
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correction: Solvation of Li+ by argon: how important are three-body forces?
Frederico V.
Prudente
 a,
Jorge M. C.
Marques
b and
Francisco B.
Pereira
cd
a,
Jorge M. C.
Marques
b and
Francisco B.
Pereira
cd
aInstituto de Física, Universidade Federal da Bahia, 40170-115 Salvador, BA, Brazil. E-mail: prudente@ufba.br
bCQC, Department of Chemistry, University of Coimbra, 3004-535 Coimbra, Portugal. E-mail: qtmarque@ci.uc.pt
cInstituto Superior de Engenharia de Coimbra Quinta da Nora, 3030-199 Coimbra, Portugal
dCentro de Informática e Sistemas da Universidade de Coimbra (CISUC), 3030-290 Coimbra, Portugal. E-mail: xico@dei.uc.pt
First published on 7th June 2018
Abstract
Correction for ‘Solvation of Li+ by argon: how important are three-body forces?’ by Frederico V. Prudente et al., Phys. Chem. Chem. Phys., 2017, 19, 25707–25716.
The authors detected an error in the code that implements eqn (8): the factor “3” multiplying C9 was missing in the Fortran program of PES I. After correction, we have searched for the global minimum structures of Li+Arn (n = 3–40) for the PES I with the evolutionary algorithm. The results lead to the following corrections in the paper:
(1) The table and figures below should replace Table 3, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7 and Fig. 8, respectively.
