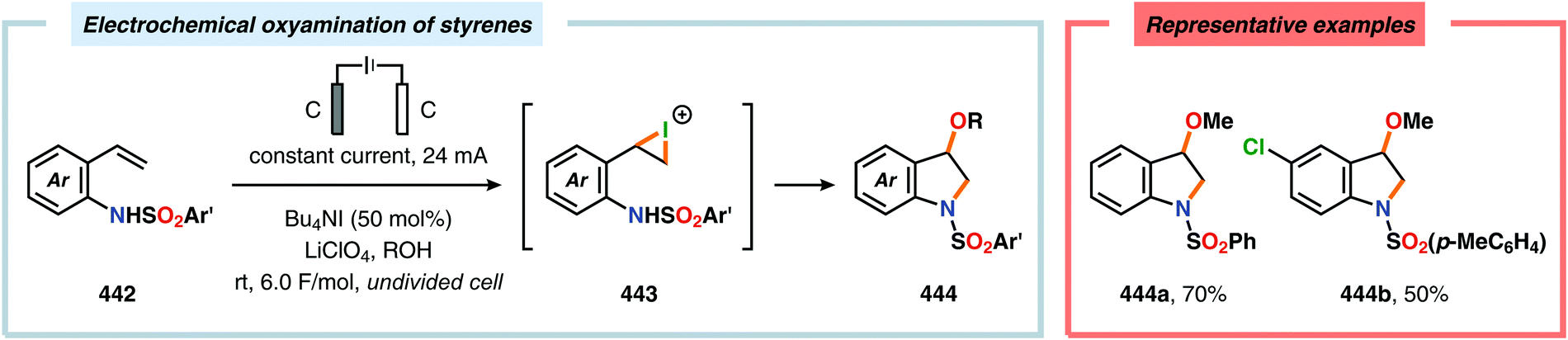
DOI:
10.1039/C7CS00619E
(Review Article)
Chem. Soc. Rev., 2018,
47, 5786-5865
Electrochemical strategies for C–H functionalization and C–N bond formation
Received
17th March 2018
First published on 18th June 2018
Abstract
Conventional methods for carrying out carbon–hydrogen functionalization and carbon–nitrogen bond formation are typically conducted at elevated temperatures, and rely on expensive catalysts as well as the use of stoichiometric, and perhaps toxic, oxidants. In this regard, electrochemical synthesis has recently been recognized as a sustainable and scalable strategy for the construction of challenging carbon–carbon and carbon–heteroatom bonds. Here, electrosynthesis has proven to be an environmentally benign, highly effective and versatile platform for achieving a wide range of nonclassical bond disconnections via generation of radical intermediates under mild reaction conditions. This review provides an overview on the use of anodic electrochemical methods for expediting the development of carbon–hydrogen functionalization and carbon–nitrogen bond formation strategies. Emphasis is placed on methodology development and mechanistic insight and aims to provide inspiration for future synthetic applications in the field of electrosynthesis.

Markus D. Kärkäs
| Markus D. Kärkäs earned his PhD degree in 2013 from Stockholm University under the direction of Prof. Björn Åkermark. His research focused on the development and mechanistic insight of artificial water oxidation catalysts. He carried out postdoctoral research in the group of Prof. Corey Stephenson at the University of Michigan working on photochemical methods for valorization of lignin. His research interests include photoredox catalysis, organic electrochemistry and artificial photosynthesis. |
1. Introduction
Synthetic chemists are constantly faced with the challenge of producing molecules, whether it be small-molecule synthesis or accessing increasingly complex target structures, in a selective and efficient manner. The methods and strategies of organic synthesis are continuously being improved and augmented in order to expand the chemical toolbox, thereby enabling new routes to solve ever-increasingly complex problems in chemical research.1–6 Over the years, the use of renewable chemical feedstocks,7 biocatalysis8 and base-metal catalysis,9 in combination with the development of cascade/tandem processes10 has become paramount for realizing atom-economic and more sustainable reaction platforms.11 Recently, research in the chemical sciences has experienced renewed interest in visible light photocatalysis12,13 and electrochemistry.14–17 These versatile synthetic platforms have the ability to promote challenging bond constructions under mild reaction conditions.
1.1. Fundamental principles and general considerations of preparative electrochemistry
Electrolysis is carried out in an electrochemical cell, a reactor that is comprised of an electroactive species/substrate, electrolyte, solvent and (at least) two electrodes (an anode and a cathode). The anode is connected to the positive pole of a power source (galvanostat or potentiostat) and is oxidative while the cathode is connected to the negative pole and is reductive. For construction of the cell, two different options exist: undivided cells and divided cells (Fig. 1). In undivided cells the anode and cathode are not separated and are thus placed in the same compartment. This setup is preferred due to its ease of construction and allows for both reduction and oxidation to occur within the same compartment, thereby enabling the substrate to be exposed to all species present in the reaction. When employing this arrangement, it is crucial to consider the possible reactions that may occur at the auxiliary electrode.18 However, examples exist where a species produced at the auxiliary electrode interferes with the electrochemical process at the working electrode. This issue can be ameliorated using a divided cell. In this arrangement, the two (anodic and cathodic) compartments are physically separated through the use of a small, porous frit. This allows for the transfer of charge and enables the two half-reactions of the electrolysis to occur separately.19,20
 |
| | Fig. 1 Selected electrolysis setups. (a) Three-necked round-bottom flask equipped with (b) a reticulated vitreous carbon anode and a platinum plate cathode (Reprinted with permission from ref. 926. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim). (c) Undivided beaker-type glass cell equipped with an isostatic graphite anode and a platinum cathode (Reproduced from ref. 918 with permission from The Royal Society of Chemistry). (d) H-Type divided cell equipped with a reticulated vitreous carbon anode and a platinum wire cathode (Reprinted with permission from ref. 398. Copyright 2018 American Chemical Society). (e) H-Type divided cell equipped with a 4G glass filter, an anode made of carbon strings and a platinum plate cathode (Reprinted with permission from ref. 958. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim). | |
In principle, electrodes can be constructed from any inert material that enables electron transfer in solution. Examples of electrodes include carbon-based materials (graphite and glassy carbon), magnesium, platinum and stainless steel. Another appealing electrode material is reticulated vitreous carbon (RVC), a material that combines the properties of carbon with glass, which has a high surface area, high void volume, and chemical resistance. At the end of the reaction, the electrodes can simply be physically removed as long as they are stable to the applied reaction conditions.21,22 Acceptable solvents that can be employed in electrochemical processes include, but are not limited to acetonitrile, dichloromethane, methanol, or dimethylformamide. Here, the use of nonconventional organic solvents, such as trifluoroethanol,23 trifluoroacetic acid,24 ionic liquids25 and supercritical fluids,26 has also received attention. In order for the reaction solution to become conductive, a supporting electrolyte has to be added. These are typically simple salts, such as alkali metal perchlorates and tetraalkyl ammonium salts, and are mainly used for increasing the conductivity of organic systems.
The electron transfer events that occur at the anode or cathode enable one to classify the electrochemical reactions as either oxidations or reductions, respectively. They occur on the surface of the electrodes and are therefore heterogeneous processes. The resulting species that is generated after the initial electron transfer between an electrode and an organic substrate is subsequently diffused into solution, which is followed by a secondary reaction of the initially produced radical species.27–31 Here, the potential of the electrode (E)—that is the difference between the potential at the electrode of interest and the selected reference electrode, such as the saturated calomel electrode (SCE)—will determine whether a specific electron transfer process is thermodynamically feasible, and is given by eqn (1):
where Δ
G is the free energy change,
F is Faraday's constant (96
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
485 Coulombs mol
−1), and
n is the number of electrons involved in the overall reaction. An advantage of preparative electrolysis is that an initial electroanalytical evaluation using, for example, cyclic voltammetry (CV) enables convenient assessment of the reaction system. Such simple analyses provide a great deal of vital information with relatively limited experimental effort and insight into which functional groups that may be oxidized or reduced, as well as the magnitude of the free energy that is needed to promote electron transfer in the studied electrochemical process.
32
It is important to note that the electrolysis experiments can be conducted using either a controlled-current or controlled-potential approach. In a controlled-current experiment, the current is held constant while the voltage gradually increases until the potential is reached for the electroactive species. Once depleted, the potential continues to rise until a second electroactive species or solvent molecule is oxidized or reduced. It is thus critical to consider the redox potentials of the substrate as well as all other species in solution in order to assure that the initial redox reaction does not involve participation of the solvent or other additives. However, if the potential needs to be tightly controlled, a controlled-potential experiment where the potential is set to a certain value will allow for a selective redox reaction to occur.33 With this setup, it is important to have in mind that the decrease in substrate concentration is accompanied by a decrease in the current, and thus the rate of the reaction.
1.2. Historical advances in synthetic electrochemistry
Applying electrical current to affect a chemical reaction can be traced back to the 1830s when Faraday electrolyzed an acetate solution to produce hydrocarbons.34 However, the Kolbe electrolysis constitutes one of the earliest carbon–carbon (C–C) bond forming reactions that was studied in detail.35,36 Here, carboxylate anions are oxidized at an anode. The initial electron transfer triggers extrusion of CO2 to generate carbon-centered free radicals, which subsequently undergo homocoupling to produce the new C–C bond. The reaction still serves as a versatile method for C–C bond formation in contemporary organic synthesis.37 Preparative electrolysis has also received significant interest from the chemical industry.38–40 Classical examples include the chloralkali and the Hall–Héroult processes, as well as production of organic compounds, such as adiponitrile and ethylene glycol.41
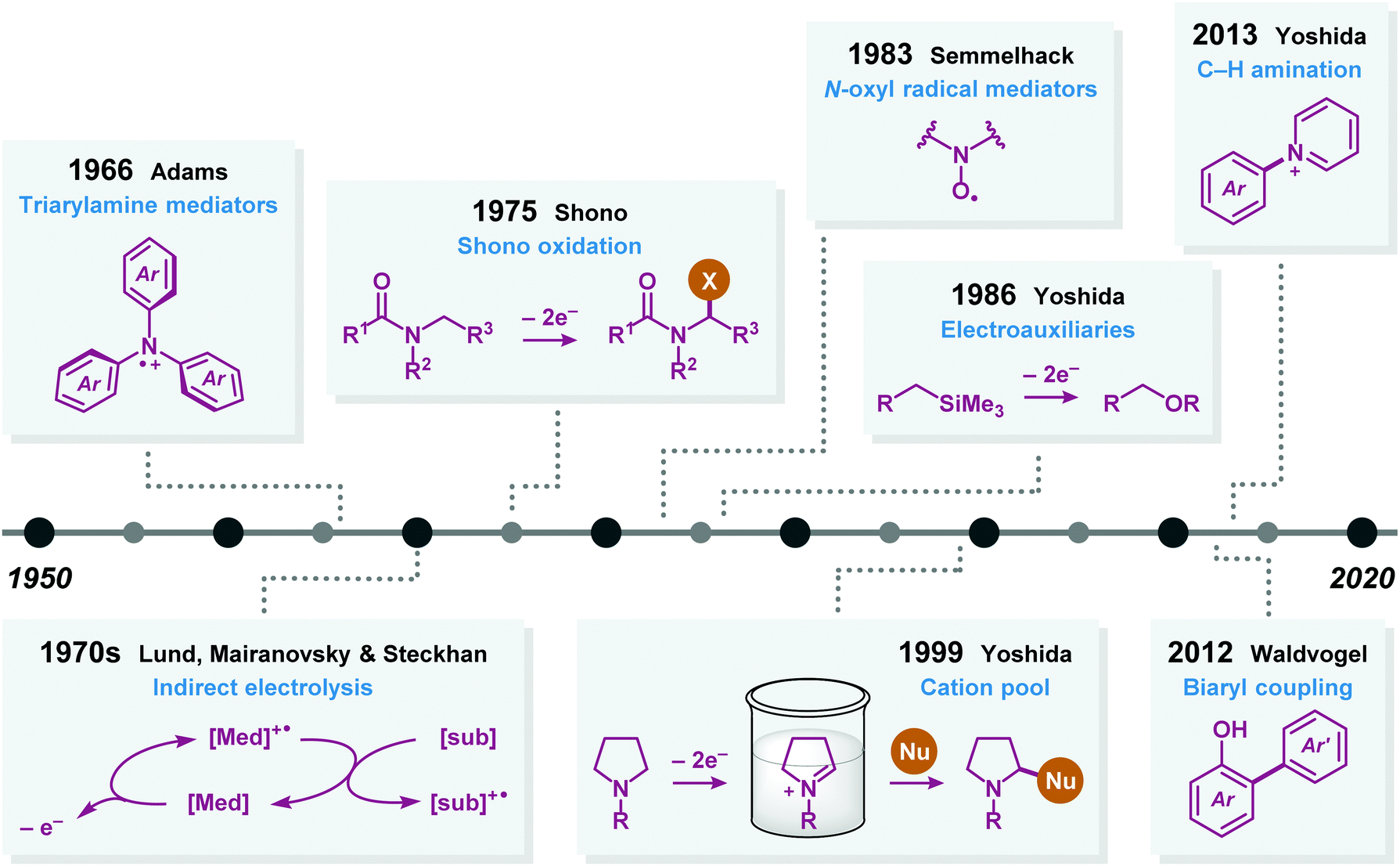
Additional advances in preparative electrolysis include the use of indirect electrolysis in which a redox mediator undergoes electron transfer at an electrode to afford an electrochemically generated reagent that triggers the reaction of interest (Fig. 2).42 This approach has a long history43 and one of the earliest industrial applications concerned the oxidation of glucose to calcium gluconate using bromide as mediator.44 During the last four decades, commonly employed organic redox mediators for anodic oxidations are triarylamines45 and nitroxyl radicals.46,47 Since the active species of the mediator is continuously regenerated at the electrode it enables the use of substoichiometric quantities. Advantages of redox-mediated electrolysis (indirect electrolysis) are that problems such as are that problems associated with heterogeneous electron transfer, such as overpotentials,48 can be avoided and that electrolysis can be conducted at lower potentials than the redox potential of the substrate, accelerating the reaction rate while affording higher selectivities by circumventing potential side reactions. It should be noted that there exist several similarities between electrochemically-mediated reactions and visible light photoredox catalysis (Fig. 3). In this visible light-mediated platform a photocatalyst absorbs photonic energy. Upon absorption of visible light, a long-lived redox-active excited state is produced, which can engage in single-electron transfer (SET) events with, for example, organic substrates.
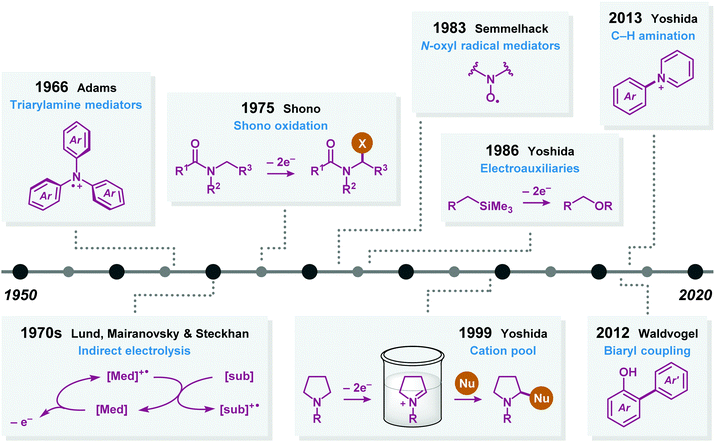 |
| | Fig. 2 Advances in organic electrochemistry. | |
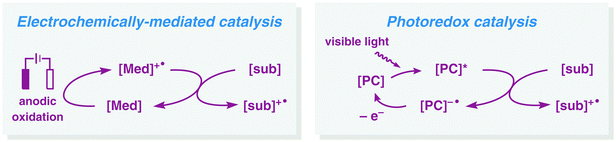 |
| | Fig. 3 Comparison of electrochemically-mediated catalysis and visible light photoredox catalysis. PC = photocatalyst. | |
In the late 1980s, Yoshida introduced the concept of using electroauxiliaries.49,50 Typical electroauxiliaries include enol ethers,51 organostannanes,52 silyl groups,53 and thioacetals.54 The electroauxiliary is functionality appended to a compound in order to aid the oxidation or reduction of the substrate by reducing its oxidation or reduction potential. This facilitates either oxidation or reduction of the starting material in the presence of the desired nucleophile and reduces the likelyhood of overoxidation or overreduction of the resultant product by promoting electron transfer in a more selective and predictable fashion. Moreover, noticeable contributions during the 21st century include Waldvogel's work on selective biaryl cross-coupling platforms55 and Yoshida's strategy for C–H amination56 of aromatic compounds using the “cation pool” method.57,58
1.3. Advantages of applying electrochemical methods in organic synthesis
The use of electrochemistry is considered a “green” and sustainable alternative to traditional, toxic redox-based methods. It has the ability to maximize atom efficiency while replacing stoichiometric redox reagents, such as NaH, OsO4 and Pb(OAc)4, with an electrical current, thereby minimizing production of reagent waste. Moreover, since the energy of the electrochemical system can be governed by the applied electrode potential, a majority of the preparative electrochemical reactions, even the ones with high activation energies, can be carried out at ambient temperature. Therefore, the application of electrochemistry in organic synthesis constitutes an appealing strategy for generating reactive radical intermediates under mild conditions.59,60
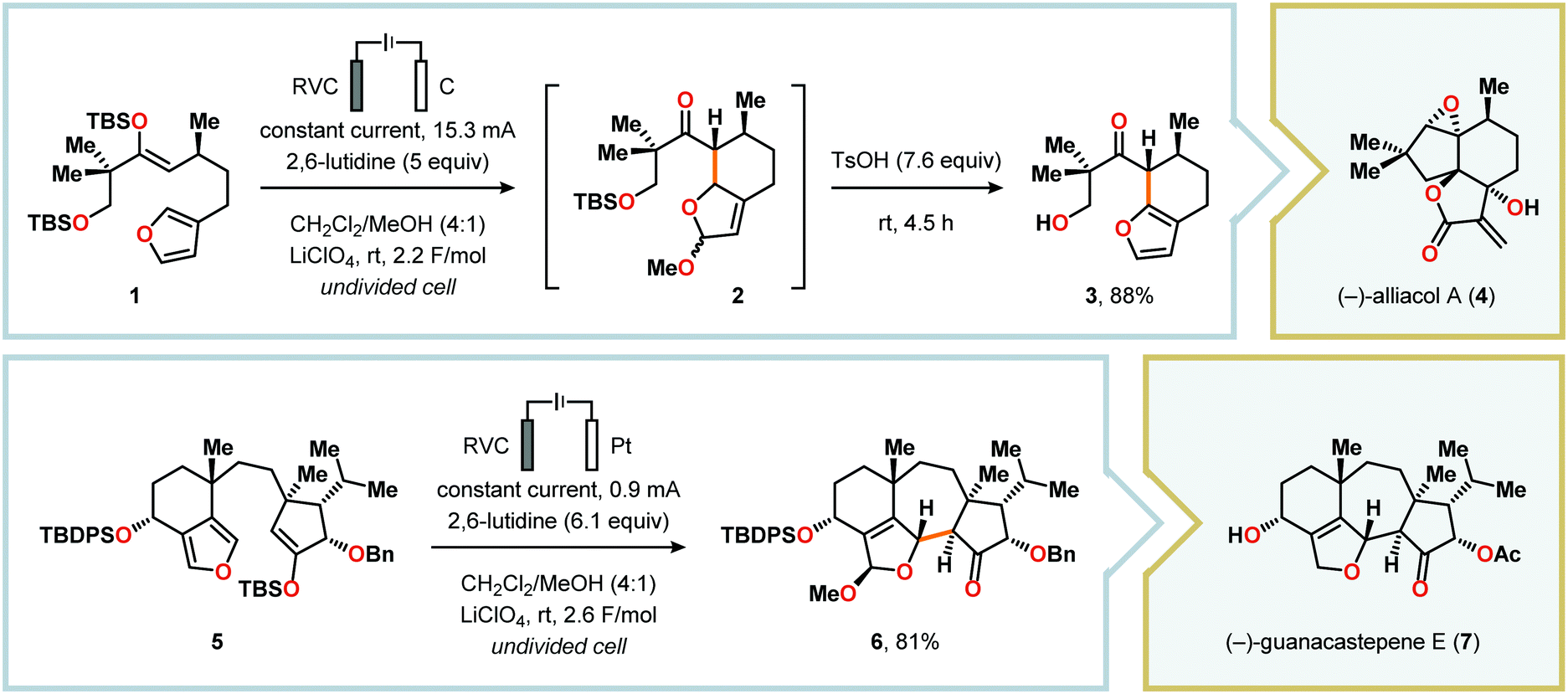
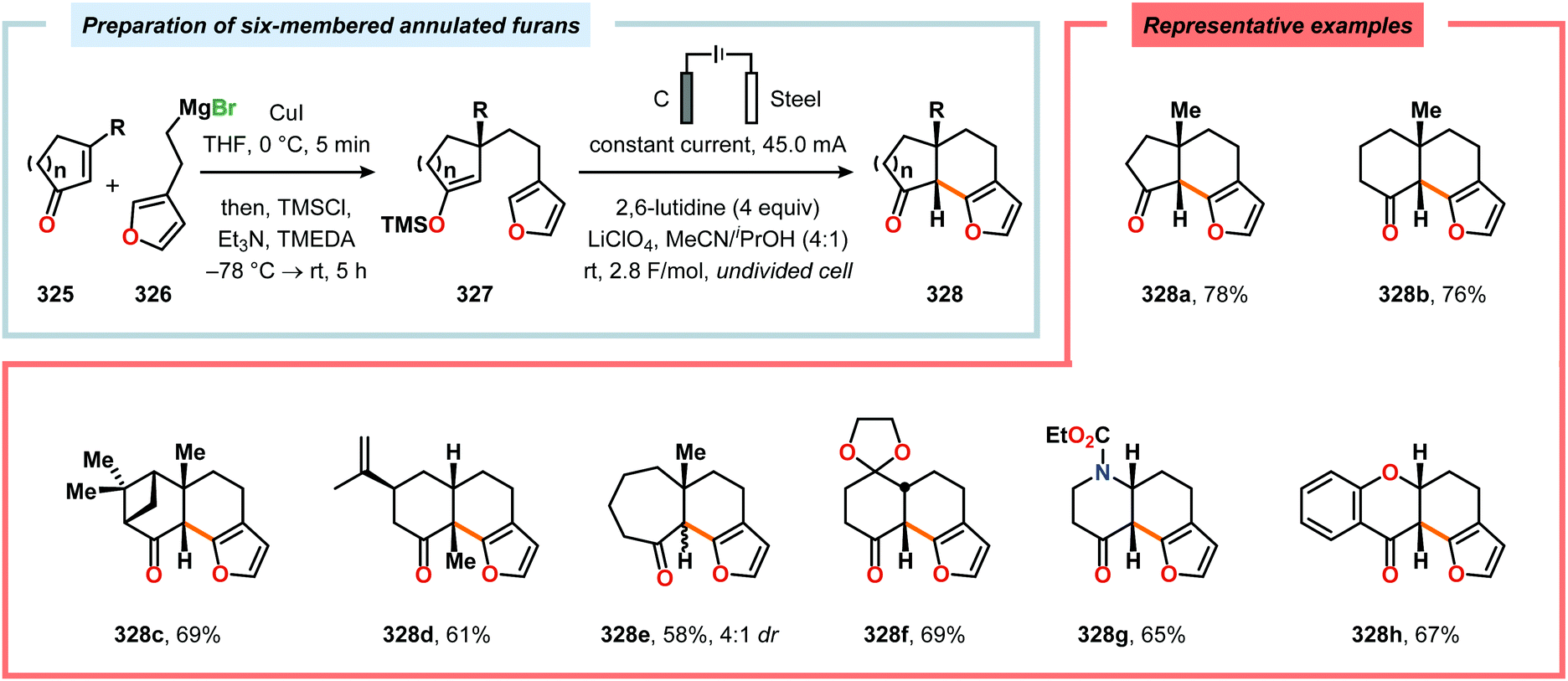
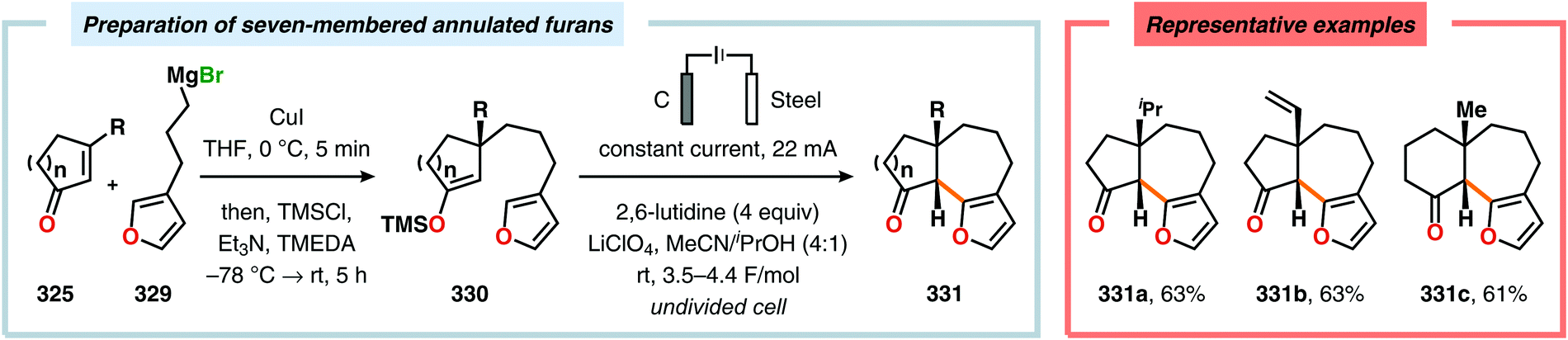
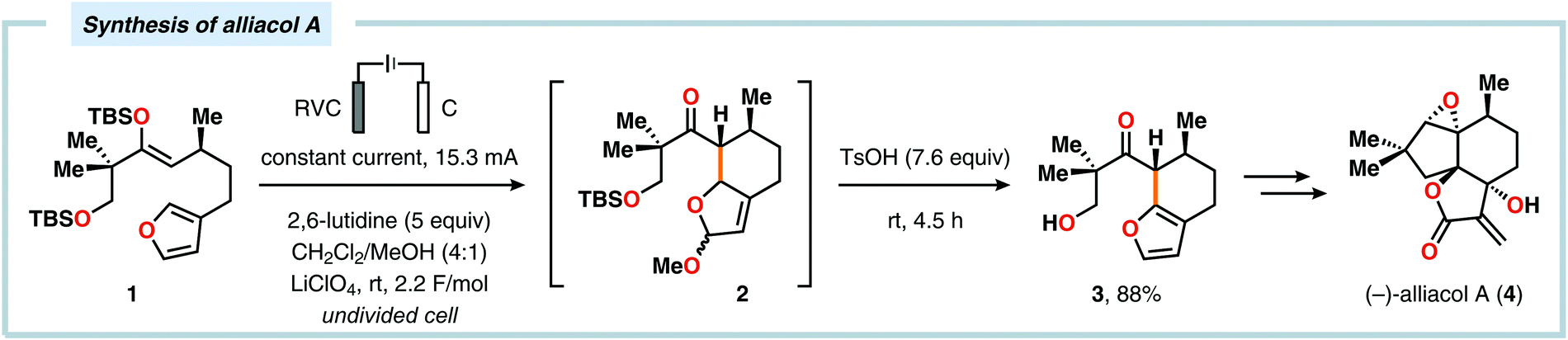
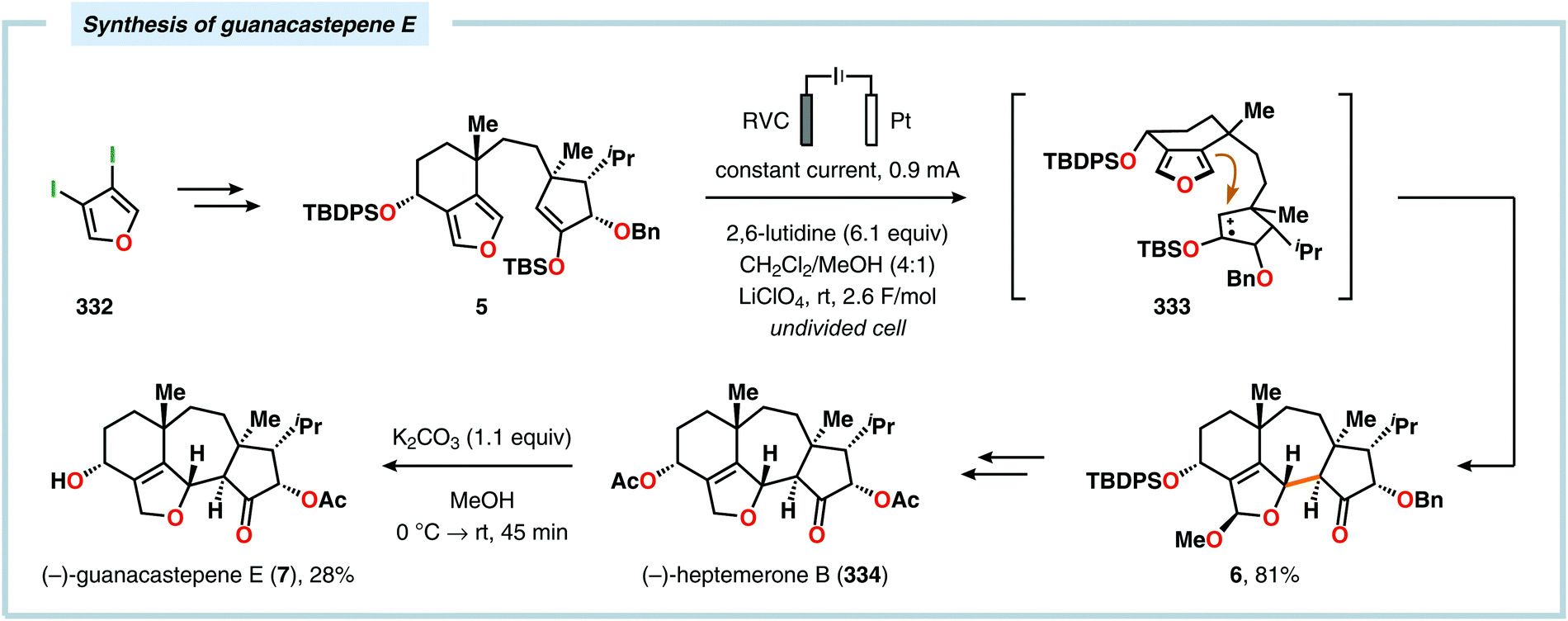
Radical cyclization reactions, for example, have typically been conducted in the presence of stoichiometric quantities of hazardous and toxic reagents, such as tin hydrides and azobis(isobutyronitrile) (AIBN). However, employing electrochemistry allows the development of reagent-free protocols that operate under mild reaction conditions, providing an environmentally friendly alternative.32,61 In addition to the aforementioned advantages of applying electrochemistry in organic synthesis, another feature of electrochemistry is that it can also be used to trigger umpolung reactions.62–64 Two elegant examples of using such a strategy can be found in Moeller's synthesis of alliacol A (4)65,66 and Trauner's synthesis of guanacastepene E (7)67,68 in which silyl enol ethers are coupled with furans via intramolecular electrooxidative coupling (Scheme 1).
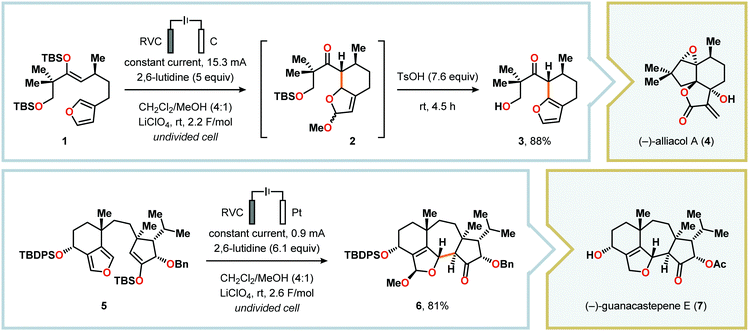 |
| | Scheme 1 Synthesis of alliacol A and guanacastepene E via anodic coupling reactions. | |
It is evident that organic electrochemistry represents an enabling technology for the organic chemistry research community for generating reactive but controllable radical intermediates under neutral reaction conditions. However, for the technique to become part of the standard chemical toolbox, the barrier to adoption needs to be eliminated. The necessity to construct your own home-made electrochemical apparatus has certainly contributed to the general lack of interest in pursuing electrochemistry, for example, as part of a total synthesis. The barrier to adoption becomes even higher if there exist alternative methods for accomplishing the desired transformation. Furthermore, the numerous reaction variables, such as cell type, experiment type, electrode material and electrolyte, might be overwhelming, especially for practicing organic chemists without any prior experience in electrochemistry. Organic electrochemistry would benefit from being presented in a more user-friendly manner. Access to a commercially available standardized device for organic electrochemistry would certainly mitigate this barrier to adoption and allow the field to develop more rapidly.69
1.4. Scope of this review
Over the years, a variety of review articles have been published that summarize the impressive advances made in the field of organic electrochemistry.70 Pioneering reviews include those by Wawzonek71 and Weinberg.72 Anodic oxidation processes have been examined by Adams,73 Eberson27,74 and Shono,75 and more recently by Boydston,76 Chiba,77 Moeller,78–81 and Schäfer.82,83 Reviews detailing both anodic oxidation and cathodic reduction processes have been summarized by Schäfer,84,85 Wright86 and Yoshida.87 Progress in indirect electrolysis has been reviewed by Nikishin,42c Steckhan,42a,b as well as Francke and Little42d while the synthetic endeavors of using electrochemistry in complex settings have been described by Baran.88 In this review an overview of the electrochemical strategies for carrying out carbon–hydrogen (C–H) functionalization as well as carbon–nitrogen (C–N) bond formation is presented with an emphasis on methods development and mechanistic insight.89 The review is organized based on the aforementioned reaction types and are grouped according to the electrochemical methods employed. While focus is on representative examples from the past two decades, early pioneering work is also highlighted. The aim of this review is to encourage researchers to explore and to adopt organic electrochemistry, a technique with considerable potential, to the general synthetic toolbox.
2. Carbon–hydrogen (C–H) functionalization under oxidative electrochemical control
Over the past decade, transition metal-catalyzed C–H functionalization has enabled the development of a variety of nontraditional and innovative bond constructions in contemporary synthetic organic chemistry. The direct functionalization of C–H bonds represents a powerful strategy for selective C–C and carbon–heteroatom (C–X) bond formation, thereby improving atom- and step economy as well as streamlining chemical synthesis. However, despite intensive efforts and increased understanding of the mechanistic aspects of the C–H functionalization reaction manifolds, the inherent difficulty of activating kinetically inert C–H bonds has limited the development of catalytic platforms that operate efficiently under mild reaction conditions.90,91 In this chapter, various electrochemical concepts to expedite the development of C–H functionalization platforms are therefore surveyed.
2.1. Shono-type anodic oxidations
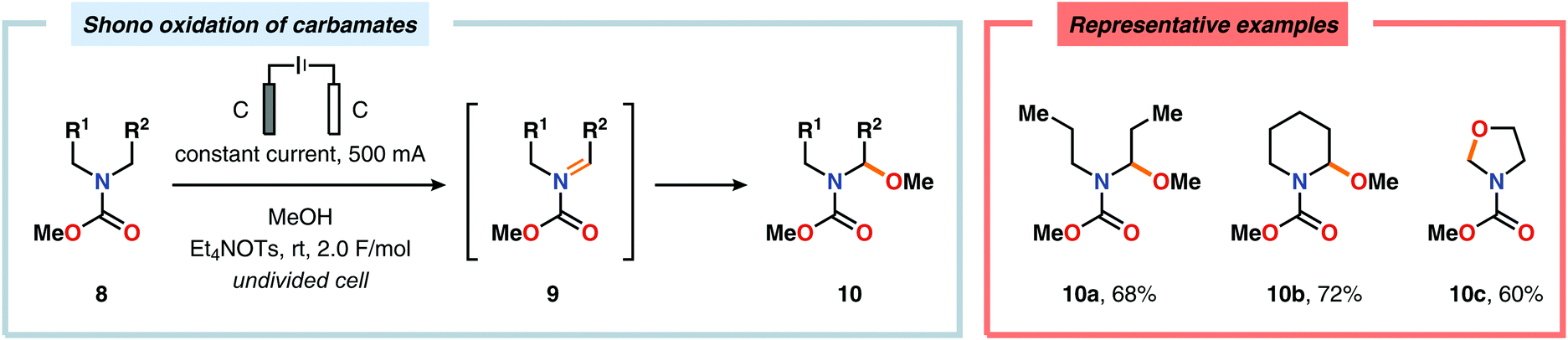
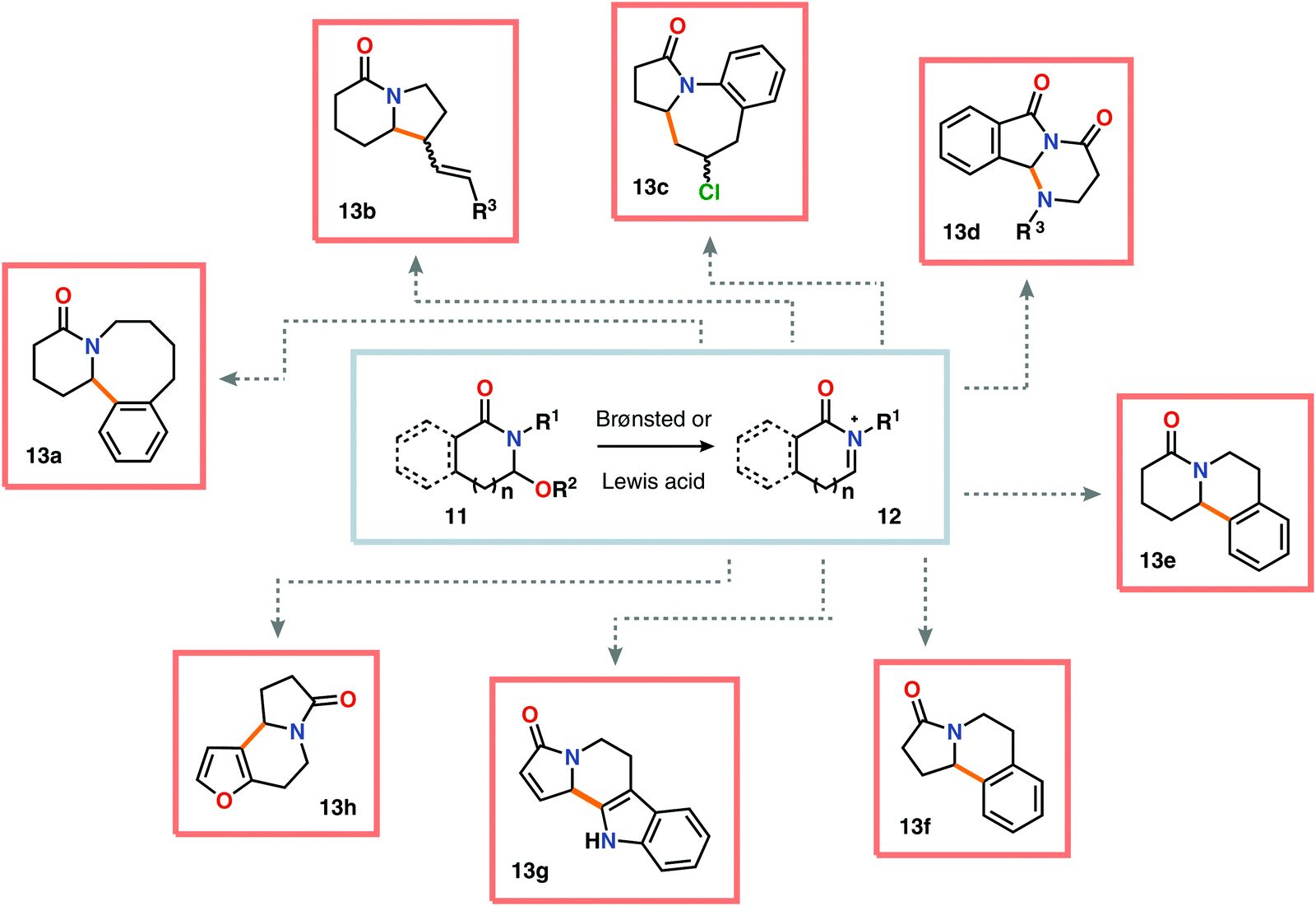
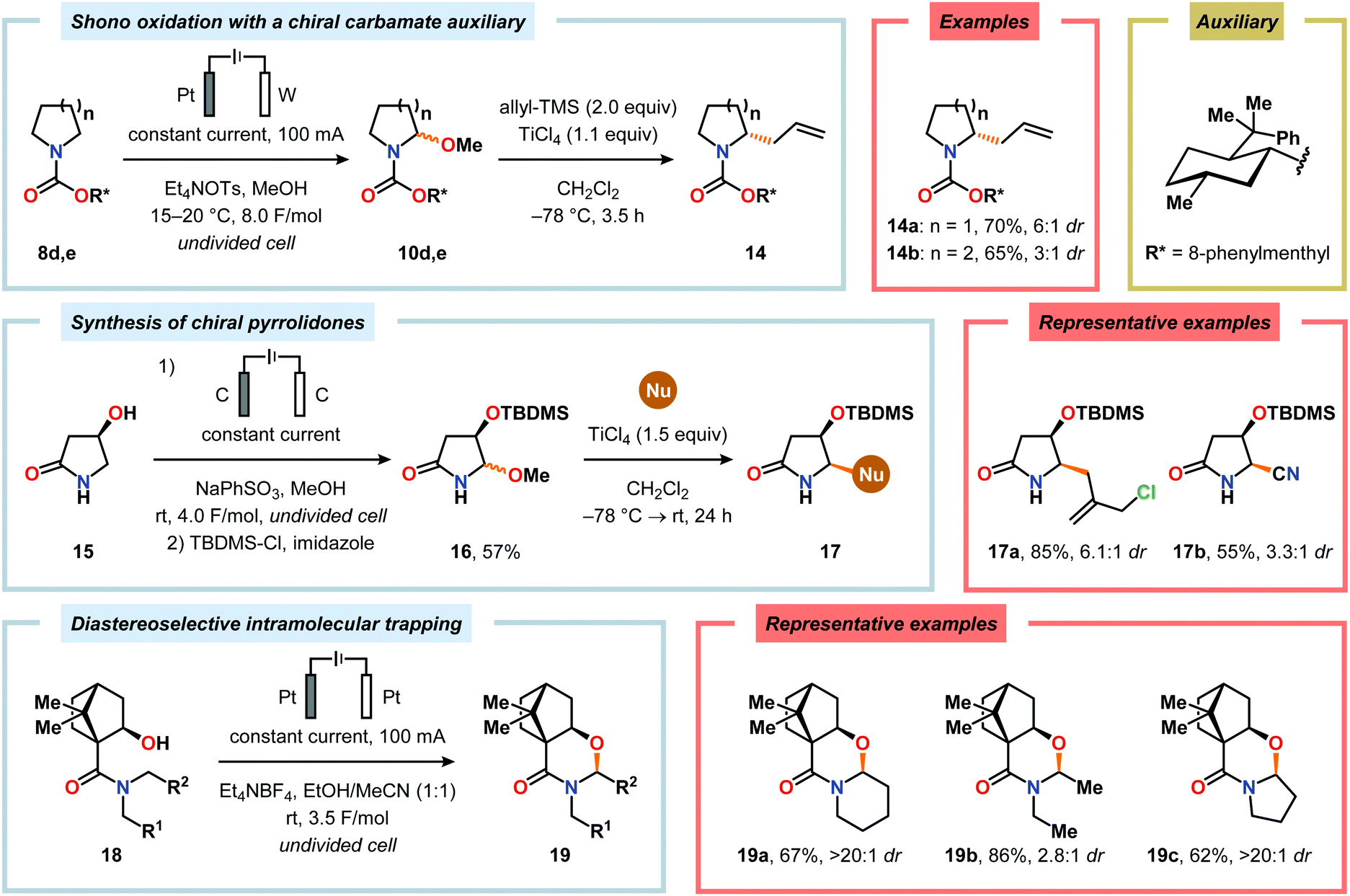
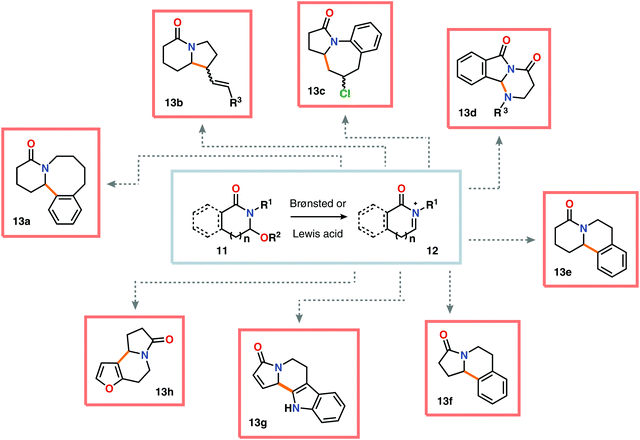
Nitrogen-based heterocycles constitute an essential structural motif and are commonly found in natural products. In 1975, Shono and coworkers disclosed an electrochemical method for oxidation of carbamates to N-carbamoyl iminium ions.92–95 The reaction proceeds through the initial formation of a nitrogen-centered radical (vide infra), which is subsequently oxidized to an iminium ion intermediate that can be trapped with an alcoholic solvent molecule.96 This allows for functionalization of the α-position adjacent to the nitrogen atom in heterocycles.97–110 For example, carrying out the anodic oxidation in methanol gives access to α-methoxylated products 10 (Scheme 2), which are established precursors to N-acyl iminium ions. If the substrate contains an alcohol moiety, intramolecular trapping produces oxazoline derivatives, such as 10c.92 Upon treatment with Brønsted or Lewis acids, the generated N,O-aminals can be converted back to the iminium ion species, which allows for trapping with allyl silanes,111–113 cyanide,114–116 fluoride,117 furans,118 isocyanides,119 silyl enol ethers,120 and trialkyl phosphites.121 Alternatively, functionalization can be carried out to yield a diversity of scaffolds as shown in Scheme 3.122 Attempts have also been focused on developing asymmetric versions of the Shono oxidation using chiral cyclic dipeptides,123–126 oxazolines,127 oxazolidinones128 and pyrrolidones129–131 as well as ketopinic acid132 and carbamate-,133–136 phosphorus-137,138 and sulfur-based139 chiral auxiliaries to give poor to high stereoselectivities in the alkylation step (Scheme 4).
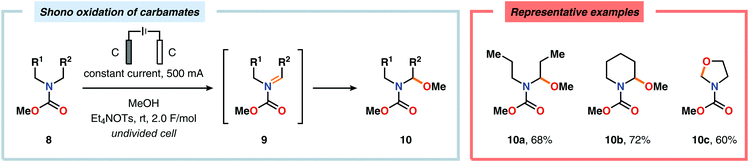 |
| | Scheme 2 Shono oxidation of carbamates. | |
 |
| | Scheme 3 Scaffold diversity from N-acyl iminium ions. | |
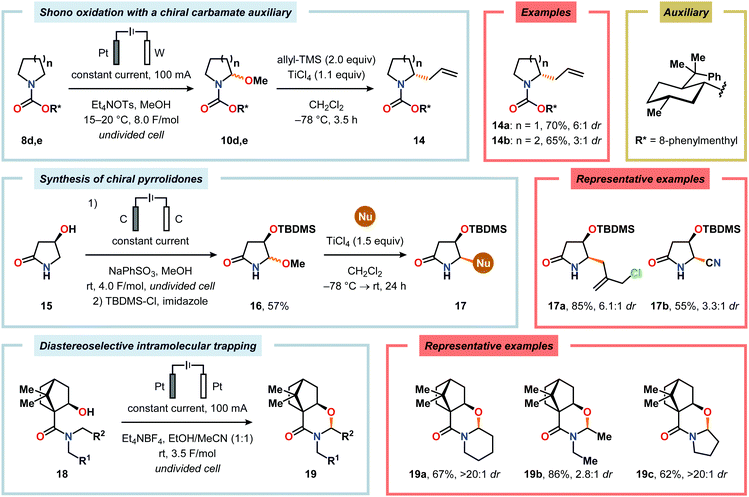 |
| | Scheme 4 Selected strategies for stereoselective functionalization of Shono oxidation products. | |
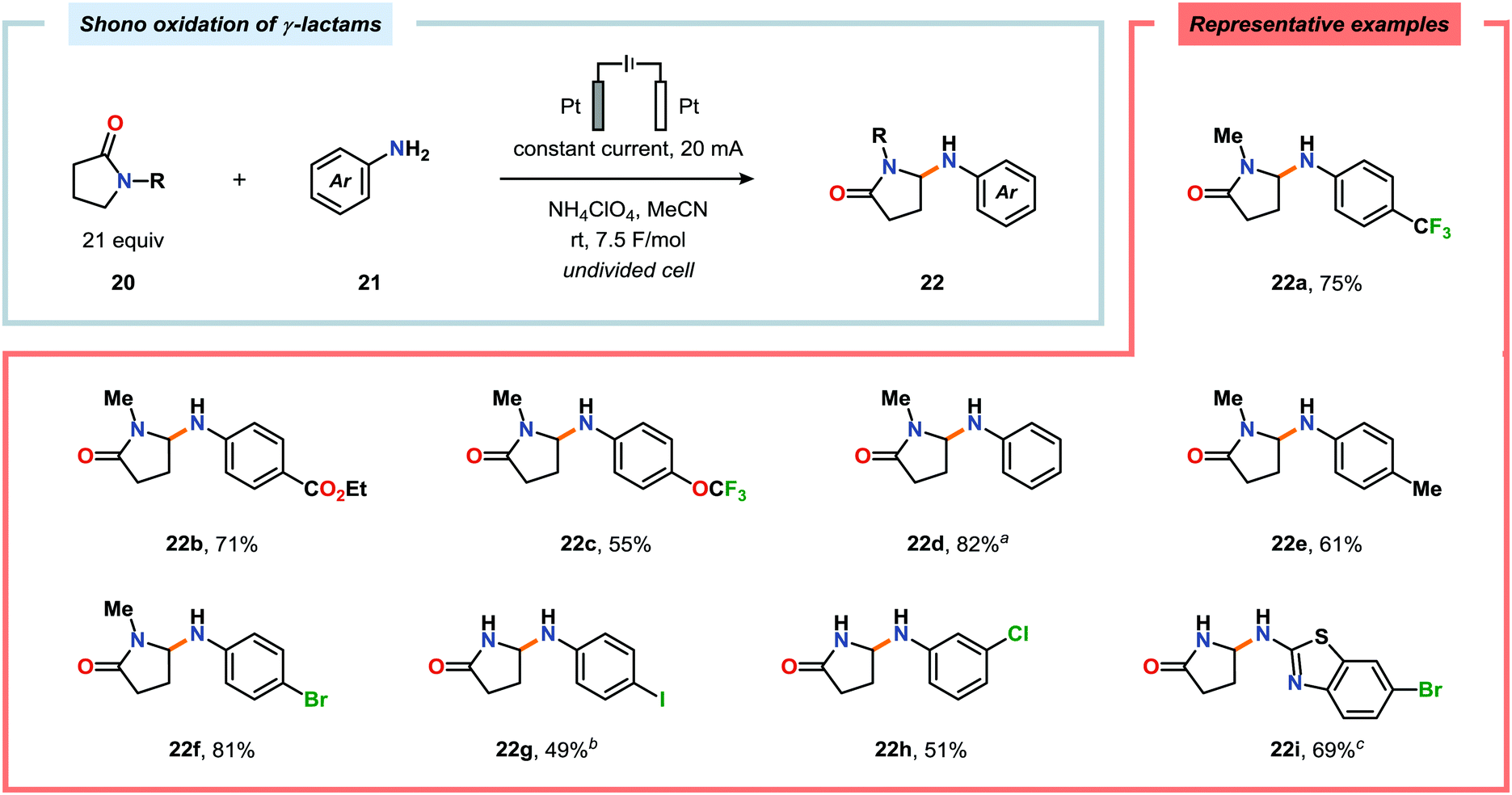
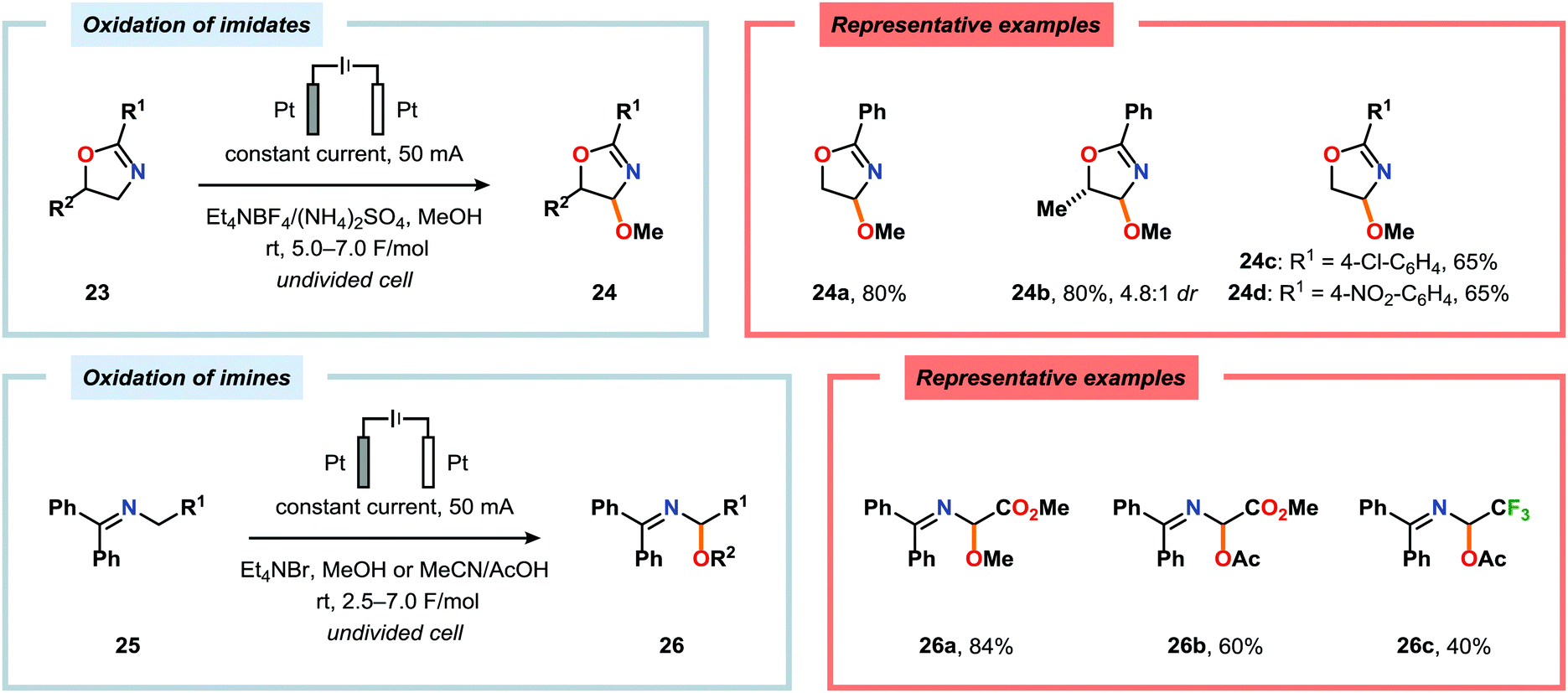
Shono-type anodic oxidations have in general been carried out with substrates and nucleophiles, such as methanol and cyanide, which possess high oxidation potentials. Although amines have lower oxidation potentials, which should facilitate the electrochemical oxidation process, protocols utilizing amines have been employed less frequently compared to their amide and carbamate counterparts. Huang and Gong recently disclosed a method for the electrochemical oxidation of γ-lactams. Direct trapping of the generated electrophilic acyliminium ions with anilines could be accomplished despite their low oxidation potential (Scheme 5).140 Key to achieving high yields of the substituted γ-lactam products was to carry out the oxidation in a “quasi-divided cell”141,142 with a Pt wire anode and a Pt foil cathode in which the large difference in surface areas of the working and counter electrodes provided high current density at the anode, thereby favoring oxidation of the lactam. The protocol was applicable to a wide range of anilines containing electron-withdrawing groups, such as CF3, CO2Et, and OCF3, as well as electron-donating groups, such as Me and OMe. Halogen-containing anilines were also effective substrates for the developed reaction, which provides an opportunity for further synthetic elaborations.140 Fuchigami and Baba have also demonstrated anodic oxidation of imidates and imines to yield α-methoxylated or α-acetoxylated products (Scheme 6).143
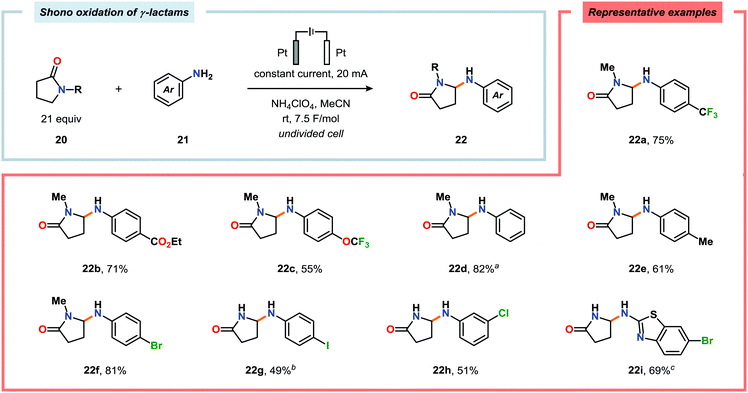 |
| | Scheme 5 Shono oxidation of γ-lactams using anilines as the trapping nucleophiles. aCarried out in MeCN/EtOAc (3:2), Et3N (1.7 equiv.), constant current (10 mA) and 10.5 h. bAfter 1.5 h. cAfter 5 h. | |
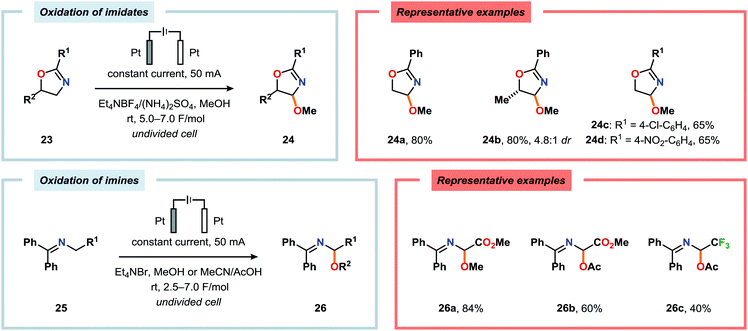 |
| | Scheme 6 Anodic oxidation of imidates and imines. | |
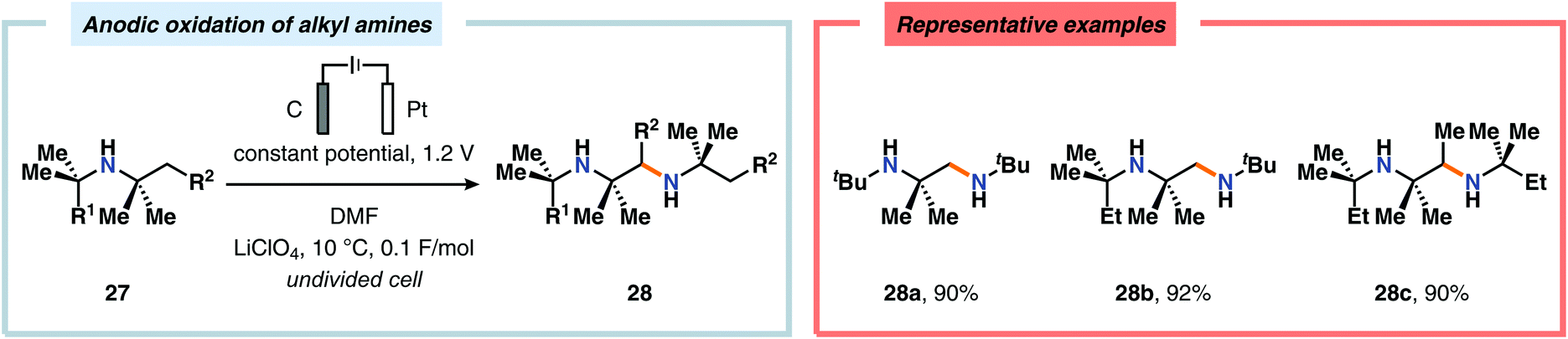
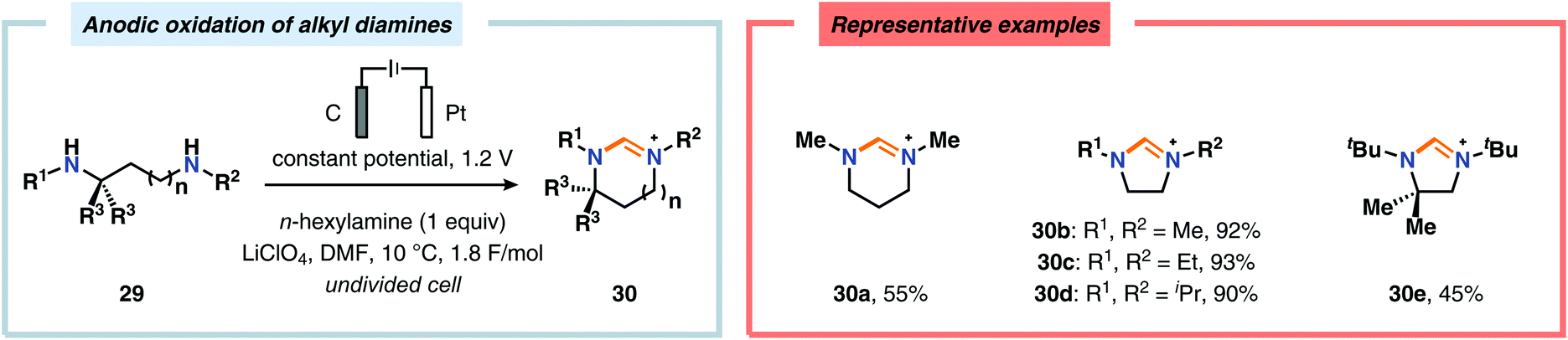
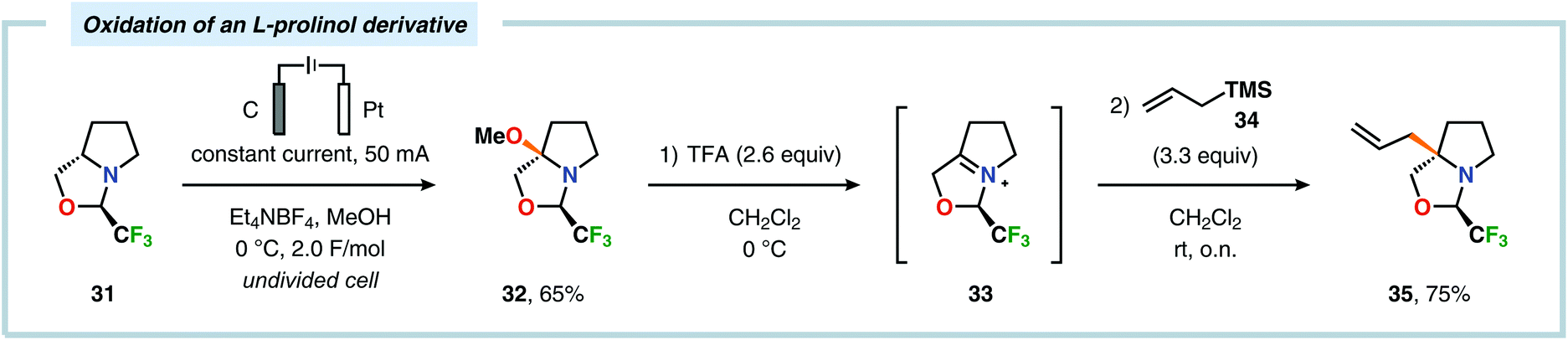
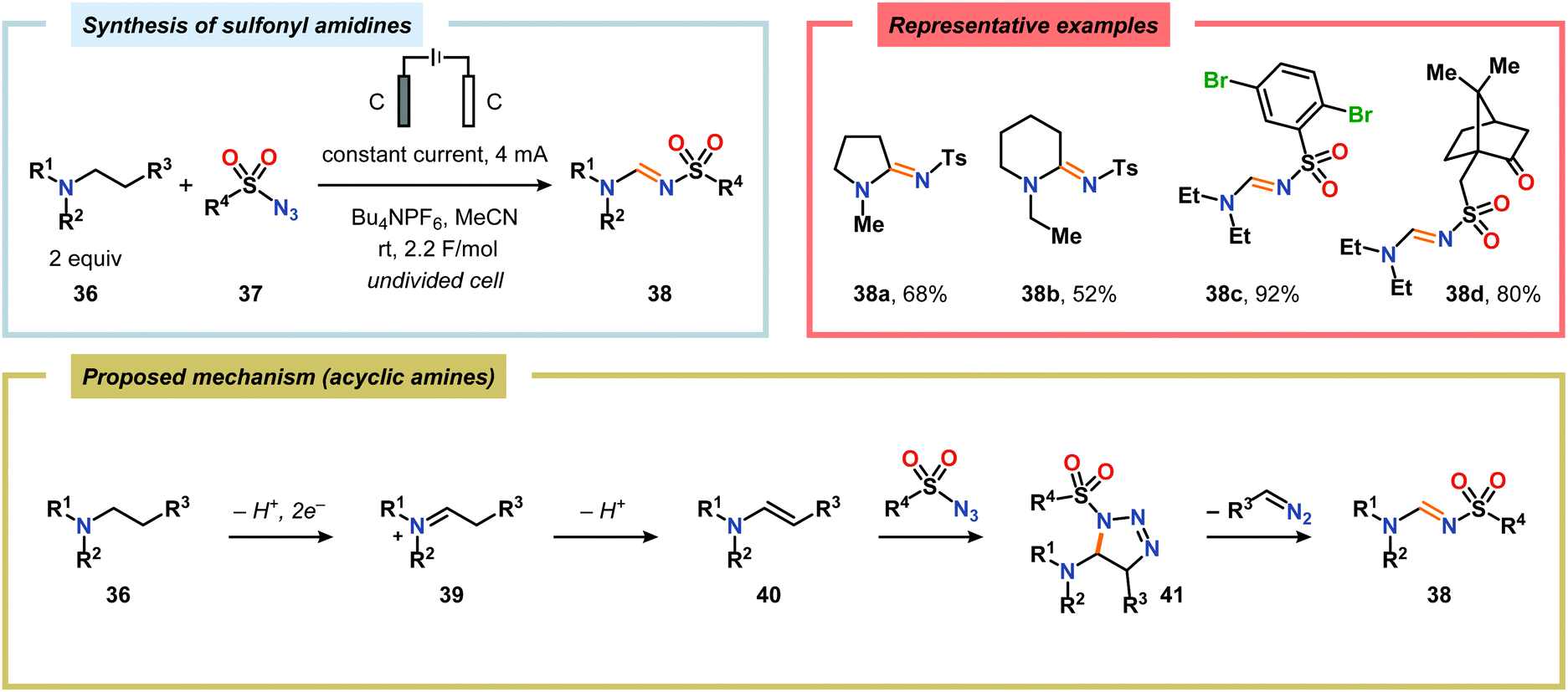
Of the limited protocols that exist that rely on using amines as substrates,144,145 Gallardo and Vilà reported that hindered secondary alkyl diamines 28 could be produced from anodic oxidation of hindered secondary alkyl amines 27 (Scheme 7).146–148 They subsequently demonstrated an electrosynthetic procedure to afford imidazolium and tetrahydropyrimidinium derivatives 30 from secondary alkyl diamines 29 (Scheme 8).149 The Matsumura group has also shown that the introduction of a nucleophile to the α-position of an L-prolinol derivative (31) upon Shono-type oxidation is viable (Scheme 9).150 Furthermore, Hurvois and coworkers prepared an α-amino nitrile through anodic oxidation in a synthesis of (+)-myrtine.151 Shono-type oxidation of aliphatic amines can also be extended to the synthesis of sulfonyl amidines.152 Different tertiary amines (36) were initially evaluated and were shown to efficiently afford the sulfonyl amidine derivatives 38 upon reaction with sulfonyl azides 37 (Scheme 10). Employing Et3N as the amine these transformations presumably include anodic oxidation of the aliphatic amine to produce an iminium intermediate (39), which tautomerizes into the corresponding enamine (40). Subsequent 1,3-dipolar cycloaddition between the generated enamine and tosyl azide furnishes cycloadduct 41. Finally, extrusion of CH2N2 delivers the desired amidine product 38. The synthesis of amidine derivatives was also accomplished using secondary and primary aliphatic amines.
