
Photoelectron spectroscopy of solvated dicarboxylate and alkali metal ion clusters, M+[O2C(CH2)2CO2]2− [H2O]n (M = Na, K; n = 1–6)†
Ren-Zhong
Li‡
ab,
Shihu H. M.
Deng
b,
Gao-Lei
Hou
 b,
Marat
Valiev
*c and
Xue-Bin
Wang
*b
b,
Marat
Valiev
*c and
Xue-Bin
Wang
*b
aCollege of Environmental and Chemical Engineering, Xi’an Polytechnic University, Xi’an, 710048, China
bPhysical Sciences Division, Pacific Northwest National Laboratory, P. O. Box 999, MS K8-88, Richland, Washington 99352, USA. E-mail: xuebin.wang@pnnl.gov
cEnvironmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, 902 Battelle Boulevard, P.O. Box 999, Richland, Washington 99352, USA. E-mail: marat.valiev@pnnl.gov
First published on 2nd November 2018
Abstract
We present results of combined experimental photoelectron spectroscopy and theoretical modeling studies of solvated dicarboxylate species (−O2C(CH2)2CO2−) in complex with Na+ and K+ metal cations. These ternary clusters serve as simple models for the investigation of aqueous ion/solute specific effects that play an important role in biological systems. The experimental characterization of these systems was performed in the presence of up to six solvating waters. In both Na+ and K+ cases, we observe the presence of one major broad band that gradually shifts to higher electron binding energy (EBE) with an increasing number of waters. In the Na+ case further detailed analysis of experimental spectra was performed using ab initio calculations. In particular, we have identified the structures of the lowest energy clusters whose EBE values match well the major band in the experimental spectra. Our results show that evolution of an aqueous solvation shell emphasizes the coordination of the negatively charged carboxylate groups accompanied by simultaneous interaction with metal cations. Calculations also indicate that in the solvation range investigated experimentally (up to 6 waters), Na+ retains direct contact with the dicarboxylate species, i.e. a contact ion-pair (CIP) complex. Preliminary modeling studies show evidence of an alternative solvent separated ion-pair complex once the solvation range approaches 8 waters, however its energy still remains above that of (∼7–8 kcal/ mol−1) the CIP complex. At a higher number of waters (n = 3 for Na+ and n = 5 for K+), the experimental spectra also show the development of a weak low energy band. Its origin cannot be precisely identified. Our calculations in the Na+ case point out the existence of a quaternary complex consisting of Na+, H2O, OH− and a singly protonated dicarboxylate anion (HO2C(CH)2CO2−). Such a complex appears to be stabilized in the solvation range corresponding to the appearance of the low EBE band and does match its peak, even though the energy of such a complex is fairly high compared to the ternary structure.
1. Introduction
Carboxylic acids and their derivatives represent an important class of organic molecules, playing a critical role in many biological and environmental processes. As a major source of negatively charged groups in biological systems they play a prominent role as a mediator in ion-specific interactions in processes involving ion channels,1 protein stability,2etc. Of particular interest are interactions between carboxylate groups and monovalent ions, such as sodium and potassium,3–8 which may provide critical insight into the salt effects.9,10 For example, the binding of smaller sodium ions to carboxylates can shift the equilibrium from the folded to the unfolded state, but such an effect was not observed for potassium ions.11The specific ion effects are non-trivial and involve a complicated interplay of the two effects – direct ion pair interaction as well as indirect effects, which involves restructuring of the water network.12–14 The balance between two forces can be influenced by a number of different factors (salt concentration, the nature of the protein, pH, etc.), which necessitates molecular level analysis.15 While much research has been done to address ion pairing of alkali metal cations with the carboxylate anions in solution,16–18 relatively little is known about their behavior in a reduced solvation environment. The latter may be directly relevant to biological systems and can provide insight into the forces that drive a local solvation structure, which has been suggested to play an important role in governing Hofmeister effects.19
At the core of our approach is the experimental characterization based on negative ion photoelectron spectroscopy (NIPES). The latter provides a sensitive probe of a local solvation structure and has been proven to be a powerful technique for cluster systems.20,21 Previously, our group reported a gas phase study of the interactions between alkali metal cations (M+) and dicarboxylate dianions (−O2C(CH2)nCO2−, DCn2−) by combined NIPES and ab initio electronic structure calculations on [(M+)(DCn2−)]− (M = Li, Na, K; n = 2, 4, 6), and suggested that the electrostatic interaction and size matching dominate the structures and energetics of these gaseous clusters, and play important roles in ion specificity and selectivity in solutions as well.22 The investigation presented here extends our previous work by including an aqueous solvation environment, with a focus on understanding the evolution of solvation structures and molecular interactions among cations, anions, and waters.
