
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Temperature dependence of dynamic, tunnelling and kinetic isotope effects in formate dehydrogenase†
Maite
Roca
 *a,
J. Javier
Ruiz-Pernía
b,
Raquel
Castillo
a,
Mónica
Oliva
a and
Vicent
Moliner
*a
*a,
J. Javier
Ruiz-Pernía
b,
Raquel
Castillo
a,
Mónica
Oliva
a and
Vicent
Moliner
*a
aDepartament de Química Física i Analítica, Universitat Jaume I, 12071 Castellón, Spain. E-mail: mroca@uji.es; moliner@uji.es; Fax: +34 964 728066; Tel: +34 964 728069 Tel: +34 964 728084
bDepartament de Química Física, Universitat de València, 46100 Burjassot, Spain
First published on 18th September 2018
Abstract
The origin of the catalytic power of enzymes has been a question of debate for a long time. In this regard, the possible contribution of protein dynamics in enzymatic catalysis has become one of the most controversial topics. In the present work, the hydride transfer step in the formate dehydrogenase (FDH EC 1.2.1.2) enzyme is studied by means of molecular dynamic (MD) simulations with quantum mechanics/molecular mechanics (QM/MM) potentials in order to explore any correlation between dynamics, tunnelling effects and the rate constant. The temperature dependence of the kinetic isotope effects (KIEs), which is one of the few tests that can be studied by experiments and simulations to shed light on this debate, has been computed and the results have been compared with previous experimental data. The classical mechanical free energy barrier and the number of recrossing trajectories seem to be temperature-independent while the quantum vibrational corrections and the tunnelling effects are slightly temperature-dependent over the interval of 5–45 °C. The computed primary KIEs are in very good agreement with previous experimental data, being almost temperature-independent within the standard deviations. The modest dependence on the temperature is due to just the quantum vibrational correction contribution. These results, together with the analysis of the evolution of the collective variables such as the electrostatic potential or the electric field created by the protein on the key atoms involved in the reaction, confirm that while the protein is well preorganised, some changes take place along the reaction that favour the hydride transfer and the product release. Coordinates defining these movements are, in fact, part of the real reaction coordinate.
1. Introduction
Elucidating what happens inside an enzyme's active site that allows difficult chemical reactions to take place in timescales compatible with life has occupied the attention of experimentalist and theoretician biochemists for a long time; this is still not clear and remains under debate. Different hypotheses have been proposed to explain the amazing enhancement of the chemical kinetics of enzymes with respect to the counterpart reaction in solution. One of these hypotheses suggests that the transition state (TS) stabilization relative to the reactant state is largely due to the preorganised electrostatic environment provided by the active site of the enzyme;1–5 this proposal has been supported by many studies.6–20 Nevertheless, it has also been proposed that protein motions contribute to reducing the energy-barrier crossing through the TS toward the products.21–33 In fact, the role of protein dynamics in enzymatic catalysis is one of the most controversial topics in modern enzymology, providing a hot debate among theoreticians and experimentalists.16,17,19–21,26,28,32,34–43Protein motions are frequently observed during substrate binding and product release steps. Nevertheless, the contribution of protein motions to the chemical reaction process and how these motions may influence enzyme catalysed reactions is difficult to probe. The dynamic idea has been explored by several experimental and computational studies. Some researchers have tried to demonstrate, specifically in enzyme-catalysed hydrogen transfer reactions, that quantum mechanical hydrogen tunnelling associated with enzymatic X–H bond cleavage links to protein dynamics for achieving optimal enzyme catalysis.33,37,44–46 These studies are based on the temperature dependence of the kinetic isotope effect (KIE) method that has been used in recent years to study the nature of H-transfer enzymatic reactions and can be a powerful tool for understanding the role of dynamics in enzyme catalysis.