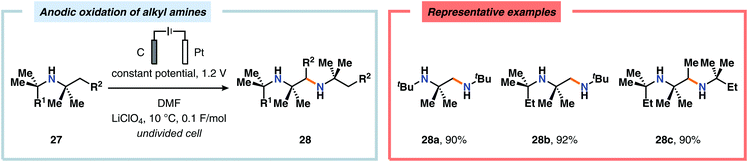 |
| | Scheme 7 Shono-type anodic oxidation of hindered secondary alkyl amines. | |
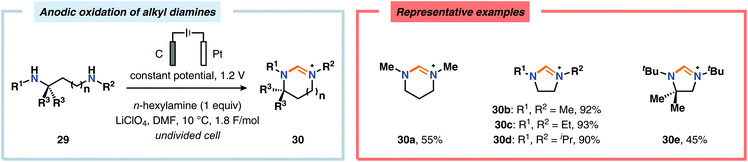 |
| | Scheme 8 Shono-type oxidation of secondary alkyl diamines. | |
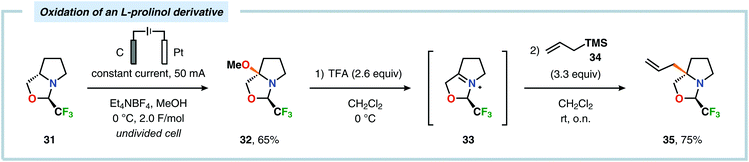 |
| | Scheme 9 Electrochemical oxidation of an L-prolinol derivative. | |
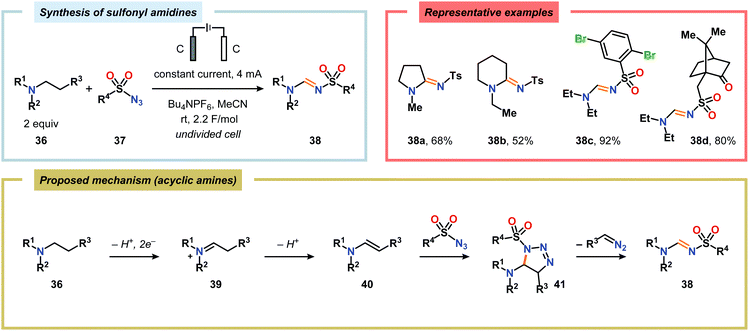 |
| | Scheme 10 Electrochemical synthesis of sulfonyl amidines from aliphatic amines and sulfonyl azides. | |
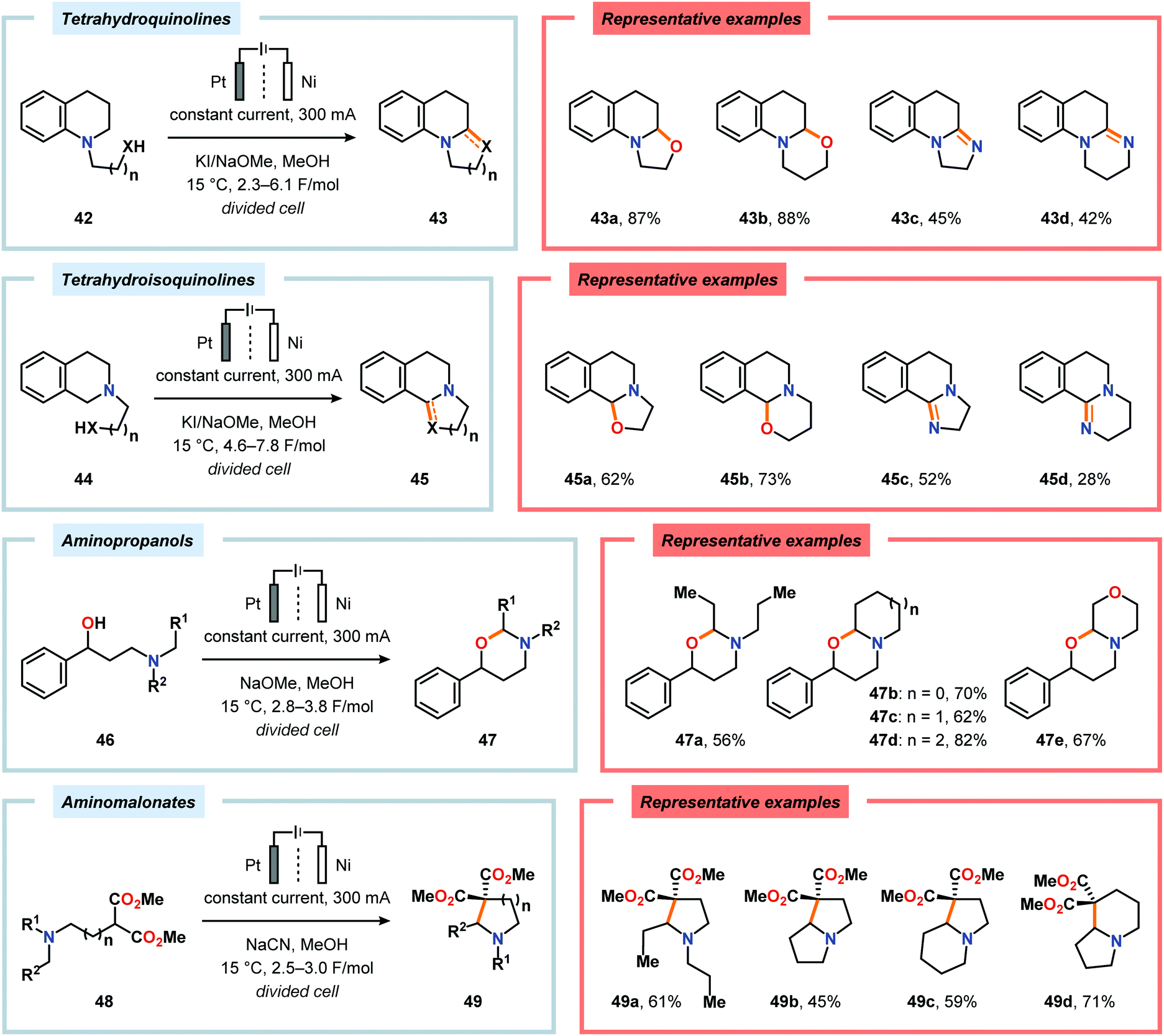
Okimoto and coworkers have also demonstrated that Shono-type oxidation of various amine-containing motifs carrying pendant nucleophiles can be employed to access a diverse set of heterocyclic compounds through intramolecular C–C or C–X bond formation processes (Scheme 11).153–159 Using a combination of NaOMe and KI as supporting electrolyte in MeOH proved to be the optimal for affecting electrochemical oxidation and subsequent intramolecular cyclization of tetrahydro(iso)quinolines 42 and 44 housing pendant alcohol or amine groups. For the tetrahydroisoquinoline-derived substrates 44a and 44b, selective oxidation at the benzylic position afforded 45a and 45b.157 The electrochemical oxidation/cyclization strategy could also be applied on 3-dialkylamino-1-phenylpropanols 46 to give the 1,3-oxazinane derivatives 47 in moderate to high yields.158 Furthermore, the authors also investigated the electrooxidation of dimethyl aminomalonates 48. Here, the use of NaCN as the supporting electrolyte resulted in the highest yields for the cyclized products 49.157,160
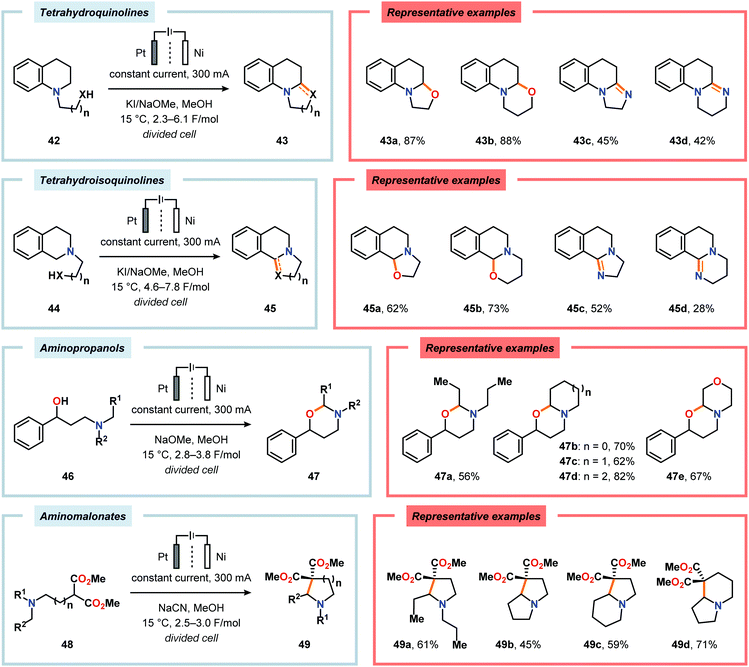 |
| | Scheme 11 Electrooxidative cyclizations of amine-containing motifs. | |
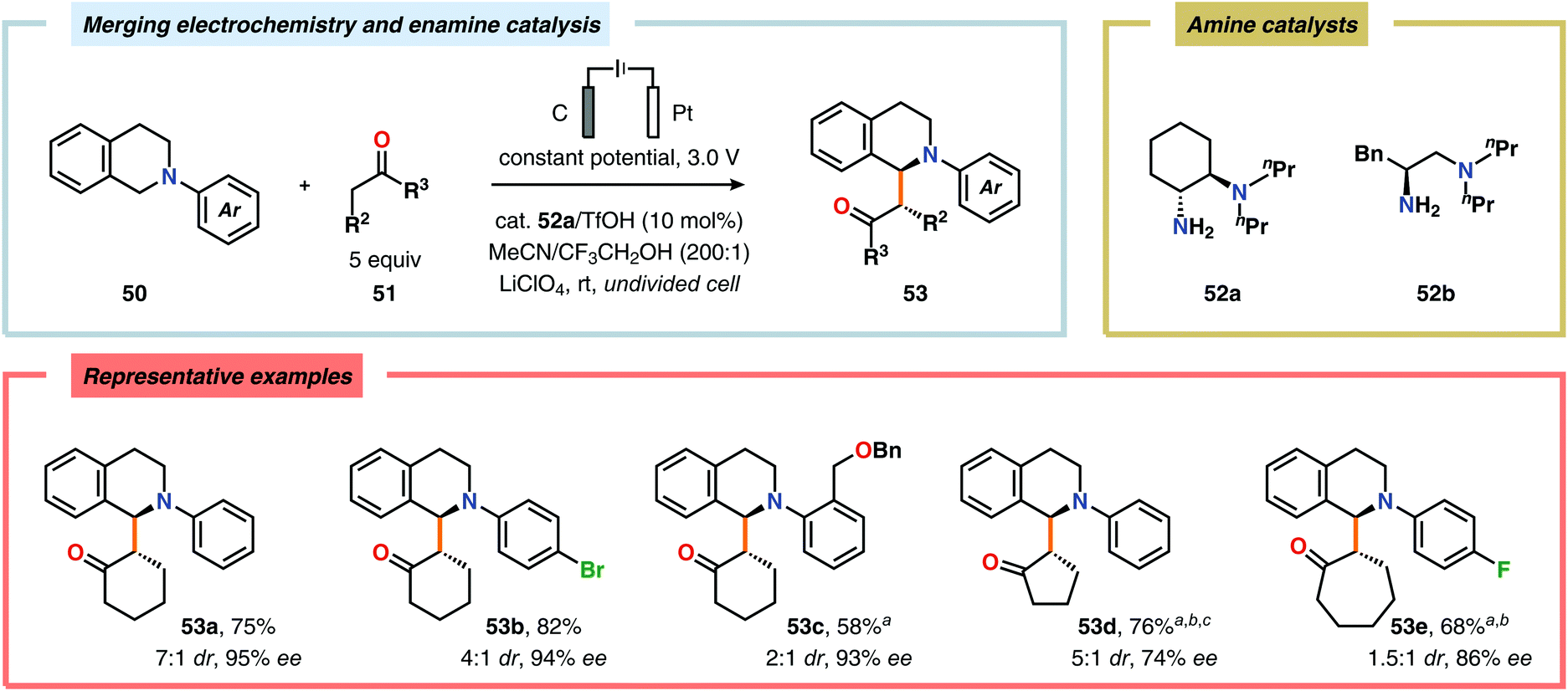
The Luo laboratory recently reported a visible light-mediated protocol for the enantioselective coupling of tetrahydroisoquinolines and simple alkyl ketones to furnish syn-Mannich-type adducts with good to excellent diastereo- and enantioselectivities. However, the process required the use of substoichiometric amounts of 3-nitrobenzoic acid as sacrificial oxidant.161 In order to improve the utility and eliminate the use of a chemical oxidant, Luo and coworkers developed a protocol based on enamine catalysis and electrochemical C–H oxidation. The authors identified the chiral primary amine 52a as the optimal catalyst and trifluoroethanol (CF3CH2OH) as the ideal additive. The established oxidant-free coupling reaction tolerated an array of N-aryl substituted tetrahydroisoquinolines and various simple ketones to provide the asymmetric oxidative coupling products 53 in good yields with good to excellent diastereo- and enantioselectivities (Scheme 12).162N-Aryl substituted tetrahydroisoquinolines are also susceptible to anodic nitro-Mannich C–C bond formation using nitromethane as the nucleophile.163 Furthermore, Shono-type oxidation of tetrahydroisoquinolines has also been used for the oxidative functionalization of benzylic C–H bonds, mediated by a dual bromide ion/2,2,6,6-tetramethylpiperidinyl-N-oxyl (TEMPO) redox catalyst system, to produce dihydro-isoquinolinones.164,165
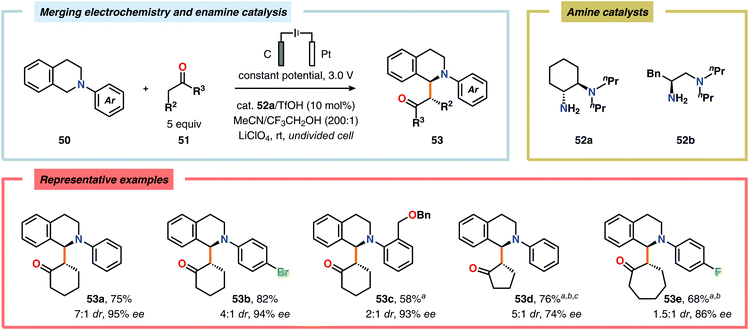 |
| | Scheme 12 Oxidative coupling of tertiary amines with simple ketones through the merger of electrochemical C–H oxidation and enamine catalysis. aHexafluoroisopropanol (HFIP) as additive. b52b as amine catalyst. cCell potential of 2.0 V. | |
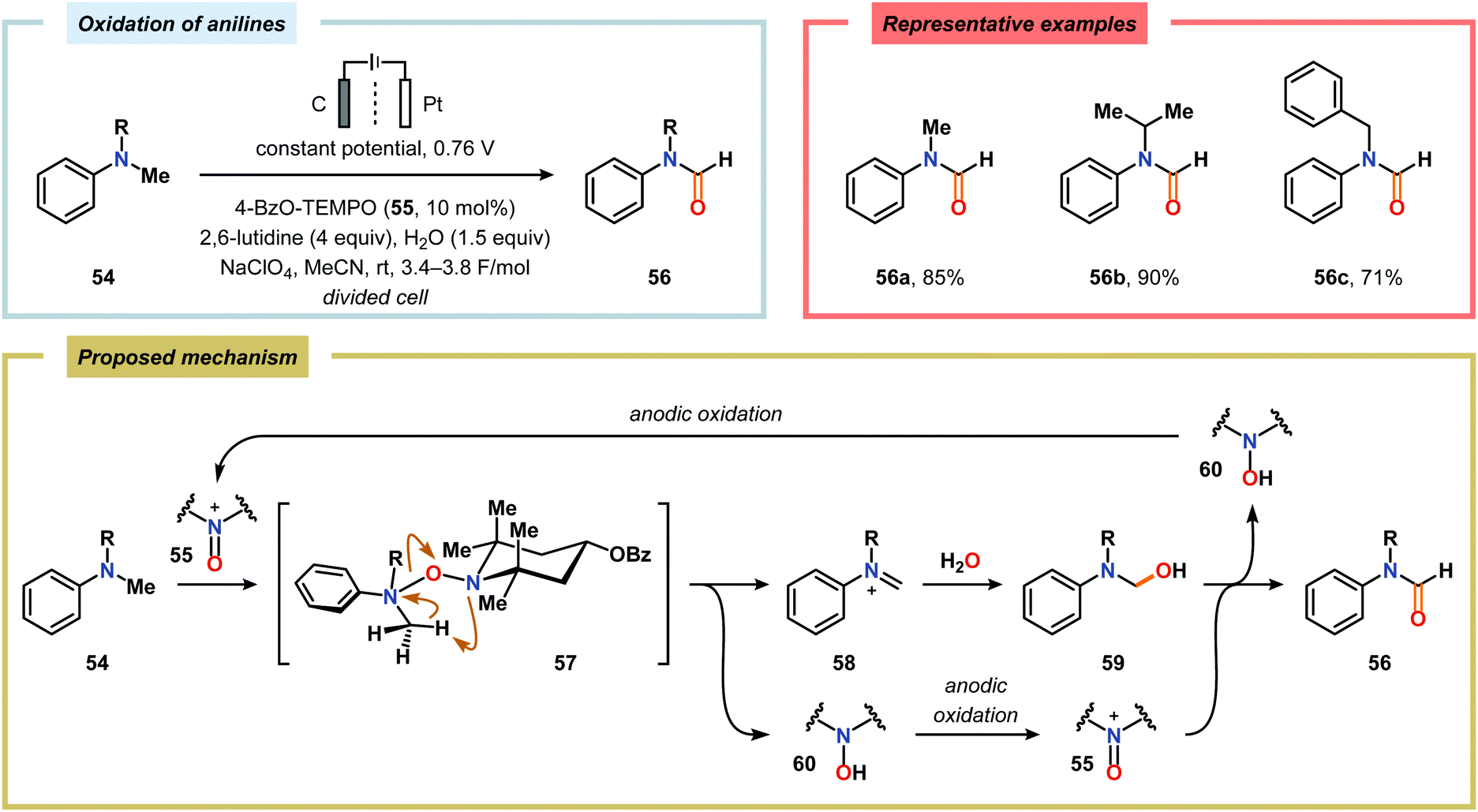
Kashiwagi and Anzai explored the electrochemical oxidation of N-alkyl-N-methylanilines (54) in aqueous media using a nitroxyl-based mediator. N-Alkylformanilides (56) were the major products and were obtained in 71–90% yield. The mechanism presumably proceeds through an iminium ion intermediate (58) which is hydrolyzed to produce hemiaminal 59. This species undergoes a second oxidation event, mediated by the electrochemically generated oxoammonium species 55, to furnish the corresponding formanilide 56 (Scheme 13).166
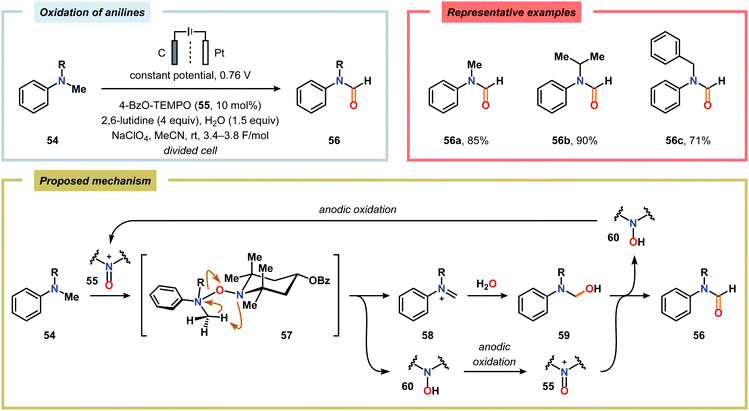 |
| | Scheme 13 Electrochemical oxidation of N-alkyl-N-methylanilines using a nitroxyl mediator. | |
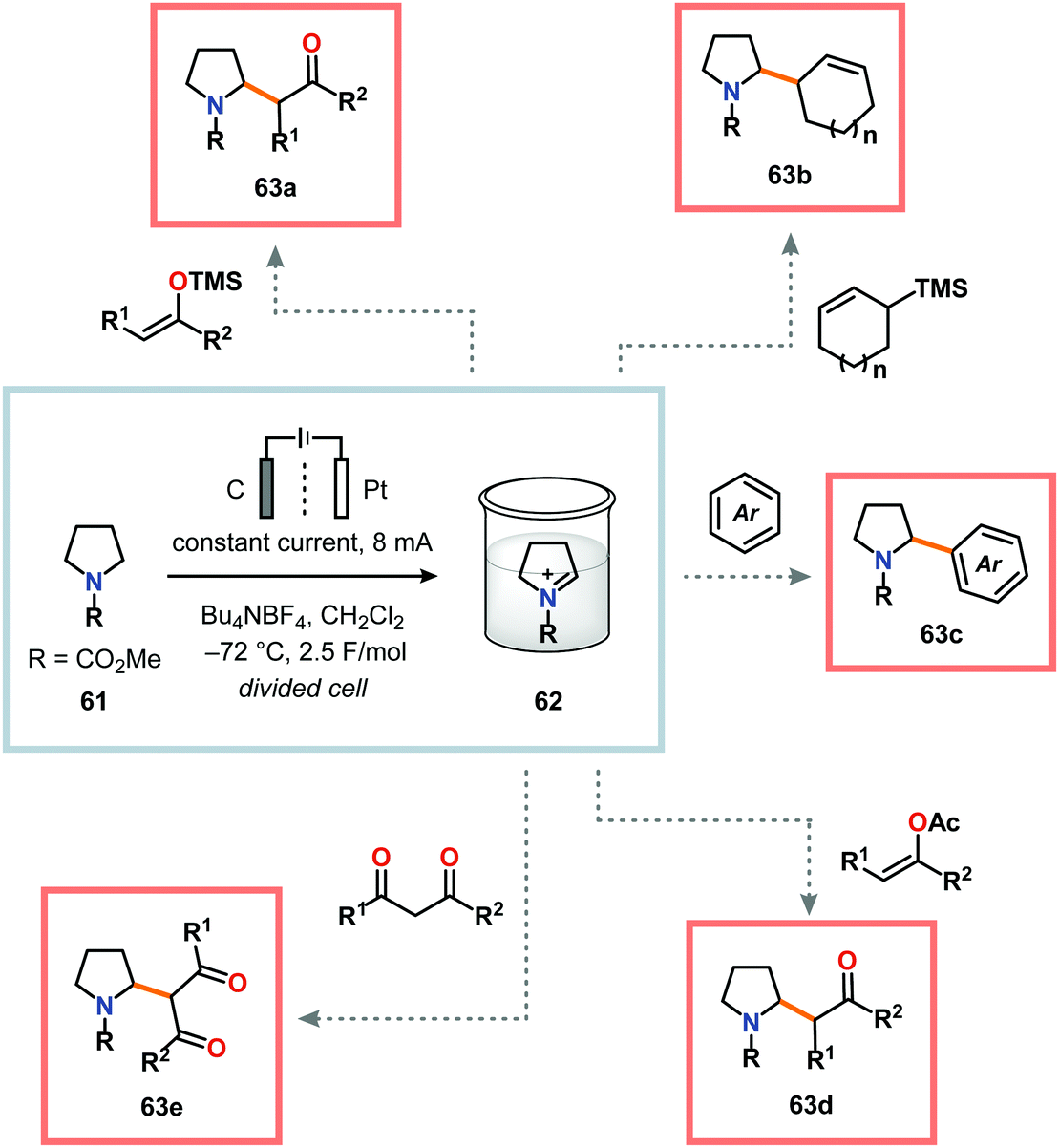
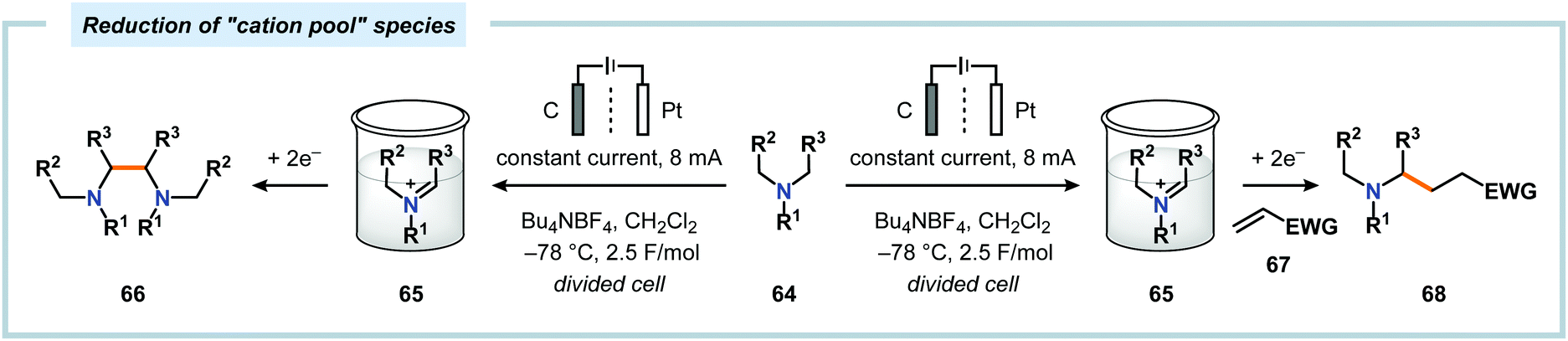
A potential strategy to expand the scope of the nucleophiles is to employ the “cation pool” method.57 The “cation pool” method involves generation and accumulation of cations, such as N-acyliminium ions, through electrolysis at low temperatures. The nucleophile is subsequently introduced to the reaction mixture under non-oxidative conditions, which allows for easily oxidized nucleophiles to be used. Early work from Yoshida and coworkers involving the generation of “cation pools” from carbamates by low-temperature electrolysis enabled the use of a collection of carbon nucleophiles, including allyl silanes, enol silyl ethers, enol acetates as well as aromatic and 1,3-dicarbonyl compounds (Scheme 14).167 Other compatible nucleophiles consist of allyl stannanes, benzyl silanes, Grignard reagents and organoaluminum compounds.168–170 These oxidative C–C bond forming reactions can also be carried out in a flow system, thus enabling continuous sequential combinatorial synthesis.171–175 The Yoshida group has also highlighted that electrochemical reduction of N-acyliminium ions, generated by the “cation pool” method, affords α-amidyl radicals. These radicals can be harnessed to produce homocoupled products or be intercepted with electron-deficient olefins in Giese-type176,177 reactions (Scheme 15).178,179 The latter transformation serves as a formal addition of C–H bonds to alkenes and offers new redox-based opportunities to manipulate free radical species.
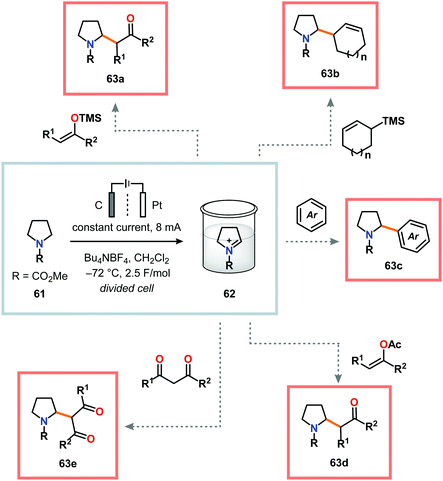 |
| | Scheme 14 Carbon–carbon bond formation of carbamates using the “cation pool” method. | |
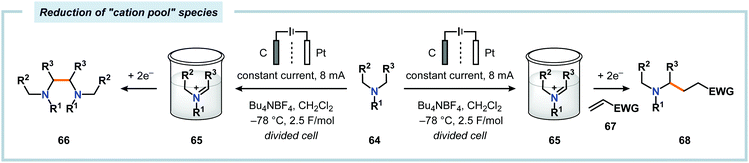 |
| | Scheme 15 Electrochemical reduction of “cation pool” generated acyliminium ions. | |
Additional strategies that allow for direct trapping of electrochemically generated N-acyliminium ions include the use of parallel laminar flow in micro-flow reactors,180,181 “site isolation”182,183 and acoustic emulsification.184–188 The Atobe group has illustrated that the direct allylation of carbamates can be accomplished through the use of micro-flow reactors in which the flow is stable and laminar, thereby enabling a stable liquid–liquid interface and mass transfer between the two streams occurs only through diffusion. This concept affords selective anodic oxidation of substrates without affecting the oxidation of nucleophile and provides effective trapping of potentially unstable cationic intermediates.180,181 Atobe and coworkers have also established the feasibility of using acoustic emulsification in Shono oxidations. Here, chemoselective anodic oxidation of the substrate is realized by selecting an electrolytic medium in which the nucleophile is insoluble in. Upon ultrasonication, the nucleophile is dispersed as submicrometer range droplets, which react with the electrogenerated carbocations when the two species are in close proximity.186,187 The Tajima laboratory has demonstrated the direct oxidative cyanation based on the concept of “site isolation”. Using a polystyrene-supported quaternary ammonium cyanide (PS-Me3N+CN−) as the heterogeneous cyanating reagent enabled the direct cyanation of anodically produced N-acyliminium ions, generated via Shono oxidation of carbamates. This highlights that the site isolation between an anode and, for example, the cyanating reagent (PS-Me3N+CN−) dramatically suppresses the undesired oxidation of CN− and allows for anodic cyanation of organic compounds, even ones that have higher oxidation potentials compared to CN−.182 Furthermore, utilizing recyclable solid-supported bases for in situ generation of electrolytes enables the development of systems where protic organic solvents serve as both the solvent and the supporting electrolyte.189–196 This allows for easy separation of the solid-supported base through filtration and provides, for example, the methoxylated Shono-type product, which can be subjected to the subsequent C–C bond forming reaction without having to replace the solvent.197,198 A complementary strategy for expanding the scope of Shono-type oxidations consists of using electroauxiliaries.50 This concept relies on the inherent properties of the electroauxiliaries to lower the oxidation potential of the substrate, thereby facilitating and providing enhanced control of the anodic oxidation of the substrate. The electroauxiliaries employed in Shono-type oxidations have typically been silyl-based,199–213 although 2,4,6-trimethoxyphenyl214 and sulfur-based215–217 electroauxiliaries have also been employed.
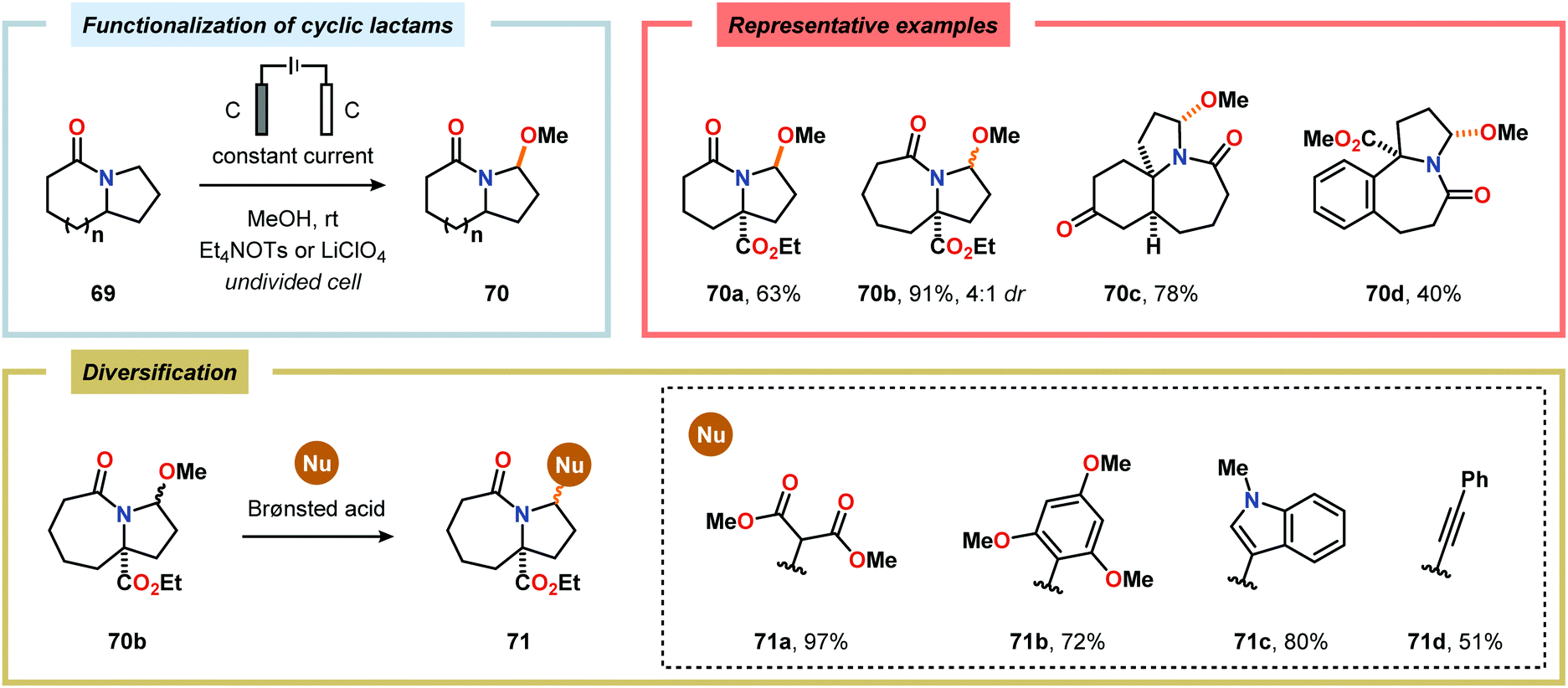
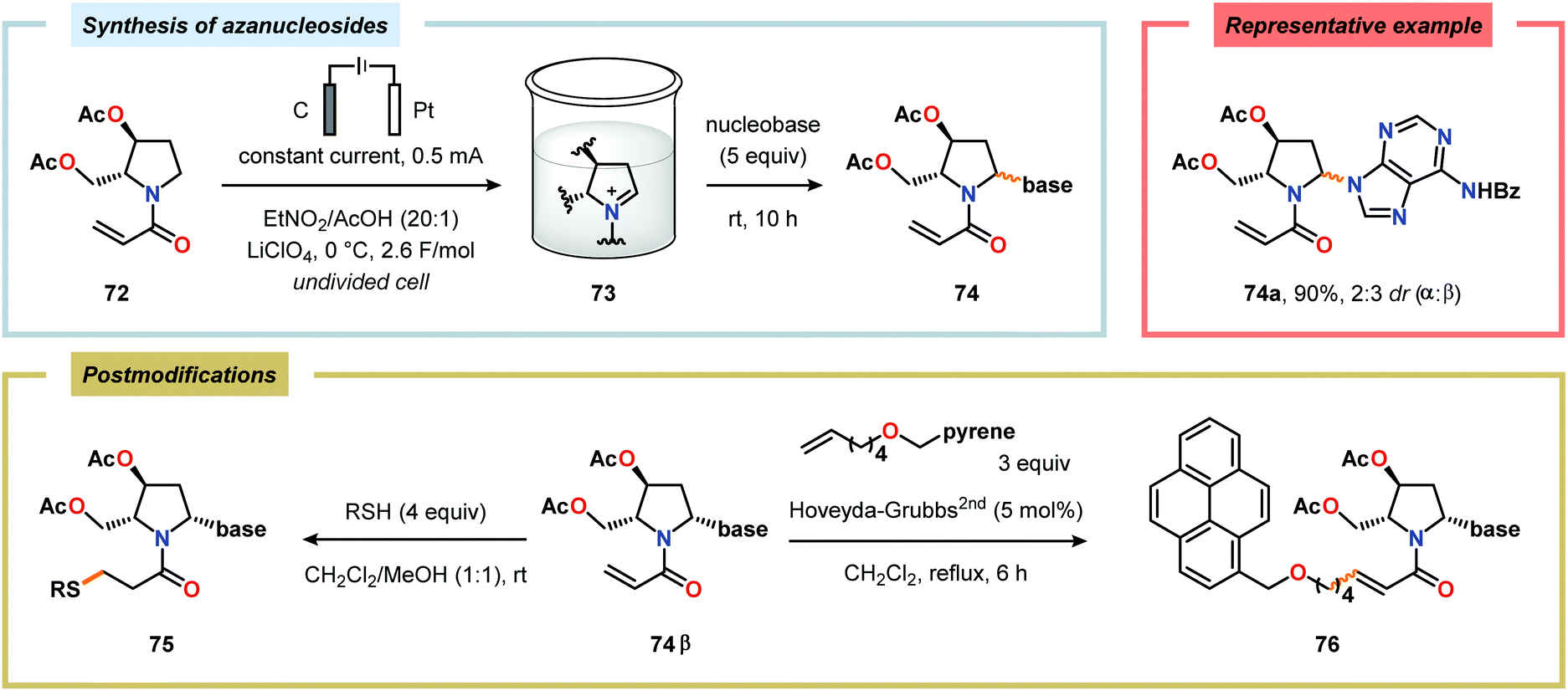
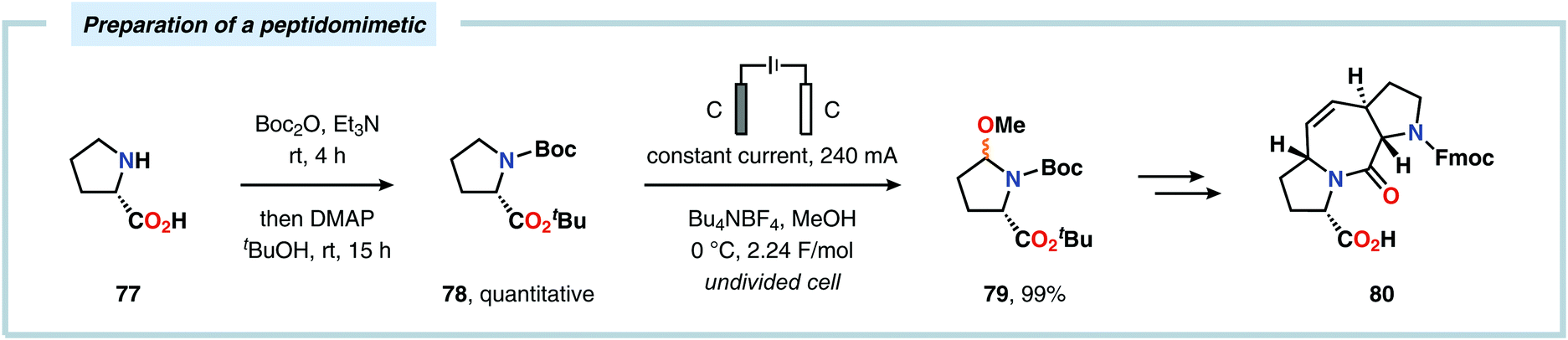
Considering the versatility of Shono-type oxidation reactions, it is perhaps unsurprising that these reactions have found numerous synthetic applications. The groups of Moeller and Aubé developed a practical two-step protocol for the late-stage functionalization of lactams by combining an intramolecular Schmidt reaction of keto azides followed by electrochemical anodic oxidation. The applicability of the Shono oxidation was exemplified across a range of ring systems. Finally, the versatility of the produced methoxyhemiaminals (70) was illustrated by synthesizing an array of functionalized lactam products (71) through the addition of various nucleophilic partners to the subsequent in situ generated N-acyliminium ions (Scheme 16).218 Azanucleosides and related analogues have been found to exhibit anticancer and antiviral activity. Furthermore, incorporation of azanucleoside motifs into oligonucleotide sequences results in increased tolerance towards degradation by nucleases.219 In this regard, the Matsumura group has exploited the Shono oxidation for preparation of azasugars,220 and Chiba and coworkers recently applied the Shono oxidation in the development of a synthetic method for accessing azanucleosides. Initial studies revealed that various nucleophiles and nucleobases could be introduced at the α-position upon the anodic modification of N-carbamate protected prolinol derivatives.221,222 Subsequent work showcased that N-acryloyl proline derivatives 72 could also be converted into the corresponding azanucleosides 74 by use of the Shono reaction. Moreover, the presence of the N-acryloyl group allowed for substrate postsynthetic modification through thiol conjugate addition or olefin cross-metathesis with a fluorescent dye (Scheme 17).223 Electrochemical oxidation has also proven to be a viable strategy towards the synthesis of γ-amino acids224 and preparing peptidomimetics.225–235 Schmalz and coworkers utilized the Shono oxidation in the context of accessing a tricyclic dipeptide mimetic (80) starting from L-proline (77). The anodic oxidation of L-proline tert-butyl ester 78 afforded gram scale quantities of the desired methoxylated product 79 in virtually quantitative yield (Scheme 18). Building block 79 was subsequently converted into diproline mimetic 80 through peptide coupling, ring-closing metathesis and protecting group adjustment.236
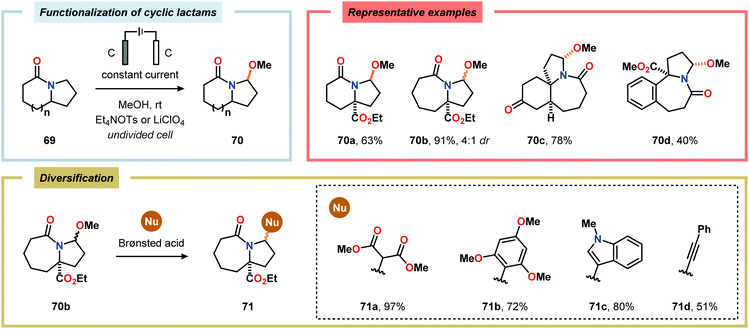 |
| | Scheme 16 Electrochemical modification and diversification of cyclic lactams. | |
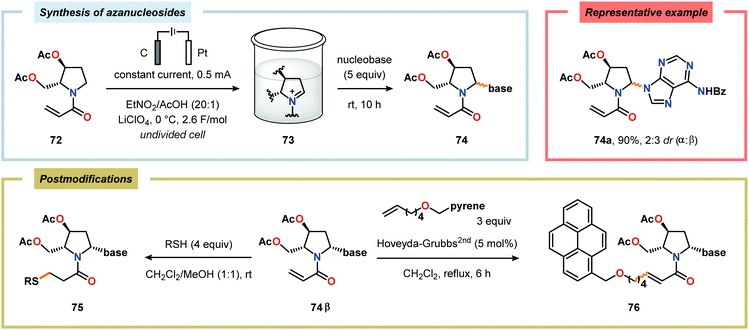 |
| | Scheme 17 Electrochemical synthesis and postmodification of N-acryloyl azanucleosides. | |
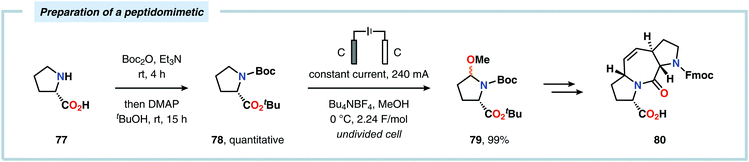 |
| | Scheme 18 Application of the Shono oxidation in the preparation of a peptidomimetic. | |
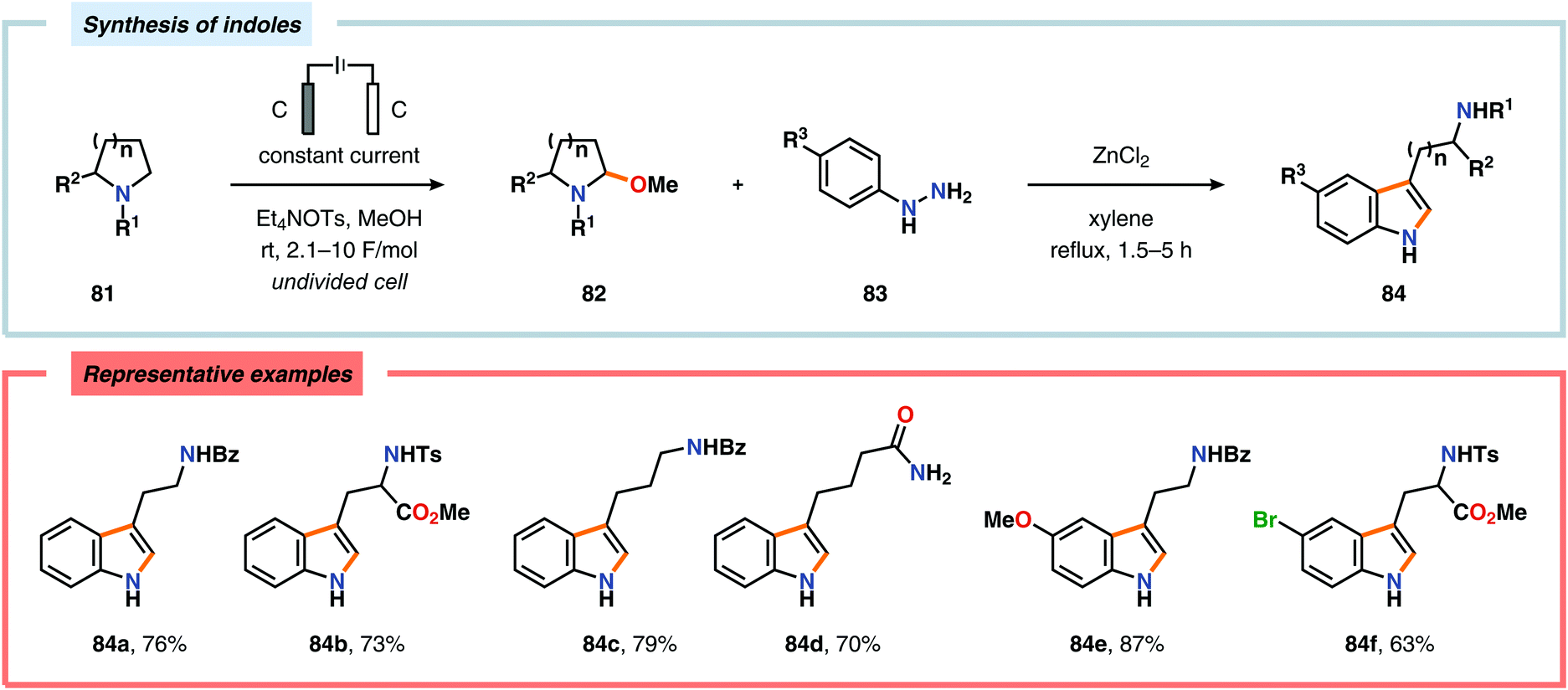
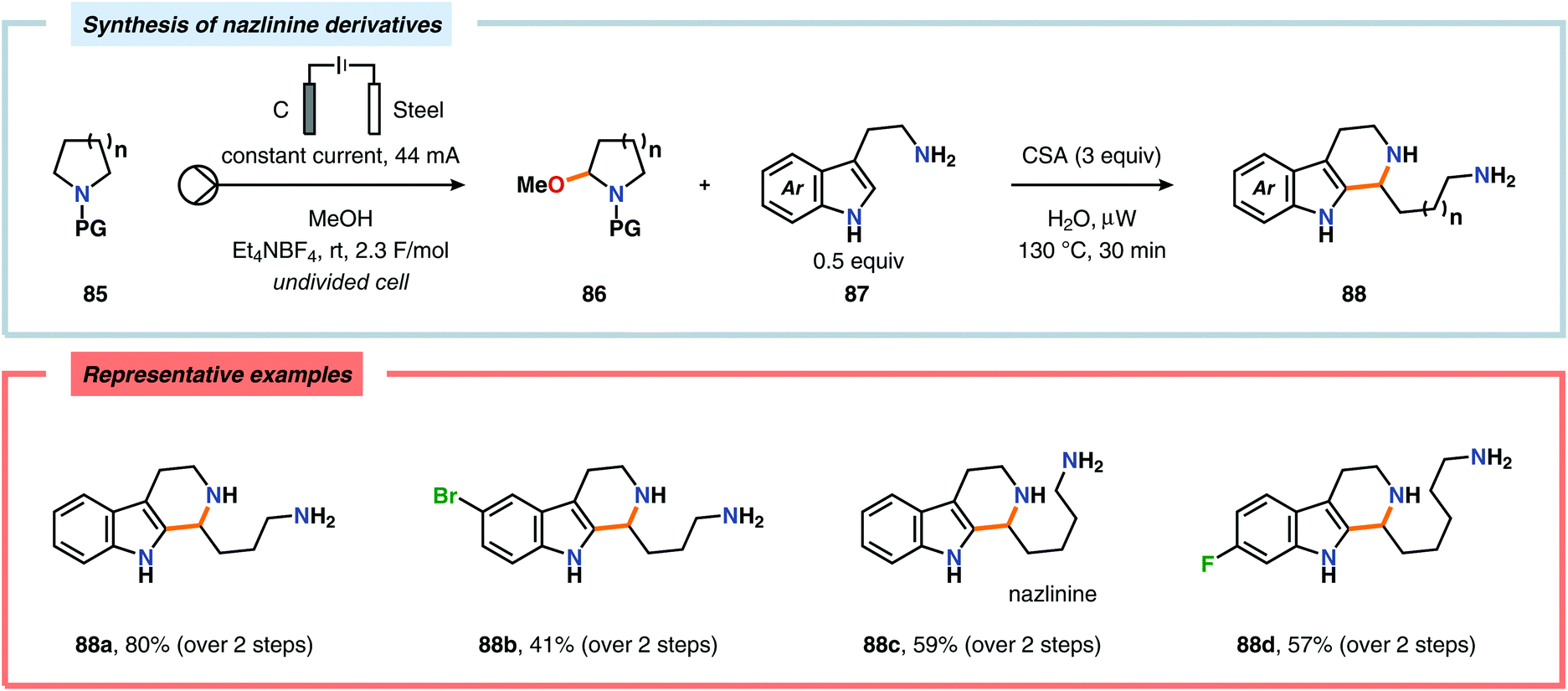
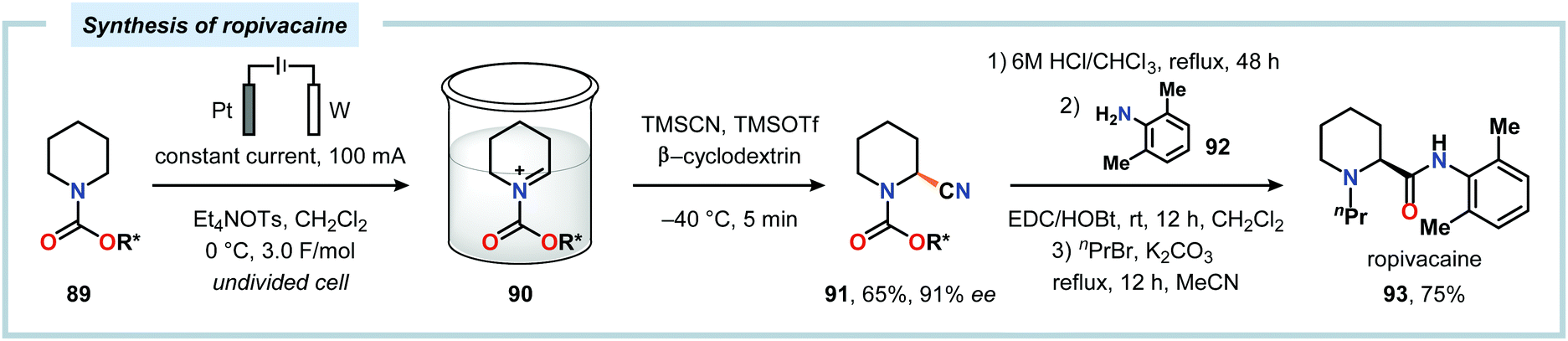
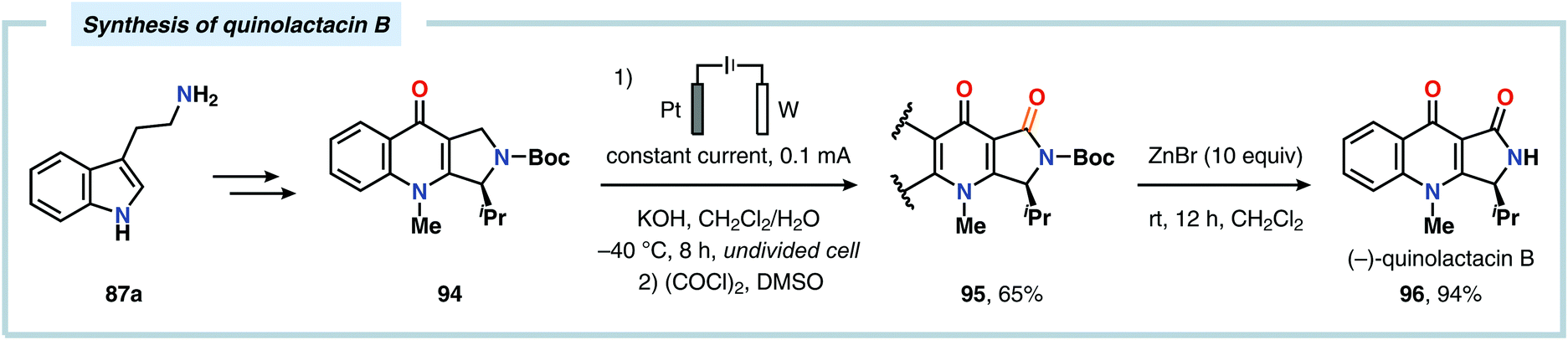
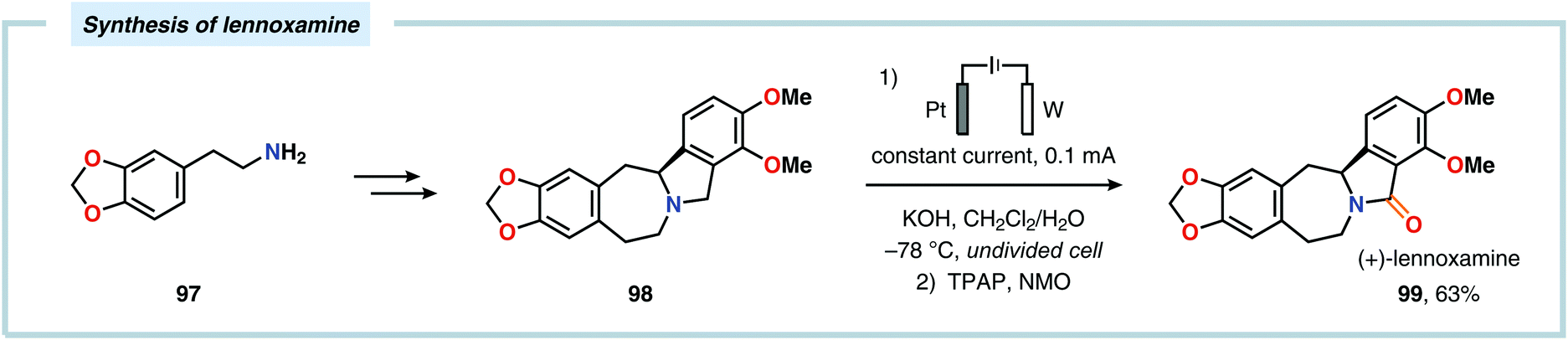
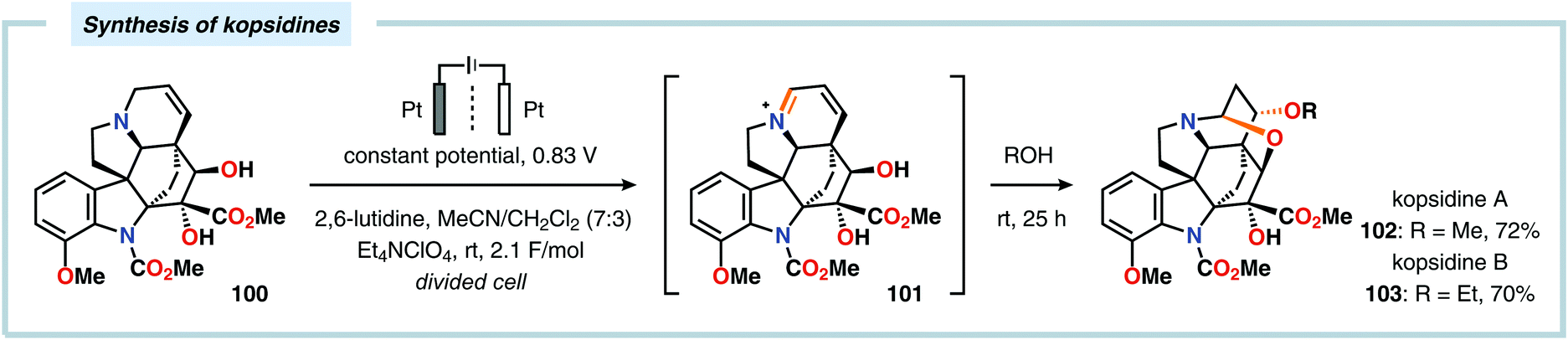
The Shono oxidation has been exploited as a general synthetic method for accessing β-substituted indoles (Scheme 19). The products derived from the Shono oxidation served as masked aminoaldehydes, which upon treatment with arylhydrazines and ZnCl2 afforded the corresponding β-substituted indoles through a Fischer indole synthesis pathway.237 Ley and coworkers identified flow electrochemistry as an enabling technology for the Shono oxidation of various N-protected cyclic amines. Here, the Shono oxidation furnished the corresponding cyclic α-methoxyamines in excellent yields. Applying a Pictet–Spengler reaction between the electrosynthesized α-methoxyamines and substituted tryptamines afforded the biologically active indole alkaloid nazlinine (88c) and numerous related unnatural congeners (Scheme 20).238 Other alkaloids that have been prepared include (−)-crispine A239 and (±)-pumiliotoxin C.240 Here, anodic cyanation furnished the desired α-amino nitriles, which upon further manipulations furnished the target alkaloids. The Santos laboratory has also demonstrated the utility of the Shono oxidation in total syntheses of ropivacaine241 (93, Scheme 21), (−)-quinolactacin B242 (96, Scheme 22) as well as (+)-lennoxamine243 (99, Scheme 23). Finally, the Kam group reported that electrochemical oxidation could be applied to aspidofractinine-type alkaloids for construction of kopsidines A and B (Scheme 24), thus highlighting the potential of carrying out late-stage functionalization of complex molecules.244 Additional synthetic applications of Shono-type oxidations include the synthesis of an angiotensin-converting enzyme inhibitor,245 preparation of methoxylated analogues of the anticancer drugs ifosfamide and cyclophosphamide,246 as well as dealkylation,247–250 deallylation and debenzylation251 of amines and amides.
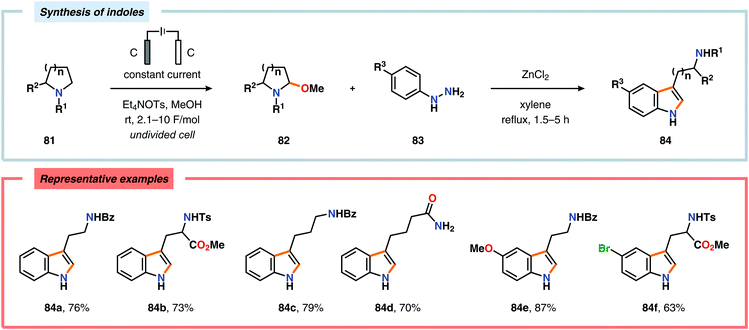 |
| | Scheme 19 Synthesis of β-substituted indoles via the Shono oxidation. | |
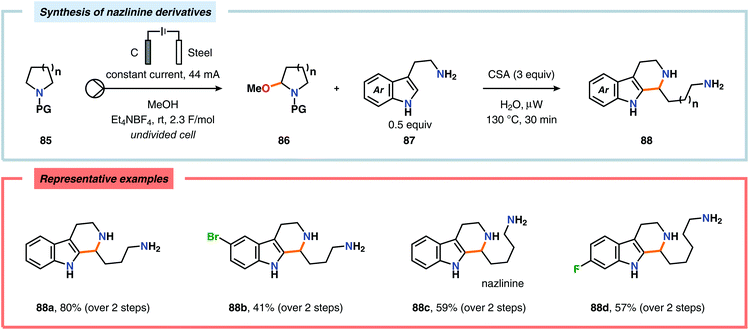 |
| | Scheme 20 Synthesis of nazlinine and unnatural congeners via a two-step method. | |
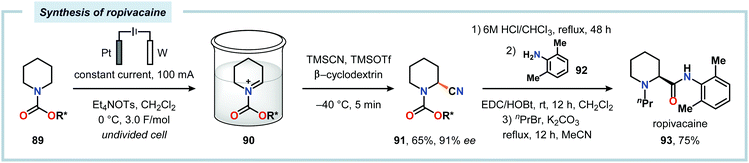 |
| | Scheme 21 Synthesis of ropivacaine employing the “cation pool” strategy. R* = 8-phenylmenthyl. | |
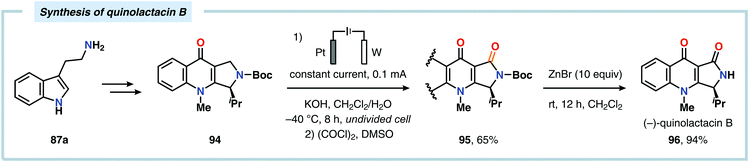 |
| | Scheme 22 Application of a late-stage Shono oxidation in the synthesis of (−)-quinolactacin B. | |
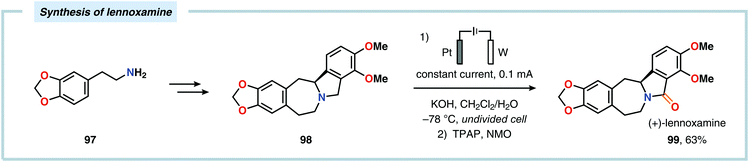 |
| | Scheme 23 Application of a late-stage Shono oxidation in the synthesis of (+)-lennoxamine. | |
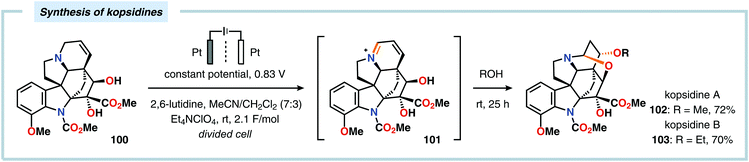 |
| | Scheme 24 Shono-type oxidation of aspidofractinine-type alkaloids. | |
2.2. Anodic halogenation
Incorporation of fluorine into organic compounds has found broad applications in medicinal,252–254 agrochemical255,256 and material sciences.257 Over the past decade, significant advances in chemo- and stereoselective fluorination of molecules with high functional and structural complexity have been made. In general, three different strategies exist for synthesis of organofluorine compounds: electrophilic,258 nucleophilic259 and radical260 fluorination.261,262
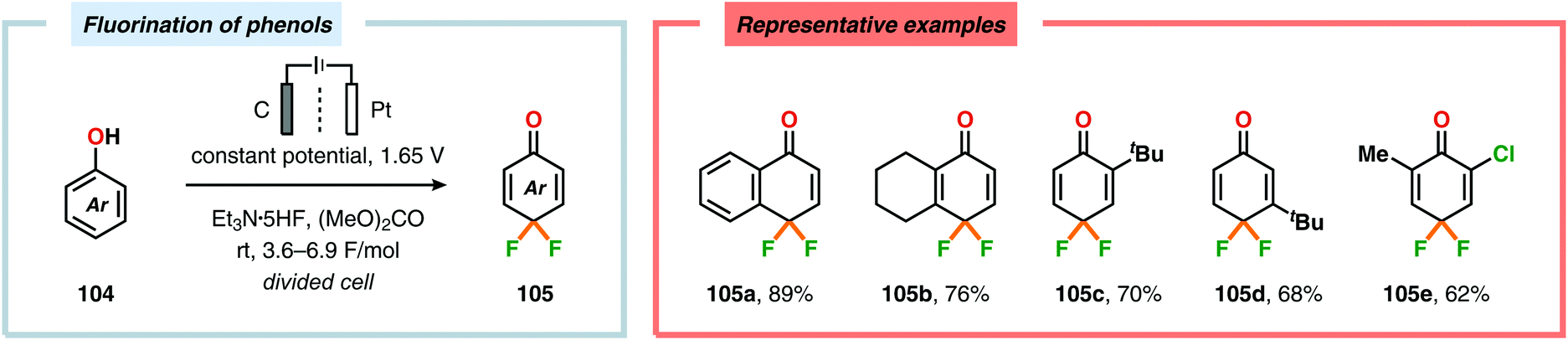
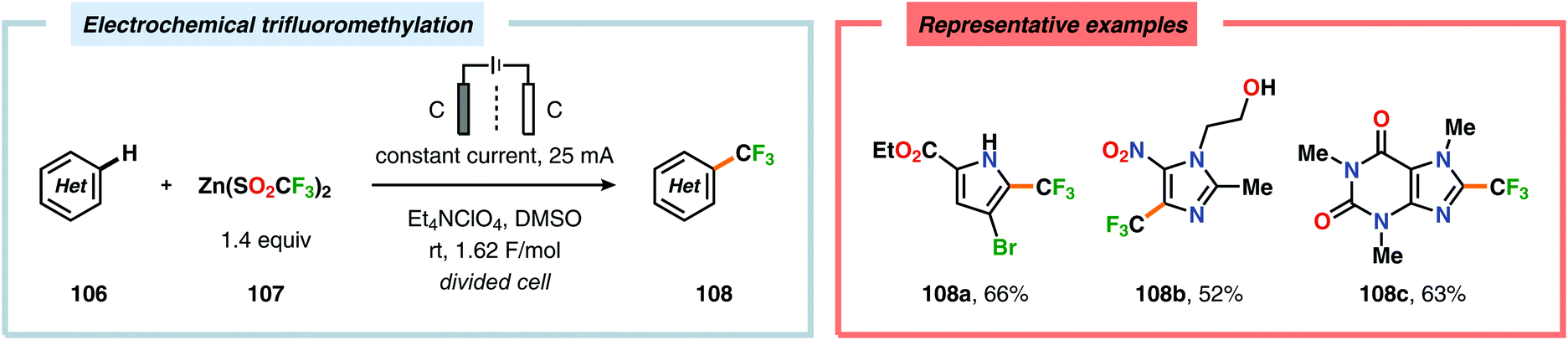
Seminal studies by Rozhkov and coworkers in 1970 highlighted that selective electrochemical fluorination of arenes could be achieved in MeCN using Et4NF·3HF as the ionic liquid, albeit in low yields. These reactions were proposed to proceed via arene radical cations, generated at the anode, that subsequently were trapped by fluoride to produce the carbon–fluorine (C–F) bond.263–265 More recently, attempts to achieve selective fluorination of simple aromatic compounds include the work by Matsuda and coworkers. However, the yields of the fluorobenzenes were typically low.266–269 Unlike unsubstituted arenes, such as benzene and naphthalene, electrochemical fluorination of toluene and related derivatives remains a challenge, as mixtures of products are formed originating from aromatic substitution, side chain substitution and addition reactions.270–275 Heteroaromatics276–283 and nitrogen-containing heterocycles284–286 have also been reported to react in a similar manner. Anodic oxidation of phenols using Et3N·5HF as the electrolyte was examined by the groups of Hara and Yoneda, and selectively afforded the corresponding 4,4-difluorocyclohexadienone derivatives in good yields (Scheme 25).287–291 Furthermore, trifluoromethanesulfinate (CF3SO2−) has recently been exploited as a source for electrochemical generation of trifluoromethyl radicals (CF3˙) in the presence of electron-rich aromatics292 and heterocycles.293 In this regard, the Baran and Blackmond groups demonstrated that electrochemical oxidation of zinc trifluoromethanesulfinate {Zn(SO2CF3)2} in DMSO in the presence of heterocycles, such as pyrazoles, pyrroles and benzothiazoles, afforded the corresponding trifluoromethylated products (Scheme 26). Moreover, the authors successfully highlighted that the developed electrochemical protocol was able to provide the functionalized heterocyclic products more effectively than conventional peroxide radical-initiated protocols for a majority of the screened substrates.293
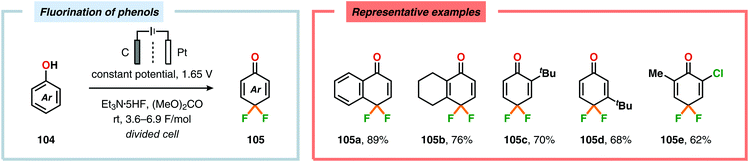 |
| | Scheme 25 Synthesis of difluorocyclohexadienones through electrochemical oxidation of phenols. | |
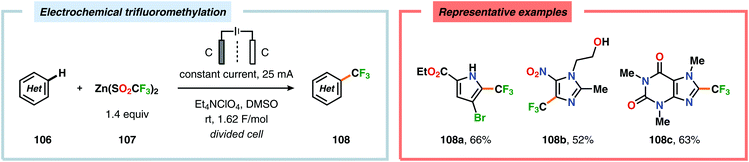 |
| | Scheme 26 Electrochemical trifluoromethylation of heterocycles. | |
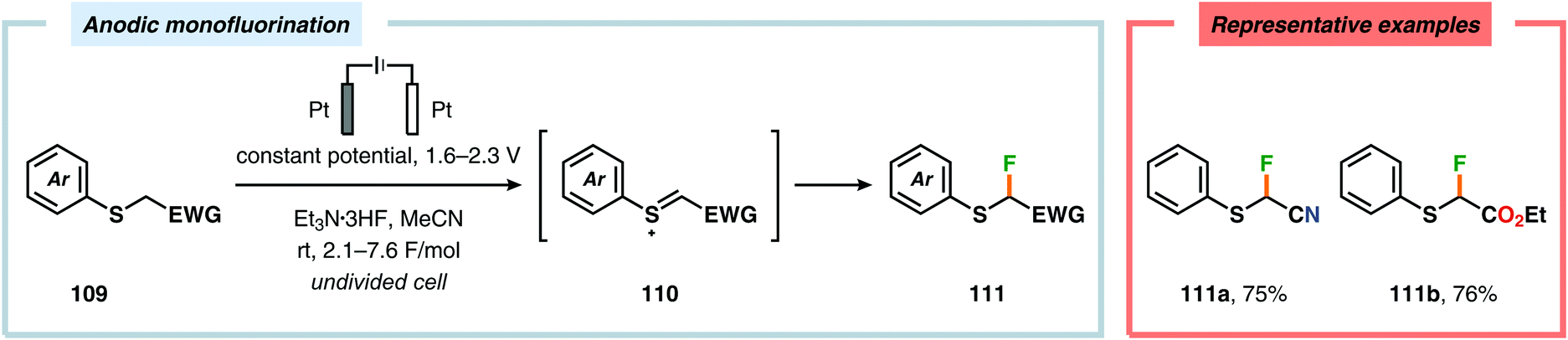
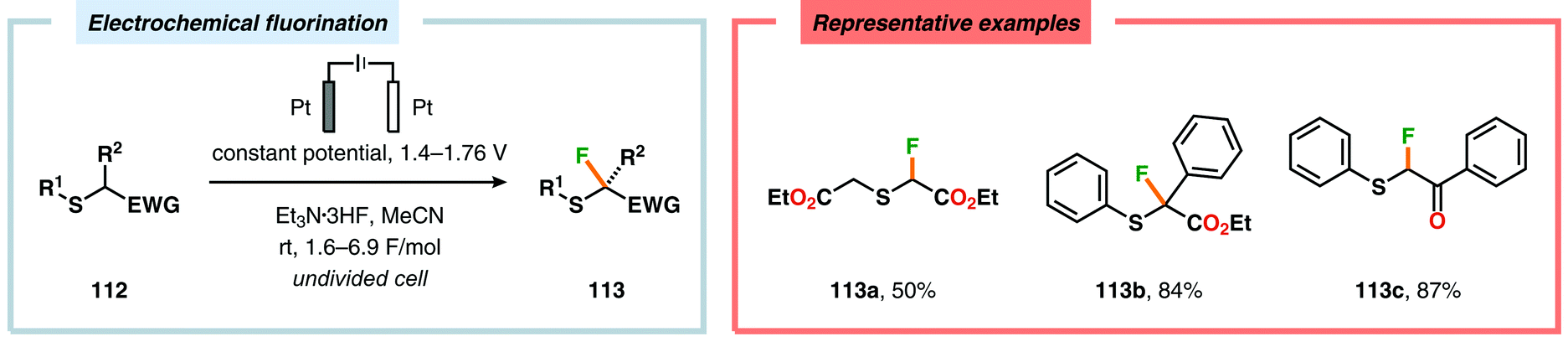
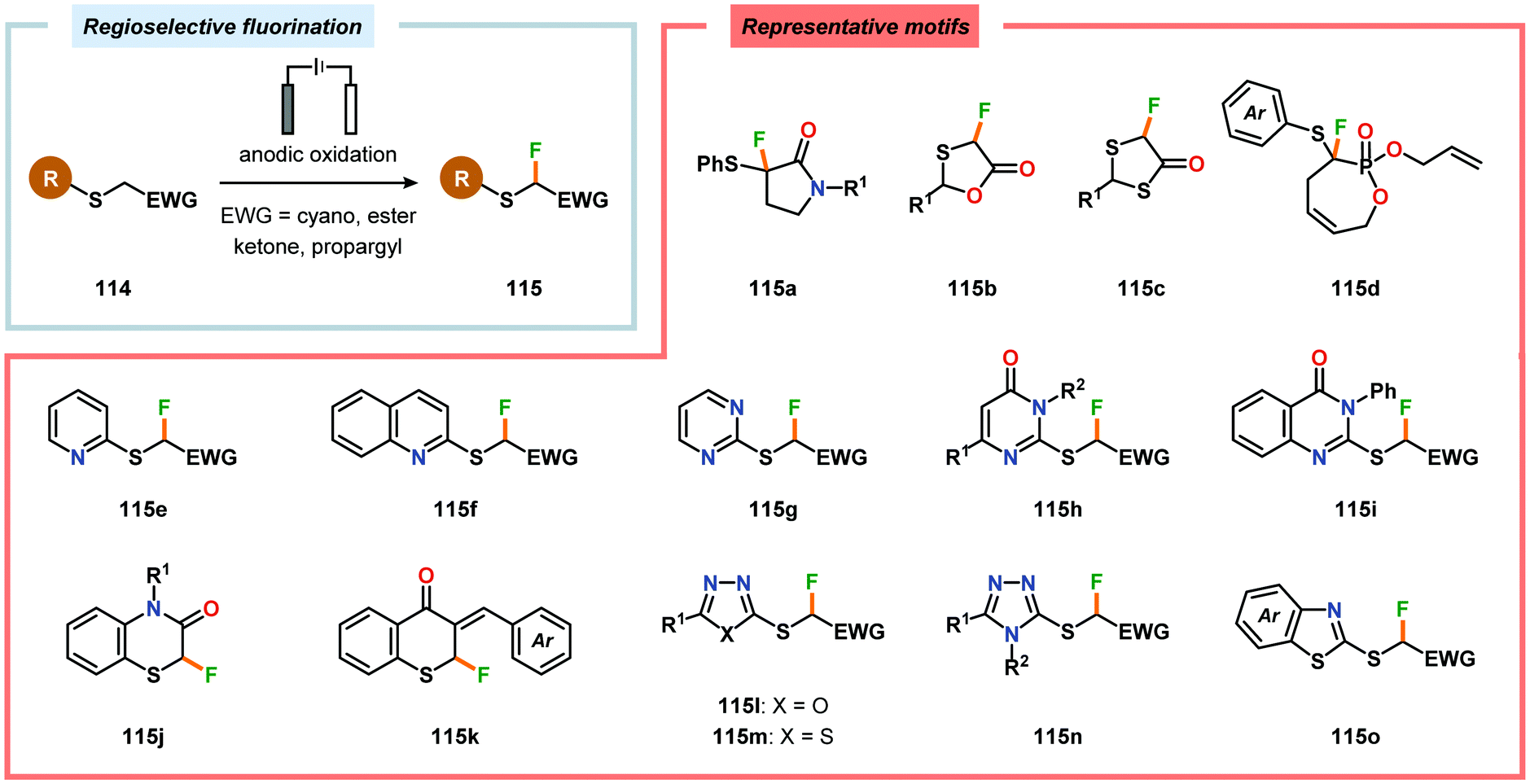
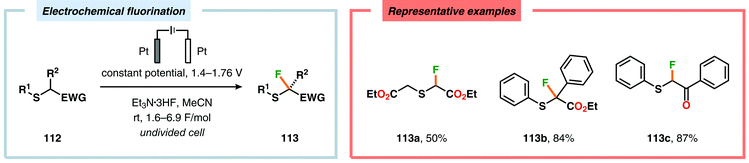
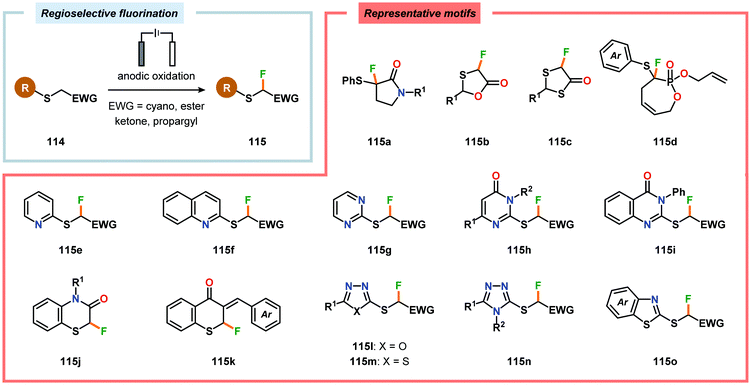
Electrochemical fluorination of arylsulfides constitutes another transformation that has been extensively studied by several research groups. Early work during the 1990s from the Fuchigami294–302 (Scheme 27) and Laurent303,304 (Scheme 28) laboratories demonstrated that the introduction of an electron-withdrawing group at the α-position to sulfur had a dramatic influence on the reactions. These α-fluorination transformations were believed to proceed through a Pummerer-type mechanism where the electron-withdrawing group facilitates deprotonation of the α-carbon to produce intermediate 110, thereby promoting fluorination.305 The same effect could also be achieved using aromatics containing strongly electron-withdrawing groups, such as p-CN, p-NO2 and p-SO2Ph, as demonstrated by Simonet and coworkers.306,307 These seminal studies constitute the first examples of selective electrochemical fluorination of chalcogen compounds. Since then, a variety of distinct sulfide-based motifs, such as 2H-benzo[b][1,4]-thiazin-3(4H)-one (114j),308 benzothiazole (114o),309–311 2,3-dihydrothiochroman-4-one (114k),312 1,3-dithiolan-4-one (114c),313 lactam (114a),308 1,3,4-oxadiazole (114l),311,314 1,3-oxathiolan-5-one (114b),315 pyrimidin-4(3H)-one (114h),316 phosphonate (114d),317,318 pyridine (114e),310,319 pyrimidine (114g),310,311,319,320 quinazolin-4(3H)-one (114i),319 quinoline (114f),310 1,3,4-thiadiazole (114m),311,314 1,2,4-triazole (114n),314 have been shown to be amenable to regioselective monofluorination (Scheme 29). Because of their good conductivity, non-flammability, non-volatility and thermal stability, ionic liquids, such as Et3N·nHF, have found widespread use as solvents in electrochemical fluorination reactions.321–326 Recently, Fuchigami and coworkers demonstrated miscellaneous approaches to improve the electrochemical fluorination. One approach makes use of ultrasonication to promote the mass transport of the substrate due to the high viscosity associated with the ionic liquids. This was shown to increase the current efficiencies, selectivities and yields in the anodic fluorination of a variety of organosulfur compounds.327 The authors have also developed an electrochemical system based on the cation-exchange reaction between alkali-metal fluorides and solid-supported acids. In the presence of 2,6-lutidine, the exchange reaction results in the in situ generation of 2,6-lutidine·HF, which functions as the fluorinating agent and supporting electrolyte, thus obviating the need to handle hazardous HF.328 The Fuchigami group has also shown that indirect anodic fluorination can be realized using a hypervalent iodoarene difluoride (ArIF2) mediator, which can be anodically produced from the corresponding iodoarene species and a HF-based ionic liquid (Et3N·3HF). Here, indirect reaction with the hypervalent iodoarene mediator proceeded efficiently to furnish the desired monofluorinated product, thus limiting anode passivation (the formation of a nonconducting polymer film on the electrode surface that suppresses faradaic current), a drawback that is often encountered when carrying out direct electrochemical fluorination.329 Attempts to develop diastereoselective fluorination protocols have also been reported using chiral auxiliaries derived from camphorsulfonic acid,330 (R)-(−)-3-chloro-1,2-propanediol,331L-cysteine332 and menthol,333 providing the monofluorinated products in modest to good diastereoselectivities.