2. Experimental and theoretical methods
2.1 Experimental methods
The NIPES experiments were conducted with an apparatus consisting of an electrospray ionization source, a cryogenic ion trap, and a magnetic-bottle time-of-flight photoelectron spectrometer.23 The metal cation–dicarboxylate dianion complexes, M+–DC22−, and their solvated clusters, M+–DC22−(H2O)n, were generated by spraying 0.1 mM aqueous acetonitrile or methanol solutions of succinic acid, titrated to pH ∼ 7 by adding a small amount of the corresponding aqueous base solutions of MOH in humid environments. It was much more difficult to generate these ternary clusters compared to our recent work on binary systems [EDTA·M(III)]−.24 The produced anions were guided by two RF-only quadruples and a 90° bender into the cryogenically controlled ion trap, where they were accumulated and cooled for 20–100 ms via collisions with cold buffer gas (20% H2 balanced in helium) at 20 K, before being pulsed out into the extraction zone of a time-of-flight mass spectrometer with a 10 Hz repetition rate. The low temperature helps to eliminate hot bands in the spectrum and populate the lowest isomers. For each NIPES experiment, the desired anions were first mass-selected and decelerated before being intercepted by a probe laser beam in the photodetachment zone. In the current experiment, only 157 nm (7.867 eV) light from an F2 excimer laser was used. The laser was operated at a 20 Hz repetition rate with the ion beam off at alternating laser shots for shot-by-shot background subtraction. Photoelectrons were collected at nearly 100% efficiency by a magnetic bottle and analyzed in a 5.2 m long electron flight tube. Time-of-flight photoelectron spectra were collected and converted to kinetic energy spectra, calibrated by the known spectra of I− and Au(CN)2−. The electron binding energy (EBE) spectra were obtained by subtracting the kinetic energy spectra from the detachment photon energy. The electron energy resolution (ΔE/E) was about 2%, that is, ∼20 meV for 1 eV kinetic energy electrons.2.2 Theoretical methods
The equilibrium structures of Na+–DC22−(H2O)n (n = 0–6) anions were optimized with density functional theory using the B3LYP25–27 exchange–correlation functional with dispersion correction28,29 and the standard Pople type 6-31++G(d,p) basis set30 without any symmetry constraints. We did an extensive geometry search, which was guided by our understanding of the major driving forces in the system – strong Na+/dicarboxylate interactions and buildup of a solvent network that maximizes the coordination of negative (carboxylates) and positive (Na+) regions in the system. We have utilized up to 30 initial distinct guess structures for each cluster. The optimized structures between 0 and 6.0 kcal mol−1 in energy were confirmed to be real minima by calculating their harmonic vibrational frequencies. The B3LYP/6-31++G(d,p) relative energies including zero-point-energy (ZPE) and free energy corrections (at both 50 and 100 K) are reported. The vertical detachment energies (VDEs), defined as the energy difference between the neutral and the anion both at the geometry of the anion, were computed using the long-range corrected Perdew–Burke–Ernzerhof (PBE) exchange functional LC-ωPBE,31–33 which has been previously shown to be suitable to study salt-water clusters.203. Photoelectron spectra
Fig. 1 and 2 show the photoelectron spectra of M+–(DC22−)(H2O)n (M = Na+ and K+, n = 0–6) clusters measured with 157 nm photons. The VDEs of these clusters measured from their spectra are summarized in Table 1. The photoelectron spectra of M+–(DC22−)(H2O)n with n > 6 could not be obtained due to the low ion intensities.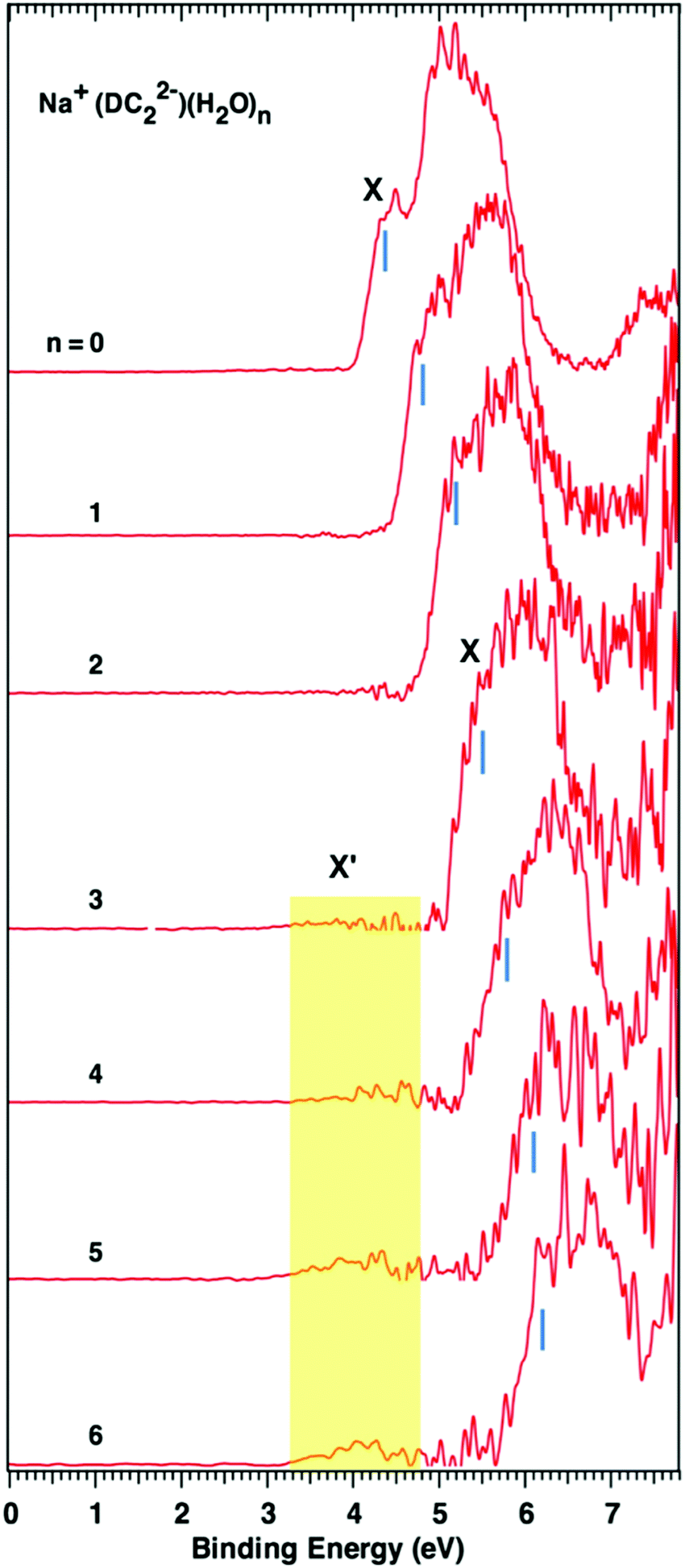 | ||
| Fig. 1 Photoelectron spectra of Na+–(DC22−)(H2O)n (n = 0–6) clusters measured with 157 nm photons. The blue vertical bars indicate the positions from which the VDEs of the main spectral bands (X) are determined. Weak features X′ show up at n ≥ 3. | ||
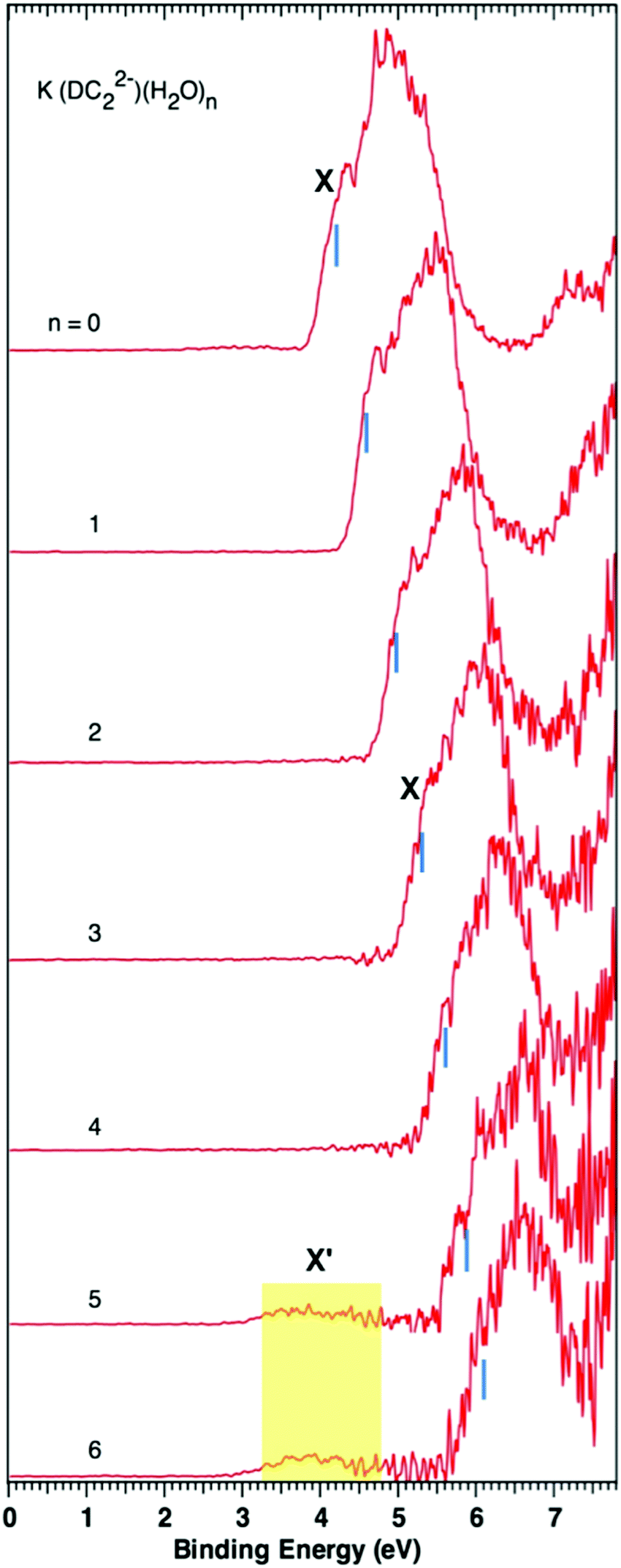 | ||
| Fig. 2 Photoelectron spectra of K+–(DC22−)(H2O)n (n = 0–6) clusters measured with 157 nm photons. The blue vertical bars indicate the positions from which the VDEs of the main spectral bands (X) are determined. Weak features X′ show up at n ≥ 5. | ||
For the bare Na+–(DC22−) complex we observe only one major broad band with a shoulder peak (X) at about 4.4 eV, which is consistent with the previous report.22 Addition of more waters shifts the peak position to higher binding energies but otherwise retains the same shape of the spectrum. Once a three water complex was approached, we have started to observe the formation of the low energy secondary band X′. It is weak compared to the main peak (X), but its intensity grows as more waters are added to the system. The VDEs of the main peak X of the experiments are determined to be 4.4, 4.8, 5.2, 5.5, 5.8, 6.1, and 6.2 eV for n = 0–6 waters, respectively, and the VDEs of band X′ are estimated to be ∼4.0 eV for n = 3 to 6 (Table 1).
