
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vibrationally induced metallisation of the energetic azide α-NaN3†
Adam A. L.
Michalchuk
 ab,
Svemir
Rudić
c,
Colin R.
Pulham
*ab and
Carole A.
Morrison
*a
ab,
Svemir
Rudić
c,
Colin R.
Pulham
*ab and
Carole A.
Morrison
*a
aEaStCHEM School of Chemistry and Centre for Science at Extreme Conditions, University of Edinburgh, Edinburgh, UK. E-mail: c.r.pulham@ed.ac.uk; carole.morrison@ed.ac.uk
bEPSRC Centre for Continuous Manufacturing and Crystallisation (CMAC), Edinburgh, UK
cISIS Neutron and Muon Source, STFC, Rutherford Appleton Laboratory, Chilton, Didcot, UK
First published on 5th November 2018
Abstract
As initiation of an energetic material requires rupture of a covalent bond, and therefore population of antibonding electronic states, consideration of the electronic band gap has dominated initiation mechanisms for solid state materials. Most prominent are models based on metallisation, where static mechanical perturbation leads to closing of the electronic band gap. This work explores an alternative mechanism for the dynamic metallisation of a model energetic material, where vibrational excitation resulting from mechanical impact is found to induce transient metallisation of α-NaN3. The normal coordinates associated with bending the azido anion close the electronic band gap, facilitating the formation of highly reactive species important for initiation of energetic materials. The DFT simulated vibrational spectrum of α-NaN3 exhibits excellent reproduction of the experimental low-temperature inelastic neutron scattering spectrum (INS).
Introduction
Amongst the most important characteristics of an energetic material (EM, explosives, propellants and pyrotechnics) is its propensity to initiate on mechanical or thermal perturbation. At present it is necessary to first synthesise a candidate EM and then experimentally probe its sensitivity. Without a priori knowledge of EM sensitivity, the preparation of these materials is associated with considerable risk to safety. It is therefore of paramount importance to develop a theoretical framework capable of rationalising and predicting the impact sensitivity of EMs. In addition, this framework is crucial to the selective and targeted design of new materials.The rapid release of energy that occurs following the initiation of an energetic material is the result of covalent bond rupture, typically C–N, N–N or N–O. From a chemical perspective, logic dictates that molecules that contain weaker energetic bonds should display a higher propensity to initiate on perturbation. Indeed, the correlation of bond dissociation energies with sensitivity has been promising in many cases.1 However, physical models based purely on the dissociation of an isolated gas-phase molecule are incapable of rationalising the notably different sensitivities displayed by e.g. different polymorphs or multicomponent materials that contain the same energetic molecule.2–4 A variety of other correlations have been explored, including the study of electrostatic potentials,50 and both empirical and semi-empirical parameterisation.1,5–7
As many energetic materials exist in the solid state, it has been suggested that initiation models should also be based on the solid state. While various correlations have been made, including non-covalent interaction lengths8 and void space,9 it is most common to rely on the study of electronic band gaps of EMs.10 These models can be rationalised by noting that bond dissociation requires population of anti-bonding states. Hence electronic excitation generally leads to a weakening of interatomic interactions. Materials that exhibit smaller band gaps can reach an excited electronic state more easily and hence are more prone to weakening of the energetic bond. This methodology is increasingly widespread, being dubbed as the ‘band gap criterion’ for energetic sensitivity.11 Moreover, a number of investigations have suggested considerable change to the band gap and dissociation energies under statically stressed solids.12–14 Another model has recently been developed, introducing the concept of pressure-induced metallisation of energetic materials.15 It was found that the pressures required for metallisation (i.e. electronic band gap narrowing to zero) typically fall between 20–100 GPa. These pressures are well above those responsible for structural phase transitions in these types of materials, and may indicate the importance of considering high-pressure phases when studying initiation and detonation phenomena. For example, high pressure studies have shown α-FOX-7 to undergo a structural phase transition at ca. 2.5 GPa (300 K), with a second transition occurring near 5.7 GPa (300 K).16,17 A number of pressure-induced phase transitions have also been observed in RDX,18 HMX,19 CL-20,20 FOX-7,17,21 and a variety of other molecular energetic materials. The inorganic azides are also known to undergo structural phase transitions at ambient temperature on compression, for example β-NaN3 → α-NaN3 → γ-NaN3 → δ-NaN3 observed at 0.5, 14.0 and 27.6 GPa, respectively.22,23
An alternative picture of impact-induced reactions was recently proposed for the inorganic azides,24 with similar models having proven successful in molecular materials.25 In this model, it is suggested that the impact sensitivity of EMs rests in understanding the up-pumping of vibrational energy.26 The input energy that results from a mechanical perturbation is initially deposited into the lowest frequency vibrational states of the solid-state material, i.e. the lattice or phonon modes. These highly excited vibrational states subsequently relax, and ‘pump’ the energy upwards towards localised molecular vibrations. If the correct vibrational modes are excited, a non-adiabatic vibronic process may permit electronic excitation by reducing the band gap between the ground and excited state. Bond dissociation can then be considered on an excited state potential surface. Thus, these models encompass all aspects of previous models from incorporation of impact energy, to the mechanically-induced electronic excitation and bond rupture.
Central to the ab initio calculation of vibrational up-pumping is an understanding of the vibrational energy landscape. Inelastic neutron scattering spectroscopy (INS) has been shown to offer a powerful means against which to validate computational modelling of vibrational structure. These data have been used to validate a broad range of systems, including a number of molecular energetic materials.17,27 However, to date, no such validation has been performed for an energetic azide.
Following from a description of the vibrational structure, the up-pumping model requires identification of the so-called target frequency, ωT.26 That is, the molecular normal coordinate responsible for the initiation of the explosophoric species. In the previous work, the identification of ωT for the azide materials was based on a study of an isolated azido anion species in the gas phase.24 While this offered a clear and rapid identification of ωT, it offers little by means of justification within a crystalline material. The present manuscript thus aims to explore whether the nature of ωT identified for a gas-phase molecule is justified once placed within a crystalline environment. This work also seeks to explore whether the structural distortions induced by the crystalline eigenvectors offer a route to induce transient metallisation, and hence provide an initiation mechanism for NaN3. Importantly, this mechanism differs from the conventional metallisation pathway, in that the stress induced by an eigenvector (and thus the existence of the metallised state) is transient, rather than static or continuous, as induced by an external pressure or shear strain.