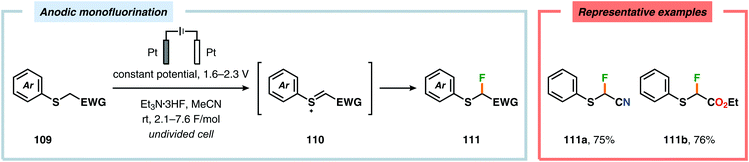 |
| | Scheme 27 Anodic monofluorination of organosulfur compounds. | |
 |
| | Scheme 28 Electrochemical fluorination of organosulfur compounds. | |
 |
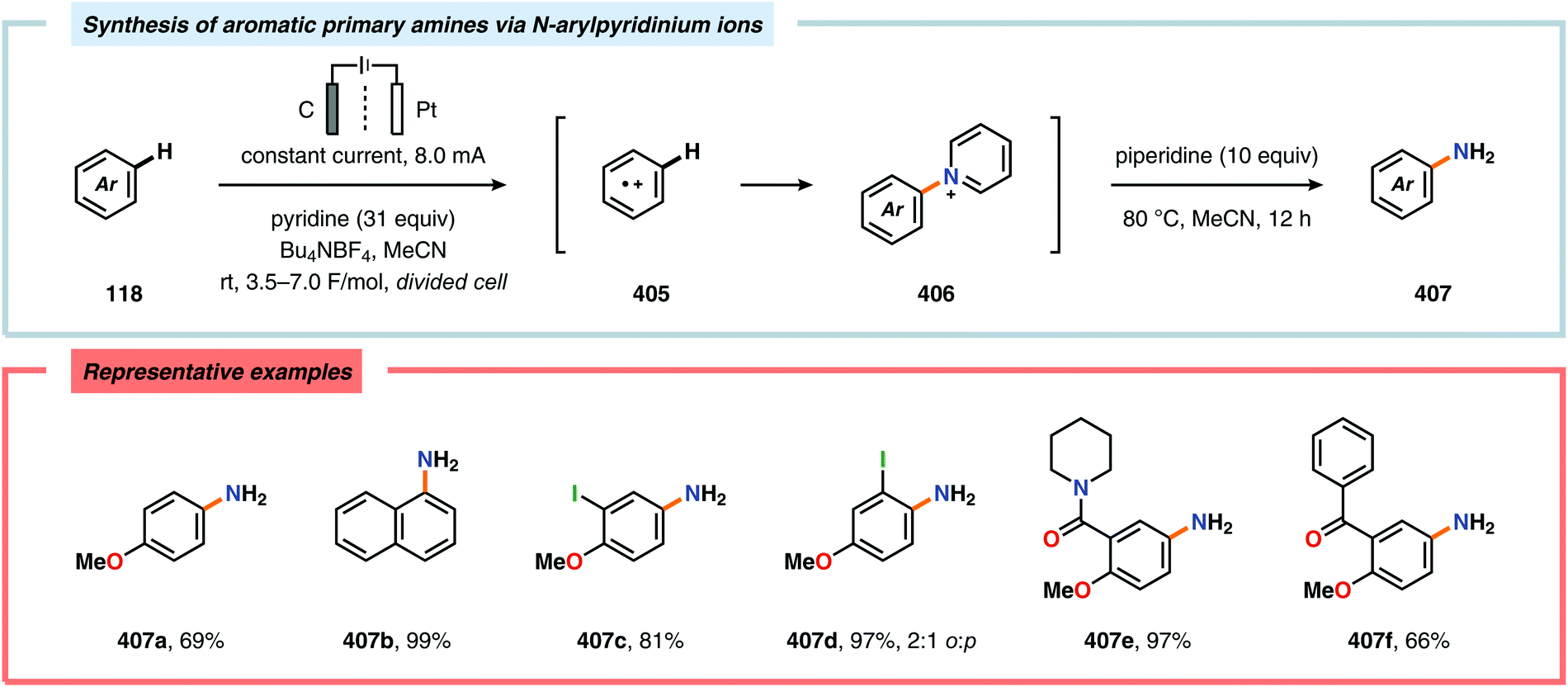
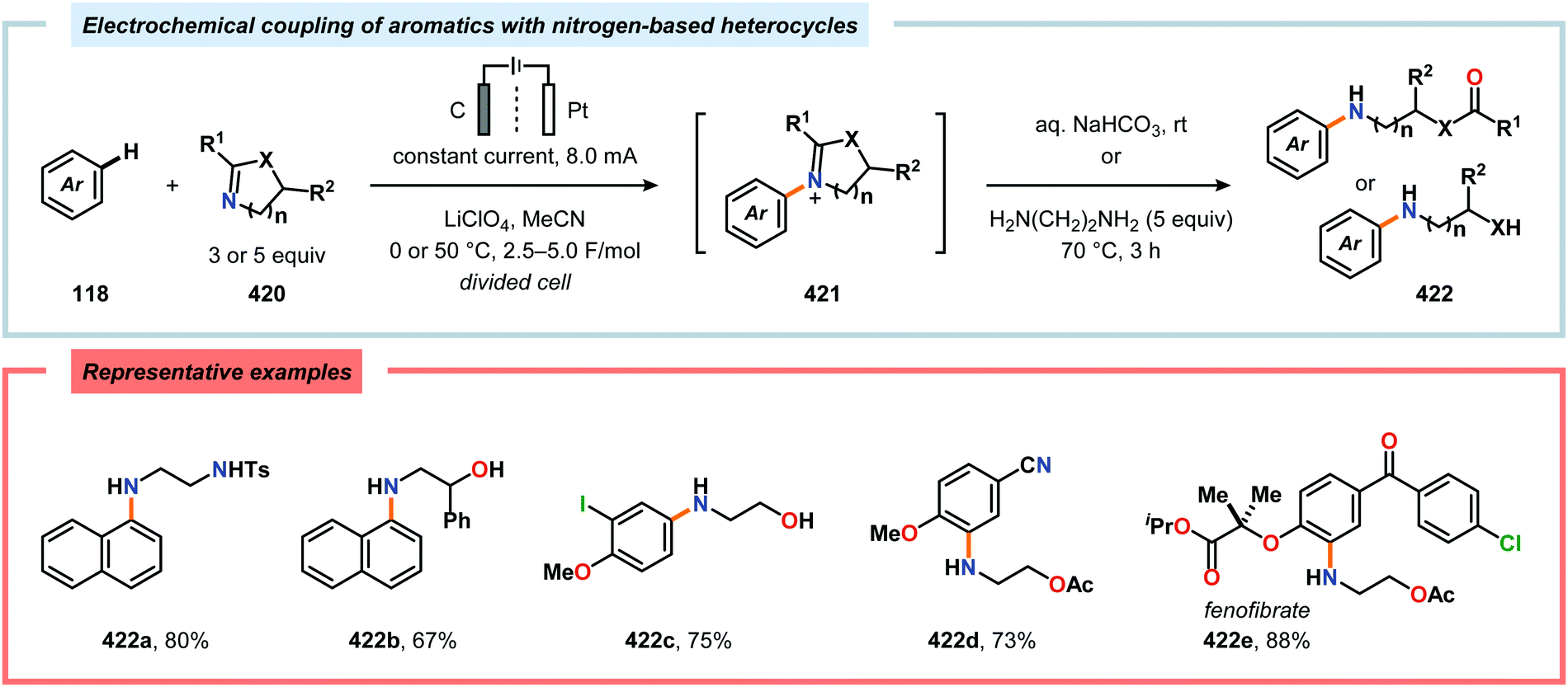
| | Scheme 29 Electrochemical regioselective fluorination of organosulfur compounds. | |
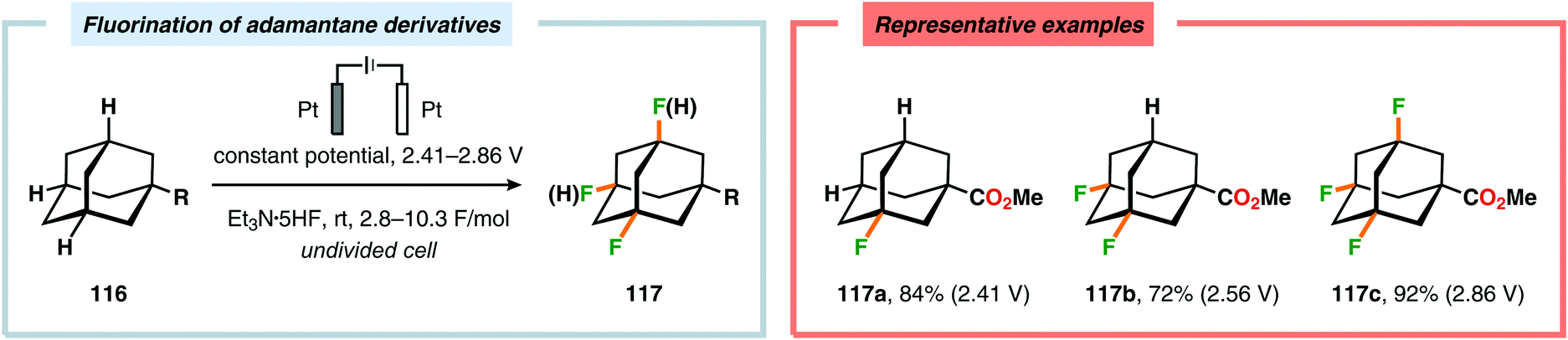
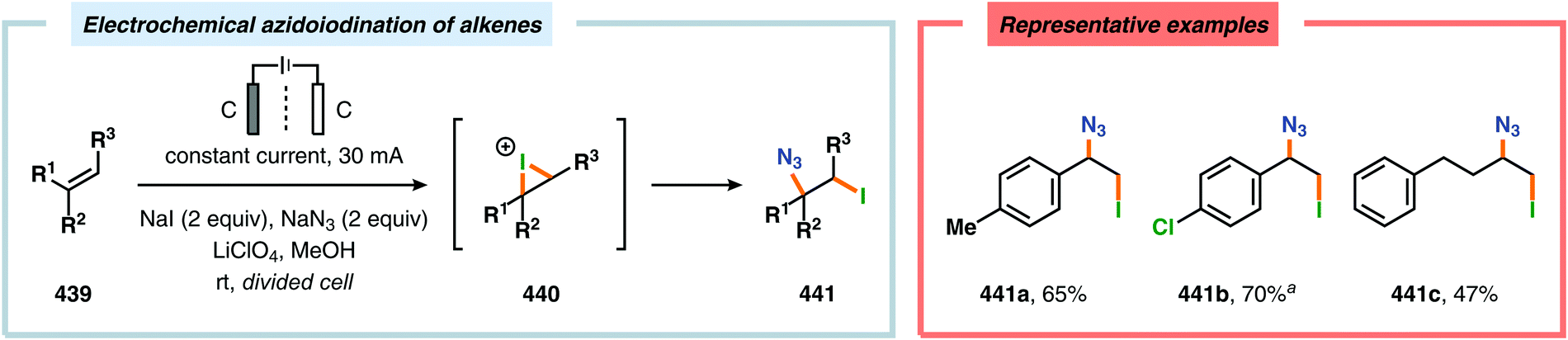
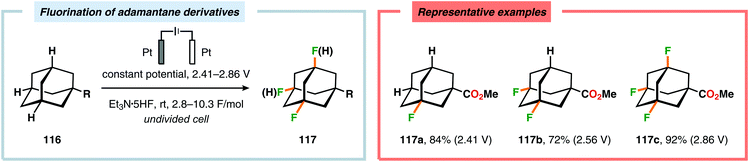
Recently, electrochemical fluorination of adamantane derivatives has been studied by Hara and coworkers. By controlling the oxidation potential, the authors were able to guide the fluorination process to selectively yield the mono-, di-, tri- or tetrafluorinated adamantane derivatives (Scheme 30).334,335 Moreover, regioselective electrochemical monofluorination has also been reported for a collection of oxygen-containing heterocycles, including carbonates,336,337 chroman-4-ones,338–340 esters,337 ethers336,337 and lactones.336,337 Additionally, electrochemical protocols for accomplishing fluorodesulfurization,341–349 difluorination,350,351 fluoro-selenylation,352 iodofluorination353 and thiofluorination354 of electron-deficient olefins as well as partial fluorination of conjugated dienes355 have been devised.356
 |
| | Scheme 30 Selective electrochemical fluorination of adamantane derivatives. | |
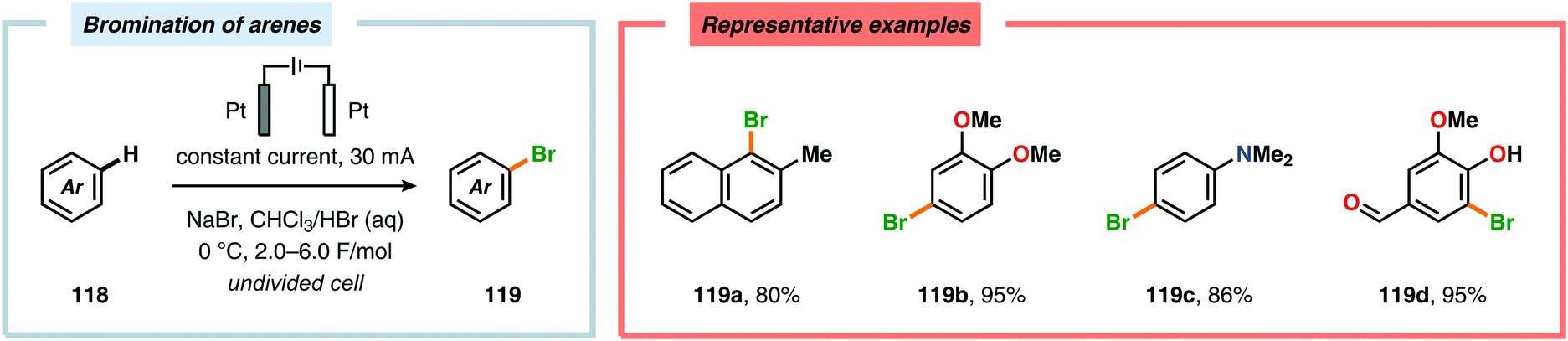
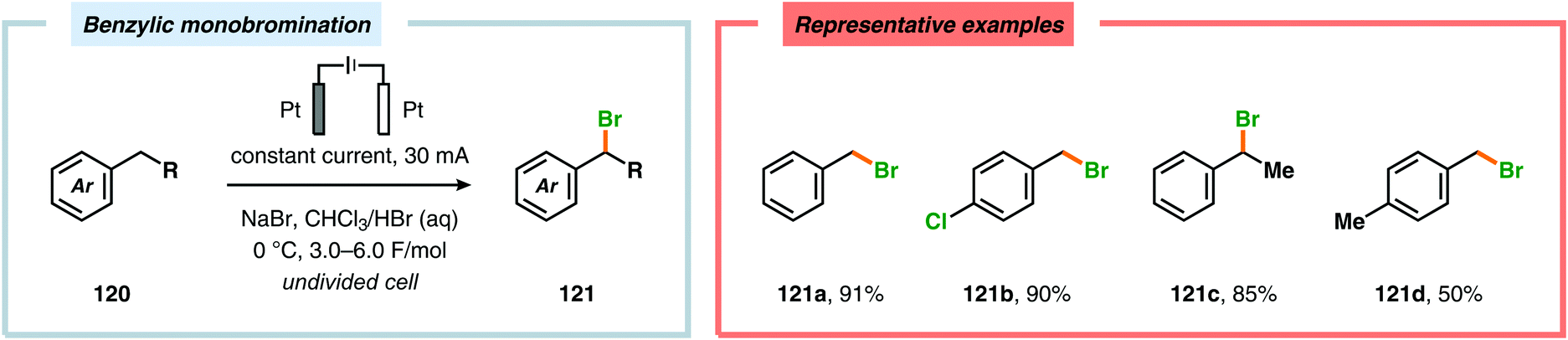
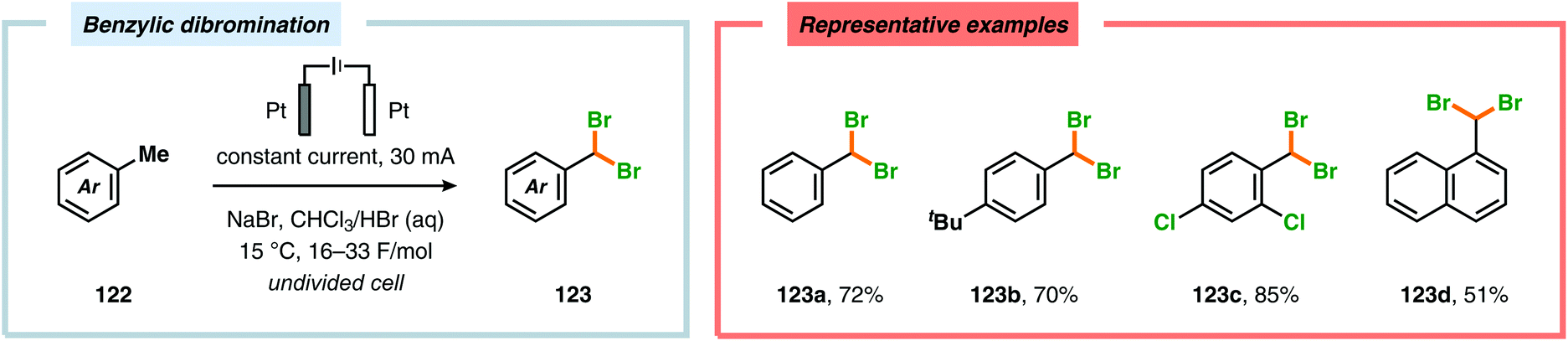
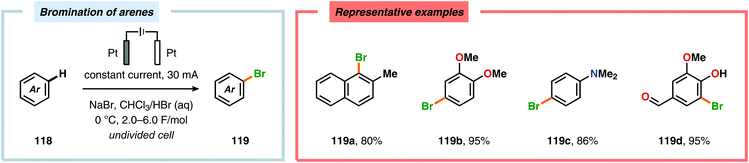
In contrast to the electrochemical fluorination strategies, the anodic oxidation protocols involving chlorides, bromides and iodides typically proceed via generation of the corresponding electrophilic molecular counterpart (X2) due to the relatively low oxidation potentials of these halogen ions.357–367 Due to the limited solubility of alkali metal halide salts in organic solvents, a biphasic system consisting of an aqueous phase and a halogenated organic solvent is commonly employed to affect chlorination of aromatics368,369 and heterocycles370 as well as bromination of toluene derivatives.371 The electrodes are placed in the upper layer of the aqueous phase, thus enabling selective oxidation of the halide. The electrochemically produced X2 or hypohalite subsequently reacts with the organic substrate at the aqueous/organic interface to ultimately produce the desired halogenated product. However, this can to some extent be circumvented using poly(ethylene glycol) as an additive, which enhances the solubility of the inorganic salts in aprotic organic solvents.372 By employing a biphasic system, bromination of various arenes373 (Scheme 31) as well as benzylic mono-374 (Scheme 32) and dibromination375 (Scheme 33) of alkyl aromatics has been achieved. In addition to these examples, electrochemical chlorination of 1,3-dicarbonyl compounds using Cu catalysis,376 azulene derivatives,377,378 olefins,379–384 phenols385 and pyrazolecarboxylic acids,386 as well as bromination of heterocycles387 and olefins388–391 have been reported.
 |
| | Scheme 31 Electrochemical bromination of arenes. | |
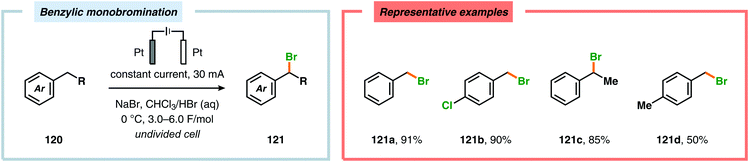 |
| | Scheme 32 Benzylic monobromination of alkyl aromatics. | |
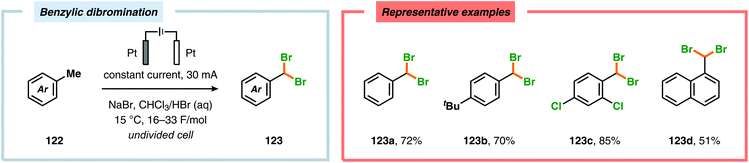 |
| | Scheme 33 Benzylic dibromination of alkyl aromatics. | |
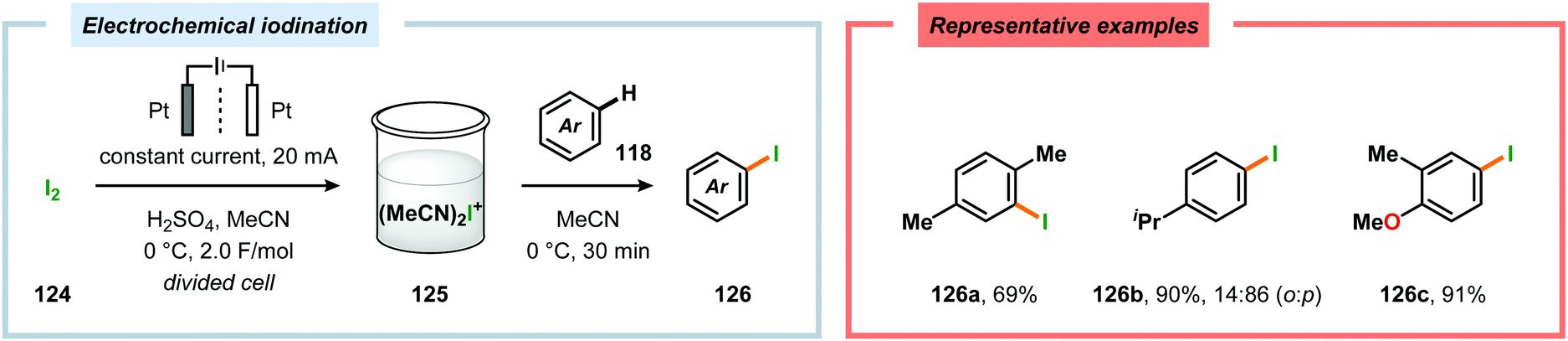
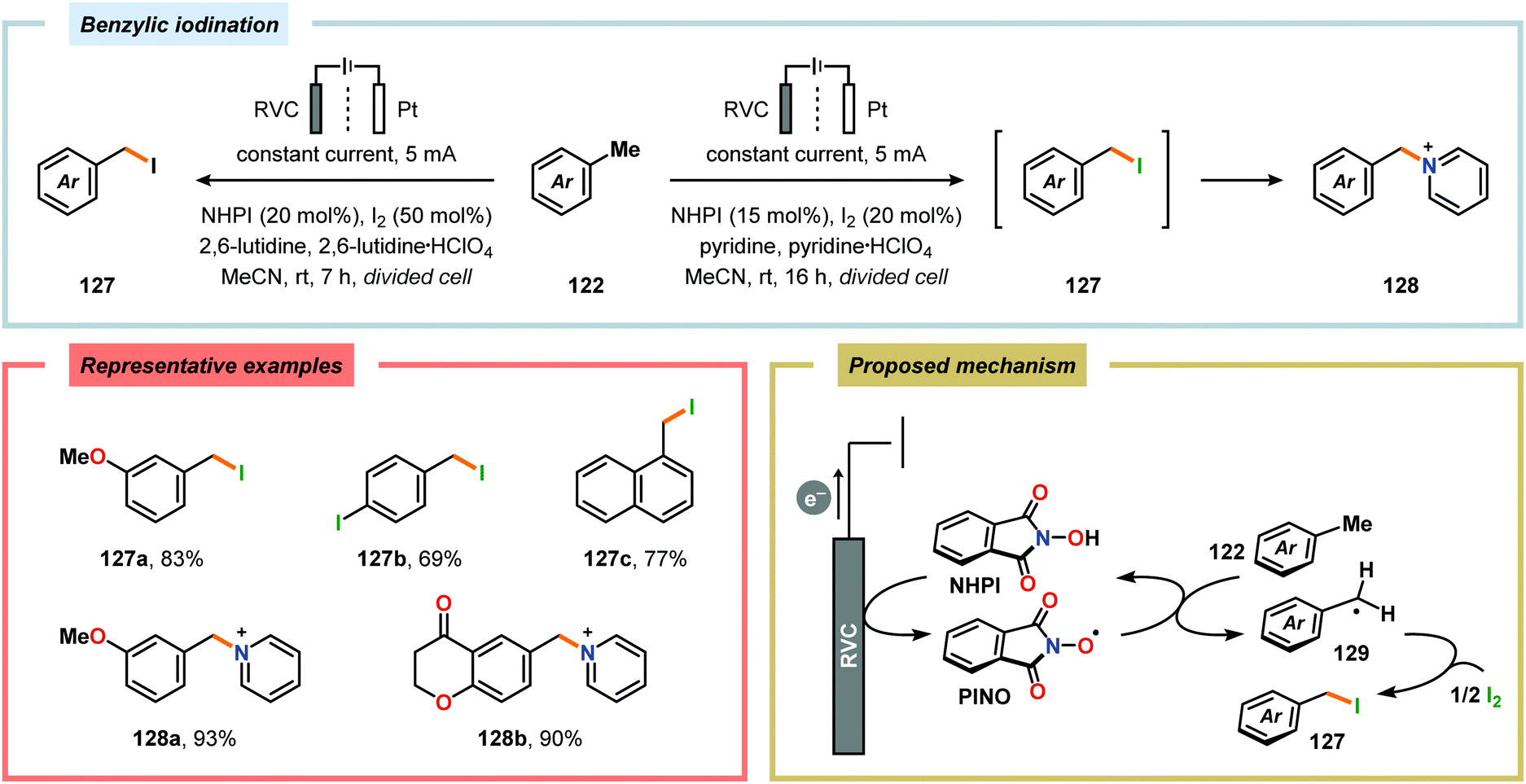
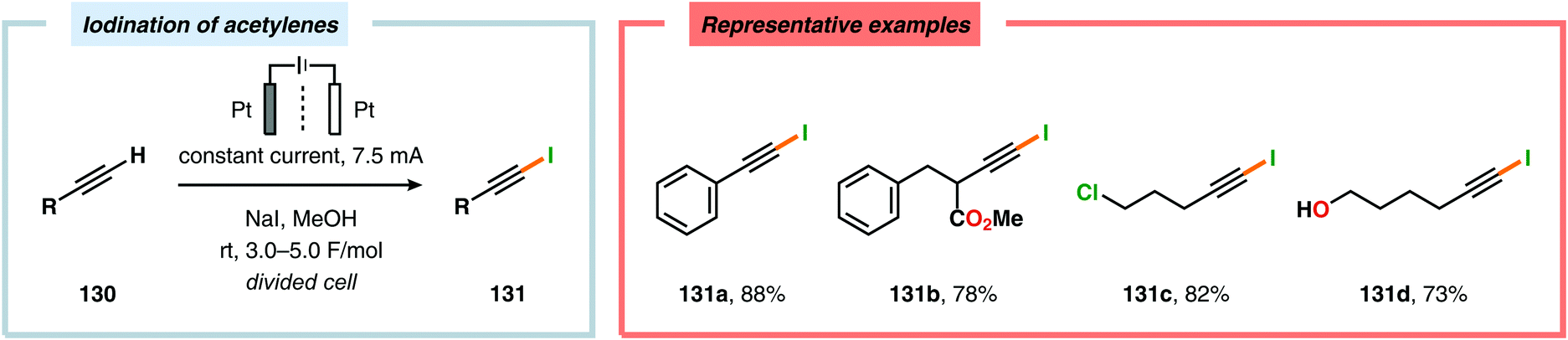
By employing a similar electrochemical strategy as used for the chlorination of pyrazolecarboxylic acids,386 Petrosyan and coworkers developed an electrochemical protocol for iodination of pyrazole and derivatives thereof. The iodination was carried out in a biphasic system consisting of water and chloroform using KI as the iodine source. As expected, the presence of electron-donating groups was found to facilitate the electrochemical iodination.392,393 The Yoshida group has elegantly demonstrated iodination of aromatic compounds using electroaccumulated I+ (Scheme 34). Initially, a “cation pool” approach was employed in which an MeCN solution containing iodine (I2) was electrolyzed for generation of the electrophilic iodination agent, which was proposed to involve (MeCN)2I+. Subsequently, the anodic solution was added to a solution containing the arene. Alternatively, running the reactions in flow was shown to both increase the selectivity for the mono- versus the diiodinated compound and the yields compared to a batch reactor. Furthermore, the flow rate had a dramatic influence on the product selectivity where lower flow rates dramatically decreased the selectivity of the monoiodo compound due to less efficient mixing.394,395 In addition, Hilt and coworkers recently revealed that electrochemical synthesis of aryl iodides can be achieved through anodic iododesilylation.396 Anodic oxidation of arenes containing alkyl side chains provides aryl radical cations, which can subsequently be further oxidized to afford benzylic cations upon release of a proton, thus presenting a strategy for carrying out benzylic functionalization.397 Recently, the Stahl group disclosed an alternative approach to benzylic C–H functionalization (Scheme 35).398,399 The developed concept consisted of employing N-hydroxyphthalimide (NHPI) as a mediator.400–402 Here, NHPI undergoes proton-coupled electron transfer at the anode to give phthalimido-N-oxyl (PINO), which subsequently mediates hydrogen atom transfer (HAT) from weak C–H bonds.403 Using toluene derivatives and in the presence of a base, such as 2,6-lutidine, Stahl and coworkers were able to generate benzylic radicals at relatively modest oxidation potentials. I2 was shown to be an effective radical trapping agent, thereby providing the corresponding benzyl iodide. The authors had to use a divided cell configuration in order to avoid reduction of the trap at the cathode. The developed benzylic C–H iodination method tolerated several functional groups, including esters, ethers, halides and ketones. In situ nucleophilic substitution using pyridine/pyridinium as the electrolyte proved to lead to efficient displacement of iodide by pyridine, leading to the corresponding benzylpyridinium product under electrochemical control. Iodide is produced in the nucleophilic displacement step, which can be reoxidized to iodine at the anode, allowing these reactions to be performed with substoichiometric amounts of iodine (20 mol%). Finally, using the in situ functionalization protocol enabled the authors to prepare three vital pharmaceutical building blocks, highlighting the concept of using toluene derivatives as alkylating agents.398 Moreover, Nishiguchi and coworkers exploited the electrochemical oxidation of terminal acetylenes using a divided cell in the presence of NaI as the supporting electrolyte. The electrochemical method efficiently provided the desired 1-iodoacetylenes in good to excellent yields (Scheme 36).404
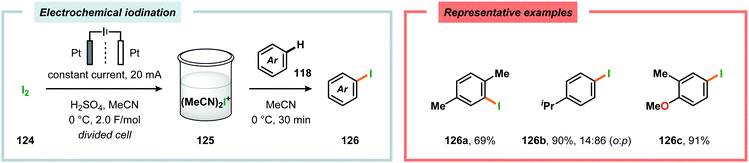 |
| | Scheme 34 Electrochemical iodination of aromatic compounds. | |
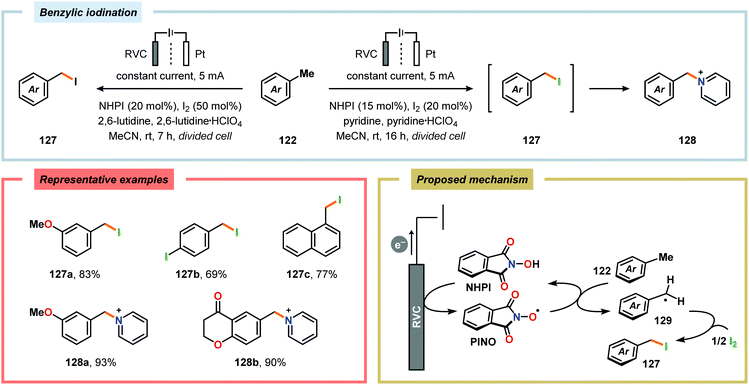 |
| | Scheme 35 Electrochemical iodination of toluene derivatives using NHPI as mediator. | |
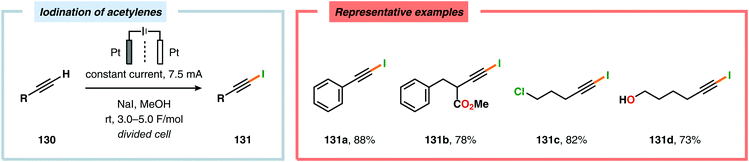 |
| | Scheme 36 Electrochemical iodination of terminal acetylenes. | |
2.3. Electrochemical C–H bond oxygenation
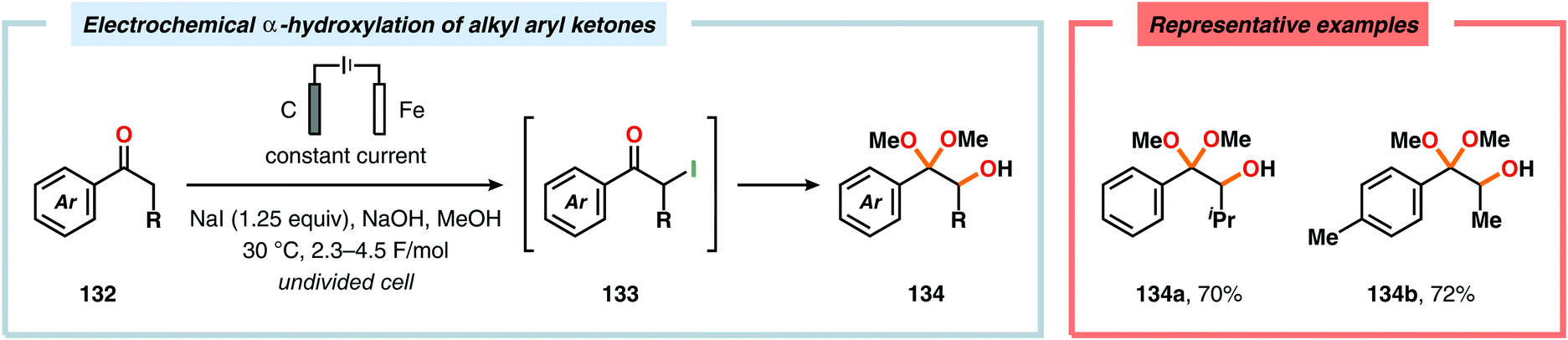
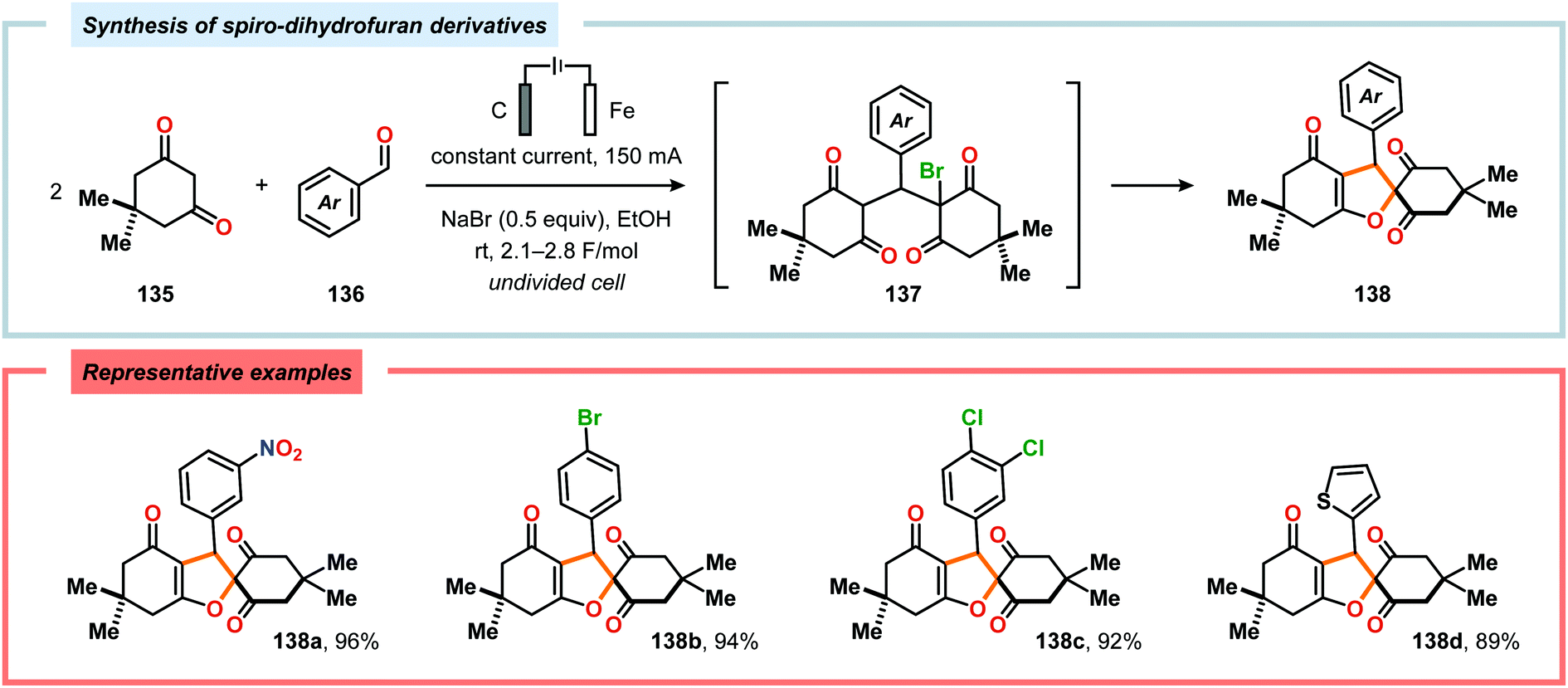
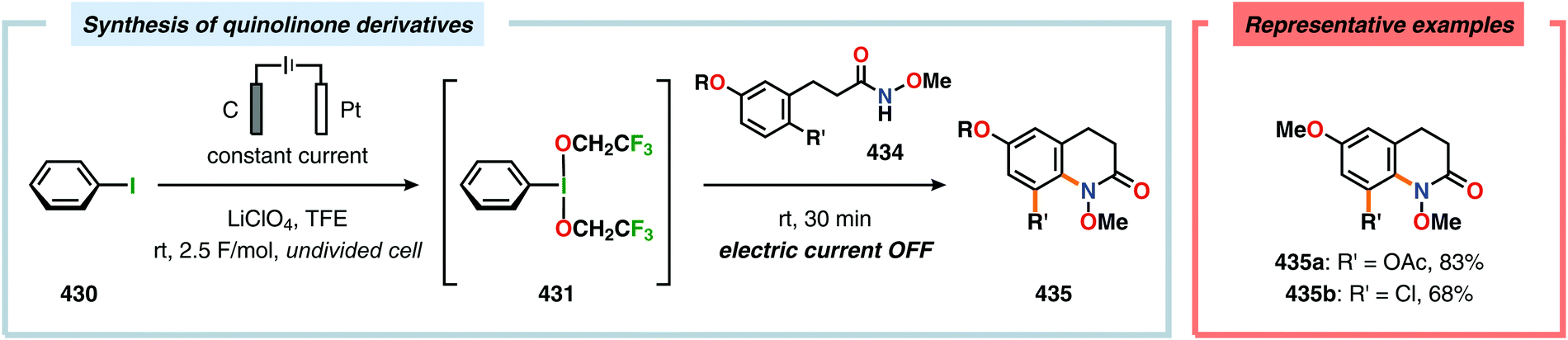
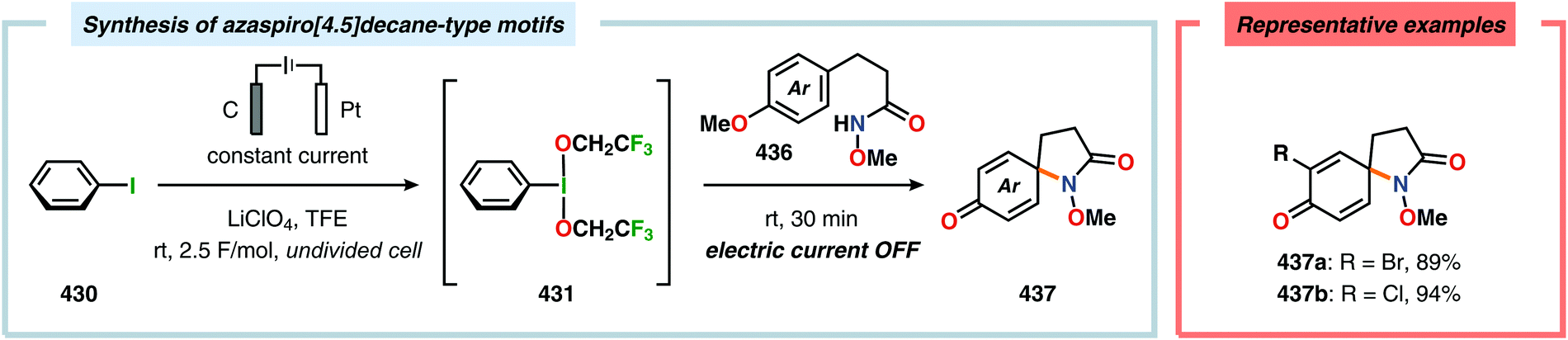
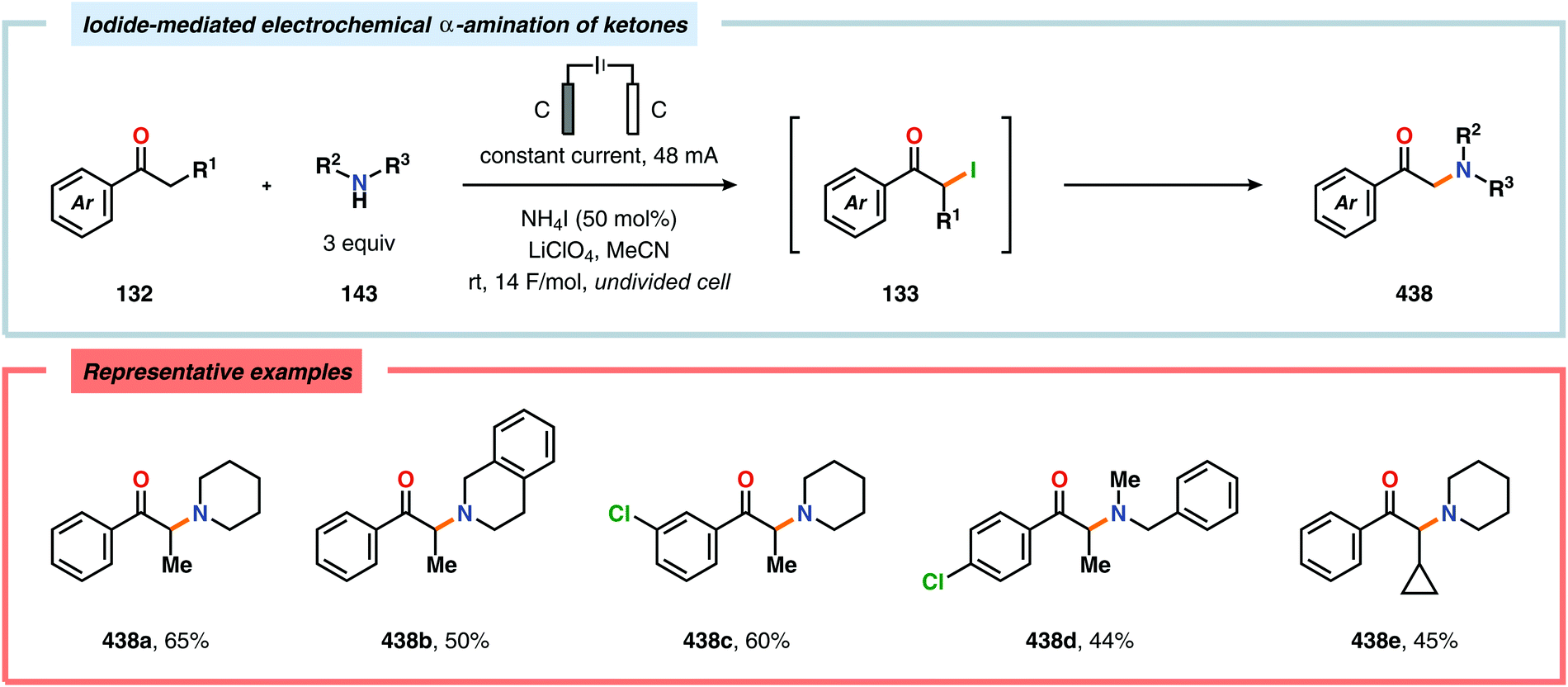
α-Oxygenation of carbonyl compounds can be accomplished using alkali metal halide salts. These halide salts can undergo anodic oxidation, generating α-halo compounds or carbon-centered α-radicals. Seminal work by Shono and coworkers demonstrated that iodide-mediated α-oxidation of aldehydes and ketones delivered the corresponding α-hydroxylated acetals or ketals.405 More recently, Elinson and coworkers have also studied the sodium halide/sodium hydroxide system for indirect electrochemical α-oxidation of carbonyl compounds.406,407 Based on these results, the authors employed the mediatory system NaI/NaOH for converting alkyl aryl ketones into the corresponding α-hydroxy ketals (Scheme 37). These reactions were proposed to proceed via α-iodoketone 133.408 The iodide-mediated oxidation system was subsequently employed for α-hydroxylation of cyclohexanones409 and piperidin-4-ones.410 Bromide-mediated α-oxidation has also been used for accessing spiro-dihydrofuran derivatives from dimedone and aldehydes (Scheme 38).411 Furthermore, both intermolecular412 and intramolecular413–428 protocols for C–C bond formation based on halide-mediated anodic oxidation of carbonyl compounds and related derivatives have also been disclosed.
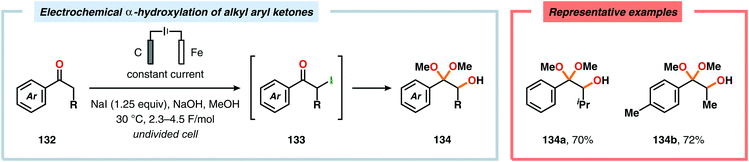 |
| | Scheme 37 Indirect electrochemical α-hydroxylation of alkyl aryl ketones. | |
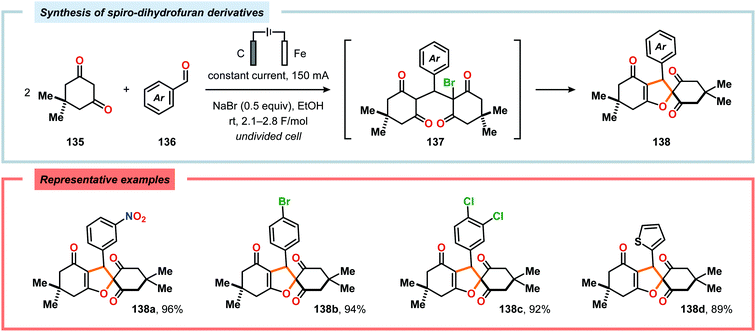 |
| | Scheme 38 Electrochemical synthesis of spiro-dihydrofuran derivatives. | |
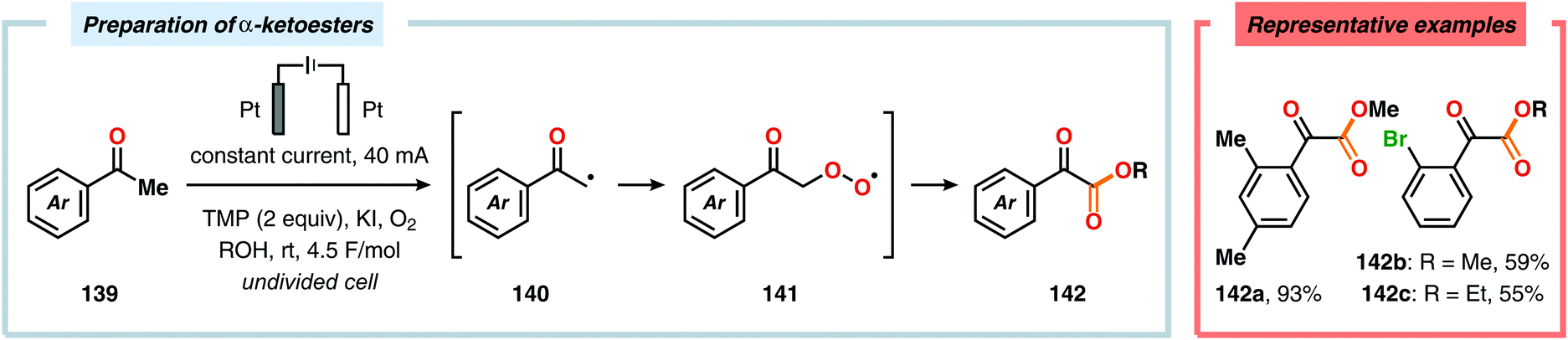
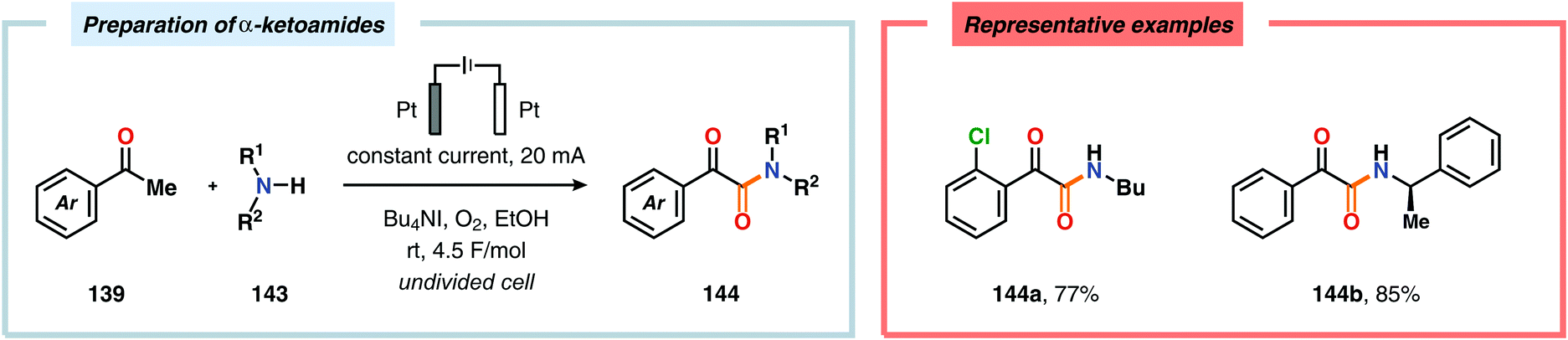
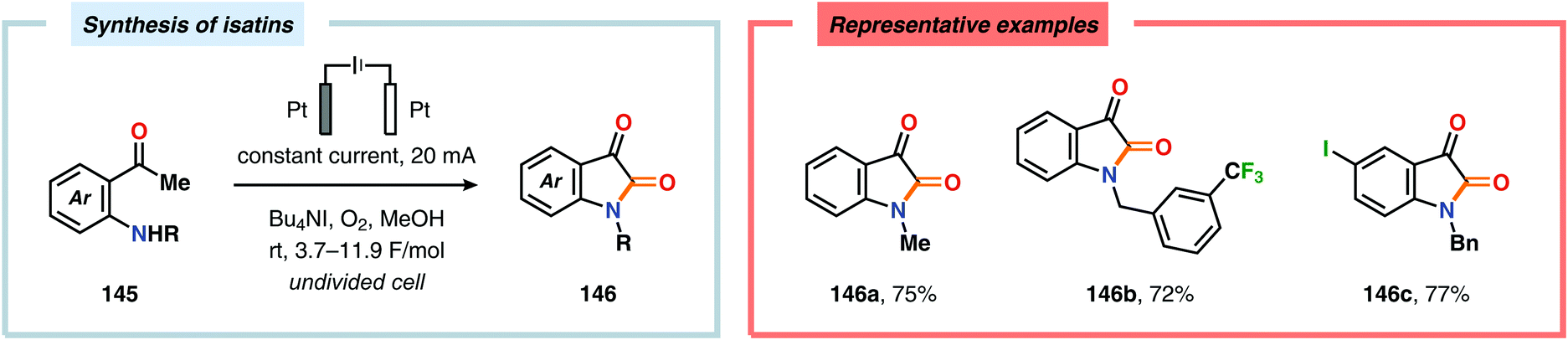
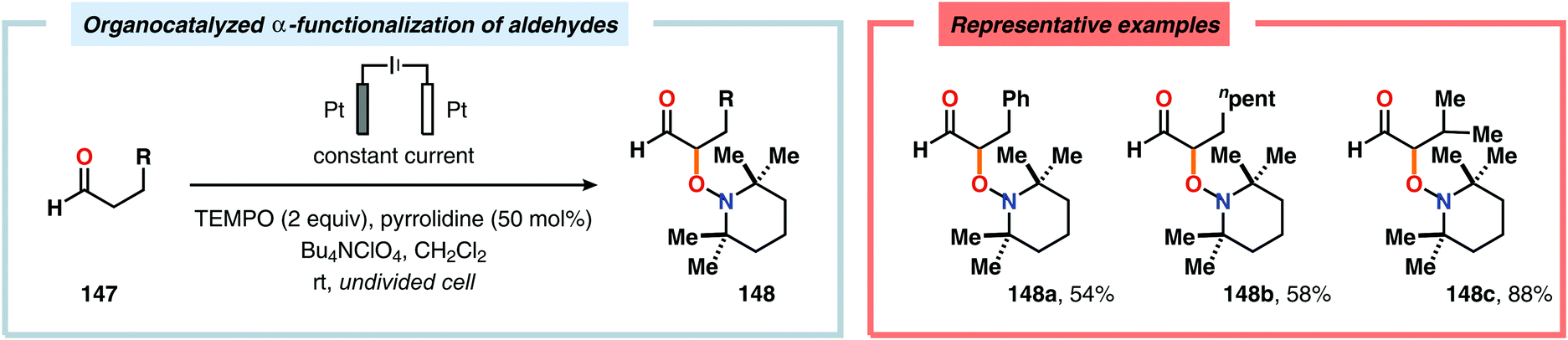
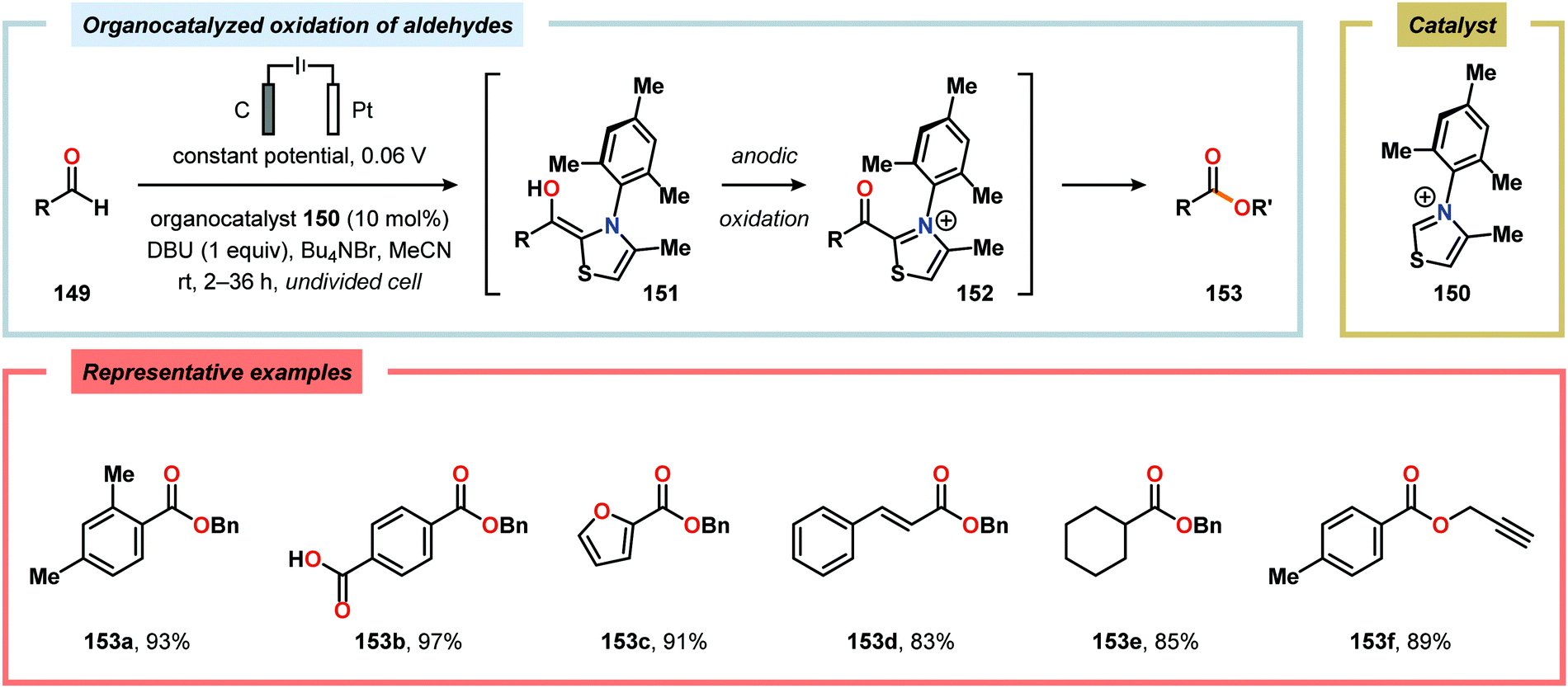
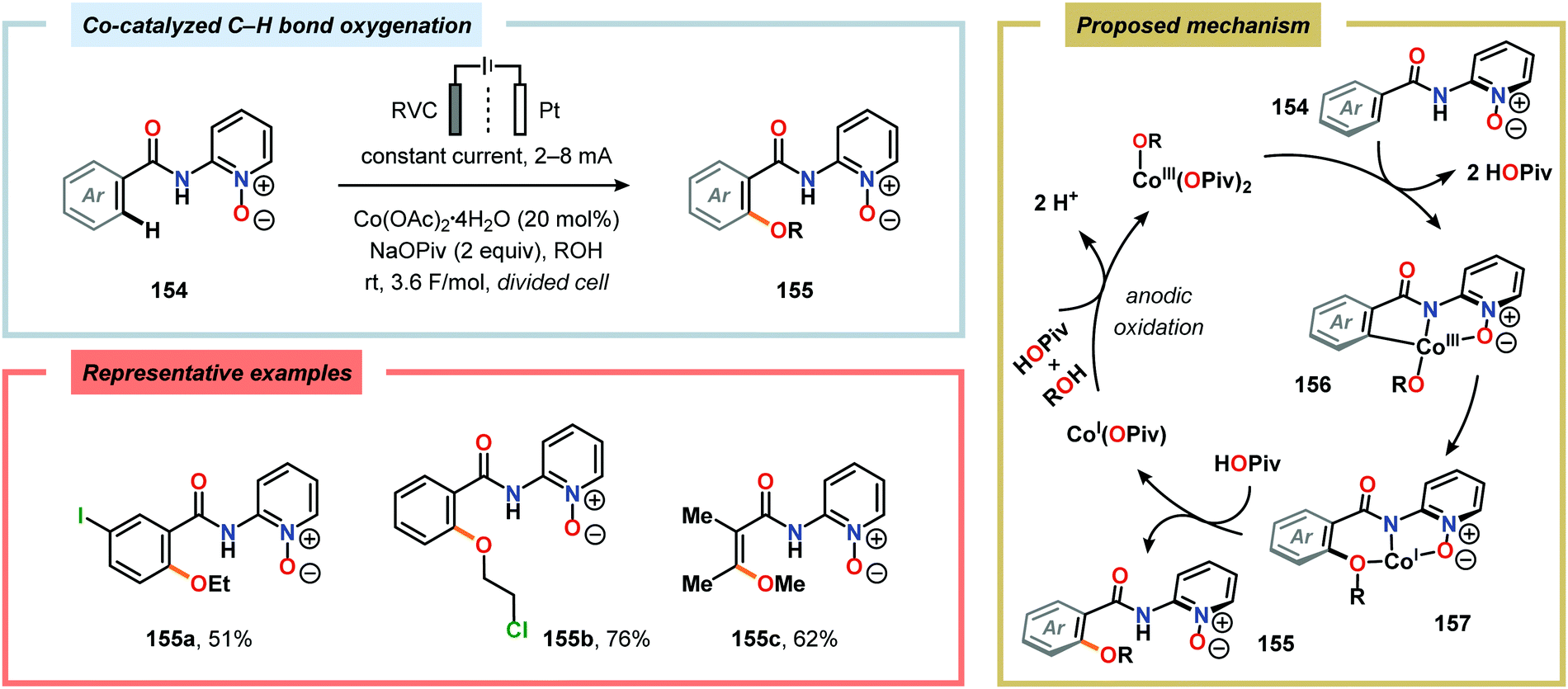
The Wang group has shown that iodide-mediated anodic oxidation of aryl methyl ketones can be exploited for synthesis of α-ketoesters (Scheme 39),429–431 α-ketoamides (Scheme 40)432 and isatins (Scheme 41).433 Here, a radical-based pathway in which an iodine radical (I˙) is proposed to undergo a HAT event with the aryl methyl ketone to deliver hydrogen iodide and an α-carbon-centered radical (140). The intermediate radical can subsequently be trapped with dioxygen to afford peroxyl radical 141, which is readily converted to the corresponding ketoaldehyde compound. Finally, nucleophilic addition to this intermediate and a further oxidation event yields the desired product. α-Oxygenation of aliphatic aldehydes has been accomplished through a dual electrochemical/organocatalytic strategy (Scheme 42).434 In this transformation, the generated α-iminyl radical cation is intercepted with TEMPO to afford the α-oxygenated product 148 upon hydrolysis of the resultant iminium ion. In addition, various chiral sec-amines were evaluated in order to develop an enantioselective protocol. However, only moderate enantioselectivities were obtained using the Hayashi–Jørgensen catalyst.435,436 The merger of electrochemistry with other organocatalytic activation modes was utilized by Boydston and coworkers for conversion of aldehydes to esters (Scheme 43). The method interfaced N-heterocyclic carbene-based organocatalysis with anodic oxidation for achieving efficient two-electron oxidation of the in situ generated Breslow intermediate (151) to produce an electrophilic 2-acylazolium species (152).437–439 Subsequently, the authors disclosed a method for the direct conversion of aldehydes to thioesters via integration of organocatalysis and electrosynthesis.440 Based on this dual catalytic activation mode, Brown and coworkers successfully demonstrated that the oxidative esterification of aldehydes441 as well as amidation of aldehydes442 could be conducted in an undivided electrochemical flow cell. Recently, the Ackermann laboratory exploited the powerful nature of dual catalytic methods for achieving C(sp2)–H oxygenation of aromatic and alkene moieties.443 Here, the combination of cobalt catalysis and electrochemical oxidation enabled C–H alkoxylation under mild reaction conditions at ambient temperature (Scheme 44). Employing electrical current as an oxidant obviated the need of stoichiometric amounts of Ag(I) salts through the use of an earth-abundant cobalt redox manifold. Furthermore, the developed protocol exhibited high levels of chemo-, regio- and diastereoselectivity. Electrooxidative C–H activation with weakly coordinating benzoic acids has also been accomplished using rhodium-444 or ruthenium catalysis.445
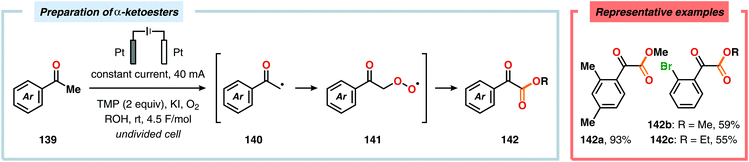 |
| | Scheme 39 Electrochemical preparation of α-ketoesters. TMP = 2,2,6,6-tetramethylpiperidine. | |
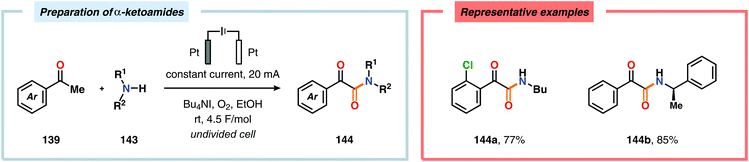 |
| | Scheme 40 Preparation of α-ketoamides through anodic oxidation. | |
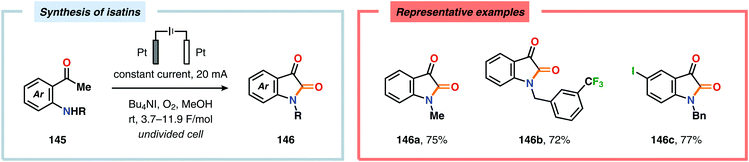 |
| | Scheme 41 Iodide-mediated synthesis of isatins. | |
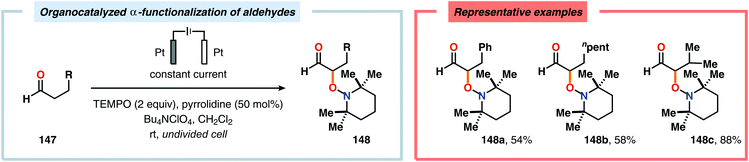 |
| | Scheme 42 Dual electrochemical/organocatalyzed α-oxygenation of aldehydes. | |
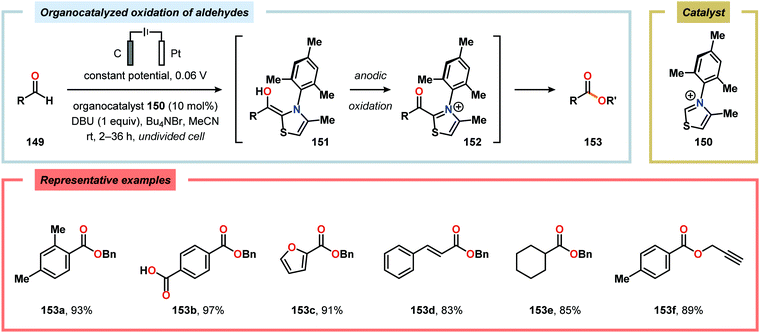 |
| | Scheme 43 Organocatalyzed anodic oxidation of aldehydes. | |
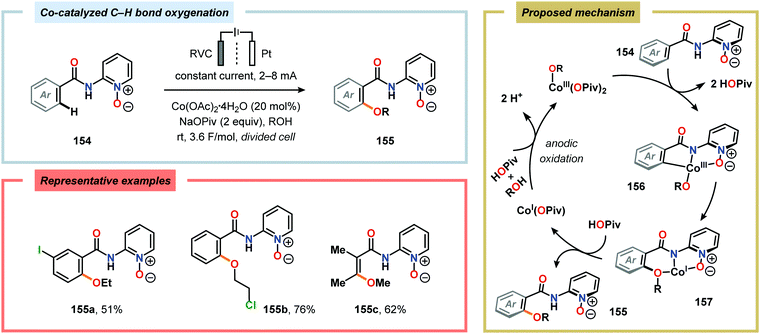 |
| | Scheme 44 Electrochemical cobalt-catalyzed C(sp2)–H alkoxylation. | |
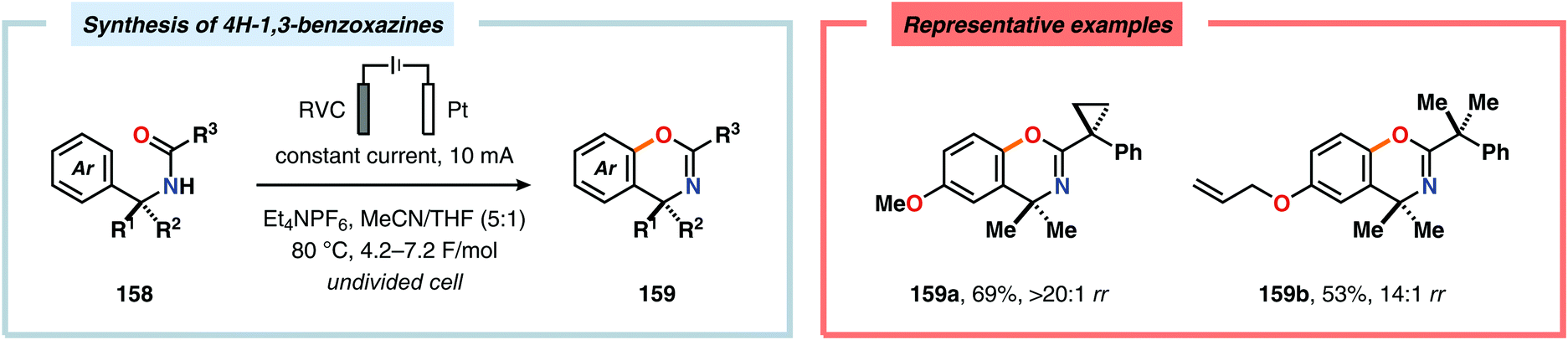
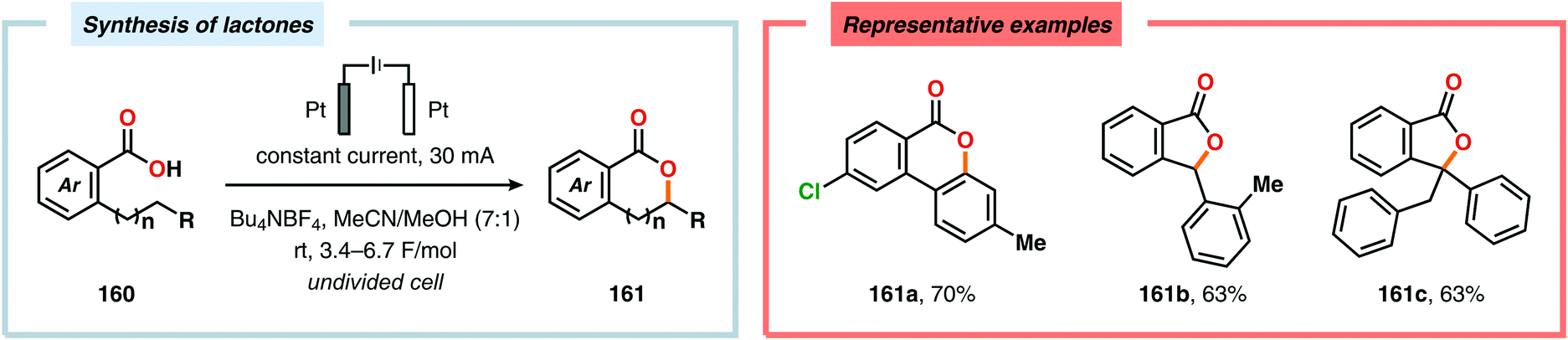
Electrochemical aromatic C(sp2)–H oxygenation has also been utilized for carrying out oxidative annulations. Xu and coworkers disclosed an electrochemical method featuring the direct electrolysis of N-benzylamides for preparing a range of substituted 4H-1,3-benzoxazines (Scheme 45). The electrolysis was proposed to facilitate anodic oxidation of the electron-rich aromatic nucleus, triggering regioselective cyclization to afford a cyclohexadienyl radical. Finally, rearomatization is realized upon abstraction of an electron and a proton, furnishing the desired benzoxazine 159.446 The Zeng group demonstrated that carboxylic acids can be anodically oxidized to generate the corresponding carboxylate radical. The generated radical could subsequently undergo intramolecular addition to electron-rich aromatic rings to produce the C–O coupled lactone product (Scheme 46).447–450 In addition to these examples, electrochemical oxygenation of furan derivatives451–453 has also been reported and has, for example, been utilized for oxygenation of hispanolone454 as well as preparation of dideoxynucleoside analogues.455
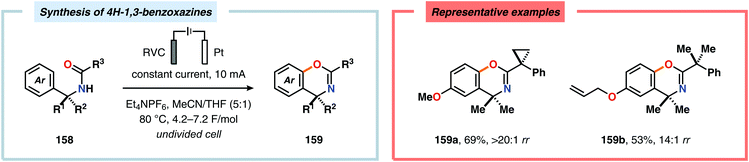 |
| | Scheme 45 Synthesis of 4H-1,3-benzoxazines through electrochemical C–H oxygenation. | |
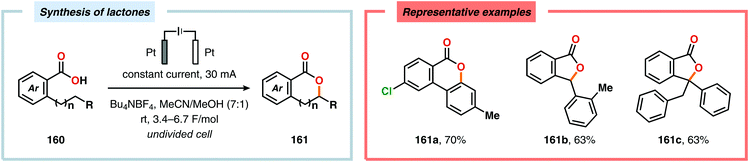 |
| | Scheme 46 Electrochemical lactonization of C–H bonds. | |
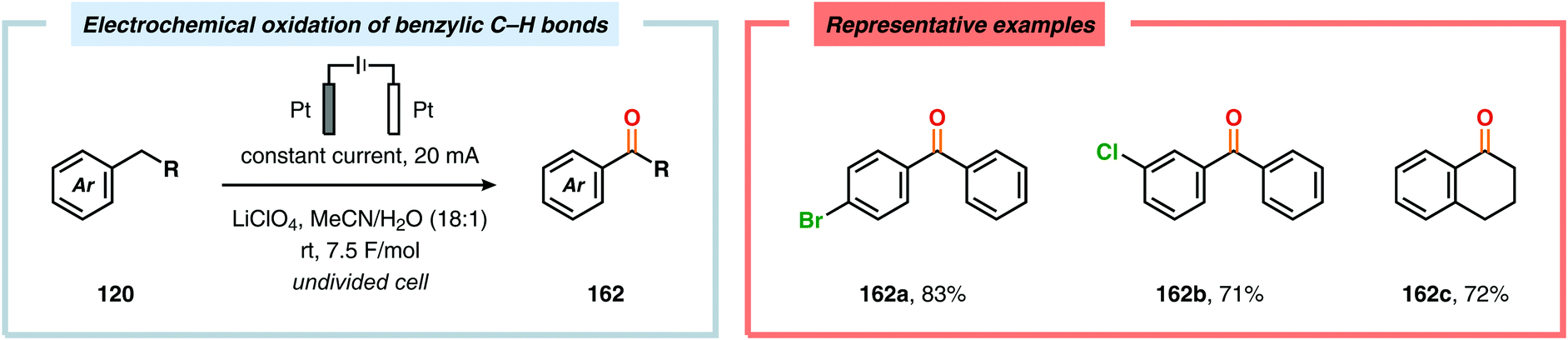
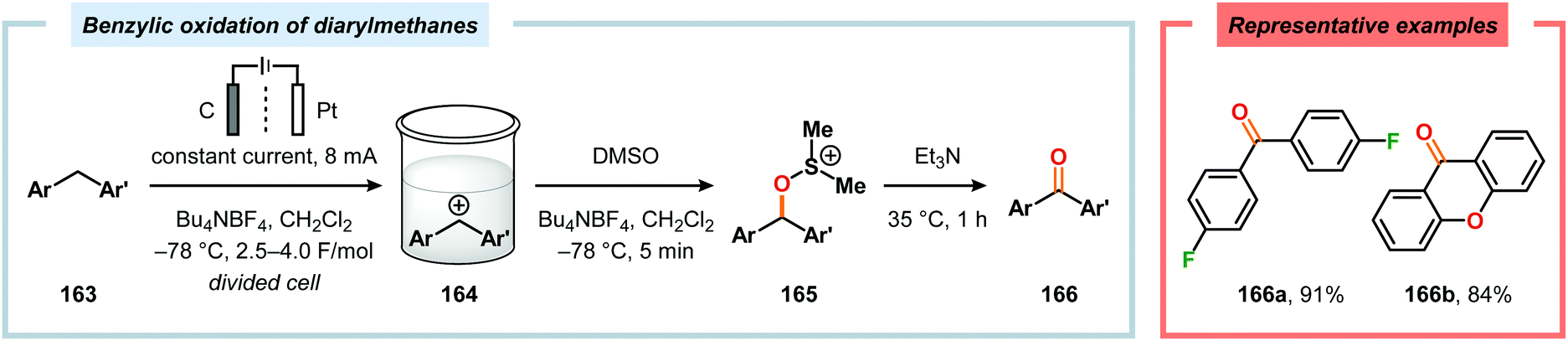
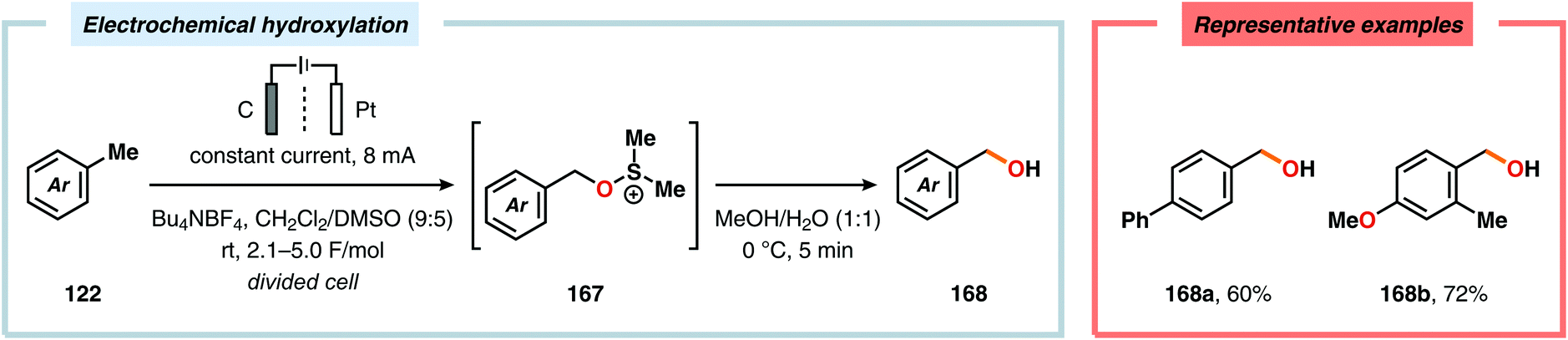
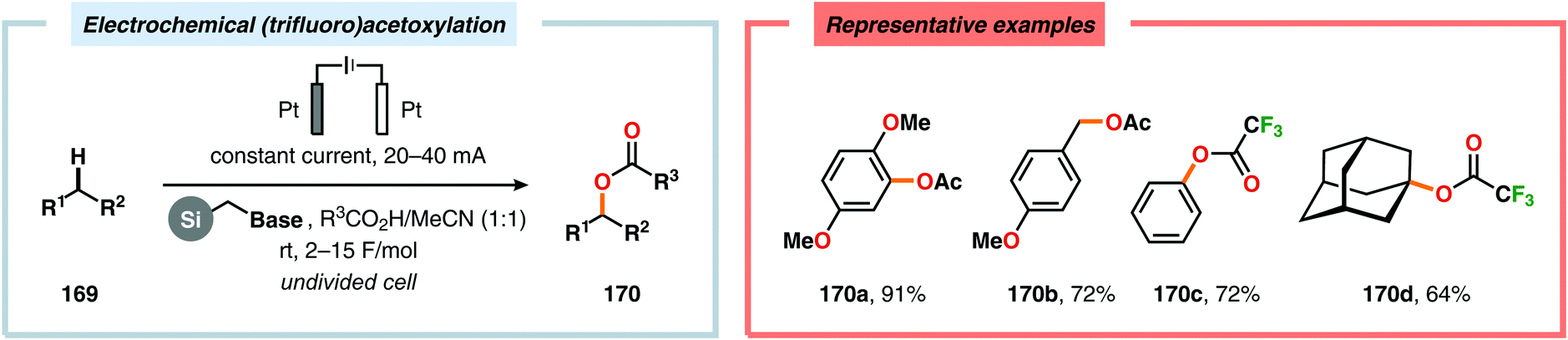
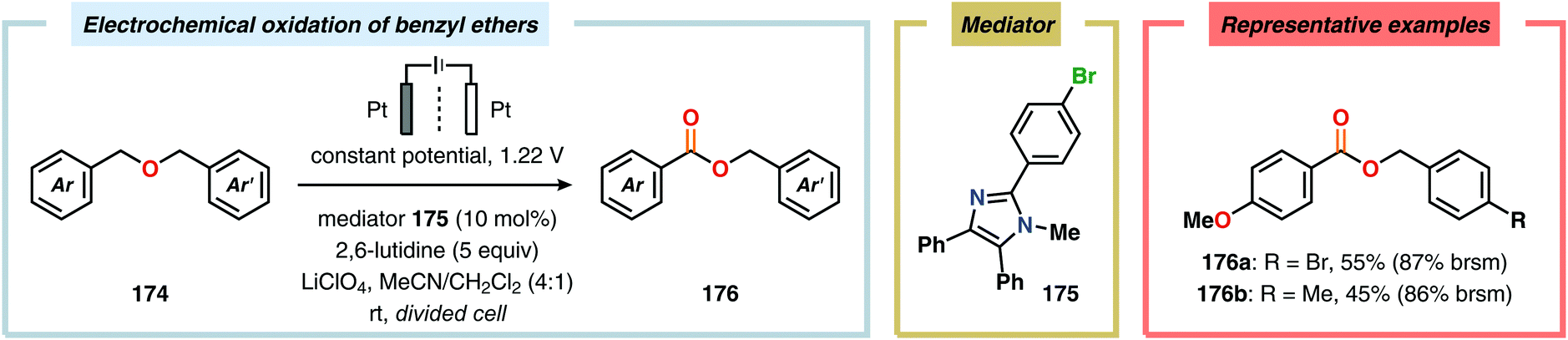
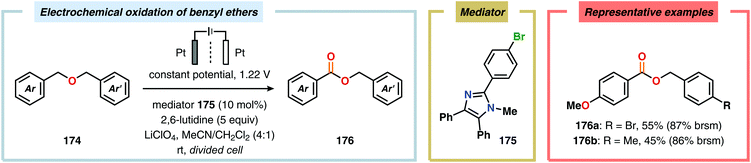
Development of strategies for oxygenation of C(sp3)–H bonds constitutes a compelling approach for delivering alcohol and carbonyl derivatives. Historically, chemists have confided to using stoichiometric, and often toxic, oxidants for carrying out these transformations, resulting in the formation of considerable amounts of waste. However, the demand for more sustainable and atom economical methodologies has gained significant attention among the scientific community.456 Electrochemical activation of toluene derivatives through initial oxidation of the aromatic core is an appealing platform for realizing C(sp3)–H oxygenation of benzylic carbons. The aryl radical cation that is generated under these reaction manifolds can subsequently undergo deprotonation and a further oxidation to furnish a benzylic cation which can be trapped with various oxygen-based nucleophiles. Such an approach was implemented by the Wang group in which the benzylic cation was intercepted with water to furnish the benzylic alcohol, which underwent additional oxidation to give the corresponding ketone (Scheme 47).457 Diarylcarbenium ions can successfully be generated and accumulated by low-temperature electrochemical oxidation using the “cation pool” method.458–466 In the presence of DMSO, alkoxysulfonium ions serve as the key intermediates. Treatment with a base, such as Et3N, or hydrolysis selectively affords the corresponding ketone467 (Scheme 48) or alcohol468 (Scheme 49). The fact that the initial electrochemical oxidation and the product-forming steps are separated implies that the products are not exposed to the electrolysis conditions, thereby limiting overoxidation. An electrochemical protocol for anodic acyloxylation based on the acid–base reactions between acetic acid or trifluoroacetic acid and a solid-supported base has also been reported (Scheme 50).469,470 It was found that the silica gel supported base was stable under the electrochemical conditions, permitting easy recovery and reuse of the solid-supported base. Indirect electrolysis using various mediators, such as TEMPO/Br−,164 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)471,472 and NHPI (Scheme 51),473 can also be harnessed for facilitating oxygenation of benzylic methylenes. In addition, Little and coworkers demonstrated that the use of triarylimidazoles474–477 as redox mediators allowed for selective benzylic oxygenation of unsymmetrical benzyl ethers (Scheme 52).478–480
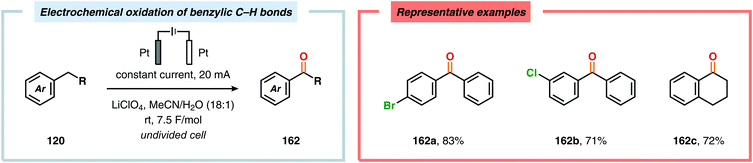 |
| | Scheme 47 Electrosynthesis of ketones through anodic oxidation of benzylic methylenes. | |
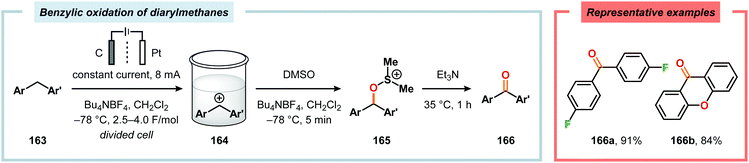 |
| | Scheme 48 Electrochemical oxygenation using alkoxysulfonium ions. | |
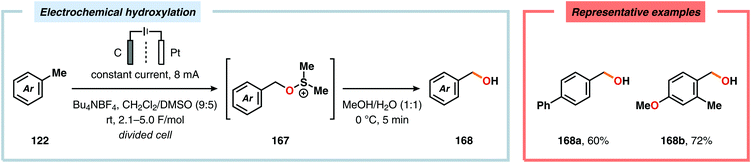 |
| | Scheme 49 Electrochemical hydroxylation using alkoxysulfonium ions. | |
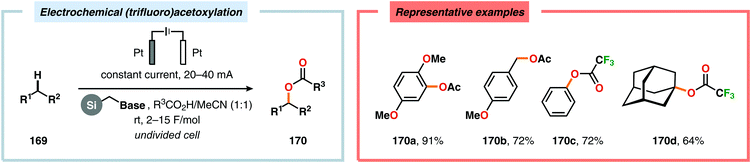 |
| | Scheme 50 Electrochemical (trifluoro)acetoxylation of C(sp2)–H and C(sp3)–H bonds. Base = morpholine. | |
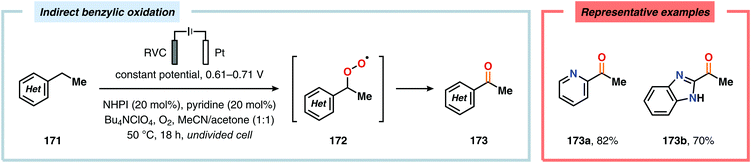 |
| | Scheme 51 Indirect electrochemical oxygenation using NHPI as mediator. | |
 |
| | Scheme 52 Benzylic oxygenation using a triarylimidazole mediator. | |
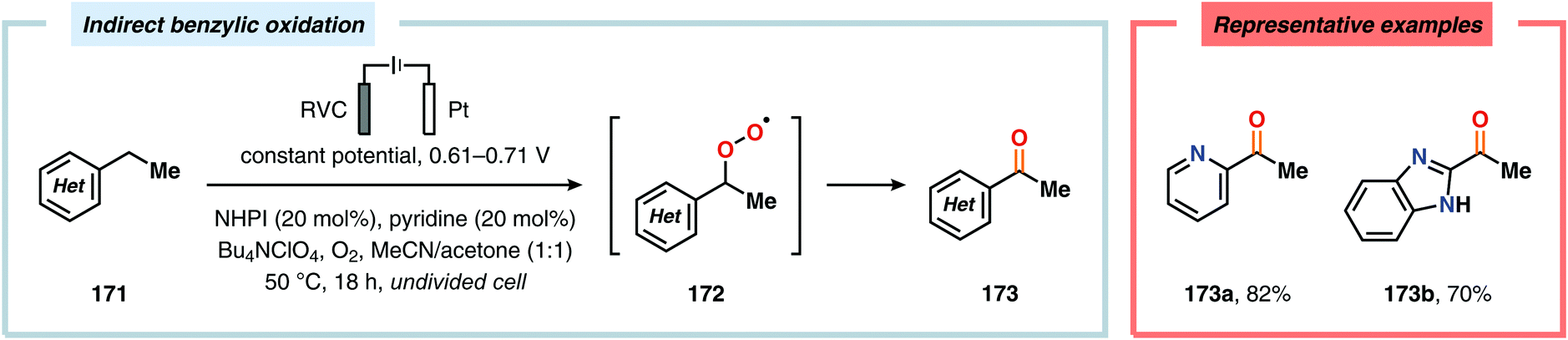
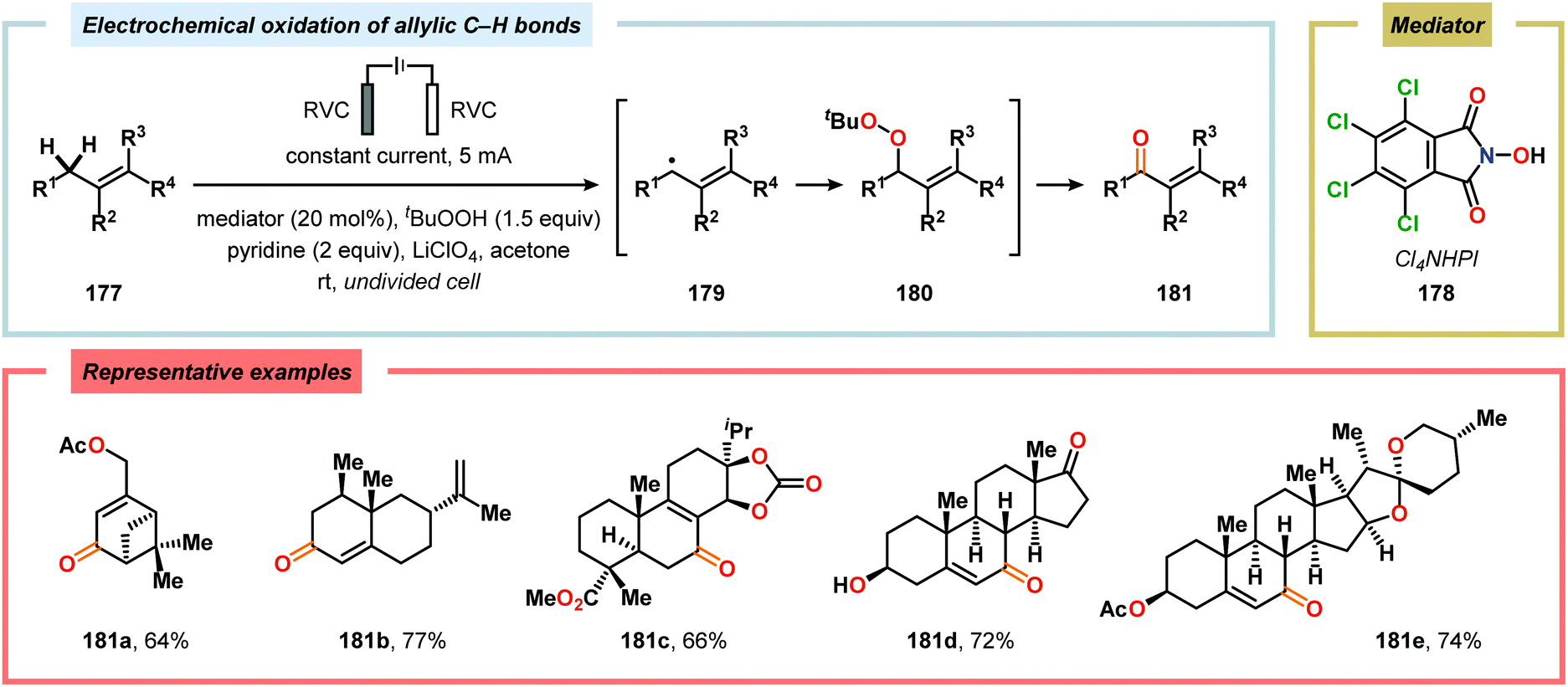
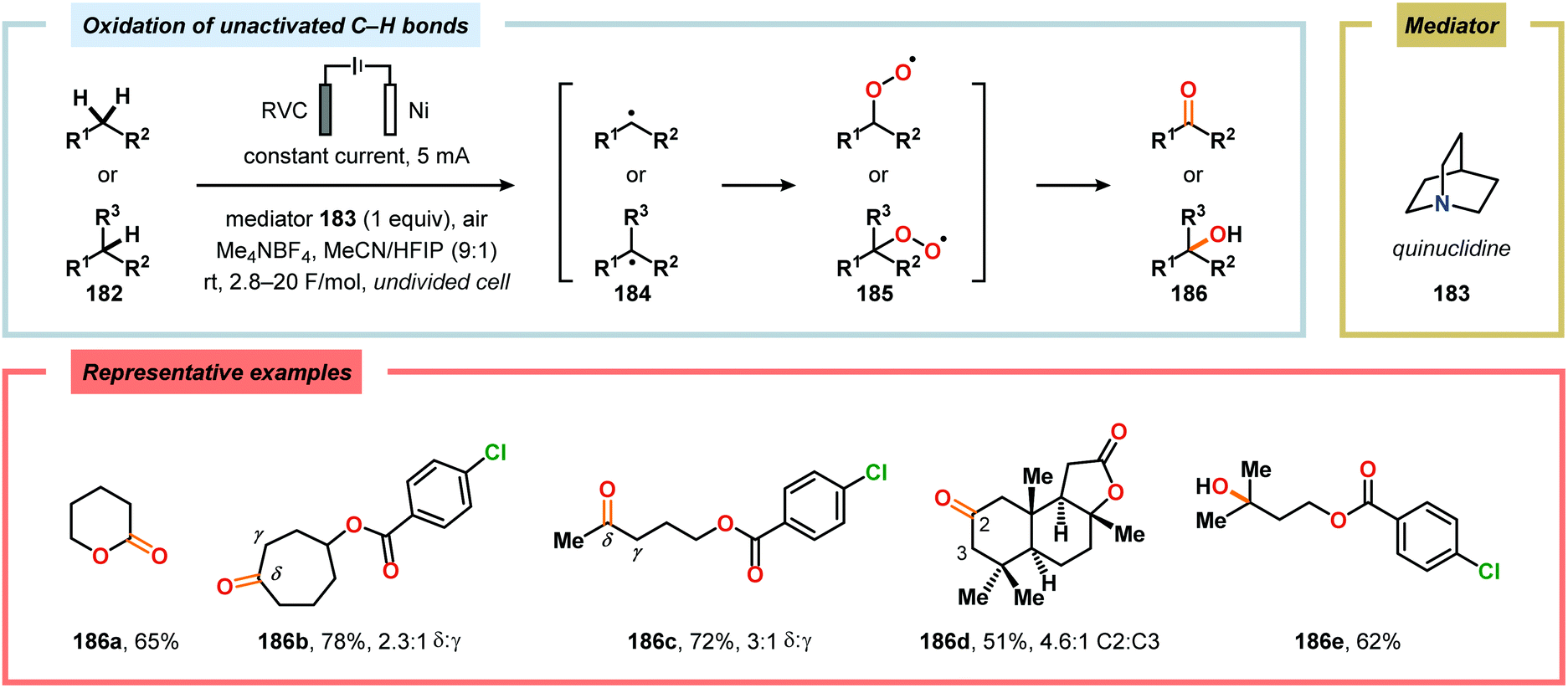
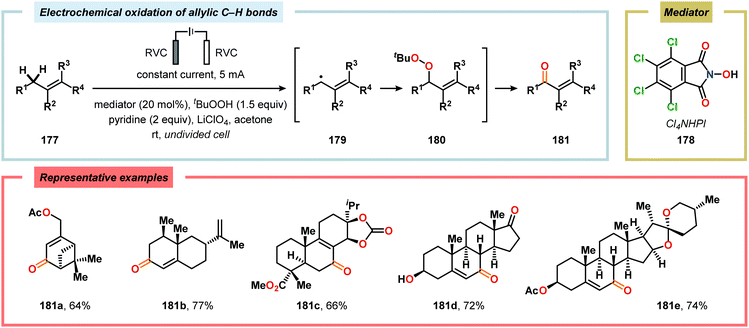
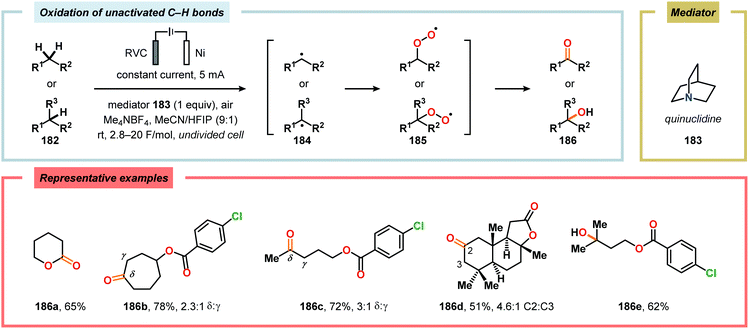
The oxidation of unactivated C–H bonds typically requires high redox potentials, thus limiting the functional group compatibility and necessitates the use of inert solvents. Allylic oxidations are generally based on the use of toxic reagents, such as chromium and selenium, or scarce transition metal catalysts, such as Pd and Rh, which causes complications in industrial settings. The Baran group has addressed the issue of oxidizing unactivated C–H bonds through indirect electrolysis. After careful evaluation of potential mediators, a NHPI derivative (Cl4NHPI) was demonstrated to be the optimal mediator for the allylic oxidation of a range of natural product inspired compounds, including sesquiterpene- and steroid-based substrates (Scheme 53).481–483 The described protocol exhibited broad substrate scope, operational simplicity and high chemoselectivity, and could even be conducted on a 100 g scale. The reaction is believed to involve hydrogen atom abstraction from the olefinic substrate to the electrochemically generated N-oxyl radical species, resulting in regeneration of Cl4NHPI (178) and an allylic radical (179). Interception of the allylic radical with electrochemically produced tBuOO˙ furnishes allylic peroxide 180, providing enone 181 upon elimination of tBuOH. Furthermore, direct electrochemical oxidation of cholesterol at the allylic position has been demonstrated by Sobkowiak and coworkers.484–489 Baran and coworkers subsequently reported a method for selective functionalization of unactivated methylene and methine moieties utilizing quinuclidine (183) as redox mediator (Scheme 54). Here, an assortment of functional groups were demonstrated to be compatible with the developed chemoselective process.490,491
 |
| | Scheme 53 Indirect electrochemical allylic C–H oxidation. | |
 |
| | Scheme 54 Indirect electrochemical oxidation of unactivated C–H bonds. | |
2.4. Palladium-catalyzed anodic C–H functionalization strategies
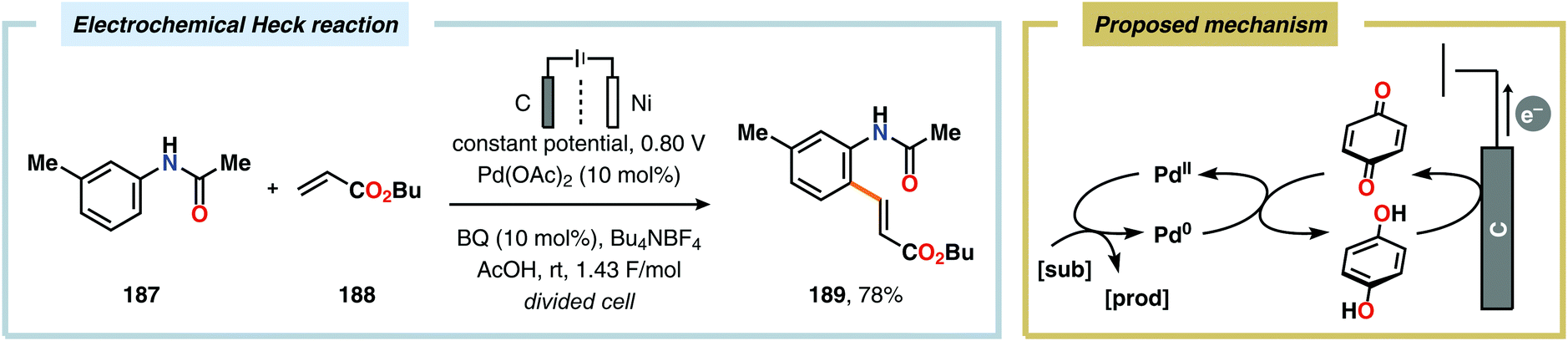
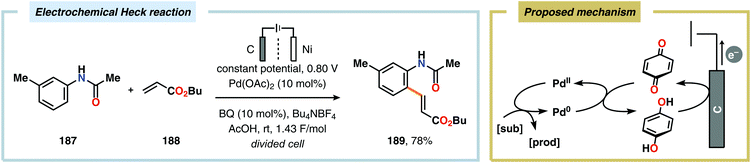
Palladium-catalyzed cross-coupling reactions have revolutionized C–C bond forming processes. Over the years, a variety of catalytic platforms, such as carbon–heteroatom coupling, α-arylation and decarboxylative coupling, based on Pd have been designed.492 It is therefore not surprising that the synergistic merger of electrochemistry and Pd catalysis has been the subject of keen interest by the synthetic community.70,493–497 Pioneering studies by Tsuji and Minato demonstrated a variant of the Wacker oxidation of terminal olefins by combining Pd-catalysis and electrochemical oxidation. In this system, benzoquinone is regenerated at the anode, which subsequently triggers the turnover-limiting reoxidation of Pd0 to PdII.498,499 More recently, related approaches have also been employed for carrying out Heck-type reactions (Scheme 55),500,501 Glaser–Hay coupling502 as well as homocoupling of arylboronic acids and derivatives thereof,503 and oxidation of alcohols under anaerobic conditions.504 Dual catalytic platforms based on Pd/TEMPO have been reported for electrooxidative synthesis of biaryls from arylboronic acids,505–507 cross-coupling between terminal alkynes and arylboronic acids508 as well as Wacker-type reactions509 and cyclizations.510 Furthermore, Moeller and coworkers have demonstrated that triarylamines are also effective mediators and have utilized this concept for site-selective functionalization of semiconducting chips.511–516