The spectra of K+–(DC22−)(H2O)n (n = 0–6) show overall similar patterns, and follow the same trend as Na+-based complexes. There is a main spectral band that gradually shifts to higher binding energy as more waters are added to the system. The corresponding VDEs are 4.2, 4.6, 5.0, 5.3, 5.6, 5.9, and 6.1 eV for n = 0–6 waters respectively (Table 1). There is also evidence of the formation of a weak low binding energy band X′, which appears at a later stage (n = 5) than the Na+ case.
4. Theoretical results and discussion
4.1 The structures of Na+–(DC22−)(H2O)n (n = 0–6)
The optimized structures of the low energy isomers of Na+–(DC22−)(H2O)n (n = 0–6) are presented in Fig. 3–6. The theoretical relative energies and VDEs of these low-lying isomers are listed in Tables 2 and 3. As shown in Tables 2 and 3, the ZPE and free energy corrections are relatively small, therefore we describe, in the following, the energy landscape of different structures using relative energies, unless otherwise noted. We expect that the K+–(DC22−)(H2O)n system should have similar structure properties given the similarity of NIPE spectral patterns for both systems.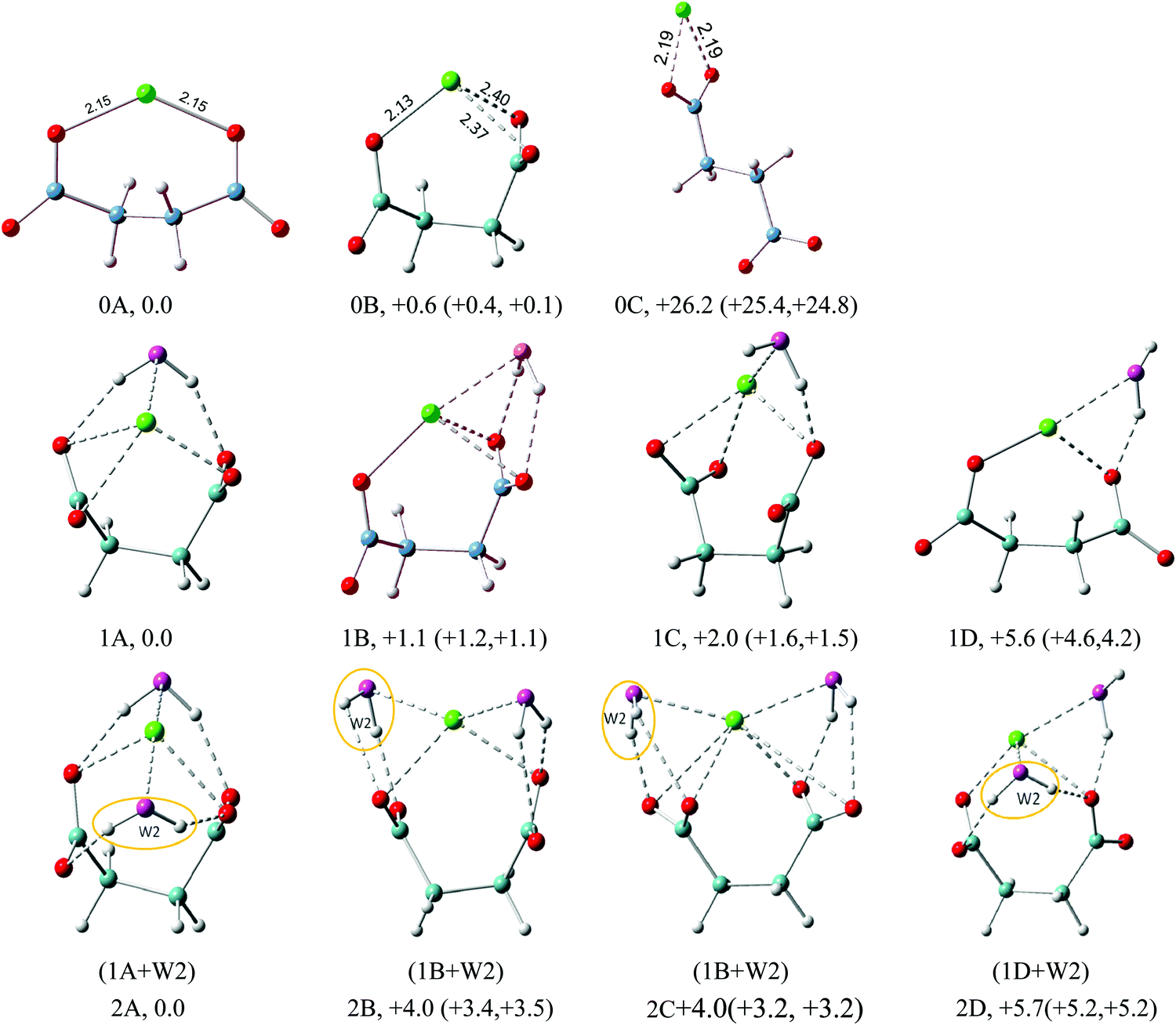 | ||
| Fig. 3 Typical low-lying isomers of Na+–(DC22−)(H2O)n (n = 0–2) clusters. The growth pattern of solvated clusters with n waters from the (n − 1) clusters is noted in parentheses with the n-th water circled. Na in green, C in light blue, H in gray, O of DC22− in red, and O of H2O in purple are used for all the structures. All of the structures are real minima based on the frequency calculations. Relative energies (kcal mol−1) including zero-point energy and free energy corrections (at 100 K) (first and second numbers in parentheses, respectively) are shown. More isomers can be found in the ESI.† | ||
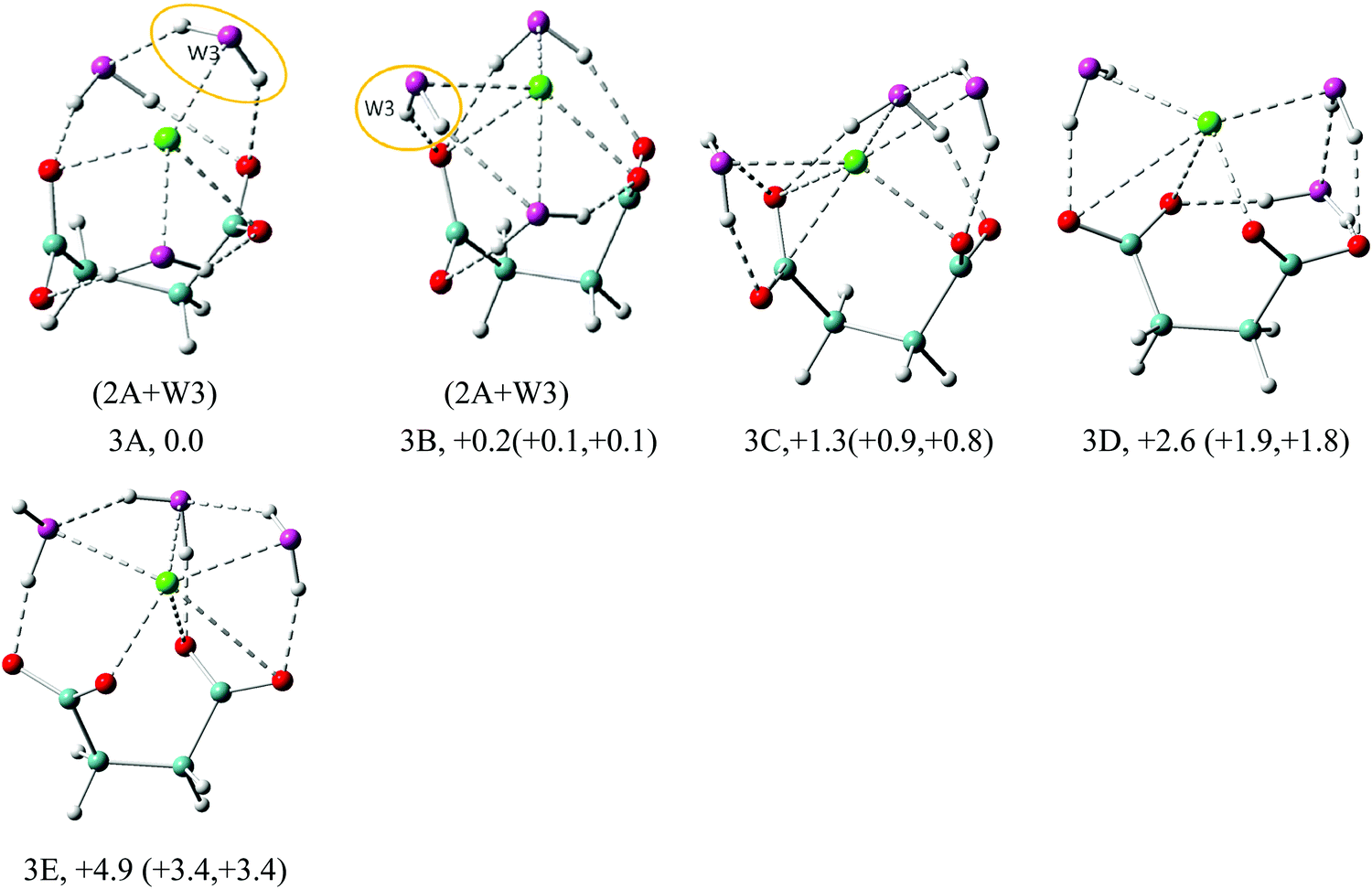 | ||
| Fig. 4 Typical low-lying isomers of Na+–(DC22−)(H2O)3 clusters. The growth pattern of solvated clusters with n waters from the (n − 1) clusters is noted in parentheses with the n-th water circled. Na in green, C in light blue, H in gray, O of DC22− in red, and O of H2O in purple are used for all the structures. All of the structures are real minima based on the frequency calculations. Relative energies (kcal mol−1) including zero-point energy and free energy corrections (at 100 K) (first and second numbers in parentheses, respectively) are shown. More isomers can be found in the ESI.† | ||
