically PEGylated AuNPs with a Y-shaped DNA duplex, in the presence of target DNA sequences, and exhibited highly sensitive concentration-dependent response due to tunable narrow interparticle distances (below 1 nm). This oriented NP aggregation strategy achieved a sensitivity of DNA detection as low as 1.0 pM, which was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold improved as compared to traditional LSPR methods. Furthermore, it offered improved long-term stability and a wider dynamic detection range (>2 orders of magnitude).
000-fold improved as compared to traditional LSPR methods. Furthermore, it offered improved long-term stability and a wider dynamic detection range (>2 orders of magnitude).
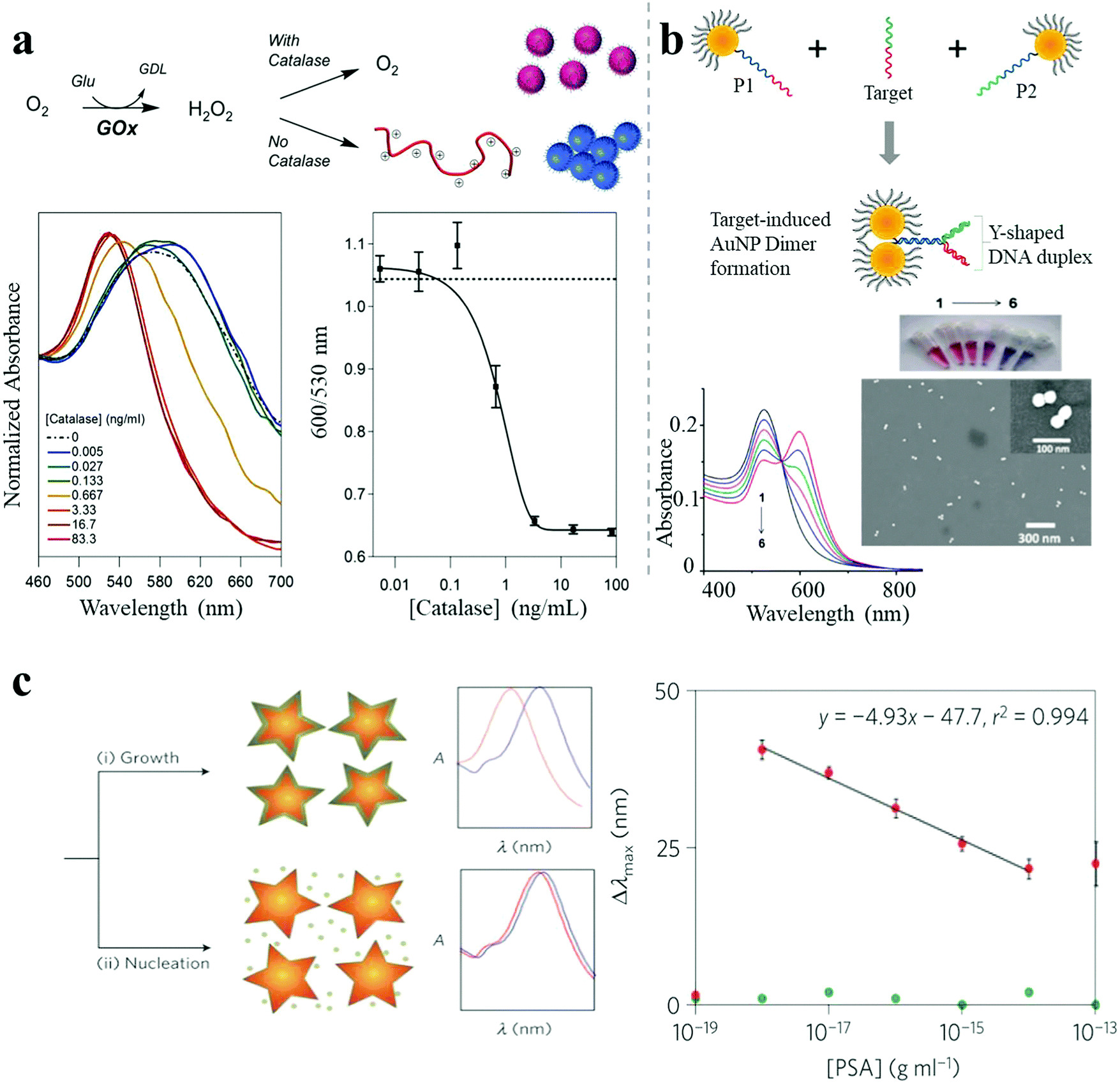 | ||
| Fig. 6 LSPR shift-based sensor. (a) A catalase-sensing inverse assay of polymerization-triggered AuNP aggregation showing catalase sensitivity down to 1 ng mL−1.38 (b) Target oligonucleotide-induced AuNP dimer formation, showing DNA sensitivity down to 1.0 pM.40 (c) Plasmonic nanosensor with inverse sensitivity by enzyme-guided coating of Ag on an Au nanostar leading to large LSPR shift. The nanosystem shows PSA sensitivity down to 10−9 ng mL−1 (4 × 10−20 M) in whole serum.30 Reproduced with permission: ref. 38 and 40 Copyright 2013, 2014 ACS; ref. 30 Copyright 2012 Macmillan Publishers Ltd. | ||
Besides the great improvements achieved in conventional methods, various studies have reported the development of new classes of LSPR sensors. Stevens and co-workers reported an ultrasensitive LSPR biosensor based on the enzyme-guided crystal growth of plasmonic NPs.30 A thin Ag shell could be coated on Au nanostars under low concentrations of Ag+ ions, resulting in a large LSPR blue shift, whereas only a smaller shift was observed for high concentrations of Ag+ ions, due to the growth of Ag nanocrystals instead of Ag shell formation (Fig. 6c). Importantly, the concentration of Ag+ ions could be reduced in a controlled manner by additional enzymes such as glucose oxidase. When Au nanostars and the enzyme glucose oxidase were modified with monoclonal antibodies against targets and secondary antibodies, a plasmonic ELISA occurred with an inverse LSPR response as the sensing signal. This was demonstrated by the successful detection of a cancer biomarker (prostate specific antigen, PSA), with a sensitivity down to 10−9 ng mL−1 (4 × 10−20 M) in whole serum. The outstanding detection performance was attributed to the inverse sensitivity regime. Importantly, LSPR sensing has completely different sensing mechanisms than other sensing techniques (e.g. SERS), so that hot spots are not so critical in LSPR sensing as in SERS sensing. LSPR sensing relies on changes due to local variations of the refractive index, which are closely related to the polarizability of the NPs or their assemblies. Therefore, nanostructures with a larger aspect ratio (NRs for example) are more sensitive to variations of the local environment, due to a strong and intrinsic polarization dependence. In general, the sensitivity will improve if the orientation of these elongated structures is matched with the polarization of the electric field.