 |
| | Scheme 55 Electrochemical Heck reaction using benzoquinone as mediator. | |
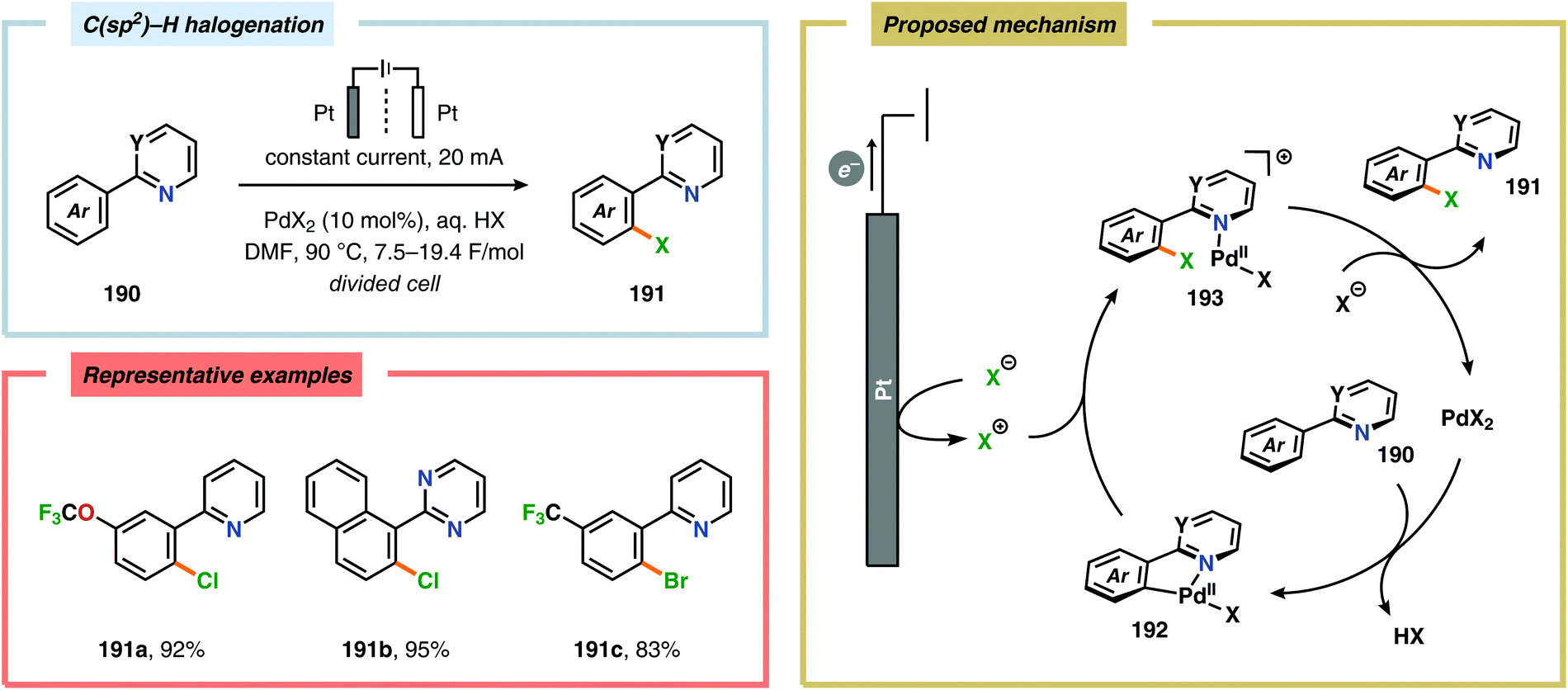
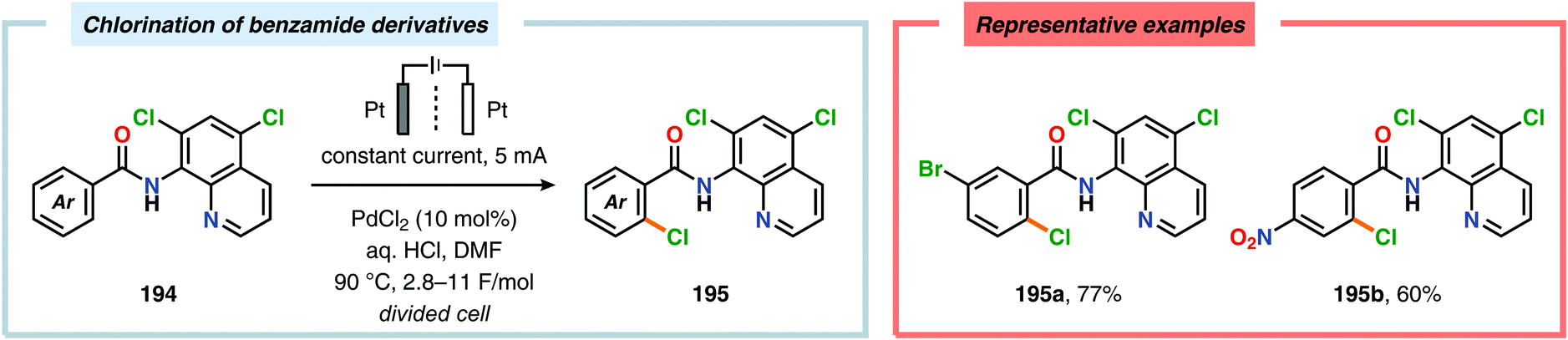
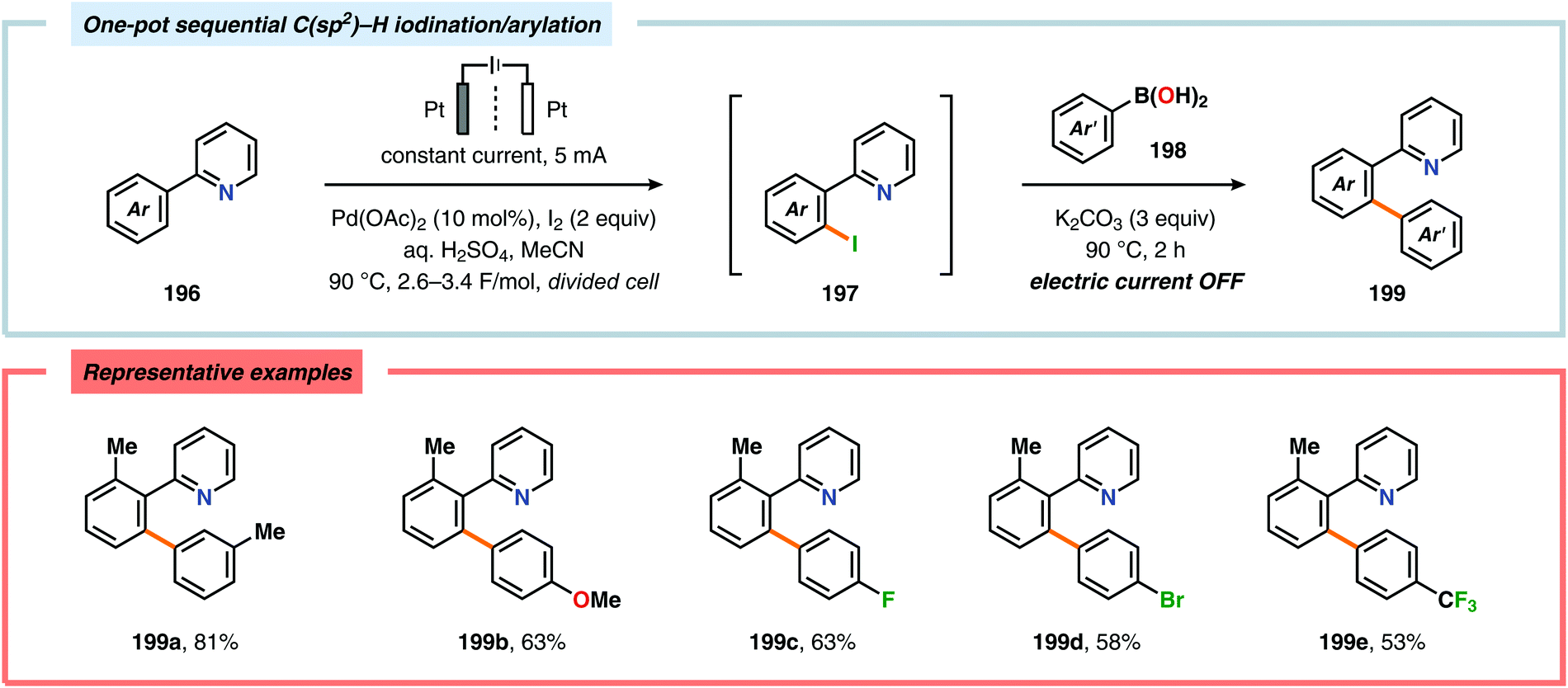
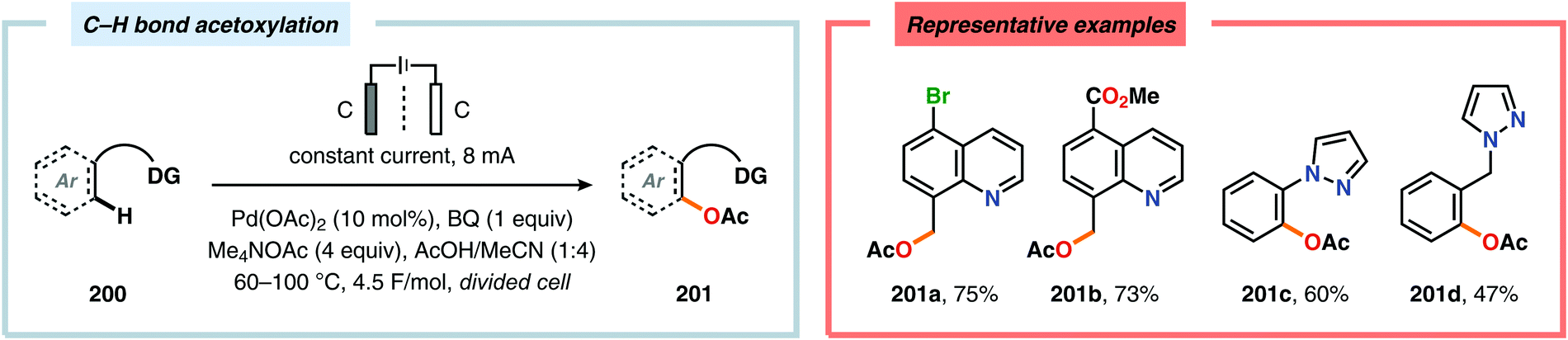
A variety of approaches have been reported for the C–H functionalization of arylpyridine derivatives via dual activation by transition-metal catalysis and electrochemical oxidation. In this regard, the Kakiuchi group has demonstrated that C(sp2)–H halogenation of aromatics using hydrogen halides can be accomplished (Schemes 56 and 57). It was proposed that anodic oxidation of the halide ion produces a halonium ion, which subsequently reacts with the generated palladacycle to provide the halogenated product while regenerating the PdII catalyst.517–520 Expanding on this concept, the Kakiuchi group disclosed a method for regioselective homocoupling of arylpyridines in the presence of I2 employing electrooxidative Pd catalysis.521 Additionally, protocols for perfluoroacetoxylation,522 perfluoroalkylation523,524 and phosphorylation525,526 of arylpyridines have been established by the Budnikova group. A procedure for sequential electrochemical C–H iodination followed by a Suzuki–Miyaura coupling has also been developed by Kakiuchi and coworkers. Here, I2 was employed as the iodine source for the electrochemical C–H iodination. Upon completion of the iodination reaction, the electric current was simply switched off whereupon the arylboronic acid was introduced to the reaction mixture to furnish the corresponding cross-coupled product (Scheme 58).527 Along similar lines, Suga and coworkers reported a dual Pd-mediated electrooxidative homocoupling of p-bromophenylacetylene with a subsequent Suzuki–Miyaura coupling. Discrimination between the two reaction sites, the terminal alkyne and the Br group, was realized by the mere on/off application of electricity.528 Furthermore, the Sanford group recently disclosed a Pd-catalyzed electrochemical method for acetoxylation of C(sp3)–H and C(sp2)–H bonds (Scheme 59). It was noted that differences in the relative rates of competing oxidation reactions could prevail under chemical versus electrochemical oxidation conditions, thereby resulting in the formation of complementary products. Additionally, the differences in reaction conditions for the chemical and electrochemical reactions, such as solvents as well as the addition of a supporting electrolyte, can translate to fundamental changes in catalyst performance and/or reaction outcomes, and will be imperative when designing new electrochemical platforms.529
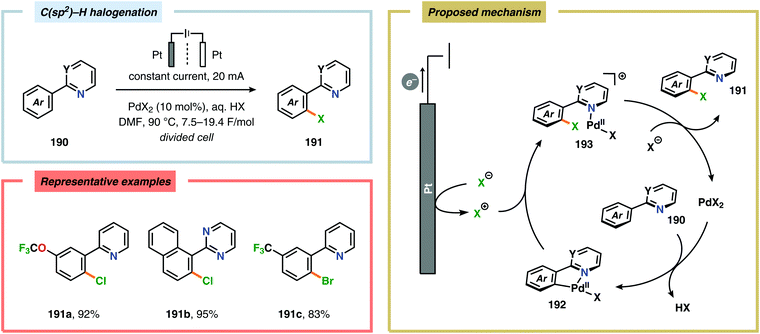 |
| | Scheme 56 Regioselective halogenation of arenes through dual Pd catalysis and electrochemical oxidation. | |
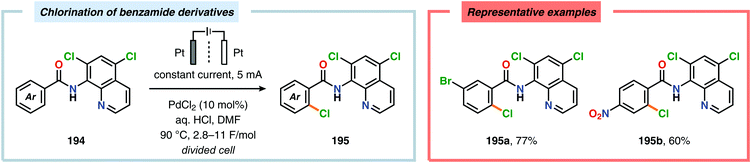 |
| | Scheme 57 Electrochemical C(sp2)–H chlorination of benzamide derivatives. | |
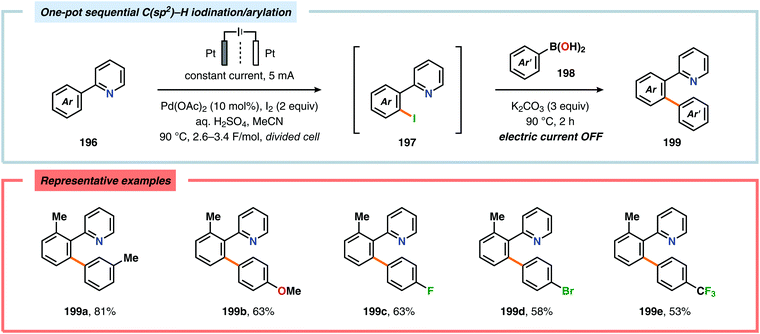 |
| | Scheme 58 One-pot C–H iodination/arylation of arylpyridines. | |
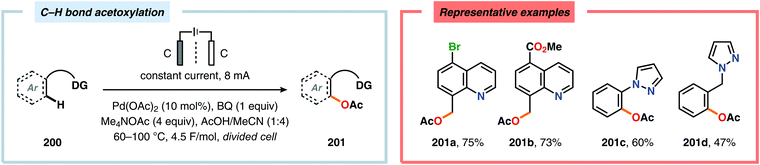 |
| | Scheme 59 Pd-catalyzed electrochemical method for acetoxylation of C(sp3)–H and C(sp2)–H bonds. | |
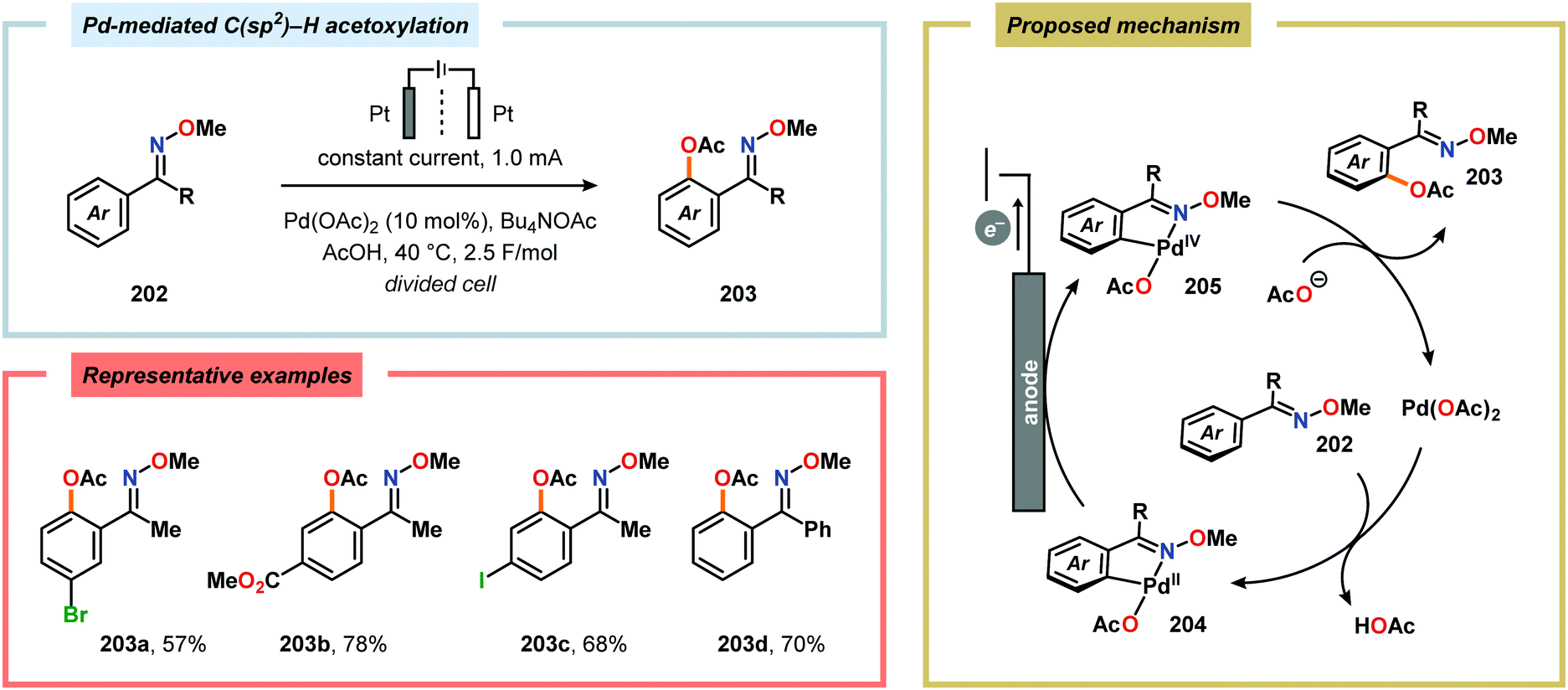
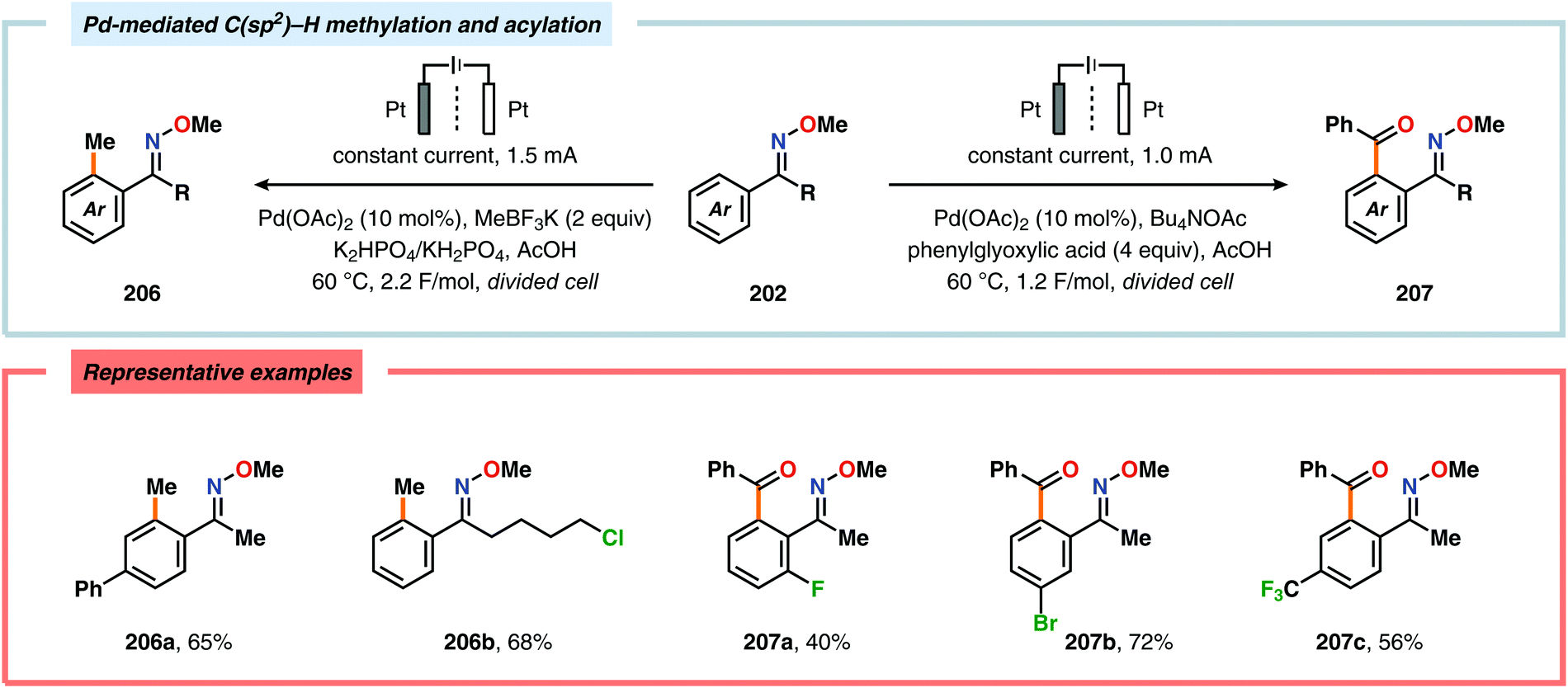
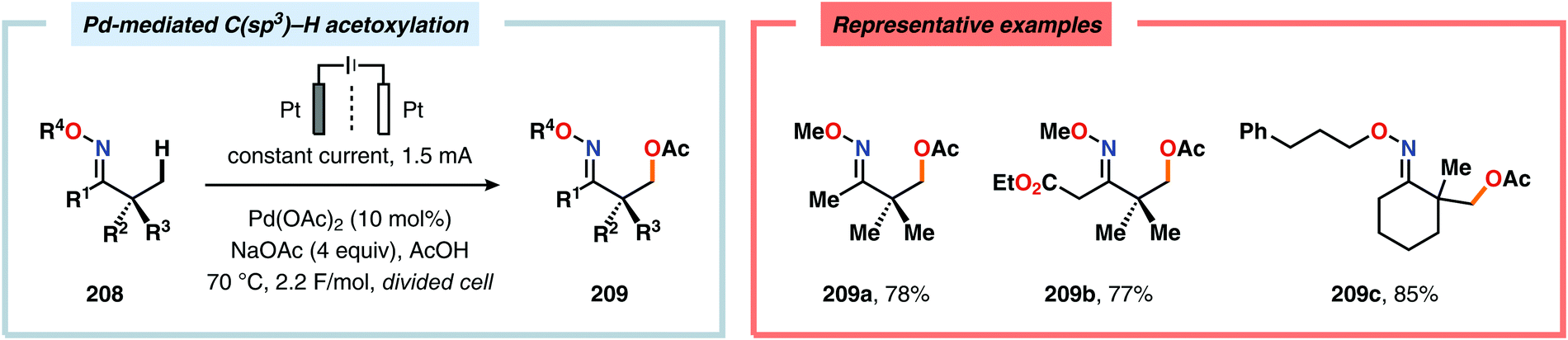
Merging Pd catalysis and electrochemical oxidation has also proven to be a successful concept for carrying out C–H functionalization on oxime-based motifs. In this regard, Mei and coworkers have examined the Pd-catalyzed oxime-directed C(sp2)–H acetoxylation employing an anodic oxidation strategy (Scheme 60). In this transformation, it was proposed that the rate-determining oxime-assisted C–H activation generates the desired palladacycle. This PdII species is subsequently oxidized at the anode to produce the corresponding PdIV intermediate, which can undergo reductive elimination to deliver the acetoxylation product, thus regenerating PdII to close the catalytic cycle.530 Shortly thereafter, the authors reported a dual Pd/electrochemical strategy to achieve C(sp2)–H methylation and acylation (Scheme 61). Here, reductive elimination was suggested to occur from a high-valent Pd intermediate, presumably PdIII or PdIV.531 Finally, functionalization of unactivated C(sp3)–H bonds has also been reported using oximes as directing groups. Along similar lines, C–H activation furnishes a PdII palladacycle, which subsequently is oxidized at the anode to give a high-valent Pd species, presumably a PdIV intermediate. Generation of the high-valent Pd center promotes the otherwise challenging C–O reductive elimination event to deliver the oxygenated product while regenerating PdII. In addition to carrying out C(sp3)–H acetoxylation (Scheme 62), the dual Pd/electrochemical strategy could also be conducted with a variety of oxyanion coupling partners, such as methoxide, tosylate and trifluoroacetate.532 The catalytic protocols described here proceed under mild reaction conditions and in the absence of exogenous ligands, representing sustainable alternatives to conventional platforms for C–H functionalization that rely on stoichiometric chemical oxidants.
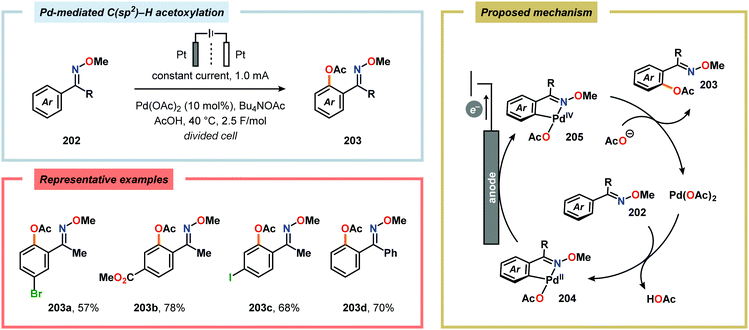 |
| | Scheme 60 Pd-catalyzed C(sp2)–H acetoxylation via anodic oxidation. | |
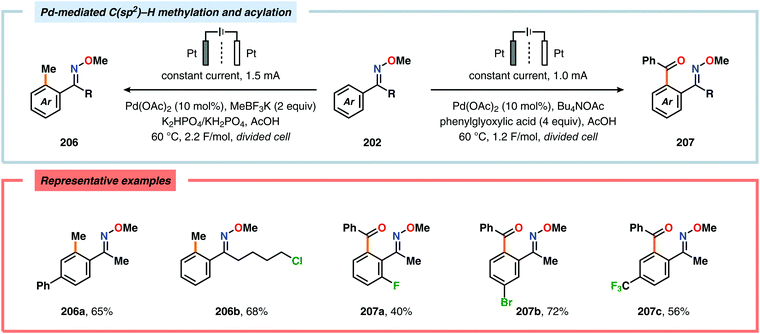 |
| | Scheme 61 Pd-mediated C(sp2)–H methylation and acylation. | |
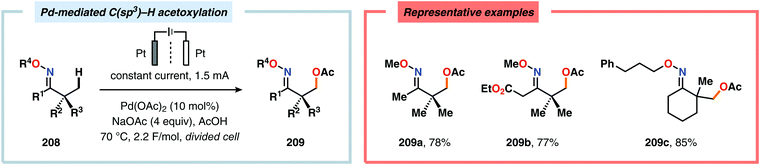 |
| | Scheme 62 Dual Pd/electrochemical C(sp3)–H functionalization. | |
2.5. Electrochemical oxidative cross-couplings
Transition metal-catalyzed cross-coupling reactions serve as an integral part of contemporary synthetic chemistry for the rapid and selective construction of complex architectures from simple building blocks. However, these catalytic bond forming platforms require the use of prefunctionalized substrates, which adds additional preceding synthetic steps. This inevitably culminates in the generation of undesired stoichiometric amounts of waste. In recent years, constructing C–C bonds through the direct coupling of two C–H coupling fragments has emerged as a highly appealing concept. This effective and straightforward strategy not only circumvents the need for using prefunctionalized reagents but also considerably enhances the atom- and step-economy. In principle, dihydrogen (H2) is the sole (by)product resulting from forging the two C–H bonds; however, typically a sacrificial oxidant is required to accept the H2, hence the name oxidative cross-coupling or cross-dehydrogenative coupling (CDC). Oxidative cross-coupling is undoubtedly an attractive catalytic platform, yet conceptually and practically challenging due to, for example, the low reactivity of the C–H bonds in combination with overcoming the limitations in site-selective functionalization.533–536 Moreover, the development of radical-mediated oxidative coupling strategies has also gained significant attention from the scientific community.537
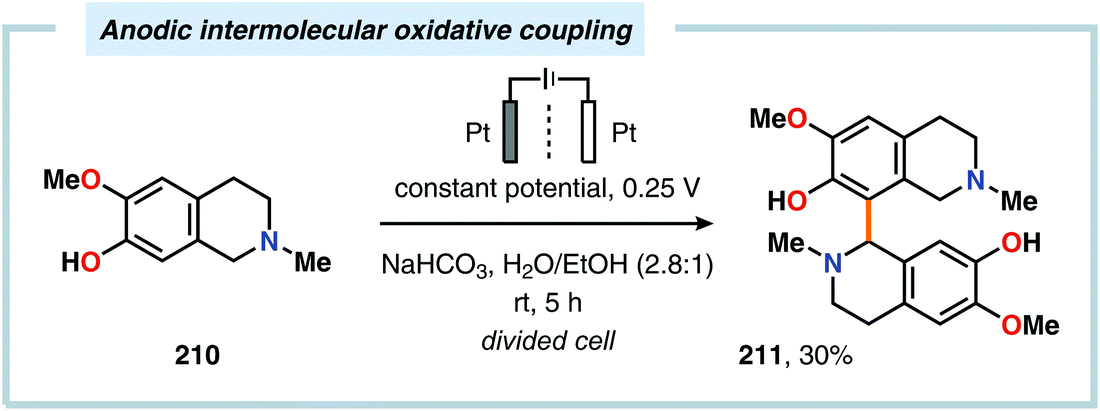
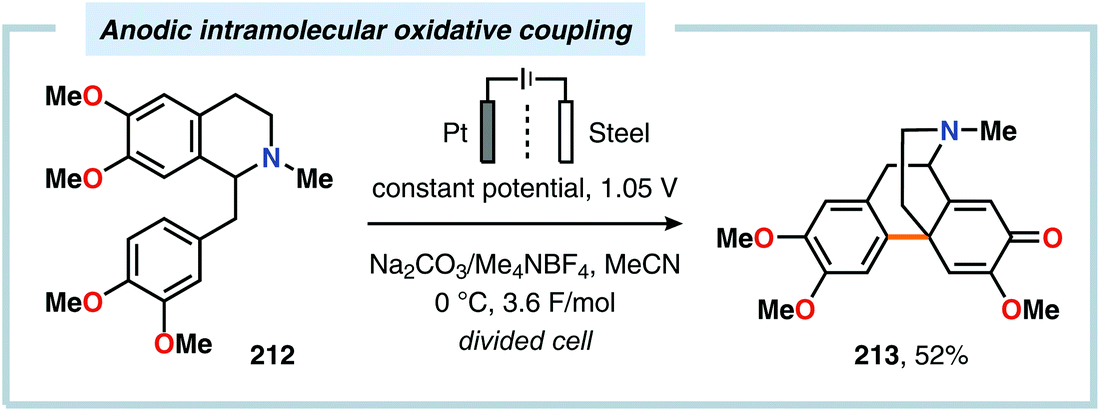
Despite the relatively simple motif, the synthesis of biphenols through oxidative cross-coupling of unprotected phenols has proven to be challenging and often relies on protocols involving expensive catalyst systems or reagents. In addition to producing the desired cross-coupled biphenolic product,538 the reaction is also accompanied by products derived from oxidative dimerization, higher molecular weight polymers as well as compounds housing C–O coupling motifs.539–543 Seminal work on the intermolecular anodic oxidative coupling of phenols demonstrated that corypalline (210) could be electrochemically oxidized to the corresponding dimer (211, Scheme 63).544–548 However, attempts to carry out the related intramolecular C–C coupling on laudanosine (212) resulted in oxidative dearomatization of one of the phenolic moieties and furnished O-methylflavinantine (213, Scheme 64).549–553 Similar reactivity has also been observed for the related alkaloids amurine, oxocrinine, oxomaritidine and pallidine.554–556
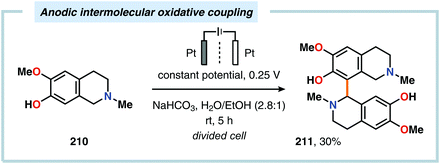 |
| | Scheme 63 Anodic intermolecular oxidative coupling of corypalline. | |
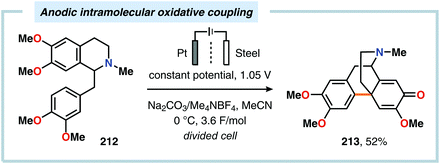 |
| | Scheme 64 Anodic intramolecular coupling of laudanosine. | |
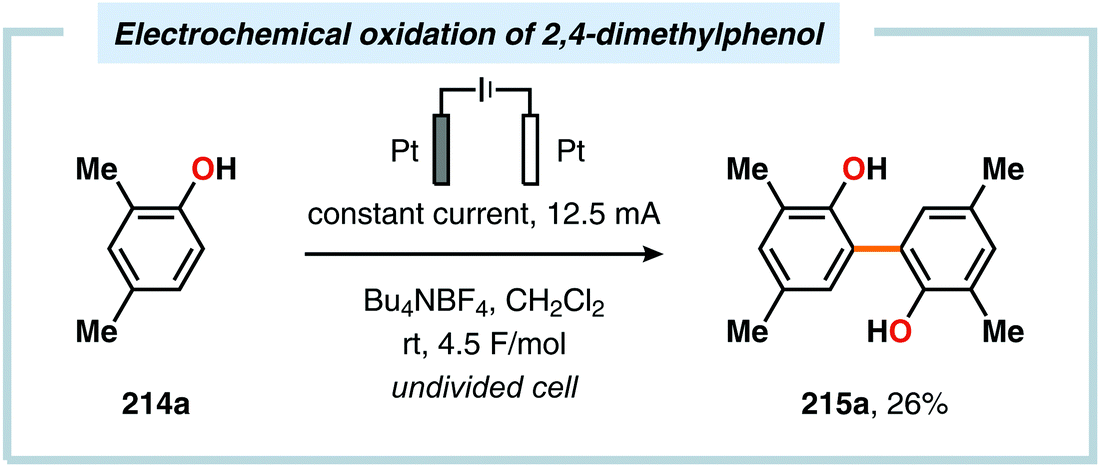
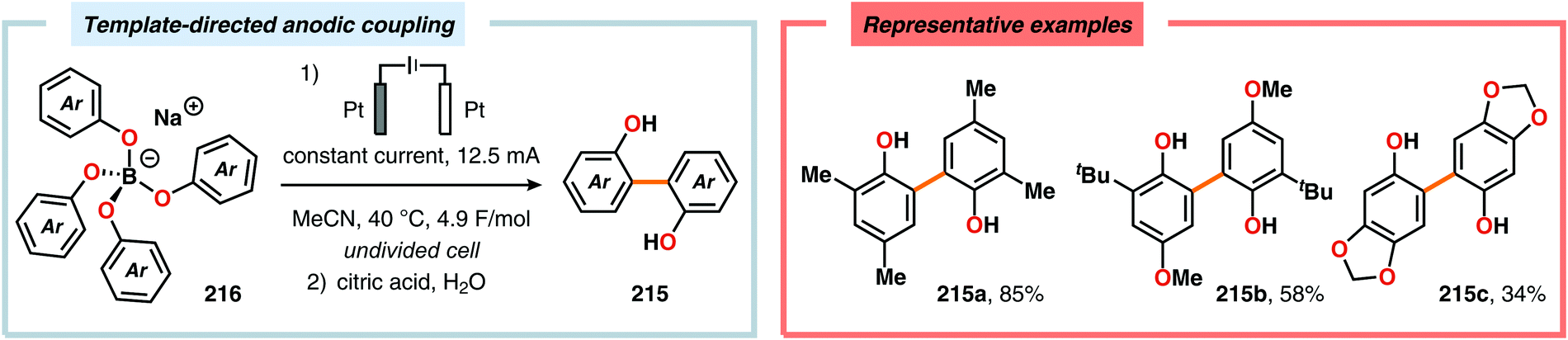
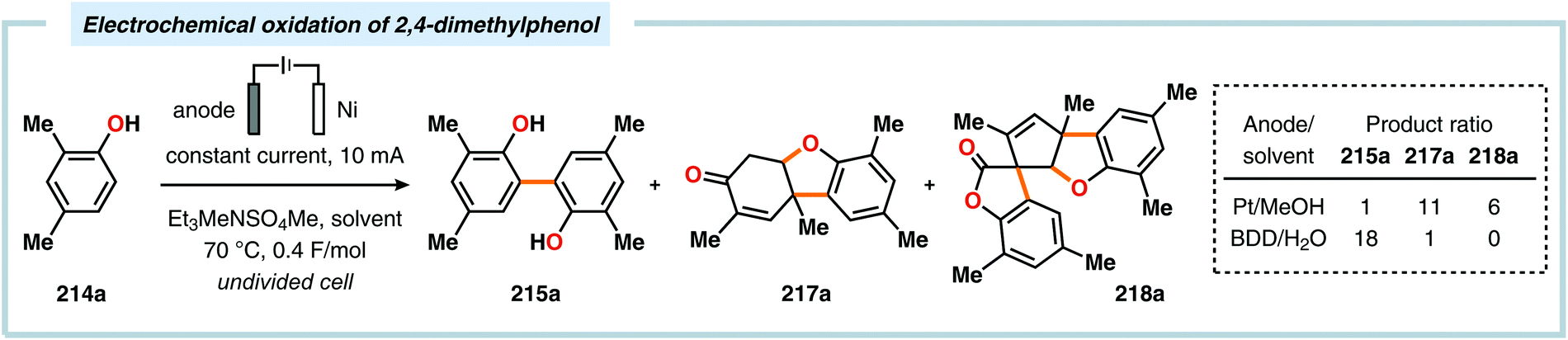
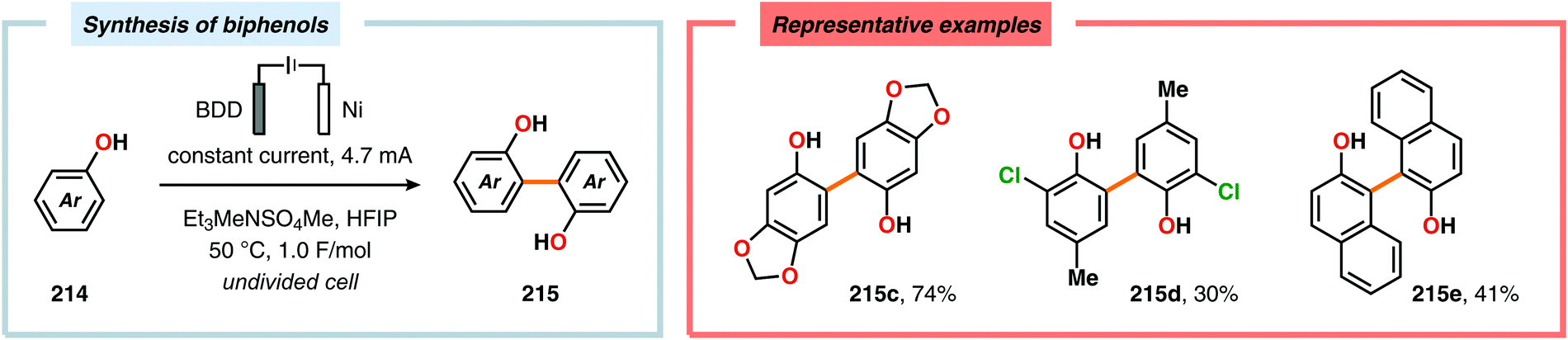
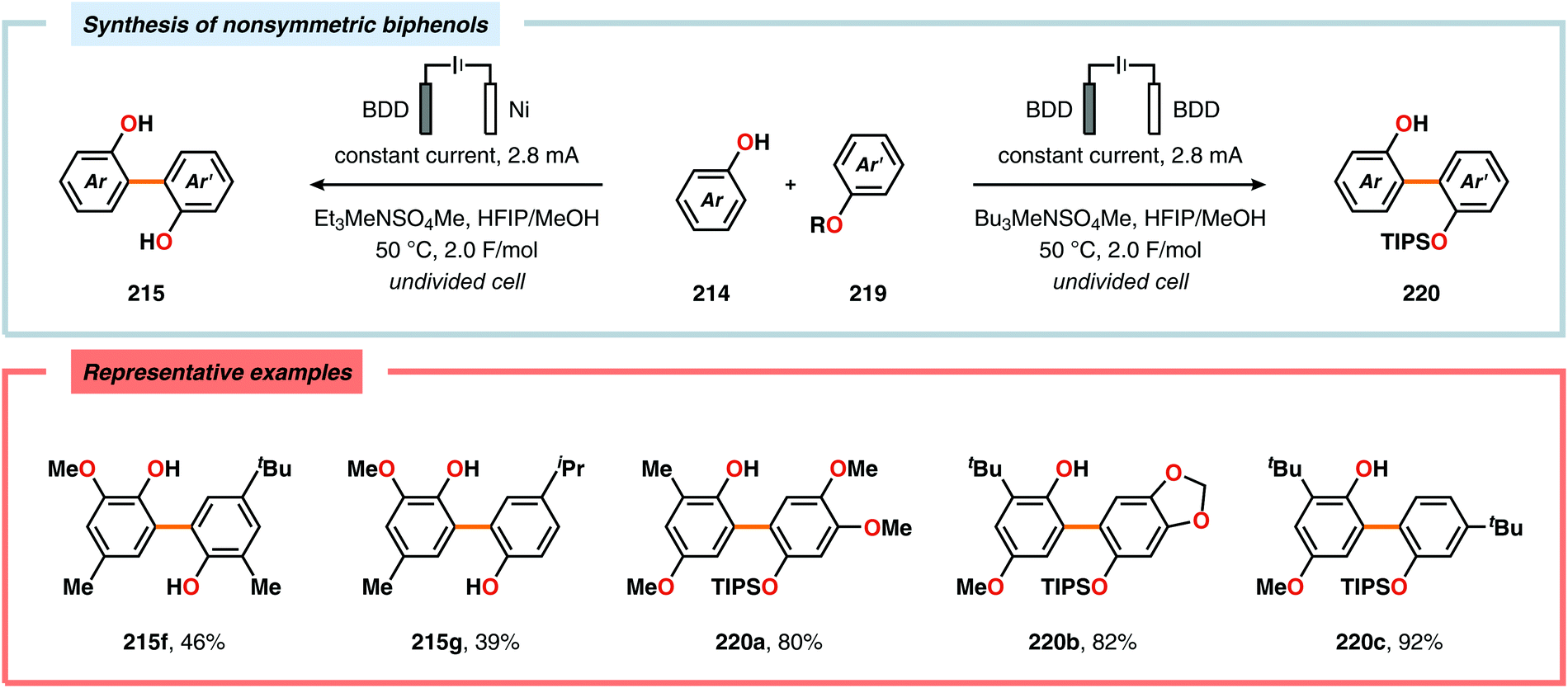
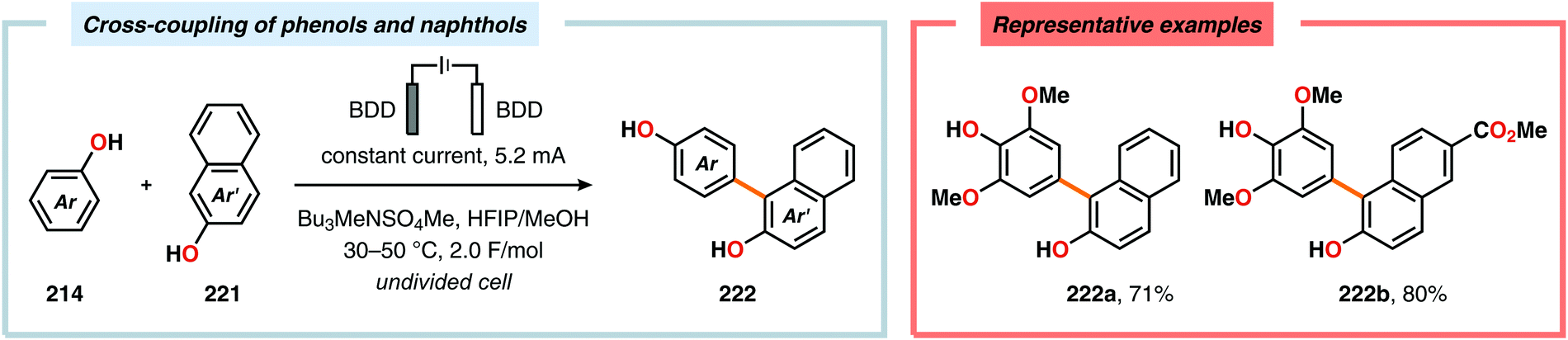
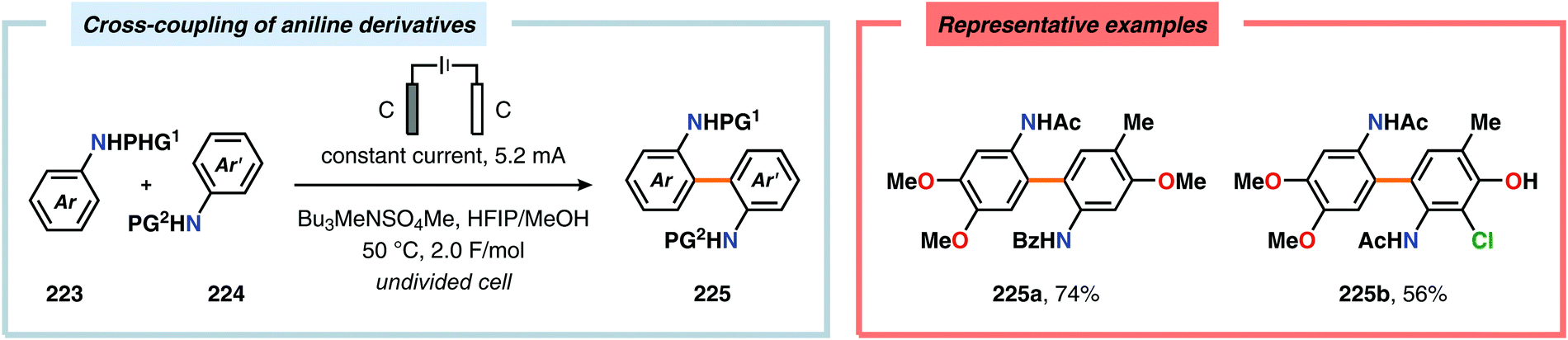
Pioneering studies by the Waldvogel group demonstrated that the direct anodic oxidation of 2,4-dimethylphenol (214a) produced the ortho,ortho-coupled biphenol 215a, albeit in rather poor yield (Scheme 65).557 The authors subsequently devised a template-directed method for the anodic coupling of substituted phenols to the corresponding 2,2′-biphenols using tetraphenoxy borate derivatives (Scheme 66). Although the developed protocol was limited to electron-rich substrates, the electrochemical synthesis could be conducted on a kilogram scale.558,559 In addition to generating the desired biphenol product, several additional coupling motifs can also be produced.560–565 To circumvent the lack of selectivity, Waldvogel and coworkers realized that boron-doped diamond (BDD) electrodes22 gave almost exclusive selectivity for the ortho-coupling product (Scheme 67).566,567 It was rationalized that the observed selectivity originated from the high chemical and electrochemical stability of the BDD electrodes in combination with the high overpotential for oxygen evolution in aqueous media and efficient formation of reactive alkoxy or hydroxyl radicals at the anode that can serve as mediators.568–570 Interestingly, by simply switching to fluorinated solvents/mediators, such as hexafluoroisopropanol (HFIP), allowed for the creation of a powerful electrolyte system and significantly expanded the scope and efficiency for the anodic coupling of simple phenols571 (Scheme 68) as well as guaiacol derivatives572–574 on BDD electrodes. Protocols for synthesis of nonsymmetric biphenols (Scheme 69),575–577 and anodic cross-coupling of phenols with naphthols578 (Scheme 70) as well as aniline derivatives579 (Scheme 71) have also been addressed.580,581
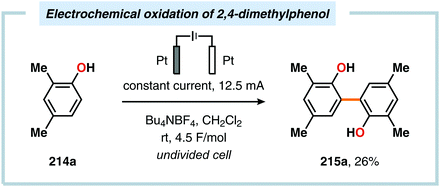 |
| | Scheme 65 Electrochemical oxidation of 2,4-dimethylphenol. | |
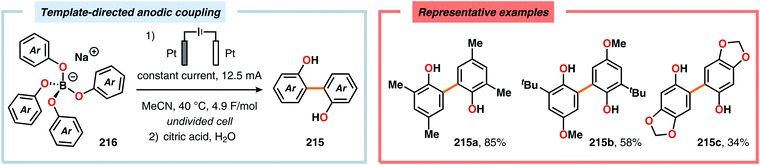 |
| | Scheme 66 Template-directed anodic coupling of phenols. | |
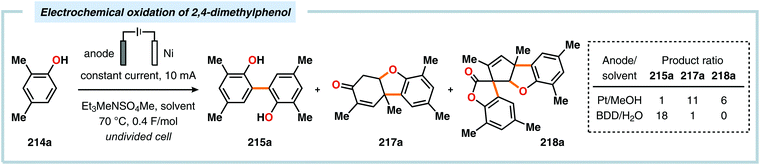 |
| | Scheme 67 Product diversity from the electrochemical oxidation of 2,4-dimethylphenol. | |
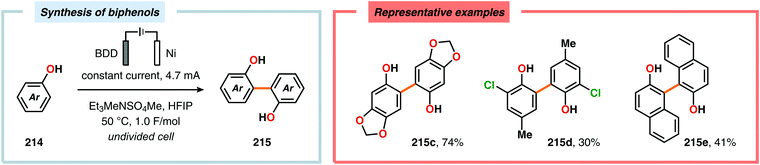 |
| | Scheme 68 Electrochemical synthesis of biphenols using a BDD anode. | |
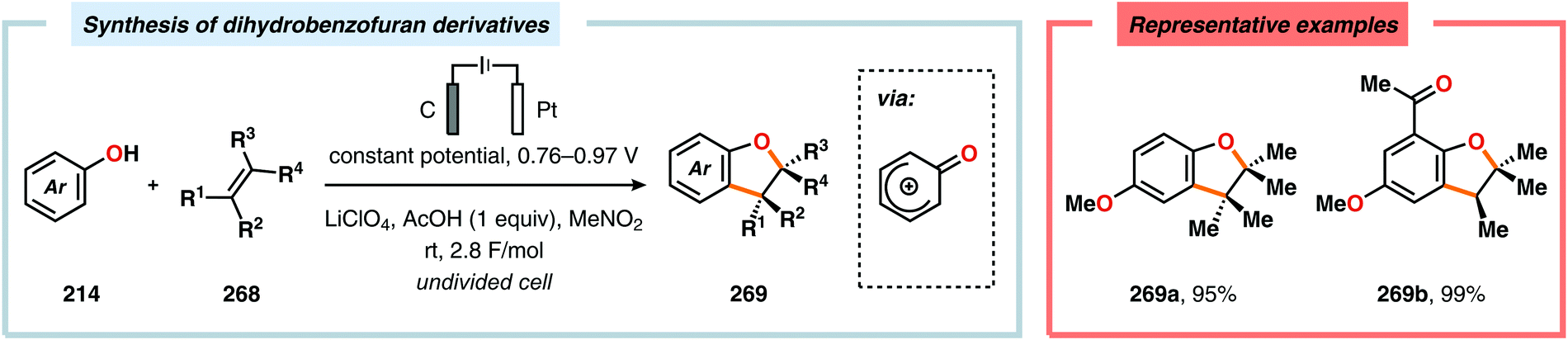
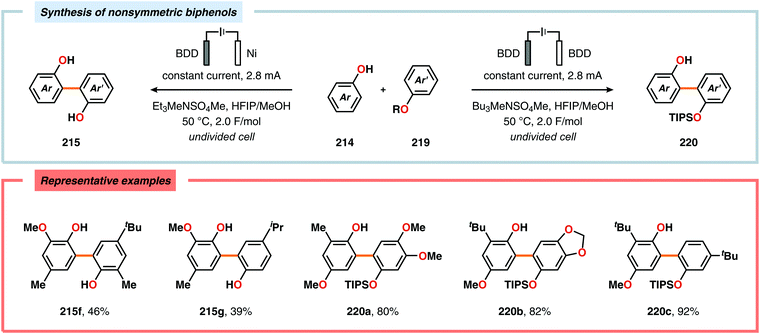 |
| | Scheme 69 Synthesis of nonsymmetric biphenols through anodic cross-coupling. | |
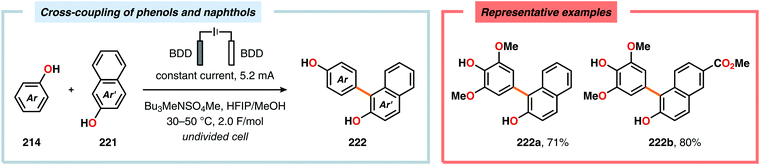 |
| | Scheme 70 Anodic cross-coupling of phenols and naphthols. | |
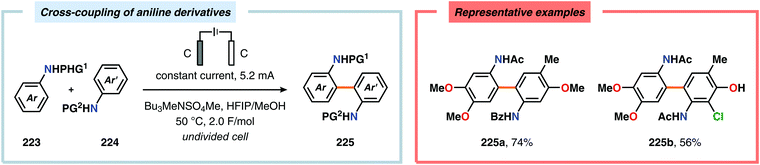 |
| | Scheme 71 Anodic cross-coupling of aniline derivatives. PG = protecting group. | |
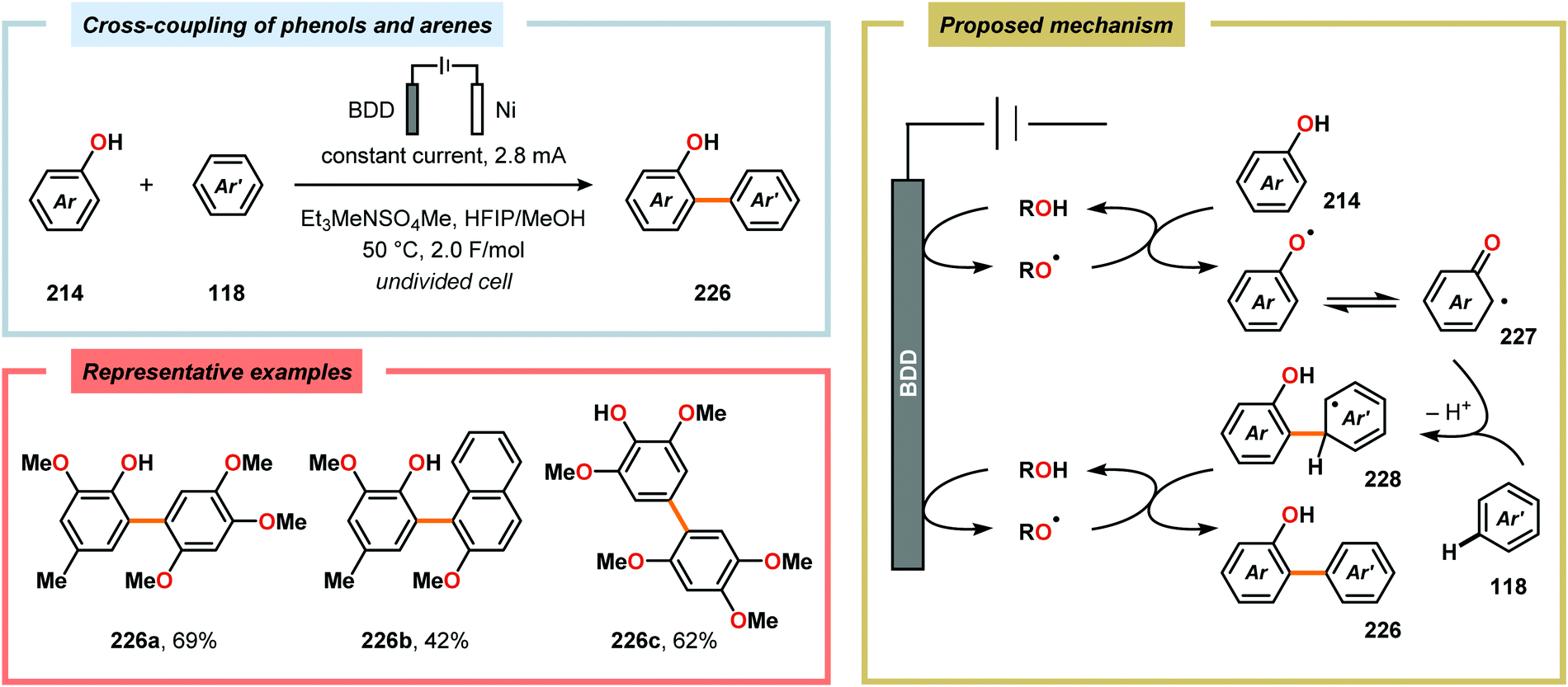
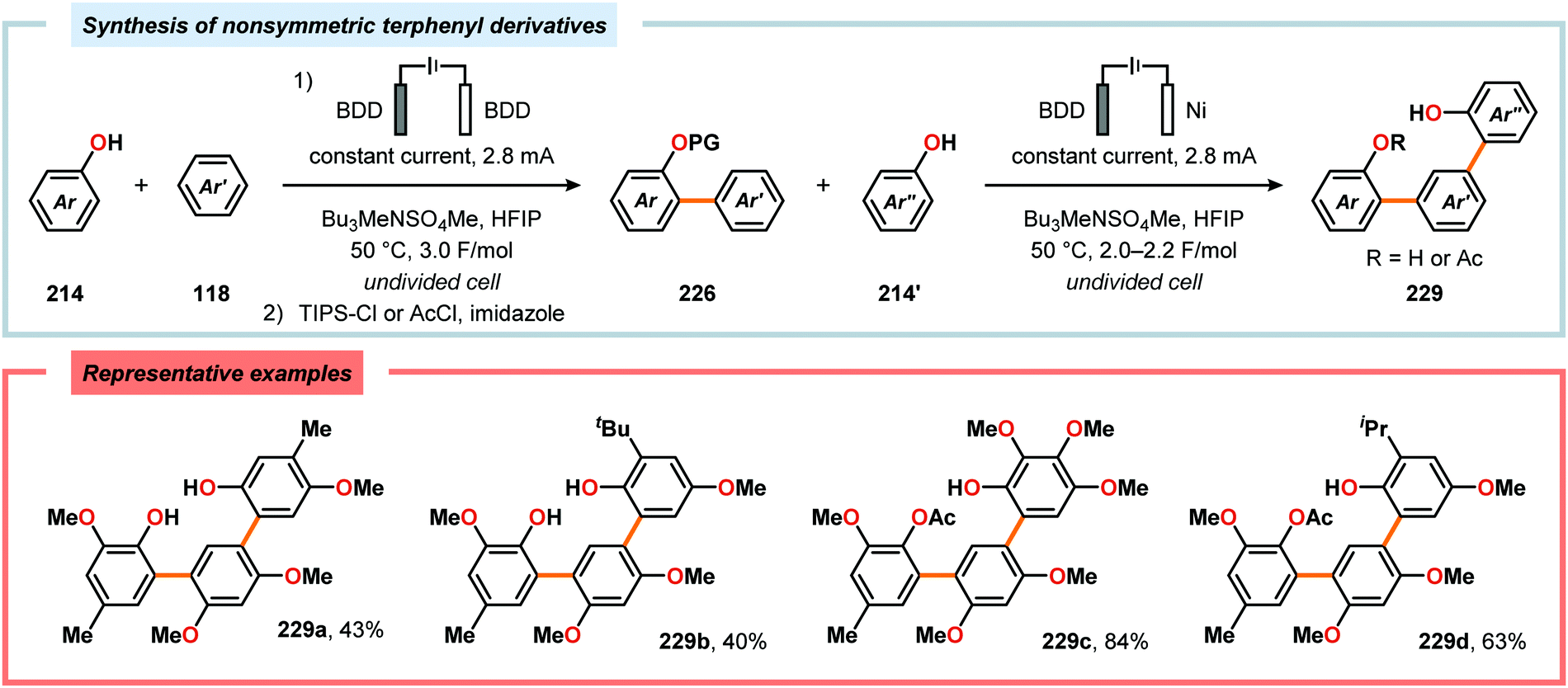
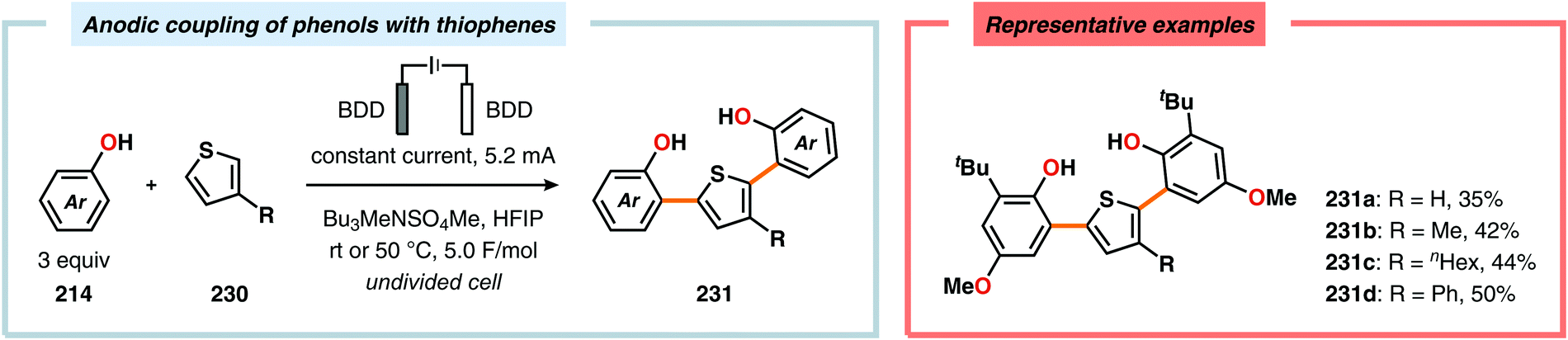
Metal-free electrochemical methodologies that facilitate anodic cross-coupling between phenols and electron-rich arenes have also been developed. The Waldvogel group discovered that this reaction could be accomplished employing a BDD anode in the presence of HFIP. It was established that HFIP had a crucial role in the reaction as no conversion was observed in the absence of HFIP but were able to rule out the possibility of HFIP acting as a redox mediator.55,582 Since the phenol and arene components have similar oxidation potentials, the high level of chemoselectivity was rationalized by oxyl–HFIP hydrogen bonding interactions, which stabilize the produced spin centers.583–585 The hydrogen-bond stabilized spin center undergoes HAT with the phenol (214), which produces the more easily oxidized phenoxyl radical 227. Radical 227 is subsequently trapped by the electron-rich arene (118), which gives intermediate 228 upon deprotonation. This is followed by a second HAT event, resulting in rearomatization and furnishes the cross-coupled biaryl 226 (Scheme 72).55,582 This strategy was subsequently extended to enable the development of anodic cross-coupling protocols for synthesis of meta-terphenyl-2,2″-diols (Scheme 73),586,587 2,5-bis(2-hydroxyphenyl)thiophenes588 (Scheme 74) and dibenzofurans.589,590
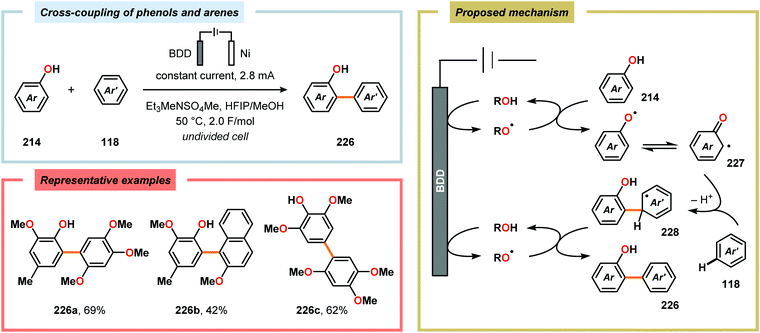 |
| | Scheme 72 Direct anodic cross-coupling of phenols and electron-rich arenes. | |
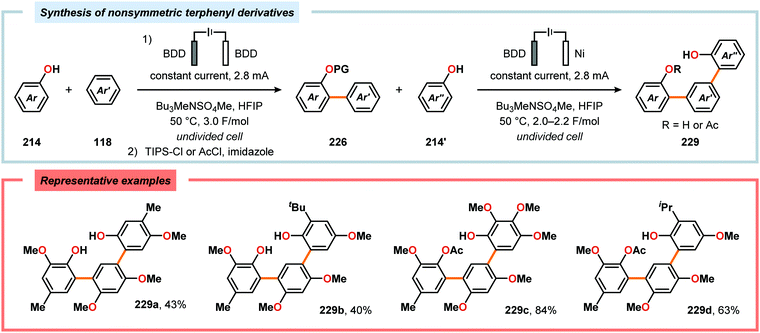 |
| | Scheme 73 Synthesis of nonsymmetric meta-terphenyl-2,2″-diols. PG = protecting group. | |
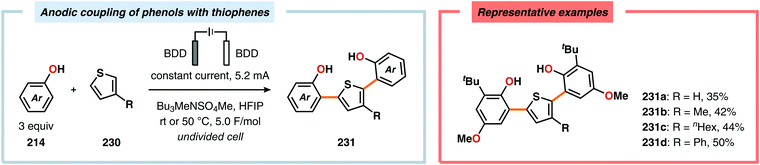 |
| | Scheme 74 Electrochemical synthesis of 2,5-bis(2-hydroxyphenyl)thiophenes. | |
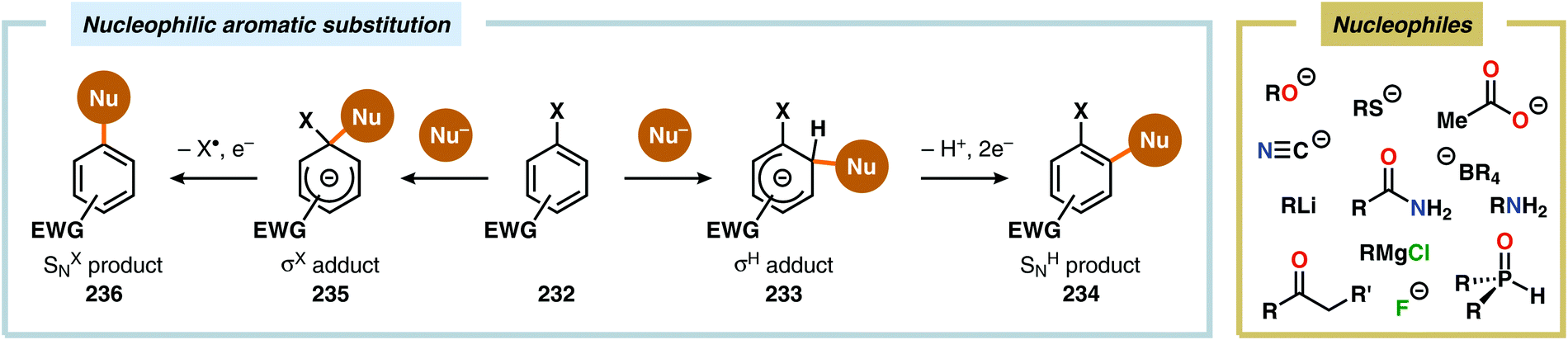
Since the high oxidation potential of electron-deficient arenes, such as nitroarenes, limits direct anodic oxidation of these species, anodic cross-coupling reactions between electron-deficient arenes and nucleophiles engage in a nucleophilic aromatic substitution (SNAr) type pathway. This two-step process can follow two mechanistically discrete pathways (Scheme 75). The first step involves nucleophilic addition to the electron-deficient arene unit, resulting in either a σH or σX adduct (Meisenheimer complex). For the σH intermediate (233), the second step follows a two-electron mechanism in which formal loss of a hydride occurs and is referred to as nucleophilic aromatic substitution of hydrogen (NASH, SNH). However, rearomatization of the σX adduct (235), generated from attack of the ipso carbon, involves an initial single-electron oxidation, which triggers radical elimination of the heteroatom (X˙) and is hence called nucleophilic aromatic substitution of a heteroatom (NASX, SNX).591–593 A range of nucleophiles, including acetate,594 alkoxides,594,595 alkyl lithium or Grignard reagents,596 amides,597 amines,594,595,597 enolates,594,598 fluoride,595 cyanide,594,599 organophosphorus compounds,600 tetraalkylborate salts601 and thiolates,595 have been utilized to affect nucleophilic aromatic substitution through electrochemical oxidation of the intermediate σ adducts. This concept was recently applied on azaaromatic scaffolds for the synthesis of asymmetrical bi(hetero)aryl motifs (Scheme 76).602,603
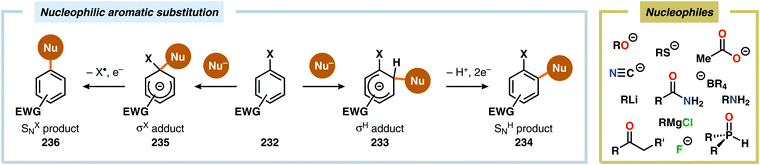 |
| | Scheme 75 Nucleophilic aromatic substitution via electrochemical oxidation. | |
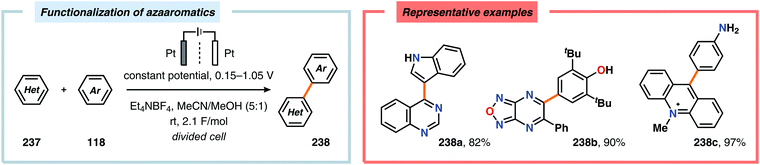 |
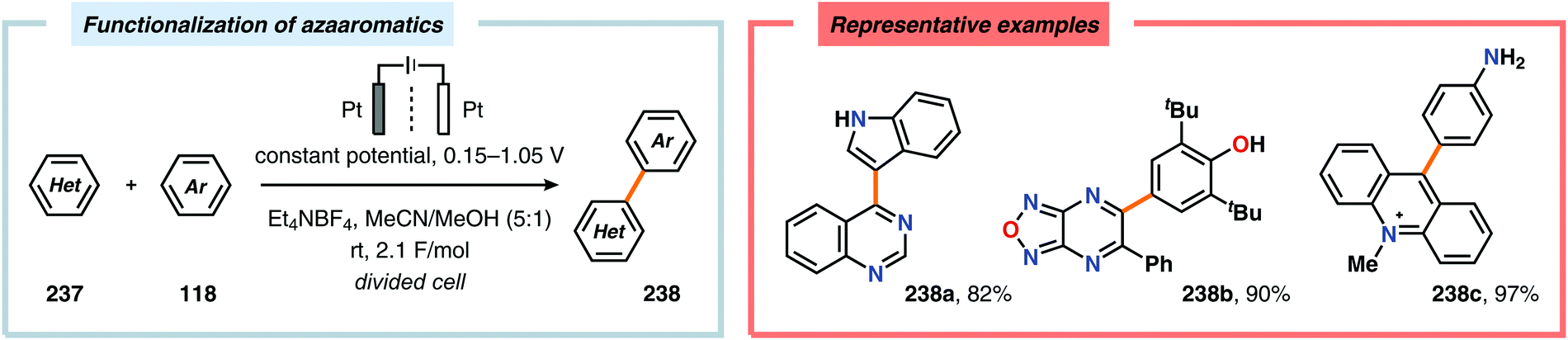
| | Scheme 76 Electrochemical functionalization of azaaromatics through nucleophilic aromatic substitution. | |
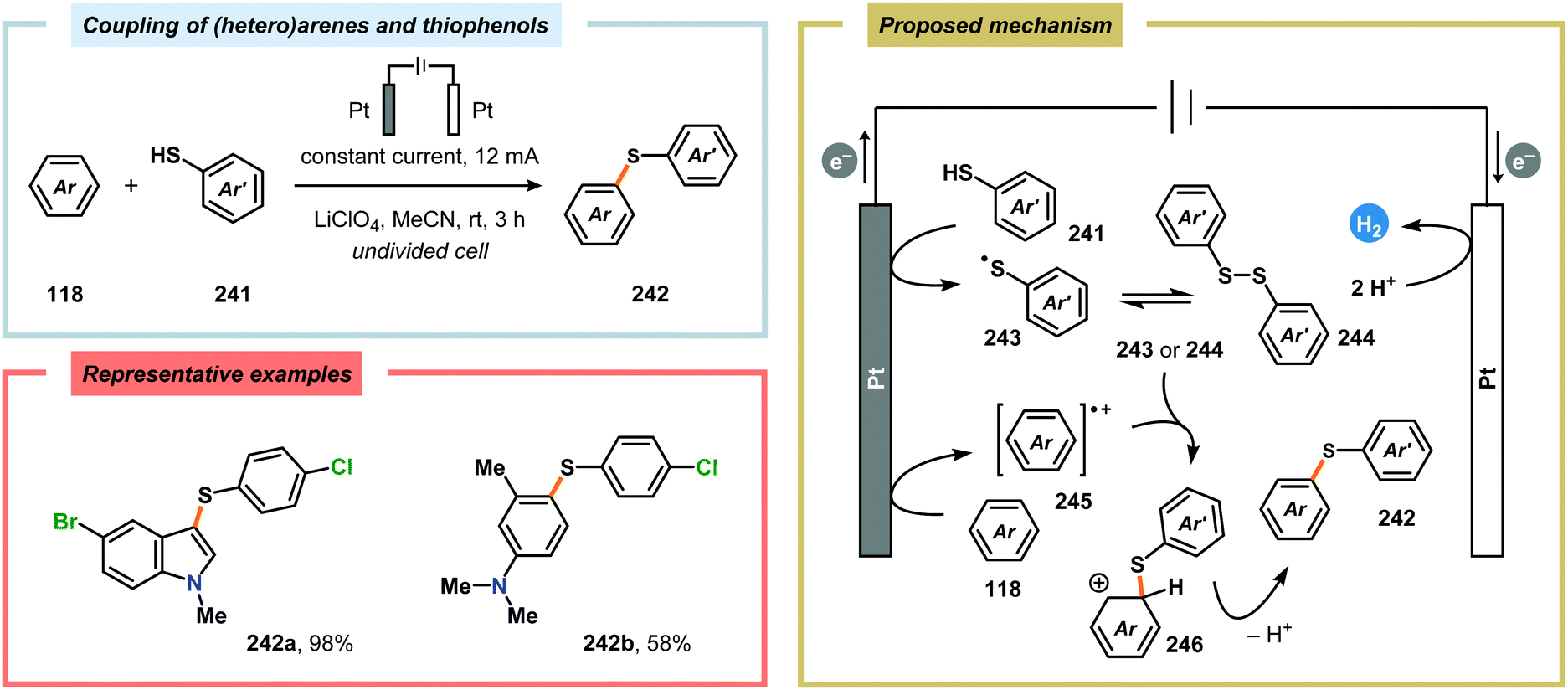
Although oxidative coupling between electron-deficient and electron-rich arenes, two moieties with distinctly different π-electronic properties, generally provides high chemoselectivity of the cross-coupled products over homocoupling as a result of the “electronic differential principle”, anodic cross-coupling of unactivated electron-rich arenes has been achieved. Recently, the Yoshida group highlighted that employing the “cation pool” method allowed the merging of two unactivated aromatic compounds in a straightforward fashion. Here, initial anodic oxidation at −78 °C of the less electron-rich arene component (naphthalene) resulted in accumulation of the corresponding radical cation species. Subsequent introduction of the other aromatic moiety under non-oxidative conditions at −90 °C avoided nonselective oxidation of the starting material and overoxidation of the biaryl product (Scheme 77). Notably, the high regioselectivity of the developed cross-coupling method can be estimated based on the spin density of the radical cation and the highest occupied molecular orbital (HOMO) coefficients of the nucleophilic aromatic partner.604,605 The use of parallel laminar flow also serves as a convenient method for preparing nonsymmetrical biaryl scaffolds as demonstrated by Atobe and coworkers.606 An electrochemical method for constructing carbon–sulfur (C–S) bonds was recently reported by the Lei group. The electrochemical reactions were conducted in an undivided cell and allowed a wide range of electron-rich arenes and heteroarenes, such as indole, pyrrole and thiophene, to be coupled with various substituted thiophenols. Mechanistic studies indicated that performing the reaction in the presence of the persistent radical TEMPO did not produce any of the desired C–S coupling product. Furthermore, the involvement of disulfides, generated from dimerization of the thiophenols, was also observed. Therefore, two mechanistically distinct pathways could explain the noted reaction outcome. Thus, product formation can occur either through direct radical–radical coupling between the arene cation radical intermediate (245) and the thiyl radical (243). Alternatively, C–S bond formation can proceed via arene radical addition to the produced disulfide (244) as depicted in Scheme 78.607,608
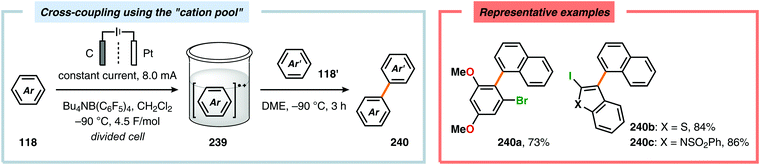 |
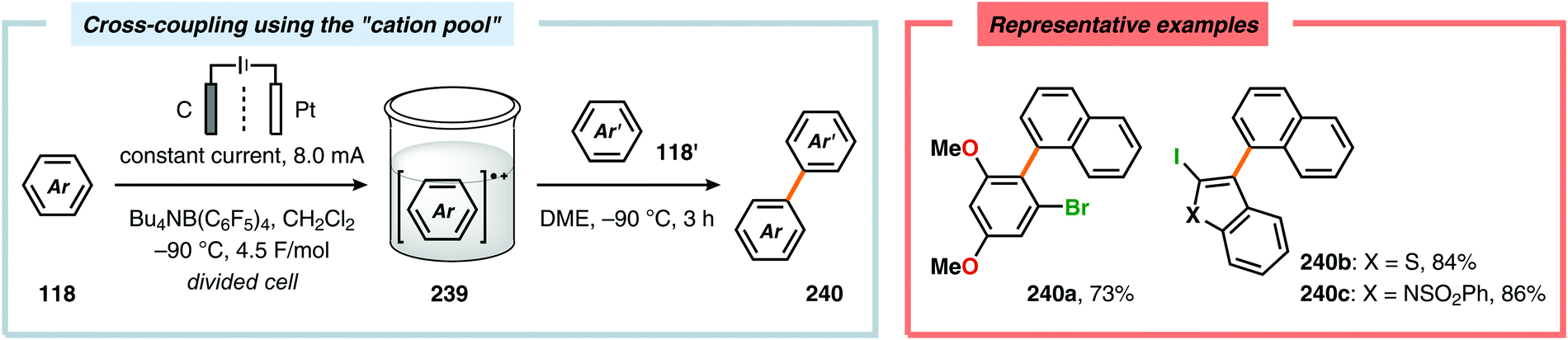
| | Scheme 77 Cross-coupling of aromatic compounds using the “cation pool” method. | |
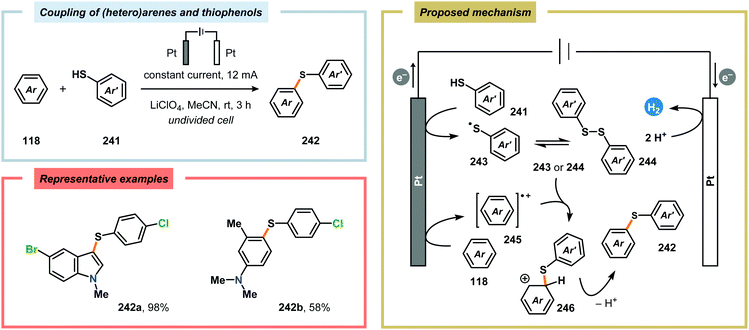 |
| | Scheme 78 Direct anodic cross-dehydrogenative coupling of (hetero)arenes and thiophenols. | |
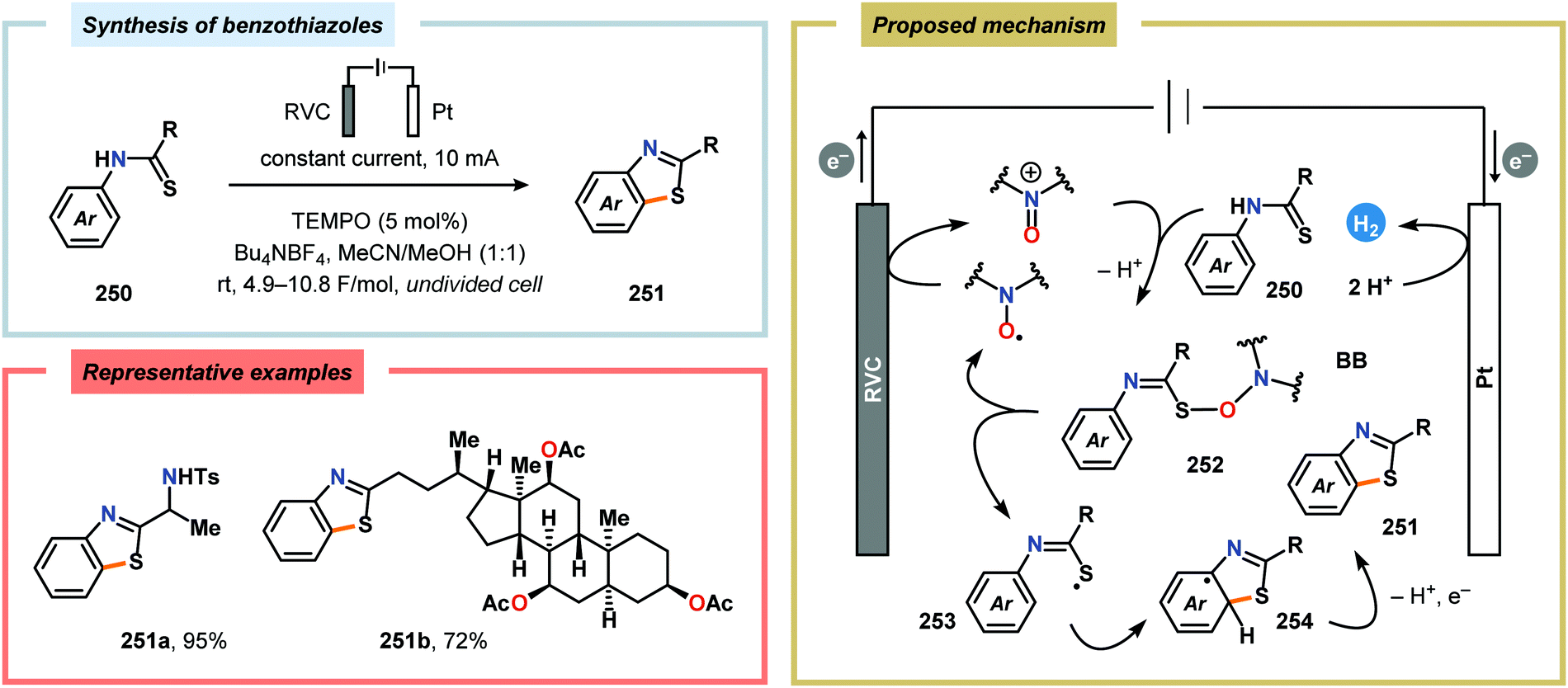
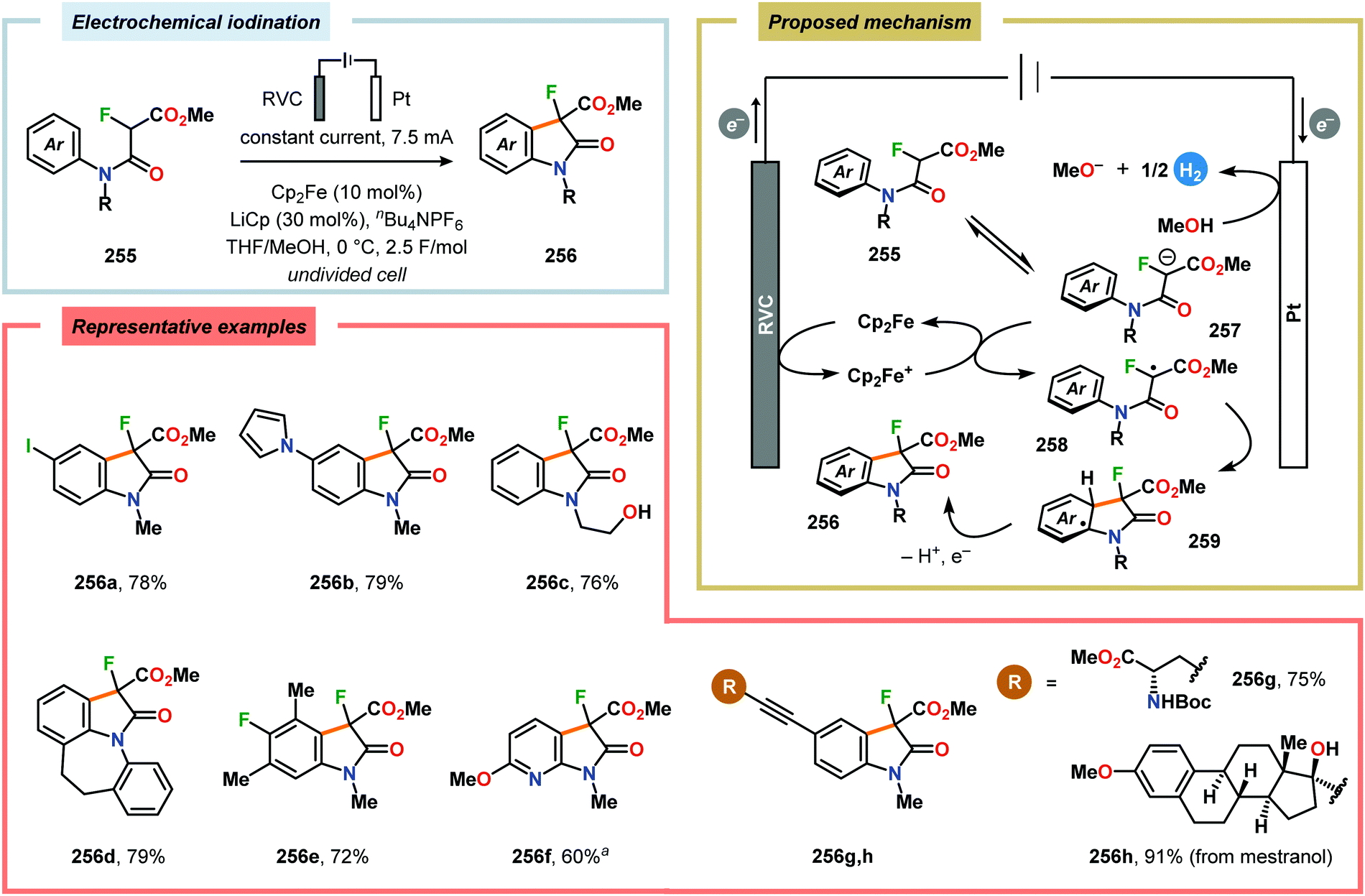
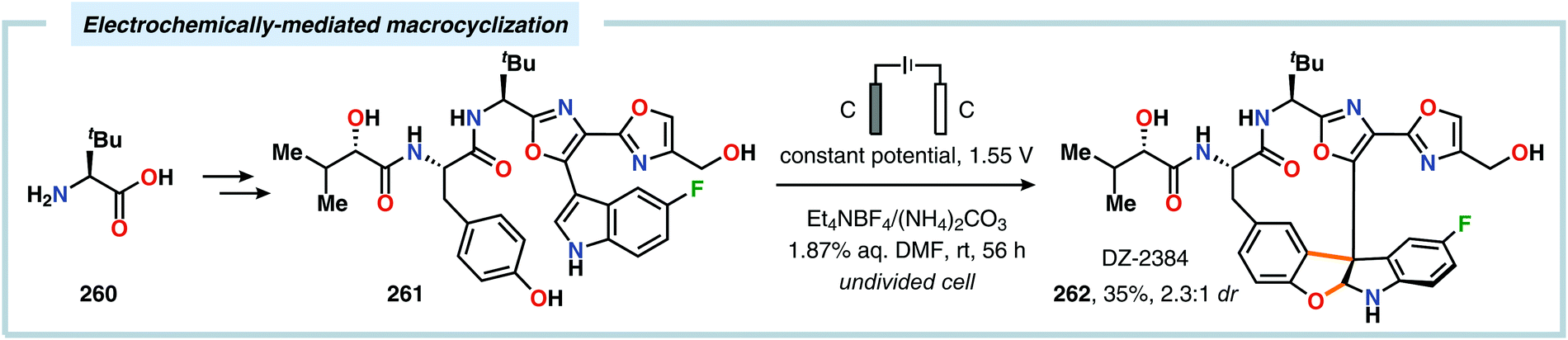
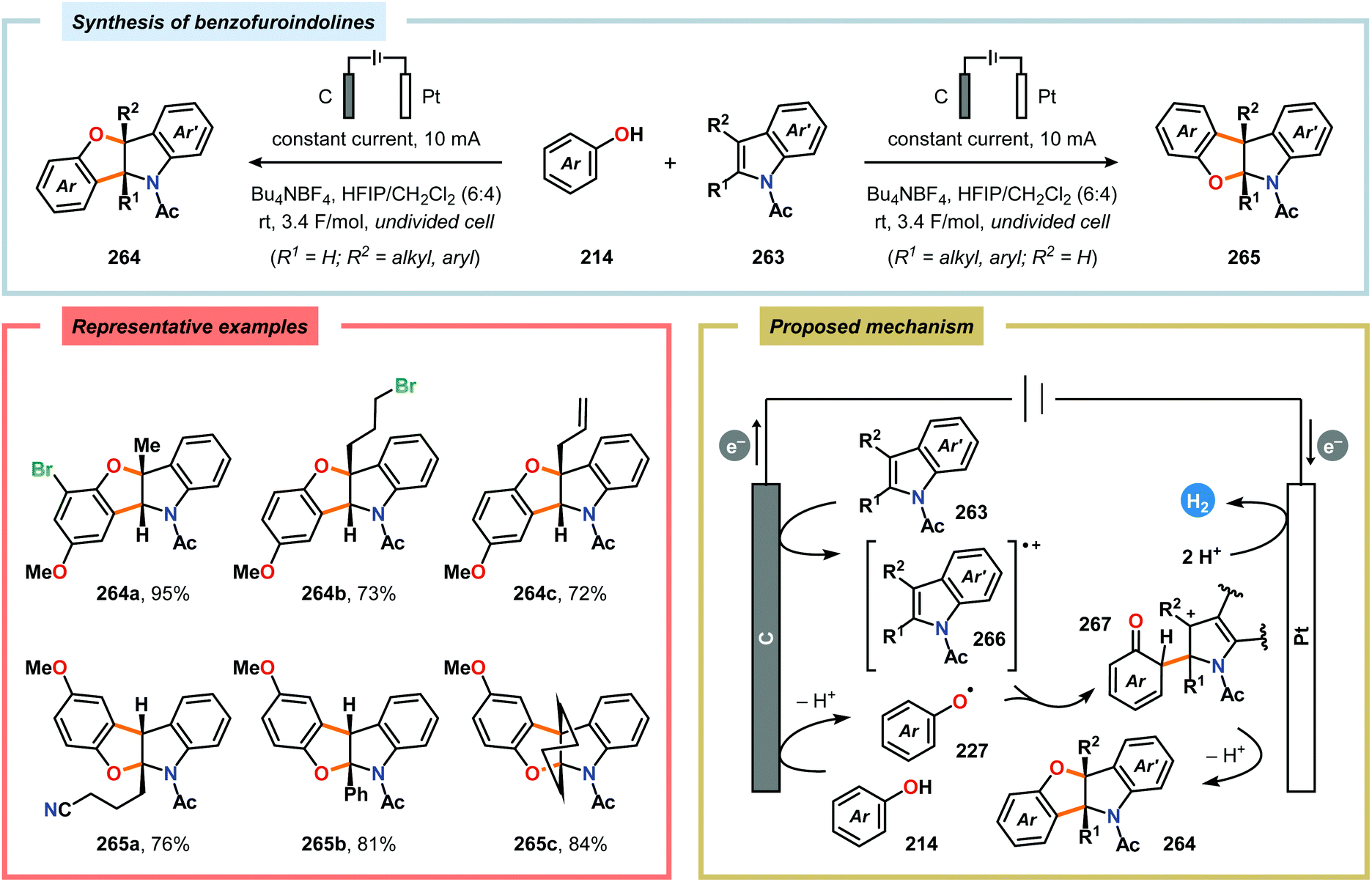
2.6. Oxidative annulations involving (hetero)arenes
Electrochemical approaches for intramolecular C–H thiolations have been reported by the Lei609 and Xu610 groups.611 Lei and coworkers’ protocol utilized aryl isothiocyanates, which were treated with amines to produce the corresponding thioamides. The in situ generated thioamides were subsequently electrolyzed at 70 °C in an undivided cell to furnish the desired benzothiazoles (Scheme 79).609 On the contrary, Xu and coworkers strategy employed TEMPO as a mild redox-mediator to enable selective C–H thiolation of a variety of N-(hetero)arylthioamides with diverse electronic properties (Scheme 80).610,612 Both C–H thiolation strategies are believed to proceed via thioamidyl radicals with concomitant formation of H2 at the cathode. Expanding on the C–H thiolation strategy, the Xu group has recently demonstrated that 3-fluorooxindoles could be accessed through cross-dehydrogenative coupling.613,614 Here, ferrocene (Cp2Fe) was selected as a redox mediator due to earlier literature precedents highlighting its capability of promoting oxidative radical cyclizations.615–618 The reactions were carried out in an undivided cell and tolerated a multitude of functional groups, including alcohols, alkynes, aminoesters, halogens, silylethers as well as redox-sensitive carbazole, N-phenyl carbamate and pyrrole groups (Scheme 81). However, products containing electron-withdrawing groups, such as CF3, OCF3 and CO2Me, were prone to base-induced decomposition and therefore had to be performed at −30 °C for optimal yields. The mechanism is presumed to involve formation of H2 at the cathode from MeOH, which results in production of MeO−. The electrochemically generated base facilitates deprotonation of the substrate (255) to furnish the corresponding enolate (257), which is subsequently oxidized by the anodically generated Cp2Fe+ to produce α-carbon radical intermediate 258. Finally, radical cyclization and rearomatization afford oxindole 256.613,619,620
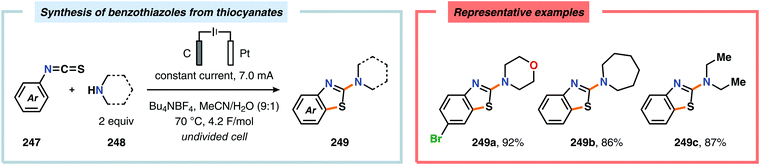 |
| | Scheme 79 Electrochemical synthesis of benzothiazoles from aryl isothiocyanates and amines. | |
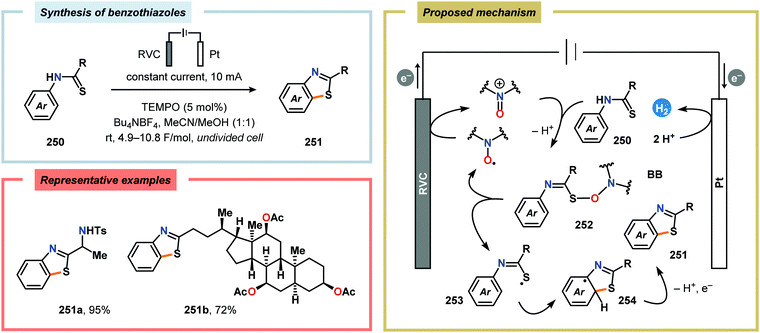 |
| | Scheme 80 Electrochemical synthesis of benzothiazoles using TEMPO as redox mediator. | |
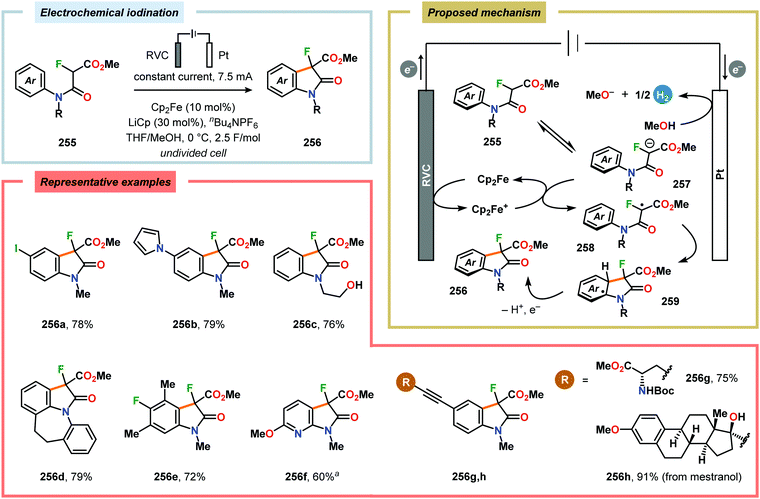 |
| | Scheme 81 Synthesis of 3-fluorooxindoles though electrochemical cross-dehydrogenative coupling using ferrocene (Cp2Fe) as redox mediator. aReaction was performed at −30 °C. | |