In other research oriented toward the study of dynamic effects, “heavy” enzymes have been created by means of the isotopic substitution of nitrogen, carbon and non-exchangeable hydrogen atoms by their heavier isotopes.36 The rate constants of these “heavy” enzymes have been obtained and compared with those of their “light” (natural isotopic abundance) counterpart.16,17,41–43,47–53 In these studies, catalysis is affected by a slight reduction in the rate constant in the heavy enzyme. From these results, some authors identify a coupling between the environmental and chemical motions, but these conformational fluctuations around the TS have a small effect on the chemical rate constant.16,17,19,20,36,41–43 Nevertheless, other authors interpret these enzyme KIEs in different ways. Thus, it has been suggested that protein promoting vibrations (PPV) are coupled with the chemical step of the catalytic process, and the catalytic power of enzymes is due to these protein motions.47–51,53 Notwithstanding these findings, there is not a definitive experimental technique that has actually established a connection between promoting motions and a decrease in the activation free energy of the chemical step.36,39
In order to computationally obtain the rate constant of the chemical step of an enzyme-catalysed reaction, the dynamic and tunnelling effects can be quantified as departures from the transition state theory (TST)54–56 by means of ensemble-averaged variational transition state theory (EA-VTST),57–59
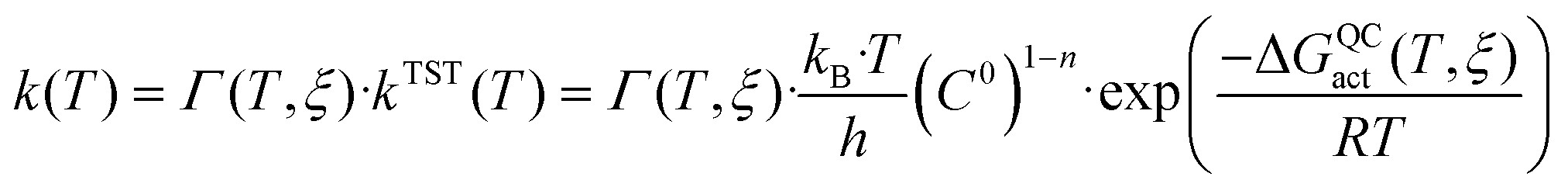 | (1) |
The quasi-classical activation free energy ΔGQCact can be obtained from the classical mechanical potential of mean force (PMF) and includes a correction for quantizing the orthogonal vibrations to the reaction coordinate (ΔW‡vib(T,ξ)):17,41
| ΔGQCact(T,ξ) = ΔG‡clas(T,ξ) + ΔW‡vib(T,ξ) | (2) |
| Γ(T,ξ) = γ(T,ξ)·κ(T) | (3) |
In order to estimate the recrossing transmission coefficient, the “positive flux” formulation,60 based on molecular dynamics (MD) trajectories starting from the TS with thermal distribution velocities or the Grote–Hynes theory61–63 can be used. Recent studies were carried out in our group to quantify the value of the recrossing transmission coefficient on different enzyme-catalysed reactions such as the methyl transfer from S-adenosylmethionine to catecholate in the catechol O-methyltransferase,9,10 or to glycine in the glycine N-methyltransferase,64,65 the Michael addition catalysed by chalcone isomerase,12 the thymidylate synthase-catalysed reaction,66 the human purine nucleoside phosphorylase-catalysed reaction17 or the hydride transfer reactions catalysed by formate dehydrogenase67 and dihydrofolate reductase.41
The tunnelling transmission coefficient κ(T), can be calculated with the small-curvature tunnelling (SCT) approximation,68,69 which includes a reaction-path curvature appropriate for enzymatic hydride transfers. This strategy has been successfully employed to estimate the quantum tunnelling effects in dihydrofolate reductase, thymidylate synthase and morphinone reductase.41,70–72
Finally, the kinetic isotope effects (KIEs) are calculated as the quotient between the light isotope rate constant (kH, if the atom to be transferred is hydrogen) and the heavy isotope rate constant (kT, if the isotope is tritium). Then, from the definition of the rate constant based on VTST, the KIE can be obtained as the product of three contributions: the KIE due to recrossings (KIE(γ)), the KIE due to tunnelling (KIE(κ)) and the KIE due to the quasi-classical activation free energy (KIE(ΔGQCact)).
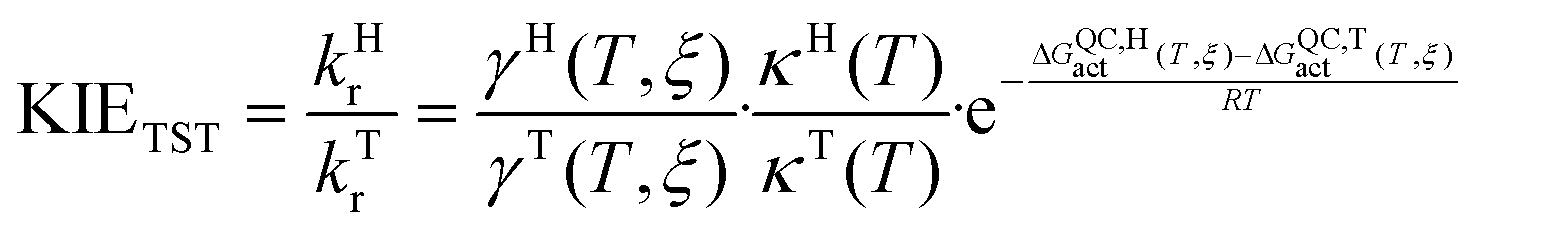 | (4) |
| KIETST = KIE(γ)·KIE(κ)·KIE(ΔGQCact) | (5) |
FDH is a ubiquitous enzyme belonging to a family of D-isomer-specific 2-hydroxy acids and plays a key role since it is one of the best enzymes for cofactor regeneration in the processes of chiral synthesis with NAD(P)+-dependent oxidoreductases.73 Moreover, FDH has the biotechnological potential for use in organic acid synthesis as a catalyst in coenzyme regeneration systems for the production of high value added pharmaceutical products, such as a biosynthetic amino acid, tert-L-leucine, which is a component of some HIV protease and matrix metalloprotease inhibitors.74–76 Site-directed mutagenesis studies on FDH have been also carried out with the goal to transform coenzyme specificity as well as to increase its turnover frequency factor or its thermal stability.77 To this end, a detailed knowledge of the catalytic mechanism of FDH at the molecular level could provide guidelines for future protein engineering on this enzyme.