Methods
Inelastic neutron scattering spectroscopy (INS)
INS spectra were collected on the TOSCA instrument28,29 at the ISIS Neutron and Muon Source. A powder sample of NaN3 (Aldrich > 99.5%, ca 1.5 g, α-NaN3 at T < ca. 17 K30) was loaded into an aluminium sachet and sealed in a flat thin-walled aluminium sample holder. The sample was cooled to <10 K and data collected to a total of 415 μA h (for approx. 3 hours). Data were processed using the Mantid software,31 and simulated spectra generated using AbINS.32Condensed matter calculations
Input unit cell geometry was taken from the experimentally determined structure, deposited in the Inorganic Crystal Structural Database (ICSD, FIZ Karlsruhe, Code 34676;33ca. 200 K). Geometry optimisation was performed using plane wave Density Functional Theory (pw-DFT) as implemented in CASTEP v16.34 The GGA functional of Perdew–Burke–Ernzerhof (PBE)35 was used, with Grimme's D2 dispersion correction,36i.e. PBE-D2. The unit cell was fully relaxed to ensure minimization of all forces below 5 × 10−4 eV Å−1, with the electronic structure expanded in plane waves up to a kinetic energy cut-off of 1800 eV and sampled on a Monkhorst-Pack37k-point grid with spacing 0.03 Å−1. Norm-conserving pseudopotentials were used throughout. The optimised structural parameters are given in Table 1. Zone-centre (Γ-point) vibrational modes were calculated using the linear response method, with the acoustic sum rule applied in reciprocal space. LO–TO splitting was not included at k = 0. The dynamical matrix was subsequently calculated across a set of 14 evenly-spaced k-points and interpolated over a fine grid.| a | b | c | α | β | γ | |
|---|---|---|---|---|---|---|
| P exp | 3.608 | 3.608 | 5.413 | 105.360 | 105.360 | 60.963 |
| P cal,D2 | 3.589 | 3.589 | 5.198 | 101.818 | 101.818 | 61.768 |
| P cal,TS | 3.610 | 3.610 | 5.149 | 103.219 | 103.219 | 60.938 |
| C exp | 6.211 | 3.658 | 5.323 | 90 | 107.225 | 90 |
| C cal,D2 | 6.161 | 3.685 | 5.198 | 90 | 103.806 | 90 |
| V(PD2)/V(exp) | +1.1% | |||||
| V(PTS)/V(exp) | −1.3% | |||||
| V(CD2)/V(exp) | −2.2% | |||||
Electronic band structures were modelled based on the same optimised geometry used for the phonon calculations. The equilibrium geometry was perturbed according to the zone-centre phonon eigenvectors. These perturbed structures were used as input for electronic band structure calculations. Further band structures were generated in CRYSTAL1738 using the HSE0639 hybrid DFT functional, which has been demonstrated to offer closer agreement with experimental band gaps for a broad range of materials compared to GGA functionals.24,40 Those presented here can therefore be regarded as accurate to within a reasonable level of confidence. Note also that as all simulations were performed as closed shell, all interpretations of the electronic band structure excitations refer to singlet channels only (that is, the spin forbidden intersystem crossing channels are not defined).
Results and discussion
Primitive cell – α-NaN3
The primitive cell setting of α-NaN3 contains only a single azido anion, and thus conforms to the chemical composition of the formula unit. The N3− anion (point group D∞h) sits on a crystallographic site with site symmetry C2h. The vibrational symmetry of the molecule therefore breaks according to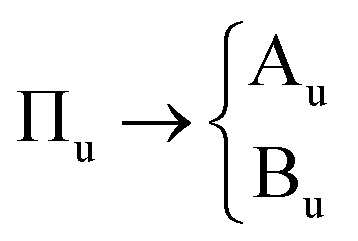
| Σg → Ag |
| Σu → Au |
There are a total of nine optical modes in the first Brillouin zone of the primitive cell, with Γoptical = 2Ag + 2Au + 1Bg + 4Bu, which manifest as the four internal (molecular) and five external (lattice) modes listed in Table 2.
| Mode | ω primitive | ω conventional | Assignment |
|---|---|---|---|
| M4 | — | 84.78 | Lattice |
| M5 | — | 88.61 | Lattice |
| M6 | 150.97 | 151.41 | Lattice |
| M7 | — | 154.47 | Lattice |
| M8 | — | 157.09 | Lattice |
| M9 | 178.00 | 184.34 | Lattice |
| M10 | 202.21 | 206.42 | Lattice |
| M11 | 214.51 | 219.05 | Lattice |
| M12 | 220.22 | 221.37 | Lattice |
| M13 | — | 221.54 | Lattice |
| M14 | — | 226.97 | Lattice |
| M15 | — | 230.22 | Lattice |
| M16 | — | 236.87 | Lattice |
| M17 | 606.28 | 610.35 | In phase δθNNN |
| M18 | 609.72 | 614.72 | In phase δθNNN |
| M19 | — | 617.49 | Out of phase δθNNN |
| M20 | — | 618.24 | Out of phase δθNNN |
| M21 | — | 1247.99 | Out of phase δRS |
| M22 | 1250.43 | 1250.25 | In phase δRS |
| M23 | — | 1929.65 | Out of phase δRA |
| M24 | 1959.81 | 1964.65 | In phase δRA |
To verify the calculated vibrational frequencies, the inelastic neutron scattering spectrum (INS) of powdered α-NaN3 was measured. This was compared with simulated INS spectra, based on the zone centre vibrational modes of the primitive cell, Fig. 1. The INS spectrum suggests that the lowest frequency modes should appear at approximately 130, 160, 196, 210 and 220 cm−1. The lowest frequency modes are Raman active, and agree with previously reported Raman spectra obtained at 100 K.41 The calculations based on PBE-D2 appear to overestimate the lowest two vibrational modes by 15–20 cm−1, although the higher frequency lattice modes are very well reproduced, differing by no more than ca. 5 cm−1. As the lowest modes are highly dependent on the underlying potential, the zone-centre frequencies were recalculated using the dispersion correction of Tkatchenko and Scheffler (TS).42 Whilst this led to a slight improvement for the lowest frequency modes (at 127 and 174 cm−1), it led to poorer agreement for the higher frequency lattice modes (at 187 cm−1, 189 cm−1 and 226 cm−1), ESI,† Section 1.
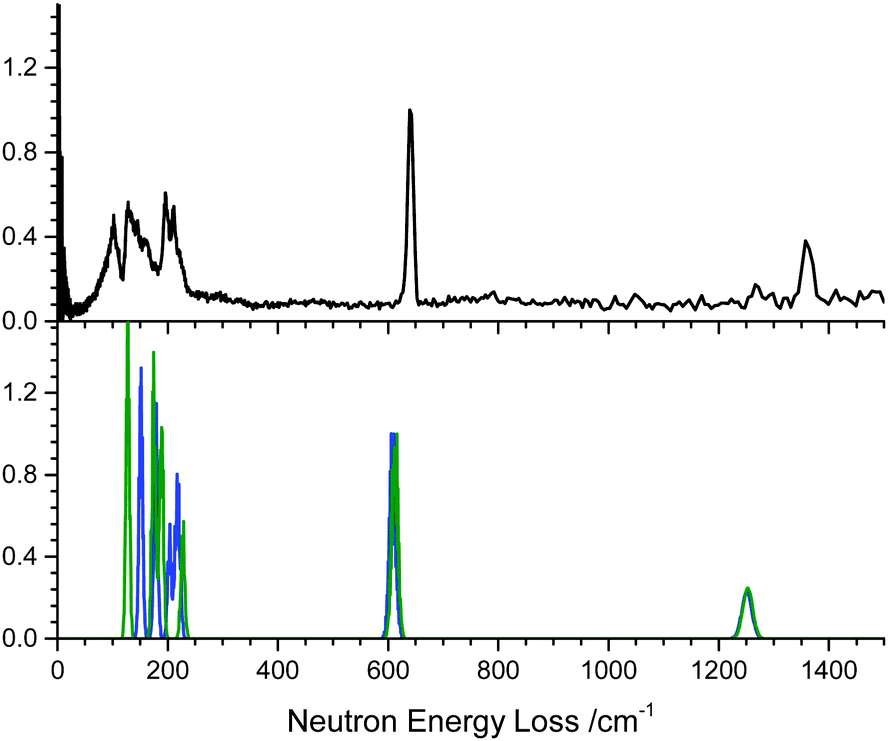 | ||
| Fig. 1 Inelastic neutron spectrum for α-NaN3. The experimental spectrum (top, recorded at 10 K) is compared with the calculated spectra (bottom) based on zone-centre calculations using the D2 (blue) and TS (green) dispersion corrections. | ||
Note there is an additional broad feature present in the INS spectrum from ca. 80–100 cm−1 which is not reproduced in any of the calculated spectra or in literature Raman spectra. Consideration of the full phonon dispersion curves for primitive α-NaN3 reveals the presence of van Hove singularities (i.e. kinks) in the acoustic branches about this frequency, Fig. 2A. This feature can therefore be ascribed to scattering from an acoustic mode away from the Brillouin zone centre and provides initial validation for the calculated dispersion curves. Interestingly, the intensity of this scattering at ca. 100 cm−1 is greater at lower momentum transfer, Q (ESI,† Section 2), which suggests complex coherence of this feature and will make for an intriguing follow-up investigation. Simulation of the INS spectrum based on the full phonon dispersion calculation leads to substantially improved reproduction of the experimental spectrum, Fig. 2B, improving significantly as the sampling of the calculated phonon dispersion curve is increased. The experimentally observed intensities become more appropriately reproduced, the broadening is well matched, and the low frequency features around 100 cm−1 are now captured. This offers validation of the general phonon dispersion structure that is produced by DFT-based calculations for this system and highlights the importance of a complete understanding of the vibrational spectrum for modelling lattice structure in systems that display high Q-dependence of ω.