Harran and coworkers recently showcased electrochemistry as a powerful strategy in organic synthesis, enabling a scalable preparation of DZ-2384 (262), a diazonamide-based drug candidate for treatment of cancer.621 Initial attempts were aimed at using PhI(OAc)2 for mediating the dehydrogenative macrocyclization between the phenol and indole components. However, this approach afforded comparable amounts of a spirocyclohexadienone, which severely limited the yield of the desired macrocyclized product and complicated its purification.622 To circumvent formation of this byproduct, an alternative method was pursued. In this regard, the use of an electrochemical method was explored as it would employ substrates derived from common amino acids. In the refined route towards DZ-2384 (262), the anodic oxidation could be performed in the final step at ambient temperature in aqueous DMF in an undivided cell open to air (Scheme 82), highlighting the practicality of the developed electrochemical macrocyclization approach.621 A related macrocyclization approach was also explored in the synthesis of azonazine.623 Subsequently, the Lei group reported a related dehydrogenative [3+2] annulation between phenol and indole derivatives for accessing benzofuroindolines using an undivided cell (Scheme 83). Here, various N-acetylindoles bearing different C-3 substituents were compatible with the electrochemical annulation and afforded the corresponding benzofuro[3,2-b]indolines (264) in high to excellent yields. However, applying 2-substituted N-acetylindoles resulted in a switch in selectivity and instead produced the corresponding benzofuro[2,3-b]indoline core (265). CV experiments of p-methoxyphenol and various N-acetylindoles revealed that the oxidation potentials were quite similar, suggesting that anodic oxidation of both coupling components might occur under the electrolytic conditions. Radical coupling between the indolyl cation radical (266) and phenoxyl radical (227) furnishes cation 267. Finally, intramolecular cyclization and deprotonation gives the target benzofuroindoline with concomitant formation of H2 at the cathode. However, a mechanism involving nucleophilic attack of the phenol moiety onto the indolyl cation radical cannot be excluded.624
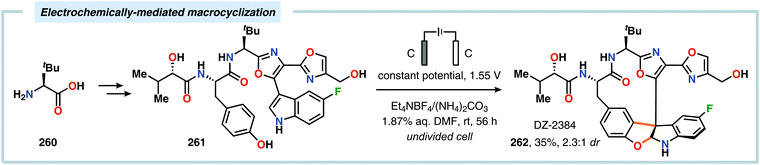 |
| | Scheme 82 Preparation of a diazonamide-based drug development candidate by employing a dehydrogenative macrocyclization approach. | |
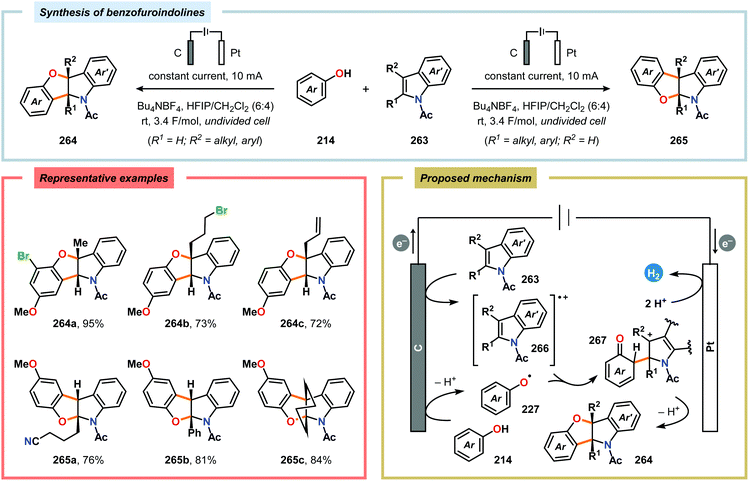 |
| | Scheme 83 Electrochemical synthesis of benzofuro[2,3-b]indolines and benzofuro[3,2-b]indolines. | |
Anodic oxidation of phenols can result in generation of phenoxonium ions upon removal of two electrons and a proton, which can react with simple nucleophiles, such as MeOH or H2O, to afford 4-substituted cyclohexa-2,5-dienones.625–630 Chiba and coworkers highlighted that the anodically generated phenoxonium ions could engage in intermolecular [3+2] cycloaddition with unactivated alkenes to afford dihydrobenzofuran derivatives (Scheme 84).631–635 The reaction has also been conducted using a temperature-controlled multiphase solution consisting of cyclohexane and LiClO4/MeNO2,636 which was shown to significantly enhance the interaction between the hydrophobic aliphatic alkenes and the polar unstable cation intermediates, thereby accelerating the desired [3+2] cycloaddition while avoiding overoxidation of the products.637 Some of these dihydrobenzofuran derivatives display fluorescent properties,638,639 a feature which has been exploited for fluorescent labeling of amino acid derivatives640 as well as homonucleosides.641 In addition, the electrochemically produced cyclohexadienone derivatives have also been reported to engage in [4+2]642–648 and [5+2]649–652 cycloadditions.653 Alternatively, incorporation of a pending nucleophile into the phenol unit allows the design of various types of intramolecular reaction manifolds for construction of spirodienone derivatives.654–661 This feature has been leveraged in the synthesis of aeroplysinin (Scheme 85),662–665 discorhabdin C (Scheme 86),666–669 gymnastatin A (Scheme 87)670 as well as heliannuol E (Scheme 88)671–673 and derivatives thereof.674
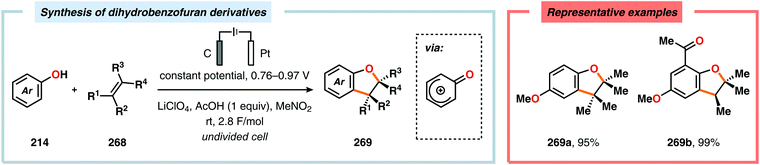 |
| | Scheme 84 Synthesis of dihydrobenzofuran derivatives via anodic intermolecular cycloaddition of phenols and unactivated alkenes. | |
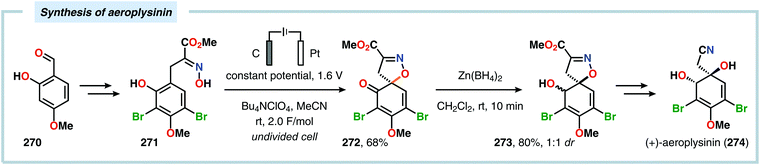 |
| | Scheme 85 Utilization of anodically generated phenoxonium ions in the synthesis of aeroplysinin. | |
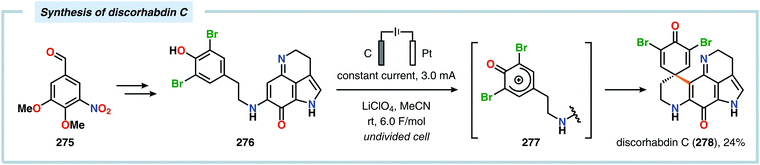 |
| | Scheme 86 Synthesis of discorhabdin C using a phenoxonium ion-based strategy. | |
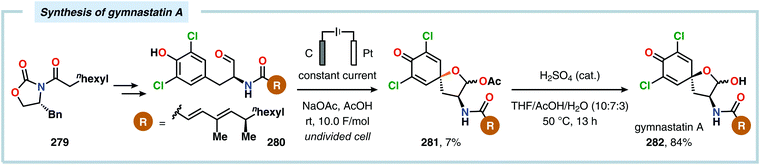 |
| | Scheme 87 Electrochemically-mediated spiro hemiacetal formation in the synthesis of gymnastatin A. | |
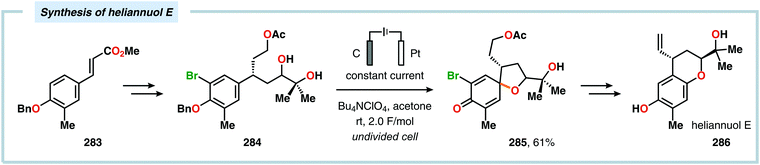 |
| | Scheme 88 Electrochemical production of a spirodienone in the synthesis of heliannuol E. | |
Catechols and related derivatives constitute another class of compounds that can undergo two-electron anodic oxidation to afford, for example, 1,2-benzoquinones.675–681 Considering their relatively low oxidation potentials, it is perhaps unsurprising that an assortment of nucleophiles are compatible with these processes without being subjected to oxidation. Here, amines,682–689 azide,690,691 enolates and other carbon nucleophiles,692–711 nitrate,712 sulfinates713–727 as well as thiols728–744 have been shown to engage in Michael additions with the anodically generated (imino)quinones. A variety of transformations for delivering annulated scaffolds have also been developed (Scheme 89). In the pursuit of this goal, a range of different benzofurans (291 and 293) have been accessed using 1,3-diketones,745–756 α-cyanoketones757 or chloranilic acid758 as the nucleophile. Additional scaffolds that have been synthesized are benzo[e][1,2,4]triazino[3,4-b][1,3,4]thiadiazine (300),759 benzoxazine (302),760,761 benzoxazole (304),762,763 coumestan (292),764,765 dihydrobenzo[e][1,2,4]triazine-3(2H)-thione (301),766 indole (294–296),767–769 phenazine (298),770,771 phenoxazine (303)772 tetrahydro-1H-benzo[b][1,4]diazepine (297)773 and thia-1,4a-diaza-fluorenone (299)774 derivatives. Furthermore, the electrochemically generated benzoquinone derivatives can also engage in [4+2] cycloaddition in the presence of cyclopentadiene,775,776 produce trimerization products777–779 or engage in transfer hydrogenation catalysis.780–782 A dual electro- and organocatalysis strategy was outlined by Jørgensen and coworkers, which enabled the enantioselective intermolecular α-arylation of aldehydes.783,784 Here, the combination of anodic oxidation and asymmetric enamine catalysis provided access to meta-alkylated anilines in good yields and excellent enantiomeric excess (Scheme 90).783
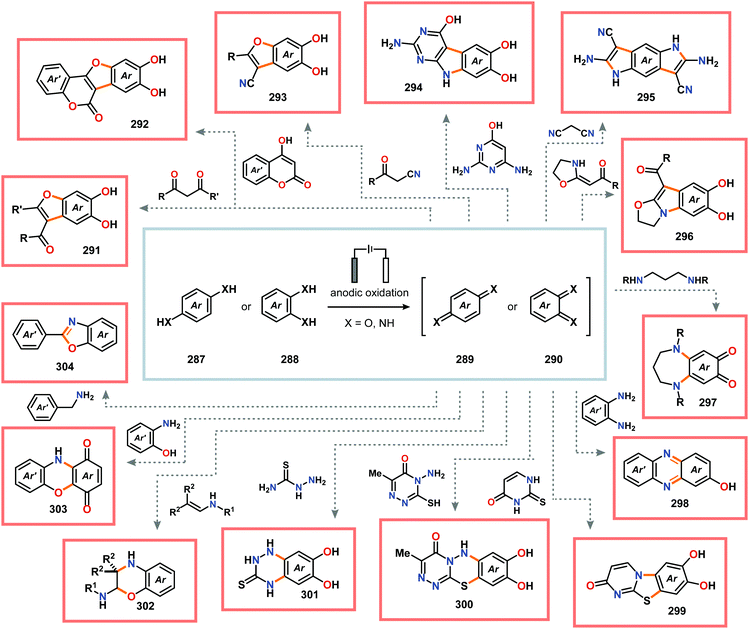 |
| | Scheme 89 Annulation of electrochemically generated benzoquinones and related derivatives. | |
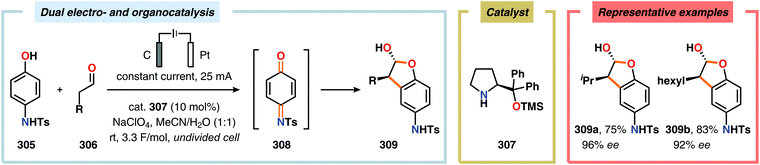 |
| | Scheme 90 α-Arylation of aldehydes through the merger of electrochemical oxidation and organocatalysis. | |
Several research groups have targeted the development of oxidative annulation protocols involving alkenes, and enol ethers or derivatives thereof. Based on earlier work that showed that enaminones785 can be anodically dimerized to yield 3,4-diketopyrroles,786,787 Schäfer and Eilenberg reported that N-benzyl- and β-phenethyl enaminones undergo intramolecular arylation to produce isoquinolines and benzazepines upon anodic oxidation using an undivided cell (Scheme 91).788 Lei and coworkers explored a related strategy for synthesis of indoles 313. The protocol relied on intramolecular dehydrogenative annulation of N-aryl enamines 312 and utilized iodide as a redox mediator.789 Mechanistic insight revealed that carrying out the annulation reaction in the absence of electricity but using N-iodosuccinimide (NIS) as an oxidant resulted in a high yield of the desired indole. Furthermore, radical inhibition experiments with TEMPO did not produce any product. Based on these results and previous observations,790 a mechanism involving a hyperiodide intermediate (I+) generated via anodic oxidation was proposed. The in situ produced I+ subsequently reacts with the N-aryl enamine to furnish an N-iodo intermediate (314), which upon N–I bond homolysis provides radical intermediate 315. Finally, intramolecular radical addition, oxidation and deprotonation afford the target product (Scheme 92).789,791,792
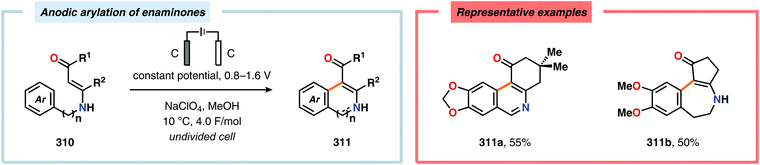 |
| | Scheme 91 Anodic intramolecular arylation of enaminones. | |
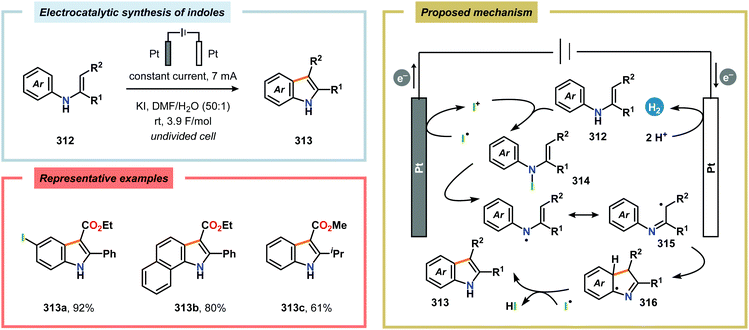 |
| | Scheme 92 Electrocatalytic synthesis of indoles using iodide as a redox mediator. | |
Pioneering studies by the Moeller group have demonstrated that alkenes, such as enol ethers, and electron-rich (hetero)arenes can be oxidatively coupled under electrochemical conditions. In these transformations, an alkene that is normally nucleophilic is initially oxidized at the anode to form a radical cation intermediate that is subsequently trapped by a second nucleophile to deliver anti-Markovnikov addition products (vide infra). Here, electron transfer confers umpolung reactivity of a functional group, thus enabling alternative retrosynthetic disconnections of complex structures through previously challenging bond disconnections.63,81 The Moeller793–795 and Wright796 groups have exploited the intramolecular anodic coupling of enol ethers or vinyl sulfides and electron-rich aromatic systems to deliver the fused, bicyclic or tricyclic ring skeletons in moderate to high yields (Schemes 93 and 94). A related approach was also utilized by the Wright group for the synthesis of the hamigeran skeleton (Scheme 95).797 Heteroaromatic rings are also compatible coupling partners in these intramolecular anodic olefin coupling reactions. Here, different tethers can be employed to afford, for example, six- (Scheme 96)794,796,798,799 or seven-membered800 (Scheme 97) annulated furan systems, which can be readily transformed into various carbo- and oxacyclic motifs.801–807 These electrochemical annulations are presumed to involve initial oxidation of the silyl enol ether moiety rather than the furyl group.800 Additional heterocycles that have been utilized consist of imidazoles,81 pyrroles794 as well as thiophenes.808–810 The applicability of the anodic olefin coupling reaction between silyl enol ethers and furans has also been exploited in syntheses of natural products. Moeller and coworkers employed the methodology as the key step in their synthesis of alliacol A65,66 (Scheme 98) while an anodic coupling strategy enabled the construction of the central seven-membered ring in Trauner's synthesis of guanacastepene E (Scheme 99).67,68 Furthermore, the arteannuin ring skeleton has been assembled through the use of a pair of anodic coupling reactions (Scheme 100). Here, both coupling reactions exploited the furan moiety as the coupling partner in which the first cyclization made use of a chiral N,O-ketene acetal initiating group to provide the desired bicyclic product in high yield and the second anodic coupling afforded the tricyclic core while generating the required quaternary stereocenter.811
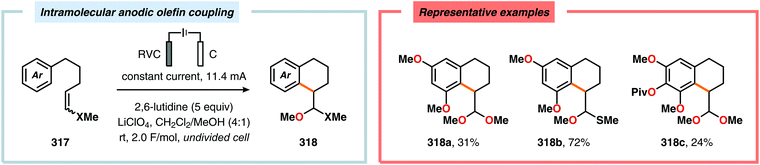 |
| | Scheme 93 Intramolecular anodic olefin coupling. | |
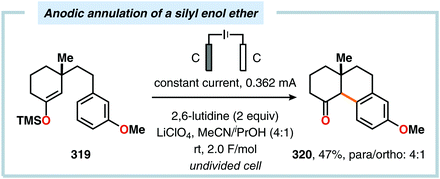 |
| | Scheme 94 Anodic annulation of a silyl enol ether and an electron-rich arene. | |
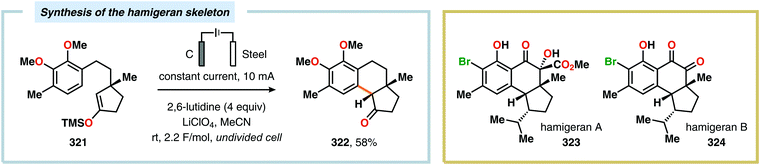 |
| | Scheme 95 Synthesis of the hamigeran skeleton through anodic coupling with an electron-rich arene. | |
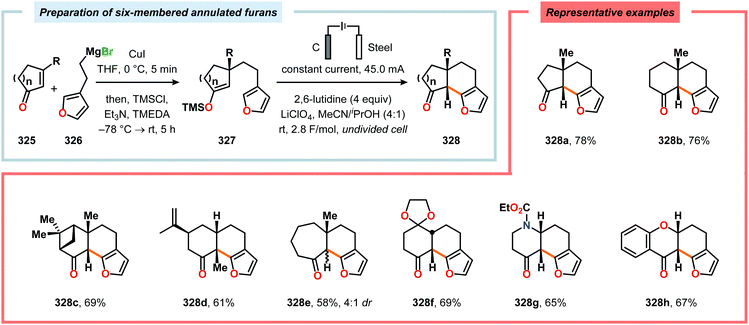 |
| | Scheme 96 Electrochemical coupling of silyl enol ethers and furans for synthesis of six-membered annulated furans. Yields are reported for two steps based on the starting enone as the limiting agent. | |
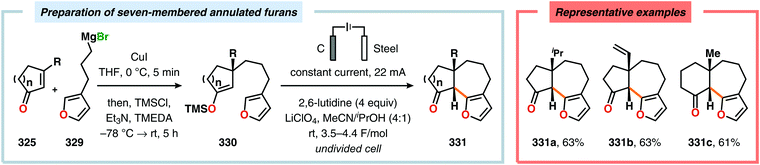 |
| | Scheme 97 Anodic coupling for synthesis of seven-membered annulated furans. Yields are reported for two steps based on the starting enone as the limiting agent. | |
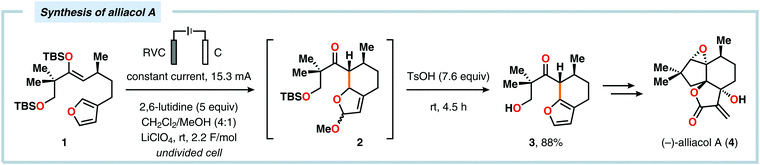 |
| | Scheme 98 Application of the anodic olefin coupling in the synthesis of (−)-alliacol A. | |
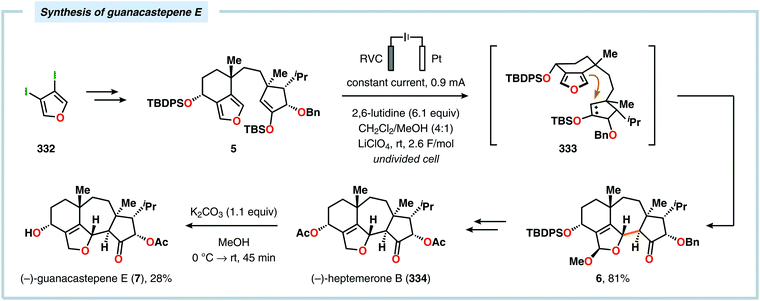 |
| | Scheme 99 Utilization of the anodic olefin coupling in the synthesis of (−)-guanacastepene E. | |
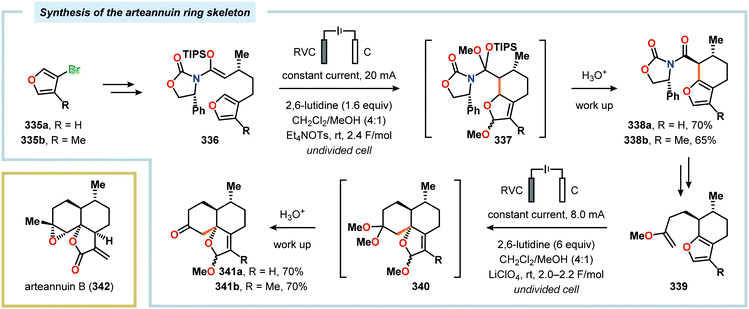 |
| | Scheme 100 Sequential anodic couplings for synthesis of the arteannuin ring skeleton. | |
Enol ether radical cations are also prone to undergo intermolecular [2+2] and [4+2] cycloaddition with another alkene or diene. Upon intermolecular C–C bond formation, the inclusion of an electron-rich arene ring in either the enol ether or alkene moiety facilitates intramolecular electron transfer from the aryl ring to form the final cycloadduct.812–822 The arene radical cation subsequently generates a second equivalent of the enol ether radical cation through intermolecular electron transfer.823 A characteristic feature of these “redox-tag”824 mediated cycloaddition reactions is that only small amounts of electricity are needed to initiate the radical chain process.825
3. Electrochemical approaches to carbon–nitrogen (C–N) bond formation
Nitrogen-containing compounds constitute essential motifs and are present in natural products, pharmaceutical agents, agrochemicals and material science. Chemists have actively targeted the development of facile and selective C–N bond formation platforms. However, despite the recent advances in C–N cross-coupling reactions, these approaches normally require elevated temperatures, prefunctionalized starting materials, such as aryl halides or pseudohalides, stoichiometric oxidants or the use of expensive catalysts. In this context, methods for direct C–H amination have emerged as efficient, step- and atom-economical alternatives, and have expanded the repertoire of reaction manifolds.826,827 In the following sections, the development of electrochemical methodologies for construction of C–N bonds and nitrogen-containing motifs is examined.
3.1. Amination of alkenes and alkynes
Considerable efforts have been devoted to the development of methods that facilitate the addition of, for example, N–H (hydroamination) and arene C–H bonds (hydroarylation) across alkenes and alkynes. Here, atom-economical and regioselective platforms for direct functionalization of double or triple bonds allow access to a range of functionalized products, thereby offering considerable opportunities for strategic chemical synthesis.828–848
In addition to the intramolecular anodic olefin coupling reactions discussed in the previous section, other carbon-849–862 as well as oxygen-based863–875 nucleophiles have also been utilized in radical cation-initiated cyclization protocols for constructing C–C and C–O bonds, respectively. Furthermore, Moeller and coworkers have designed systems for direct functionalization of alkenes using nitrogen functionalities as trapping groups. It was demonstrated that the nature of the substituents on the generated radical cation intermediate had a profound influence on its ability to react with various trapping groups.876 Thus, more polarized radical cations tend to favor C–C bond forming reactions, while less polarized radical cations are beneficial when using heteroatomic trapping groups. Employing more basic reaction conditions also had a dramatic influence on the cyclization reactions. Here, the use of a strong base, such as LiOMe, was considered to increase the nucleophilicity of the nitrogen trapping group by facilitating formation of the sulfonamide anion (Scheme 101). Although five-membered ring systems were accessible in a relatively straightforward fashion, coupling reactions leading to six-membered rings presented problems due to competing elimination of a proton from the carbon-center next to the radical cation intermediate.877,878 Ensuing mechanistic studies aimed at examining the relative reactivity of olefin-based radical cations toward various intramolecular nucleophiles. To address this, an electron-rich olefin tethered to two different nucleophiles/trapping groups, an alcohol and a sulfonamide, was exploited. Trapping of the formed radical cation with the alcohol moiety was shown to be facile and reversible, affording the kinetically favored product. In contrast, sulfonamide cyclization was demonstrated to furnish the thermodynamically favored product and can be enhanced with nonpolar reaction conditions, elevated temperatures, and low current. In these reactions, the normally nucleophilic olefin functionality is initially oxidized to a radical cation intermediate. This intermediate is subsequently trapped by the sulfonamide anion to form the desired C–N bond. However, a mechanistic portrayal involving initial anodic oxidation of the sulfonamide anion to produce a nitrogen-centered radical that is subsequently reduced through intramolecular electron transfer from the olefin unit to afford an olefinic radical cation cannot be ruled out for certain substrates.879–881 The Moeller group has also employed photovoltaic cells in which sunlight is harnessed as the power source for driving anodic olefin cyclization reactions.882 Following on from these reports, the Moeller group disclosed a potentially more practical approach that took advantage of unprotected amines as the trapping group (Scheme 102). Although the method employed a dithioketene acetal group as the electron-rich olefin, the oxidation potential of the secondary amine moiety in the product is lower than that of either functional group in the starting material, suggesting that overoxidation of the product should be of concern. However, this reasoning does not take into consideration that rapid cyclization reactions can lower the oxidation potential of the substrate. Hence, in the developed protocol the intramolecular cyclization is so rapid that it causes a 460 mV drop in the oxidation potential of substrate 348. This effect is so dramatic that the substrate potential is now significantly lower than that of the product, highlighting that a simple analysis of oxidation potentials for isolated functional groups can be deceiving when attempting to predict the success of an oxidative cyclization reaction.883 Yoshida and coworkers have demonstrated that anodic cyclization and chemical oxidation manifolds can be successfully integrated in a one-pot, sequential manner (Scheme 103). Here, anodic oxidation of the olefinic bond is followed by intramolecular cyclization by the tethered nucleophilic moiety to ultimately afford a carbocation 354 upon a subsequent one-electron oxidation. In the presence of DMSO as an external nucleophile, efficient trapping of intermediate 354 occurs to provide the corresponding alkoxysulfonium ion 355. This intermediate is sufficiently stable at low temperatures and subsequent treatment with Et3N gives a sulfur ylide (356) that can undergo an intramolecular proton transfer event, resulting in elimination of dimethylsulfide and the exocyclic ketone 351.467,884
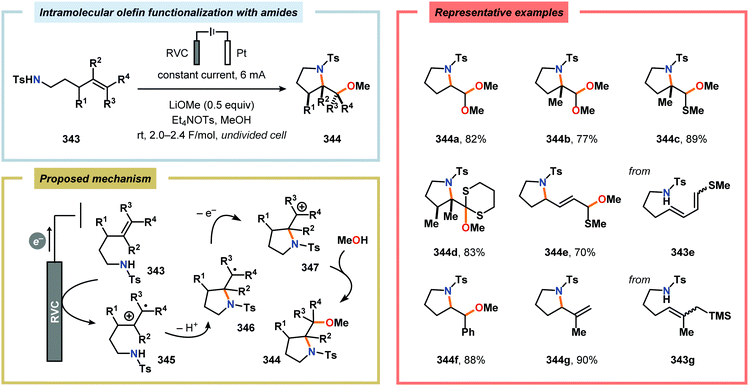 |
| | Scheme 101 Intramolecular anodic olefin coupling reactions with sulfonamides. | |
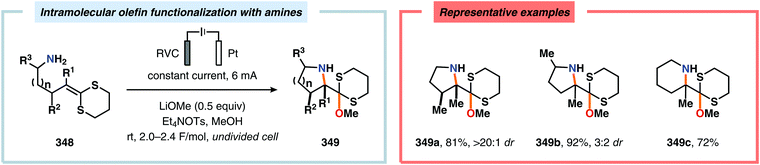 |
| | Scheme 102 Anodic coupling of amines and dithioketene acetals. | |
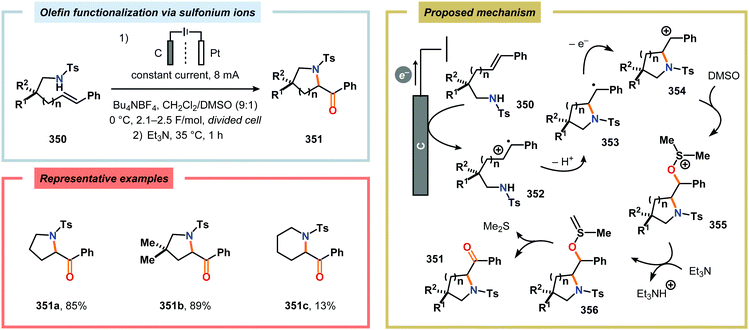 |
| | Scheme 103 Integrated electrooxidative cyclization followed by chemical oxidation of alkenes via alkoxysulfonium ions. | |
Recently, the Lin laboratory presented an electrochemical protocol for the diazidation of alkenes. The reported operationally simple and environmentally friendly method converted alkenes and NaN3 to 1,2-diazides (Scheme 104). The merger of electrochemical oxidation and an earth-abundant manganese catalyst enabled the transformation to proceed under mild reaction conditions. The system exhibited a broad substrate scope and high functional group compatibility. For example, substituents typically susceptible to nucleophilic displacement by N3−, such as epoxides, esters, alkyl halides, were tolerated by the metal-catalyzed electrochemical diazidation strategy. Furthermore, the authors showed that several 1,2-diazides, including those that contain reductively labile groups, could be chemoselectively converted to the corresponding 1,2-diamines. This can be performed consecutively and circumvents elaborate isolation of any intermediates, providing a general and operationally simple alternative for synthesis of vicinal diamine. Finally, mechanistic insight through radical clock experiments confirmed the intermediacy of radical adduct 358.885–888 Based on this platform, the Lin group subsequently reported a manganese-catalyzed electrochemical protocol for dichlorination889 as well as chlorotrifluoromethylation890 of alkenes.891 Alkyne annulations are typically limited to high reaction temperatures and/or the use of stoichiometric amounts of toxic metals as sacrificial oxidants. Recently, the Ackermann892 and Lei893 groups independently reported methods for cobalt-catalyzed electrochemical C–H/N–H annulation of alkynes, such as ethyne, as well as ethylene (Schemes 105 and 106). The cobalt-catalyzed electrochemical strategies allowed for efficient C–H functionalization of ortho-, meta- and para-substituted amides, and aryl halides while obviating undesired coupling reactions. Subsequently, the two groups independently disclosed methods for electrochemical cobalt-catalyzed C–H amination of arenes with alkylamines.894,895 Furthermore, a ruthenium-catalyzed electrochemical dehydrogenative annulation reaction of aniline derivatives and alkynes for the synthesis of indoles was recently described.896 Here, the electric current was proposed to promote reoxidation of the active ruthenium-based catalyst and facilitate H2 evolution.