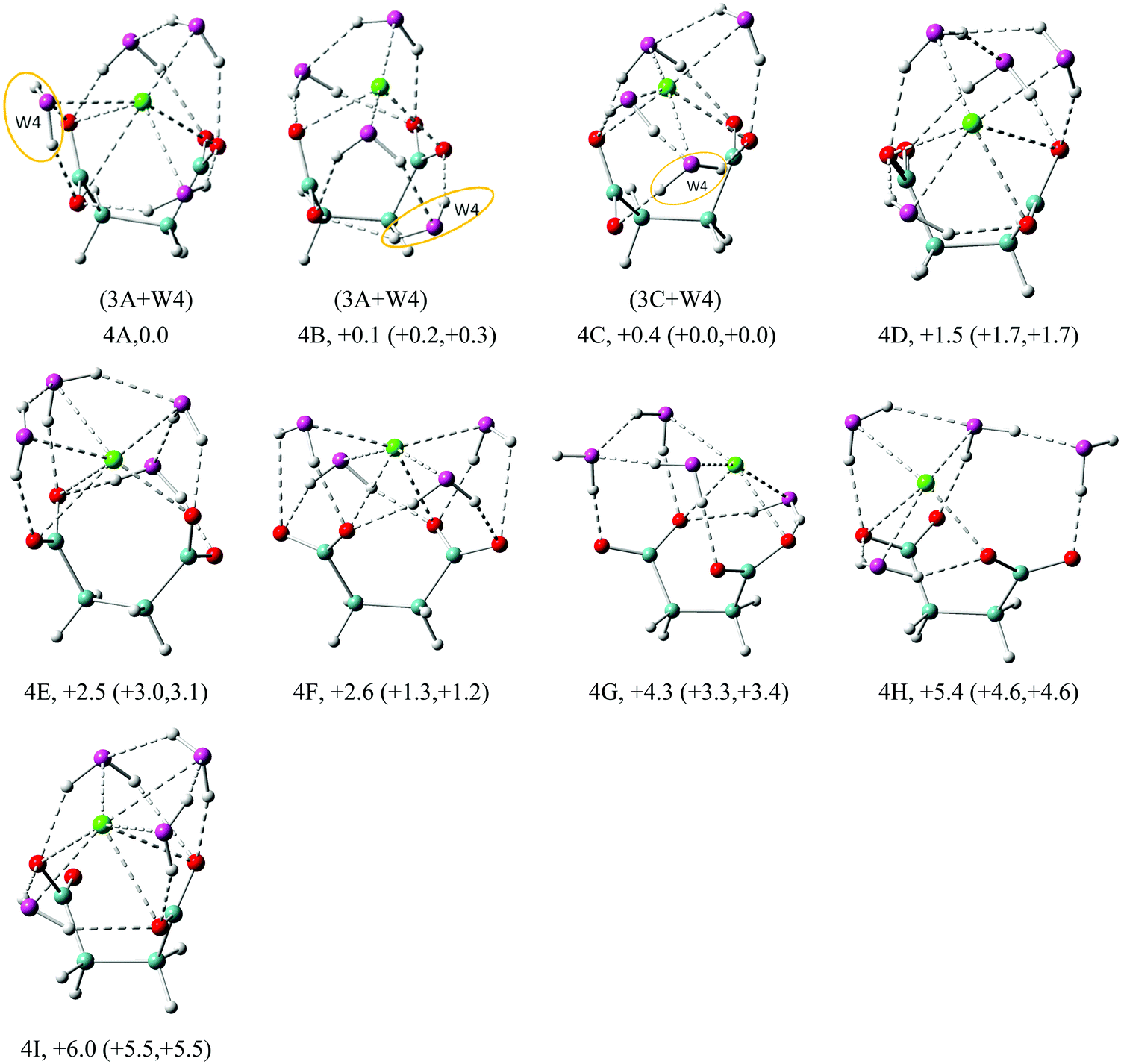 | ||
| Fig. 5 Structures of low energy isomers of Na+–(DC22−)(H2O)4 clusters. The growth pattern of solvated clusters with n waters from the (n − 1) clusters is noted in parentheses with the n-th water circled. Na in green, C in light blue, H in gray, O of DC22− in red and O of H2O in purple are used for all the structures. All of the structures are real minima based on the frequency calculations. Relative energies (kcal mol−1) including zero-point energy and free energy corrections (at 100 K) (first and second numbers in parentheses, respectively) are shown. More isomers can be found in the ESI.† | ||
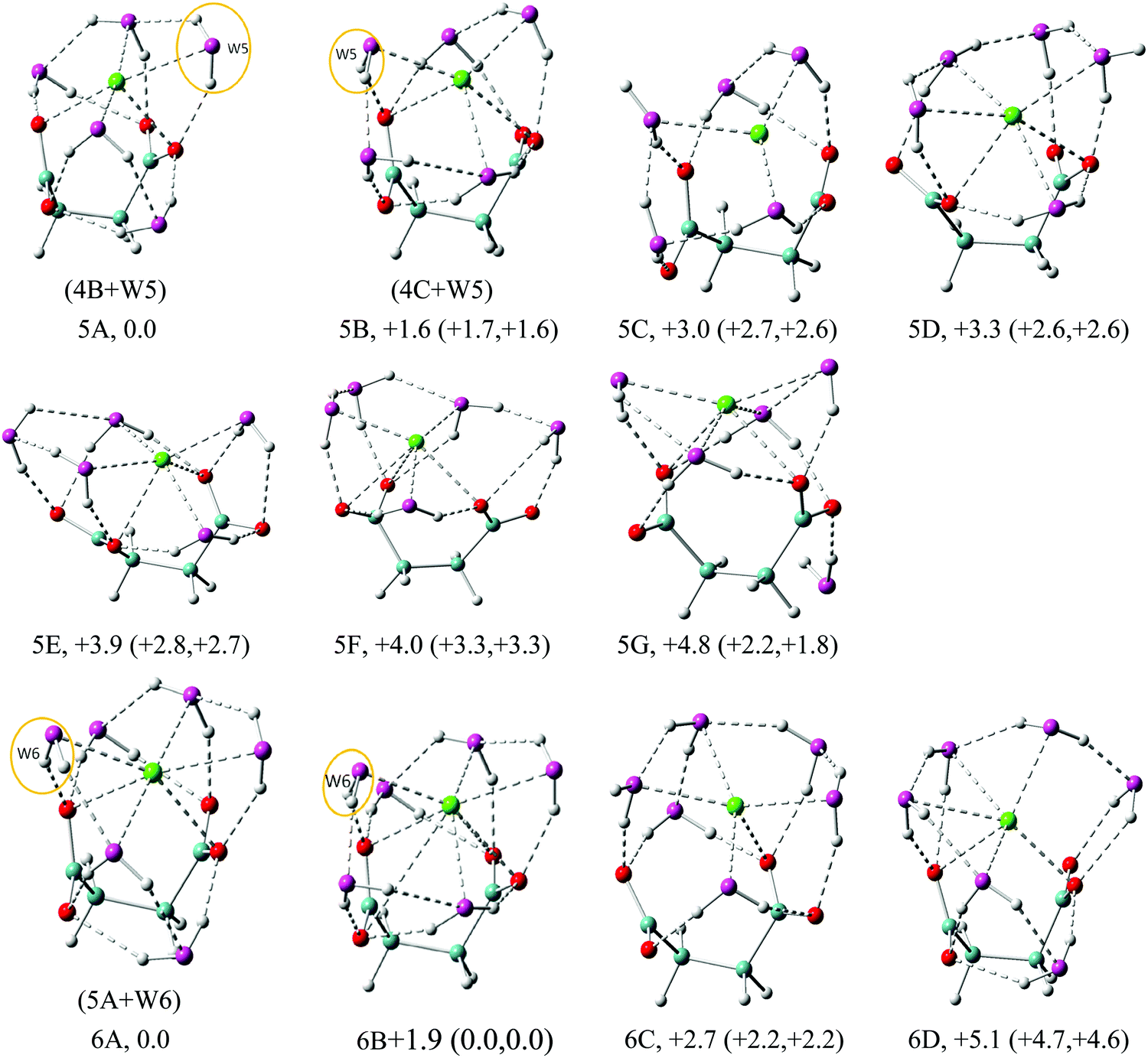 | ||
| Fig. 6 Structures of low energy isomers of Na+–(DC22−)(H2O)n (n = 5–6) clusters. The growth pattern of solvated clusters with n waters from the (n − 1) clusters is noted in parentheses with the n-th water circled. Na in green, C in light blue, H in gray, O of DC22− in red and O of H2O in purple are used for all the structures. All of the structures are real minima based on the frequency calculations. Relative energies (kcal mol−1) including zero-point energy and free energy corrections (at 100 K) (first and second numbers in parentheses, respectively) are shown. More isomers can be found in the ESI.† | ||
| Isomer | ΔE* (kcal mol−1) | VDE (eV) | ||
|---|---|---|---|---|
| Theo. | ||||
| LC-ωPBE | Expt | |||
| Na+–(DC22−) | 0A | 0.0 | 4.35 | 4.4 |
| 0B | +0.6 (+0.4, +0.1) | 4.31 | ||
| 0C | +26.2 (+25.4, +24.8) | 3.36 | ||
| Na+–(DC22−)(H2O) | 1A | 0.0 | 4.66 | 4.8 |
| 1B | +1.1 (+1.2, +1.1) | 4.65 | ||
| 1C | +2.0 (+1.6, +1.5) | 4.64 | ||
| 1D | +5.6 (+4.6, +4.2) | 4.49 | ||
| Na+–(DC22−)(H2O)2 | 2A | 0.0 | 5.11 | 5.2 |
| 2B | +4.0 (+3.4, +3.5) | 4.96 | ||
| 2C | +4.0 (+3.2, +3.2) | 4.95 | ||
| 2D | +5.7 (+5.2, +5.2) | 4.93 | ||
| Na+–(DC22−)(H2O)3 | 3A | 0.0 | 5.37 | 5.5 |
| 3B | +0.2 (+0.1, +0.1) | 5.41 | ||
| 3C | +1.3 (+0.9, +0.8) | 5.35 | ||