| n | PES I | PES II | RMSD | ||
|---|---|---|---|---|---|
| V GM | SYM | V GM | SYM | ||
| 1 | −10.6156 | C ∞v | −10.6156 | C ∞v | 0.00 |
| 2 | −20.4677 | D ∞h | −21.6866 | C 2v | 1.66 |
| 3 | −29.3372 | D 3h | −33.2131 | C 3v | 0.93 |
| 4 | −37.1950 | T d | −45.1235 | T d | 0.10 |
| 5 | −42.2282 | C 4v | −55.3408 | C 4v | 0.16 |
| 6 | −47.4114 | O h | −66.3792 | O h | 0.20 |
| 7 | −49.2123 | C 3v | −69.0324 | C 3v | 0.23 |
| 8 | −51.0488 | C 2v | −71.8400 | C 2v | 0.29 |
| 9 | −52.8727 | C 3v | −74.5812 | C s | 2.36 |
| 10 | −55.0185 | C s | −77.4174 | C 4v | 1.86 |
| 11 | −57.1545 | C s | −80.1327 | C s | 2.26 |
| 12 | −59.2801 | C 3v | −82.9402 | C 2v | 2.93 |
| 13 | −61.5426 | C s | −85.7355 | C 3v | 2.79 |
| 14 | −63.8410 | C 2v | −88.6286 | O h | 2.88 |
| 15 | −66.0022 | C s | −91.2832 | C 3v | 2.44 |
| 16 | −68.1649 | C 2v | −94.1454 | C 3v | 2.66 |
| 17 | −70.2635 | C s | −96.7194 | C s | 2.60 |
| 18 | −72.3542 | C 2v | −99.6969 | C 2v | 2.70 |
| 19 | −74.6213 | C s | −102.2157 | C s | 2.89 |
| 20 | −76.8875 | C s | −105.2226 | C 3v | 2.85 |
| 21 | −79.0391 | C 1 | −107.7581 | C s | 2.55 |
| 22 | −81.2321 | C 1 | −110.6664 | C s | 2.23 |
| 23 | −83.6009 | C s | −113.2250 | C 1 | 2.16 |
| 24 | −85.9783 | C 3 | −116.1347 | C s | 2.63 |
| 25 | −88.4916 | C 3 | −118.7100 | C s | 2.28 |
| 26 | −90.5702 | C 1 | −121.6112 | C 3v | 2.12 |