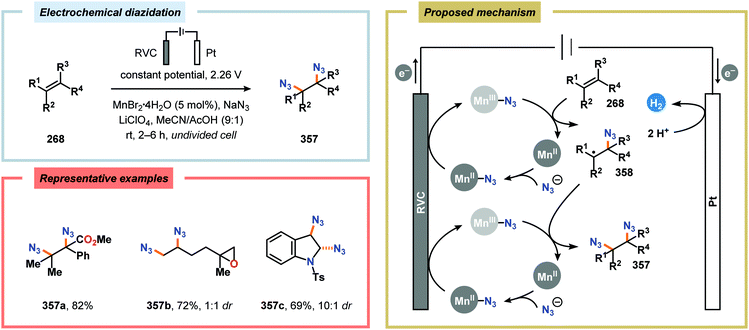 |
| | Scheme 104 Metal-catalyzed electrochemical diazidation of alkenes. | |
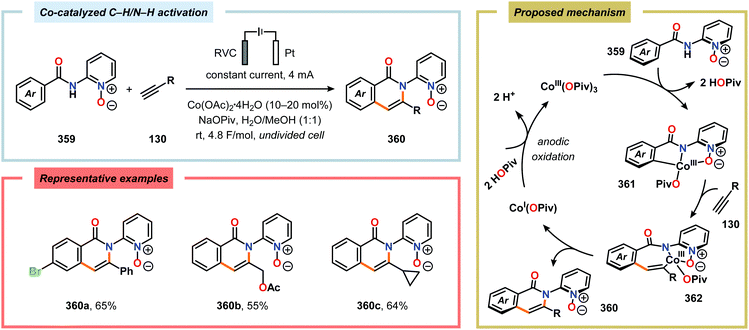 |
| | Scheme 105 Cobalt-catalyzed electrochemical C–H/N–H annulation of alkynes. | |
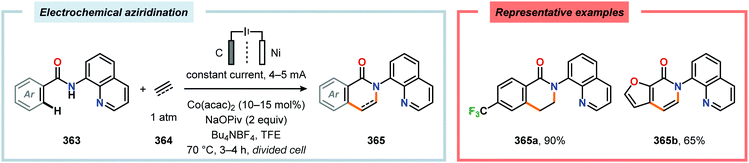 |
| | Scheme 106 Cobalt-catalyzed electrochemical C–H/N–H annulation of ethylene or ethyne. | |
3.2. Electrochemical generation of nitrogen-centered radicals
The term nitrogen-centered radical refers to species where the generated radical is localized on a nitrogen atom. In contrast to carbon-centered radicals, nitrogen-centered radicals have historically been relatively underutilized from a synthetic viewpoint. This was mainly due to a lack of mild and reliable methods for generating nitrogen-centered radicals, which constrained their applicability in academic and industrial settings.897–900 However, strategies based on nitrogen-centered radicals have recently been shown to expedite C–N bond construction and have therefore received considerable attention from the scientific community. Nitrogen-centered radicals have, for example, been demonstrated to add to alkenes, alkynes, dienes and engage in C–H functionalization, making them versatile intermediates in contemporary organic synthesis.13m,901–908
Early work from the Yudin group demonstrated that electrochemical aziridination of olefins could be achieved in the presence of N-aminophthalimide (366), a reaction that typically is hampered by the use of stoichiometric amounts of toxic PbIV oxidants. In this reaction, nitrene-transfer from N-aminophthalimide to olefins occurs under mild conditions on platinum electrodes (Scheme 107). Using the developed approach, both electron-rich and electron-poor olefins were effectively converted to aziridines in good to excellent yields. Although some olefins displayed similar oxidation potentials as the nitrene-transfer reagent, the crucial factor that accounts for the high levels of chemoselectivity in the transformations is the overpotential. It was noted that the nature of the electrode material was found to be critical for the reaction to proceed. For example, replacing the platinum electrodes with carbon-based ones resulted in termination of the nitrene-transfer reactions. Thus, under particular conditions various substrates possess different overpotentials depending on, for example, the electrode material. This highlights that reactive species can be selectively generated by maximizing the difference in overpotentials between the substrate and the reagent, thereby avoiding detrimental background reactions.909–911 Likewise, under similar reaction conditions a variety of sulfoxides could be chemoselectively converted into the corresponding sulfoximines.912
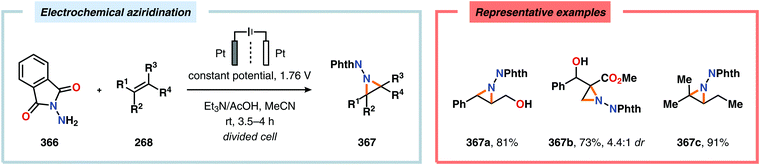 |
| | Scheme 107 Electrochemical aziridination of olefins. | |
The Moeller group developed a protocol for synthesis of γ- and δ-lactams using O-benzyl hydroxamates or N-phenyl amides.913 Here, the anodically generated nitrogen-centered amidyl radicals 371 underwent cyclization reactions with electron-rich olefins to form carbon-centered radicals 372 that are subsequently oxidized to furnish carbocations 373. These carbocations can then be trapped with nucleophiles, such as MeOH, to provide the target lactams 369 (Scheme 108). In general, ketene dithioacetal motifs were found to be the most effective coupling partners for the developed cyclization strategy. While the O-benzyl hydroxamate derivatives efficiently underwent 6-exo-trig radical cyclization to produce the six-membered ring products, the related N-phenyl amides produced a complex mixture of products. Furthermore, attempts to induce 7-exo-trig radical cyclizations were unsuccessful and instead resulted in dimerization to afford the corresponding hydrazide. Thus, the success of these amidyl radical cyclization reactions is influenced by the energetics of the cyclization relative to the competing pathways, such as radical–radical dimerization and hydrogen atom abstraction. Further advances in this field were made by Xu and coworkers and enabled the intramolecular oxidative amination of tri- and tetrasubstituted alkenes (Scheme 109).914 A wide range of amide, carbamate and urea derivatives carrying various trisubstituted or sterically demanding tetrasubstituted alkenyl moieties were all viable substrates. The protocol was compatible with a variety of (hetero)aryl-, alkyl- and alkynyl-substituted olefins and a range of functional groups were tolerated, including alcohols, alkynes, aryl bromides, esters, pyridines, silyl ethers, thiazoles and thiophenes. Upon radical cyclization, proton elimination occurred regioselectively at the distal carbon center, providing the cyclized products 375 in good to high diastereoselectivity. Alternatively, the generated carbon-centered radical can also engage in hydrogen atom abstraction to provide hydroamination products915via ferrocene-mediated generation of amidyl radicals (Scheme 110) or be trapped with N-oxyl radicals, such as TEMPO, to give oxyamination products (Scheme 111).916 The latter reaction has also been conducted using an electrochemical flow microreactor (Scheme 112), making the process less costly and enabling easier purification.917 Here, TEMPO has a dual function, being the mediator as well as the oxygen source.
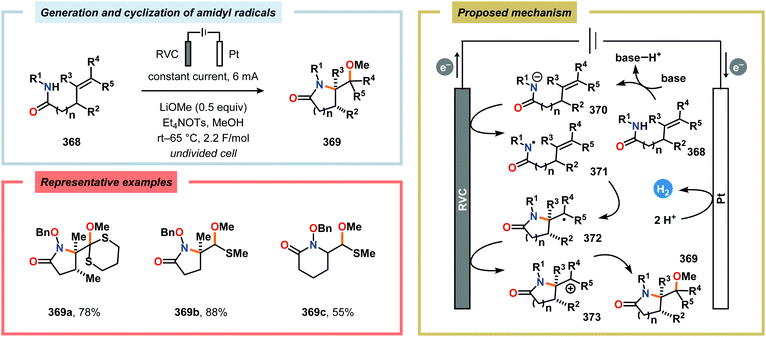 |
| | Scheme 108 Electrochemical generation of amidyl radicals for synthesis of γ- and δ-lactams. | |
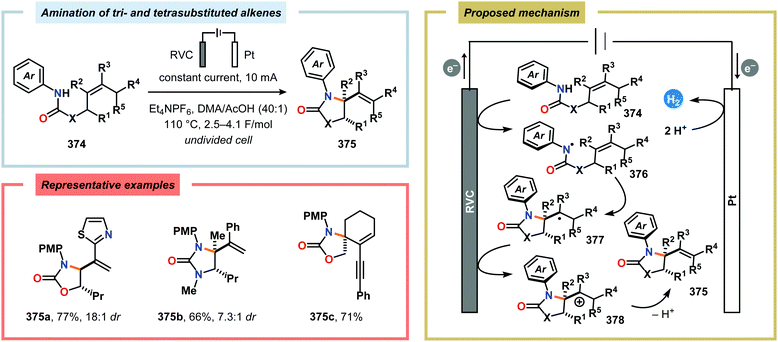 |
| | Scheme 109 Oxidative amination of tri- and tetrasubstituted alkenes. | |
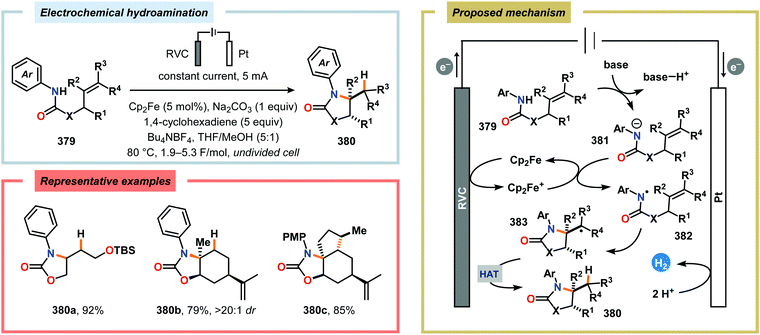 |
| | Scheme 110 Electrochemical hydroamination of alkenes via ferrocene-mediated generation of amidyl radicals. | |
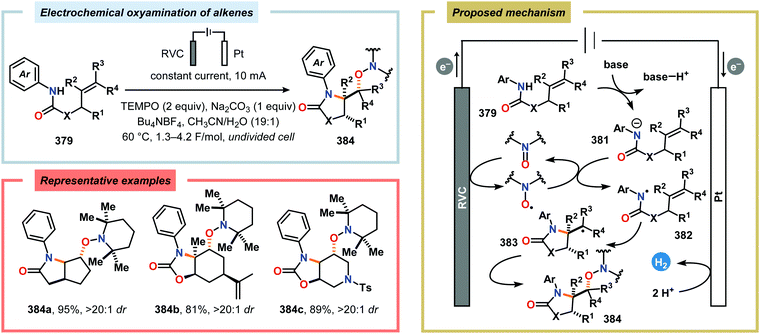 |
| | Scheme 111 Electrochemical oxyamination of alkenes via TEMPO-mediated generation of amidyl radicals. | |
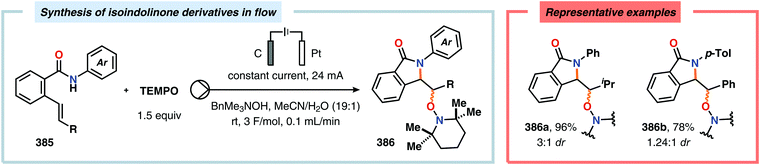 |
| | Scheme 112 Synthesis of isoindolinones using an electrochemical flow microreactor. | |
The Waldvogel and Moeller groups have exploited the electrochemical generation of amidyl radicals from anilides, enabling the direct synthesis of benzoxazoles (Scheme 113).918 Although methanol has been commonly applied in electrochemical transformations involving amidyl radicals, this proved to be incompatible, leading to degradation of anilides 387. This observation suggested that the intramolecular cyclization process was slow. Of the screened solvents, only HFIP was found to efficiently stabilize the electrochemically generated radical intermediates, affording the desired benzoxazoles 389 in good to high yields. The authors also demonstrated that the aromatic moiety connected to the produced amidyl radicals was of substantial radical nature as the para-position of the N-substituted arene ring had to be substituted in order to prevent coupling reactions at this position.
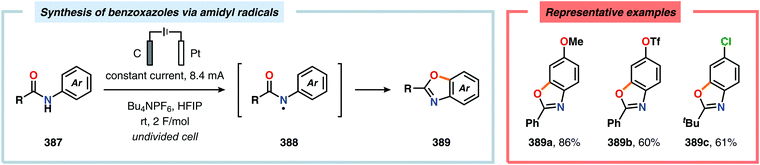 |
| | Scheme 113 Electrochemical synthesis of benzoxazoles from anilides. | |
The Xu group has recently made significant contributions through the design of distinct reaction manifolds that capitalize on nitrogen-centered radicals. Various mediator-based radical cyclization manifolds using (hetero)arylamines containing tethered alkynes have been developed. In this context, functionalized (aza)indoles 391 were accessed through an oxidative alkyne annulation reaction (Scheme 114).919,920 The broad functional group tolerance of the electrochemical protocol was illustrated by the preparation of (aza)indoles housing assorted substituents, such as acetals, alcohols, aldehydes, N-aryl carbamates, dipeptides, halides, ketones, (ortho)esters and sulfonamides. The reaction is believed to be initiated with the anodic oxidation of [Cp2Fe] to [Cp2Fe]+. Subsequent SET between [Cp2Fe]+ and the deprotonated urea derivative results in regeneration of [Cp2Fe] and furnishes a nitrogen-centered radical. This radical is suggested to engage in 6-exo-dig cyclization, which is followed by a second cyclization with the aryl moiety. Finally, rearomatization of the generated arene radical through oxidation and loss of a proton provides (aza)indoles 391. A related mediator-based radical cyclization cascade approach was applied to the preparation of imidazo-fused N-heteroaromatic compounds (395), including imidazo[1,2-a]pyridines, imidazo[1,2-b]pyridazines and imidazo[1,2-a]pyrazines (Scheme 115).921,922 However, here tetraarylhydrazine 393 was found to be the optimal redox catalyst. Nitrogen-centered radicals have also been harnessed for synthesis of nitrogen-doped polycyclic aromatic hydrocarbons (Scheme 116).923–925 In the presence of ferrocene, the urea-tethered diynes 396 underwent efficient radical cyclization to afford a variety of electron-rich polycyclic aromatic hydrocarbons (398). The use of a mild redox catalyst to facilitate the cascade cyclization instead of carrying out direct electrolysis was crucial to avoid overoxidation of the products as the diynes 396 are oxidized at a higher potential than that of products 398. Strategies relying on direct electrolysis for generation of nitrogen-centered radicals have also been achieved. Based on this concept, amidinyl and iminyl radicals have been exploited for preparing polycyclic imidazole derivatives926,927 (Scheme 117) and assorted pyridine-fused aromatic motifs928 (Scheme 118), respectively. In addition to using nitrogen-centered radicals in C–N bond coupling manifolds, nitrogen–nitrogen (N–N) bond formation has also been accomplished. These intermediates have, for example, been explored for N–N dimerization929 in the synthesis of dixiamycin B,930,931 phthalazin-1,4-diones932 and pyrazolidin-3,5-diones.933,934
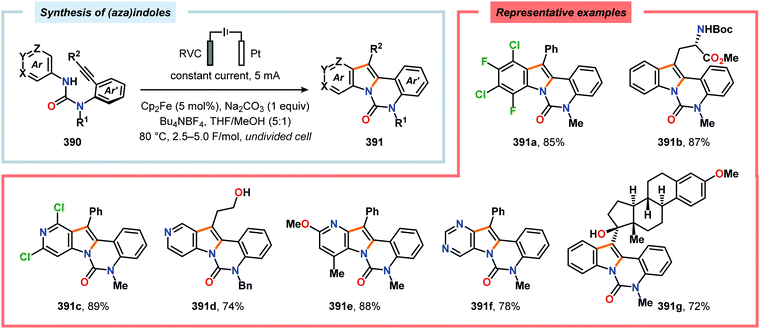 |
| | Scheme 114 Synthesis of (aza)indoles using ferrocene as mediator. | |
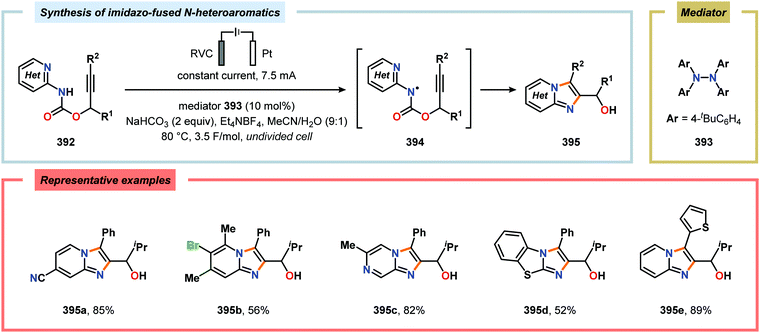 |
| | Scheme 115 Synthesis of imidazo-fused N-heteroaromatic compounds through a radical cyclization cascade. | |
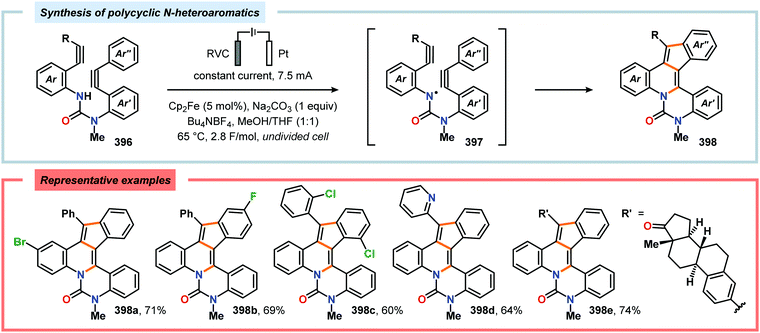 |
| | Scheme 116 Electrochemical synthesis of nitrogen-doped polycyclic aromatic hydrocarbons through radical cyclization of diynes. | |
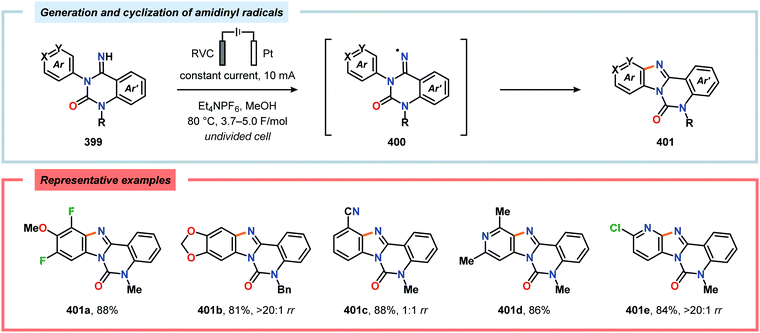 |
| | Scheme 117 Electrochemical generation of amidinyl radicals for synthesis of tetracyclic benzimidazoles and pyridoimidazoles. | |
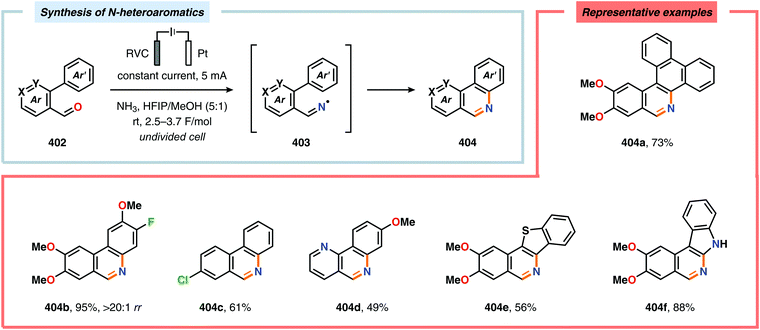 |
| | Scheme 118 Synthesis of pyridine-fused polycyclic aromatic motifs via iminyl radicals. | |
3.3. Pyridine and related nitrogen-containing heterocycles as nitrogen sources
Seminal work by Lund during the 1950s highlighted that aromatic hydrocarbons could be oxidized at a Pt electrode in acetonitrile solutions containing pyridine and NaClO4 to afford the corresponding pyridinium perchlorate species.935 Although related electrochemical amination reactions were studied by other research groups,936–943 it took more than five decades until Yoshida and coworkers disclosed an efficient and practical method for C–H amination of aromatic compounds based on electrochemical oxidation of aromatic compounds in the presence of pyridine (Scheme 119).56,944,945 Conducting the electrochemical oxidation in an H-type divided cell at ambient temperature furnished N-arylpyridinium ions (406), so called Zincke intermediates,946–948 which upon treatment with an alkylamine resulted in primary amines 407.56 Here, the high oxidation potential of pyridine enables selective oxidation of the aromatic compound in the presence of pyridine. Furthermore, the high nucleophilicity of pyridine allows for efficient trapping of the generated arene radical cation and the introduction of multiple amino groups is avoided due to the strong electron-withdrawing effect of the pyridinium moiety in 406, thereby suppressing overoxidation. However, one drawback was that only electron-rich and activated substrates, such as anisole derivatives, were viable substrates. Subsequently, the Waldvogel group demonstrated that the use of BDD anodes allowed for amination of less activated substrates949 as well as diamination.950 Furthermore, a protocol for the anodic C–H amination of phenoxy acetates was shown to provide efficient access to 1,4-benzoxazin-3-ones (Scheme 120).951 Alternatively, rendering the C–H amination intramolecular through the use of arenes containing a tethered 2-pyrimidyl moiety provided access to 2-aminobenzoxazoles and 2-aminobenzothiazoles (Scheme 121).952,953
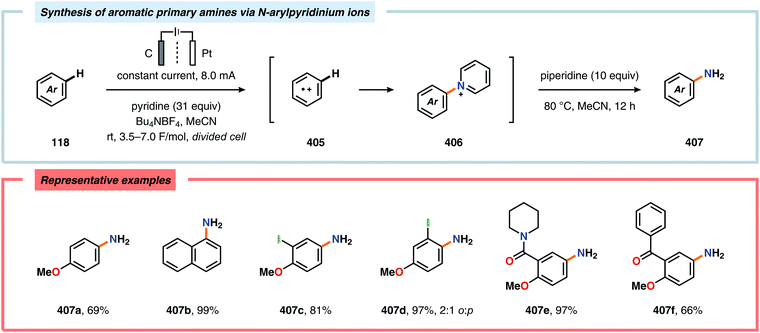 |
| | Scheme 119 Electrochemical synthesis of aromatic primary amines via N-arylpyridinium ions. | |
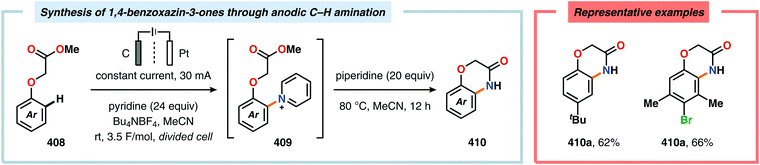 |
| | Scheme 120 Synthesis of 1,4-benzoxazin-3-ones via anodic C–H amination. | |
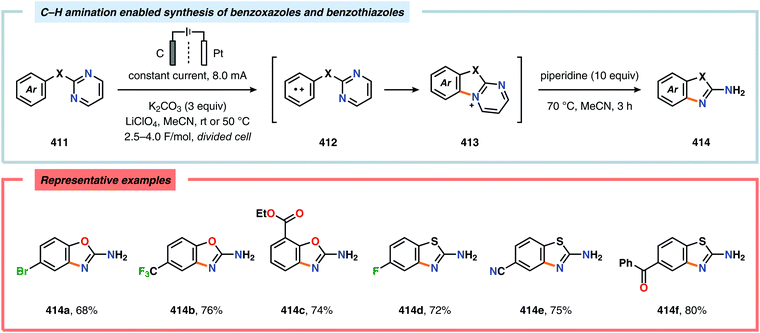 |
| | Scheme 121 Preparation of 2-aminobenzoxazoles and 2-aminobenzothiazoles via electrochemical intramolecular C–H amination. | |
Work on expanding the scope of the nitrogen nucleophiles revealed that N-protected imidazole derivatives were suitable trapping agents. The choice of an appropriate protecting group was crucial to the success of the developed C–H amination method. After an initial examination of various protecting groups, the use of N-methylsulfonyl (Ms) protected imidazole afforded the C–N coupled product in the highest yield. The developed protocol was applicable not only to aromatic but also to benzylic C–H amination. Here, the initial N-aryl or N-benzylimidazolium cations can be subjected to non-oxidative removal of the protecting group, resulting in cleavage of the sulfonyl protecting group to produce the corresponding N-aryl or N-benzylimidazoles (Scheme 122). Similar to N-arylpyridinium ions 406, the intermediate imidazolium ions 416 and 417 are electrochemically inactive, thus preventing overoxidation and allow these intermediates to be converted to the corresponding imidazole products 418 and 419.954–956 Subsequently, the Yoshida group reported a mechanistically similar reaction for the coupling of aromatics and masked primary alkyl amines bearing additional oxygen or nitrogen functionalities (Scheme 123). The masked primary amines 420 were prepared by treatment of the amines with nitriles or derivatives thereof to afford the target five- or six-membered heterocycle. Under electrochemical conditions, these heterocycles undergo coupling with arene radical cations to give cationic intermediates 421, which can be chemically converted to the desired cross-coupling products 422 under non-oxidative conditions.957 The same group also developed an electrochemical approach for C–H amination of toluene derivatives employing N-tosyldiphenylsulfilimine 423 as the amination reagent (Scheme 124). This enabled the preparation of N-tosylbenzylaminosulfonium ions 424, which upon treatment with an iodide source, such as Bu4NI, under non-electrolytic conditions results in N–S bond cleavage to yield the corresponding N-tosylbenzylamines 425.958 Alternatively, the generated benzylaminosulfonium ions 424 can react with added aromatic nucleophiles to give the corresponding cross-coupling products 426 (Scheme 125).959 In these transformations, the high oxidation potential of N-tosyldiphenylsulfilimine 423 allows a wide range of toluene derivatives to be oxidized and functionalized. Finally, Lei and coworkers have developed an electrochemical strategy for amination of C(sp3)–H bonds. Here, C(sp3)–H bonds adjacent to heteroatoms, such as nitrogen, oxygen and sulfur, could be functionalized with various amines to give the corresponding C–N coupled products 429 in moderate to good yields (Scheme 126).960,961
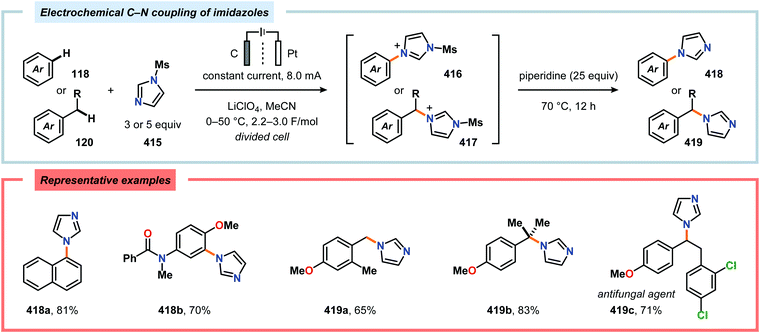 |
| | Scheme 122 Direct C–N coupling of imidazoles with aromatic and benzylic compounds. | |
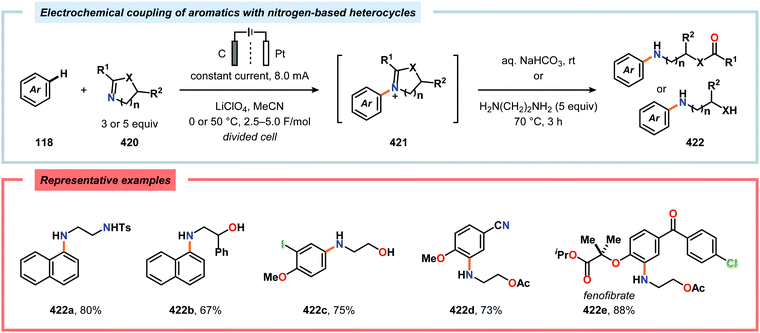 |
| | Scheme 123 C–N bond coupling of aromatic compounds and nitrogen-containing heterocycles. | |
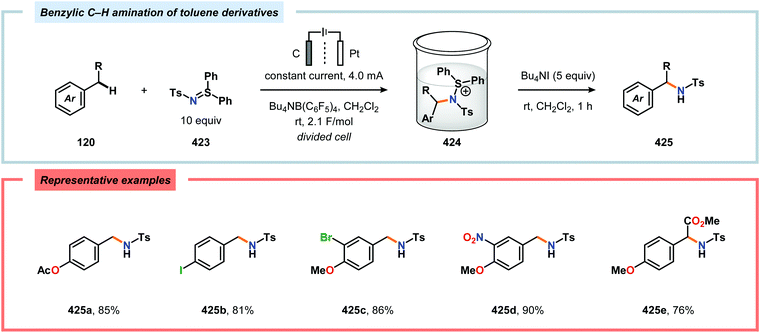 |
| | Scheme 124 C–H amination via electrochemically generated benzylaminosulfonium ions. | |
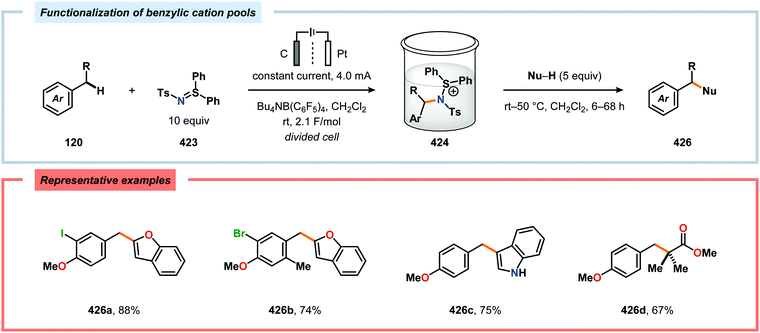 |
| | Scheme 125 Benzylic cross-coupling via electrochemically generated benzylaminosulfonium ions. | |
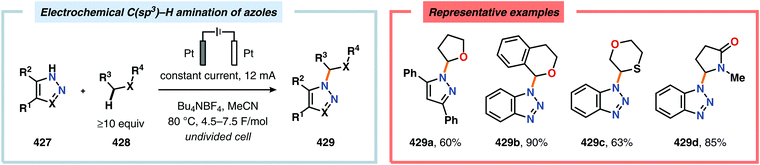 |
| | Scheme 126 Electrochemical C(sp3)–H amination of azoles with heterocycles. | |
3.4. Halide-mediated approaches to C–N bond coupling
Hypervalent iodine compounds have been extensively employed in organic synthesis as selective metal-free oxidants and environmentally friendly reagents. Synthetic applications of these versatile reagents include, for example, aminations, C–C bond-forming reaction manifolds, halogenations, oxidations, rearrangements, and various dual catalytic activation modes.962–969 In this context, hypervalent iodine species electrochemically generated from the corresponding iodoarenes have been applied for the synthesis of various heterocyclic frameworks, such as carbazoles970 (Scheme 127), pyrroloindoles,971 quinolinone derivatives972 (Scheme 128) and spirocycles (Scheme 129).973,974 The utility of electrochemically generated hypervalent iodine reagents has also been demonstrated in the synthesis of glycozoline970 and tetrahydropyrroloiminoquinone alkaloids.975,976 The supporting electrolyte has also been merged with the iodine mediator through tethering of the redox-active iodophenyl moiety to an alkylammonium moiety. The ionically tagged iodophenyl motif was applied to several oxidative C–N bond coupling scenarios and allowed for straightforward recovery and reuse of the ionic mediator.977,978 A related approach was also employed for the synthesis of benzoxazoles.979 Additionally, various oxidative amination strategies for preparing 2-substituted benzoxazoles using iodide as a redox mediator have also been reported.980–982
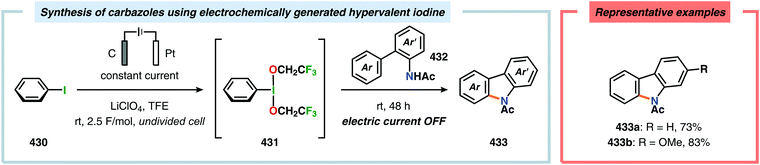 |
| | Scheme 127 Synthesis of carbazoles using an electrochemically generated hypervalent iodine oxidant. | |
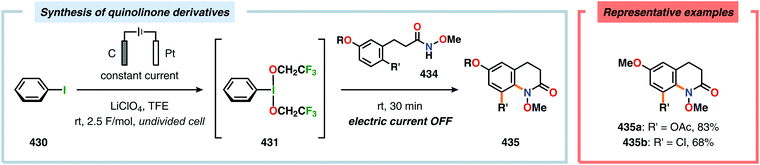 |
| | Scheme 128 Preparation of quinolinone derivatives through oxidative cyclization and concomitant rearrangement of the functional group. | |
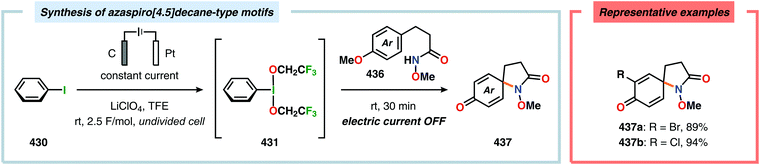 |
| | Scheme 129 Hypervalent iodine-mediated synthesis of azaspiro[4.5]decane-type motifs. | |
An electrochemical protocol for α-amination of ketones was recently reported using iodide as a redox catalyst (Scheme 130).983 Here, NH4I was found to be the optimal redox catalyst for facilitating the coupling of ketones and secondary amines at ambient temperature. The reactions were conveniently carried out in a simple undivided beaker-type cell and were believed to proceed via initial α-iodination of the ketones (132) with the anodically generated I2, followed by nucleophilic substitution of the amines (143), thus avoiding isolation of the key α-iodo ketone intermediate 133. It was noted that the addition of radical scavangers, such as TEMPO, terminated the α-amination process and did not yield any product. Related halide-mediated procedures for cleavage of β-O-4 lignin model compounds,984–986 thiocyanation987 as well as synthesis of 3-amino-2-thiocyanato-α,β-unsaturated carbonyl derivatives988 and β-keto sulfones989 have also been developed.990
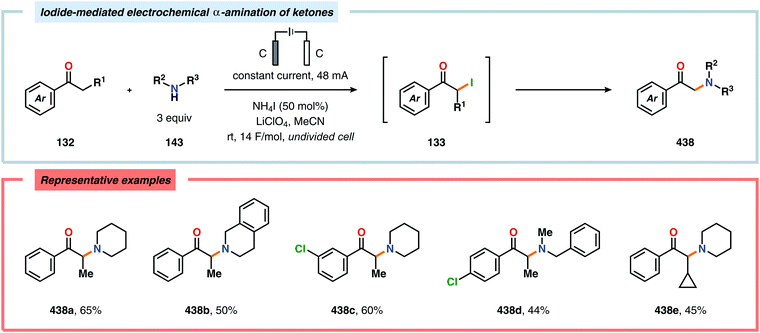 |
| | Scheme 130 Electrochemical α-amination of ketones using NH4I as redox catalyst. | |
Halide-mediated protocols for C–N bond coupling with alkenes have also been targeted. Zeng, Little and coworkers demonstrated an efficient approach for aziridination of alkenes Bu4NI as a redox catalyst and N-aminophthalimide (366) as the source of electrophilic nitrogen (cf. Scheme 107). The process was carried out at a constant current in an undivided cell and was proposed to follow a radical mechanism where hydrogen atom abstraction from N-aminophthalimide by I˙ generates a nitrogen-centered radical that is subsequently trapped by the alkene.991 Based on earlier precendent,992 the Zeng group presented an indirect electrochemical method for synthesis of vicinal iodoazides (Scheme 131). In the developed protocol, IN3 is electrochemically generated in situ, thus avoiding the use of external oxidants or corrosive I2. Furthermore, the reaction proceeds via a cyclic iodonium intermediate to afford regioselective azidoiodination products in a Markovnikov-type fashion.993 Finally, an electrochemical strategy for oxyamination of styrenes for accessing indoline derivatives was recently achieved (Scheme 132).994 The electrochemical reactions were conducted under constant current conditions in a simple undivided cell using Bu4NI as a redox catalyst. This paired electrolysis process avoids the use of external bases and oxidants, representing an appealing and environmentally benign route for synthesis of these heterocyclic compounds. Mechanistic insight suggested that the reaction commences with the anodic oxidation of iodide to generate I2, which subsequently reacts with alkene 442 to afford the corresponding iodonium intermediate 443. Finally, intramolecular cyclization with the pendant sulfonamide followed by nucleophilic substitution by alkoxide yields the indoline product 444. The examples discussed here highlight that halide ions are versatile mediators capable of promoting a collection of different transformations.
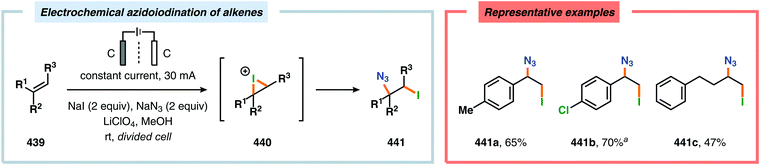 |
| | Scheme 131 Electrochemical regioselective azidoiodination of alkenes via in situ generation of IN3. aCarried out in MeOH/H2O (5:1). | |
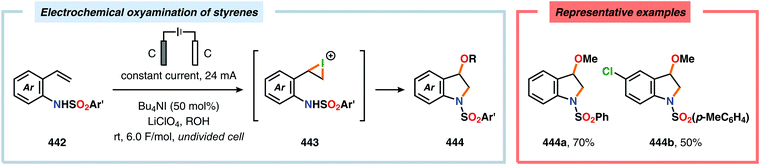 |
| | Scheme 132 Electrochemical oxyamination of styrenes for synthesis of indolines. | |
4. Conclusions and outlook
During the last few decades, the imperative to develop and adopt more sustainable and atom economical methodologies for synthetic purposes has not been overlooked by the scientific community. In this regard, electrochemistry serves as an appealing platform since only electrons serve as reagents,995 thereby minimizing production of reagent waste while allowing the reactions to be carried out at milder conditions.996,997 Organic electrochemistry can be classified into two categories, direct and indirect electrolysis. Here, the initial electron transfer between the electrode and a substrate to produce a radical intermediate does not generally pose a problem. Instead, it is crucial to control the reactive intermediates that are subsequently generated throughout the process. Although related activated species can be accessed using traditional organic reagents, there is a dramatic difference in the nature of these species.998 While the activated species are uniformly distributed throughout the solution in conventional organic reactions, these species are only produced at the surface of the electrode in electrochemical reactions. This difference in distribution of the reactive species will thus affect the chemical behavior as well as reactivity and selectivity.
For an unexperienced electrochemist it might seem that there are a wide range of experimental variables that need to be optimized, such as cell type, electrode composition and electrolyte.999 However, this should be seen as an opportunity for accessing unique selectivity and expediting unconventional reaction pathways for challenging bond construction, thus enabling novel and strategic bond disconnections in synthetic endeavors. Since organic chemists are rather unfamiliar with the concept of using electric current to facilitate chemical reactions, the purpose of the present review is to highlight the potential of applying organic electrochemistry to organic synthesis. Here, an overview of the development of electrosynthetic reaction manifolds for C–H bond functionalization and C–N bond formation is provided. The electrochemical strategies and methods discussed in this review clearly showcase the diversity of transformations that can be facilitated through organic electrosynthesis. Furthermore, the refinement and expansion of reaction engineering methods and wider commercial availability of electrochemical reactors provide a multifaceted and powerful toolbox for efficient generation of reactive intermediates from an ensemble of organic substrates. These include, among others, the development of dual catalytic platforms (Scheme 133),1000–1007 paired electrolysis1008–1012 (Scheme 134) and electrochemical flow cells,1013–1019 which have contributed to advancing the field of electrosynthesis. Based on these unique features and the current renaissance in synthetic electrochemistry, it is anticipated that this field will soon be considered as a reliable and versatile platform by the broader scientific community for nontraditional bond construction in organic chemistry.
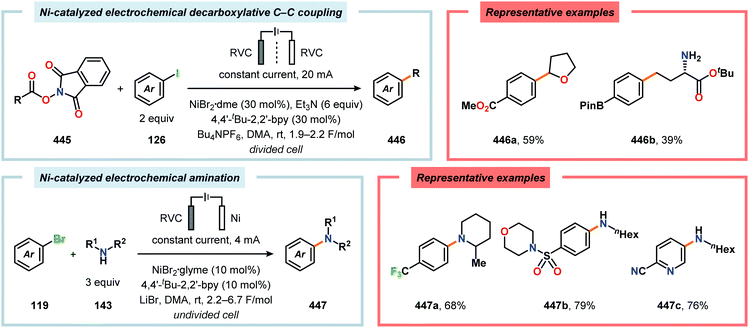 |
| | Scheme 133 Ni-catalyzed electrochemical coupling platforms. | |
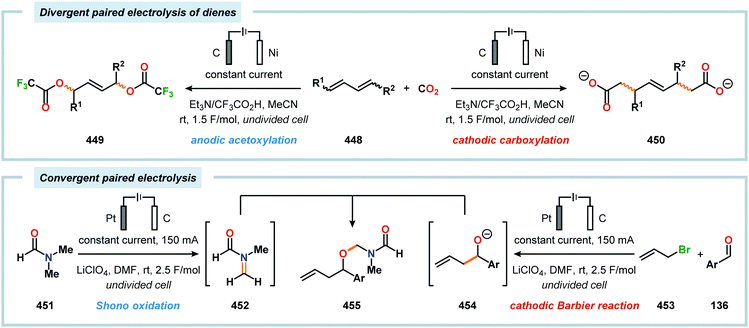 |
| | Scheme 134 Divergent and convergent paired electrolysis. | |
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Financial support from the Swedish Research Council (637-2013-7314) is gratefully acknowledged. The author would also like to thank Dr Bryan S. Matsuura (New York University, New York) for helpful discussions and feedback on this manuscript.
References
- G. M. Whitesides, Isr. J. Chem., 2018, 58, 142–150 CrossRef.
- R. Noyori, Nat. Chem., 2009, 1, 5–6 CrossRef PubMed.
- G. M. Whitesides, Angew. Chem., Int. Ed., 2015, 54, 3196–3209 CrossRef PubMed.
-
Organic chemistry – Breakthroughs and perspectives, ed. K. Ding and L.-X. Dai, Wiley-VCH Verlag & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
-
P. Wyatt and S. Warren, Organic synthesis: Strategy and control, John Wiley and Sons Ltd., Chichester, England, 2007 Search PubMed.
- For selected reviews, see:
(a) K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef;
(b) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef PubMed;
(c) I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193–205 CrossRef PubMed;
(d) K. C. Morrison and P. J. Hergenrother, Nat. Prod. Rep., 2014, 31, 6–14 RSC;
(e) X.-W. Li and B. Nay, Nat. Prod. Rep., 2014, 31, 533–549 RSC;
(f) A. M. Armaly, Y. C. DePorre, E. J. Groso, P. S. Riehl and C. S. Schindler, Chem. Rev., 2015, 115, 9232–9276 CrossRef PubMed;
(g) R. Ardkhean, D. F. J. Caputo, S. M. Morrow, H. Shi, Y. Xiong and E. A. Anderson, Chem. Soc. Rev., 2016, 45, 1557–1569 RSC;
(h) C. Cabrele and O. Reiser, J. Org. Chem., 2016, 81, 10109–10125 CrossRef PubMed.
- For selected reviews on the use of renewable chemical feedstocks, see:
(a) C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef PubMed;
(b) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed;
(c) G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC;
(d) A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 RSC;
(e) Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- For selected reviews on biocatalysis, see:
(a) U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef PubMed;
(b) M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef PubMed;
(c) M. Hönig, P. Sondermann, N. J. Turner and E. M. Carreira, Angew. Chem., Int. Ed., 2017, 56, 8942–8973 CrossRef PubMed;
(d) R. A. Sheldon and P. C. Pereira, Chem. Soc. Rev., 2017, 46, 2678–2691 RSC;
(e) F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef PubMed;
(f) R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef PubMed.
- For discussions and applications of base-metal catalysis, see:
(a) J. R. Ludwig and C. S. Schindler, Chem, 2017, 2, 313–316 CrossRef;
(b) A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed;
(c) J. E. Zweig, D. E. Kim and T. R. Newhouse, Chem. Rev., 2017, 117, 11680–11752 CrossRef PubMed;
(d) P. J. Chirik, Angew. Chem., Int. Ed., 2017, 56, 5170–5181 CrossRef PubMed.
- For selected reviews on cascade/tandem reactions in organic synthesis, see:
(a) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef PubMed;
(b) K. C. Nicolaou and J. S. Chen, Chem. Soc. Rev., 2009, 38, 2993–3009 RSC;
(c) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef PubMed;
(d) Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- Q.-L. Zhou, Angew. Chem., Int. Ed., 2016, 55, 5352–5353 CrossRef PubMed.
- B. König, Eur. J. Org. Chem., 2017, 1979–1981 CrossRef.
- For recent reviews on visible light photocatalysis, see:
(a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef PubMed;
(b) D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49, 2295–2306 CrossRef PubMed;
(c) M. D. Levin, S. Kim and F. D. Toste, ACS Cent. Sci., 2016, 2, 293–301 CrossRef PubMed;
(d) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef PubMed;
(e) E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef PubMed;
(f) T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937–1945 CrossRef PubMed;
(g) M. D. Kärkäs, J. A. Porco, Jr. and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed;
(h) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef PubMed;
(i) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef PubMed;
(j) N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC;
(k) J.-R. Chen, D.-M. Yan, Q. Wei and W.-J. Xiao, ChemPhotoChem, 2017, 1, 148–158 CrossRef;
(l) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef;
(m) M. D. Kärkäs, ACS Catal., 2017, 7, 4999–5022 CrossRef.
- K. D. Moeller, Electrochemistry, 2006, 74, 583 CrossRef.
- A. V. Shchepochkin, O. N. Chupakhin, V. N. Charushin and V. A. Petrosyan, Russ. Chem. Rev., 2013, 82, 747–771 CrossRef.
- V. V. Turygin and A. P. Tomilov, Russ. J. Electrochem., 2015, 51, 999–1020 CrossRef.
- K. Chiba and Y. Okada, Curr. Opin. Electrochem., 2017, 2, 53–59 CrossRef.
- The auxiliary electrode is the anode for a reduction and cathode for an oxidation reaction.
- O. R. Luca, J. L. Gustafson, S. M. Maddox, A. Q. Fenwick and D. C. Smith, Org. Chem. Front., 2015, 2, 823–848 RSC.
- C. Gütz, B. Klöckner and S. R. Waldvogel, Org. Process Res. Dev., 2016, 20, 26–32 CrossRef.
- For a review on electrode materials, see: R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef PubMed.
- For a review on electrochemical synthesis on boron-doped diamond electrode materials, see: S. R. Waldvogel and B. Elsler, Electrochim. Acta, 2012, 82, 434–443 CrossRef.
-
(a) J. Y. Becker, B. E. Smart and T. Fukunaga, J. Org. Chem., 1988, 53, 5714–5720 CrossRef;
(b) Y. Matsumura, M. Yamada, N. Kise and M. Fujiwara, Tetrahedron, 1995, 51, 6411–6418 CrossRef.
- K. Fujimoto, Y. Tokuda, H. Maekawa, Y. Matsubara, T. Mizuno and I. Nishiguchi, Tetrahedron, 1996, 52, 3889–3896 CrossRef.
- For reviews, see:
(a) H. Liu, Y. Liu and J. Li, Phys. Chem. Chem. Phys., 2010, 12, 1685–1697 RSC;
(b) M. Kathiresan and D. Velayutham, Chem. Commun., 2015, 51, 17499–17516 RSC;
(c) M. Alvarez-Guerra, J. Albo, E. Alvarez-Guerra and A. Irabien, Energy Environ. Sci., 2015, 8, 2574–2599 RSC . For selected examples, see;
(d) M. Mellah, J. Zeitouny, S. Gmouh, M. Vaultier and V. Jouikov, Electrochem. Commun., 2005, 7, 869–874 CrossRef;
(e) A. C. Herath and J. Y. Becker, Electrochim. Acta, 2008, 53, 4324–4330 CrossRef.
- For recent reviews, see:
(a) K. E. Toghill, M. A. Méndez and P. Voyame, Electrochem. Commun., 2014, 44, 27–30 CrossRef;
(b) J. A. Branch and P. N. Bartlett, Philos. Trans. R. Soc., A, 2015, 373, 20150007 CrossRef PubMed.
- L. Eberson and K. Nyberg, Tetrahedron, 1976, 32, 2185–2206 CrossRef.
- L. Eberson, J. Mol. Catal., 1983, 20, 27–52 CrossRef.
- For reviews on radical cations, see:
(a) A. Ledwith, Acc. Chem. Res., 1972, 5, 133–139 CrossRef;
(b) H. D. Roth, Acc. Chem. Res., 1987, 20, 343–350 CrossRef;
(c) N. L. Bauld, D. J. Bellville, B. Harirchian, K. T. Lorenz, R. A. Pabon, Jr., D. W. Reynolds, D. D. Wirth, H. S. Chiou and B. K. Marsh, Acc. Chem. Res., 1987, 20, 371–378 CrossRef;
(d) N. L. Bauld, Tetrahedron, 1989, 45, 5307–5363 CrossRef;
(e) A. Albini, E. Fasani and N. d'Alessandro, Coord. Chem. Rev., 1993, 125, 269–281 CrossRef;
(f) M. Schmittel and A. Burghart, Angew. Chem., Int. Ed. Engl., 1997, 36, 2550–2589 CrossRef;
(g) A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551–1594 CrossRef PubMed.
- For reviews on radical anions, see:
(a) L. M. Dorfman, Acc. Chem. Res., 1970, 3, 224–230 CrossRef;
(b) N. L. Holy and J. D. Marcum, Angew. Chem., Int. Ed. Engl., 1971, 10, 115–124 CrossRef;
(c) N. L. Holy, Chem. Rev., 1974, 74, 243–277 CrossRef;
(d) F. Gerson and W. Huber, Acc. Chem. Res., 1987, 20, 85–90 CrossRef.
- For reviews on the applications of radical reactions in organic synthesis, see:
(a) E. Godineau and Y. Landais, Chem. – Eur. J., 2009, 15, 3044–3055 CrossRef PubMed;
(b) L. J. Sebren, J. J. Devery III and C. R. J. Stephenson, ACS Catal., 2014, 4, 703–716 CrossRef PubMed;
(c) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef PubMed;
(d) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef PubMed;
(e) A. J. Clark, Eur. J. Org. Chem., 2016, 2231–2243 CrossRef;
(f) H. Miyabe, A. Kawashima, E. Yoshioka and S. Kohtani, Chem. – Eur. J., 2017, 23, 6225–6236 CrossRef PubMed;
(g) M. P. Plesniak, H.-M. Huang and D. J. Procter, Nat. Rev. Chem., 2017, 1, 0077 CrossRef;
(h) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef PubMed;
(i) H. Miyabe, Eur. J. Org. Chem., 2017, 3302–3310 CrossRef.
-
A. J. Fry, Synthetic organic electrochemistry, John Wiley & Sons, New York, 1989 Search PubMed.
- Throughout this review redox potentials are given in volts (V) versus the saturated calomel electrode (SCE). Occasionally the referenced potentials have been determined using different electrodes and have been converted to SCE for the purpose of comparison. For conversion constants, see: V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef.
- M. Faraday, Ann. Phys., 1834, 109, 433–451 CrossRef.
-
(a) H. Kolbe, Ann. Chem. Pharm., 1848, 64, 339–341 CrossRef;
(b) H. Kolbe, Ann. Chem. Pharm., 1849, 69, 257–294 CrossRef.
- For reviews on the Kolbe electrolysis, see:
(a) A. K. Vijh and B. E. Conway, Chem. Rev., 1967, 67, 623–664 CrossRef;
(b) H.-J. Schäfer, Top. Curr. Chem., 1990, 152, 91–151 CrossRef;
(c) H. J. Schäfer, Chem. Phys. Lipids, 1979, 24, 321–333 CrossRef.
- For synthetic applications of the Kolbe electrolysis, see:
(a) J. Knolle and H. J. Schäfer, Angew. Chem., Int. Ed. Engl., 1975, 14, 758 CrossRef;
(b) H. Klünenberg and H. J. Schäfer, Angew. Chem., Int. Ed. Engl., 1978, 17, 47–48 CrossRef;
(c) U. Jensen and H. J. Schäfer, Chem. Ber., 1981, 114, 292–297 CrossRef;
(d) L. Becking and H. J. Schäfer, Tetrahedron Lett., 1988, 29, 2797–2800 CrossRef;
(e) J. Weiguny and H. J. Schäfer, Liebigs Ann. Chem., 1994, 235–242 CrossRef;
(f) A. Matzeit, H. J. Schäfer and C. Amatore, Synthesis, 1995, 1432–1444 CrossRef.
- D. Pletcher, Rev. Port. Quim., 1985, 27, 449–458 Search PubMed.
- C. A. C. Sequeira and D. M. F. Santos, J. Braz. Chem. Soc., 2009, 20, 387–406 CrossRef.
- D. S. P. Cardoso, B. Šljukić, D. M. F. Santos and C. A. C. Sequeira, Org. Process Res. Dev., 2017, 21, 1213–1226 CrossRef.
- For a discussion on the historical advances in organic electrochemistry, see: H. Lund, J. Electrochem. Soc., 2002, 149, S21–S33 CrossRef.
- For reviews and applications of indirect electrolysis reactions, see:
(a) E. Steckhan, Angew. Chem., Int. Ed. Engl., 1986, 28, 683–701 CrossRef;
(b) E. Steckhan, Top. Curr. Chem., 1987, 142, 1–69 CrossRef;
(c) Y. N. Ogibin, M. N. Elinson and G. I. Nikishin, Russ. Chem. Rev., 2009, 78, 89–140 CrossRef;
(d) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
-
F. Fichter, Organische Elektrochemie, Steinkopf, Dresden, 1942, pp. 13 and 75 Search PubMed.
-
C. Jackson and A. T. Kuhn, Industrial electrochemical processes, Elsevier, Amsterdam, 1971, pp. 513–519 Search PubMed.
- For early examples, see:
(a) E. T. Seo, R. F. Nelson, J. M. Fritsch, L. S. Marcoux, D. W. Leedy and R. N. Adams, J. Am. Chem. Soc., 1966, 88, 3498–3503 CrossRef;
(b) L. Papouchado, R. N. Adams and S. W. Feldberg, J. Electroanal. Chem. Interfacial Electrochem., 1969, 21, 408–410 CrossRef;
(c) W. Schmidt and E. Steckhan, J. Electroanal. Chem., 1978, 89, 215–220 CrossRef;
(d) W. Schmidt and E. Steckhan, Angew. Chem., Int. Ed. Engl., 1978, 17, 673–674 CrossRef.
- For seminal work, see: M. F. Semmelhack, C. S. Chou and D. A. Cortes, J. Am. Chem. Soc., 1983, 105, 4492–4494 CrossRef.
- For recent applications of N-oxyl radicals as redox mediators, see:
(a) M. Rafiee, K. C. Miles and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 14751–14757 CrossRef PubMed;
(b) D. P. Hickey, D. A. Schiedler, I. Matanovic, P. V. Doan, P. Atanassov, S. D. Minteer and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 16179–16186 CrossRef PubMed;
(c) I. Bosque, G. Magallanes, M. Rigoulet, M. D. Kärkäs and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef PubMed;
(d) T. Li, Y. Cao, J. He and C. P. Berlinguette, ACS Cent. Sci., 2017, 3, 778–783 CrossRef PubMed.
- By definition, overpotential is the additional potential (beyond the thermodynamic requirement) needed to drive a reaction at a certain rate.
- For seminal work, see:
(a) J.-i. Yoshida, T. Murata and S. Isoe, Tetrahedron Lett., 1986, 27, 3373–3376 CrossRef;
(b) J.-i. Yoshida and S. Isoe, Tetrahedron Lett., 1987, 28, 6621–6624 CrossRef.
- For reviews on the use of electroauxiliaries, see:
(a) J.-i. Yoshida and K. Nishiwaki, J. Chem. Soc., Dalton Trans., 1998, 2589–2596 RSC;
(b) J.-i. Yoshida, Top. Curr. Chem., 1994, 170, 39–81 CrossRef.
- For representative examples, see:
(a) F. Tang and K. D. Moeller, Tetrahedron, 2009, 65, 10863–10875 CrossRef;
(b) J. M. Campbell, J. A. Smith, L. Gonzalez and K. D. Moeller, Tetrahedron Lett., 2015, 56, 3595–3599 CrossRef.
- For selected examples, see:
(a) J.-i. Yoshida, K. Takada, Y. Ishichi and S. Isoe, J. Chem. Soc., Chem. Commun., 1994, 2361–2362 RSC;
(b) M. Jinno, Y. Kitano, M. Tada and K. Chiba, Org. Lett., 1999, 1, 435–438 CrossRef;
(c) J.-i. Yoshida, M. Watanabe, H. Toshioka, M. Imagawa and S. Suga, J. Electroanal. Chem., 2001, 507, 55–65 CrossRef.
- For representative examples, see:
(a) S. Suga, M. Watanabe and J.-i. Yoshida, J. Am. Chem. Soc., 2002, 124, 14824–14825 CrossRef PubMed;
(b) D. Kesselring, K. Maurer and K. D. Moeller, Org. Lett., 2008, 10, 2501–2504 CrossRef PubMed.
- For selected examples, see:
(a) J.-i. Yoshida, M. Sugawara and N. Kise, Tetrahedron Lett., 1996, 37, 3157–3160 CrossRef;
(b) S. Suzuki, K. Matsumoto, K. Kawamura, S. Suga and J.-i. Yoshida, Org. Lett., 2004, 6, 3755–3758 CrossRef PubMed.
- A. Kirste, B. Elsler, G. Schnakenburg and S. R. Waldvogel, J. Am. Chem. Soc., 2012, 134, 3571–3576 CrossRef PubMed.
- T. Morofuji, A. Shimizu and J.-i. Yoshida, J. Am. Chem. Soc., 2013, 135, 5000–5003 CrossRef PubMed.
- For reviews on the “cation pool” method, see:
(a) J.-i. Yoshida and S. Suga, Chem. – Eur. J., 2002, 8, 2650–2658 CrossRef;
(b) K. Matsumoto, S. Suga and J.-i. Yoshida, Org. Biomol. Chem., 2011, 9, 2586–2596 RSC;
(c) J.-i. Yoshida, A. Shimizu, Y. Ashikari, T. Morofuji, R. Hayashi, T. Nokami and A. Nagaki, Bull. Chem. Soc. Jpn., 2015, 88, 763–775 CrossRef;
(d) J.-i. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702–4730 CrossRef PubMed.
- For a review on the preparation of “glycosyl triflate pools”, see: T. Nokami, K. Saito and J.-i. Yoshida, Carbohydr. Res., 2012, 363, 1–6 CrossRef PubMed.
- For a review discussing the sustainability of organic electrosynthesis, see: B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 Search PubMed.
- J. Utley, Chem. Soc. Rev., 1997, 26, 157–167 RSC.
- R. Francke, Beilstein J. Org. Chem., 2014, 10, 2858–2873 CrossRef PubMed.
- For representative reviews on umpolung reactions, see:
(a) D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239–258 CrossRef;
(b) A. B. Smith, III and C. M. Adams, Acc. Chem. Res., 2004, 37, 365–377 CrossRef PubMed;
(c) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef PubMed;
(d) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC;
(e) J. Streuff, Synthesis, 2013, 281–307 CrossRef;
(f) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef PubMed.
- For reviews on electrochemical umpolung reactions, see:
(a) R. D. Little and K. D. Moeller, Electrochem. Soc. Interface, 2002, 11, 36–42 Search PubMed;
(b) F. Tang, C. Chen and K. D. Moeller, Synthesis, 2007, 3411–3420 Search PubMed;
(c) K. D. Moeller, Synlett, 2009, 1208–1218 CrossRef.
- For synthetic applications, see:
(a) D. L. Wright, C. R. Whitehead, E. H. Sessions, I. Ghiviriga and D. A. Frey, Org. Lett., 1999, 1, 1535–1538 CrossRef PubMed;
(b) S. Duan and K. D. Moeller, Org. Lett., 2001, 3, 2685–2688 CrossRef PubMed.
- J. Mihelcic and K. D. Moeller, J. Am. Chem. Soc., 2003, 125, 36–37 CrossRef PubMed.
- J. Mihelcic and K. D. Moeller, J. Am. Chem. Soc., 2004, 126, 9106–9111 CrossRef PubMed.
- C. C. Hughes, A. K. Miller and D. Trauner, Org. Lett., 2005, 7, 3425–3428 CrossRef PubMed.
- A. K. Miller, C. C. Hughes, J. J. Kennedy-Smith, S. N. Gradl and D. Trauner, J. Am. Chem. Soc., 2006, 128, 17057–17062 CrossRef PubMed.
- For a detailed discussion on the ideal electrochemical device, see: M. Yan, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 4149–4155 CrossRef PubMed.
- For a review on elucidating the mechanisms of transition metal-catalyzed reactions using electrochemistry, see: A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef PubMed.
- S. Wawzonek, Science, 1967, 155, 39–44 CrossRef PubMed.
- N. L. Weinberg and H. R. Weinberg, Chem. Rev., 1968, 68, 449–523 CrossRef.
- R. N. Adams, Acc. Chem. Res., 1969, 2, 175–180 CrossRef.
- L. Eberson and K. Nyberg, Acc. Chem. Res., 1973, 6, 106–112 CrossRef.
- T. Shono, Tetrahedron, 1984, 40, 811–850 CrossRef.
- K. A. Ogawa and A. J. Boydston, Chem. Lett., 2015, 44, 10–16 CrossRef.
- K. Chiba and S. Kim, Electrochemistry, 2009, 77, 21–29 CrossRef.
- K. D. Moeller, Top. Curr. Chem., 1997, 185, 49–86 CrossRef.
- K. D. Moeller, Tetrahedron, 2000, 56, 9527–9554 CrossRef.
- K. D. Moeller, Electrochem. Soc. Interface, 2016, 25, 53–59 CrossRef.
- R. Feng, J. A. Smith and K. D. Moeller, Acc. Chem. Res., 2017, 50, 2346–2352 CrossRef PubMed.
- H. J. Schäfer, M. Harenbrock, E. Klocke, M. Plate and A. Weiper-Idelmann, Pure Appl. Chem., 2007, 79, 2047–2057 Search PubMed.
- H. J. Schäfer, ChemCatChem, 2014, 6, 2792–2795 CrossRef.
- H. J. Schäfer, Angew. Chem., Int. Ed. Engl., 1981, 20, 911–934 CrossRef.
- H. J. Schäfer, C. R. Chim., 2011, 14, 745–765 CrossRef.
- J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605–621 RSC.
- J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef PubMed.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef PubMed.
- During the preparation of this review the following review articles on organic electrochemistry were published:
(a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef PubMed;
(b) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef PubMed;
(c) K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef PubMed;
(d) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef PubMed;
(e) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed;
(f) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef.
- For recent reviews on C–H activation/functionalization, see:
(a) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC;
(b) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef;
(c) X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef PubMed;
(d) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef PubMed;
(e) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef PubMed;
(f) M. Parasram and V. Gevorgyan, Acc. Chem. Res., 2017, 50, 2038–2053 CrossRef PubMed;
(g) R. Das, G. S. Kumar and M. Kapur, Eur. J. Org. Chem., 2017, 5439–5459 CrossRef;
(h) M. T. Mihai, G. R. Genov and R. J. Phipps, Chem. Soc. Rev., 2018, 47, 149–171 RSC.
- For a review on late stage functionalization of drug-like molecules, see: T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- T. Shono, H. Hamaguchi and Y. Matsumura, J. Am. Chem. Soc., 1975, 97, 4264–4268 CrossRef.
- T. Shono, Y. Matsumura, K. Tsubata, Y. Sugihara, S.-i. Yamane, T. Kanazawa and T. Aoki, J. Am. Chem. Soc., 1982, 104, 6697–6703 CrossRef.
- For early contributions, see:
(a) S. D. Ross, M. Finkelstein and R. C. Petersen, J. Org. Chem., 1966, 31, 128–133 CrossRef;
(b) S. D. Ross, M. Finkelstein and R. C. Petersen, J. Am. Chem. Soc., 1966, 88, 4657–4660 CrossRef.
- For reviews on Shono-type anodic oxidations, see:
(a) T. Shono, Top. Curr. Chem., 1988, 148, 131–162 CrossRef;
(b) A. M. Jones and C. E. Banks, Beilstein J. Org. Chem., 2014, 10, 3056–3072 CrossRef PubMed.
- P. Alfonso-Súarez, A. V. Kolliopoulos, J. P. Smith, C. E. Banks and A. M. Jones, Tetrahedron Lett., 2015, 56, 6863–6867 CrossRef.
- P. D. Palasz, J. H. P. Utley and J. D. Hardstone, J. Chem. Soc., Perkin Trans. 2, 1984, 807–813 RSC.
- T. Shone, Y. Matsumura, K. Uchida and H. Kobayashi, J. Org. Chem., 1985, 50, 3243–3245 CrossRef.
- T. Shono, Y. Matsumura and K. Tsubata, Org. Synth., 1985, 63, 206–211 CrossRef.
- T. Shono, Y. Matsumura and K. Inoue, J. Org. Chem., 1983, 48, 1388–1389 CrossRef.
- M. Thaning and L.-G. Wistrand, Acta Chem. Scand., 1989, 43, 290–295 CrossRef.
- K.-D. Ginzel, P. Brungs and E. Steckhan, Tetrahedron, 1989, 45, 1691–1701 CrossRef.
- K. D. Moeller, P. W. Wang, S. Tarazi, M. R. Marzabadi and P. L. Wong, J. Org. Chem., 1991, 56, 1058–1067 CrossRef.
- M. Thaning and L.-G. Wistrand, Acta Chem. Scand., 1992, 46, 194–199 CrossRef.
- G. Butora, J. W. Reed, T. Hudlicky, L. E. Brammer, Jr., P. I. Higgs, D. P. Simmons and N. E. Heard, J. Am. Chem. Soc., 1997, 119, 7694–7701 CrossRef.
- M. K. S. Vink, C. A. Schortinghuis, J. Luten, J. H. van Maarseveen, H. E. Schoemaker, H. Hiemstra and F. P. J. T. Rutjes, J. Org. Chem., 2002, 67, 7869–7871 CrossRef PubMed.
- M. David and H. Dhimane, Synlett, 2004, 1029–1033 Search PubMed.
- M. R. Faust, G. Höfner, J. Pabel and K. T. Wanner, Eur. J. Med. Chem., 2010, 45, 2453–2466 CrossRef PubMed.
- T. Golub and J. Y. Becker, Electrochim. Acta, 2015, 173, 408–415 CrossRef.
- For a review on strategies for the direct α-functionalization of saturated cyclic amines, see: E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem. – Eur. J., 2012, 18, 10092–10142 CrossRef PubMed.
- T. Shono, Y. Matsumura, O. Onomura and M. Sato, J. Org. Chem., 1988, 53, 4118–4121 CrossRef.
- Y. Demizu, H. Shiigi, H. Mori, K. Matsumoto and O. Onomura, Tetrahedron: Asymmetry, 2008, 19, 2659–2665 CrossRef.
- X. Wang, J. Li, R. A. Saporito and N. Toyooka, Tetrahedron, 2013, 69, 10311–10315 CrossRef.
- S. S. Libendi, Y. Demizu, Y. Matsumura and O. Onomura, Tetrahedron, 2008, 64, 3935–3942 CrossRef.
- S. S. Libendi, Y. Demizu and O. Onomura, Org. Biomol. Chem., 2009, 7, 351–356 RSC.
- A. D. Tereshchenko, J. S. Myronchuk, L. D. Leitchenko, I. V. Knysh, G. O. Tokmakova, O. O. Litsis, A. Tolmachev, K. Liubchak and P. Mykhailiuk, Tetrahedron, 2017, 73, 750–757 CrossRef.
- Y. Cao, K. Suzuki, T. Tajima and T. Fuchigami, Tetrahedron, 2005, 61, 6854–6859 CrossRef.
- T. Shono, Y. Matsumura, K. Tsubata and J. Takata, Chem. Lett., 1981, 1121–1124 CrossRef.
- T. Shono, Y. Matsumura and K. Tsubata, Tetrahedron Lett., 1981, 22, 2411–2412 CrossRef.
- T. Shono, Y. Matsumura and K. Tsubata, J. Am. Chem. Soc., 1981, 103, 1172–1176 CrossRef.
- T. Shono, Y. Matsumura and K. Tsubata, Tetrahedron Lett., 1981, 22, 3249–3252 CrossRef.
- For synthetic applications of N-acyl iminium ions, see:
(a) B. E. Maryanoff, H.-C. Zhang, J. H. Cohen, I. J. Turchi and C. A. Maryanoff, Chem. Rev., 2004, 104, 1431–1628 CrossRef PubMed;
(b) P. Wu and T. E. Nielsen, Chem. Rev., 2017, 117, 7811–7856 CrossRef PubMed.
- A. Papadopoulos, J. Heyer, K.-D. Ginzel and E. Steckhan, Chem. Ber., 1989, 122, 2159–2164 CrossRef.
- A. Papadopoulos, B. Lewall, E. Steckhan, K.-D. Ginzel, F. Knoch and M. Nieger, Tetrahedron, 1991, 47, 563–572 CrossRef.
- G. Kardassis, P. Brungs and E. Steckhan, Tetrahedron, 1998, 54, 3471–3478 CrossRef.
- G. Kardassis, P. Brungs, C. Nothhelfer and E. Steckhan, Tetrahedron, 1998, 54, 3479–3488 CrossRef.
- Y. Kanda, O. Onomura, T. Maki and Y. Matsumura, Chirality, 2003, 15, 89–94 CrossRef PubMed.
- K. Schierle-Arndt, D. Kolter, K. Danielmeier and E. Steckhan, Eur. J. Org. Chem., 2001, 2425–2433 CrossRef.
- M. Lennartz, M. Sadakane and E. Steckhan, Tetrahedron, 1999, 55, 14407–14420 CrossRef.
- M. Lennartz and E. Steckhan, Synlett, 2000, 319–322 Search PubMed.
- M. Lennartz and E. Steckhan, Tetrahedron, 2001, 57, 675–680 CrossRef.
- D.-S. Lee, Tetrahedron: Asymmetry, 2009, 20, 2014–2020 CrossRef.
- M. G. M. D’Oca, R. A. Pilli and I. Vencato, Tetrahedron Lett., 2000, 41, 9709–9712 CrossRef.
- Y. Matsumura, Y. Kanda, K. Shirai, O. Onomura and T. Maki, Tetrahedron, 2000, 56, 7411–7422 CrossRef.
- M. G. M. D’Oca, R. A. Pilli, V. L. Pardini, D. Curi and F. C. M. Comninos, J. Braz. Chem. Soc., 2001, 12, 507–513 Search PubMed.
- M. Zelgert, M. Nieger, M. Lennartz and E. Steckhan, Tetrahedron, 2002, 58, 2641–2646 CrossRef.
- E. Sierecki, S. Turcaud, T. Martens and J. Royer, Synthesis, 2006, 3199–3208 Search PubMed.
- E. Sierecki, G. Errasti, T. Martens and J. Royer, Tetrahedron, 2010, 66, 10002–10007 CrossRef.
- S. Turcaud, T. Martens, E. Sierecki, J. Pérard-Viret and J. Royer, Tetrahedron Lett., 2005, 46, 5131–5134 CrossRef.