The crystal structures of FDH have been solved for three species: the methylotrophic bacteria Pseudomonas sp. 101 (PsFDH)78 and Moraxella C-1 (MorFDH)79 and the yeast Candida boidinii (CbFDH).80 FDH catalyses the oxidation of formate ion to carbon dioxide via hydride transfer to the C4 carbon of the nicotinamide ring of NAD+ to obtain NADH (see Scheme 1). This hydride transfer reaction is partly a rate-limiting step in the catalytic mechanism of FDH with an observed rate constant of 7.3 s−1.81–83 This value of the rate constant shows that FDH is a low efficiency enzyme, compared to other NAD+-dependent dehydrogenases that show much higher rate constants, probably due to the strong interactions between the charged substrate in the reactant state and several residues of the active site.84
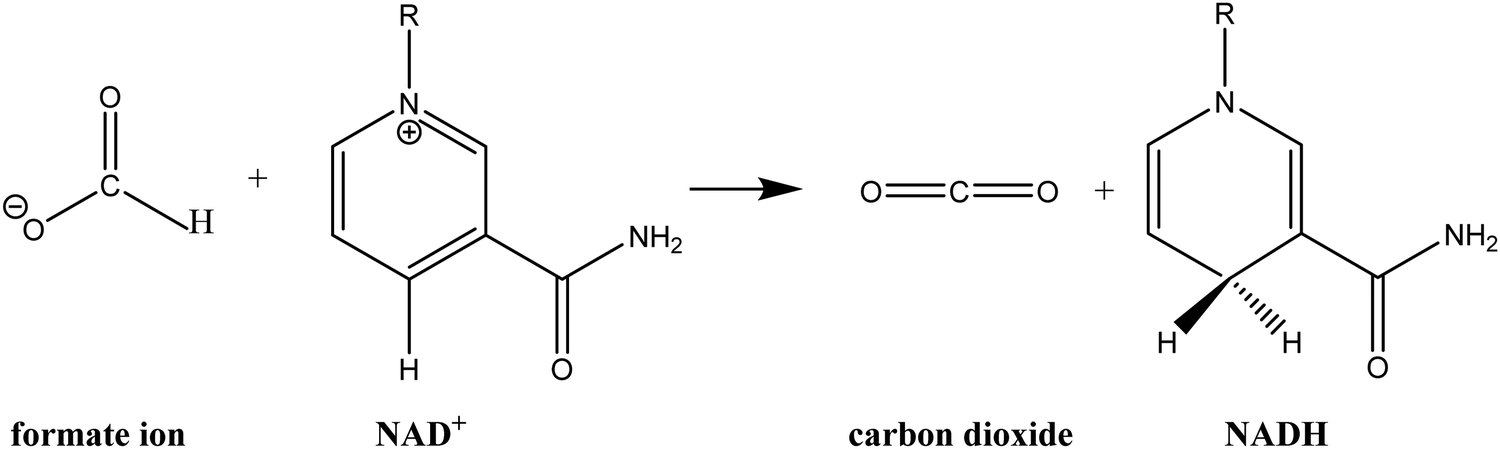 | ||
| Scheme 1 Schematic representation of the hydride transfer reaction from formate ion to NAD+ catalysed by FDH. | ||
Theoretical studies of the FDH-catalysed hydride transfer reaction of PsFDH were carried out by Major and co-workers86 using hybrid QM/MM simulations developing a specific reaction parameter (SRP) Hamiltonian by reparameterizing the semiempirical AM1 method. The PMFs were obtained as a function of the antisymmetric coordinate (bond breaking and bond forming distances) as well as an orbital rehybridization coordinate and a donor–acceptor distance coordinate, showing an early TS and that the chemical and environmental coordinates are not fully synchronized. The activation free energy barrier when the AM1-SRP/MM Hamiltonian was employed (ΔG = 15.1 kcal mol−1) was almost identical to that obtained when the QM atoms were treated with the standard AM1 potential. However, it is worth mentioning that differences in the reaction free energies obtained with both methods were more remarkable (being around 8.0 kcal mol−1 more exergonic for the standard AM1/MM PMF). Quantum PMFs were obtained for hydride, deuteride and triteride transfer reactions in order to compute kinetic isotope effects using a mass-perturbation-based path-integral approach.87 Using QM(AM1-SPR)/MM, the inclusion of nuclear quantum effects in the simulations lowered the computed free energies of activation for the protium, deuterium, and tritium transfer by 1.9, 1.4 and 1.1 kcal mol−1, respectively. In this way, the resulting activation quantum free energy was 13.2 kcal mol−1,86 a quite similar value to that obtained by us previously (12.4 kcal mol−1).13,67
Recently, structural, kinetic and spectroscopic studies of FDH from recombinant Candida boidinii were carried out80 to study the dynamic effects in CbFDH.44,85,88 The studies on recombinant CbFDH found temperature-independent intrinsic KIEs,80 as previously reported for commercial CbFDH (a mixture of isozymes).44 This temperature independence of KIEs has been interpreted as a well-organized donor–acceptor distance for the H transfer and an optimal “fine-tuning” of the transition state for the H transfer step in FDH.44,80 These results indicate that the donor–acceptor distance is narrowly distributed in both recombinant enzyme and in the isozyme mixture.80 QM/MM free energy simulations on the recombinant CbFDH enzyme yielded a free energy profile in agreement with the experimental data and it is very similar to the one obtained from PsFDH.86
Recently, two-dimensional infrared spectroscopy measurements on the ternary complex of FDH with cofactor and the azide as the transition state analogue, carried out by Kohen and co-workers in CbFDH,46 revealed structural fluctuations on the picosecond to femtosecond timescale, which could be relevant to catalysis. The measurements of the frequency–frequency correlation function for the azide complexed to FDH with NAD+ showed underdamped oscillatory frequency fluctuations on the picosecond timescale. The authors suggested that the active site insulates the reacting molecules from the thermal noise of the surrounding solvent. These oscillations have been attributed to motions of the charged nicotinamide ring of the NAD+ cofactor since these oscillations were essentially gone in the complex with the reduced cofactor.
In our previous theoretical study of the hydride transfer reaction catalysed by the PsFDH, the analysis of free downhill molecular dynamic trajectories allowed us to explore the dynamic coupling between the reacting fragments and the environment, as well as an estimation of the dynamic effect contribution to the catalytic effect from the calculation of the recrossing transmission coefficient in the enzyme and in solution at 300 K. The obtained recrossing transmission coefficients for the chemical reaction in the enzyme and in solution were 0.46 ± 0.04 and 0.20 ± 0.03, respectively. These values represent that the contribution of the dynamic effects to the catalysis, computed in the framework of the VTST, is 0.50 ± 0.15 kcal mol−1, which is small but not negligible, keeping in mind the low efficiency of FDH. The analysis of the reactive trajectories revealed how relative movements of some amino acids, mainly His332 and Arg284, precede the formation of the TS. Additionally, the time-dependent evolution of the electric field created by the enzyme on the key atoms of the reaction revealed a permanent field. The conclusion from this previous study was that the catalytic effect is due to substrate preorganization and TS stabilization with respect to the aqueous solution, thus reducing the activation barrier. Finally, application of Grote–Hynes theory allowed the identification of the modes responsible for the substrate–environment coupling, showing how some protein motions take place simultaneously with the reaction. Thus, the equilibrium approach between the distinguished reaction coordinate (the antisymmetric combination of the distances defining the H transfer) and the environment provides an overestimation of the catalysed rate constant in this system.