| 3D | +2.6 (+1.9, +1.8) | 5.34 | ||
| 3E | +4.9 (+3.4, +3.4) | 5.10 | ||
| Isomer | ΔE* (kcal mol−1) | VDE (eV) | ||
|---|---|---|---|---|
| Theo. | ||||
| LC-ωPBE | Expt | |||
| Na+–(DC22−)(H2O)4 | 4A | 0.0 | 5.67 | 5.8 |
| 4B | +0.1 (+0.2, +0.3) | 5.77 | ||
| 4C | +0.4 (0.0, 0.0) | 5.69 | ||
| 4D | +1.5 (+1.7, +1.7) | 5.66 | ||
| 4E | +2.5 (+3.0, +3.1) | 5.59 | ||
| 4F | +2.6 (+1.3, +1.2) | 5.71 | ||
| 4G | +4.3 (+3.3, +3.4) | 5.41 | ||
| 4H | +5.4 (+4.6, +4.6) | 5.48 | ||
| 4I | +6.0 (+5.5, +5.5) | 5.50 | ||
| Na+–(DC22−)(H2O)5 | 5A | 0.0 | 6.17 | 6.1 |
| 5B | +1.6 (+1.7, +1.6) | 6.00 | ||
| 5C | +3.0 (+2.7, +2.6) | 5.74 | ||
| 5D | +3.3 (+2.6, +2.6) | 5.58 | ||
| 5E | +3.9 (+2.8, +2.7) | 6.11 | ||
| 5F | +4.0 (+3.3, +3.3) | 5.89 | ||
| 5G | +4.8 (+2.2, +1.8) | 6.04 | ||
| Na+–(DC22−)(H2O)6 | 6A | 0.0 | 6.29 | 6.2 |
| 6B | +1.9 (+0.0, +0.0) | 6.24 | ||
| 6C | +2.7 (+2.2, +2.2) | 5.88 | ||
| 6D | +5.1 (+4.8, +4.6) | 5.96 | ||
The geometry optimizations show that the lowest minimum of Na+(DC22−), the 0A isomer, has a C2 symmetry with Na+ interacting directly with only one oxygen atom of each carboxylate group. The Na+–O distance is 2.15 Å in agreement with the previous report.22 These interactions prevent DC22− from reaching a fully unfolded configuration, with a dihedral backbone angle (–C–C–C–C–) of DC22− of 136°. The next lowest energy isomer (0B) exhibits a more folded configuration of DC22−, with a backbone angle of 65°. This enables higher 3-fold coordination Na+, with a total energy of only 0.6 kcal mol−1 above 0A (Fig. 3). The theoretical VDEs of 0A and 0B isomers are calculated to be 4.35 and 4.31 eV, close to the experimental value of 4.4 eV measured in this work. The third isomer 0C represents an example of an unfolded structure, with a backbone angle of DC22− of 180°. In this case Na+ interacts only with one of the carboxylate groups, leaving the other group uncoordinated. The loss of coordination results in significant energy penalties, raising the energy of the 0C isomer of 26 kcal above the ground state. Consistent with this we find that the VDE of isomer 0C (3.36 eV) is significantly lower than those of the other two isomers.