| 27 | −92.7125 | C 1 | −124.0709 | C 1 | 2.36 |
| 28 | −95.2238 | C 3 | −126.9316 | C 2 | 2.72 |
| 29 | −97.3301 | C 1 | −129.5222 | C 1 | 2.63 |
| 30 | −99.6171 | D 2d | −132.4044 | D 2d | 0.24 |
| 31 | −101.8398 | C 1 | −134.9826 | C s | 2.07 |
| 32 | −104.4660 | C 2 | −137.8239 | C 2v | 1.22 |
| 33 | −106.6944 | C 1 | −140.4135 | C s | 1.27 |
| 34 | −109.3778 | T | −143.4439 | T | 0.26 |
| 35 | −111.6685 | D 3 | −146.0222 | D 3 | 0.35 |
| 36 | −113.6036 | C 2 | −148.2719 | C 2 | 0.34 |
| 37 | −116.0264 | C 3v | −150.8371 | C 3v | 2.80 |
| 38 | −118.8373 | O h | −154.1266 | O h | 0.24 |
| 39 | −120.8533 | C 4v | −156.2772 | C 4v | 0.24 |
| 40 | −122.8692 | C 2v | −158.4278 | C 2v | 0.24 |
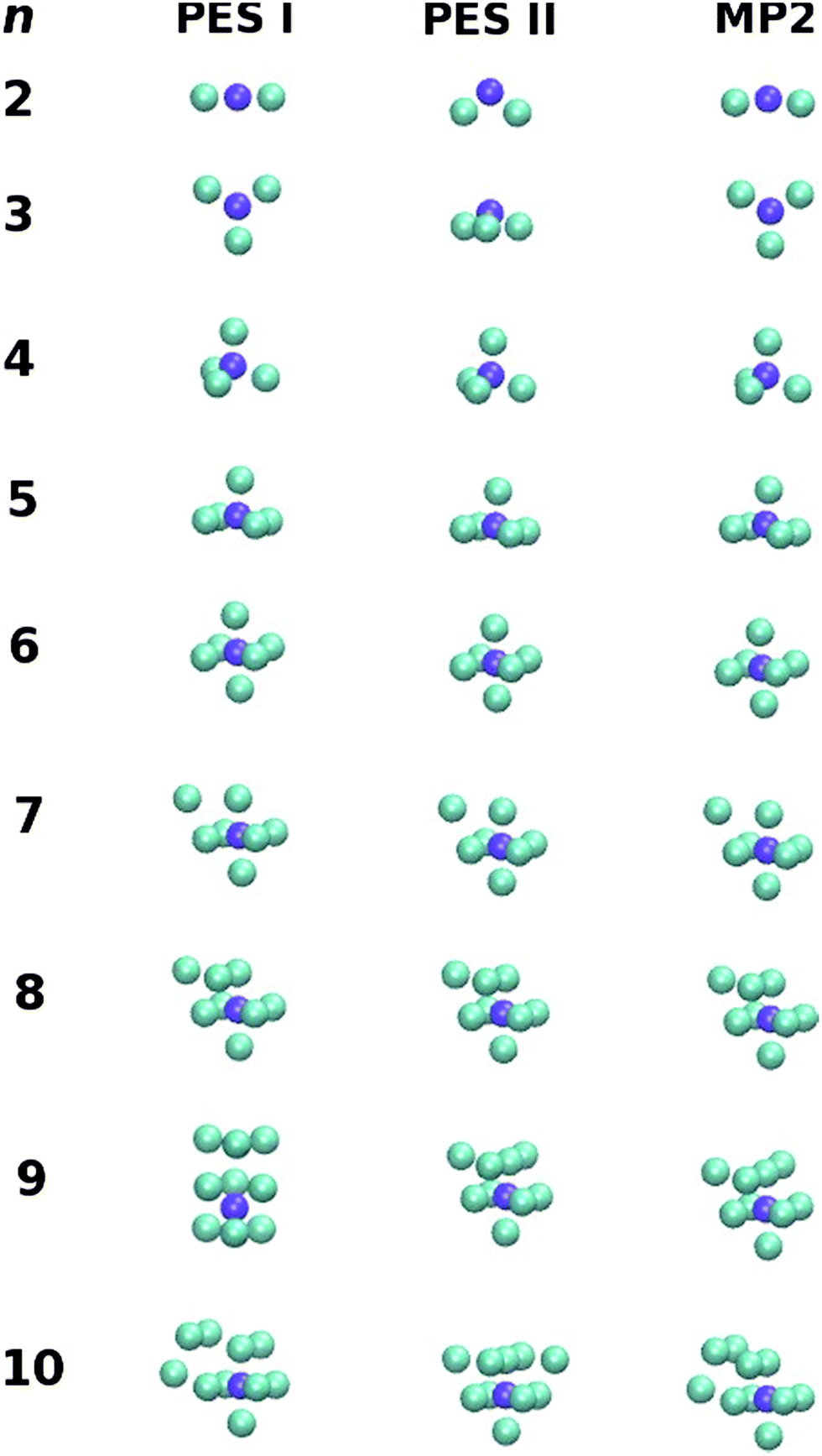 | ||
| Fig. 3 Putative global minimum structures for PES I, PES II and MP2 up to n = 10. | ||
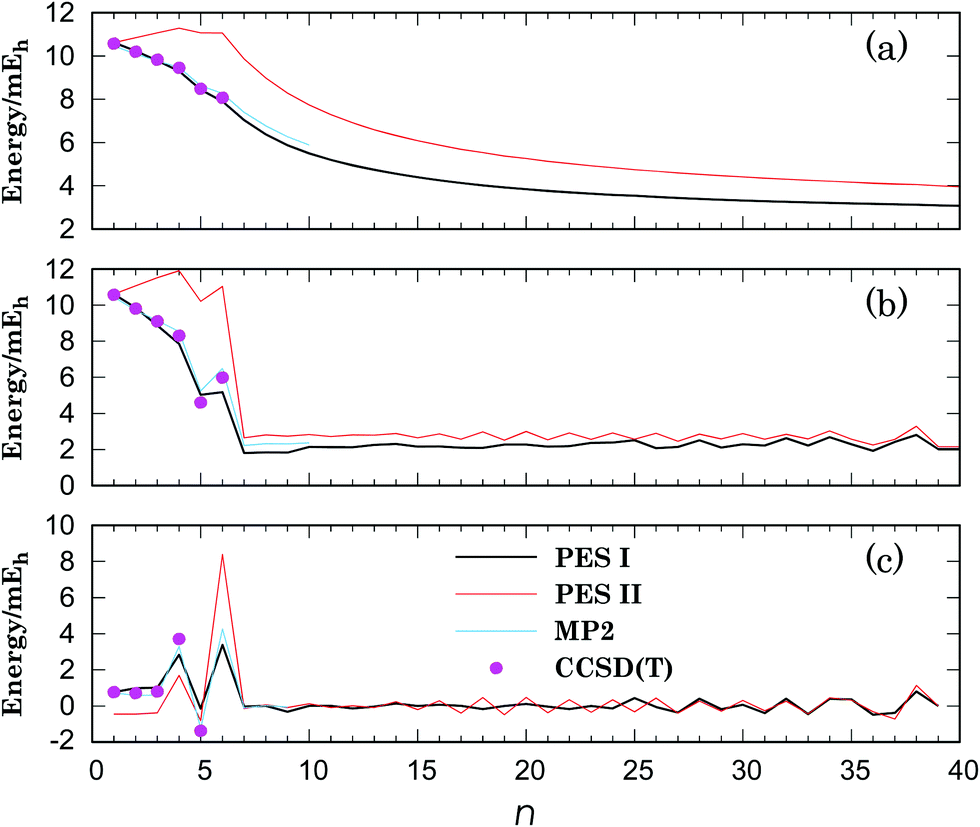 | ||
| Fig. 4 Binding energy (a), sublimation energy (b) and second energy-difference (c) for Li+Arn (n = 1–40) clusters. The key for symbols are in panel (c): PES I (black line); PES II (red line); MP2 optimization (light-blue line); CCSD(T) single-point calculation (magenta dots). Plotted values are obtained from the putative global minima at each level of theory. | ||
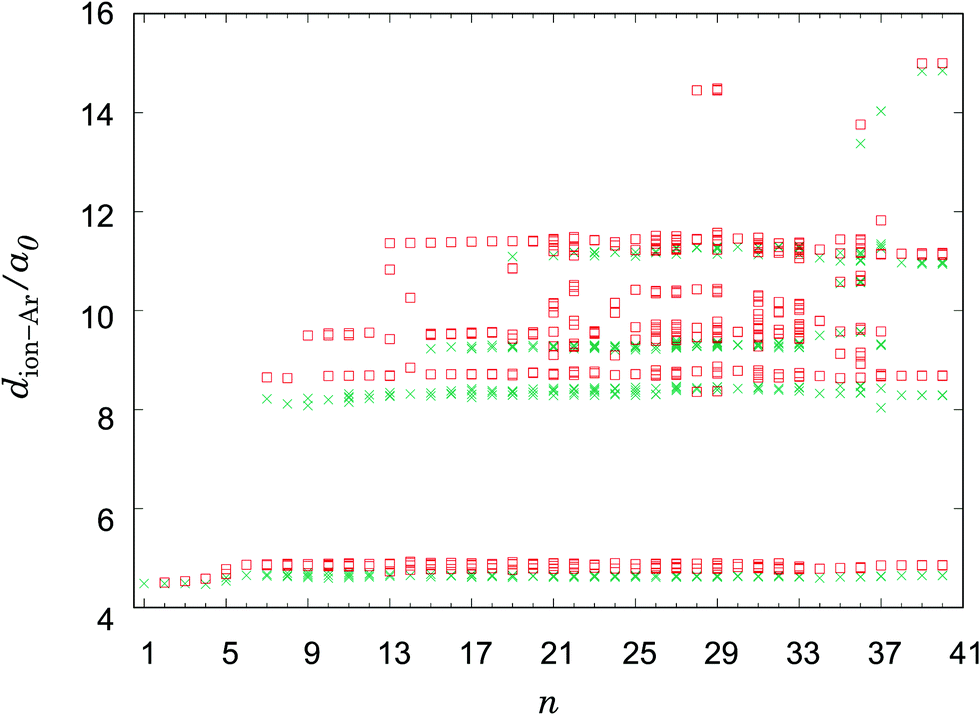 | ||