The present study aims to get a deeper insight into the dynamic and tunnelling effects on the reaction catalysed by FDH. We will estimate the contribution of the dynamic and tunnelling effects to the rate constant enhancement from recrossing and tunnelling transmission coefficient calculations at different temperatures. The selected temperatures covered the range of temperatures used experimentally by Kohen and co-workers44 and by Tishkov and co-workers.83 The optimal temperature for this enzyme is 337 K,83 but the kinetic experiments of Tishkov and co-workers were done at 303 K, thus justifying the selection of 300 K as one of our simulation temperatures to carry out proper comparisons.
It is important to point out that the temperature dependency of KIEs has been suggested to be a proper way to estimate the dynamic behavior of enzymes, even for the relatively narrow range of temperatures explored in the present study. In fact, a similar range of temperatures has been explored in previous studies of this and other enzymes, either by computational and experimental studies.44,86,89–91 In the present study, our predicted temperature dependence of the KIEs was analysed and compared with experimental and computational data obtained by Kohen and co-workers and Major and co-workers, respectively.44,86 The study of dynamic effects can also provide information to reveal the origin of the catalysis and for further mutational studies or transition-state analogue design.
2. Methodology
2.1. Potential of mean force
The initial coordinates of FDH were taken from the X-ray crystal structure of the Pseudomonas sp. complexed with azide inhibitor in the formate binding site with PDB code 2NAD.78 The azide molecule was replaced by the product of the FDH-catalysed reaction, CO2. The PROPKA3 semiempirical program92–95 was used to estimate the pKa values of the titratable protein residues to verify their protonation states at a pH of 7. According to the results, hydrogen atoms were added and assigned protonation states for all ionisable residues using the fDYNAMO library,96,97 assuming pH 7. The system was solvated by a cubic box of water molecules of side dimension 80 Å, centred on the C4 atom of the cofactor of subunit B. Because of the size of the system, all the residues further than 38 Å from C4 of the cofactor were removed, and those found further than 18 Å from the same atom were kept frozen.The one-dimensional potential of mean force (1D-PMF) of the reaction catalysed by PsFDH was obtained by means of QM/MM MD simulations using fDYNAMO96,97 at three different temperatures (278, 300 and 318 K), after previous equilibration running MD simulations at the three temperatures. The QM region consists of the formate anion together with the nicotinamide and ribose rings of the nicotinamide adenine dinucleotide (NAD+) cofactor (33 atoms, as depicted in Fig. 1), which is described using the AM1 Hamiltonian.98 To saturate the valence of the QM/MM frontier we used the link atom procedure.99,100 The MM subsystem, consisting of the protein and the solvent water molecules, was then described using the OPLS-AA101,102 and TIP3P103 force fields, respectively, as implemented in fDYNAMO. To treat the nonbonding interactions, a switch function with a cutoff distance in the range of 8–12 Å was used. The 1D-PMFs were obtained using the antisymmetric combination of the distances describing the breaking and forming bonds, i.e., d(C–H)–d(H–C4) (see Fig. 1) as the distinguished reaction coordinate.13 The Langevin–Verlet integrator with a time a step of 0.5 fs was employed, using the thermodynamic ensemble NVT at the temperatures of 278, 300 and 318 K. A total of 76 simulation windows changing the reference value of the reaction coordinate by 0.04 Å were run. Each window consisted of 10 ps of equilibration followed by 20 ps of production. The umbrella sampling approach104 was used to constrain the system to a particular value of the reaction coordinate by means of the addition of a harmonic potential with a force constant of 2000 kJ mol−1 Å−2. The probability distributions obtained from MD simulations within each individual window were combined by means of the weighted histogram analysis method (WHAM)105 to obtain the whole PMF.
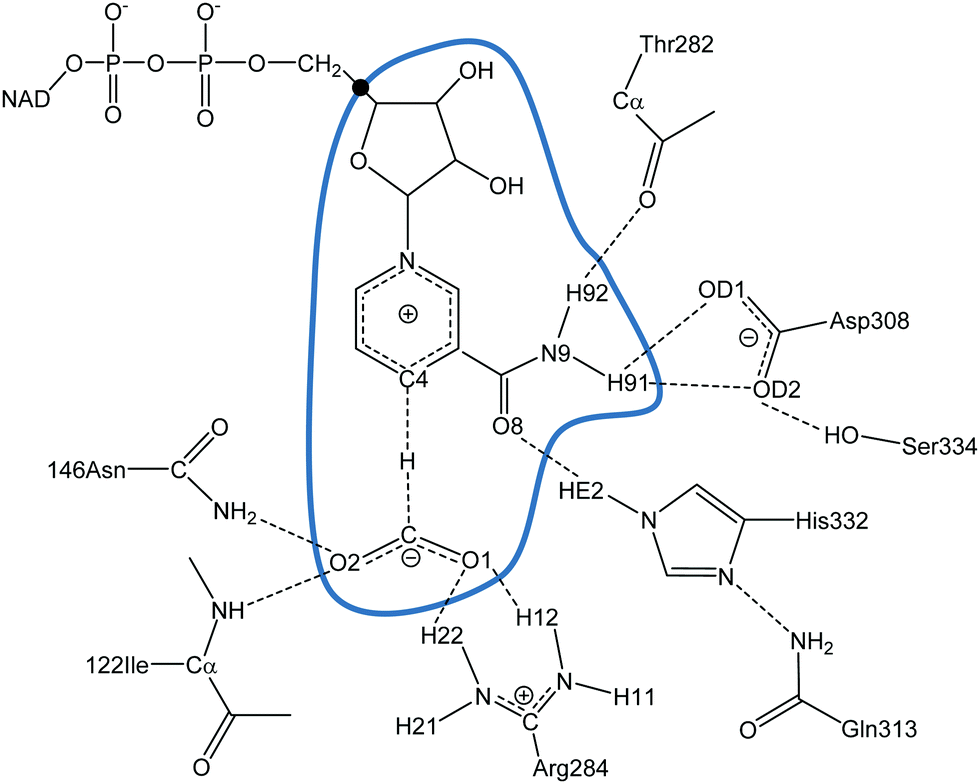 | ||
| Fig. 1 Details of the PsFDH active site. The atoms inside the blue curve are described quantum mechanically and the link atom is displayed as ●. | ||
From the maxima of each 1D-PMF at each temperature, a structure was selected to trace the AM1/MM refined 1D-PMFs at the three temperatures (278, 300 and 318 K) for only a small range of the reaction coordinate values close to the free-energy maxima found in the previous 1D-PMFs. The reaction coordinate was changed by only 0.01 Å and 21 simulation windows were run applying a force constant to the reaction coordinate of 2000 kJ mol−1 Å−2. As in the 1D-PMFs previously obtained, each window consisted of 10 ps of equilibration followed by 20 ps of production.