4.2 SERS-based sensors
As described above, SERS spectroscopy is a powerful sensing technique, extensively used for non-invasive and ultrasensitive detection of a wide range of molecules. Recent advances in SERS detection include the reports by Tian et al. of shell-isolated NP-enhanced Raman spectroscopy (SHINERS), with amplified Raman signal induced by AuNPs coated with ultrathin silica or alumina layers.6 Importantly, the ultrathin coating enabled a shell-isolated mode, which exhibited stronger hot-spots and reduced agglomeration of NPs. Simple “smart dust” (shell-isolated NPs) spreading over the probed surface improved the sensitivity and reproducibility of SERS biosensing. This was demonstrated by successful practical applications including in situ probing of hydrogen adsorption on a Pt/Si surface, living yeast cells and pesticide residues on food or fruits (Fig. 7a).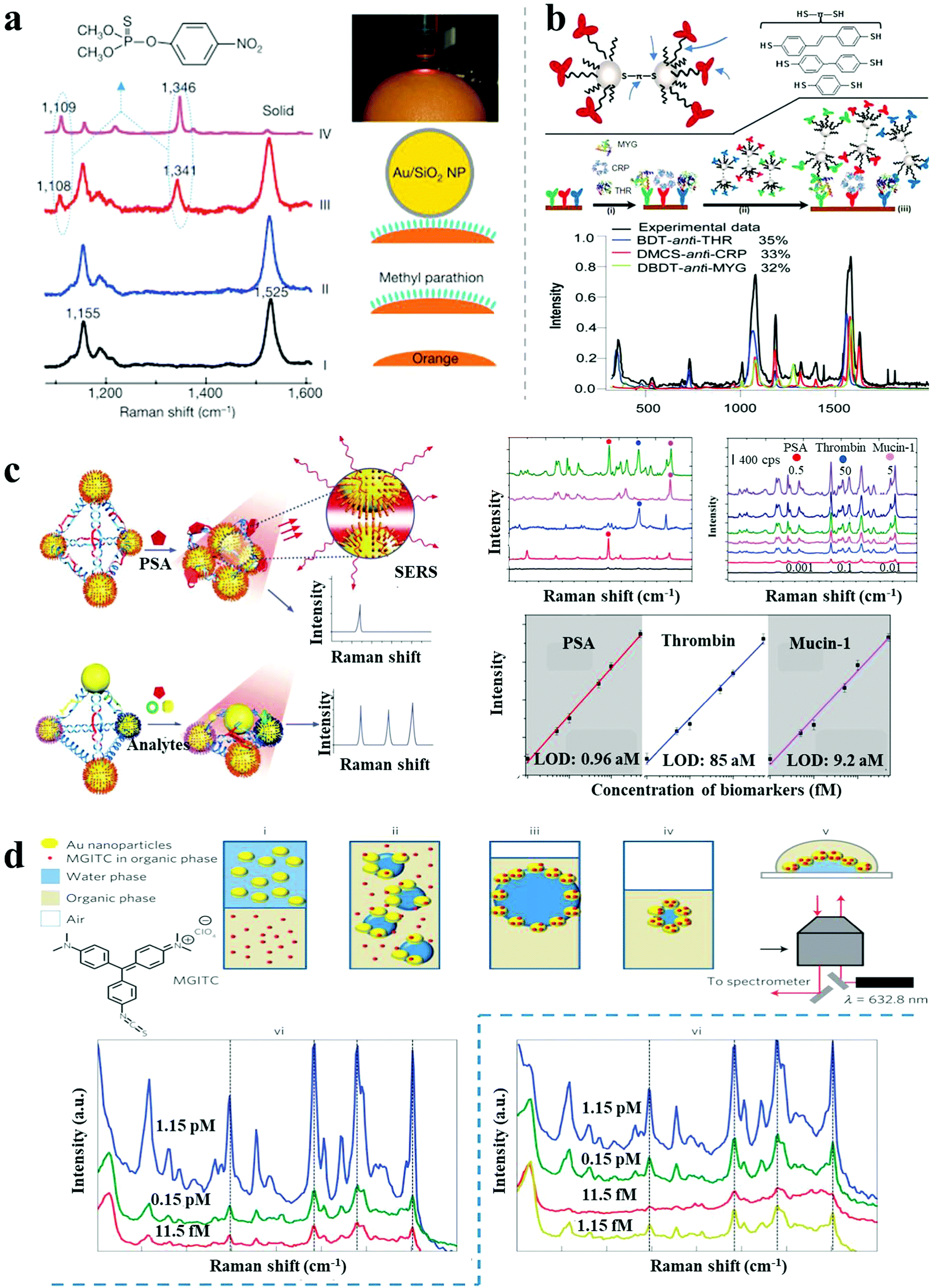 | ||
| Fig. 7 SERS-based sensors. (a) In situ probing of pesticide residues on food/fruit by SHINERS.6 (b) Scheme and SERS spectra of single-spot multiplex protein detection based on SERS-active AgNP dimer “antitag”.43 (c) Ag pyramids SERS sensor for attomolar multiplexed disease biomarker analysis.13 (d) SERS encoded AuNP arrays assembled at liquid/liquid interface for trace dual-phase-dual-analyte detection.44 Reproduced with permission: ref. 6 and 44 Copyright 2010, 2013 Macmillan Publishers Ltd; ref. 13 and 43 Copyright 2013, 2015 Wiley-VCH Verlag. | ||
Recently, a variety of approaches were reported for multiplex SERS biosensing based on plasmonic nanoassemblies. Bazan et al. demonstrated multiplex protein detection by SERS-encoded AgNP dimers.43 As shown in Fig. 7b, AgNP dimers were assembled by dithiolated Raman-active molecules, and subsequently modified with monoclonal antibodies against a protein target, serving both as the capture probes and Raman-sensing probes in a sandwich immunoassay. By simply coating with different Raman reporters and related antibodies, the AgNP dimer tags enabled single-spot simultaneous detection of multiplex protein targets with 100 pM sensitivity. SERS-active Ag pyramids were also reported for multiplexed cancer biomarker analysis.13 The plasmonic nanopyramids were assembled by AgNPs based on a well-designed DNA framework, embedding multiplex biomarker-specific aptamers in the DNA frame and coating different Raman reporters on the corresponding AgNPs. In the presence of target proteins, increased Raman intensities were observed because of stronger hot-spots generated from the shorter gaps, showing attomolar detection of proteins (0.96 aM for PSA, 85 aM for thrombin and 9.2 aM for mucin-1) in one pot (Fig. 7c).