| Fig. 5 Scatter plot of the distances between the Li+-ion and the argon atoms: PES I (open squares); PES II (crosses). | ||
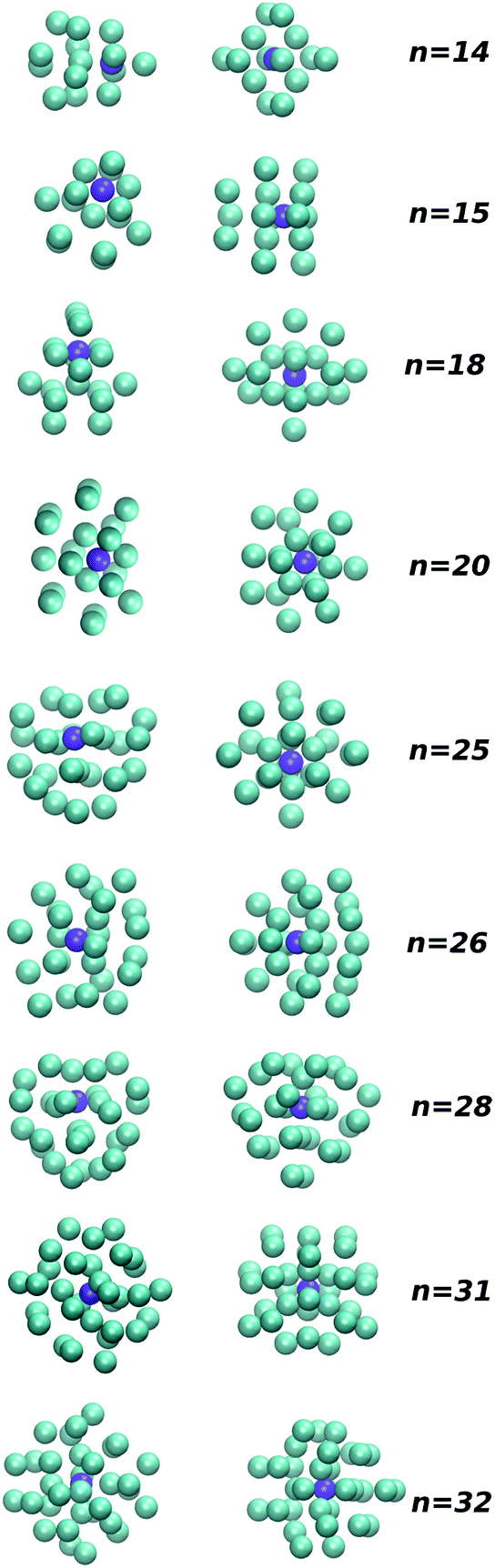 | ||
| Fig. 6 Putative global minimum structures for PES I (left) and PES II (right) for n = 14, 15, 18, 20, 25, 26, 28, 31 and 32. | ||
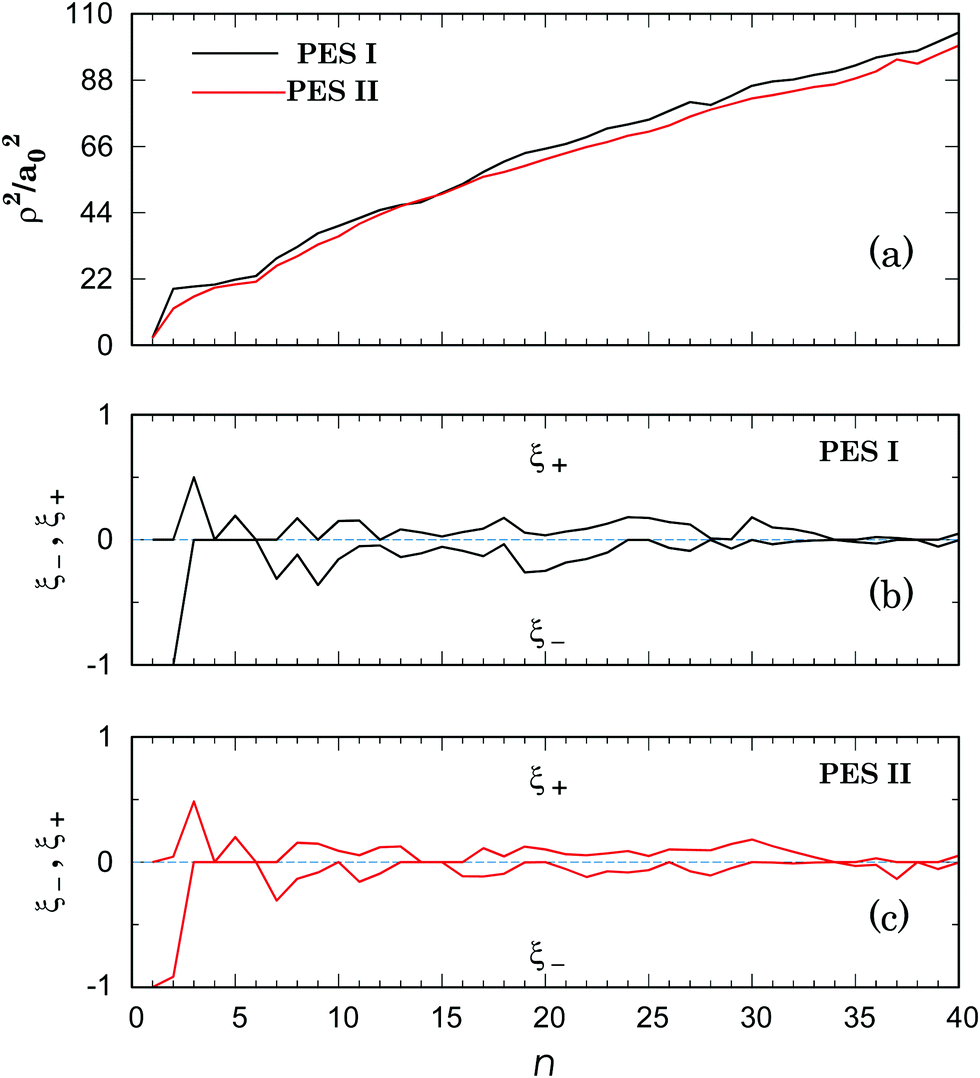 | ||
| Fig. 7 Square of the hyperradius [panel (a)] and deformation indices [panel (b) for PES I and panel (c) for PES II] of the global minimum structures of the Li+Arn (n = 1–40) clusters. In panel (a), the black curve is for PES I, while the red one refers to PES II. See the text. | ||
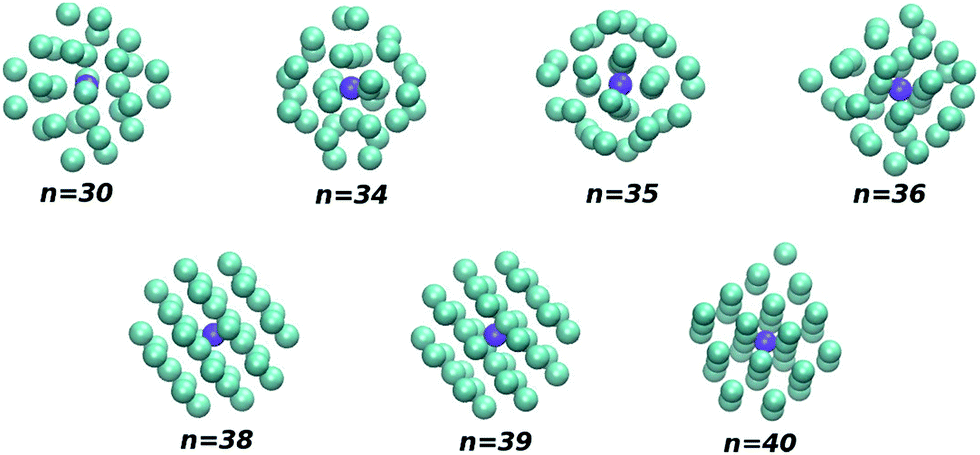 | ||
| Fig. 8 Global minimum structures for large clusters that are similar in both PES I and PES II. The shapes of the clusters are oblate (n = 30 and 40), spherical (n = 34, 35 and 38) and prolate (n = 36 and 39). | ||
(2) On the bottom of the right column on page 25711, the sentence beginning as “The energy difference…” (first paragraph of Section 3.2) must be replaced by: “The energy difference between the two potentials increases from ∼1.2 mEh (∼6%) for n = 2 to ∼35.6 mEh (∼29%) for n = 40.”
(3) On the bottom of the left column and on the top of the right column on page 25712 (i.e., the end of the second paragraph of Section 3.2), the text should read: “Although most of the global minima are similar