- M. Gong and J.-M. Huang, Chem. – Eur. J., 2016, 22, 14293–14296 CrossRef PubMed.
-
Organic electrochemistry, fourth edition, revised and expanded, ed. O. Hammerich and H. Lund, Marcel Dekker, Inc., New York, 4th edn, 2001 Search PubMed.
-
(a) B. Wermeckes and F. Beck, Electrochim. Acta, 1985, 30, 1491–1500 CrossRef;
(b) K. Danielmeier, K. Schierle and E. Steckhan, Tetrahedron, 1996, 52, 9743–9754 CrossRef;
(c) H. Senboku, K. Nagakura, T. Fukuhara and S. Hara, Tetrahedron, 2015, 71, 3850–3856 CrossRef.
- D. Baba and T. Fuchigami, Tetrahedron Lett., 2003, 44, 3133–3136 CrossRef.
- S. Andreades and E. W. Zahnow, J. Am. Chem. Soc., 1969, 91, 4181–4190 CrossRef.
- T. Chiba and Y. Takata, J. Org. Chem., 1977, 42, 2973–2977 CrossRef.
- I. Gallardo and N. Vilà, J. Org. Chem., 2008, 73, 6647–6656 CrossRef PubMed.
- An electrochemical approach to afford phosphinic amides has also been developed, see: Y. Wang, P. Qian, J.-H. Su, Y. Li, M. Bi, Z. Zha and Z. Wang, Green Chem., 2017, 19, 4769–4773 RSC.
- For early studies on electrochemical oxidation of aliphatic amines, see:
(a) R. F. Dapo and C. K. Mann, Anal. Chem., 1963, 35, 677–680 CrossRef;
(b) C. K. Mann, Anal. Chem., 1964, 36, 2424–2426 CrossRef;
(c) K. K. Barnes and C. K. Mann, J. Org. Chem., 1967, 32, 1474–1479 CrossRef;
(d) P. J. Smith and C. K. Mann, J. Org. Chem., 1968, 33, 316–317 CrossRef.
- I. Gallardo and N. Vilà, J. Org. Chem., 2010, 75, 680–689 CrossRef PubMed.
- O. Onomura, Y. Ishida, T. Maki, D. Minato, Y. Demizu and Y. Matsumura, Electrochemistry, 2006, 74, 645–648 CrossRef.
- V. H. Vu, F. Louafi, N. Girard, R. Marion, T. Roisnel, V. Dorcet and J.-P. Hurvois, J. Org. Chem., 2014, 79, 3358–3373 CrossRef PubMed.
- L. Zhang, J.-H. Su, S. Wang, C. Wan, Z. Zha, J. Du and Z. Wang, Chem. Commun., 2011, 47, 5488–5490 RSC.
- K. D. Moeller, S. Tarazi and M. R. Marzabadi, Tetrahedron Lett., 1989, 30, 1213–1216 CrossRef.
- K. D. Moeller, S. L. Rothfus and P. L. Wang, Tetrahedron, 1991, 47, 583–592 CrossRef.
- M. A. Endoma, G. Butora, C. D. Claeboe, T. Hudlicky and K. A. Abboud, Tetrahedron Lett., 1997, 38, 8833–8836 CrossRef.
- M. Okimoto, T. Yoshida, M. Hoshi, K. Hattori, M. Komata, K. Numata and K. Tomozawa, Heterocycles, 2006, 68, 2563–2570 CrossRef.
- M. Okimoto, T. Yoshida, M. Hoshi, K. Hattori, M. Komata, K. Numata and K. Tomozawa, Aust. J. Chem., 2007, 60, 236–242 CrossRef.
- M. Okimoto, K. Ohashi, H. Yamamori, S. Nishikawa, M. Hoshi and T. Yoshida, Synthesis, 2012, 1315–1322 CrossRef.
- For electrochemical oxidation of benzylic amines to imines, see: M. Okimoto, Y. Takahashi, K. Numata, Y. Nagata and G. Sasaki, Synth. Commun., 2005, 35, 1989–1995 CrossRef.
- For anodic cyanation of tetrahydroisoquinolines, see: F. Louafi, J.-P. Hurvois, A. Chibani and T. Roisnel, J. Org. Chem., 2010, 75, 5721–5724 CrossRef PubMed.
- Q. Yang, L. Zhang, C. Ye, S. Luo, L.-Z. Wu and C.-H. Tung, Angew. Chem., Int. Ed., 2017, 56, 3694–3698 CrossRef PubMed.
- N. Fu, L. Li, Q. Yang and S. Luo, Org. Lett., 2017, 19, 2122–2125 CrossRef PubMed.
- O. Baslé, N. Borduas, P. Dubois, J. M. Chapuzet, T.-H. Chan, J. Lessard and C.-J. Li, Chem. – Eur. J., 2010, 16, 8162–8166 CrossRef PubMed.
- C. Li, C.-C. Zeng, L.-M. Hu, F.-L. Yang, S. J. Yoo and R. D. Little, Electrochim. Acta, 2013, 114, 560–566 CrossRef.
- For acceptorless dehydrogenation of N-heterocycles using TEMPO as mediator, see: Y. Wu, H. Yi and A. Lei, ACS Catal., 2018, 8, 1192–1196 CrossRef.
- Y. Kashiwagi and J.-i. Anzai, Chem. Pharm. Bull., 2001, 49, 324–326 CrossRef PubMed.
- J.-i. Yoshida, S. Suga, S. Suzuki, N. Kinomura, A. Yamamoto and K. Fujiwara, J. Am. Chem. Soc., 1999, 121, 9546–9549 CrossRef.
- S. Suga, M. Okajima and J.-i. Yoshida, Tetrahedron Lett., 2001, 42, 2173–2176 CrossRef.
- S. Suga, T. Nishida, D. Yamada, A. Nagaki and J.-i. Yoshida, J. Am. Chem. Soc., 2004, 126, 14338–14339 CrossRef PubMed.
- T. Maruyama, Y. Mizuno, I. Shimizu, S. Suga and J.-i. Yoshida, J. Am. Chem. Soc., 2007, 129, 1902–1903 CrossRef PubMed.
- S. Suga, M. Okajima, K. Fujiwara and J.-i. Yoshida, J. Am. Chem. Soc., 2001, 123, 7941–7942 CrossRef PubMed.
- S. Suga, M. Okajima, K. Fujiwara and J.-i. Yoshida, QSAR Comb. Sci., 2005, 24, 728–741 CrossRef.
- S. Suga, D. Yamada and J.-i. Yoshida, Chem. Lett., 2010, 39, 404–406 CrossRef.
- J. Kuleshova, J. T. Hill-Cousins, P. R. Birkin, R. C. D. Brown, D. Pletcher and T. J. Underwood, Electrochim. Acta, 2011, 56, 4322–4326 CrossRef.
- K. Watts, W. Gattrell and T. Wirth, Beilstein J. Org. Chem., 2011, 7, 1108–1114 CrossRef PubMed.
- B. Giese and J. Dupuis, Angew. Chem., Int. Ed. Engl., 1983, 22, 622–623 CrossRef.
- For reviews on Giese-type reactions, see:
(a) B. Giese, Angew. Chem., Int. Ed. Engl., 1983, 22, 753–764 CrossRef;
(b) W. Zhang, Tetrahedron, 2001, 57, 7237–7262 CrossRef;
(c) G. S. C. Srikanth and S. L. Castle, Tetrahedron, 2005, 61, 10377–10441 CrossRef.
- S. Suga, S. Suzuki and J.-i. Yoshida, J. Am. Chem. Soc., 2002, 124, 30–31 CrossRef PubMed.
- S. Suga, S. Suzuki, T. Maruyama and J.-i. Yoshida, Bull. Chem. Soc. Jpn., 2004, 77, 1545–1554 CrossRef.
- D. Horii, T. Fuchigami and M. Atobe, J. Am. Chem. Soc., 2007, 129, 11692–11693 CrossRef PubMed.
- D. Horii, F. Amemiya, T. Fuchigami and M. Atobe, Chem. – Eur. J., 2008, 14, 10382–10387 CrossRef PubMed.
- T. Tajima and A. Nakajima, J. Am. Chem. Soc., 2008, 130, 10496–10497 CrossRef PubMed.
- For seminal studies on the concept of “site isolation”, see:
(a) B. J. Cohen, M. A. Kraus and A. Patchornik, J. Am. Chem. Soc., 1977, 99, 4165–4167 CrossRef;
(b) B. J. Cohen, M. A. Kraus and A. Patchornik, J. Am. Chem. Soc., 1981, 103, 7620–7629 CrossRef.
- R. Asami, M. Atobe and T. Fuchigami, J. Am. Chem. Soc., 2005, 127, 13160–13161 CrossRef PubMed.
- R. Asami, T. Fuchigami and M. Atobe, Langmuir, 2006, 22, 10258–10263 CrossRef PubMed.
- R. Asami, T. Fuchigami and M. Atobe, Chem. Commun., 2008, 244–246 RSC.
- R. Asami, T. Fuchigami and M. Atobe, Org. Biomol. Chem., 2008, 6, 1938–1943 RSC.
- M. Atobe, S. Ikari, K. Nakabayashi, F. Amemiya and T. Fuchigami, Langmuir, 2010, 26, 9111–9115 CrossRef PubMed.
- T. Tajima and T. Fuchigami, Angew. Chem., Int. Ed., 2005, 44, 4760–4763 CrossRef PubMed.
- H. Kurihara, T. Tajima and T. Fuchigami, Electrochemistry, 2006, 74, 615–617 CrossRef.
- T. Tajima, H. Kurihara and T. Fuchigami, J. Am. Chem. Soc., 2007, 129, 6680–6681 CrossRef PubMed.
- H. Kurihara, T. Fuchigami and T. Tajima, J. Org. Chem., 2008, 73, 6888–6890 CrossRef PubMed.
- T. Tajima and H. Kurihara, Chem. Commun., 2008, 5167–5169 RSC.
- T. Tajima, S. Ishino and H. Kurihara, Chem. Lett., 2008, 37, 1036–1037 CrossRef.
- T. Tajima and A. Nakajima, Chem. Lett., 2009, 38, 160–161 CrossRef.
- T. Tajima, H. Kurihara, S. Shimizu and H. Tateno, Electrochemistry, 2013, 81, 353–355 CrossRef.
- T. Tajima and T. Fuchigami, J. Am. Chem. Soc., 2005, 127, 2848–2849 CrossRef PubMed.
- T. Tajima and T. Fuchigami, Chem. – Eur. J., 2005, 11, 6192–6196 CrossRef PubMed.
- K. Suds, K. Hotoda, J.-i. Watanabe, K. Shiozawa and T. Takanami, J. Chem. Soc., Perkin Trans. 1, 1992, 1283–1284 Search PubMed.
- K. Suda, K. Hotoda, F. Iemuroe and T. Takanami, J. Chem. Soc., Perkin Trans. 1, 1993, 1553–1555 RSC.
- E. Le Gall, J.-P. Hurvois and S. Sinbandhit, Eur. J. Org. Chem., 1999, 2645–2653 CrossRef.
- S. Suga, M. Watanabe and J.-i. Yoshida, J. Am. Chem. Soc., 2002, 124, 14824–14825 CrossRef PubMed.
- T. Kamada and A. Oku, J. Chem. Soc., Perkin Trans. 1, 2002, 1105–1110 RSC.
- S. Suga, A. Nagaki, Y. Tsutsui and J.-i. Yoshida, Org. Lett., 2003, 5, 945–947 CrossRef PubMed.
- A. Nagaki, K. Kawamura, S. Suga, T. Ando, M. Sawamoto and J.-i. Yoshida, J. Am. Chem. Soc., 2004, 126, 14702–14703 CrossRef PubMed.
- S. Suga, Y. Kageyama, G. Babu, K. Itami and J.-i. Yoshida, Org. Lett., 2004, 6, 2709–2711 CrossRef PubMed.
- T. Maruyama, S. Suga and J.-i. Yoshida, J. Am. Chem. Soc., 2005, 127, 7324–7325 CrossRef PubMed.
- A. Nagaki, M. Togai, S. Suga, N. Aoki, K. Mae and J.-i. Yoshida, J. Am. Chem. Soc., 2005, 127, 11666–11675 CrossRef PubMed.
- S. Suga, Y. Tsutsui, A. Nagaki and J.-i. Yoshida, Bull. Chem. Soc. Jpn., 2005, 78, 1206–1217 CrossRef.
- T. Maruyama, S. Suga and J.-i. Yoshida, Tetrahedron, 2006, 62, 6519–6525 CrossRef.
- S. Suga, M. Watanabe, C.-H. Song and J.-i. Yoshida, Electrochemistry, 2006, 74, 672–679 CrossRef.
- A. Nagaki, T. Iwasaki, K. Kawamura, D. Yamada, S. Suga, T. Ando, M. Sawamoto and J.-i. Yoshida, Chem. – Asian J., 2008, 3, 1558–1567 CrossRef PubMed.
- Y. Ashikari, Y. Kiuchi, T. Takeuchi, K. Ueoka, S. Suga and J.-i. Yoshida, Chem. Lett., 2014, 43, 210–212 CrossRef.
- T. Shoji, S. Kim, K. Yamamoto, T. Kawai, Y. Okada and K. Chiba, Org. Lett., 2014, 16, 6404–6407 CrossRef PubMed.
- M. Sugawara, K. Mori and J.-i. Yoshida, Electrochim. Acta, 1997, 42, 1995–2003 CrossRef.
- T. Fuchigami, M. Tetsu, T. Tajima and H. Ishii, Synlett, 2001, 1269–1271 CrossRef.
- S. Kim, K. Hayashi, Y. Kitano, M. Tada and K. Chiba, Org. Lett., 2002, 4, 3735–3737 CrossRef PubMed.
- K. J. Frankowski, R. Liu, G. L. Milligan, K. D. Moeller and J. Aubé, Angew. Chem., Int. Ed., 2015, 54, 10555–10558 CrossRef PubMed.
- For leading reviews on azanucleosides and azanucleotides, see:
(a) C. Nájera and J. M. Sansano, Org. Biomol. Chem., 2009, 7, 4567–4581 RSC;
(b) G. Romeo, U. Chiacchio, A. Corsaro and P. Merino, Chem. Rev., 2010, 110, 3337–3370 CrossRef PubMed;
(c) B. L. Stocker, E. M. Dangerfield, A. L. Win-Mason, G. W. Haslett and M. S. M. Timmer, Eur. J. Org. Chem., 2010, 1615–1637 CrossRef;
(d) D. Hernández and A. Boto, Eur. J. Org. Chem., 2014, 2201–2220 CrossRef.
- S. Furukubo, N. Moriyama, O. Onomura and Y. Matsumura, Tetrahedron Lett., 2004, 45, 8177–8181 CrossRef.
- S. Kim, T. Shoji, Y. Kitano and K. Chiba, Chem. Commun., 2013, 49, 6525–6527 RSC.
- T. Shoji, S. Haraya, S. Kim and K. Chiba, Electrochim. Acta, 2016, 200, 290–295 CrossRef.
- T. Shoji, S. Kim and K. Chiba, Angew. Chem., Int. Ed., 2017, 56, 4011–4014 CrossRef PubMed.
- K. Bodmann, T. Bug, S. Steinbeisser, R. Kreuder and O. Reiser, Tetrahedron Lett., 2006, 47, 2061–2064 CrossRef.
- F. Cornille, U. Slomczynska, M. L. Smythe, D. D. Beusen, K. D. Moeller and G. R. Marshall, J. Am. Chem. Soc., 1995, 117, 909–917 CrossRef.
- U. Slomczynska, D. K. Chalmers, F. Cornille, M. L. Smythe, D. D. Beusen, K. D. Moeller and G. R. Marshall, J. Org. Chem., 1996, 61, 1198–1204 CrossRef.
- W. Li and K. D. Moeller, J. Am. Chem. Soc., 1996, 118, 10106–10112 CrossRef.
- W. Chu and K. D. Moeller, Tetrahedron Lett., 1999, 40, 7939–7943 CrossRef.
- L. M. Beal, B. Liu, W. Chu and K. D. Moeller, Tetrahedron, 2000, 56, 10113–10125 CrossRef.
- H. Sun and K. D. Moeller, Org. Lett., 2002, 4, 1547–1550 CrossRef PubMed.
- H. Sun and K. D. Moeller, Org. Lett., 2003, 5, 3189–3192 CrossRef PubMed.
- H. Sun, C. Martin, D. Kesselring, R. Keller and K. D. Moeller, J. Am. Chem. Soc., 2006, 128, 13761–13771 CrossRef PubMed.
- For selected reviews on synthetic approaches towards peptidomimetic design and applications, see:
(a) A. Giannis and T. Kolter, Angew. Chem., Int. Ed. Engl., 1993, 32, 1244–1267 CrossRef;
(b) I. Avan, C. D. Hall and A. R. Katritzky, Chem. Soc. Rev., 2014, 43, 3575–3594 RSC;
(c) M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef PubMed;
(d) A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC;
(e) D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef PubMed;
(f) O. Van der Poorten, A. Knuhtsen, D. Sejer Pedersen, S. Ballet and D. Tourwé, J. Med. Chem., 2016, 59, 10865–10890 CrossRef PubMed;
(g) A. Mizuno, K. Matsui and S. Shuto, Chem. – Eur. J., 2017, 23, 14394–14409 CrossRef PubMed;
(h) N. Qvit, S. J. S. Rubin, T. J. Urban, D. Mochly-Rosen and E. R. Gross, Drug Discovery Today, 2017, 22, 454–462 CrossRef PubMed.
- For a review on radical reactions in the synthesis of α-amino acids and related derivatives, see: C. J. Easton, Chem. Rev., 1997, 97, 53–82 CrossRef PubMed.
- For synthesis and applications of silicon-containing α-amino acids, see: M. Mortensen, R. Husmann, E. Veri and C. Bolm, Chem. Soc. Rev., 2009, 38, 1002–1010 RSC.
- C. Reuter, P. Huy, J.-M. Neudörfl, R. Kühne and H.-G. Schmalz, Chem. – Eur. J., 2011, 17, 12037–12044 CrossRef PubMed.
- T. Shono, Y. Matsumura and T. Kanazawa, Tetrahedron Lett., 1983, 24, 1259–1262 CrossRef.
- M. A. Kabeshov, B. Musio, P. R. D. Murray, D. L. Browne and S. V. Ley, Org. Lett., 2014, 16, 4618–4621 CrossRef PubMed.
- F. Louafi, J. Moreau, S. Shahane, S. Golhen, T. Roisnel, S. Sinbandhit and J.-P. Hurvois, J. Org. Chem., 2011, 76, 9720–9732 CrossRef PubMed.
- N. Girard, J.-P. Hurvois, C. Moinet and L. Toupet, Eur. J. Org. Chem., 2005, 2269–2280 CrossRef.
- N. Shankaraiah, R. A. Pilli and L. S. Santos, Tetrahedron Lett., 2008, 49, 5098–5100 CrossRef.
- N. Shankaraiah, W. A. da Silva, C. K. Z. Andrade and L. S. Santos, Tetrahedron Lett., 2008, 49, 4289–4291 CrossRef.
- Y. Mirabal-Gallardo, J. Piérola, N. Shankaraiah and L. S. Santos, Tetrahedron Lett., 2012, 53, 3672–3675 CrossRef.
- T.-S. Kam, T.-M. Lim and G.-H. Tan, J. Chem. Soc., Perkin Trans. 1, 2001, 1594–1604 RSC.
- K. D. Moeller and P. L. Wong, Bioorg. Med. Chem. Lett., 1992, 2, 739–742 CrossRef.
- A. Paci, T. Martens and J. Royer, Bioorg. Med. Chem. Lett., 2001, 11, 1347–1349 CrossRef PubMed.
- P. J. Smith and C. K. Mann, J. Org. Chem., 1969, 34, 1821–1826 CrossRef.
- L. C. Portis, V. V. Bhat and C. K. Mann, J. Org. Chem., 1970, 35, 2175–2178 CrossRef.
- S. D. Ross, Tetrahedron Lett., 1973, 14, 1237–1240 CrossRef.
- L. R. Hall, R. T. Iwamoto and R. P. Hanzlik, J. Org. Chem., 1989, 54, 2446–2451 CrossRef.
- P. G. Kirira, M. Kuriyama and O. Onomura, Chem. – Eur. J., 2010, 16, 3970–3982 CrossRef PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef PubMed.
- D. E. Yerien, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 8398–8427 RSC.
- P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef PubMed.
- T. Fujiwara and D. O’Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- For selected reviews on electrophilic fluorination, see:
(a) G. S. Lal, G. P. Pez and R. G. Syvret, Chem. Rev., 1996, 96, 1737–1756 CrossRef PubMed;
(b) S. D. Taylor, C. C. Kotoris and G. Hum, Tetrahedron, 1999, 55, 12431–12477 CrossRef;
(c) S. Barata-Vallejo, B. Lantaño and A. Postigo, Chem. – Eur. J., 2014, 20, 16806–16829 CrossRef PubMed;
(d) T. Koike and M. Akita, J. Fluorine Chem., 2014, 167, 30–36 CrossRef;
(e) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef PubMed.
- For selected reviews on nucleophilic fluorination, see:
(a) R. P. Singh and J. M. Shreeve, Synthesis, 2002, 2561–2578 Search PubMed;
(b) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160–171 CrossRef PubMed;
(c) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC;
(d) J.-W. Lee, M. T. Oliveira, H. B. Jang, S. Lee, D. Y. Chi, D. W. Kim and C. E. Song, Chem. Soc. Rev., 2016, 45, 4638–4650 RSC;
(e) S. V. Kohlhepp and T. Gulder, Chem. Soc. Rev., 2016, 45, 6270–6288 RSC;
(f) S. Liang, G. B. Hammond and B. Xu, Chem. – Eur. J., 2017, 23, 17850–17861 CrossRef PubMed;
(g) C. N. Neumann and T. Ritter, Acc. Chem. Res., 2017, 50, 2822–2833 CrossRef PubMed.
- For selected reviews on radical fluorination, see:
(a) C. Chatalova-Sazepin, R. Hemelaere, J.-F. Paquin and G. M. Sammis, Synthesis, 2015, 2554–2569 Search PubMed;
(b) M. P. Sibi and Y. Landais, Angew. Chem., Int. Ed., 2013, 52, 3570–3572 CrossRef PubMed;
(c) X. Pan, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 1163–1185 RSC;
(d) B. Lantaño and A. Postigo, Org. Biomol. Chem., 2017, 15, 9954–9973 RSC;
(e) T. Koike and M. Akita, Chem, 2018, 4, 409–437 CrossRef.
- For general reviews on synthetic approaches for the synthesis of organofluorine compounds, see:
(a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef PubMed;
(b) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef PubMed;
(c) C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216–3221 CrossRef PubMed;
(d) D. L. Orsi and R. A. Altman, Chem. Commun., 2017, 53, 7168–7181 RSC;
(e) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. – Eur. J., 2017, 23, 14676–14701 CrossRef PubMed.
- For a review on electrochemical fluoroalkylation, see: Y. B. Dudkina, M. N. Khrizanforov, T. V. Gryaznova and Y. H. Budnikova, J. Organomet. Chem., 2014, 751, 301–305 CrossRef.
- I. L. Knunyants, I. N. Rozhkov, A. V. Bukhtiarov, M. M. Gol'din and R. V. Kudryavtsev, Izv. Akad. Nauk SSSR, Ser. Khim., 1970, 1207–1208 Search PubMed.
- I. N. Rozhkov, A. B. Bukhtiarov, N. D. Kuleshova and I. L. Knunyants, Dokl. Akad. Nauk SSSR, 1970, 193, 1322–1325 Search PubMed.
- I. N. Rozhkov, Russ. Chem. Rev., 1976, 45, 615–629 CrossRef.
- K. Momota, M. Morita and Y. Matsuda, Electrochim. Acta, 1993, 38, 619–624 CrossRef.
- K. Momota, M. Morita and Y. Matsuda, Electrochim. Acta, 1993, 38, 1123–1130 CrossRef.
- K. Momota, K. Kato, M. Morita and Y. Matsuda, Electrochim. Acta, 1994, 39, 41–49 CrossRef.
- K. Momota, H. Horio, K. Kato, M. Morita and Y. Matsuda, Electrochim. Acta, 1995, 40, 233–240 CrossRef.
- K. Momota, K. Kato, M. Morita and Y. Matsuda, Electrochim. Acta, 1994, 39, 681–690 CrossRef.
- K. Momota, T. Yonezawa, Y. Hayakawa, K. Kato, M. Morita and Y. Matsuda, J. Appl. Electrochem., 1995, 25, 651–658 CrossRef.
- K. Momota, K. Mukai, K. Kato and M. Morita, Electrochim. Acta, 1998, 43, 2503–2514 CrossRef.
- K. Momota, T. Yonezawa, K. Mukai and M. Morita, J. Fluorine Chem., 1998, 87, 173–178 CrossRef.
- T. Tajima, H. Ishii and T. Fuchigami, Electrochem. Commun., 2002, 4, 589–592 CrossRef.
- N. Ilayaraja and M. Noel, J. Electroanal. Chem., 2010, 638, 39–45 CrossRef.
- T. Tajima, H. Ishii and T. Fuchigami, Electrochem. Commun., 2001, 3, 467–471 CrossRef.
- T. Tajima, H. Ishii and T. Fuchigami, Tetrahedron Lett., 2001, 42, 4857–4860 CrossRef.
- M. R. Shaaban and T. Fuchigami, Synlett, 2001, 1644–1646 CrossRef.
- S. M. Riyadh, H. Ishii and T. Fuchigami, Tetrahedron, 2002, 58, 9273–9278 CrossRef.
- K. M. Dawood and T. Fuchigami, J. Org. Chem., 2004, 69, 5302–5306 CrossRef PubMed.
- T. Tajima, A. Nakajima and T. Fuchigami, J. Org. Chem., 2006, 71, 1436–1441 CrossRef PubMed.
- B. Yin, L. Wang, S. Inagi and T. Fuchigami, Tetrahedron, 2010, 66, 6820–6825 CrossRef.
- B. Yin, S. Inagi and T. Fuchigami, Synlett, 2011, 1313–1317 Search PubMed.
- S. M. Riyadh and T. Fuchigami, J. Org. Chem., 2002, 67, 9379–9383 CrossRef PubMed.
- M. R. Shaaban and T. Fuchigami, Tetrahedron Lett., 2002, 43, 273–276 CrossRef.
- M. R. Shaaban, S. Inagi and T. Fuchigami, Electrochim. Acta, 2009, 54, 2635–2639 CrossRef.
- T. Fukuhara, M. Sawaguchi and N. Yoneda, Electrochem. Commun., 2000, 2, 259–261 CrossRef.
- M. Sawaguchi, T. Fukuhara and N. Yoneda, J. Electroanal. Chem., 2001, 507, 66–70 CrossRef.
- T. Fukuhara, Y. Akiyama, N. Yoneda, T. Tada and S. Hara, Tetrahedron Lett., 2002, 43, 6583–6585 CrossRef.
- For electrochemical reduction of aromatic systems, see: M. Ishifune, H. Yamashita, Y. Kera, N. Yamashita, K. Hirata, H. Murase and S. Kashimura, Electrochim. Acta, 2003, 48, 2405–2409 CrossRef.
- For electrochemical reduction of pyridine derivatives, see: Y. Kita, H. Maekawa, Y. Yamasaki and I. Nishiguchi, Tetrahedron, 2001, 57, 2095–2102 CrossRef.
- J.-B. Tommasino, A. Brondex, M. Médebielle, M. Thomalla, B. R. Langlois and T. Billard, Synlett, 2002, 1697–1699 CrossRef.
- A. G. O’Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868–11871 CrossRef PubMed.
- T. Fuchigami, M. Shimojo, A. Konno and K. Nakagawa, J. Org. Chem., 1990, 55, 6074–6075 CrossRef.
- A. Konno, K. Nakagawa and T. Fuchigami, J. Chem. Soc., Chem. Commun., 1991, 1027–1029 RSC.
- T. Fuchigami, H. Yano and A. Konno, J. Org. Chem., 1991, 56, 6731–6733 CrossRef.
- T. Fuchigami, A. Konno, K. Nakagawa and M. Shimojo, J. Org. Chem., 1994, 59, 5937–5941 CrossRef.
- A. Konno and T. Fuchigami, J. Appl. Electrochem., 1995, 25, 173–175 CrossRef.
- T. Fuchigami, M. Shimojo and A. Konno, J. Org. Chem., 1995, 60, 3459–3464 CrossRef.
- A. Konno and T. Fuchigami, J. Org. Chem., 1997, 62, 8579–8581 CrossRef PubMed.
- S. Higashiya, T. Sato and T. Fuchigami, J. Fluorine Chem., 1998, 87, 203–208 CrossRef.
- See also: T. Fuchigami, K. Yamamoto and A. Konno, Tetrahedron, 1991, 47, 625–634 CrossRef.
- T. Brigaud and E. Laurent, Tetrahedron Lett., 1990, 31, 2287–2290 CrossRef.
- T. Brigaud, A. Laurent and E. Laurent, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 59, 153–156 CrossRef.
- For discussions on the parameters affecting fluorination, see:
(a) V. Suryanarayanan and M. Noel, J. Fluorine Chem., 1998, 91, 153–157 CrossRef;
(b) K. M. Dawood and T. Fuchigami, J. Org. Chem., 1999, 64, 138–143 CrossRef PubMed;
(c) K. Suzuki, S. Inagi and T. Fuchigami, Electrochim. Acta, 2009, 54, 961–965 CrossRef.
- P. Baroux, R. Tardivel and J. Simonet, Tetrahedron Lett., 1995, 36, 3851–3852 CrossRef.
- P. Baroux, R. Tardivel and J. Simonet, J. Electrochem. Soc., 1997, 144, 841–846 CrossRef.
- A. Konno, W. Naito and T. Fuchigami, Tetrahedron Lett., 1992, 33, 7017–7020 CrossRef.
- Y. Hou, S. Higashiya and T. Fuchigami, J. Org. Chem., 1997, 62, 9173–9176 CrossRef.
- S. M. Riyadh, H. Ishii and T. Fuchigami, Tetrahedron, 2001, 57, 8817–8821 CrossRef.
- S. Kuribayashi, N. Shida, S. Inagi and T. Fuchigami, Tetrahedron, 2016, 72, 5343–5349 CrossRef.
- K. M. Dawood, H. Ishii and T. Fuchigami, J. Org. Chem., 2001, 66, 7030–7034 CrossRef PubMed.
- T. Fuchigami, S. Narizuka, A. Konno and K. Momota, Electrochim. Acta, 1998, 43, 1985–1989 CrossRef.
- M. R. Shaaban, H. Ishii and T. Fuchigami, J. Org. Chem., 2001, 66, 5633–5636 CrossRef PubMed.
- S. Higashiya, S. Narizuka, A. Konno, T. Maeda, K. Momota and T. Fuchigami, J. Org. Chem., 1999, 64, 133–137 CrossRef PubMed.
- M. R. Shaaban, H. Ishii and T. Fuchigami, J. Org. Chem., 2000, 65, 8685–8689 CrossRef PubMed.
- Y. Cao, A. Hidaka, T. Tajima and T. Fuchigami, J. Org. Chem., 2005, 70, 9614–9617 CrossRef PubMed.
- B. Zagipa, A. Hidaka, Y. Cao and T. Fuchigami, J. Fluorine Chem., 2006, 127, 552–557 CrossRef.
- K. M. Dawood, S. Higashiya, Y. Hou and T. Fuchigami, J. Org. Chem., 1999, 64, 7935–7939 CrossRef.
- S. Inagi, T. Sawamura and T. Fuchigami, Electrochem. Commun., 2008, 10, 1158–1160 CrossRef.
- M. Noel and V. Suryanarayanan, J. Appl. Electrochem., 2004, 34, 357–369 CrossRef.
- K. M. Dawood, Tetrahedron, 2004, 60, 1435–1451 CrossRef.
- T. Fuchigami and T. Tajima, J. Fluorine Chem., 2005, 126, 181–187 CrossRef.
- T. Fuchigami, J. Fluorine Chem., 2007, 128, 311–316 CrossRef.
- T. Fuchigami and S. Inagi, Chem. Commun., 2011, 47, 10211–10223 RSC.
- Z. U. H. Khan, D. Kong, Y. Chen, N. Muhammad, A. U. Khan, F. U. Khan, K. Tahir, A. Ahmad, L. Wang and P. Wan, J. Ind. Eng. Chem., 2015, 31, 26–38 CrossRef.
- T. Sunaga, M. Atobe, S. Inagi and T. Fuchigami, Chem. Commun., 2009, 956–958 RSC.
- T. Tajima, A. Nakajima, Y. Doi and T. Fuchigami, Angew. Chem., Int. Ed., 2007, 46, 3550–3552 CrossRef PubMed.
- T. Sawamura, S. Kuribayashi, S. Inagi and T. Fuchigami, Org. Lett., 2010, 12, 644–646 CrossRef PubMed.
- D. Baba, Y.-J. Yang, B.-J. Uang and T. Fuchigami, J. Fluorine Chem., 2003, 121, 93–96 CrossRef.
- K. Suzuki and T. Fuchigami, J. Org. Chem., 2004, 69, 1276–1282 CrossRef PubMed.
- D. Baba, H. Ishii, S. Higashiya, K. Fujisawa and T. Fuchigami, J. Org. Chem., 2001, 66, 7020–7024 CrossRef PubMed.
- S. Narizuka, H. Koshiyama, A. Konno and T. Fuchigami, J. Fluorine Chem., 1995, 73, 121–127 CrossRef.
- M. Aoyama, T. Fukuhara and S. Hara, J. Org. Chem., 2008, 73, 4186–4189 CrossRef PubMed.
- M. Monoi and S. Hara, J. Fluorine Chem., 2012, 140, 28–30 CrossRef.
- M. Hasegawa, H. Ishii and T. Fuchigami, Tetrahedron Lett., 2002, 43, 1503–1505 CrossRef.
- M. Hasegawa, H. Ishii, Y. Cao and T. Fuchigami, J. Electrochem. Soc., 2006, 153, D162–D166 CrossRef.
- See also: K. M. Dawood and T. Fuchigami, J. Org. Chem., 2001, 66, 7691–7695 CrossRef PubMed.
- K. M. Dawood and T. Fuchigami, Tetrahedron Lett., 2001, 42, 2513–2515 CrossRef.
- K. M. Dawood and T. Fuchigami, J. Org. Chem., 2005, 70, 7537–7541 CrossRef PubMed.
- H. Ishii, N. Yamada and T. Fuchigami, Chem. Commun., 2000, 1617–1618 RSC.
- H. Ishii, N. Yamada and T. Fuchigami, Tetrahedron, 2001, 57, 9067–9072 CrossRef.
- M. Hasegawa, H. Ishii and T. Fuchigami, Green Chem., 2003, 5, 512–515 RSC.
- M. Hasegawa and T. Fuchigami, Electrochim. Acta, 2004, 49, 3367–3372 CrossRef.
- T. Sawamura, S. Kuribayashi, S. Inagi and T. Fuchigami, Adv. Synth. Catal., 2010, 352, 2757–2760 CrossRef.
- T. Sawamura, K. Takahashi, S. Inagi and T. Fuchigami, Angew. Chem., Int. Ed., 2012, 51, 4413–4416 CrossRef PubMed.
- B. Yin, S. Inagi and T. Fuchigami, Beilstein J. Org. Chem., 2015, 11, 85–91 CrossRef PubMed.
- For fluorodesulfurization of polymers, see:
(a) S. Inagi, S. Hayashi and T. Fuchigami, Chem. Commun., 2009, 1718–1720 RSC;
(b) S. Inagi and T. Fuchigami, Macromol. Rapid Commun., 2014, 35, 854–867 CrossRef PubMed.
- For the use of boron derivatives as electroauxiliaries, see:
(a) M. Tanigawa, Y. Kuriyama, S. Inagi and T. Fuchigami, Electrochim. Acta, 2016, 199, 314–318 CrossRef;
(b) J. Suzuki, N. Shida, S. Inagi and T. Fuchigami, Electroanalysis, 2016, 28, 2797–2801 CrossRef.
- Y. Hou, S. Higashiya and T. Fuchigami, J. Org. Chem., 1999, 64, 3346–3349 CrossRef PubMed.
- V. Dinoiu, K. Kanno, T. Fukuhara and N. Yoneda, J. Fluorine Chem., 2005, 126, 753–758 CrossRef.
- H. Nagura, S. Inagi and T. Fuchigami, Tetrahedron, 2009, 65, 1559–1566 CrossRef.
- H. Nagura, S. Kuribayashi, Y. Ishiguro, S. Inagi and T. Fuchigami, Tetrahedron, 2010, 66, 183–186 CrossRef.
- S. Fujie, K. Matsumoto, S. Suga and J.-i. Yoshida, Chem. Lett., 2009, 38, 1186–1187 CrossRef.
- V. Dinoiu, T. Fukuhara, S. Hara and N. Yoneda, J. Fluorine Chem., 2000, 103, 75–80 CrossRef.
- For an electrochemical approach to hydrodefluorination of fluoroaromatic compounds, see: W.-B. Wu, M.-L. Li and J.-M. Huang, Tetrahedron Lett., 2015, 56, 1520–1523 CrossRef.
- G. Faita, M. Fleischmann and D. Pletcher, J. Electroanal. Chem., 1970, 25, 455–459 CrossRef.
- L. Appelbaum, D. Danovich, G. Lazanes, M. Michman and M. Oron, J. Electroanal. Chem., 2001, 499, 39–47 CrossRef.
- For anodic chlorination of aniline, see: Y. Matsuda and T. Nishiki, Electrochim. Acta, 1984, 29, 35–39 CrossRef.
- G. Casalbore, M. Mastragostino and S. Valcher, J. Electroanal. Chem., 1978, 87, 411–418 CrossRef.
- I. Taniguchi, M. Yano, H. Yamaguchi and K. Yasukouchi, J. Electroanal. Chem., 1982, 132, 233–245 CrossRef.
- M. Mastragostino, S. Valcher and M. Biserni, J. Electroanal. Chem., 1983, 158, 369–373 CrossRef.
- C. Visy and M. Novák, Electrochim. Acta, 1987, 32, 1757–1759 CrossRef.
- G. D. Allen, M. C. Buzzeo, I. G. Davies, C. Villagrán, C. Hardacre and R. G. Compton, J. Phys. Chem. B, 2004, 108, 16322–16327 CrossRef.
- E. I. Rogers, D. S. Silvester, L. Aldous, C. Hardacre and R. G. Compton, J. Phys. Chem. C, 2008, 112, 6551–6557 CrossRef.
- For a review on electrochemical halogenation of organic compounds, see: B. V. Lyalin and V. A. Petrosyan, Russ. J. Electrochem., 2013, 49, 497–529 CrossRef.
- For a review on halogenation of organic compounds using continuous flow and microreactor technology, see: D. Cantillo and C. O. Kappe, React. Chem. Eng., 2017, 2, 7–19 RSC.
- S. R. Ellis, D. Pletcher, W. N. Brooks and K. P. Healy, J. Appl. Electrochem., 1983, 13, 735–741 CrossRef.
- S. R. Forsyth, D. Pletcher and K. P. Healy, J. Appl. Electrochem., 1987, 17, 905–913 CrossRef.
- B. V. Lyalin, V. A. Petrosyan and B. I. Ugrak, Russ. J. Electrochem., 2008, 44, 1320–1326 CrossRef.
- T. Raju, G. Kalpana Devi and K. Kulangiappar, Electrochim. Acta, 2006, 51, 4596–4600 CrossRef.
- Takahiro Sawamura, Kohta Takahashi, Shinsuke Inagi and Toshio Fuchigami, Electrochemistry, 2013, 81, 365–367 CrossRef.
- T. Raju, K. Kulangiappar, M. Anbu Kulandainathan, U. Uma, R. Malini and A. Muthukumaran, Tetrahedron Lett., 2006, 47, 4581–4584 CrossRef.
- T. Raju, K. Kulangiappar, M. Anbu Kulandainathan and A. Muthukumaran, Tetrahedron Lett., 2005, 46, 7047–7050 CrossRef.
- K. Kulangiappar, G. Karthik and M. Anbu Kulandainathan, Synth. Commun., 2009, 39, 2304–2309 CrossRef.
- K. Tsuchida, T. Kochi and F. Kakiuchi, Asian J. Org. Chem., 2013, 2, 935–937 CrossRef.
- E. M. Ungureanu, A. C. Razus, L. Birzan, G. Buica, M. Cretu and E. Cristian, Electrochim. Acta, 2006, 52, 794–803 CrossRef.
- For an electrochemical study of azulene derivatives, see: E.-M. Ungureanu, A. C. Razus, L. Birzan, M.-S. Cretu and G.-O. Buica, Electrochim. Acta, 2008, 53, 7089–7099 CrossRef.
- M. Verniette, C. Daremon and J. Simonet, Electrochim. Acta, 1978, 23, 929–934 CrossRef.
- M. Novák, C. Visy and K. Bodor, Electrochim. Acta, 1982, 27, 1293–1295 CrossRef.
- M. Novák and C. Visy, Electrochim. Acta, 1983, 28, 507–510 CrossRef.
- M. Novák and C. Visy, Electrochim. Acta, 1983, 28, 511–513 CrossRef.
- C. Visy and M. Novák, J. Electroanal. Chem., 1988, 252, 91–97 CrossRef.
- C. Visy and M. Novák, J. Electroanal. Chem., 1990, 296, 571–581 CrossRef.
- D. Stevanović, I. Damljanović, M. Vukićević, N. Manojlović, N. S. Radulović and R. D. Vukićević, Helv. Chim. Acta, 2011, 94, 1406–1415 CrossRef.
- B. V. Lyalin, V. A. Petrosyan and B. I. Ugrak, Russ. Chem. Bull., 2009, 58, 291–296 CrossRef.
- Z. Tan, Y. Liu, R. Helmy, N. R. Rivera, D. Hesk, S. Tyagarajan, L. Yang and J. Su, Tetrahedron Lett., 2017, 58, 3014–3018 CrossRef.
- P. Pouillen, R. Minko, M. Verniette, P. Martinet and A. M. Martre, Electrochim. Acta, 1979, 24, 1189–1194 CrossRef.
- Y. Ashikari, A. Shimizu, T. Nokami and J.-i. Yoshida, J. Am. Chem. Soc., 2013, 135, 16070–16073 CrossRef PubMed.
- A. Shimizu, R. Hayashi, Y. Ashikari, T. Nokami and J.-i. Yoshida, Beilstein J. Org. Chem., 2015, 11, 242–248 CrossRef PubMed.
- K. Kulangiappar, M. Ramaprakash, D. Vasudevan and T. Raju, Synth. Commun., 2016, 46, 145–153 CrossRef.
- B. V. Lyalin, V. A. Petrosyan and B. I. Ugrak, Russ. Chem. Bull., 2010, 59, 1549–1555 CrossRef.
- For electrochemical synthesis of pyrazoles, see: X. Wei and B. Speiser, Electrochim. Acta, 1997, 42, 73–79 CrossRef.
- K. Kataoka, Y. Hagiwara, K. Midorikawa, S. Suga and J.-i. Yoshida, Org. Process Res. Dev., 2008, 12, 1130–1136 CrossRef.
- K. Midorikawa, S. Suga and J.-i. Yoshida, Chem. Commun., 2006, 3794–3796 RSC.
- R. Möckel, J. Hille, E. Winterling, S. Weidemüller, T. M. Faber and G. Hilt, Angew. Chem., Int. Ed., 2018, 57, 442–445 CrossRef PubMed.
- Y. Zhu, Y. Zhu, H. Zeng, Z. Chen, R. D. Little and C. Ma, J. Electroanal. Chem., 2015, 751, 105–110 CrossRef.
- M. Rafiee, F. Wang, D. P. Hruszkewycz and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 22–25 CrossRef PubMed.
- For selective electrochemical generation of benzylic radicals using ferrocene-based mediators, see: A. J. J. Lennox, J. E. Nutting and S. S. Stahl, Chem. Sci., 2018, 9, 356–361 RSC.
- M. Masui, S. Hara, T. Ueshima, T. Kawaguchi and S. Ozaki, Chem. Pharm. Bull., 1983, 31, 4209–4211 CrossRef.
- M. Masui, T. Kawaguchi and S. Ozaki, J. Chem. Soc., Chem. Commun., 1985, 1484–1485 RSC.
- For reviews on NHPI-mediated reactions, see:
(a) Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393–427 CrossRef;
(b) R. A. Sheldon and I. W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef;
(c) F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800–3842 CrossRef PubMed;
(d) S. Coseri, Catal. Rev., 2009, 51, 218–292 CrossRef;
(e) S. Wertz and A. Studer, Green Chem., 2013, 15, 3116–3134 RSC;
(f) L. Melone and C. Punta, Beilstein J. Org. Chem., 2013, 9, 1296–1310 CrossRef PubMed.
- The bond dissociation energy (BDE) of NHPI has been determined to 88 kcal mol−1 and the BDE of toluene derivatives ranges from 88–90 kcal mol−1. See:
(a) T. Fox and P. A. Kollman, J. Phys. Chem., 1996, 100, 2950–2956 CrossRef;
(b) G. P. Bean, Tetrahedron, 2002, 58, 9941–9948 CrossRef;
(c) R. Amorati, M. Lucarini, V. Mugnaini, G. F. Pedulli, F. Minisci, F. Recupero, F. Fontana, P. Astolfi and L. Greci, J. Org. Chem., 2003, 68, 1747–1754 CrossRef PubMed;
(d) N. Hirai, N. Sawatari, N. Nakamura, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2003, 68, 6587–6590 CrossRef PubMed;
(e) D. A. Pratt, G. A. DiLabio, P. Mulder and K. U. Ingold, Acc. Chem. Res., 2004, 37, 334–340 CrossRef PubMed.
- I. Nishiguchi, O. Kanbe, K. Itoh and H. Maekawa, Synlett, 2000, 89–91 Search PubMed.
- T. Shono, Y. Matsumura, K. Inoue and F. Lwasaki, J. Chem. Soc., Perkin Trans. 1, 1986, 73–77 RSC.
- F. Barba, M. N. Elinson, J. Escudero and S. K. Feducovich, Tetrahedron, 1997, 53, 4427–4436 CrossRef.
- F. Barba, M. N. Elinson, J. Escudero, M. Guirado and S. K. Feducovich, Electrochim. Acta, 1998, 43, 973–976 CrossRef.
- M. N. Elinson, S. K. Feducovich, A. S. Dorofeev, A. N. Vereshchagin and G. I. Nikishin, Tetrahedron, 2000, 56, 9999–10003 CrossRef.
- M. N. Elinson, S. K. Feducovich, D. E. Dmitriev, A. S. Dorofeev, A. N. Vereshchagin and G. I. Nikishin, Tetrahedron Lett., 2001, 42, 5557–5559 CrossRef.
- M. N. Elinson, A. S. Dorofeev, S. K. Feducovich, R. F. Nasybullin, E. F. Litvin, M. V. Kopyshev and G. I. Nikishin, Tetrahedron, 2006, 62, 8021–8028 CrossRef.
- C. Yao, Y. Wang, T. Li, C. Yu, L. Li and C. Wang, Tetrahedron, 2013, 69, 10593–10597 CrossRef.
- H. Gao, Z. Zha, Z. Zhang, H. Ma and Z. Wang, Chem. Commun., 2014, 50, 5034–5036 RSC.
- M. N. Elinson, S. K. Feducovich, S. G. Bushuev, A. A. Zakharenkov, D. V. Pashchenko and G. I. Nikishin, Mendeleev Commun., 1998, 8, 15–16 CrossRef.
- M. N. Elinson, S. K. Feducovich, T. L. Lizunova and G. I. Nikishin, Tetrahedron, 2000, 56, 3063–3069 CrossRef.
- M. N. Elinson, S. K. Feducovich, Z. A. Starikova, O. S. Olessova, A. N. Vereshchagin and G. I. Nikishin, Tetrahedron Lett., 2000, 41, 4937–4941 CrossRef.
- M. N. Elinson, S. K. Feducovich and G. I. Nikishin, ARKIVOC, 2001, 31–36 Search PubMed.
- O. Mitsuhiro, T. Yukio and K. Toyoji, Bull. Chem. Soc. Jpn., 2003, 76, 207–208 CrossRef.
- M. N. Elinson, S. K. Fedukovich, T. A. Zaimovskaya, A. N. Vereshchagin and G. I. Nikishin, Russ. Chem. Bull., 2003, 52, 2241–2246 CrossRef.
- M. N. Elinson, S. K. Feducovich, Z. A. Starikova, A. N. Vereshchagin, S. V. Gorbunov and G. I. Nikishin, Tetrahedron Lett., 2005, 46, 6389–6393 CrossRef.
- M. N. Elinson, S. K. Feducovich, Z. A. Starikova, A. N. Vereshchagin, P. A. Belyakov and G. I. Nikishin, Tetrahedron, 2006, 62, 3989–3996 CrossRef.
- A. N. Vereshchagin, M. N. Elinson, T. A. Zaimovskaya and G. I. Nikishin, Tetrahedron, 2008, 64, 9766–9770 CrossRef.
- D. Minato, S. Mizuta, M. Kuriyama, Y. Matsumura and O. Onomura, Tetrahedron, 2009, 65, 9742–9748 CrossRef.
- M. Okimoto, H. Yamamori, K. Ohashi, S. Nishikawa, M. Hoshi and T. Yoshida, Synlett, 2012, 2544–2548 CrossRef.
- M. Okimoto, H. Yamamori, K. Ohashi, M. Hoshi and T. Yoshida, Synlett, 2013, 1568–1572 CrossRef.
- A. N. Vereshchagin, M. N. Elinson, E. O. Dorofeeva, D. V. Demchuk, I. S. Bushmarinov, A. S. Goloveshkin and G. I. Nikishin, Tetrahedron, 2013, 69, 5234–5241 CrossRef.
- M. Okimoto, H. Yamamori, M. Hoshi and T. Yoshida, Tetrahedron Lett., 2014, 55, 1299–1302 CrossRef.
- A. N. Vereshchagin, M. N. Elinson, E. O. Dorofeeva, R. F. Nasybullin, I. S. Bushmarinov, A. S. Goloveshkin and M. P. Egorov, Electrochim. Acta, 2015, 165, 116–121 CrossRef.
- M. N. Elinson, E. O. Dorofeeva, A. N. Vereshchagin, R. F. Nasybullin and M. P. Egorov, Catal. Sci. Technol., 2015, 5, 2384–2387 RSC.
- Z. Zhang, J. Su, Z. Zha and Z. Wang, Chem. – Eur. J., 2013, 19, 17711–17714 CrossRef PubMed.
- For electrochemical C−H acylation of electron-deficient N-heteroarenes with α-keto acids, see: Q.-Q. Wang, K. Xu, Y.-Y. Jiang, Y.-G. Liu, B.-G. Sun and C.-C. Zeng, Org. Lett., 2017, 19, 5517–5520 CrossRef PubMed.
- For preparation of benzimidazoles through decarboxylation of α-keto acids, see: H.-B. Wang and J.-M. Huang, Adv. Synth. Catal., 2016, 358, 1975–1981 CrossRef.
- Z. Zhang, J. Su, Z. Zha and Z. Wang, Chem. Commun., 2013, 49, 8982–8984 RSC.
- P. Qian, J.-H. Su, Y. Wang, M. Bi, Z. Zha and Z. Wang, J. Org. Chem., 2017, 82, 6434–6440 CrossRef PubMed.
- N.-N. Bui, X.-H. Ho, S.-i. Mho and H.-Y. Jang, Eur. J. Org. Chem., 2009, 5309–5312 CrossRef.
-
(a) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794–797 CrossRef PubMed;
(b) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef PubMed.
- For reviews on diarylprolinol silyl ethers as organocatalysts, see:
(a) K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248–264 CrossRef PubMed;
(b) L. Klier, F. Tur, P. H. Poulsen and K. A. Jørgensen, Chem. Soc. Rev., 2017, 46, 1080–1102 RSC.
- E. E. Finney, K. A. Ogawa and A. J. Boydston, J. Am. Chem. Soc., 2012, 134, 12374–12377 CrossRef PubMed.
- For electrochemical characterization of azolium salts, see: K. A. Ogawa and A. J. Boydston, Chem. Lett., 2014, 43, 907–909 CrossRef.
- For recent reviews on the oxidative esterification and amidation of aldehydes, see:
(a) K. Ekoue-Kovi and C. Wolf, Chem. – Eur. J., 2008, 14, 6302–6315 CrossRef PubMed;
(b) S. Gaspa, A. Porcheddu and L. De Luca, Tetrahedron Lett., 2016, 57, 3433–3440 CrossRef.
- K. A. Ogawa and A. J. Boydston, Org. Lett., 2014, 16, 1928–1931 CrossRef PubMed.
- R. A. Green, D. Pletcher, S. G. Leach and R. C. D. Brown, Org. Lett., 2015, 17, 3290–3293 CrossRef PubMed.
- R. A. Green, D. Pletcher, S. G. Leach and R. C. D. Brown, Org. Lett., 2016, 18, 1198–1201 CrossRef PubMed.
- N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef PubMed.
- Y. Qiu, W.-J. Kong, J. Struwe, N. Sauermann, T. Rogge, A. Scheremetjew and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5828–5832 CrossRef PubMed.
- Y. Qiu, C. Tian, L. Massignan, T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5818–5822 CrossRef PubMed.
- F. Xu, X.-Y. Qian, Y.-J. Li and H.-C. Xu, Org. Lett., 2017, 19, 6332–6335 CrossRef PubMed.
- S. Zhang, L. Li, H. Wang, Q. Li, W. Liu, K. Xu and C. Zeng, Org. Lett., 2018, 20, 252–255 CrossRef PubMed.
- For a related approach, see: L. Zhang, Z. Zhang, J. Hong, J. Yu, J. Zhang and F. Mo, J. Org. Chem., 2018, 83, 3200–3207 CrossRef PubMed.
- For a related electrocatalytic dehydrogenative esterification strategy, see: S. Zhang, F. Lian, M. Xue, T. Qin, L. Li, X. Zhang and K. Xu, Org. Lett., 2017, 19, 6622–6625 CrossRef PubMed.
- For electrochemical synthesis of phthalides, see: D. Hayrapetyan, V. Shkepu, O. T. Seilkhanov, Z. Zhanabil and K. Lam, Chem. Commun., 2017, 53, 8451–8454 RSC.
- S. Nad and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 2297–2299 CrossRef PubMed.
- S. Nad, S. Roller, R. Haag and R. Breinbauer, Org. Lett., 2006, 8, 403–406 CrossRef PubMed.
- C. Laurencé, M. Rivard, I. Lachaise, J. Bensemhoun and T. Martens, Tetrahedron, 2011, 67, 9518–9521 CrossRef.
- E. Nieto-Mendoza, J. A. Guevara-Salazar, M. T. Ramírez-Apan, B. A. Frontana-Uribe, J. A. Cogordan and J. Cárdenas, J. Org. Chem., 2005, 70, 4538–4541 CrossRef PubMed.
- M. Albert, D. De Souza, P. Feiertag and H. Hönig, Org. Lett., 2002, 4, 3251–3254 CrossRef PubMed.
- For issues associated with the use of oxidants in the pharmaceutical industry, see: S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef PubMed.
- L. Meng, J. Su, Z. Zha, L. Zhang, Z. Zhang and Z. Wang, Chem. – Eur. J., 2013, 19, 5542–5545 CrossRef PubMed.
- M. Okajima, K. Soga, T. Nokami, S. Suga and J.-i. Yoshida, Org. Lett., 2006, 8, 5005–5007 CrossRef PubMed.
- T. Nokami, K. Ohata, M. Inoue, H. Tsuyama, A. Shibuya, K. Soga, M. Okajima, S. Suga and J.-i. Yoshida, J. Am. Chem. Soc., 2008, 130, 10864–10865 CrossRef PubMed.
- M. Okajima, K. Soga, T. Watanabe, K. Terao, T. Nokami, S. Suga and J.-i. Yoshida, Bull. Chem. Soc. Jpn., 2009, 82, 594–599 CrossRef.
- K. Terao, T. Watanabe, T. Suehiro, T. Nokami and J.-i. Yoshida, Tetrahedron Lett., 2010, 51, 4107–4109 CrossRef.
- T. Nokami, T. Watanabe, N. Musya, T. Morofuji, K. Tahara, Y. Tobe and J.-i. Yoshida, Chem. Commun., 2011, 47, 5575–5577 RSC.
- T. Nokami, T. Watanabe, N. Musya, T. Suehiro, T. Morofuji and J.-i. Yoshida, Tetrahedron, 2011, 67, 4664–4671 CrossRef.
- T. Nokami, T. Watanabe, K. Terao, K. Soga, K. Ohata and J.-i. Yoshida, Electrochemistry, 2013, 81, 399–401 CrossRef.
- T. Nokami, N. Musya, T. Morofuji, K. Takeda, M. Takumi, A. Shimizu and J.-i. Yoshida, Beilstein J. Org. Chem., 2014, 10, 3097–3103 CrossRef PubMed.
- For generation of benzylic cations through carbon–carbon bond cleavage, see: M. Okajima, S. Suga, K. Itami and J.-i. Yoshida, J. Am. Chem. Soc., 2005, 127, 6930–6931 CrossRef PubMed.
- Y. Ashikari, T. Nokami and J.-i. Yoshida, J. Am. Chem. Soc., 2011, 133, 11840–11843 CrossRef PubMed.
- Y. Ashikari, T. Nokami and J.-i. Yoshida, Org. Lett., 2012, 14, 938–941 CrossRef PubMed.
- T. Tajima, Y. Kishi and A. Nakajima, Electrochim. Acta, 2009, 54, 5959–5963 CrossRef.
- For early work on anodic acetoxylation, see:
(a) L. Eberson and K. Nyberg, J. Am. Chem. Soc., 1966, 88, 1686–1691 CrossRef;
(b) L. Eberson, J. Am. Chem. Soc., 1967, 89, 4669–4677 CrossRef.
- J. H. P. Utley and G. G. Rozenberg, J. Appl. Electrochem., 2003, 33, 525–532 CrossRef.
- For electrochemical synthesis of benzoxazoles using DDQ as a redox catalyst, see: L.-S. Kang, H.-l. Xiao, C.-C. Zeng, L.-M. Hu and R. D. Little, J. Electroanal. Chem., 2016, 767, 13–17 CrossRef.
- D. P. Hruszkewycz, K. C. Miles, O. R. Thiel and S. S. Stahl, Chem. Sci., 2017, 8, 1282–1287 RSC.
- N.-t. Zhang, C.-c. Zeng, C. M. Lam, R. K. Gbur and R. D. Little, J. Org. Chem., 2013, 78, 2104–2110 CrossRef PubMed.
- R. Francke and R. D. Little, J. Am. Chem. Soc., 2014, 136, 427–435 CrossRef PubMed.
- N.-n. Lu, S. J. Yoo, L.-J. Li, C.-C. Zeng and R. D. Little, Electrochim. Acta, 2014, 142, 254–260 CrossRef.
- K.-Y. Zhang, N.-n. Lu, S. J. Yoo, L.-M. Hu, R. D. Little and C.-C. Zeng, Electrochim. Acta, 2016, 199, 357–365 CrossRef.
- C.-c. Zeng, N.-t. Zhang, C. M. Lam and R. D. Little, Org. Lett., 2012, 14, 1314–1317 CrossRef PubMed.
- See also: S. J. Yoo, L.-J. Li, C.-C. Zeng and R. D. Little, Angew. Chem., Int. Ed., 2015, 54, 3744–3747 CrossRef PubMed.
- For arylative ring-opening of epoxides using triarylimidazole redox mediators, see: N.-n. Lu, N.-t. Zhang, C.-C. Zeng, L.-M. Hu, S. J. Yoo and R. D. Little, J. Org. Chem., 2015, 80, 781–789 CrossRef PubMed.
- E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef PubMed.
- S. R. Waldvogel and M. Selt, Angew. Chem., Int. Ed., 2016, 55, 12578–12580 CrossRef PubMed.
- For seminal work on electrochemical allylic C–H oxidation using NHPI as mediator, see: M. Masui, K. Hosomi, K. Tsuchida and S. Ozaki, Chem. Pharm. Bull., 1985, 33, 4798–4802 CrossRef.
- J. Kowalski, J. Płoszyńska, A. Sobkowiak, J. W. Morzycki and A. Z. Wilczewska, J. Electroanal. Chem., 2005, 585, 275–280 CrossRef.
- J. Kowalski, Z. Łotowski, J. W. Morzycki, J. Płoszyńska, A. Sobkowiak and A. Z. Wilczewska, Steroids, 2008, 73, 543–548 CrossRef PubMed.
- Y.-Y. Hosokawa, H. Hakamata, T. Murakami, S. Aoyagi, M. Kuroda, Y. Mimaki, A. Ito, S. Morosawa and F. Kusu, Electrochim. Acta, 2009, 54, 6412–6416 CrossRef.
- Y.-Y. Hosokawa, H. Hakamata, T. Murakami and F. Kusu, Tetrahedron Lett., 2010, 51, 129–132 CrossRef.
- For electrochemical synthesis of glycoconjugates, see: A. M. Tomkiel, J. Kowalski, J. Płoszyńska, L. Siergiejczyk, Z. Łotowski, A. Sobkowiak and J. W. Morzycki, Steroids, 2014, 82, 60–67 CrossRef PubMed.
- For electrochemical synthesis of glycoconjugates using nonactivated sugars, see: A. M. Tomkiel, K. Brzezinski, Z. Łotowski, L. Siergiejczyk, P. Wałejko, S. Witkowski, J. Kowalski, J. Płoszyńska, A. Sobkowiak and J. W. Morzycki, Tetrahedron, 2013, 69, 8904–8913 CrossRef.
- Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef PubMed.
- C. Sambiagio, H. Sterckx and B. U. W. Maes, ACS Cent. Sci., 2017, 3, 686–688 CrossRef PubMed.
- For general reviews on palladium-catalyzed reactions, see:
(a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef PubMed;
(b) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef PubMed;
(c) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555–1564 CrossRef PubMed;
(d) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef PubMed;
(e) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC;
(f) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed;
(g) H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef;
(h) P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497–5508 CrossRef;
(i) R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888 RSC;
(j) M. M. Lorion, K. Maindan, A. R. Kapdi and L. Ackermann, Chem. Soc. Rev., 2017, 46, 7399–7420 RSC;
(k) N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 0025 CrossRef PubMed.
- K. D. Moeller, Organometallics, 2014, 33, 4607–4616 CrossRef.
- Y. B. Dudkina, T. V. Gryaznova, O. G. Sinyashin and Y. H. Budnikova, Russ. Chem. Bull., 2015, 64, 1713–1725 CrossRef.
- K.-J. Jiao, C.-Q. Zhao, P. Fang and T.-S. Mei, Tetrahedron Lett., 2017, 58, 797–802 CrossRef.
- F. Kakiuchi and T. Kochi, Isr. J. Chem., 2017, 57, 953–963 CrossRef.
- For an example on the use of electrosynthesis for generation of catalytic Pd intermediates, see: V. A. Zinovyeva, C. Luo, S. Fournier, C. H. Devillers, H. Cattey, H. Doucet, J.-C. Hierso and D. Lucas, Chem. – Eur. J., 2011, 17, 9901–9906 CrossRef PubMed.
- J. Tsuji and M. Minato, Tetrahedron Lett., 1987, 28, 3683–3686 CrossRef.
- See also: K. Mitsudo, S. Fukunaga, T. Fujita, H. Mandai, S. Suga and H. Tanaka, Electrochemistry, 2013, 81, 347–349 CrossRef.
- C. Amatore, C. Cammoun and A. Jutand, Adv. Synth. Catal., 2007, 349, 292–296 CrossRef.
- For electrochemically-mediated Heck reactions that rely on cathodic reduction of PdII to Pd0, see:
(a) J. Tian, K. Maurer, E. Tesfu and K. D. Moeller, J. Am. Chem. Soc., 2005, 127, 1392–1393 CrossRef PubMed;
(b) J. Tian and K. D. Moeller, Org. Lett., 2005, 7, 5381–5383 CrossRef PubMed;
(c) L. Hu, M. Stuart, J. Tian, K. Maurer and K. D. Moeller, J. Am. Chem. Soc., 2010, 132, 16610–16616 CrossRef PubMed.
- K. Mitsudo, N. Kamimoto, H. Murakami, H. Mandai, A. Wakamiya, Y. Murata and S. Suga, Org. Biomol. Chem., 2012, 10, 9562–9569 RSC.
- C. Amatore, C. Cammoun and A. Jutand, Eur. J. Org. Chem., 2008, 4567–4570 CrossRef.
- C. Amatore, C. Cammoun and A. Jutand, Synlett, 2007, 2173–2178 CrossRef.
- K. Mitsudo, T. Shiraga and H. Tanaka, Tetrahedron Lett., 2008, 49, 6593–6595 CrossRef.
- K. Mitsudo, T. Shiraga, D. Kagen, D. Shi, J. Y. Becker and H. Tanaka, Tetrahedron, 2009, 65, 8384–8388 CrossRef.
- or electroreductive coupling of aryl bromides using a Pd/viologen double mediatory system, see: M. Kuroboshi, T. Shiba and H. Tanaka, Tetrahedron Lett., 2013, 54, 3666–3668 CrossRef.
- K. Mitsudo, T. Shiraga, J.-i. Mizukawa, S. Suga and H. Tanaka, Chem. Commun., 2010, 46, 9256–9258 RSC.
- K. Mitsudo, T. Kaide, E. Nakamoto, K. Yoshida and H. Tanaka, J. Am. Chem. Soc., 2007, 129, 2246–2247 CrossRef PubMed.
- K. Mitsudo, T. Ishii and H. Tanaka, Electrochemistry, 2008, 76, 859–861 CrossRef.
- E. Tesfu, K. Maurer, S. R. Ragsdale and K. D. Moeller, J. Am. Chem. Soc., 2004, 126, 6212–6213 CrossRef PubMed.
- E. Tesfu, K. Maurer and K. D. Moeller, J. Am. Chem. Soc., 2006, 128, 70–71 CrossRef PubMed.
- E. Tesfu, K. Roth, K. Maurer and K. D. Moeller, Org. Lett., 2006, 8, 709–712 CrossRef PubMed.
- B. H. Nguyen, D. Kesselring, E. Tesfu and K. D. Moeller, Langmuir, 2014, 30, 2280–2286 CrossRef PubMed.
- For a review on he site-selective functionalization of electrode surfaces, see: M. D. Graaf and K. D. Moeller, Langmuir, 2015, 31, 7697–7706 CrossRef PubMed.
- For seminal work on electrochemical regeneration of PdII using triarylamines as recyclable mediators, see: T. Inokuchi, L. Ping, F. Hamaue, M. Izawa and S. Torii, Chem. Lett., 1994, 121–124 CrossRef.
- F. Kakiuchi, T. Kochi, H. Mutsutani, N. Kobayashi, S. Urano, M. Sato, S. Nishiyama and T. Tanabe, J. Am. Chem. Soc., 2009, 131, 11310–11311 CrossRef PubMed.
- M. Konishi, K. Tsuchida, K. Sano, T. Kochi and F. Kakiuchi, J. Org. Chem., 2017, 82, 8716–8724 CrossRef PubMed.
- K. Sano, N. Kimura, T. Kochi and F. Kakiuchi, Asian J. Org. Chem. DOI:10.1002/ajoc.201800202.
- For Pd-catalyzed electroreductive cross-coupling of alkyl and allylic halides, see: Y.-L. Lai and J.-M. Huang, Org. Lett., 2017, 19, 2022–2025 CrossRef PubMed.
- F. Saito, H. Aiso, T. Kochi and F. Kakiuchi, Organometallics, 2014, 33, 6704–6707 CrossRef.
- Y. B. Dudkina, D. Y. Mikhaylov, T. V. Gryaznova, A. I. Tufatullin, O. N. Kataeva, D. A. Vicic and Y. H. Budnikova, Organometallics, 2013, 32, 4785–4792 CrossRef.
- Y. B. Dudkina, D. Y. Mikhaylov, T. V. Gryaznova, O. G. Sinyashin, D. A. Vicic and Y. H. Budnikova, Eur. J. Org. Chem., 2012, 2114–2117 CrossRef.
- See also:
(a) D. Y. Mikhaylov, Y. H. Budnikova, T. V. Gryaznova, D. V. Krivolapov, I. A. Litvinov, D. A. Vicic and O. G. Sinyashin, J. Organomet. Chem., 2009, 694, 3840–3843 CrossRef;
(b) D. Mikhaylov, T. Gryaznova, Y. Dudkina, M. Khrizanphorov, S. Latypov, O. Kataeva, D. A. Vicic, O. G. Sinyashin and Y. Budnikova, Dalton Trans., 2012, 41, 165–172 RSC.
- T. V. Grayaznova, Y. B. Dudkina, D. R. Islamov, O. N. Kataeva, O. G. Sinyashin, D. A. Vicic and Y. H. Budnikova, J. Organomet. Chem., 2015, 785, 68–71 CrossRef.
- See also: S. O. Strekalova, M. N. Khrizanforov, T. V. Gryaznova, V. V. Khrizanforova and Y. H. Budnikova, Russ. Chem. Bull., 2016, 65, 1295–1298 CrossRef.
- H. Aiso, T. Kochi, H. Mutsutani, T. Tanabe, S. Nishiyama and F. Kakiuchi, J. Org. Chem., 2012, 77, 7718–7724 CrossRef PubMed.
- N. Kamimoto, N. Nakamura, A. Tsutsumi, H. Mandai, K. Mitsudo, A. Wakamiya, Y. Murata, J.-y. Hasegawa and S. Suga, Asian J. Org. Chem., 2016, 5, 373–379 CrossRef.
- A. Shrestha, M. Lee, A. L. Dunn and M. S. Sanford, Org. Lett., 2018, 20, 204–207 CrossRef PubMed.
- Y.-Q. Li, Q.-L. Yang, P. Fang, T.-S. Mei and D. Zhang, Org. Lett., 2017, 19, 2905–2908 CrossRef PubMed.
- C. Ma, C.-Q. Zhao, Y.-Q. Li, L.-P. Zhang, X.-T. Xu, K. Zhang and T.-S. Mei, Chem. Commun., 2017, 53, 12189–12192 RSC.
- Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293–3298 CrossRef PubMed.
- For reviews on metal-catalyzed oxidative cross-couplings, see:
(a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef PubMed;
(b) C. J. Scheuermann, Chem. – Asian J., 2010, 5, 436–451 CrossRef PubMed;
(c) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540–548 RSC;
(d) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef PubMed;
(e) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC;
(f) K. Hirano and M. Miura, Chem. Commun., 2012, 48, 10704–10714 RSC;
(g) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC;
(h) X. Shang and Z.-Q. Liu, Chem. Soc. Rev., 2013, 42, 3253–3260 RSC;
(i) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886–896 RSC;
(j) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef PubMed;
(k) Y. Wu, J. Wang, F. Mao and F. Y. Kwong, Chem. – Asian J., 2014, 9, 26–47 CrossRef PubMed;
(l) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef PubMed;
(m) X.-H. Yang, R.-J. Song, Y.-X. Xie and J.-H. Li, ChemCatChem, 2016, 8, 2429–2445 CrossRef;
(n) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef PubMed.
- For a review on oxidative cross-coupling for C–O bond formation, see: I. B. Krylov, V. A. Vil’ and A. O. Terent’ev, Beilstein J. Org. Chem., 2015, 11, 92–146 CrossRef PubMed.
- For recent reviews on metal-free oxidative cross-coupling reactions, see:
(a) R. Samanta, K. Matcha and A. P. Antonchick, Eur. J. Org. Chem., 2013, 5769–5804 CrossRef;
(b) K. Morimoto, T. Dohi and Y. Kita, Synlett, 2017, 1680–1694 Search PubMed.
- For a recent review on electrochemical oxidative cross-couplings, see: S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 Search PubMed.
- For recent reviews on radical cross-coupling reactions, see:
(a) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464–3484 RSC;
(b) C. Liu, D. Liu and A. Lei, Acc. Chem. Res., 2014, 47, 3459–3470 CrossRef PubMed;
(c) X. Zhu and S. Chiba, Chem. Soc. Rev., 2016, 45, 4504–4523 RSC;
(d) S.-r. Guo, P. S. Kumar and M. Yang, Adv. Synth. Catal., 2017, 359, 2–25 CrossRef;
(e) K. Liang and C. Xia, Chin. J. Chem., 2017, 35, 255–270 CrossRef;
(f) J.-R. Zhang, L. Xu, Y.-Y. Liao, J.-C. Deng and R.-Y. Tang, Chin. J. Chem., 2017, 35, 271–279 CrossRef.
- K. M. Johnston, Tetrahedron Lett., 1967, 8, 837–839 Search PubMed.
- P. Kočovský, Š. Vyskočil and M. Smrčina, Chem. Rev., 2003, 103, 3213–3246 CrossRef PubMed.
- M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC.
- H. Wang, Chirality, 2010, 22, 827–837 CrossRef PubMed.
- A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef PubMed.
- M. C. Kozlowski, Acc. Chem. Res., 2017, 50, 638–643 CrossRef PubMed.
- J. M. Bobbitt, J. T. Stock, A. Marchand and K. H. Weisgraber, Chem. Ind., 1966, 2127–2128 Search PubMed.
- G. F. Kirkbright, J. T. Stock, R. D. Pugliese and J. M. Bobbitt, J. Electrochem. Soc., 1969, 116, 219–221 CrossRef.
- J. M. Bobbitt, K. H. Weisgraber, A. S. Steinfeld and S. G. Weiss, J. Org. Chem., 1970, 35, 2884–2888 CrossRef.
- J. M. Bobbitt, H. Yagi, S. Shibuya and J. T. Stock, J. Org. Chem., 1971, 36, 3006–3010 CrossRef.
- For anodic oxidation of p-cresol, see: T. Kametani, K. Ohkubo and S. Takano, Chem. Pharm. Bull., 1968, 16, 1095–1097 CrossRef PubMed.
- L. L. Miller, F. R. Stermitz and J. R. Falck, J. Am. Chem. Soc., 1971, 93, 5941–5942 CrossRef.
- L. L. Miller, F. R. Stermitz and J. R. Falck, J. Am. Chem. Soc., 1973, 95, 2651–2656 CrossRef.
- T. Kametani, K. Shishido and S. Takano, J. Heterocycl. Chem., 1975, 12, 305–307 CrossRef.
- J. B. Kerr, T. C. Jempty and L. Miller, J. Am. Chem. Soc., 1979, 101, 7338–7346 CrossRef.
- Anodic oxidation of phenolic compounds has also been reported to produce C–O coupled products, see: J. M. Bobbitt and R. C. Hallcher, J. Chem. Soc. D, 1971, 543–544 RSC.
- E. Kotani, N. Takeuchi and S. Tobinaga, J. Chem. Soc., Chem. Commun., 1973, 550–551 RSC.
- E. Kotani and S. Tobinaga, Tetrahedron Lett., 1973, 14, 4759–4762 CrossRef.
- For a related report on the intramolecular oxidative coupling of phenols, see: M. A. Schwartz, R. A. Holton and S. W. Scott, J. Am. Chem. Soc., 1969, 91, 2800 CrossRef.
- I. M. Malkowsky, C. E. Rommel, K. Wedeking, R. Fröhlich, K. Bergander, M. Nieger, C. Quaiser, U. Griesbach, H. Pütter and S. R. Waldvogel, Eur. J. Org. Chem., 2006, 241–245 CrossRef.