Most examples of SERS biosensing have been performed in a single phase, either on solid substrates or in solution. Recently, Edel et al. demonstrated multiphase SERS detection for trace analytes.44 Hydrophilic AuNPs in the water phase and Raman active target analytes in an organic phase under gentle agitation resulted in a thin AuNP layer incorporated with analytes that self-assembled at the liquid/liquid interface. Upon transfer onto a coverslip for SERS measurements, a LOD of ∼10 fM was achieved for the fluorophore malachite green isothiocyanate (Fig. 7d). Under similar conditions, when the analytes were initially dissolved in the water phase, the LOD was further improved to 1.15 fM, owing to direct binding to the NPs. This multiphase SERS sensor can achieve simultaneous dual-analyte detection or be used at a liquid/air interface, widely expanding the scope of the SERS biosensing platform. In a recent report by Liz-Marzán and co-workers, in situ and ultrasensitive detection of a quorum-sensing signalling metabolite (pyocyanin) in growing Pseudomonas aeruginosa biofilms and microcolonies was achieved by surface-enhanced resonance Raman scattering (SERRS) spectroscopy.45 Pyocyanin, produced by P. aeruginosa, presents a broad absorption band in the visible-NIR region (550–900 nm), which exhibits resonant Raman scattering under laser excitation at 633 nm or 785 nm. Therefore, the recording of SERRS, rather than the more common SERS, enabled higher enhancement factors for extremely sensitive biosensing. Micropatterned Au@SiO2 supercrystal arrays were fabricated as the enhancing substrate, comprising AuNRs organized in micrometre-sized pedestal-like structures coated with mesoporous silica. The supercrystals showed a high density of efficient hotspots and collective plasmonic modes, enabling in situ detection of pyocyanin down to 10−14 M, thus being a powerful analytical approach for studying intercellular communication on the basis of secreted molecules as signals.
4.3 Chiroplasmonic sensors
CD spectroscopy is an accurate method for the determination of higher order hierarchical structures of chiral biomolecules and secondary/tertiary structure of proteins. However, the low sensitivity and the need for UV excitation have greatly limited its applicability to biosensing. Chiroplasmonic nanoassemblies with new CD bands in the visible or near-infrared wavelength ranges make it possible to develop CD-based biodetection with high sensitivity. Chiroplasmonic side-by-side AuNR assemblies based on the PCR method demonstrated ultrasensitive biosensing of long DNA strands.16 The side-by-side assembly of NRs resulted in strong bisignate CD signals in the plasmonic region (500–800 nm), attributed to the twisted conformation of NRs (7–9 degree twist between the NR axes). The amplitude of the CD signals showed a positive relation to the AuNR numbers in the assembly (n < 10), and the g-factor increased from 1.6 × 10−3 to 2.3 × 10−3 for 2–10 PCR cycles. This CD response was used for the biodetection of oligonucleotides by employing reactant DNA templates within the PCR process, with a low LOD of 3.7 aM (Fig. 8a). This was considerably more sensitive than typical PCR (0.1 fM), reverse transcription PCR (156 aM) or SERS (1.14 fM). Importantly, the chiroplasmonic method was less dependent on the gap distance, as compared to the conventional methods wherein hot-spots are employed (such as LSPR, SERS), making it particularly useful for the analysis of larger analytes (>2 nm). Subsequently, Au–Ag NP heterodimers demonstrated expanded bioanalytical applications for other biological compounds with improved LODs.4 