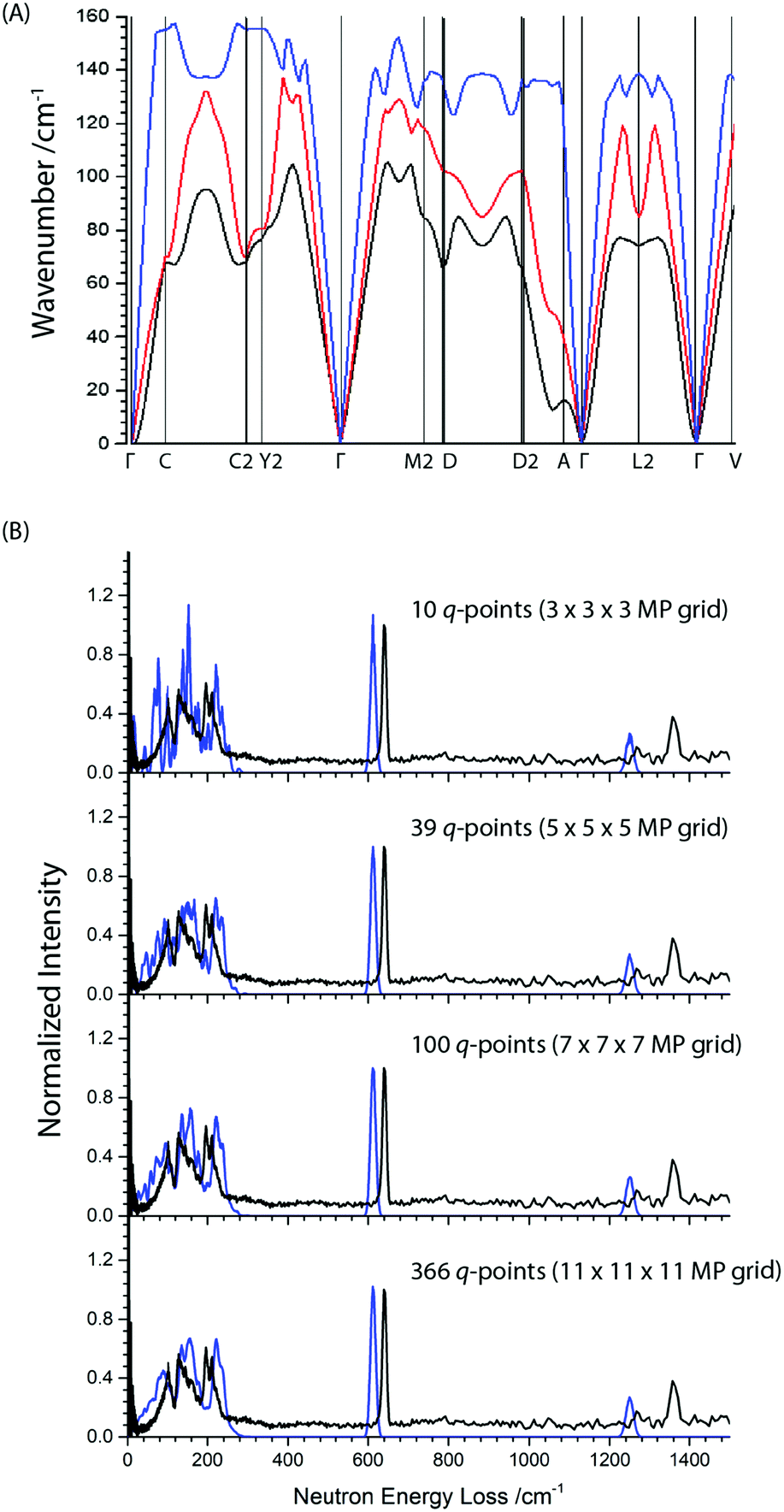 | ||
| Fig. 2 (A) Simulated phonon dispersion curve for the three acoustic branches in primitive α-NaN3. (B) The effect of increasing phonon grid size (blue curves) on the simulation of the INS spectrum for α-NaN3. | ||
The next vibrational band observed in the experimental spectrum is the N3− bend at 640 cm−1, which is ca. 30 cm−1 higher than predicted computationally. This <5% underestimation is consistent with expected performance for pw-DFT(PBE) vibrational frequency calculations, including those reported previously for this material.22 The next mode is the symmetric stretch which at 1370 cm−1 in the INS spectrum is also underestimated by the PBE-D2 calculation, but this time by as much as 100 cm−1. The INS frequency is consistent with literature Raman measurements,43 suggesting the mis-match is due to limitations in the computational model. Previous attempts at calculating zone-centre frequencies with pw-DFT(PBE) also report underestimating this mode by the same amount.44 While the reason for the disagreement is unclear, we can conclude that that the model used in this study represents the experimental structure as well as could be expected. The asymmetric stretching mode (calculated at 1959 cm−1 without LO–TO and 2037 cm−1 with LO–TO) does not appear in the INS spectrum. This peak absence is attributed to a negligible incoherent scattering cross-section for 14N combining with a low vibrational amplitude. However, Raman spectroscopy has suggested this mode occurs around 2040 cm−1,43 and hence our calculations underestimate this mode by less than 1%.
Conventional cell of α-NaN3
The conventional unit cell of α-NaN3 is the result of doubling the primitive unit cell contents, and thus comprises two azido anions and two sodium cations. By doubling the cell size, and hence halving the size of the Brillouin zone, a total of 24 vibrational modes (8 internal and 16 external) can be expected. These 24 modes correspond to the set of 12 modes that are the previously discussed zone-centre vibrational modes, and a further set of 12 modes in which the eigenvectors of each of the neighbouring primitive cells are directly out of phase with each other. A comparison of the calculated vibrational frequencies for the primitive and conventional cell is given in Table 2.In order to consider the effect of molecular perturbations on the electronic structure of the crystalline solid, we have chosen to impose perturbations of the normal coordinates on the conventional unit cell. This permits study of the edge of the first Brillouin zone, where the motions of the atoms in the two neighbouring primitive unit cells are out of phase, as well as the centre of the Brillouin zone, where the motions of the atoms are in phase, commensurate with the eigenvectors as expressed within the primitive unit cell.