for the three potentials, it is worth noting that the outcome provided by PES I disagrees with the MP2 one for n = 9, whereas PES II shows different structures for n = 2, 3 and 10. In addition, we have also performed MP2 re-optimizations departing from the lowest-energy structures of PES II for n = 2, 3 and 9. The geometries so obtained for n = 2 and n = 3 are similar to those from PES I, which is a further indication of the importance of three-body interactions to reach structural accuracy of the global minima. In contrast for n = 9, departing from the global minimum geometry of either PES I or PES II, the MP2 re-optimization leads to different structures and the one corresponding to PES II has the lowest energy (cf.Fig. 3).”
(4) The two last sentences of the right column on page 25714 must be changed to read: “Moreover, Fig. 7 and Table 3 show that, besides the above-mentioned small clusters (namely for n = 1, 4–8), there are other larger clusters with identical structures for both PES I and PES II. Such structures arise in the size range 30 ≤ n ≤ 40 and are depicted in Fig. 8.”
(5) On the left column of page 25715, fourth and fifth sentences before the end of the first paragraph of Section 4 must be replaced by: “The Li+Arn clusters obtained from PES I had a good agreement with the corresponding ones optimized at the ab initio level for n = 1–8 and 10. Although the PES II global minimum structure coincides with that from MP2 optimization for n = 9, this potential function leads to distinct geometries for Li+Ar2, Li+Ar3 and Li+Ar10, and fails to represent the main energetic features up to n = 10.”
Although the energies of the global minima for PES I are, obviously, different from the values in the original paper, the corresponding geometries only vary for n = 9, 19, 20, 31 and 40 (see Table 3). For n = 9, the lowest-energy structure at the MP2 level of theory coincides with the global minimum for PES II (see Fig. 3); actually, the corresponding lowest-energy structure from the PES I is a local minimum at the MP2 level of theory. In turn, the average binding energy, the sublimation energy and the second difference in energy keep essentially the same pattern (see Fig. 4). Finally, we should emphasize that these results confirm the main conclusions of the paper about the importance of three-body interactions for the title system.
Ab initio energies and coordinates of all clusters in this work can now be found in the Electronic Supplementary Information file available alongside the original article.
The Royal Society of Chemistry apologises for these errors and any consequent inconvenience to authors and readers.
| This journal is © the Owner Societies 2018 |