The energetic preference for the folded DC22− persists throughout the solvated complexes analyzed in this work. The internal structure of the underlying Na+(DC22−) complex remains fairly similar, and can be closely identified with the folded 0B isomer, with the backbone angle staying in the range of 53–68°. The sodium cation maintains its 2 or 3-fold coordination with DC22−. Unlike the monomolecular anion case, our particular system has three separate localized charge centers of different sign, positive metal cations and two negative carboxylate groups (Na+)(−O2C(CH2)2CO2−). Thus the early stages of the solvation process are driven by balancing the coordination of both of the negative and positive regions in the complex.
The geometry of DC22− is such that a single water molecule is able to form simultaneous contacts with two carboxylate oxygen atoms. The latter can belong to the different carboxylate groups – we will refer to such a solvation pattern as cross-linked. Alternatively, the interaction can also be formed with oxygens located on the same carboxylate group – we will refer to such a structure as lateral. The two solvation patterns are prominent in the n = 1 case. As shown in Fig. 3, the lowest energy isomer 1A features a cross-linked solvation. The enhanced stability can be easily rationalized, since it allows solvation of both negative charge centers in our complex. In the next isomer 1B, we observe an example of lateral solvation. The latter involves interaction with only one negative charge center, which makes it weaker than the cross-linked configuration, resulting in a slight elevation of the total energy (+1.1 kcal mol−1). We have located three low energy isomers within the range of ∼2 kcal mol−1. Isomer 1C represents another example of lateral solvation. However, in this case only one hydrogen bond is formed resulting in a further increase of the energy (+2.0 kcal mol−1). All three isomers feature direct contact between metal cations and water oxygen, with an average distance of ∼2.4 Å. The calculated VDEs of isomers 1A, 1B, and 1C are very close to each other, 4.66, 4.65, and 4.64 eV correspondingly, matching well the main experimental peak (X) (4.8 eV). Isomer 1D is very similar to 1C, but involves a more unfolded configuration of DC22− with a backbone angle of 137°. Its energy is 5.6 kcal mol−1 higher above 1A and its calculated VDE (4.49 eV) is much lower than the experimental value.
The structures of the Na+–(DC22−)(H2O)2 complex are all closely related to those found in the n = 1 case. The lowest energy isomer 2A can be obtained from 1A by adding water in the equivalent cross-linked position on the other side of DC22−. The overall structure is compact and maximizes a number of solute–solvent interactions, with 4-fold coordination of DC22− and 2-fold coordination of Na+. This is consistent with much more pronounced energy separation to the next lowest energy isomer. The calculated VDE for the 2A isomer is 5.11 eV, which agrees well with the experimental value. The next isomer 2B is derived from the 1B isomer, with additional lateral solvation on the vacant side of DC22−. The third isomer 2C is characterized by the same solvation structure as 2B but has different orientations of carboxylate groups. The fourth isomer, 2D, is related to 1D, with additional water attached in a cross-linked configuration. Overall the higher energies of 2B, 2C and 2D isomers (4.0, 4.0, and 5.7 kcal mol−1) are consistent with the trends observed in the n = 1 case, and suggest that a cross-linked solvation motif offers the best stabilization of DC22−.
In the n = 3 case (Fig. 4) we start observing the formation of the solvent–solvent network. The first two isomers, 3A and 3B, evolve from the parent 2A structure through the simple addition of water on top of the existing double cross-linked structure. The water molecule is added laterally to one of the carboxylate groups of DC22−, simultaneously creating contact with Na+ and existing cross-linked water. In 3A the resulting water dimer has a common hydrogen bond to the same carboxylate oxygen atom. In 3B the two cross-linked waters coordinate different oxygen atoms on the same carboxylate group, which leads to a larger backbone angle (64° in 3B vs. 56° in 3A). The 3B structure achieves the highest overall coordination of Na+ (5 in 3B vs. 4 in 3A), however the energies of isomers 3A and 3B are nearly the same. The third isomer 3C emphasizes a lateral solvation pattern, with only one water participating in cross-linked solvation of DC22−. This leads to a slight elevation of the energy (∼1 kcal mol−1), which is consistent with our observation in the n = 1, 2 cases. Despite its higher energy, isomer 3C has the highest 6-fold coordination of Na+. One can view that the evolution of solvation at these early stages described above is driven by the solvation of DC22−. Isomer 3D is similar to 3C but has different orientation of carboxylate groups. Its energy slightly elevated and is 1.3 kcal mol−1 higher in energy than 3C. All four isomers have fairly similar VDEs (5.37 eV for 3A, 5.41 eV for 3B, 5.35 eV for 3C, and 5.34 eV for 3D) and match well the main experimental peak at 5.5 eV. Isomer 3E switches completely to a lateral solvation mode. Its energy is higher than that of 3A by 4.9 kcal mol−1, and has a much lower calculated VDE than the experimental value.