For the purpose of the following discussion, it is convenient to define a perturbative term associated with ‘walking’ along each eigenvector, such that
| Ui = αεiReqm | (1) |
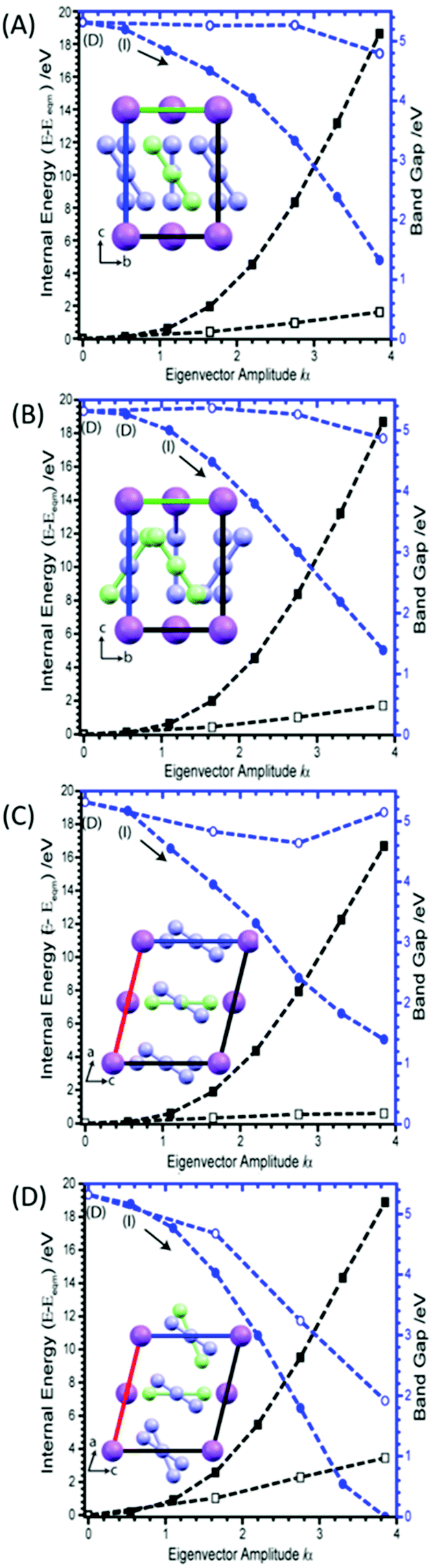 | ||
| Fig. 3 Tilting of the azido anion molecule along the crystallographic b-axis, showing (A) M6 (B) M8, (C) M9 and (D) M16. Closed symbols are derived from rectilinear application of eigenvectors, and open symbols result after correcting N–N bond lengths to equilibrium values. Internal (black) and band gap (blue) energies plotted on left and right Y-axes, respectively. The band gap is initially a direct band gap (D) but becomes indirect on perturbation (I). Inset graphics show the distortion (green molecule) with respect to the unperturbed (purple) molecular position. | ||
Considering first the tilting modes along the b-axis (M6 and M8), Fig. 3A and B, it was found that an enormous reduction in the band gap is observed if either of the two eigenvectors are followed. However, this appears to be an artefact of the rectilinear nature of the imposed eigenvectors. Indeed, if the N–N bond lengths are corrected to their equilibrium values, this trend towards metallisation is lost. Only a small reduction in the band gap is observed at very large perturbations from the equilibrium geometry. However, the large difference observed between rectilinear and corrected distortions clearly shows that the addition of internal molecular modes may be a promising mechanism to induce transient metallisation.
The normal mode M9 is akin to the zone centre azide tilt along the crystallographic a-axis. If the associated eigenvector is followed, the band gap is seen to decrease very slightly before returning towards its equilibrium band gap at higher distortions, Fig. 3C. In stark contrast, the eigenvector of M16 is associated with a considerable decrease in the band gap, Fig. 3D. Analysis of the canonical orbitals suggests that this results from overlap of the out-of-phase π non-bonding orbitals of the two N3− anions. Imposing large perturbations along this eigenvector (to a factor of α = 8) decreases the band gap further to ca. 0.15 eV (with increased internal energy of ca. 5.5 eV), suggesting potential metallization when vibrational modes at the edge of the Brillouin zone are considered (ESI,† Section 3).
Interestingly, however, analysis of the band structure shows that this metallisation does not result from a closing of the azido anion band gap, but rather a reduction in the N⋯Na band gap, Fig. 4A. This can be found to result largely from an increase in energy of the N valence bands by approximately 3.5 eV. Metallisation in this manner suggests oxidation of the azido anion may occur under extreme excitations of the lattice modes in α-NaN3, leading to transient formation of the neutral N3 radical. This is particularly interesting given previous suggestions45 that azide decomposition occurs via
| 2N3− + M+ →2N3˙ + M0 |
| 2N3˙ → 3N2 |
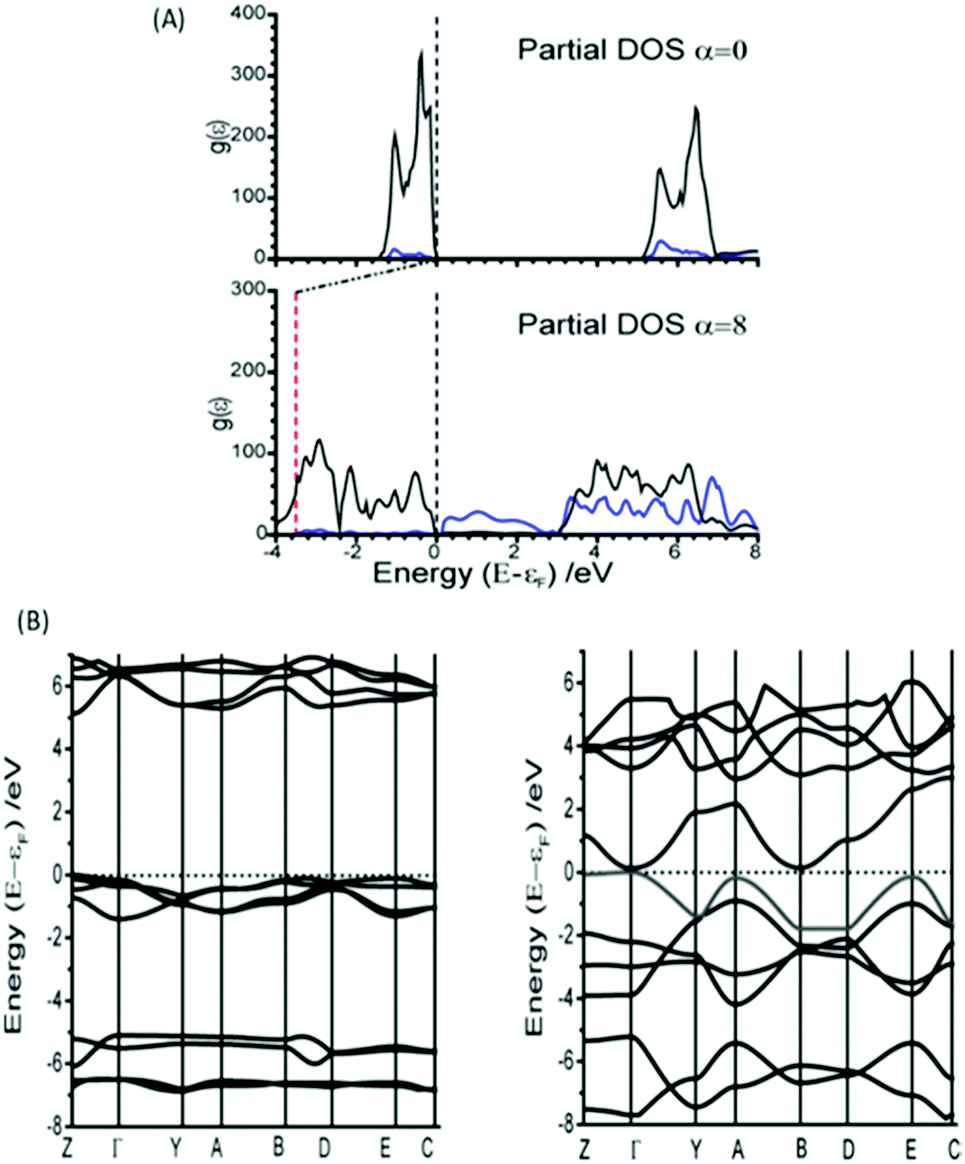 | ||
| Fig. 4 Comparison of the electronic band gap at equilibrium geometry and after imposing an α = 8 fold perturbation of M16. (A) The partial DOS is given for (black) N and (blue) Na species. The vertical dotted line on (A) is given to indicate the Fermi surface (εF), and the red vertical line shows the energy of εF relative to the unperturbed structure. (B) The electronic band structure plotted along high symmetry lines in the Brillouin zone for (left) unperturbed and (right) α = 8 structures. | ||
Mechanical perturbation of a solid leads to large excitation of phonon modes. Because the phonon heat capacity is much lower than the total heat capacity of the material, this same input energy leads to lower populations of the internal vibrational modes upon equilibration.26 Consequently, displacement along the external mode eigenvectors exceeds that of the internal modes, and hence metallisation via M16 can be expected to occur more readily than by internal modes. However, NaN3 is well known to be insensitive to initiation by impact.46 Thus, despite the ease with which M16 can induce metallisation, we suggest that this mode is unlikely to be responsible for decomposition of NaN3 by dynamic impact initiation. However, it may be important for equilibrium decomposition via a thermal mechanism. Analysis of the band structure shown in Fig. 4B shows that while the eigenvector for M16 does result in an indirect band gap, the minimum energy gap spans a very narrow range of k-space, and hence results in very few excitation channels. Excitation across an indirect band gap is known to be very slow.47 Thus as the impact-induced vibrational energy transfer processes are expected to occur on the sub-nanosecond timescale (picosecond for phonon–phonon dissipation), it is reasonable to suggest that, while metallisation may occur, electronic excitation along eigenvector M16 is simply too slow to be considered here as a route to detonation. Further work on the explicit consideration of electron dynamics is however required.