- I. M. Malkowsky, C. E. Rommel, R. Fröhlich, U. Griesbach, H. Pütter and S. R. Waldvogel, Chem. – Eur. J., 2006, 12, 7482–7488 CrossRef PubMed.
- I. M. Malkowsky, R. Fröhlich, U. Griesbach, H. Pütter and S. R. Waldvogel, Eur. J. Inorg. Chem., 2006, 1690–1697 CrossRef.
- J. Barjau, P. Königs, O. Kataeva and S. R. Waldvogel, Synlett, 2008, 2309–2312 Search PubMed.
- A. Kirste, S. Hayashi, G. Schnakenburg, I. M. Malkowsky, F. Stecker, A. Fischer, T. Fuchigami and S. R. Waldvogel, Chem. – Eur. J., 2011, 17, 14164–14169 CrossRef PubMed.
- J. Barjau, G. Schnakenburg and S. R. Waldvogel, Synthesis, 2011, 2054–2061 Search PubMed.
- M. Mirion, L. Andernach, C. Stobe, J. Barjau, D. Schollmeyer, T. Opatz, A. Lützen and S. R. Waldvogel, Eur. J. Org. Chem., 2015, 4876–4882 CrossRef.
- For diversity oriented synthesis of polycyclic scaffolds by modification of these motifs, see: J. Barjau, G. Schnakenburg and S. R. Waldvogel, Angew. Chem., Int. Ed., 2011, 50, 1415–1419 CrossRef PubMed.
- Anodic oxidation of ortho-halogenated phenols has been reported to produce diaryl ethers, see:
(a) S. Nishiyama, M. H. Kim and S. Yamamura, Tetrahedron Lett., 1994, 35, 8397–8400 CrossRef;
(b) K. Mori, M. Takahashi, S. Yamamura and S. Nishiyama, Tetrahedron, 2001, 57, 5527–5532 CrossRef;
(c) T. Tanabe, F. Doi, T. Ogamino and S. Nishiyama, Tetrahedron Lett., 2004, 45, 3477–3480 CrossRef;
(d) Y. Naito, T. Tanabe, Y. Kawabata, Y. Ishikawa and S. Nishiyama, Tetrahedron Lett., 2010, 51, 4776–4778 CrossRef;
(e) K. Uno, T. Tanabe and S. Nishiyama, ECS Trans., 2010, 25, 91–95 Search PubMed;
(f) Y. Kawabata, Y. Naito, T. Saitoh, K. Kawa, T. Fuchigami and S. Nishiyama, Eur. J. Org. Chem., 2014, 99–104 CrossRef.
- I. M. Malkowsky, U. Griesbach, H. Pütter and S. R. Waldvogel, Eur. J. Org. Chem., 2006, 4569–4572 CrossRef.
- For dehydrogenative coupling reactions using a molybdenum-based anode, see: S. B. Beil, T. Müller, S. B. Sillart, P. Franzmann, A. Bomm, M. Holtkamp, U. Karst, W. Schade and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 2450–2454 CrossRef PubMed.
- B. Marselli, J. Garcia-Gomez, P.-A. Michaud, M. A. Rodrigo and C. Comninellis, J. Electrochem. Soc., 2003, 150, D79–D83 CrossRef.
- R. Fardel, U. Griesbach, H. Pütter and C. Comninellis, J. Appl. Electrochem., 2006, 36, 249–253 CrossRef.
- Alkoxy radicals generated through anodic oxidation have also been reported to add to bisdimethoxybenzene and related derivatives, see:
(a) M. G. Dolson and J. S. Swenton, J. Am. Chem. Soc., 1981, 103, 2361–2371 CrossRef;
(b) J. S. Swenton, Acc. Chem. Res., 1983, 16, 74–81 CrossRef;
(c) M. C. Carreño and M. Ribagorda, J.
Org. Chem., 2000, 65, 1231–1234 CrossRef;
(d) C.-C. Zeng and J. Y. Becker, J. Org. Chem., 2004, 69, 1053–1059 CrossRef PubMed;
(e) S. Quideau, I. Fabre and D. Deffieux, Org. Lett., 2004, 6, 4571–4573 CrossRef PubMed;
(f) T. Saitoh, E. Suzuki, A. Takasugi, R. Obata, Y. Ishikawa, K. Umezawa and S. Nishiyama, Bioorg. Med. Chem. Lett., 2009, 19, 5383–5386 CrossRef PubMed;
(g) S. Yajima, T. Saitoh, K. Kawa, K. Nakamura, H. Nagase, Y. Einaga and S. Nishiyama, Tetrahedron, 2016, 72, 8428–8435 CrossRef.
- A. Kirste, M. Nieger, I. M. Malkowsky, F. Stecker, A. Fischer and S. R. Waldvogel, Chem. – Eur. J., 2009, 15, 2273–2277 CrossRef PubMed.
- A. Kirste, G. Schnakenburg and S. R. Waldvogel, Org. Lett., 2011, 13, 3126–3129 CrossRef PubMed.
- T. Yamamoto, B. Riehl, K. Naba, K. Nakahara, A. Wiebe, T. Saitoh, S. R. Waldvogel and Y. Einaga, Chem. Commun., 2018, 54, 2771–2773 RSC.
- Guaiacol derivatives are potentially attainable aromatic monomers from depolymerization of lignin. For recent reviews on approaches to valorization of lignin, see:
(a) C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef PubMed;
(b) M. D. Kärkäs, B. S. Matsuura, T. M. Monos, G. Magallanes and C. R. J. Stephenson, Org. Biomol. Chem., 2016, 14, 1853–1914 RSC;
(c) M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef PubMed;
(d) R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef PubMed;
(e) R. E. Key and J. J. Bozell, ACS Sustainable Chem. Eng., 2016, 4, 5123–5135 CrossRef;
(f) M. D. Kärkäs, ChemSusChem, 2017, 10, 2111–2115 CrossRef PubMed;
(g) S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Łukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2014, 53, 5210–5213 Search PubMed.
- A. Wiebe, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 11801–11805 CrossRef PubMed.
- B. Elsler, A. Wiebe, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Chem. – Eur. J., 2015, 21, 12321–12325 CrossRef PubMed.
- B. Riehl, K. M. Dyballa, R. Franke and S. R. Waldvogel, Synthesis, 2017, 49, 252–259 Search PubMed.
-
(a) L. Schulz, M. Enders, B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 4877–4881 CrossRef PubMed;
(b) L. Schulz, R. Franke and S. R. Waldvogel, ChemElectroChem DOI:10.1002/celc.201800422.
- H. J. Schäfer, Angew. Chem., Int. Ed., 2017, 56, 15502–15503 CrossRef PubMed.
- For dehydrogenative cross-coupling of naphthylamines with phenols, see: B. Dahms, R. Franke and S. R. Waldvogel, ChemElectroChem, 2018, 5, 1249–1252 CrossRef.
- A. Kirste, G. Schnakenburg, F. Stecker, A. Fischer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2010, 49, 971–975 CrossRef PubMed.
- L. Eberson, M. P. Hartshorn and O. Persson, Angew. Chem., Int. Ed. Engl., 1995, 34, 2268–2269 CrossRef.
- L. Eberson, M. P. Hartshorn and O. Persson, J. Chem. Soc., Perkin Trans. 2, 1995, 1735–1744 RSC.
- O. Hollóczki, A. Berkessel, J. Mars, M. Mezger, A. Wiebe, S. R. Waldvogel and B. Kirchner, ACS Catal., 2017, 7, 1846–1852 CrossRef.
- S. Lips, A. Wiebe, B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 10872–10876 CrossRef PubMed.
- A. Wiebe, B. Riehl, S. Lips, R. Franke and S. R. Waldvogel, Sci. Adv., 2017, 3, eaao3920 CrossRef PubMed.
- A. Wiebe, S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 14727–14731 CrossRef PubMed.
- B. Elsler, D. Schollmeyer and S. R. Waldvogel, Faraday Discuss., 2014, 172, 413–420 Search PubMed.
- For additional electrochemical strategies for synthesis of benzofuran derivatives, see:
(a) A. R. Fakhari, H. Ahmar, S. S. Hosseiny Davarani, A. Shaabani, S. Nikjah and A. Maleki, Synth. Commun., 2011, 41, 561–568 CrossRef;
(b) M. Ameri, A. Asghari, A. Amoozadeh, M. Bakherad and D. Nematollahi, J. Electrochem. Soc., 2014, 161, G75–G80 CrossRef.
- G. Moutiers, J. Pinson, F. Terrier and R. Goumont, Chem. – Eur. J., 2001, 7, 1712–1719 CrossRef.
- I. Gallardo and G. Guirado, Eur. J. Org. Chem., 2008, 2463–2472 CrossRef.
- For reviews on nucleophilic aromatic substitution involving electron-deficient arenes, see:
(a) M. Mąkosza and K. Wojciechowski, Chem. Rev., 2004, 104, 2631–2666 CrossRef PubMed;
(b) M. Mąkosza, Chem. Soc. Rev., 2010, 39, 2855–2868 RSC;
(c) A. V. Gulevskaya and I. N. Tyaglivaya, Russ. Chem. Bull., 2012, 61, 1321–1341 CrossRef;
(d) M. Mąkosza, Chem. – Eur. J., 2014, 20, 5536–5545 CrossRef PubMed;
(e) M. Mąkosza and K. Wojciechowski, Heterocycles, 2014, 88, 75–101 CrossRef;
(f) O. N. Chupakhin and V. N. Charushin, Tetrahedron Lett., 2016, 57, 2665–2672 CrossRef;
(g) O. N. Chupakhin and V. N. Charushin, Pure Appl. Chem., 2017, 89, 1195–1208 Search PubMed.
- H. Cruz, I. Gallardo and G. Guirado, Green Chem., 2011, 13, 2531–2542 RSC.
- I. Gallardo, G. Guirado and J. Marquet, J. Org. Chem., 2002, 67, 2548–2555 CrossRef PubMed.
- I. Gallardo, G. Guirado and J. Marquet, J. Org. Chem., 2003, 68, 631–633 CrossRef PubMed.
- I. Gallardo, G. Guirado and J. Marquet, Eur. J. Org. Chem., 2002, 251–259 CrossRef.
- I. Gallardo, G. Guirado and J. Marquet, Eur. J. Org. Chem., 2002, 261–267 CrossRef.
- I. Gallardo, G. Guirado and J. Marquet, Chem. – Eur. J., 2001, 7, 1759–1765 CrossRef.
- H. Cruz, I. Gallardo and G. Guirado, Eur. J. Org. Chem., 2011, 7378–7389 CrossRef.
- I. Gallardo, G. Guirado and J. Marquet, J. Org. Chem., 2003, 68, 7334–7341 CrossRef PubMed.
- A. V. Shchepochkin, O. N. Chupakhin, V. N. Charushin, D. V. Steglenko, V. I. Minkin, G. L. Rusinov and A. I. Matern, RSC Adv., 2016, 6, 77834–77840 RSC.
- O. N. Chupakhin, A. V. Shchepochkin and V. N. Charushin, Green Chem., 2017, 19, 2931–2935 RSC.
- T. Morofuji, A. Shimizu and J.-i. Yoshida, Angew. Chem., Int. Ed., 2012, 51, 7259–7262 CrossRef PubMed.
- For trapping of arene radical cations with N-vinylamides using indirect electrolysis, see: L.-J. Li, Y.-Y. Jiang, C. M. Lam, C.-C. Zeng, L.-M. Hu and R. D. Little, J. Org. Chem., 2015, 80, 11021–11030 CrossRef PubMed.
- T. Arai, H. Tateno, K. Nakabayashi, T. Kashiwagi and M. Atobe, Chem. Commun., 2015, 51, 4891–4894 RSC.
- P. Wang, S. Tang, P. Huang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009–3013 CrossRef PubMed.
- For indirect electrochemical arylation of pyrroles, see: G. Sun, S. Ren, X. Zhu, M. Huang and Y. Wan, Org. Lett., 2016, 18, 544–547 CrossRef PubMed.
- P. Wang, S. Tang and A. Lei, Green Chem., 2017, 19, 2092–2095 RSC.
- X.-Y. Qian, S.-Q. Li, J. Song and H.-C. Xu, ACS Catal., 2017, 7, 2730–2734 CrossRef.
- For early studies on electrochemically-mediated cyclization of N-arylthioamides, see:
(a) M. Laćan, K. Jakopčić, V. Rogić, S. Damoni, O. Rogić and I. Tabaković, Synth. Commun., 1974, 4, 219–224 CrossRef;
(b) I. Tabaković, M. Trkovnik, M. Batušić and K. Tabaković, Synthesis, 1979, 590–592 CrossRef.
- For a related electrochemical approach in flow, see: A. A. Folgueiras-Amador, X.-Y. Qian, H.-C. Xu and T. Wirth, Chem. – Eur. J., 2018, 24, 487–491 CrossRef PubMed.
- Z.-J. Wu and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 4734–4738 CrossRef PubMed.
- Z.-J. Wu, S.-R. Li, H. Long and H.-C. Xu, Chem. Commun., 2018, 54, 4601–4604 RSC.
- U. Jahn and P. Hartmann, Chem. Commun., 1998, 209–210 RSC.
- U. Jahn, M. Müller and S. Aussieker, J. Am. Chem. Soc., 2000, 122, 5212–5213 CrossRef.
- U. Jahn, P. Hartmann and E. Kaasalainen, Org. Lett., 2004, 6, 257–260 CrossRef PubMed.
- F. Kafka, M. Holan, D. Hidasová, R. Pohl, I. Císařová, B. Klepetářová and U. Jahn, Angew. Chem., Int. Ed., 2014, 53, 9944–9948 CrossRef PubMed.
- For an alternative electrochemical approach to access oxindoles, see: Y.-Y. Jiang, S. Liang, C.-C. Zeng, L.-M. Hu and B.-G. Sun, Green Chem., 2016, 18, 6311–6319 RSC.
- For synthesis of oxindoles involving cathodic reduction, see: R. Munusamy, K. S. Dhathathreyan, K. K. Balasubramanian and C. S. Venkatachalam, J. Chem. Soc., Perkin Trans. 2, 2001, 1154–1166 RSC.
- H. Ding, P. L. DeRoy, C. Perreault, A. Larivée, A. Siddiqui, C. G. Caldwell, S. Harran and P. G. Harran, Angew. Chem., Int. Ed., 2015, 54, 4818–4822 CrossRef PubMed.
- A. W. G. Burgett, Q. Li, Q. Wei and P. G. Harran, Angew. Chem., Int. Ed., 2003, 42, 4961–4966 CrossRef PubMed.
- J.-C. Zhao, S.-M. Yu, H.-B. Qiu and Z.-J. Yao, Tetrahedron, 2014, 70, 3197–3210 CrossRef.
- K. Liu, S. Tang, P. Huang and A. Lei, Nat. Commun., 2017, 8, 775 CrossRef PubMed.
- A. Ronlán and V. D. Parker, J. Chem. Soc. C, 1971, 3214–3218 RSC.
- A. Ronlán, J. Chem. Soc. D, 1971, 1643–1645 RSC.
- J. A. Richards, P. E. Whitson and D. H. Evans, J. Electroanal. Chem., 1975, 63, 311–327 CrossRef.
- A. Nilsson, U. Palmquist, T. Pettersson and A. Ronlán, J. Chem. Soc., Perkin Trans. 1, 1978, 696–707 RSC.
- K. Ponsold and H. Kasch, Tetrahedron Lett., 1979, 20, 4465–4466 CrossRef.
- H. Eickhoff, G. Jung and A. Rieker, Tetrahedron, 2001, 57, 353–364 CrossRef.
- K. Chiba, M. Fukuda, S. Kim, Y. Kitano and M. Tada, J. Org. Chem., 1999, 64, 7654–7656 CrossRef.
- See also: Y. Imada, Y. Yamaguchi, N. Shida, Y. Okada and K. Chiba, Chem. Commun., 2017, 53, 3960–3963 RSC.
- For seminal studies on the [3+2] cycloaddition between phenoxonium ions and electron-rich alkenes through anodic oxidation, see:
(a) Y. Shizuri, K. Nakamura and S. Yamamura, J. Chem. Soc., Chem. Commun., 1985, 530–531 RSC;
(b) B. D. Gates, P. Dalidowicz, A. Tebben, S. Wang and J. S. Swenton, J. Org. Chem., 1992, 57, 2135–2143 CrossRef.
- For a seminal study on the [3+2] cycloaddition between phenoxonium ions and vinyl sulfides through anodic oxidation, see: M. L. Kerns, S. M. Conroy and J. S. Swenton, Tetrahedron Lett., 1994, 35, 7529–7532 CrossRef.
- For a related [3+2] cycloaddition of anodically generated radical intermediates, see:
(a) A. Nishiyama, H. Eto, Y. Terada, M. Iguchi and S. Yamamura, Chem. Pharm. Bull., 1983, 31, 2834–2844 CrossRef;
(b) T. Sumi, T. Saitoh, K. Natsui, T. Yamamoto, M. Atobe, Y. Einaga and S. Nishiyama, Angew. Chem., Int. Ed., 2012, 51, 5443–5446 CrossRef PubMed.
- K. Chiba, M. Jinno, R. Kuramoto and M. Tada, Tetrahedron Lett., 1998, 39, 5527–5530 CrossRef.
- S. Kim, S. Noda, K. Hayashi and K. Chiba, Org. Lett., 2008, 10, 1827–1829 CrossRef PubMed.
- Y. Okada, M. Sugai and K. Chiba, J. Org. Chem., 2016, 81, 10922–10929 CrossRef PubMed.
- H. Wakamatsu, Y. Okada, M. Sugai, S. R. Hussaini and K. Chiba, Asian J. Org. Chem., 2017, 6, 1584–1588 CrossRef.
- S. Kim, K. Hirose, J. Uematsu, Y. Mikami and K. Chiba, Chem. – Eur. J., 2012, 18, 6284–6288 CrossRef PubMed.
- Y. Okada, K. Shimada, Y. Kitano and K. Chiba, Eur. J. Org. Chem., 2014, 1371–1375 CrossRef.
- M. Iguchi, A. Nishiyama, Y. Terada and S. Yamamura, Chem. Lett., 1978, 451–454 CrossRef.
- A. Nishiyama, H. Eto, Y. Terada, M. Iguchi and S. Yamamura, Chem. Pharm. Bull., 1983, 31, 2820–2833 CrossRef.
- Y. Shizuri and S. Yamamura, Tetrahedron Lett., 1983, 24, 5011–5012 Search PubMed.
- S. Yamamura, Y. Shizuri, H. Shigemori, Y. Okuno and M. Ohkubo, Tetrahedron, 1991, 47, 635–644 CrossRef.
- D. Deffieux, I. Fabre, C. Courseille and S. Quideau, J. Org. Chem., 2002, 67, 4458–4465 CrossRef PubMed.
- D. Deffieux, I. Fabre, A. Titz, J.-M. Léger and S. Quideau, J. Org. Chem., 2004, 69, 8731–8738 CrossRef PubMed.
- L. Rycek, J. J. Hayward, M. A. Latif, J. Tanko, R. Simionescu and T. Hudlicky, J. Org. Chem., 2016, 81, 10930–10941 CrossRef PubMed.
- H. Takakura and S. Yamamura, Tetrahedron Lett., 1998, 39, 3717–3720 CrossRef.
- H. Takakura and S. Yamamura, Tetrahedron Lett., 1999, 40, 299–302 CrossRef.
- H. R. El-Seedi, S. Yamamura and S. Nishiyama, Tetrahedron, 2002, 58, 7485–7489 CrossRef.
- H. R. El-Seedi, S. Yamamura and S. Nishiyama, Tetrahedron Lett., 2002, 43, 3301–3304 CrossRef.
- S. Yamamura and S. Nishiyama, Synlett, 2002, 533–543 CrossRef.
- U. Palmquist, A. Ronlán and V. D. Parker, Acta Chem. Scand., Ser. B, 1974, 28, 267–269 CrossRef.
- I. G. C. Coutts, N. J. Culbert, M. Edwards, J. A. Hadfield, D. R. Musto, V. H. Pavlidis and D. J. Richards, J. Chem. Soc., Perkin Trans. 1, 1985, 1829–1836 RSC.
- G. W. Morrow and J. S. Swenton, Tetrahedron Lett., 1987, 28, 5445–5448 CrossRef.
- G. W. Morrow, Y. Chen and J. S. Swenton, Tetrahedron, 1991, 47, 655–664 CrossRef.
- J. S. Swenton, K. Carpenter, Y. Chen, M. L. Kerns and G. W. Morrow, J. Org. Chem., 1993, 58, 3308–3316 CrossRef.
- J. S. Swenton, A. Callinan, Y. Chen, J. J. Rohde, M. L. Kerns and G. W. Morrow, J. Org. Chem., 1996, 61, 1267–1274 CrossRef.
- A. Hutinec, A. Ziogas, M. El-Mobayed and A. Rieker, J. Chem. Soc., Perkin Trans. 1, 1998, 2201–2208 RSC.
- K. Mori, S. Yamamura and S. Nishiyama, Tetrahedron, 2001, 57, 5533–5542 CrossRef.
- T. Ogamino and S. Nishiyama, Tetrahedron, 2003, 59, 9419–9423 CrossRef.
- T. Ogamino, Y. Ishikawa and S. Nishiyama, Heterocycles, 2003, 61, 73–78 CrossRef.
- T. Ogamino, R. Obata and S. Nishiyama, Tetrahedron Lett., 2006, 47, 727–731 CrossRef.
- See also: T. Ogamino and S. Nishiyama, Tetrahedron Lett., 2005, 46, 1083–1086 CrossRef.
- J.-F. Cheng, S. Nishiyama and S. Yamamura, Chem. Lett., 1990, 1591–1594 CrossRef.
- S. Nishiyama, J.-F. Cheng, X. L. Tao and S. Yamamura, Tetrahedron Lett., 1991, 32, 4151–4154 CrossRef.
- X. L. Tao, J.-F. Cheng, S. Nishiyama and S. Yamamura, Tetrahedron, 1994, 50, 2017–2028 CrossRef.
- Y. Wada, H. Fujioka and Y. Kita, Mar. Drugs, 2010, 8, 1394–1416 CrossRef PubMed.
- T. Ogamino, S. Ohnishi, Y. Ishikawa, T. Sugai, R. Obata and S. Nishiyama, Sci. Technol. Adv. Mater., 2006, 7, 175–183 CrossRef.
- F. Doi, T. Ogamino, T. Sugai and S. Nishiyama, Tetrahedron Lett., 2003, 44, 4877–4880 CrossRef.
- F. Doi, T. Ogamino, T. Sugai and S. Nishiyama, Synlett, 2003, 411–413 Search PubMed.
- F. Doi, T. Ohara, T. Ogamino, K. Higashinakasu, K. Hasegawa and S. Nishiyama, Bull. Chem. Soc. Jpn., 2004, 77, 2257–2263 CrossRef.
- F. Doi, T. Ohara, T. Ogamino, T. Sugai, K. Higashinakasu, K. Yamada, H. Shigemori, K. Hasegawa and S. Nishiyama, Phytochemistry, 2004, 65, 1405–1411 CrossRef PubMed.
- A. Maleki and D. Nematollahi, Org. Lett., 2011, 13, 1928–1931 CrossRef PubMed.
- D. Nematollahi and B. Dadpou, Chin. Chem. Lett., 2011, 22, 1067–1070 CrossRef.
- F. Varmaghani and D. Nematollahi, J. Phys. Org. Chem., 2012, 25, 511–514 CrossRef.
- A. Maleki, D. Nematollahi, F. Rasouli and A. Zeinodini-Meimand, Green Chem., 2016, 18, 672–675 RSC.
- D. Nematollahi, F. Ghasemi, M. Sharafi-Kolkeshvandi and F. Varmaghani, J. Electroanal. Chem., 2016, 775, 299–305 CrossRef.
- V. M. Shanmugam, K. Kulangiappar, M. Ramaprakash, D. Vasudevan, R. S. Kumar, D. Velayutham and T. Raju, Tetrahedron Lett., 2017, 58, 2294–2297 CrossRef.
- S. B. H. Lang, D. Nematollahi, A. R. Massah, M. Sharafi-Kolkeshvandi and H. Alizadeh, J. Electrochem. Soc., 2017, 164, G87–G91 CrossRef.
- D. Nematollahi and M. Hesari, J. Electroanal. Chem., 2005, 577, 197–203 CrossRef.
- S. Osati, S. S. Hosseiny Davarani, N. Safari and M. H. Banitaba, Electrochim. Acta, 2011, 56, 9426–9432 CrossRef.
- D. Nematollahi, Z. Zohdijamil and H. Salehzadeh, J. Electroanal. Chem., 2014, 720–721, 156–161 CrossRef.
- S. Khazalpour, D. Nematollahi and M. R. Pajohi-Alamoti, New J. Chem., 2015, 39, 6734–6737 RSC.
- M. Sharafi-Kolkeshvandi, D. Nematollahi, F. Nikpour, M. Bayat and E. Soltani, Electrochim. Acta, 2016, 222, 845–855 CrossRef.
- D. Nematollahi, H. Hesari, H. Salehzadeh, M. Hesari and S. Momeni, C. R. Chim., 2016, 19, 357–362 CrossRef.
- M. Jamshidi and D. Nematollahi, ACS Sustainable Chem. Eng., 2017, 5, 9423–9430 CrossRef.
- S. Dadkhah-Tehrani, M. Shabani-Nooshabadi and M. Moradian, J. Iran. Chem. Soc., 2018, 15, 171–179 CrossRef.
- D. Nematollahi, A. Afkhami, E. Tammari, T. Shariatmanesh, M. Hesariac and M. Shojaeifard, Chem. Commun., 2007, 162–164 RSC.
- D. Nematollahi and H. Khoshsafar, Tetrahedron, 2009, 65, 4742–4750 CrossRef.
- D. Nematollahi and E. Tammari, Electrochim. Acta, 2005, 50, 3648–3654 CrossRef.
- A. B. Moghaddam, M. R. Ganjali, P. Norouzi and M. Latifi, Chem. Pharm. Bull., 2006, 54, 1391–1396 CrossRef PubMed.
- M. Rafiee and D. Nematollahi, Chem. Pharm. Bull., 2007, 55, 915–917 CrossRef PubMed.
- D. Nematollahi, M. Alimoradi and M. Rafiee, J. Phys. Org. Chem., 2007, 20, 49–54 CrossRef.
- D. Nematollahi, M. Rafiee and H. R. Khavasi, Bull. Chem. Soc. Jpn., 2008, 81, 1505–1511 CrossRef.
- D. Nematollahi and H. Shayani-jam, J. Org. Chem., 2008, 73, 3428–3434 CrossRef PubMed.
- D. Nematollahi, S. Dehdashtian and A. Niazi, J. Electroanal. Chem., 2008, 616, 79–86 CrossRef.
- D. Nematollahi and S. Dehdashtian, Tetrahedron Lett., 2008, 49, 645–649 CrossRef.
- S. S. Hosseiny Davarani, N. Sheijooni Fumani, H. Arvin-Nezhad and F. Moradi, Tetrahedron Lett., 2008, 49, 710–714 CrossRef.
- C.-C. Zeng, D.-W. Ping, Y.-S. Xu, L.-M. Hu and R.-G. Zhong, Eur. J. Org. Chem., 2009, 5832–5840 CrossRef.
- S. S. Hosseiny Davarani, N. Sheijooni Fumani, S. Vahidi, M.-A. Tabatabaei and H. Arvin-Nezhad, J. Heterocycl. Chem., 2010, 47, 40–45 Search PubMed.
- C.-C. Zeng, D.-W. Ping, L.-M. Hu, X.-Q. Song and R.-G. Zhong, Org. Biomol. Chem., 2010, 8, 2465–2472 RSC.
- Y.-X. Bai, D.-W. Ping, R. D. Little, H.-Y. Tian, L.-M. Hu and C.-C. Zeng, Tetrahedron, 2011, 67, 9334–9341 CrossRef.
- H. Salehzadeh, D. Nematollahi and M. Rafiee, J. Electroanal. Chem., 2011, 650, 226–232 CrossRef.
- N.-T. Zhang, X.-G. Gao, C.-C. Zeng, L.-M. Hu, H.-Y. Tian and Y.-B. She, RSC Adv., 2012, 2, 298–306 RSC.
- A. Amani, S. Khazalpour and D. Nematollahi, J. Electroanal. Chem., 2012, 670, 36–41 CrossRef.
- D. Nematollahi, S. S. Hosseiny Davarani and P. Mirahmadpour, ACS Sustainable Chem. Eng., 2014, 2, 579–583 CrossRef.
- M. Ameri, A. Asghari, A. Amoozadeh, M. Bakherad and D. Nematollahi, Chin. Chem. Lett., 2014, 25, 1607–1610 CrossRef.
- D. Nematollahi, S. Momeni and S. Khazalpour, Electrochim. Acta, 2014, 147, 310–318 CrossRef.
- A. Asghari, M. Ameri, A. A. Ziarati, S. Radmannia, A. Amoozadeh, B. Barfi and L. Boutorabi, Chin. Chem. Lett., 2015, 26, 681–684 CrossRef.
- E. Salahifar, D. Nematollahi, M. Bayat, A. Mahyari and H. Amiri Rudbari, Org. Lett., 2015, 17, 4666–4669 CrossRef PubMed.
- C.-C. Zeng, C.-F. Liu, J. Zeng and R.-G. Zhong, J. Electroanal. Chem., 2007, 608, 85–90 CrossRef.
- D. Nematollahi and F. Varmaghani, Electrochim. Acta, 2008, 53, 3350–3355 CrossRef.
- D. Nematollahi and R. Esmaili, Tetrahedron Lett., 2010, 51, 4862–4865 CrossRef.
- H.-L. Xiao, C.-W. Yang, N.-T. Zhang, C.-C. Zeng, L.-M. Hu, H.-Y. Tian and R. D. Little, Tetrahedron, 2013, 69, 658–663 CrossRef.
- H. Salehzadeh, M. Rafiee, D. Nematollahi and H. Beiginejad, Curr. Org. Chem., 2013, 17, 848–852 CrossRef.
- H. Beiginejad and D. Nematollahi, J. Org. Chem., 2014, 79, 6326–6329 CrossRef PubMed.
- A. Alizadeh, M. M. Khodaei, M. Fakhari and M. Shamsuddin, RSC Adv., 2014, 4, 20781–20788 RSC.
- S. Kaihani, H. Salehzadeh and D. Nematollahi, Electrochim. Acta, 2015, 157, 166–174 CrossRef.
- E. Salahifar and D. Nematollahi, New J. Chem., 2015, 39, 3852–3858 RSC.
- S. Khazalpour and D. Nematollahi, Green Chem., 2015, 17, 3508–3514 RSC.
- M. Sharafi-Kolkeshvandi, D. Nematollahi, F. Nikpour and E. Salahifar, New J. Chem., 2016, 40, 5442–5447 RSC.
- M. Jamshidi, D. Nematollahi, M. Bayat and E. Salahifar, J. Electrochem. Soc., 2016, 163, G211–G218 CrossRef.
- D. Nematollahi, S. Khazalpour, M. Ranjbar and S. Momeni, Sci. Rep., 2017, 7, 4436 CrossRef PubMed.
- S. Momeni and D. Nematollahi, Sci. Rep., 2017, 7, 41963 CrossRef PubMed.
- M. Sharafi-Kolkeshvandi, D. Nematollahi and F. Nikpour, Synthesis, 2017, 1555–1560 Search PubMed.
- D. Nematollahi and E. Tammari, J. Org. Chem., 2005, 70, 7769–7772 CrossRef PubMed.
- M. Shamsipur, S. S. Hosseiny Davarani, M. Nasiri-Aghdam and D. Nematollahi, Electrochim. Acta, 2006, 51, 3327–3331 CrossRef.
- C.-C. Zeng, D.-W. Ping, S.-C. Zhang, R.-G. Zhong and J. Y. Becker, J. Electroanal. Chem., 2008, 622, 90–96 CrossRef.
- F.-J. Liu, C.-C. Zeng, D.-W. Ping, Y.-L. Cai and R.-G. Zhong, Chin. J. Chem., 2008, 26, 1651–1655 CrossRef.
- D. Nematollahi, J. Azizian, M. Sargordan-Arani, M. Hesari, S. Jameh-Bozorghi, A. Alizadeh, L. Fotouhi and B. Mirza, Chem. Pharm. Bull., 2008, 56, 1562–1566 CrossRef PubMed.
- M. M. Khodaei, A. Alizadeh and N. Pakravan, J. Org. Chem., 2008, 73, 2527–2532 CrossRef PubMed.
- A. R. Fakhari, S. S. Hosseiny Davarani, H. Ahmar and S. Makarem, J. Appl. Electrochem., 2008, 38, 1743–1747 CrossRef.
- P. R. Jamaat, S. S. Hosseiny Davarani, N. Sheijooni Fumani, L. Masoumi and N. Safari, J. Porphyrins phthalocyanines, 2008, 12, 85–93 CrossRef.
- C.-C. Zeng, F.-J. Liu, D.-W. Ping, L.-M. Hu, Y.-L. Cai and R.-G. Zhong, Tetrahedron, 2009, 65, 4505–4512 CrossRef.
- C.-C. Zeng, F.-J. Liu, D.-W. Ping, Y.-L. Cai, R.-G. Zhong and J. Y. Becker, J. Electroanal. Chem., 2009, 625, 131–137 CrossRef.
- A. R. Fakhari, S. S. Hosseiny Davarani, H. Ahmar, K. Hasheminasab and H. R. Khavasi, J. Heterocycl. Chem., 2009, 46, 443–446 CrossRef.
- D. Nematollahi, J. Azizian, M. Sargordan-Arani, M. Hesari and B. Mirza, J. Heterocycl. Chem., 2009, 46, 1000–1002 CrossRef.
- A. Amani and D. Nematollahi, J. Org. Chem., 2012, 77, 11302–11306 CrossRef PubMed.
- M. Ameri, A. Asghari, A. Amoozadeh, H. Daneshinejad and D. Nematollahi, Chin. Chem. Lett., 2014, 25, 797–801 CrossRef.
- D. Nematollahi, S. Mahdinia, P. Karimi, H. Salehzadeh and S. Kaihani, RSC Adv., 2015, 5, 29209–29213 RSC.
- E. Tammari, A. Nezhadali and S. Lotfi, Electroanalysis, 2015, 27, 1693–1698 CrossRef.
- O. Ghaderi, A. Asghari, M. Ameri and M. Rajabi, Chem. Lett., 2016, 45, 430–432 CrossRef.
- I. Tabaković, Z. Grujić and Z. Bejtović, J. Heterocycl. Chem., 1983, 20, 635–638 CrossRef.
- D. Nematollahi, D. Habibi, M. Rahmati and M. Rafiee, J. Org. Chem., 2004, 69, 2637–2640 CrossRef PubMed.
- D. Nematollahi and M. Rafiee, Green Chem., 2005, 7, 638–644 RSC.
- S. S. Hosseiny Davarani, D. Nematollahi, M. Shamsipur, N. M. Najafi, L. Masoumi and S. Ramyar, J. Org. Chem., 2006, 71, 2139–2142 CrossRef PubMed.
- S. S. Hosseiny Davarani, N. Mashkouri Najafi, S. Ramyar, L. Masoumi and M. Shamsipur, Chem. Pharm. Bull., 2006, 54, 959–962 CrossRef.
- M. Shamsipur, S. S. Hosseiny Davarani and D. Nematollahi, J. Heterocycl. Chem., 2006, 43, 1673–1677 CrossRef.
- A. Bayandori Moghaddam, F. Kobarfard, S. S. Hosseiny Davarani, D. Nematollahi, M. Shamsipur and A. R. Fakhari, J. Electroanal. Chem., 2006, 586, 161–166 CrossRef.
- D. Nematollahi, A. Amani and E. Tammari, J. Org. Chem., 2007, 72, 3646–3651 CrossRef PubMed.
- S. S. Hosseiny Davarani, M. Shamsipur, D. Nematollahi, S. Ramyar and L. Masoumi, Chem. Pharm. Bull., 2007, 55, 1198–1202 CrossRef.
- S. Osati, N. Safari, M. Kalate-Bojdi and S. S. Hosseiny Davarani, J. Electroanal. Chem., 2011, 655, 120–127 CrossRef.
- M. Ameri, A. Asghari, A. Amoozadeh, M. Bakherad and D. Nematollahi, Tetrahedron Lett., 2015, 56, 2141–2144 CrossRef.
- N. Pakravan, D. Habibi, F. Varmaghani and M. Rahmati, J. Electroanal. Chem., 2017, 801, 206–214 CrossRef.
- D. Nematollahi, A. R. Atlasi-Pak and R. Esmaili, Helv. Chim. Acta, 2012, 95, 1605–1612 CrossRef.
- S. S. Hosseiny Davarani, A. R. Fakhari, N. Sheijooni Fumani and M. Kalate-bojdi, Electrochem. Commun., 2008, 10, 1765–1768 CrossRef.
- L. Fotouhi, M. Mosavi, M. M. Heravi and D. Nematollahi, Tetrahedron Lett., 2006, 47, 8553–8557 CrossRef.
- E. Blattes, M.-B. Fleury and M. Largeron, Electrochim. Acta, 2005, 50, 4902–4910 CrossRef.
- D. Xu, A. Chiaroni, M.-B. Fleury and M. Largeron, J. Org. Chem., 2006, 71, 6374–6381 CrossRef PubMed.
- H. Salehzadeh, D. Nematollahi and H. Hesari, Green Chem., 2013, 15, 2441–2446 RSC.
- See also: Y. Shih, C. Ke, C. Pan and Y. Huang, RSC Adv., 2013, 3, 7330–7336 RSC.
- Z. Grujić, I. Tabaković and M. Trkovnik, Tetrahedron Lett., 1976, 17, 4823–4824 CrossRef.
- S. M. Golabi and D. Nematollahi, J. Electroanal. Chem., 1997, 420, 127–134 CrossRef.
- H. Ghadimi, A. S. M. Ali, N. Mohamed and S. Ab Ghani, J. Electrochem. Soc., 2012, 159, E127–E131 CrossRef.
- C.-C. Zeng, F.-J. Liu, D.-W. Ping, L.-M. Hu, Y.-L. Cai and R.-G. Zhong, J. Org. Chem., 2009, 74, 6386–6389 CrossRef PubMed.
- A. Asghari, M. Ameri, S. Radmannia, M. Rajabi, M. Bakherad and D. Nematollahi, J. Electroanal. Chem., 2014, 733, 47–52 CrossRef.
- M. Shabani-Nooshabadi, M. Moradian and S. Dadkhah-Tehrani, Bull. Chem. Soc. Jpn., 2017, 90, 68–73 CrossRef.
- S. S. Hosseiny Davarani, A. R. Fakhari, A. Shaabani, H. Ahmar, A. Maleki and N. Sheijooni Fumani, Tetrahedron Lett., 2008, 49, 5622–5624 CrossRef.
- M. Sharafi-Kolkeshvandi, D. Nematollahi, F. Nikpour, B. Dadpou and H. Alizadeh, Electrochim. Acta, 2016, 214, 147–155 CrossRef.
- S. S. Hosseiny Davarani, M. Kalate Bojdi and A. Mehdinia, Chem. Pharm. Bull., 2011, 59, 1209–1213 CrossRef PubMed.
- D. Habibi, D. Nematollahi, H. M. Pordanjani and M. R. Sadeghi, J. Heterocycl. Chem., 2015, 52, 197–200 CrossRef.
- S. Shahrokhian and A. Hamzehloei, Electrochem. Commun., 2003, 5, 706–710 CrossRef.
- D. Nematollahi, M. S. Workentin and E. Tammari, Chem. Commun., 2006, 1631–1633 RSC.
- D. Nematollahi and A. Ghorbani, J. Electroanal. Chem., 2008, 624, 310–314 CrossRef.
- K. Bechgaard and V. D. Parker, J. Am. Chem. Soc., 1972, 94, 4749–4750 CrossRef.
- J.-M. Chapuzet and J. Simonet, Tetrahedron, 1991, 47, 791–798 CrossRef.
- S. R. Waldvogel and D. Mirk, Tetrahedron Lett., 2000, 41, 4769–4772 CrossRef.
- M. Largeron, A. Neudorffer and M.-B. Fleury, Angew. Chem., Int. Ed., 2003, 42, 1026–1029 CrossRef PubMed.
- M. Largeron, A. Chiaroni and M.-B. Fleury, Chem. – Eur. J., 2008, 14, 996–1003 CrossRef PubMed.
- M. Largeron and M.-B. Fleury, Org. Lett., 2009, 11, 883–886 CrossRef PubMed.
- K. L. Jensen, P. T. Franke, L. T. Nielsen, K. Daasbjerg and K. A. Jørgensen, Angew. Chem., Int. Ed., 2010, 49, 129–133 CrossRef PubMed.
- For a related enantioselective approach for α-alkylation of aldehydes through the merger of electro- and organocatalysis, see: X.-H. Ho, S.-i. Mho, H. Kang and H.-Y. Jang, Eur. J. Org. Chem., 2010, 4436–4441 Search PubMed.
- For electrochemical synthesis of enaminones, see:
(a) K. Xu, Z. Zhang, P. Qian, Z. Zha and Z. Wang, Chem. Commun., 2015, 51, 11108–11111 RSC;
(b) Y. Li, H. Gao, Z. Zhang, P. Qian, M. Bi, Z. Zha and Z. Wang, Chem. Commun., 2016, 52, 8600–8603 RSC.
- D. Koch and H. Schäfer, Angew. Chem., Int. Ed. Engl., 1973, 12, 245–246 CrossRef.
- See also:
(a) Ž. Saničanin and I. Tabaković, Electrochim. Acta, 1988, 33, 1601–1604 CrossRef;
(b) I. Tabaković, Electrochim. Acta, 1995, 40, 2809–2813 CrossRef.
- W. Eilenberg and H. J. Schäfer, Tetrahedron Lett., 1984, 25, 5023–5026 CrossRef.
- S. Tang, X. Gao and A. Lei, Chem. Commun., 2017, 53, 3354–3356 RSC.
- Z. He, W. Liu and Z. Li, Chem. – Asian J., 2011, 6, 1340–1343 CrossRef PubMed.
- For preparation of polycyclic indoles through anodic oxidation, see:
(a) X.-G. Gao, N.-T. Zhanga, C.-C. Zeng, Y.-D. Liu, L.-M. Hu and H.-Y. Tian, Curr. Org. Synth., 2014, 11, 141–148 CrossRef;
(b) M. Ameri, A. Asghari, A. Amoozadeh and M. Bakherad, Chin. Chem. Lett., 2017, 28, 1031–1034 CrossRef.
- For synthetic approaches to indoles involving electroreduction, see:
(a) S. C. Mishra and R. A. Mishra, J. Electrochem. Soc. India, 1990, 39, 51–53 Search PubMed;
(b) F. LeStrat, J. A. Murphy and M. Hughes, Org. Lett., 2002, 4, 2735–2738 CrossRef PubMed;
(c) L. Bouchard, I. Marcotte, J. M. Chapuzet and J. Lessard, Can. J. Chem., 2003, 81, 1108–1118 CrossRef;
(d) P. Du and D. G. Peters, J. Electrochem. Soc., 2010, 157, F167–F172 CrossRef;
(e) P. Du, J. L. Brosmer and D. G. Peters, Org. Lett., 2011, 13, 4072–4075 CrossRef PubMed.
- K. D. Moeller and D. G. New, Tetrahedron Lett., 1994, 35, 2857–2860 CrossRef.
- D. G. New, Z. Tesfai and K. D. Moeller, J. Org. Chem., 1996, 61, 1578–1598 CrossRef PubMed.
- D. A. Frey, N. Wu and K. D. Moeller, Tetrahedron Lett., 1996, 37, 8317–8320 CrossRef.
- C. R. Whitehead, E. H. Sessions, I. Ghiviriga and D. L. Wright, Org. Lett., 2002, 4, 3763–3765 CrossRef PubMed.
- J. B. Sperry and D. L. Wright, Tetrahedron Lett., 2005, 46, 411–414 CrossRef.
- J. B. Sperry, C. R. Whitehead, I. Ghiviriga, R. M. Walczak and D. L. Wright, J. Org. Chem., 2004, 69, 3726–3734 CrossRef PubMed.
- A. Redden and K. D. Moeller, Org. Lett., 2011, 13, 1678–1681 CrossRef PubMed.
- J. B. Sperry and D. L. Wright, J. Am. Chem. Soc., 2005, 127, 8034–8035 CrossRef PubMed.
- I. Kuwajima and H. Urabe, Tetrahedron Lett., 1981, 22, 5191–5194 CrossRef.
- A. Padwa and S. S. Murphree, Org. Prep. Proced. Int., 1991, 23, 545–568 CrossRef.
- C. O. Kappe, S. S. Murphree and A. Padwa, Tetrahedron, 1997, 53, 14179–14233 CrossRef.
- D. L. Wright, C. R. Whitehead, E. H. Sessions, I. Ghiviriga and D. A. Frey, Org. Lett., 1999, 1, 1535–1538 CrossRef PubMed.
- D. L. Wright, Prog. Heterocycl. Chem., 2005, 17, 1–32 Search PubMed.
- M. A. Battiste, P. M. Pelphrey and D. L. Wright, Chem. – Eur. J., 2006, 12, 3438–3447 CrossRef PubMed.
- M. Harmata, Chem. Commun., 2010, 46, 8904–8922 RSC.
- J. B. Sperry and D. L. Wright, Tetrahedron, 2006, 62, 6551–6557 CrossRef.
- J. B. Sperry, I. Ghiviriga and D. L. Wright, Chem. Commun., 2006, 194–196 RSC.
- See also: C. Adouama, R. Keyrouz, G. Pilet, C. Monnereau, D. Gueyrard, T. Noël and M. Médebielle, Chem. Commun., 2017, 53, 5653–5656 RSC.
- H. Wu and K. D. Moeller, Org. Lett., 2007, 9, 4599–4602 CrossRef PubMed.
- K. Chiba, T. Miura, S. Kim, Y. Kitano and M. Tada, J. Am. Chem. Soc., 2001, 123, 11314–11315 CrossRef PubMed.
- T. Miura, S. Kim, Y. Kitano, M. Tada and K. Chiba, Angew. Chem., Int. Ed., 2006, 45, 1461–1463 CrossRef PubMed.
- M. Arata, T. Miura and K. Chiba, Org. Lett., 2007, 9, 4347–4350 CrossRef PubMed.
- Y. Okada, R. Akaba and K. Chiba, Org. Lett., 2009, 11, 1033–1035 CrossRef PubMed.
- Y. Okada and K. Chiba, Electrochim. Acta, 2010, 55, 4112–4119 CrossRef.
- Y. Okada and K. Chiba, Electrochim. Acta, 2011, 56, 1037–1042 CrossRef.
- Y. Okada, A. Nishimoto, R. Akaba and K. Chiba, J. Org. Chem., 2011, 76, 3470–3476 CrossRef PubMed.
- Y. Yamaguchi, Y. Okada and K. Chiba, J. Org. Chem., 2013, 78, 2626–2638 CrossRef PubMed.
- J. C. Moore, E. S. Davies, D. A. Walsh, P. Sharma and J. E. Moses, Chem. Commun., 2014, 50, 12523–12525 RSC.
- Y. Okada, Y. Yamaguchi, A. Ozaki and K. Chiba, Chem. Sci., 2016, 7, 6387–6393 RSC.
- A. Ozaki, Y. Yamaguchi, Y. Okada and K. Chiba, ChemElectroChem, 2017, 4, 1852–1855 CrossRef.
- Y. Okada, R. Akaba and K. Chiba, Tetrahedron Lett., 2009, 50, 5413–5416 CrossRef.
- For a review on redox-tag processes, see: Y. Okada and K. Chiba, Chem. Rev., 2018, 118, 4592–4630 CrossRef PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- For an overview on metal-catalyzed C–N bond forming cross-coupling reactions, see:
(a) J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC;
(b) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC;
(c) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef PubMed;
(d) S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef PubMed;
(e) J. Schranck and A. Tlili, ACS Catal., 2018, 8, 405–418 CrossRef.
- For recent reviews on catalytic methods for C–H amination, see:
(a) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef PubMed;
(b) J. Jiao, K. Murakami and K. Itami, ACS Catal., 2016, 6, 610–633 CrossRef;
(c) H. Kim and S. Chang, ACS Catal., 2016, 6, 2341–2351 CrossRef;
(d) Y. Zhou, J. Yuan, Q. Yang, Q. Xiao and Y. Peng, ChemCatChem, 2016, 8, 2178–2192 CrossRef;
(e) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef PubMed;
(f) H. Kim and S. Chang, Acc. Chem. Res., 2017, 50, 482–486 CrossRef PubMed.
- R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef PubMed.
- T. J. Donohoe, C. K. A. Callens, A. Flores, A. R. Lacy and A. H. Rathi, Chem. – Eur. J., 2011, 17, 58–76 CrossRef PubMed.
- S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938–10953 CrossRef PubMed.
- S. Pan and T. Shibata, ACS Catal., 2013, 3, 704–712 CrossRef.
- S. R. Chemler and M. T. Bovino, ACS Catal., 2013, 3, 1076–1091 CrossRef PubMed.
- M. Chiarucci and M. Bandini, Beilstein J. Org. Chem., 2013, 9, 2586–2614 CrossRef PubMed.
- A. M. Beauchemin, Org. Biomol. Chem., 2013, 11, 7039–7050 RSC.
- J. C.-H. Yim and L. L. Schafer, Eur. J. Org. Chem., 2014, 6825–6840 CrossRef.
- L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596–2697 CrossRef PubMed.
- E. Bernoud, C. Lepori, M. Mellah, E. Schulz and J. Hannedouche, Catal. Sci. Technol., 2015, 5, 2017–2037 RSC.
- S. M. Coman and V. I. Parvulescu, Org. Process Res. Dev., 2015, 19, 1327–1355 CrossRef.
- V. Rodriguez-Ruiz, R. Carlino, S. Bezzenine-Lafollée, R. Gil, D. Prim, E. Schulz and J. Hannedouche, Dalton Trans., 2015, 44, 12029–12059 RSC.
- M. T. Pirnot, Y.-M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48–57 CrossRef PubMed.
- J. R. Coombs and J. P. Morken, Angew. Chem., Int. Ed., 2016, 55, 2636–2649 CrossRef PubMed.
- G. E. M. Crisenza and J. F. Bower, Chem. Lett., 2016, 45, 2–9 CrossRef.
- Z. Sorádová and R. Šebesta, ChemCatChem, 2016, 8, 2581–2588 CrossRef.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef PubMed.
- M. Patel, R. K. Saunthwal and A. K. Verma, Acc. Chem. Res., 2017, 50, 240–254 CrossRef PubMed.
- X.-W. Lan, N.-X. Wang and Y. Xing, Eur. J. Org. Chem., 2017, 5821–5851 CrossRef.
- C. Lepori and J. Hannedouche, Synthesis, 2017, 1158–1167 Search PubMed.
- P. Kalck and M. Urrutigoïty, Chem. Rev., 2018, 118, 3833–3861 CrossRef PubMed.
- K. D. Moeller, M. R. Marzabadi, D. G. New, M. Y. Chiang and S. Keith, J. Am. Chem. Soc., 1990, 112, 6123–6124 CrossRef.
- C. M. Hudson, M. R. Marzabadi, K. D. Moeller and D. G. New, J. Am. Chem. Soc., 1991, 113, 7372–7385 CrossRef.
- K. D. Moeller and L. V. Tinao, J. Am. Chem. Soc., 1992, 114, 1033–1041 CrossRef.
- L. V. Tinao-Wooldridge, K. D. Moeller and C. M. Hudson, J. Org. Chem., 1994, 59, 2381–2389 CrossRef.
- D. A. Frey, S. H. Krishna Reddy and K. D. Moeller, J. Org. Chem., 1999, 64, 2805–2813 CrossRef PubMed.
- S. H. Krishna Reddy, K. Chiba, Y. Sun and K. D. Moeller, Tetrahedron, 2001, 57, 5183–5197 CrossRef.
- K. D. Moeller and C. M. Hudson, Tetrahedron Lett., 1991, 32, 2307–2310 CrossRef.
- D. A. Frey, J. A. Marx and K. D. Moeller, Electrochim. Acta, 1997, 42, 1967–1970 CrossRef.
- S. H. Krishna Reddy and K. D. Moeller, Tetrahedron Lett., 1998, 39, 8027–8030 CrossRef.
- Y. Sun and K. D. Moeller, Tetrahedron Lett., 2002, 43, 7159–7161 CrossRef.
- Y.-t. Huang and K. D. Moeller, Org. Lett., 2004, 6, 4199–4202 CrossRef PubMed.
- Y.-t. Huang and K. D. Moeller, Tetrahedron, 2006, 62, 6536–6550 CrossRef.
- F. Tang and K. D. Moeller, J. Am. Chem. Soc., 2007, 129, 12414–12415 CrossRef PubMed.
- A. Redden, R. J. Perkins and K. D. Moeller, Angew. Chem., Int. Ed., 2013, 52, 12865–12868 CrossRef PubMed.
- A. Sutterer and K. D. Moeller, J. Am. Chem. Soc., 2000, 122, 5636–5637 CrossRef.
- Y. Sun, B. Liu, J. Kao, D. A. d’Avignon and K. D. Moeller, Org. Lett., 2001, 3, 1729–1732 CrossRef PubMed.
- B. Liu and K. D. Moeller, Tetrahedron Lett., 2001, 42, 7163–7165 CrossRef.
- S. Duan and K. D. Moeller, J. Am. Chem. Soc., 2002, 124, 9368–9369 CrossRef PubMed.
- B. Liu, S. Duan, A. C. Sutterer and K. D. Moeller, J. Am. Chem. Soc., 2002, 124, 10101–10111 CrossRef PubMed.
- J. D. Brandt and K. D. Moeller, Org. Lett., 2005, 7, 3553–3556 CrossRef PubMed.
- J. D. Brandt and K. D. Moeller, Heterocycles, 2006, 67, 621–628 CrossRef.
- H.-C. Xu, J. D. Brandt and K. D. Moeller, Tetrahedron Lett., 2008, 49, 3868–3871 CrossRef.
- G. Xu and K. D. Moeller, Org. Lett., 2010, 12, 2590–2593 CrossRef PubMed.
- J. A. Smith and K. D. Moeller, Org. Lett., 2013, 15, 5818–5821 CrossRef PubMed.
- R. J. Perkins, H.-C. Xu, J. M. Campbell and K. D. Moeller, Beilstein J. Org. Chem., 2013, 9, 1630–1636 CrossRef PubMed.
- J. A. Smith, G. Xu, R. Feng, J. W. Janetka and K. D. Moeller, Electroanalysis, 2016, 28, 2808–2817 CrossRef.
- For a different approach to electrochemical oxyfunctionalization of alkenes, see: P. Röse, S. Emge, J.-i. Yoshida and G. Hilt, Beilstein J. Org. Chem., 2015, 11, 174–183 CrossRef PubMed.
- H.-C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2008, 130, 13542–13543 CrossRef PubMed.
- H.-C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2010, 132, 2839–2844 CrossRef PubMed.
- For an approach to access six-membered ring systems, see: H.-C. Xu and K. D. Moeller, Org. Lett., 2010, 12, 5174–5177 CrossRef PubMed.
- H.-C. Xu and K. D. Moeller, Org. Lett., 2010, 12, 1720–1723 CrossRef PubMed.
- J. M. Campbell, H.-C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2012, 134, 18338–18344 CrossRef PubMed.
- For early work on kinetic studies of enol ether radical cations, see: J. H. Horner, E. Taxil and M. Newcomb, J. Am. Chem. Soc., 2002, 124, 5402–5410 CrossRef PubMed.
- L. A. Anderson, A. Redden and K. D. Moeller, Green Chem., 2011, 13, 1652–1654 RSC.
- H.-C. Xu and K. D. Moeller, Angew. Chem., Int. Ed., 2010, 49, 8004–8007 CrossRef PubMed.
- Y. Ashikari, T. Nokami and J.-i. Yoshida, Org. Biomol. Chem., 2013, 11, 3322–3331 RSC.
- N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef PubMed.
- J. B. Parry, N. Fu and S. Lin, Synlett, 2018, 257–265 Search PubMed.
- M. Zanda, SYNFORM, 2017, A202–A204 Search PubMed.
- For seminal work on electrochemical diazidation of alkenes, see: H. Schäfer, Angew. Chem., Int. Ed. Engl., 1970, 9, 158–159 CrossRef.
- N. Fu, G. S. Sauer and S. Lin, J. Am. Chem. Soc., 2017, 139, 15548–15553 CrossRef PubMed.
- K.-Y. Ye, G. Pombar, N. Fu, G. S. Sauer, I. Keresztes and S. Lin, J. Am. Chem. Soc., 2018, 140, 2438–2441 CrossRef PubMed.
- For a recent review on radical difunctionalization of alkenes, see: G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175–5187 CrossRef.
- C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383–2387 CrossRef PubMed.
- S. Tang, D. Wang, Y. Liu, L. Zeng and A. Lei, Nat. Commun., 2018, 9, 798 CrossRef PubMed.
- N. Sauermann, R. Mei and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5090–5094 CrossRef PubMed.
- X. Gao, P. Wang, L. Zeng, S. Tang and A. Lei, J. Am. Chem. Soc., 2018, 140, 4195–4199 CrossRef PubMed.
- F. Xu, Y.-J. Li, C. Huang and H.-C. Xu, ACS Catal., 2018, 8, 3820–3824 CrossRef.
- M. E. Wolff, Chem. Rev., 1963, 63, 55–64 CrossRef.
- R. S. Neale, Synthesis, 1971, 1–15 CrossRef.
- L. Stella, Angew. Chem., Int. Ed. Engl., 1983, 22, 337–350 CrossRef.
- G. Majetich and K. Wheless, Tetrahedron, 1995, 51, 7095–7129 CrossRef.
- S. Z. Zard, Synlett, 1996, 1148–1154 CrossRef.
- W. R. Bowman, A. J. Fletcher and G. B. S. Potts, J. Chem. Soc., Perkin Trans. 1, 2002, 2747–2762 RSC.
- S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603–1618 RSC.
- J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092–4106 RSC.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC.
- R. K. Rit, M. Shankar and A. K. Sahoo, Org. Biomol. Chem., 2017, 15, 1282–1293 RSC.
- K. Murakami, G. J. P. Perry and K. Itami, Org. Biomol. Chem., 2017, 15, 6071–6075 RSC.
- T. Siu and A. K. Yudin, J. Am. Chem. Soc., 2002, 124, 530–531 CrossRef PubMed.
- T. Siu, C. J. Picard and A. K. Yudin, J. Org. Chem., 2005, 70, 932–937 CrossRef PubMed.
- For electrochemical aziridination proceeding via initial alkene oxidation, see: J. Li, W. Huang, J. Chen, L. He, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2018, 57, 5695–5698 CrossRef PubMed.
- T. Siu and A. K. Yudin, Org. Lett., 2002, 4, 1839–1842 CrossRef PubMed.
- H.-C. Xu, J. M. Campbell and K. D. Moeller, J. Org. Chem., 2014, 79, 379–391 CrossRef PubMed.
- P. Xiong, H.-H. Xu and H.-C. Xu, J. Am. Chem. Soc., 2017, 139, 2956–2959 CrossRef PubMed.
- L. Zhu, P. Xiong, Z.-Y. Mao, Y.-H. Wang, X. Yan, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 2226–2229 CrossRef PubMed.
- F. Xu, L. Zhu, S. Zhu, X. Yan and H.-C. Xu, Chem. – Eur. J., 2014, 20, 12740–12744 CrossRef PubMed.
- A. A. Folgueiras-Amador, K. Philipps, S. Guilbaud, J. Poelakker and T. Wirth, Angew. Chem., Int. Ed., 2017, 56, 15446–15450 CrossRef PubMed.
- T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, Chem. Commun., 2017, 53, 2974–2977 RSC.
- Z.-W. Hou, Z.-Y. Mao, H.-B. Zhao, Y. Y. Melcamu, X. Lu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 9168–9172 CrossRef PubMed.
- Z.-W. Hou, Z.-Y. Mao and H.-C. Xu, Synlett, 2017, 1867–1872 Search PubMed.
- Z.-W. Hou, Z.-Y. Mao, Y. Y. Melcamu, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2018, 57, 1636–1639 CrossRef PubMed.
- For electrochemical homo-coupling of imidazopyridine-based heterocycles, see: Y. Gao, Y. Wang, J. Zhou, H. Mei and J. Han, Green Chem., 2018, 20, 583–587 RSC.
- Z.-W. Hou, Z.-Y. Mao, J. Song and H.-C. Xu, ACS Catal., 2017, 7, 5810–5813 CrossRef.
- For ferrocene-mediated electrochemical difluoromethylarylation of alkynes, see: P. Xiong, H.-H. Xu, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 2460–2464 CrossRef PubMed.
- For ferrocene-mediated intermolecular dehydrogenative cross-coupling of N-alkyloxyamides with xanthenes, see: M.-Y. Lin, K. Xu, Y.-Y. Jiang, Y.-G. Liu, B.-G. Sun and C.-C. Zeng, Adv. Synth. Catal., 2018, 360, 1665–1672 CrossRef.
- H.-B. Zhao, Z.-W. Hou, Z.-J. Liu, Z.-F. Zhou, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 587–590 CrossRef PubMed.
- For synthesis of benzimidazoles through the merger of electrochemistry and Co catalysis, see: Y.-L. Lai, J.-S. Ye and J.-M. Huang, Chem. – Eur. J., 2016, 22, 5425–5429 CrossRef PubMed.
- H.-B. Zhao, Z.-J. Liu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 12732–12735 CrossRef PubMed.
- K. Subramanian, S. L. Yedage and B. M. Bhanage, J. Org. Chem., 2017, 82, 10025–10032 CrossRef PubMed.
- B. R. Rosen, E. W. Werner, A. G. O’Brien and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5571–5574 CrossRef PubMed.
- S. R. Waldvogel and B. Janza, Angew. Chem., Int. Ed., 2014, 53, 7122–7123 CrossRef PubMed.
- A. Kehl, T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Chem. – Eur. J., 2018, 24, 590–593 CrossRef PubMed.
- T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 9437–9440 CrossRef PubMed.
- T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, J. Am. Chem. Soc., 2017, 139, 12317–12324 CrossRef PubMed.
- H. Lund, Acta Chem. Scand., 1957, 11, 1323–1330 CrossRef.
- G. Manning, V. D. Parker and R. N. Adams, J. Am. Chem. Soc., 1969, 91, 4584–4585 CrossRef.
- C. V. Ristagno and H. J. Shine, J. Org. Chem., 1971, 36, 4050–4055 CrossRef.
- H. N. Blount, J. Electroanal. Chem., 1973, 42, 271–274 CrossRef.
- M. Masui, H. Ohmori, H. Sayo, A. Ueda and C. Ueda, J. Chem. Soc., Perkin Trans. 2, 1976, 1180–1183 RSC.
- J. F. Evans and H. N. Blount, J. Org. Chem., 1976, 41, 516–519 CrossRef.
- B. Reitstöen and V. D. Parker, Acta Chem. Scand., 1992, 46, 464–468 CrossRef.
- Y. Li, S. Asaoka, T. Yamagishi and T. Iyoda, Electrochemistry, 2004, 72, 171–174 Search PubMed.
- See also: C. J. Schlesener and J. K. Kochi, J. Org. Chem., 1984, 49, 3142–3150 CrossRef.
- S. R. Waldvogel and S. Möhle, Angew. Chem., Int. Ed., 2015, 54, 6398–6399 CrossRef PubMed.
- For copper-promoted electrochemical amination of boronic acids, see: H.-L. Qi, D.-S. Chen, J.-S. Ye and J.-M. Huang, J. Org. Chem., 2013, 78, 7482–7487 CrossRef PubMed.
- T. Zincke, Justus Liebigs Ann. Chem., 1904, 330, 361–374 CrossRef.
- T. Zincke, G. Heuser and W. Möller, Justus Liebigs Ann. Chem., 1904, 333, 296–345 CrossRef.
- T. Zincke and W. Würker, Justus Liebigs Ann. Chem., 1904, 338, 107–141 CrossRef.
- S. Herold, S. Möhle, M. Zirbes, F. Richter, H. Nefzger and S. R. Waldvogel, Eur. J. Org. Chem., 2016, 1274–1278 CrossRef.
- S. Möhle, S. Herold, F. Richter, H. Nefzger and S. R. Waldvogel, ChemElectroChem, 2017, 4, 2196–2210 CrossRef.
- L. J. Wesenberg, S. Herold, A. Shimizu, J.-i. Yoshida and S. R. Waldvogel, Chem. – Eur. J., 2017, 23, 12096–12099 CrossRef PubMed.
- T. Morofuji, A. Shimizu and J.-i. Yoshida, Chem. – Eur. J., 2015, 21, 3211–3214 CrossRef PubMed.
- For a related dehydrogenative intramolecular C–N cross-coupling reaction, see: Z. Ye, M. Ding, Y. Wu, Y. Li, W. Hua and F. Zhang, Green Chem., 2018, 20, 1732–1737 RSC.
- T. Morofuji, A. Shimizu and J.-i. Yoshida, J. Am. Chem. Soc., 2014, 136, 4496–4499 CrossRef PubMed.
- For synthesis of pyren-1-yl azoliums via electrochemical oxidative C–N coupling, see: G. de Robillard, O. Makni, H. Cattey, J. Andrieu and C. H. Devillers, Green Chem., 2015, 17, 4669–4679 RSC.
- For an example on anodic flow oxidation of toluene derivatives to benzyl amides, see: M. A. Kabeshov, B. Musio and S. V. Ley, React. Chem. Eng., 2017, 2, 822–825 RSC.
- T. Morofuji, A. Shimizu and J.-i. Yoshida, J. Am. Chem. Soc., 2015, 137, 9816–9819 CrossRef PubMed.
- R. Hayashi, A. Shimizu, Y. Song, Y. Ashikari, T. Nokami and J.-i. Yoshida, Chem. – Eur. J., 2017, 23, 61–64 CrossRef PubMed.
- R. Hayashi, A. Shimizu and J.-i. Yoshida, J. Am. Chem. Soc., 2016, 138, 8400–8403 CrossRef PubMed.
- J. Wu, Y. Zhou, Y. Zhou, C.-W. Chiang and A. Lei, ACS Catal., 2017, 7, 8320–8323 CrossRef.
- For electrochemical C(sp3)–H functionalization of ethers with indoles, see: K.-S. Du and J.-M. Huang, Org. Lett., 2018, 20, 2911–2915 CrossRef PubMed.
- E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef PubMed.
- F. V. Singh and T. Wirth, Chem. – Asian J., 2014, 9, 950–971 CrossRef PubMed.
- G. Maertens, C. L’Homme and S. Canesi, Front. Chem., 2015, 2, 115 Search PubMed.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef PubMed.
- Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436–4454 CrossRef PubMed.
- L. Wang and J. Liu, Eur. J. Org. Chem., 2016, 1813–1824 CrossRef.
- A. Yoshimura, M. S. Yusubov and V. V. Zhdankin, Org. Biomol. Chem., 2016, 14, 4771–4781 RSC.
- F. C. Sousa e Silva, A. F. Tierno and S. E. Wengryniuk, Molecules, 2017, 22, 780 CrossRef PubMed.
- D. Kajiyama, K. Inoue, Y. Ishikawa and S. Nishiyama, Tetrahedron, 2010, 66, 9779–9784 CrossRef.
- D. Kajiyama, T. Saitoh, S. Yamaguchi and S. Nishiyama, Synthesis, 2012, 1667–1671 Search PubMed.
- Y. Amano, K. Inoue and S. Nishiyama, Synlett, 2008, 134–136 Search PubMed.
- Y. Amano and S. Nishiyama, Tetrahedron Lett., 2006, 47, 6505–6507 CrossRef.
- Y. Amano and S. Nishiyama, Heterocycles, 2008, 75, 1997–2003 CrossRef.
- K. Inoue, Y. Ishikawa and S. Nishiyama, Org. Lett., 2010, 12, 436–439 CrossRef PubMed.
- See also: D. Kajiyama, T. Saitoh and S. Nishiyama, Electrochemistry, 2013, 81, 319–324 CrossRef.
- T. Broese and R. Francke, Org. Lett., 2016, 18, 5896–5899 CrossRef PubMed.
- For the use of soluble polymediators and polyelectrolytes in electrosynthesis, see: B. Schille, N. O. Giltzau and R. Francke, Angew. Chem., Int. Ed., 2018, 57, 422–426 CrossRef PubMed.
- O. Koleda, T. Broese, J. Noetzel, M. Roemelt, E. Suna and R. Francke, J. Org. Chem., 2017, 82, 11669–11681 CrossRef PubMed.
- W.-C. Li, C.-C. Zeng, L.-M. Hu, H.-Y. Tian and R. D. Little, Adv. Synth. Catal., 2013, 355, 2884–2890 CrossRef.
- W.-J. Gao, W.-C. Li, C.-C. Zeng, H.-Y. Tian, L.-M. Hu and R. D. Little, J. Org. Chem., 2014, 79, 9613–9618 CrossRef PubMed.
- See also: E. O. Yurko, T. V. Gryaznova, K. V. Kholin, V. V. Khrizanforova and Y. H. Budnikova, Dalton Trans., 2018, 47, 190–196 RSC.
- S. Liang, C.-C. Zeng, H.-Y. Tian, B.-G. Sun, X.-G. Luo and F.-z. Ren, J. Org. Chem., 2016, 81, 11565–11573 CrossRef PubMed.
- W.-J. Gao, C. M. Lam, B.-G. Sun, R. D. Little and C.-C. Zeng, Tetrahedron, 2017, 73, 2447–2454 CrossRef.
- For synthesis of potential building blocks from lignin using electrochemistry, see: B. H. Nguyen, R. J. Perkins, J. A. Smith and K. D. Moeller, J. Org. Chem., 2015, 80, 11953–11962 CrossRef PubMed.
- See also: D. T. Rensing, B. H. Nguyen and K. D. Moeller, Org. Chem. Front., 2016, 3, 1236–1240 RSC.
- S. Liang, C.-C. Zeng, H.-Y. Tian, B.-G. Sun, X.-G. Luo and F.-z. Ren, Adv. Synth. Catal., 2018, 360, 1444–1452 CrossRef.
- L.-S. Kang, M.-H. Luo, C. M. Lam, L.-M. Hu, R. D. Little and C.-C. Zeng, Green Chem., 2016, 18, 3767–3774 RSC.
- X.-J. Pan, J. Gao and G.-Q. Yuan, Tetrahedron, 2015, 71, 5525–5530 CrossRef.
- For a review on iodine-catalyzed oxidative coupling reactions, see: D. Liu and A. Lei, Chem. – Asian J., 2015, 10, 806–823 CrossRef PubMed.
- J. Chen, W.-Q. Yan, C. M. Lam, C.-C. Zeng, L.-M. Hu and R. D. Little, Org. Lett., 2015, 17, 986–989 CrossRef PubMed.
- K. Fujimoto, Y. Tokuda, Y. Matsubara, H. Maekawa, T. Mizuno and I. Nishiguchi, Tetrahedron Lett., 1995, 36, 7483–7486 CrossRef.
- W.-q. Yan, M.-y. Lin, R. D. Little and C.-C. Zeng, Tetrahedron, 2017, 73, 764–770 CrossRef.
- S. Liang, C.-C. Zeng, X.-G. Luo, F.-z. Ren, H.-Y. Tian, B.-G. Sun and R. D. Little, Green Chem., 2016, 18, 2222–2230 RSC.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef PubMed.
- B. H. Nguyen, A. Redden and K. D. Moeller, Green Chem., 2014, 16, 69–72 RSC.
- B. H. Nguyen, R. J. Perkins, J. A. Smith and K. D. Moeller, Beilstein J. Org. Chem., 2015, 11, 280–287 CrossRef PubMed.
- K. N. Singh and A. K. Shukla, J. Sci. Ind. Res., 1999, 58, 327–331 Search PubMed.
- D. Pletcher, Electrochem. Commun., 2018, 88, 1–4 CrossRef.
- K.-P. Ho, T. H. Chan and K.-Y. Wong, Green Chem., 2006, 8, 900–905 RSC.
- S. Sengmany, A. Vitu-Thiebaud, E. Le Gall, S. Condon, E. Léonel, C. Thobie-Gautier, M. Pipelier, J. Lebreton and D. Dubreuil, J. Org. Chem., 2013, 78, 370–379 CrossRef PubMed.
- S. Sengmany, S. Vasseur, A. Lajnef, E. Le Gall and E. Léonel, Eur. J. Org. Chem., 2016, 4865–4871 CrossRef.
- H. Shimakoshi, Z. Luo, T. Inaba and Y. Hisaeda, Dalton Trans., 2016, 45, 10173–10180 RSC.
- C. Li, Y. Kawamata, H. Nakamura, J. C. Vantourout, Z. Liu, Q. Hou, D. Bao, J. T. Starr, J. Chen, M. Yan and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 13088–13093 CrossRef PubMed.
- R. J. Perkins, D. J. Pedro and E. C. Hansen, Org. Lett., 2017, 19, 3755–3758 CrossRef PubMed.
- H. Li, C. P. Breen, H. Seo, T. F. Jamison, Y.-Q. Fang and M. M. Bio, Org. Lett., 2018, 20, 1338–1341 CrossRef PubMed.
- For a discussion on electrobiorefineries, see: F. Harnisch and C. Urban, Angew. Chem., Int. Ed. DOI:10.1002/anie.201711727.
- All electrochemical reactions can be divided into two half-reactions, oxidation and reduction. It is thus possible to combine two desirable half-reactions, allowing these reactions to be carried out simultaneously.
- For a review on paired electrolysis, see: C. A. Paddon, M. Atobe, T. Fuchigami, P. He, P. Watts, S. J. Haswell, G. J. Pritchard, S. D. Bull and F. Marken, J. Appl. Electrochem., 2006, 36, 617–634 CrossRef.
- For the use of paired electrolysis for reduction of CO2, see: M. J. Llorente, B. H. Nguyen, C. P. Kubiak and K. D. Moeller, J. Am. Chem. Soc., 2016, 138, 15110–15113 CrossRef PubMed.
- For synthesis of diacids and diols in a divergent fashion using paired electrolysis, see: R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, ChemElectroChem, 2015, 2, 73–76 CrossRef.
- For synthesis of protected homoallylic alcohols by combining a cathodic Barbier reaction with the Shono oxidation using convergent paired electrolysis, see: G. Hilt, Angew. Chem., Int. Ed., 2003, 42, 1720–1721 CrossRef PubMed.
- J.-i. Yoshida, Chem. Commun., 2005, 4509–4516 RSC.
- F. J. del Campo, Electrochem. Commun., 2014, 45, 91–94 CrossRef.
- M. Atobe, H. Tateno and Y. Matsumura, Chem. Rev., 2018, 118, 4541–4572 CrossRef PubMed.
- D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573–4591 CrossRef PubMed.
- M. Atobe, Curr. Opin. Electrochem., 2017, 2, 1–6 CrossRef.
- M. Navarro, Curr. Opin. Electrochem., 2017, 2, 43–52 CrossRef.
- C. Gütz, A. Stenglein and S. R. Waldvogel, Org. Process Res. Dev., 2017, 21, 771–778 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a