Targeted peptides or proteins induced heterodimer assemblies with ultrastrong and linear decrease or increase in CD intensity based on competitive recognition or sandwich immunoassay patterns. The determined LOD was 0.8 pM for an environmental toxin (microcystin-LR) and 15 zM for a cancer biomarker (PSA) (Fig. 8b). Importantly, DNA, peptides or proteins modified on the surface of NPs/NRs may serve as recognition probes, which drive the self-assembly between the NPs or NRs. As their biological functions are retained in the assemblies, the robustness of performance can even be improved as compared to discrete conditions. Recently, Kadodwala et al. demonstrated an improved LOD in the bioanalysis of higher order hierarchical protein structures based on chiral plasmonic nano-metamaterials.46 Left-handed and right-handed plasmonic “shuriken” nanostructures fabricated using a top-down method exhibited bisignate CD signals in the 700–750 nm spectral region. The enhanced sensitivity of superchiral evanescent fields from the shuriken-like structures enabled picomolar detection of protein higher order structures (tertiary/quaternary) by CD spectroscopy (Fig. 8c).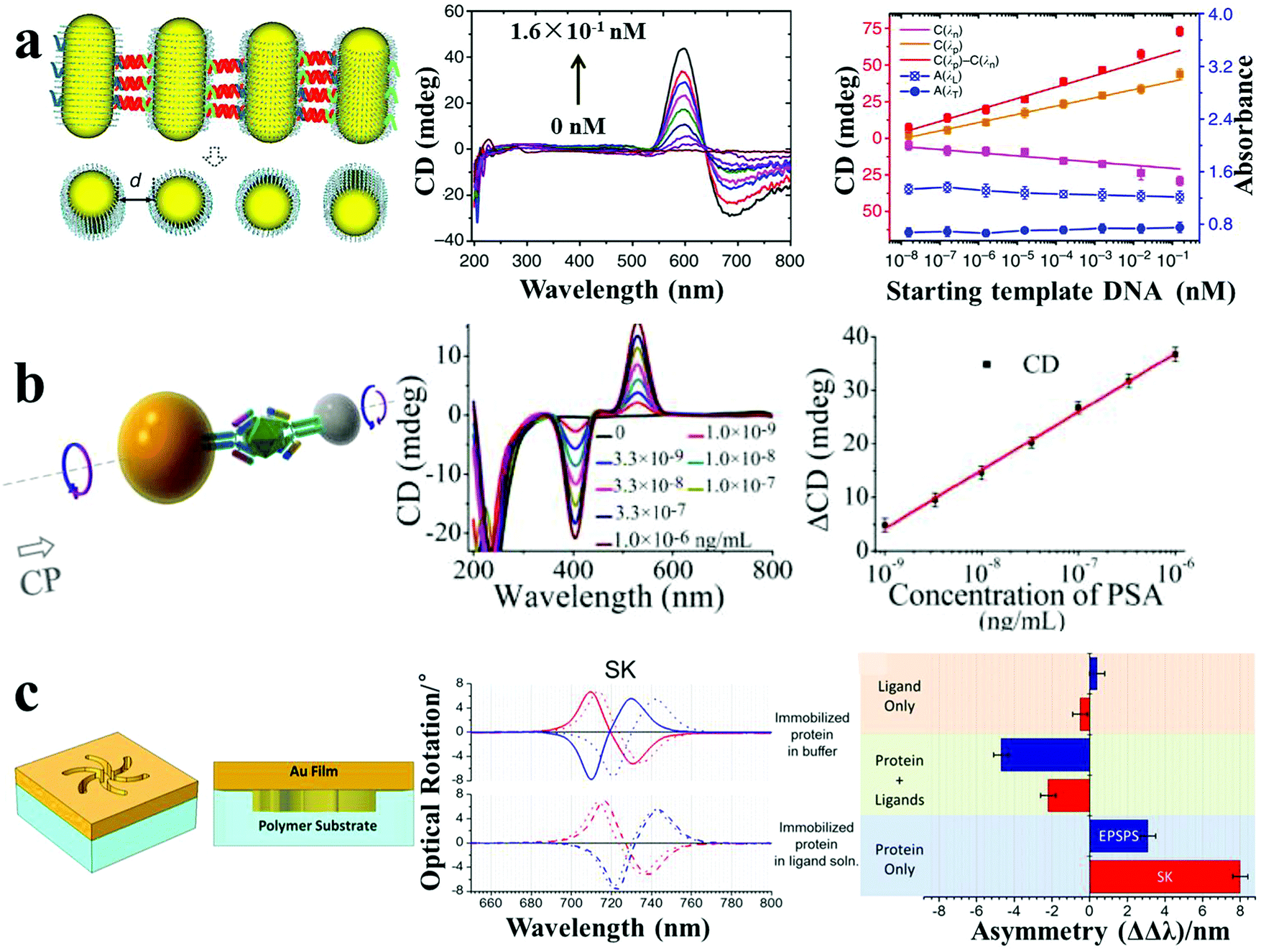 | ||
| Fig. 8 Chiroplasmonic sensors. (a) PCR-based SBS AuNR assemblies and CD response for attomolar DNA analysis.16 (b) Sandwich immunoassay-guided Au–Ag heterodimer assembly and resulting CD response for ultrasensitive PSA detection.4 (c) Left-handed chiroplasmonic Shuriken nanostructures and CD response for detection of the higher order hierarchical structure of immobilized proteins. ΔΔλ values were obtained from the collected optical rotatory dispersion spectra, where ΔΔλ = ΔλR − ΔλL (ΔλR and ΔλL are resonance wavelength shifts measured on right-handed and left-handed Shuriken nanosubstrates, respectively).46 Reproduced with permission: ref. 16 Copyright 2013 Macmillan Publishers Ltd; ref. 4 and 46 Copyright 2013, 2015 ACS. | ||
4.4 Photoluminescence-based sensors
Photoluminescence-based detection is another popular sensing method, widely used in the field of biotechnology and life sciences, mainly relying on strategies including the quenching and recovery of luminescence via NSET, plasmon-enhanced luminescence, and FRET response. Spherical nucleic acids, also called nanoflares, were reported by Mirkin's group for single-cell level detection of intracellular mRNA in live cells.47 The nanoflares were composed of 13 nm spherical AuNPs densely modified with a monolayer of antisense single-stranded DNA, serving as the recognition sequence. A reporter flare sequence containing a dye was hybridized to the nanoflares, leading to fluorescence quenching. When the complementary mRNA target was present, it displaced the reporter flare and bound to the recognition sequence, resulting in recovery of the fluorescence signal. Coupling with flow cytometry enabled nanoflares for detection of genetic markers of circulating tumor cells with as few as 100 live cancer cells per milliliter of blood. Recently, hybrid nanopyramid NPs with dual luminescent response were reported for intracellular dual microRNAs biodetection.14 The hybrid pyramids were assembled with one UCNP, two Ag2S NPs, and one Au–Cu9S5 NP, with the direction of a well-defined DNA frame. Two sensing DNA sequences were embedded in the DNA skeleton that could be respectively recognized by two target microRNAs (miR-21 and miR-203b). In the presence of miR-21, the Ag2S NPs separated from the pyramids, leading to the recovery of the quenched luminescence. When miR-203b was present, a similar situation occurred for the UCNPs. Due to the NIR excitation