The remaining nine external modes all exhibit a mixture of N and Na displacement (see eigenvectors in ESI,† Section 4). It is reassuring to find that none of these modes lead to any notable decrease in the band gap, Fig. 5. There is typically no more than ca. 1 eV decrease in band gap along any of these eigenvectors, with M12 (the out of phase translation of Na and N3− species along the crystallographic c-axis), actually leading to an overall increase in band gap. The only exception is M15, which at large distortions appears to reduce the band gap quite drastically. However, it must be noted that this is associated with an increase in energy of around 60 eV, due to the manifestation of short contacts between Na atoms for this eigenvector.
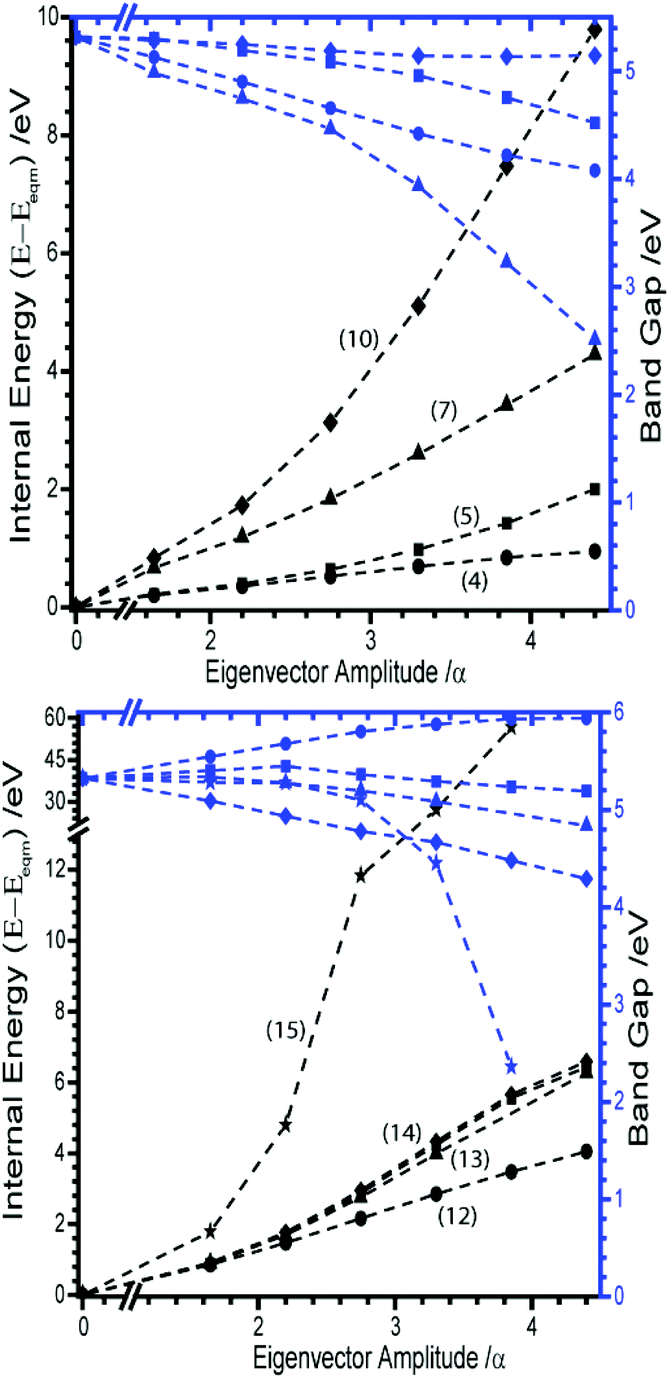 | ||
| Fig. 5 Effect of external vibration normal coordinates on the energies and band gap of α-NaN3. Internal (black) and band gap (blue) energies plotted on left and right Y-axes, respectively. Modes are identified as, (Top) M4 (-●-), M5 (-■-), M7 (-▲-), M10 (-✦-), (bottom) M11 (-■-), M12 (-●-), M13 (-▲-), M14 (-✦-), M15 (-★-). | ||
Internal vibrational modes
The next four vibrational modes (M17–M20) correspond to the internal bending motion of the azido anions. M17 and M18 are the zone-centre (i.e. in phase) bending of the azido anions perpendicular to and along the crystallographic b-axis, respectively. M19 and M20 are the corresponding out of phase bending modes.For the purpose of following the effects of M17–M20 on the electronic band gap, the rectilinear perturbation was applied according to eqn (1). In doing so, however, artificial elongation of N–N bond lengths was introduced. To ensure that only the isolated angle bend normal coordinate was investigated the N–N bond lengths were therefore restored to equilibrium geometries. Further, to offer a comparison to previous gas-phase studies,24 and to represent the energy per primitive cell, the internal energy is normalised by the number of azido anions in the conventional cell.
As the azido anion was perturbed along the bending mode, θNNN, the band gap was found to steadily decrease, with the band gap reaching approximately half of its original value at θNNN ≈ 130°. By 110° the band gap drops to 0 eV, Fig. 6.