Two-dimensional potentials of the mean force (2D-PMF) of the reaction catalysed by the PsFDH enzyme at the AM1/MM level at the three different temperatures were performed as a function of the antisymmetric reaction coordinate (ξ1) and the donor–acceptor distance (ξ2) treated independently. A total of 1491 simulation windows were run by changing the reference value of ξ1 and ξ2 by 0.04 Å and 0.07 Å, respectively. The force constants applied for the reaction coordinate and donor–acceptor distance were 2000 kJ mol−1 Å−2 and 2500 kJ mol−1 Å−2, respectively. Each window consisted of 10 ps of relaxation and 20 ps of production with a time step of 0.5 fs.
2.2. Rare event trajectories
From the maxima of the refined 1D-PMFs, a structure was chosen with the corresponding value of the ξ1 coordinate of the TS for each temperature. Starting from these structures, three QM/MM MD simulations of 1 ns with a time step of 0.5 ps were run at three different temperatures (278, 300 and 318 K) restraining ξ1 with a force constant of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kJ mol−1 Å−2. From these simulations, a total of 118, 112 and 113 configurations at three different temperatures, 278, 300 and 318 K, respectively, with a reaction coordinate about ±0.005 Å around the TS positions were selected to compute free downhill trajectories. The velocity associated with the reaction coordinate is not properly thermalized in these configurations. Thus, following a procedure similar to that used by Gao and co-workers106 and used in our previous studies,9,12,41 we selectively removed the projection of the velocity on the reaction coordinate and we added a random value taken from a Maxwell–Boltzmann distribution just for the atoms involved in the description of the antisymmetric coordinate ξ1.
000 kJ mol−1 Å−2. From these simulations, a total of 118, 112 and 113 configurations at three different temperatures, 278, 300 and 318 K, respectively, with a reaction coordinate about ±0.005 Å around the TS positions were selected to compute free downhill trajectories. The velocity associated with the reaction coordinate is not properly thermalized in these configurations. Thus, following a procedure similar to that used by Gao and co-workers106 and used in our previous studies,9,12,41 we selectively removed the projection of the velocity on the reaction coordinate and we added a random value taken from a Maxwell–Boltzmann distribution just for the atoms involved in the description of the antisymmetric coordinate ξ1.
For each of the selected configurations with their modified velocities, free NVE simulations were performed, integrating the equations of motion forward and backward, just changing the sign of the velocity components. These simulations were run at the three temperatures (278, 300 and 318 K) and using protium and tritium as the transferring species. The downhill trajectories were propagated from −8 to +8 ps by using a time step of 0.5 fs. The trajectories obtained were classified as reactive trajectories when the reactant state connected to the product state (RP) and nonreactive trajectories when either reactant state connected to reactant state (RR) or product state connected to product state (PP). All trajectories may exhibit recrossings in the TS dividing surface. To compute the recrossing transmission coefficient (γ(T,ξ)), we used the ratio between the reactive (RP) and total (RR, RP and PP) trajectories.
The changes in the environment as the reaction proceeds, such as distances, charges, electrostatic potential and electric field created by the environment, were analysed by means of detailed inspection of the reactive trajectories at the three temperatures (278, 300 and 318 K) and using protium and tritium as the transferred atoms.