and distinct emission spectral regions (NIR-II emission of Ag2S NP and visible emission of UCNPs), the assembled nanopyramids showed excellent photostability and high signal-to-noise ratio for intracellular detection. The LOD was 0.09 fM/10 μgRNA for miR-203b and 0.23 fM/10 μgRNA for miR-21 (Fig. 9a). The developed method could distinguish between normal cells (primary uterine fibroblast cells, low microRNA expression) and cancer cells (human epithelial cancer cells, high microRNA expression). Moreover, the expression levels of miR-21 and miR-203b, which are difficult to measure with conventional methods, were accurately quantified for the cancer cells using this technique.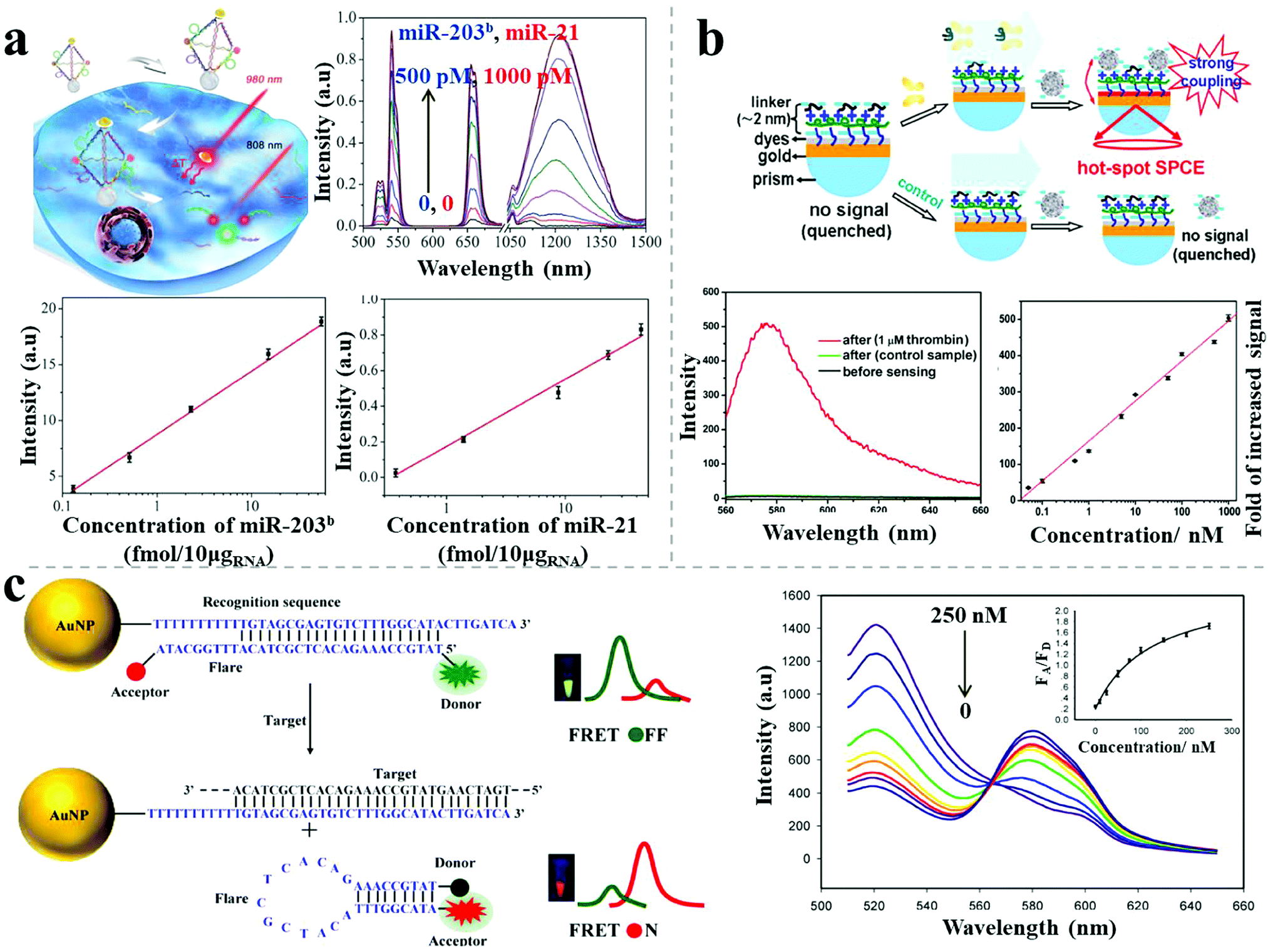 | ||
| Fig. 9 Photoluminescence-based sensors. (a) Plasmonic hybrid NP pyramids and luminescent recovery effect for intracellular dual microRNA quantitation.14 (b) AgNP–dyes–Au film assemblies and hot-spot induced emission for thrombin detection.48 (c) AuNP-based FRET nanoflares and fluorescence response for intracellular mRNA detection.49 Reproduced with permission: ref. 14 Copyright 2017 Wiley-VCH Verlag GmbH & Co.; ref. 48 and 49 Copyright 2014, 2015 ACS. | ||
Recently, Li et al. reported a sensitive label-free aptamer sensor based on directional surface plasmon coupled emission in nanoscale assemblies.48 As shown in Fig. 9b, a layer of dye-coated Au nanofilm was fabricated onto a quartz substrate, with efficiently quenched fluorescence because of NSET. An ultrathin linker layer, composed of cationic polymers and abundant negative-charged aptamers, was employed on the surface of the dye-coated Au nanofilm, serving both as the linker for subsequent NP connection and as the sensing layer for the targets. In the presence of targets, the introduced AgNPs assembled on the surface of the linker layer due to electrostatic attraction, leading to fluorescence emission via significant plasmon coupling. In contrast, in the absence of targets, the AgNPs did not undergo assembly on the surface due to strong electrostatic repulsion, resulting in undetectable fluorescence. A linear calibration curve of thrombin sensing was established, showing a LOD of 33 pM. The selectivity of the sensor was tested, showing no obvious influence by either negatively charged proteins (bovine serum albumin) or positively charged proteins (lysozyme), even at 100-fold excess of the target proteins. The use of ultrathin layers triggered the capture of the target protein (aptamers as the recognition probes) whereas the intense plasmon-enhanced fluorescence emission guaranteed high specificity and sensitivity for the biosensor.
To avoid false positive signals, Wang et al. reported upgraded FRET nanoflares for intracellular mRNA detection.49 The FRET nanoprobe was composed of an AuNP, a recognition sequence and a donor–acceptor labeled flare. In the absence of targets, low FRET efficiency was observed because of the separation of the labeled donor and acceptor on the flare. When the target DNA was present, the flares dissociated and formed hairpin structures, resulting in high FRET efficiency because of the proximity of the donor and acceptor (Fig. 9c). Through this upgraded FRET method, the expression levels of TK1 mRNA were successfully determined in different cancer cells (human liver hepatocellular carcinoma cell line, breast cancer cell line) and various tumor progression stages of cancer cells. Importantly, no false positive was observed for the FRET nanoflares in contrast to the conventional single-dye nanoflares, which was attributed to minimized thermodynamic fluctuations and to chemical anti-interference.