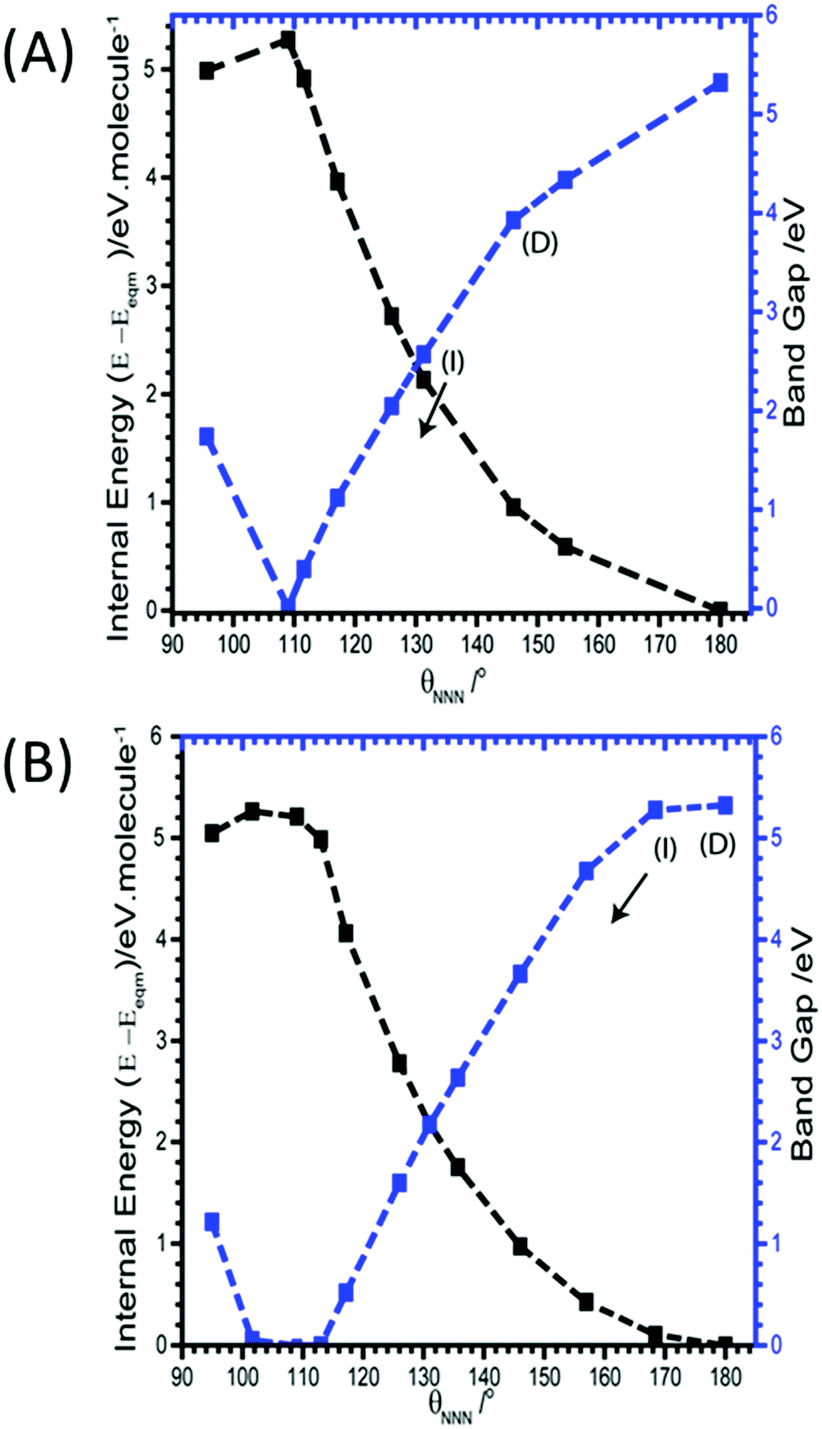 | ||
| Fig. 6 Effect of (A) M17 and (B) M19 (B) on the band gaps and energies of α-NaN3. Band gap momentum conservation is indicated as direct (D) or indirect (I). Internal (black) and band gap (blue) energies plotted on left and right Y-axes, respectively. The arrow indicates continuation of an indirect band gap. M18 and M20 are given in ESI,† Section 5. | ||
Unlike for M16, the narrowing in the band gap that is observed on imposing normal coordinate perturbations 17–20 are not the result of N⋯Na band gap closing. Instead, metallization occurs by a closing of the N3− conduction/valence band gap, Fig. 7. The full band structure diagram also suggests that, in contrast to M16, very little increase in band dispersion occurs as metallization approaches. Thus whilst the band gap does switch from direct to indirect, the shortest crossing points persist across broad ranges of k-space, indicating very many more excitation channels exist across the Brillouin zone, in contrast to M16.
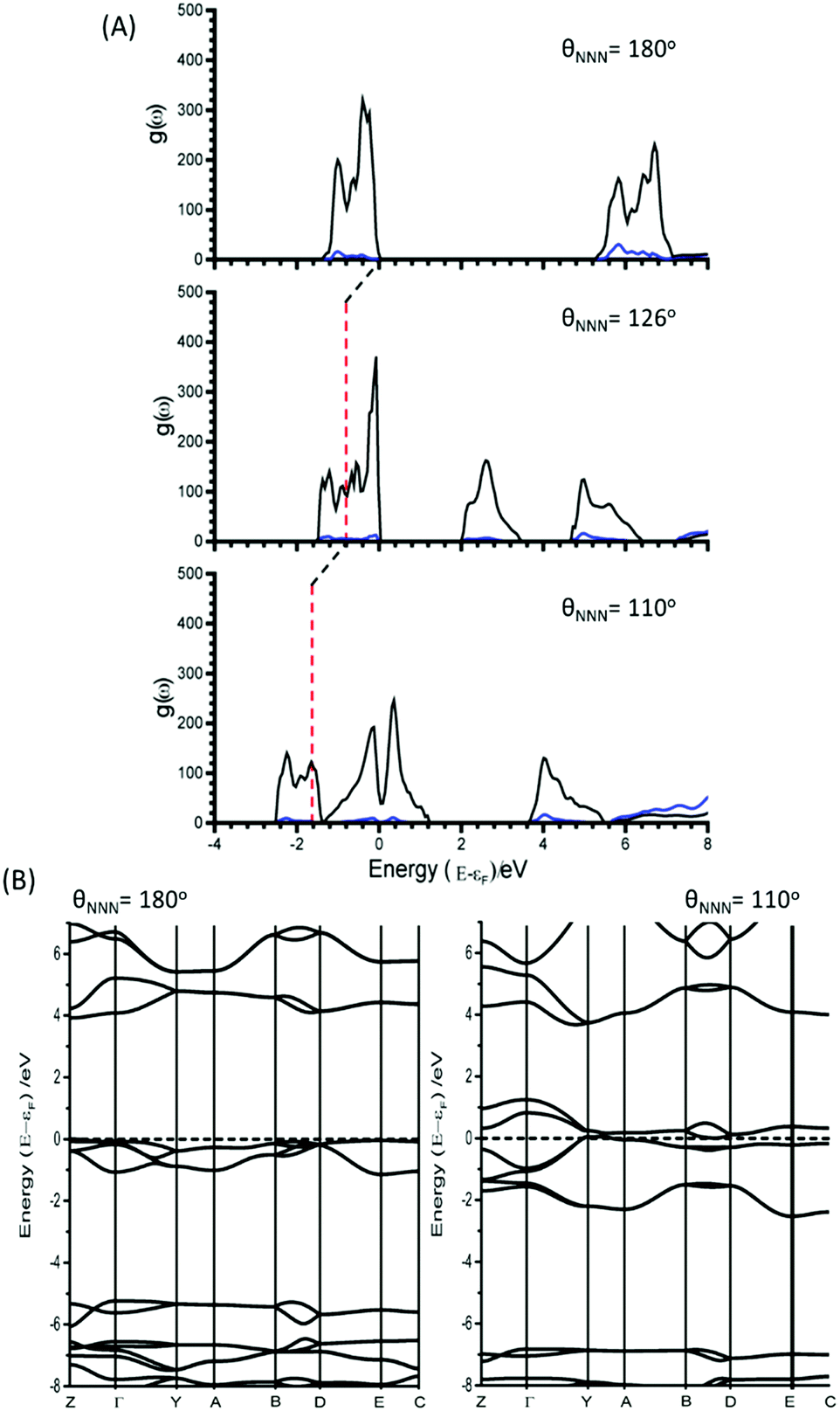 | ||
| Fig. 7 (A) Partial electronic density of states as a function of M17, with contributions from N3− (black) and Na+ (blue). The relative energy of εF at θNNN = 180° is shown as a red vertical line in the perturbed structures. (B) Electronic band structures for NaN3 with θNNN = 180° and θNNN = 110°. εF is marked with a dotted line. | ||
Note the behavior of the azide anion in the crystal lattice contrasts markedly with what has been reported for the same ion in the gas phase,24 where molecular perturbations of the vibrational eigenvectors could only locate conical intersections between S0/T1 electronic states, i.e. spin ‘forbidden’ channels. Thus the periodicity of the crystalline lattice has led not only to a notable (and expected) reduction in the molecular HOMO–LUMO gap but has also opened up more probable singlet channels for electronic excitation.