2.3. Quantum mechanical tunnelling effects and rate constants
As mentioned in the Introduction section, TST deviations from the quantum tunnelling effects can be estimated by means of EA-VTST.57–59 In this section, the combination of fDYNAMO96,97 and POLYRATE107 has been used to obtain the tunnelling transmission coefficient, κ, for protium and tritium transfer at the three different temperatures (278, 300 and 318 K). The SCT approximation was employed to compute κi, since the reaction path curvature was found to be small over the region important for tunnelling. Pu et al.89 showed that only a 2% smaller difference in the small-curvature with respect to the large-curvature tunnelling paths was found for hydride transfer (both for protium and tritium) if calculations were limited to small curvature tunnelling paths and the transmission coefficient was computed microcanonically. This strategy has been successfully employed in our laboratory in previous theoretical studies of hydride transfer reactions catalysed by thymidylate synthase, lactate dehydrogenase, morphinone reductase or dihydrofolate reductase.41–43,70–72,108–110 The final tunnelling contribution was then obtained as the average over the reaction paths of 10 TS structures. The Hessian was numerically determined by a central difference scheme with a step size of 0.01 Bohr, and the generalized normal modes were calculated in rectilinear coordinates. During the calculation of the minimum energy path, all the systems inside of a sphere of 18 Å around the QM region were minimized in each step. When calculating generalized normal mode frequencies, the reaction coordinates were projected out of the Hessian.111 The calculated frequencies for the various configurations were averaged over all the configurations that belonged to a given bin of width 0.01 Å, and the quantum vibrational correction of the PMF as the difference of the sum of the frequencies between TS and reactants, ΔWvib(T,ξ), was calculated for the localised and characterised transition structures and reactants using protium and tritium as transferred particles at the three temperatures.2.4. Kinetic isotope effects
The KIEs were obtained from eqn (4) as far as the global transmission coefficient and the quasi-classical free energy of activation were computed for two different isotopes labelled compounds. In our case, protium and tritium are used as the transferring species in order to obtain the primary intrinsic KIEs. The temperature dependence was studied at 278, 300 and 318 K. These values were compared with the experimental or computational results obtained by Kohen and co-workers and Major and co-workers, respectively, for the same hydride transfer step.44,863. Results and discussion
3.1. Potential of mean force
The 1D-PMFs for the hydride transfer reaction catalysed by PsFDH at the three different temperatures computed at the AM1/MM level are shown in Fig. 2. The free energy barriers obtained as the difference between the TS and the reactant state, are 12.2, 11.8 and 11.9 kcal mol−1 at 278 K, 300 K and 318 K, respectively. This result shows that the classical mechanical activation free energy barriers (ΔG‡clas(T,ξ)) are temperature-independent.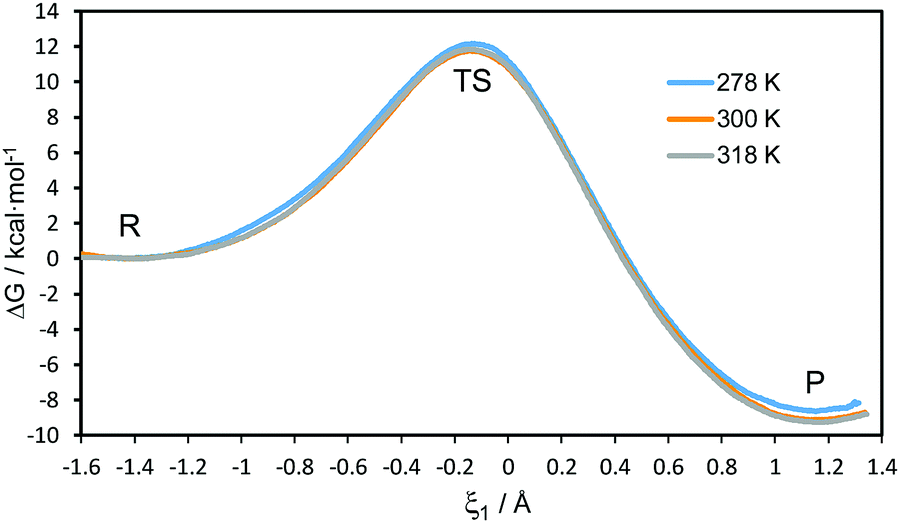 | ||
| Fig. 2 AM1/MM 1D-PMFs for the hydride transfer reaction catalysed by PsFDH at 278, 300 and 310 K. The reaction coordinate (ξ1) is defined as the antisymmetric combination of the distances describing the breaking and forming of bonds, d(C–H) and d(C4–H), respectively (see Fig. 1 for atom numbering). | ||
From the maxima of the 1D-PMFs, structures were selected to perform the refined AM1/MM 1D-PMFs in the vicinity of the TS at the three temperatures. From these refined 1D-PMFs, a good estimation of the TS position in terms of the antisymmetric reaction coordinate (ξ1) was obtained at each temperature. The values of ξ1 at the TS positions are located at −0.145, −0.137 and −0.146 Å at 278, 300 and 318 K, respectively. No temperature dependence appeared in the position of the TS along the 1D-PMFs.
In order to have a better description of the donor–acceptor distance along the reaction, 2D-PMFs were performed using the antisymmetric reaction coordinate (ξ1) and the distance between the donor and acceptor carbon atoms (ξ2). As is shown in Fig. 3, the three 2D-PMFs are almost equal, displaying similar activation free energy barriers of 13.2, 13.2 and 13.1 kcal mol−1 at 278, 300 and 318 K, respectively. This result supports the temperature-independence derived from the 1D-PMFs. Nevertheless, a difference in the three 2D-PMFs can be detected if focusing on the difference between reactants and products. Thus, the reaction is slightly more exergonic at 300 K and 318 K than at 278 K. Comparison of the free energy barriers from the 2D-PMF, that can be considered as more precise than those derived from the 1D-PMFs, to the value estimated from the experimental rate constant reported in the literature81,82 (16.6 kcal mol−1) is in good agreement, keeping in mind that our theoretical results correspond exclusively to a single step rather than a complex multistep process that comprises the experimental rate constant.