4.5 Combined methods
Apart from the single-mode sensors described above, combined methods comprising different kinds of individual sensors have been reported, with improved sensitivity or specificity, due to the combined merits of the individual modes. Jiang et al. reported a dual readout (colorimetric and fluorometric) assay for highly sensitive acetylcholinesterase detection based on AuNP assemblies.50 Dispersed rhodamine B-modified AuNPs aggregated under the influence of thiocholine generated from the hydrolysis of acetylthiocholine catalyzed by the presence of the acetylcholinesterase target, resulting in simultaneous fluorescence recovery and colorimetric responses. The ratios of the UV-vis absorption spectral areas (aggregated/dispersed area under the absorption peak) and fluorescence intensity were calculated to monitor the acetylcholinesterase level in the cerebrospinal fluid of transgenic mice suffering from Alzheimer's disease. The LOD was 1.0 mU mL−1 for the colorimetric method and 0.1 mU mL−1 for the fluorescence method, much lower than other AuNP-based acetylcholinesterase biosensors (Fig. 10a).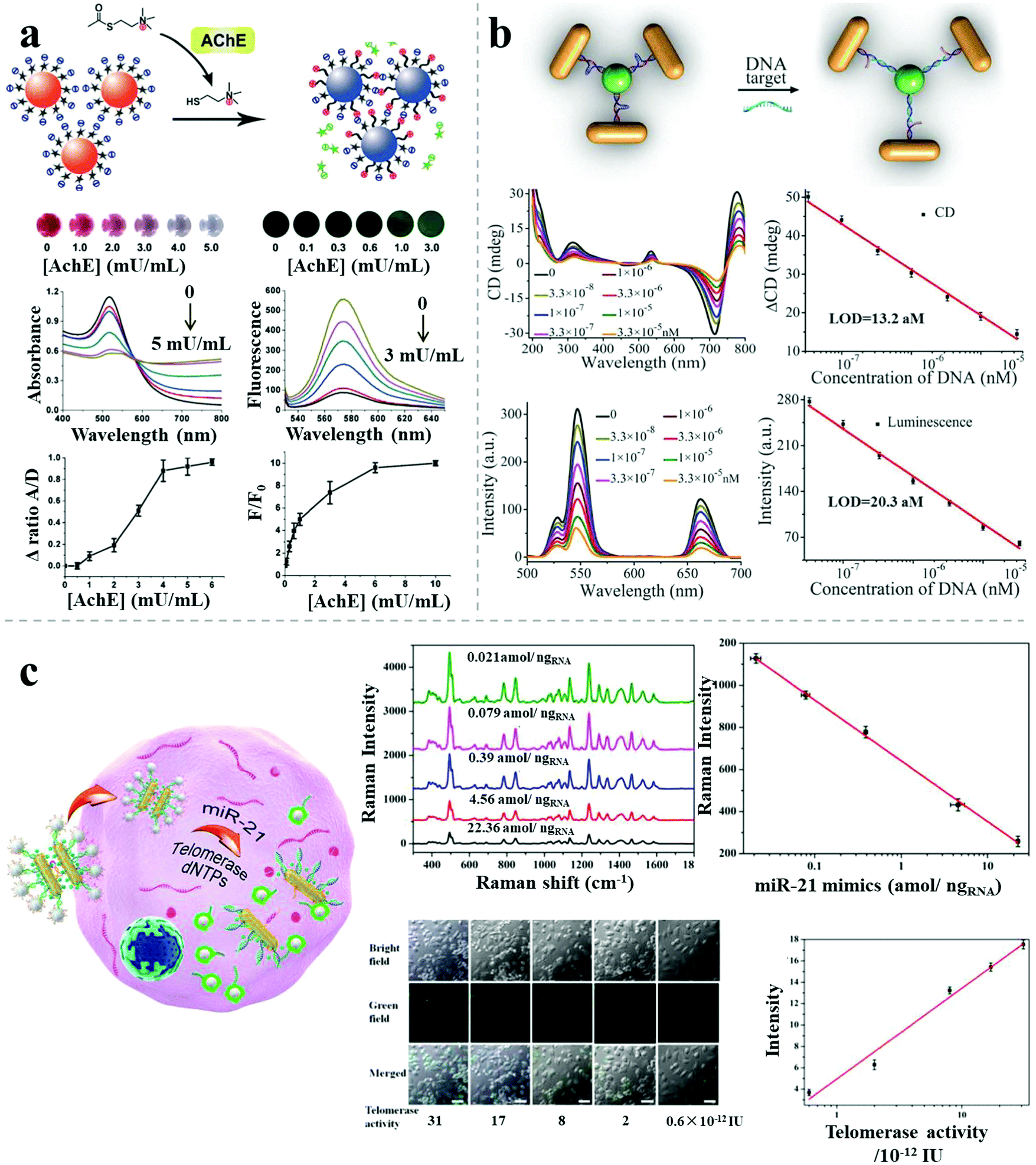 | ||
| Fig. 10 Combined methods for sensing. (a) Rhodamine B–AuNP assemblies with colorimetric and fluorimetric responses for acetylcholinesterase detection.50 (b) Propeller-like NR–UCNP assemblies with chiroptical and plasmonic luminescence enhancement responses for DNA detection.12 (c) AuNR dimer–UCNP core–satellite assemblies for intracellular dual-analyte detection based on SERS effect and luminescent recovery.11 Reproduced with permission: ref. 12 and 50 Copyright 2012, 2016 Wiley-VCH Verlag GmbH & Co.; ref. 11 Copyright 2017 ACS. | ||
Recently, Xu demonstrated a bimodal DNA cancer biomarker detection method with improved reliability and versatility based on propeller-like NR–UCNP plasmonic tetramers.12 The assembled tetramers, embedded with hairpin-like recognition DNA sequences, possessed distance-dependent chiroplasmonic activity and enhanced upconversion luminescent emission. In the presence of DNA targets (Vall7 polyprotein gene from the hepatitis A virus), the tetramers adopted longer distances because of the extended conformation of the hairpin-like DNA strands, resulting in linearly reduced intensities of upconversion luminescence emission as well as of the CD signal. The determined LOD was 20.3 aM by the luminescent signal and 13.2 aM by the CD signal. The dual signals showed good agreement in analysis of DNA from a complex biological matrix, which enabled more accurate quantitative detection of cancer biomarkers for early disease diagnosis (Fig. 10b).
Besides the improved sensitivity, specificity or accuracy, combined methods can also pave the way for high-throughput biosensing. A recent report demonstrated dual target simultaneous intracellular biodetection by AuNR dimer–UCNP core–satellite assemblies.11 As shown in Fig. 10c, SERS-active AuNR dimers initially assembled with Raman dye molecules-modified miR-21 recognition sequences, but disassembled in the presence of the miR-21 target, leading to reduced SERS response. UCNPs were then assembled around the AuNR dimers through mismatched telomerase sequences, which exhibited quenched luminescence. When the telomerase targets were present, the quenched luminescence was recovered by weak NSET for larger interparticle distances between the plasmonic NRs and UCNPs. Therefore, dual detection was achieved based on the independent, simultaneous SERS and luminescence responses, with a LOD of 3.2 × 10−13 IU for intracellular telomerase and 0.011 aM ngRNA−1 for intracellular miR-21.