The n = 4 case in Fig. 5 continues to build the solvent coordination around DC22−. The first isomer 4A evolves from 3A, with the additional water forming a bidentate complex with carboxylate, coordinating both oxygen atoms. This maximizes solvation of DC22− to 7-fold, but creates no additional solvent–solvent hydrogen bonds. We observe 6-fold coordination of Na+, which is equally split between solvent and DC22− interactions. In the next isomer 4B, the bidentate carboxylate solvation is switched to a single one, enabling the formation of an additional solvent–solvent hydrogen bond. Similar to 3A, the resulting water dimer shares interaction with the same carboxylate oxygen. The coordination of Na+ in the 4B isomer is only 4-fold – two from solvent and two from DC22−. Isomer 4C is akin to 4B, but in this case the two interacting waters coordinate different carboxylate oxygens. Similar to 4A, isomer 4C is characterized by 6-fold coordination of Na+, but now four of those come from solvent. Despite all these differences, all three isomers are nearly degenerate in energy. Isomer 4D has a 6-fold coordination of Na+, with 1.5 kcal mol−1 more energy than 4A. All of the first four isomers are characterized by similar VDEs (5.67 eV for 4A, 5.77 eV for 4B, 5.69 eV for 4C, and 5.66 eV for 4D) that match the experimental value of 5.8 eV. Isomers 4E–4I are higher in energy than 4A by 2.5, 2.6, 4.3, 5.4, and 6.0 kcal mol−1, respectively.
In the n = 5 case (Fig. 6) we observe the creation of additional solvent–solvent hydrogen bonds. We also start detecting a separation between coordination shells of DC22− and Na+. In particular, in the lowest energy isomer 5A the two cross-linked waters no longer coordinate Na+ directly. The Na+ coordination is actually lowered to 5-fold compared to 6-fold in the 4A isomer, while DC22− remains unchanged at 7-fold. This separation is less pronounced in the next isomer 5B, which is located 1.6 kcal mol−1 above 5A. The third isomer 5C is located ∼3 kcal mol−1 above, and is missing the double cross-linked solvation pattern present in 5A and 5B. All three isomers are characterized by three solvent–solvent hydrogen bonds coming from disjoint dimer and trimer chain water clusters. Comparison of VDE values identifies isomers 5A (6.17 eV) and 5B (6.00 eV) as likely candidates contributing to the main experimental peak at 6.1 eV. The higher energy isomers 5D–5G can also be characterized by the formation of lateral or cross-linked solvation patterns with the water attached to the DC22− unit.
In the n = 6 case, as shown in Fig. 6, the lowest energy isomer 6A is very similar to 5A with additional water interacting with Na+, DC22−, and neighboring water. With this arrangement every oxygen atom on DC22− now forms two hydrogen bonds to a solvent. The calculated VDE of 6.29 eV agrees well with the experimental value of 6.2 eV. Isomer 6B is higher in energy than 6A by 1.9 kcal mol−1, but becomes degenerate after ZPE and free energy corrections, with a similar structure to 6A except the slight difference of bond lengths and bond angles. The calculated VDE of 6.24 matches well with the experimental value. The next two isomers 6C and 6D are located 2.7 and 5.1 kcal mol−1 above 6A. The VDEs (5.88 eV for 6C and 5.96 eV for 6D) also differ from experimentally observed signals. We note that in isomer 6C the Na+ ion shows significantly less interaction with DC22−, exhibiting essentially only one contact with DC22−. This may be viewed as an early indication of the solvent-separated complex.
More low-lying isomers for Na+–(DC22−)(H2O)n (n = 0–6) clusters are shown in Fig. S1–S6 in the ESI† of this manuscript. It is worth noting that for solvated clusters, in particular with a flexible solute, as in the current case, various solvation motifs exist, and can contribute to the experiments. As shown in Tables 2 and 3, multiple isomers with the lowest energies and free energies are identified to contribute to the experimental spectrum for each cluster. In reality, considering the fluxionality and dynamic nature of hydrogen bond networks, our experiments should be viewed to sample many isomeric structures that are inter-converted and governed by free energy changes.
4.2 Ion separation in Na+–(DC22−)(H2O)n
Throughout the experimental solvation range of n = 0–6 for Na+–(DC22−)(H2O)n clusters, the cation Na+ and anion DC22− are always in contact with each other, forming what is commonly referred to be a contact ion pair (CIP). However, addition of waters clearly weakens interactions between the two species as evidenced by the increase of the distance between Na+ and carboxylate oxygen (2.15 Å in the bare cluster to 2.45 Å in the n = 6 cluster). One would expect for such a trend to continue, eventually leading to the appearance of low energy solvent separated ion-pair (SSIP) complexes. To get more insight into this process, we performed a preliminary exploration of the next few larger cluster sizes, n = 7–10. The lack of experimental VDE data and increased complexity make the identification of ground state structures much more uncertain, but still should provide useful qualitative information.The computational protocol utilized in these calculations was similar to that utilized for smaller clusters, however in addition to manual construction we also utilized ab initio molecular dynamics (MD) to diversify a pool of initial guess structures. Similar to optimization, MD simulations were based on density functional theory using the B3LYP25–27 exchange–correlation functional with dispersion correction28,29 with the 6-31++G(d,p) basis set.30 The actual process consisted of choosing suitable configurations from approximately 1 ps MD simulation performed at T = 300 K temperature. The final optimized lowest energy isomers are presented in Fig. S7–S10 in the ESI† of this manuscript. All of the optimized structures are verified to be local minima by calculating the harmonic vibrational frequencies.
Similar to smaller clusters, the ground state structures of n = 7–10 clusters continue to maintain their CIP-like character, with Na+ interacting with both carboxylate groups. Interestingly the distance between Na+ and carboxylate oxygen for n = 7–10 remains essentially the same (∼2.45 Å) as in the n = 6 cluster, suggesting that solvent coordination of CIP complexes reaches the saturation point. No SSIP structures were observed for the n = 7 cluster. For n = 8, the lowest energy SSIP structure (8M) was ∼8.7 kcal above the ground state, for n = 9 at 7.9 kcal mol−1 (see 9G), and for n = 10 at 7.4 kcal mol−1 (see 10J). Overall, based on these, admittedly limited results, one can conjecture that the n = 8 cluster may mark the onset of SSIP like structures, albeit still higher in energy than the ground state CIP complex.