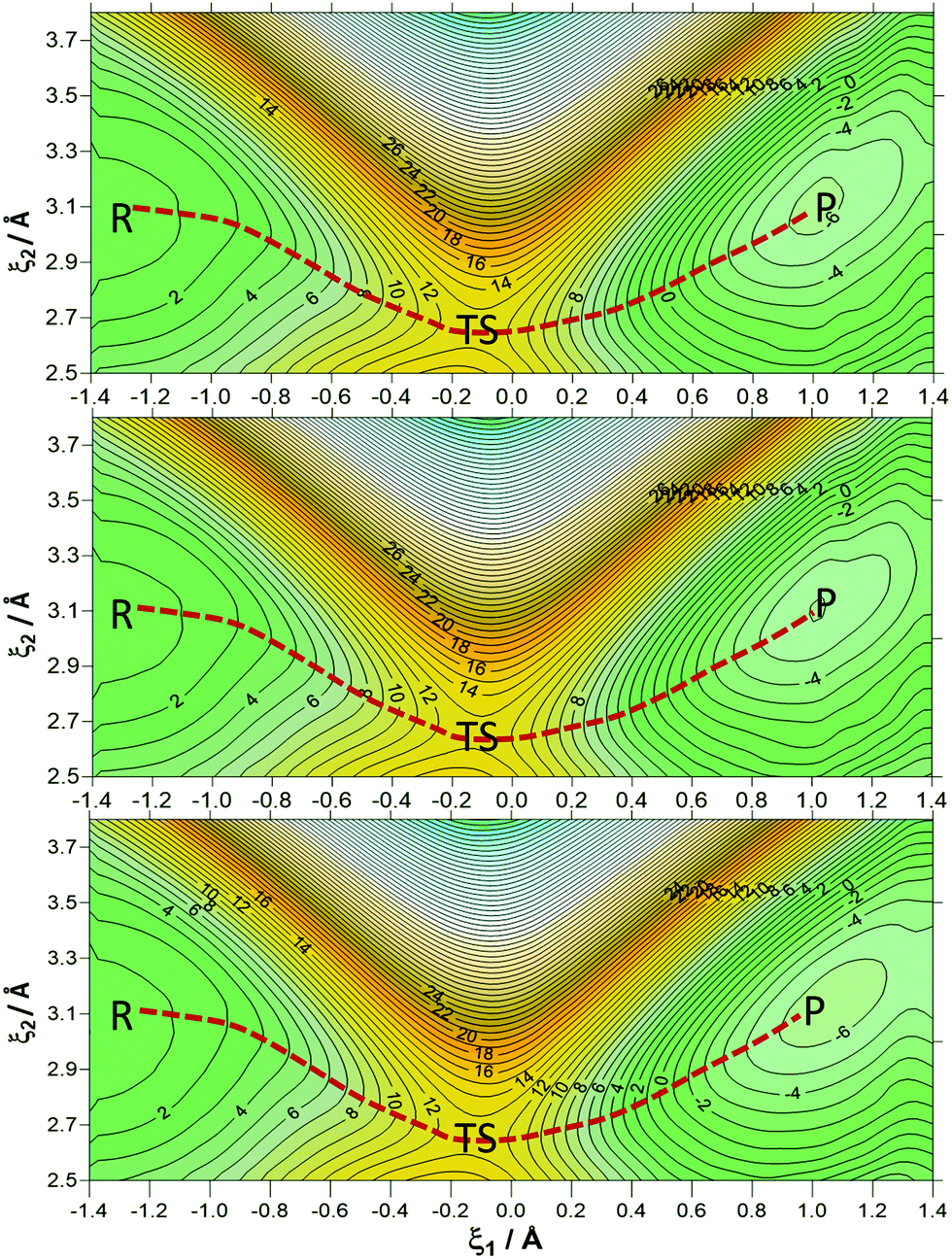 | ||
| Fig. 3 2D-PMFs of the hydride transfer reaction catalysed by PsFDH at 278 (top panel), 300 K (middle panel) and 310 K (bottom panel). On the abscissa, the antisymmetric reaction coordinate (ξ1) is displayed, while the distance between the donor and acceptor carbon atoms (ξ2) is on the ordinate axis. Distances are shown in Å and the isoenergetic lines are displayed at 1 kcal mol−1. | ||
Table 1 contains average values of the donor–acceptor distance (d(C–C4)), the distances describing the breaking and forming bonds (d(C–H) and d(C4–H)) and the antisymmetric reaction coordinate (ξ1) with their standard deviations for reactants (R), TS and products (P) obtained at the 1D-PMFS and 2D-PMFs at 278 K (a), 300 K (b) and 318 K (c). If we compare these key distances in the R and TS structures from the 1D-PMFs and 2D-PMFS, all the distances are similar and the small deviations are within the standard deviations. Meanwhile, the breaking bond distance (d(C–H)) and the donor–acceptor distance (d(C–C4)) for products are quite different for the averaged values obtained from the 1D-PMFs and 2D-PMFs. Keeping in mind that this reaction consists of charged species that are transformed into neutral ones, the neutral carbon dioxide molecule in P has more freedom to move in the active site than the formate anion in R. Nevertheless, if we compare the distances of the stationary structures at different temperatures, there is no significant difference in the results. Thus, from the geometrical point of view, the reaction catalysed by FDH must also be considered as temperature-independent.