5. Concluding remarks and perspectives
In summary, rapid developments have been achieved in plasmonic nanoassemblies and detection applications, making great progress in the design and construction of nanostructures, manipulation of plasmonic properties, and sensing applications. Precisely assembled nanostructures with specific spacing, angle and configuration have been fabricated, even for multi-component, hierarchical and sophisticated superstructures such as single/double helices and superlattice arrays. Further, a variety of synergistically enhanced and tailorable plasmonic properties (SERS, plasmon-enhanced luminescence, plasmon-induced CD) have been realized, and diverse, novel multi-modal responses achieved (multiplex luminescence/Raman, LSPR-luminescence, Raman-CD, luminescence-Raman). In the realm of sensing applications, plasmonic assemblies enable extremely high sensitivity down to the single particle or even single molecule level, which are difficult to achieve by conventional methods. The simultaneous multi-modal response characteristics of the engineered assemblies enable precise and reliable quantitative analysis for various target analytes in vitro and in vivo, including ions, heavy metals (Pb, Hg), small molecules (biotoxins, antibiotics, pesticides, narcotics, explosives), biomacromolecules (peptides, proteins, enzymes, DNAs, RNAs), as well as for the sensitive determination of the secondary/tertiary structure of proteins, and enantiomer discrimination.Plasmonic nanoassemblies have achieved remarkable sensitivities, breaking through the limits of conventional detection methods. However, further improvements and developments are still needed. First of all, some of the current assemblies are based on expensive and complex templates such as origami constructions. It will be desirable to simplify their synthetic protocols to allow broader utilization in clinical practice. Second, more advanced assembly strategies must be developed for the fabrication of high-yield and high-throughput nanostructures. Third, the stability and plasmonic properties of the nanostructures, along with their high sensitivity, should be preserved when in complex systems such as in the cell matrix. Further research is required for the exploration of intracellular retention, toxicity, biodegradability and tissue penetration of plasmonic assemblies. Fourth, NP constructs for plasmon-mediated SERS require high stability and reproducibility in the cell matrix, high penetration in vivo, and in situ visualization. Fifth, chiroplasmonic nanoprobes have reached attomolar sensitivity, even in cells. Hence, CD instruments with capabilities of multiplexed imaging and portable chiroptical instruments are highly desirable. Expansion to higher hierarchical structures or other biomolecules can be explored with new CD instruments with IR capabilities. Sixth, plasmon-enhanced luminescence has been reported for the detection of various targets, however, better synthetic methods and regulatory strategies are required toward addressing the difficulties related to precise manipulation. Seventh, current plasmonic nanosensors are mainly focused on the proof of concept, and are far from practical applications. There is still a relatively long way to address the barriers of entry into the mainstream market. This includes long-duration testing for the stability and reproducibility of the nanoscale assemblies in complicated matrices, especially under high concentrations of protein or fatty acids in physiological conditions. Overall, addressing some of the above challenges can lead to miniaturized devices based on plasmonic assemblies that display fast response, extremely high sensitivity and specificity, robustness and simplicity of use.
Conflicts of interest
Other authors declare no competing financial interest.Acknowledgements
XW acknowledges funding from Natural Science Foundation of Jiangsu Province (BK20150138). L. M. L.-M. acknowledges funding from the Spanish Ministry of Economy and Competitiveness (Project MAT2017-86659-R). NAK acknowledges the support of Department of Army project Reconfigurable Matter from Programmable Colloids (W911NF-10-1-0518) and NSF project “Energy- and Cost-Efficient Manufacturing Employing Nanoparticles” (1463474). JK acknowledges financial support from the European Commission under Marie Sklodowska-Curie Program H2020-MSCA-IF-2015 (Grant 708321, CINMAB). This work is financially supported by the National Natural Science Foundation of China (21771090, 21631005, 21673104, 21522102, 21503095, 31771084).References
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS PubMed
.
- S. Tanwar, K. K. Haldar and T. Sen, J. Am. Chem. Soc., 2017, 139, 17639–17648 CrossRef CAS PubMed
- G. P. Acuna, F. M. Möller, P. Holzmeister, S. Beater, B. Lalkens and P. Tinnefeld, Science, 2012, 338, 506–510 CrossRef CAS PubMed
- X. Wu, L. Xu, L. Liu, W. Ma, H. Yin, H. Kuang, L. Wang, C. Xu and N. A. Kotov, J. Am. Chem. Soc., 2013, 135, 18629–18636 CrossRef CAS PubMed
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E.-M. Roller, A. Högele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS PubMed
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, Y. WuDe, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed
- P. Kühler, E.-M. Roller, R. Schreiber, T. Liedl, T. Lohmüller and J. Feldmann, Nano Lett., 2014, 14, 2914–2919 CrossRef PubMed
- A. Kuzyk, R. Schreiber, H. Zhang, A. O. Govorov, T. Liedl and N. Liu, Nat. Mater., 2014, 13, 862–866 CrossRef CAS PubMed
- E.-M. Roller, L. V. Besteiro, C. Pupp, L. K. Khorashad, A. O. Govorov and T. Liedl, Nat. Phys., 2017, 13, 761–766 CrossRef CAS PubMed
- M. Pilo-Pais, A. Watson, S. Demers, T. H. LaBean and G. Finkelstein, Nano Lett., 2014, 14, 2099–2104 CrossRef CAS PubMed