Within the confines of symmetry, metallisation permits electron transfer from the valence bands of the metal cation to the azido anion, leading to potential, albeit chemically unlikely, transient formation of N32−. The ability to lower the conduction band to such considerable degrees also offers pathways to the role of local electron defects within these structures. Such processes have previously been suggested as being crucial to the initiation of energetic compounds, although no mechanism for their athermal influence was proposed.48,49 It therefore appears that coupling normal mode behavior with electronic excitation offers a route to explaining the formation of highly unstable species in energetic materials during the detonation process.
M21 is the out of phase symmetric stretching mode. Due to the contraction of one set of N–N bonds over this normal coordinate it was not possible to follow this mode beyond a N–N stretch of 1.4 Å. By this limit, the band gap was found to decrease only slightly, from ca. 5.2 to 3.3 eV. It is therefore unlikely that this mode provides a route to metallization. The in-phase (zone centre) symmetric stretching mode is expressed as M22. By the same change in internal energy (ca. +5 eV molecule−1) at which the bending modes led to metallisation, the band gap from the symmetric stretching mode had reduced to only ca. 2 eV. Further extension of this normal coordinate unsurprisingly leads to metallisation as the N–N bonds are ruptured. However, this occurs with an increase of internal energy >12 eV molecule−1 (Fig. 8A). The final two normal coordinates, M23 and M24, are the in- and out-of-phase asymmetric stretching modes, respectively and behave in the same manner. For the same reason described for M21, there is a physical limitation on the maximum Ui that can be applied to these modes. This limit was reached with an energy penalty of ca. 13 eV molecule−1, by which point the band gap was found to reduce to only 3.8 eV (Fig. 8B). Thus, metallisation can only be attained via the bending normal coordinate, consistent with findings for the gas phase N3− molecule.
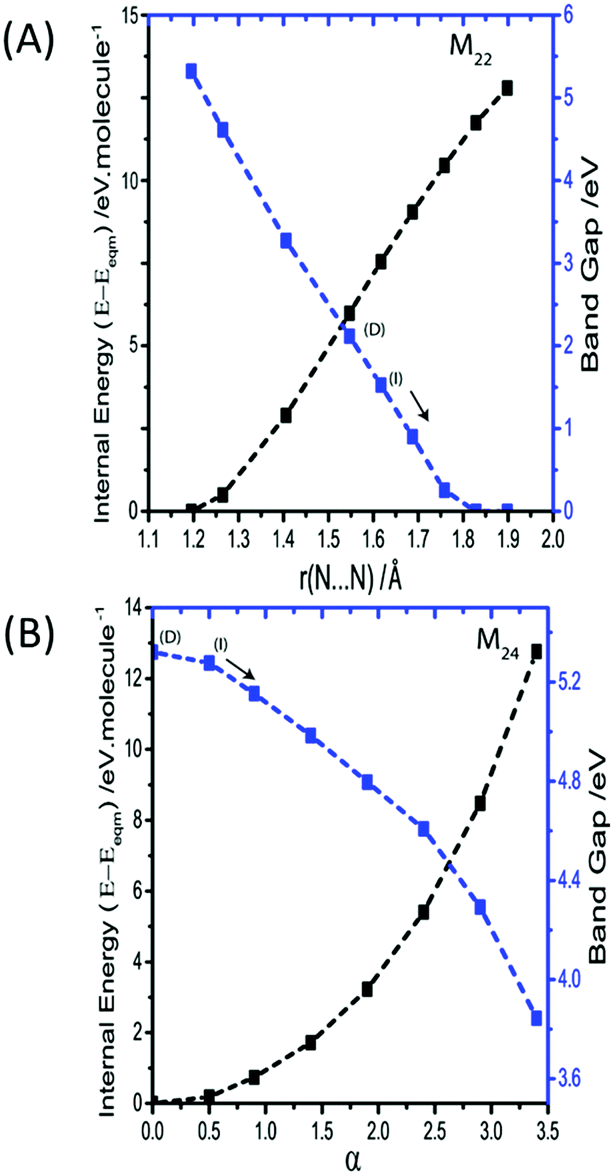 | ||
| Fig. 8 Effect of M22 and M24 (i.e. the symmetric and asymmetric stretches, respectively) on the electronic band gap. The latter is plotted as α, defined as r = (reqm + α/10), where reqm is the equilibrium bond length. Band gaps are defined as direct (D) or indirect (I) and the arrow indicates that the indirect band gap continues. To reflect perturbation of two azido anions (in the conventional cell), energy is given per molecule. | ||
Conclusions
Impact sensitivity is a crucial parameter in understanding the performance, safety and application of energetic materials. To date, little has been understood concerning the physical parameters that govern the sensitivity of these materials. It has recently been demonstrated that a vibrational up-pumping model offers a powerful base upon which to understand this phenomenon. Within this model, the azide materials were suggested to initiate via the bending normal coordinate of the N3− species.The present manuscript demonstrates the ability of density functional theory to faithfully reproduce most components of the vibrational spectrum of the well-known inorganic azide αNaN3. This was done by comparison with low temperature experimental inelastic neutron scattering spectra. The high levels of vibrational dispersion exhibited by this crystalline system required complete modelling of vibrations across the Brillouin zone in order to fully reproduce the INS spectrum. As the normal coordinates were followed away from the equilibrium structure, the electronic band gap was calculated for the conventional unit cell. For the thirteen external optical modes, only one was found to induce an indirect band gap reduction (between N and Na bands) to give viable excitation channels but over a restricted range of k-space. Whilst excitation across an indirect band gap may be expected to be slow, it is possible that this crossing may offer a mechanism for formation of radical N3 in the solid. However as NaN3 is well known to be insensitive to impact initiation it is unlikely that the detonation model is dependent on a readily excitable low energy phonon mode. A more thorough understanding of why this external mode does not lead to initiation is therefore required.
Consideration of the internal modes clearly demonstrates that vibrationally-induced, transient metallisation of the azido anion within the solid state can be readily achieved through the bending mode eigenvector. Narrowing the angle in the azide anion by about 60°, at the cost of around 5 eV per molecular unit, results in a closing of the valence and conduction band. This suggests that an S0/S1 conical intersection may be available in the solid state which is absent in the gas phase. The electronic band gap narrowing may account for the formation of the highly reactive N32− species, which has been suggested previously to be crucial in the initiation of energetic compounds. The up-pumping model for impact induced initiation of energetic materials therefore holds when the effect of the crystalline environment is considered. We suggest that the concept of vibrationally induced metallisation, resulting in transient band gap narrowing, can lead to the creation of reactive species in crystalline solids, and may be key to understanding reactivity under the highly vibrationally excited states that are associated with shocked materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
INS Spectra were collected on the TOSCA instrument at the ISIS Neutron and Muon Facility (RB 1710382). The authors thank EPSRC CMAC EP/I033459/1 (AM) for funding. Thanks to the UK Materials and Molecular Modelling Hub for computational resources, partially funded by EPSRC (EP/P020194/1). Additional thanks to the Edinburgh Compute and Data Facilities (ECDF) for further computational resources. Final thanks to Prof Adam Cumming and Dr Stewart Parker for many helpful discussions.Notes and references
- D. Mathieu and T. Alaime, J. Phys. Chem. A, 2014, 118, 9720–9726 CrossRef CAS PubMed.