4.3 The origin of the weak band (X′) of M+–DC22−(H2O)n (M = Na, K)
As mentioned earlier, the experimental data indicate the presence of a weak low energy band (X′) centered at ∼4.0 eV in the n = 3 and higher solvated clusters in the Na+ case. A similar phenomenon is also observed for n = 5 and 6 for K+–DC22−(H2O)n clusters. The precise origin of this band is not entirely clear. Our calculations offer no evidence that this signal may be attributed to any of the low-lying states of Na+–DC22−(H2O)n complexes. The remaining options consist of possible photo-fragmented species or other chemically different clusters that have the same mass-to-charge ratio. Excited state calculations performed at the B3LYP level for the solvated anion clusters do indicate the presence of broad absorption bands that would be accessible with 157 nm photon energy used in the experiment (Fig. S11, ESI†). This could, in principle, lead to photo-induced fragment daughter anions, which may undergo electron detachment through the absorption of a second photon, and in principle, could generate signals at lower electron binding energies. However, in our experiment, the 157 nm photon flux is fairly low, and may not be enough to sustain this type of two-photon process. Regarding the second option, in the course of our calculations we did observe an unusual stable quaternary complex that involved OH− and protonated dicarboxylate (HO2C(CH)2CO2−, HDC2−) species. An example, of such a complex for the n = 3 case [Na+(HDC2)−(OH)−(H2O)2] is shown in Fig. S12 in the ESI† of this manuscript. The calculated VDE of this particular species is ∼4 eV, consistent with the low binding energy band in the NIPE spectra. The possibility of such species in aqueous electrolyte solutions was previously suggested by Robinson and Harned,34 where it was argued that the field from anions has the capacity to induce the so-called localized hydrolysis. It has also been reported34 that the dissociation of water in sodium salt solutions is greater than that in potassium salt solutions, which would be consistent with the later onset of the low energy band in the K+ clusters. The main problem of this hypothesis is that our calculations place the quaternary complex at a relatively high energy of ∼22 kcal mol−1 above the ground state of ternary clusters for n = 3. However, it needs to be pointed out that all component ions in both quaternary and ternary complexes [Na+, HDC2−, OH−, H2O, DC22−] exist in our sprayed solution, and previous studies have shown that higher energy isomers can often exist even at low temperatures due to kinetic trapping.35–39 Therefore the existence of such a quaternary system remains a possibility, which may contribute to the weak low binding energy band.5. Conclusions
We have presented combined experimental and theoretical analysis of solvated dicarboxylate Na+/K+ metal ion complexes. In both cases, the experimental photoelectron spectroscopy data show the presence of a well-defined main spectral peak that gradually shifts to higher energies with an increasing number of waters. The experimental VDE value of each solvated K+ cluster is lower than that of its Na+ counterpart by ∼0.2 eV pointing at slightly lower binding energies of K+. In the Na+ case, based on ab initio calculations we have identified the ground state structures that provide a good match to photoelectron spectroscopy data. For the solvation range of up to six waters studied in this work, the core of the cluster, Na+(DC22−) complex, essentially remains unchanged maintaining two direct contacts between Na+ and respective carboxylate groups. The evolution of the solvation structure appears to be dominated by building coordination to DC22− with opportunistic coordination of Na+. The initial coordination of DC22− appears to be completed at n = 6, whereby every oxygen atom on DC22− now forms two hydrogen bonds to a solvent. Preliminary theoretical analysis of larger n = 7–10 clusters shows that the SSIP structure model starts to appear, albeit higher in energy than the CIP structure when the water number increases to 8. The origin of the experimental low energy band observed starting at n = 3 for Na+ (n = 5 for K+) cannot be conclusively identified at this point. We tentatively assign it to a higher energy quaternary [Na+, HDC2−, OH−, H2O] complex, hinging on the hypothesis that such a complex may be kinetically trapped.35–39Conflicts of interest
The authors declare no competing interest.Acknowledgements
This research was supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences and Biosciences (X.-B. W. and M. V.) and was performed at the EMSL, a national scientific user facility sponsored by DOE's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory. This work is also in part supported (to R.-Z. L.) by the National Science Foundation of China (Grant No. 21301134) and the China Scholarship Council. We are grateful to Dr Niranjan Govind for valuable discussions.References
- B. Hille, Ion channels of excitable membranes, Sinauer Sunderland, MA, USA, 3rd edn, 2001 Search PubMed.
- T. Alber, Annu. Rev. Biochem., 1989, 58, 765 CrossRef CAS PubMed.
- L. Vrbka, J. Vondrášek, B. Jagoda-Cwiklik, R. Vácha and P. Jungwirth, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15440 CrossRef CAS PubMed.
- J. S. Uejio, C. P. Schwartz, A. M. Duffin, W. S. Drisdell, R. C. Cohen and R. J. Saykally, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6809 CrossRef CAS PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2010, 132, 2321 CrossRef CAS PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2009, 131, 8092 CrossRef CAS PubMed.
- C. Y. Tang and H. C. Allen, J. Phys. Chem. A, 2009, 113, 7383 CrossRef CAS PubMed.
- E. F. Aziz, N. Ottosson, S. Eisebitt, W. Eberhardt, B. Jagoda-Cwiklik, R. Vácha, P. Jungwirth and B. Winter, J. Phys. Chem. B, 2008, 112, 12567 CrossRef CAS PubMed.
- K. D. Collins, Methods, 2004, 34, 300 CrossRef CAS PubMed.
- K. D. Collins, Biophys. J., 1997, 72, 65 CrossRef CAS PubMed.
- J. Dzubiella, J. Am. Chem. Soc., 2008, 130, 14000 CrossRef CAS PubMed.
- C. Dedonder-Lardeux, G. Grégoire, C. Jouvet, S. Martrenchard and D. Solgadi, Chem. Rev., 2000, 100, 4023 CrossRef CAS PubMed.
- T. Wende, N. Heine, T. I. Yacovitch, K. R. Asmis, D. M. Neumark and L. Jiang, Phys. Chem. Chem. Phys., 2016, 18, 267 RSC.
- T. Jin, B. Zhang, J. Song, L. Jiang, Y. Qiu and W. Zhuang, J. Phys. Chem. A, 2014, 118, 9157 CrossRef CAS PubMed.
- L. Vrbka, P. Jungwirth, P. Bauduin, D. Touraud and W. Kunz, J. Phys. Chem. B, 2006, 110, 7036 CrossRef CAS PubMed.
- B. Hess and N. F. van der Vegt, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13296 CrossRef CAS PubMed.
- T. B. Robertson, J. Biol. Chem., 1911, 9, 303 CAS.
- S. Maldonado, M. P. Irún, L. A. Campos, J. A. Rubio, A. Luquita, A. Lostao, R. Wang, E. García-Moreno and J. Sancho, Protein Sci., 2002, 11, 1260 CrossRef CAS PubMed.
- W. Kunz, J. Henle and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 19 CrossRef CAS.
- R.-Z. Li, C.-W. Liu, Y. Q. Gao, H. Jiang, H.-G. Xu and W.-J. Zheng, J. Am. Chem. Soc., 2013, 135, 5190 CrossRef CAS PubMed.
- X.-B. Wang, J. Phys. Chem. A, 2017, 121, 1389 CrossRef CAS PubMed.
- G. Murdachaew, M. Valiev, S. M. Kathmann and X.-B. Wang, J. Phys. Chem. A, 2012, 116, 2055 CrossRef CAS PubMed.
- X.-B. Wang and L.-S. Wang, Rev. Sci. Instrum., 2008, 79, 073108 CrossRef PubMed.
- Q. Yuan, X.-T. Kong, G.-L. Hou, L. Jiang and X.-B. Wang, Phys. Chem. Chem. Phys., 2018, 20, 19458 RSC.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463 CrossRef CAS PubMed.
- G. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081 CrossRef CAS.
- O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed.
- O. A. Vydrov, J. Heyd, A. V. Krukau and G. E. Scuseria, J. Chem. Phys., 2006, 125, 074106 CrossRef PubMed.
- O. A. Vydrov, G. E. Scuseria and J. P. Perdew, J. Chem. Phys., 2007, 126, 154109 CrossRef PubMed.
- R. A. Robinson and H. S. Harned, Chem. Rev., 1941, 28, 419 CrossRef CAS.
- G.-L. Hou, X.-T. Kong, M. Valiev, L. Jiang and X.-B. Wang, Phys. Chem. Chem. Phys., 2016, 18, 3628 RSC.
- V. Brites, A. Cimas, R. Spezia, N. Sieffert, J. M. Lisy and M. P. Gaigeot, J. Chem. Theory Comput., 2015, 11, 871 CrossRef CAS PubMed.
- B. Yang and M. T. Rodgers, Phys. Chem. Chem. Phys., 2014, 16, 16110 RSC.
- G. Papadopoulos, A. Svendsen, O. V. Boyarkin and T. R. Rizzo, J. Am. Soc. Mass Spectrom., 2012, 23, 1173 CrossRef CAS PubMed.
- D. J. Goebbert, T. Wende, L. Jiang, G. Meijer, A. Sanov and K. R. Asmis, J. Phys. Chem. Lett., 2010, 1, 2465 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp03896a |
| ‡ Visiting scholar supported by the PNNL alternate sponsored fellowship. |
| This journal is © the Owner Societies 2018 |