- W. Ma, P. Fu, M. Sun, L. Xu, H. Kuang and C. Xu, J. Am. Chem. Soc., 2017, 139, 11752–11759 CrossRef CAS PubMed
- X. Wu, L. Xu, W. Ma, L. Liu, H. Kuang, N. A. Kotov and C. Xu, Adv. Mater., 2016, 28, 5907–5915 CrossRef CAS PubMed
- L. Xu, W. Yan, W. Ma, H. Kuang, X. Wu, L. Liu, Y. Zhao, L. Wang and C. Xu, Adv. Mater., 2015, 27, 1706–1711 CrossRef CAS PubMed
- S. Li, L. Xu, M. Sun, X. Wu, L. Liu, H. Kuang and C. Xu, Adv. Mater., 2017, 29, 1606086 CrossRef PubMed
- M. J. Urban, P. K. Dutta, P. Wang, X. Duan, X. Shen, B. Ding, Y. Ke and N. Liu, J. Am. Chem. Soc., 2016, 138, 5495–5498 CrossRef CAS PubMed
- W. Ma, H. Kuang, L. Xu, L. Ding, C. Xu, L. Wang and N. A. Kotov, Nat. Commun., 2013, 4, 2689 Search PubMed
- A. D. Merg, J. C. Boatz, A. Mandal, G. Zhao, S. Mokashi-Punekar, C. Liu, X. Wang, P. Zhang, P. C. van der Wel and N. L. Rosi, J. Am. Chem. Soc., 2016, 138, 13655–13663 CrossRef CAS PubMed
- C. Song, M. G. Blaber, G. Zhao, P. Zhang, H. C. Fry, G. C. Schatz and N. L. Rosi, Nano Lett., 2013, 13, 3256–3261 CrossRef CAS PubMed
- X. Lan, Z. Su, Y. Zhou, T. Meyer, Y. Ke, Q. Wang, W. Chiu, N. Liu, S. Zou, H. Yan and Y. Liu, Angew. Chem., Int. Ed., 2017, 56, 14632–14636 CrossRef CAS PubMed
- A. L. Feng, M. L. You, L. Tian, S. Singamaneni, M. Liu, Z. Duan, T. J. Lu, F. Xu and M. Lin, Sci. Rep., 2015, 5, 7779–7788 CrossRef CAS PubMed
- X. Zhang, C. A. Marocico, M. Lunz, V. A. Gerard, Y. K. Gun'ko, V. Lesnyak, N. Gaponik, A. S. Susha, A. L. Rogach and A. L. Bradley, ACS Nano, 2012, 6, 9283–9290 CrossRef CAS PubMed
- A. Querejeta-Fernández, G. Chauve, M. Methot, J. Bouchard and E. Kumacheva, J. Am. Chem. Soc., 2014, 136, 4788–4793 CrossRef PubMed
- K. L. Gurunatha, A. C. Fournier, A. Urvoas, M. Valerio-Lepiniec, V. Marchi, P. Minard and E. Dujardin, ACS Nano, 2016, 10, 3176–3185 CrossRef CAS PubMed
- E. Pazos, E. Sleep, C. M. Rubert Pérez, S. S. Lee, F. Tantakitti and S. I. Stupp, J. Am. Chem. Soc., 2016, 138, 5507–5510 CrossRef CAS PubMed
- T. Udayabhaskararao, T. Altantzis, L. Houben, M. Coronado-Puchau, J. Langer, R. Popovitz-Biro, L. M. Liz-Marzán, L. Vuković, P. Král, S. Bals and R. Klajn, Science, 2017, 358, 514–518 CrossRef CAS PubMed
- Y. Kim, B. Yeom, O. Arteaga, S. Jo Yoo, S.-G. Lee, J.-G. Kim and N. A. Kotov, Nat. Mater., 2016, 15, 461–468 CrossRef CAS PubMed
- M. Chirumamilla, A. Toma, A. Gopalakrishnan, G. Das, R. P. Zaccaria, R. Krahne, E. Rondanina, M. Leoncini, C. Liberale, F. De Angelis and E. Di Fabrizio, Adv. Mater., 2014, 26, 2353–2358 CrossRef CAS PubMed
- D.-K. Lim, K.-S. Jeon, H. M. Kim, J.-M. Nam and Y. D. Suh, Nat. Mater., 2010, 9, 60–67 CrossRef CAS PubMed
- P. Bai, S. Yang, W. Bao, J. Kao, K. Thorkelsson, M. Salmeron, X. Zhang and T. Xu, Nano Lett., 2017, 17, 6847–6854 CrossRef CAS PubMed
- L. Rodríguez-Lorenzo, R. de La Rica, R. A. Álvarez-Puebla, L. M. Liz-Marzán and M. M. Stevens, Nat. Mater., 2012, 11, 604–607 CrossRef PubMed
- K. L. Young, M. B. Ross, M. G. Blaber, M. Rycenga, M. R. Jones, C. Zhang, A. J. Senesi, B. Lee, G. C. Schatz and C. A. Mirkin, Adv. Mater., 2014, 26, 653–659 CrossRef CAS PubMed
- B. J. Kim, J. J. Chiu, G. R. Yi, D. J. Pine and E. J. Kramer, Adv. Mater., 2005, 17, 2618–2622 CrossRef CAS
- H. Kang, F. A. Detcheverry, A. N. Mangham, M. P. Stoykovich, K. C. Daoulas, R. J. Hamers, M. Müller, J. J. de Pablo and P. F. Nealey, Phys. Rev. Lett., 2008, 100, 148303 CrossRef PubMed
- L. A. Lane, X. Qian and S. Nie, Chem. Rev., 2015, 115, 10489–10529 CrossRef CAS PubMed
- J. Song, F. Wang, X. Yang, B. Ning, M. G. Harp, S. H. Culp, S. Hu, P. Huang, L. Nie, J. Chen and X. Chen, J. Am. Chem. Soc., 2016, 138, 7005–7015 CrossRef CAS PubMed
- K.-S. Kim, J.-H. Kim, H. Kim, F. Laquai, E. Arifin, J.-K. Lee, S. I. Yoo and B.-H. Sohn, ACS Nano, 2012, 6, 5051–5059 CrossRef CAS PubMed
- Y. Xianyu, Y. Xie, N. Wang, Z. Wang and X. Jiang, Small, 2015, 11, 5510–5514 CrossRef CAS PubMed
- A. J. Gormley, R. Chapman and M. M. Stevens, Nano Lett., 2014, 14, 6368–6373 CrossRef CAS PubMed
- Y. Jiang, M. Shi, Y. Liu, S. Wan, C. Cui, L. Zhang and W. Tan, Angew. Chem., Int. Ed., 2017, 56, 11916–11920 CrossRef CAS PubMed
- L. Guo, Y. Xu, A. R. Ferhan, G. Chen and D.-H. Kim, J. Am. Chem. Soc., 2013, 135, 12338–12345 CrossRef CAS PubMed
- J. Lee, A. O. Govorov, J. Dulka and N. A. Kotov, Nano Lett., 2004, 4, 2323–2330 CrossRef CAS
- J. Lee, P. Hernandez, J. Lee, A. O. Govorov and N. A. Kotov, Nat. Mater., 2007, 6, 291–295 CrossRef CAS PubMed
- N. Guarrotxena and G. C. Bazan, Adv. Mater., 2014, 26, 1941–1946 CrossRef CAS PubMed
- M. P. Cecchini, V. A. Turek, J. Paget, A. A. Kornyshev and J. B. Edel, Nat. Mater., 2013, 12, 165–171 CrossRef CAS PubMed
- G. Bodelón, V. Montes-García, V. López-Puente, E. H. Hill, C. Hamon, M. N. Sanz-Ortiz, S. Rodal-Cedeira, C. Costas, S. Celiksoy, I. Pérez-Juste, L. Scarabelli, A. La Porta, J. Pérez-Juste, I. Pastoriza-Santos and L. M. Liz-Marzán, Nat. Mater., 2016, 15, 1203–1213 CrossRef PubMed
- R. Tullius, A. S. Karimullah, M. Rodier, B. Fitzpatrick, N. Gadegaard, L. D. Barron, V. M. Rotello, G. Cooke, A. Lapthorn and M. Kadodwala, J. Am. Chem. Soc., 2015, 137, 8380–8383 CrossRef CAS PubMed
- T. L. Halo, K. M. McMahon, N. L. Angeloni, Y. Xu, W. Wang, A. B. Chinen, D. Malin, E. Strekalova, V. L. Cryns, C. Cheng, C. A. Mirkin and C. S. Thaxton, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17104–17109 CrossRef CAS PubMed
- S.-H. Cao, W.-P. Cai, Q. Liu, K.-X. Xie, Y.-H. Weng, S.-X. Huo, Z.-Q. Tian and Y.-Q. Li, J. Am. Chem. Soc., 2014, 136, 6802–6805 CrossRef CAS PubMed
- Y. Yang, J. Huang, X. Yang, K. Quan, H. Wang, L. Ying, N. Xie, M. Ou and K. Wang, J. Am. Chem. Soc., 2015, 137, 8340–8343 CrossRef CAS PubMed
- D. Liu, W. Chen, Y. Tian, S. He, W. Zheng, J. Sun, Z. Wang and X. Jiang, Adv. Healthcare Mater., 2012, 1, 90–95 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2018 |