- B. W. Asay, B. F. Henson, L. B. Smilowitz and P. M. Dickson, J. Energ. Mater., 2003, 21, 223–235 CrossRef CAS.
- K. B. Landenberger, O. Bolton and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 5074–5079 CrossRef CAS PubMed.
- S. R. Kennedy and C. R. Pulham, in Co-crystals: Preparation, Characterization and Applications, ed. C. B. Aakeröy and A. S. Sinha, Royal Society of Chemistry, Cambridge, UK, 2018, pp. 231–266 Search PubMed.
- D. Mathieu, Ind. Eng. Chem. Res., 2017, 56, 8191–8201 CrossRef CAS.
- N. R. Badders, C. Wei, A. A. Aldeeb, W. J. Rogers and M. S. Mannan, J. Energ. Mater., 2006, 24, 17–33 CrossRef CAS.
- R. V. Tsyshevsky, O. Sharia and M. M. Kuklja, Molecules, 2016, 21, 236 CrossRef PubMed.
- M. Cartwright and J. Wilkinson, Propellants, Explos., Pyrotech., 2010, 35, 326–332 CrossRef CAS.
- P. Politzer and J. S. Murray, J. Mol. Model., 2014, 20, 2233 CrossRef PubMed.
- W. Zhu, J. Xiao and H. Xiao, J. Phys. Chem. B, 2006, 110, 9856–9862 CrossRef CAS PubMed.
- H. Zhang, F. Cheung, F. Zhao and X. Cheng, Int. J. Quantum Chem., 2009, 109, 1547–1552 CrossRef CAS.
- M. M. Kuklja, S. N. Rashkeev and F. J. Zerilli, Appl. Phys. Lett., 2006, 89, 071904 CrossRef.
- M. M. Kuklja and S. N. Rashkeev, Appl. Phys. Lett., 2007, 90, 2005–2008 CrossRef.
- M. M. Kuklja and S. N. Rashkeev, Appl. Phys. Lett., 2007, 90, 151913 CrossRef.
- S. V. Bondarchuk, J. Phys. Chem. A, 2018, 122, 5455–5463 CrossRef CAS PubMed.
- M. M. Bishop, N. Velisavljevic, R. Chellappa and Y. K. Vohra, J. Phys. Chem. A, 2015, 119, 9739–9747 CrossRef CAS PubMed.
- S. Hunter, P. L. Coster, A. J. Davidson, D. I. A. Millar, S. F. Parker, W. G. Marshall, R. I. Smith, C. A. Morrison and C. R. Pulham, J. Phys. Chem. C, 2015, 119, 2322–2334 CrossRef CAS.
- I. D. H. Oswald, D. I. A. Millar, A. J. Davidson, D. J. Francis, W. G. Marshall, C. R. Pulham, A. Cumming, A. R. Lennie and J. E. Warren, High Pressure Research, 2010, vol. 30, pp. 280–291 Search PubMed.
- F. Goetz, T. Brill and J. Ferraro, J. Phys. Chem., 1978, 82, 1912–1917 CrossRef CAS.
- J. Li and T. B. Brill, Propellants, Explos., Pyrotech., 2007, 32, 326–330 CrossRef CAS.
- Z. A. Dreger, Y. Tao and Y. M. Gupta, Chem. Phys. Lett., 2013, 584, 83–87 CrossRef CAS.
- D. Li, P. Zhu, J. Jiang, D. Xu, R. Liu, X. Wang, Q. Cui and H. Zhu, J. Phys. Commun., 2017, 1, 025002 CrossRef.
- D. I. A. Millar, C. Barry, W. G. Marshall and C. R. Pulham, Z. Kristallogr., 2014, 229, 259–275 Search PubMed.
- A. A. L. Michalchuk, P. T. Fincham, P. Portius, C. R. Pulham and C. A. Morrison, J. Phys. Chem. C, 2018, 122, 19395–19408 CrossRef CAS.
- J. Bernstein, J. Chem. Phys., 2018, 148, 084502 CrossRef PubMed.
- D. D. Dlott and M. D. Fayer, J. Chem. Phys., 1990, 92, 3798–3812 CrossRef CAS.
- S. Hunter, T. Sutinen, S. F. Parker, C. A. Morrison, D. M. Williamson, S. Thompson, P. J. Gould and C. R. Pulham, J. Phys. Chem. C, 2013, 117, 8062–8071 CrossRef CAS.
- S. F. Parker, F. Fernandez-Alonso, A. J. Ramirez-Cuesta, J. Tomkinson, S. Rudic, R. S. Pinna, G. Gorini and J. Fernández Castañon, J. Phys. Conf. Ser., 2014, 554, 012003 CrossRef.
- R. S. Pinna, S. Rudić, S. F. Parker, J. Armstrong, M. Zanetti, G. Škoro, S. P. Waller, D. Zacek, C. A. Smith, M. J. Capstick, D. J. McPhail, D. E. Pooley, G. D. Howells, G. Gorini and F. Fernandez-Alonso, Nucl. Instrum. Methods Phys. Res., Sect. A, 2018, 896, 68–74 CrossRef CAS.
- S. R. Aghdaee and A. I. M. Rae, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 214–218 CrossRef.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- K. Dymkowski, S. F. Parker, F. Fernandez-Alonso and S. Mukhopadhyay, Phys. B, 2018, 1–6 Search PubMed , in Press.
- G. E. Pringle and D. E. Noakes, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 262–269 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- J. D. Pack and H. J. Monkhorst, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D’Arco, Y. Noël, M. Causà, M. Rérat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287–1317 CrossRef CAS.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- A. J. Garza and G. E. Scuseria, J. Phys. Chem. Lett., 2016, 7, 5–10 Search PubMed.
- Z. Iqbal, J. Chem. Phys., 1973, 59, 1769–1774 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 6–9 CrossRef PubMed.
- J. I. Bryant, J. Chem. Phys., 1964, 40, 3195–3203 CrossRef CAS.
- K. Ramesh Babu, C. Bheema Lingam, S. P. Tewari and G. Vaitheeswaran, J. Phys. Chem. A, 2011, 115, 4521–4529 CrossRef CAS PubMed.
- W. Zhu, X. Xu and H. Xiao, J. Phys. Chem. Solids, 2007, 68, 1762–1769 CrossRef CAS.
- H. D. Fair and R. F. Walker, Energetic Materials 1: Physics and Chemistry of the Inoganic Azides, Plenum Press, New York, 1977 Search PubMed.
- T. Kirchartz and U. Rau, J. Phys. Chem. Lett., 2017, 8, 1265–1271 CrossRef CAS PubMed.
- B. P. Aduev, E. D. Aluker, V. G. Kriger and Y. A. Zakharov, Solid State Ionics, 1997, 101–103, 33–36 CrossRef CAS.
- B. P. Aduev, É. D. Aluker, G. M. Belokurov, A. N. Drobchik, Y. A. Zakharov, A. G. Krechetov and A. Y. Mitrofanov, Combust., Explos. Shock Waves, 2000, 36, 622–632 CrossRef.
- P. Politzer and J. Murray, J. Mol. Struct., 1996, 376, 419–424 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp06161k |
| This journal is © the Owner Societies 2018 |