| d(C–C4) | d(C4–H) | d(C–H) | ξ 1 | ||
|---|---|---|---|---|---|
| a Distances are shown in Å. | |||||
| (a) T = 278 K | |||||
| 1D-PMF | R | 3.04 ± 0.15 | 2.53 ± 0.04 | 1.13 ± 0.03 | −1.40 ± 0.03 |
| TS | 2.67 ± 0.07 | 1.42 ± 0.04 | 1.30 ± 0.03 | −0.12 ± 0.04 | |
| P | 3.20 ± 0.09 | 1.13 ± 0.03 | 2.29 ± 0.04 | 1.16 ± 0.03 | |
| 2D-PMF | R | 3.07 ± 0.17 | 2.50 ± 0.04 | 1.12 ± 0.04 | −1.38 ± 0.03 |
| TS | 2.66 ± 0.05 | 1.43 ± 0.03 | 1.31 ± 0.04 | −0.12 ± 0.05 | |
| P | 3.10 ± 0.08 | 1.10 ± 0.05 | 2.12 ± 0.03 | 1.02 ± 0.02 | |
| (b) T = 300 K | |||||
| 1D-PMF | R | 3.01 ± 0.14 | 2.53 ± 0.04 | 1.12 ± 0.03 | −1.41 ± 0.03 |
| TS | 2.67 ± 0.07 | 1.42 ± 0.04 | 1.31 ± 0.04 | −0.12 ± 0.04 | |
| P | 3.21 ± 0.08 | 1.13 ± 0.03 | 2.33 ± 0.04 | 1.20 ± 0.03 | |
| 2D-PMF | R | 3.07 ± 0.15 | 2.52 ± 0.05 | 1.14 ± 0.03 | −1.38 ± 0.04 |
| TS | 2.65 ± 0.06 | 1.41 ± 0.02 | 1.29 ± 0.03 | −0.12 ± 0.03 | |
| P | 3.11 ± 0.08 | 1.12 ± 0.03 | 2.14 ± 0.04 | 1.02 ± 0.03 | |
| (c) T = 318 K | |||||
| 1D-PMF | R | 3.11 ± 0.19 | 2.53 ± 0.04 | 1.13 ± 0.03 | −1.40 ± 0.03 |
| TS | 2.66 ± 0.07 | 1.41 ± 0.04 | 1.30 ± 0.04 | −0.11 ± 0.04 | |
| P | 3.20 ± 0.09 | 1.13 ± 0.03 | 2.29 ± 0.04 | 1.15 ± 0.03 | |
| 2D-PMF | R | 3.08 ± 0.20 | 2.53 ± 0.05 | 1.15 ± 0.02 | −1.38 ± 0.04 |
| TS | 2.66 ± 0.04 | 1.41 ± 0.06 | 1.28 ± 0.04 | −0.13 ± 0.05 | |
| P | 3.11 ± 0.10 | 1.13 ± 0.04 | 2.15 ± 0.04 | 1.02 ± 0.02 | |
3.2. Recrossing effects
After randomizing the reaction coordinate velocity for each of the saved configurations, as described in the computing methods section, free hybrid QM/MM MD trajectories were run, allowing the system to evolve downhill from the TS. The rare event trajectories were run at the three different temperatures and using protium and tritium as transferring atoms. The number and the proportions of reactive trajectories (RP type) and non-reactive trajectories (RR type and PP type) at the different temperatures for protium and tritium are given in Table S1 of the ESI.† Analysis of the trajectories indicates that similar proportions of reactive and non-reactive trajectories were obtained at the different temperatures. When comparing the protium and tritium trajectories, the proportion of reactive trajectories was vaguely larger for tritium than for protium. As explained in a previous section, these values were used to compute the recrossing coefficients.The transmission coefficients at the three temperatures for each isotope-labelled compound are shown in Table 2. As deduced from the results, it seems that the recrossing transmission coefficient, γ(T,ξ), is slightly larger for tritium compared to protium at any of the three temperatures. This result indicates that the slower transfer of the heavier particle, tritium, allows the protein environment to better equilibrate and, consequently, a lower amount of recrossing trajectories is obtained for this system compared to the case of the transfer of the lighter particle, protium. Nevertheless, it must be pointed out that the differences between protium and tritium can be considered within the uncertainty of the standard deviation and are temperature-independent.
| κ(T) | γ(T,ξ) | Γ(T,ξ) | ΔW‡vib(T,ξ) | ΔGQCact(T,ξ) | ΔGeff(T) | KIE | |
|---|---|---|---|---|---|---|---|
| T = 278 K | |||||||
| H | 6.3 ± 0.6 | 0.45 ± 0.05 | 2.8 ± 0.6 | −2.83 ± 0.11 | 10.38 ± 0.11 | 9.81 ± 0.20 | 4.5 ± 0.4 |
| T | 5.8 ± 0.6 | 0.48 ± 0.05 | 2.8 ± 0.6 | −2.01 ± 0.09 | 11.20 ± 0.09 | 10.64 ± 0.18 | |
| T = 300 K | |||||||
| H | 5.0 ± 0.9 | 0.42 ± 0.05 | 2.1 ± 0.6 | −2.76 ± 0.11 | 10.47 ± 0.11 | 10.03 ± 0.26 | 3.9 ± 0.6 |
| T | 4.3 ± 0.8 | 0.48 ± 0.05 | 2.1 ± 0.6 | −1.96 ± 0.11 | 11.27 ± 0.11 | 10.84 ± 0.25 | |
| T = 318 K | |||||||
| H | 4.3 ± 0.8 | 0.42 ± 0.05 | 1.8 ± 0.5 | −2.72 ± 0.11 | 10.41 ± 0.11 | 10.04 ± 0.27 | 3.4 ± 0.6 |
| T | 3.9 ± 0.7 | 0.49 ± 0.05 | 1.9 ± 0.6 | −1.92 ± 0.11 | 11.21 ± 0.11 | 10.80 ± 0.27 | |
3.3. Geometrical analysis of rare-event trajectories
The changes in the chemical system and in the environment as the reaction proceeds were analysed by means of the detailed inspection of the reactive trajectories at the three temperatures (278, 300 and 318 K). Fig. 4 shows the evolution of the key interatomic distances involved in the hydride transfer at the three temperatures averaged over the reactive trajectories. In this figure, t = 0 indicates the passage of the system over the TS; negative times correspond to the evolution of the system towards the reactant valley and positive times towards the product. The corresponding plots for the transfer of tritium, which are qualitatively equivalent, are shown in Fig. S1 (see ESI†). How the reaction proceeds can be observed by the approaching of the donor and acceptor carbon atoms, showing a minimum distance in the TS region while the breaking bond (C–H) distance is increasing and the forming bond (C4–H) distance is decreasing. The same behaviour can be predicted from the analysis based on the 2D-PMFs reported in Fig. 3, computed under the equilibrium approximation, and in our previous study performed at 300 K.67 The evolution of the angle described by the donor atom, the transferring particle and the acceptor atom is also depicted in Fig. 4; it becomes more linear in the TS region, in agreement with previous studies.9,66 Interestingly, the behaviour of the analysed geometrical parameters is perfectly reproduced at all different temperatures for the transfer of the two different species, protium and tritium.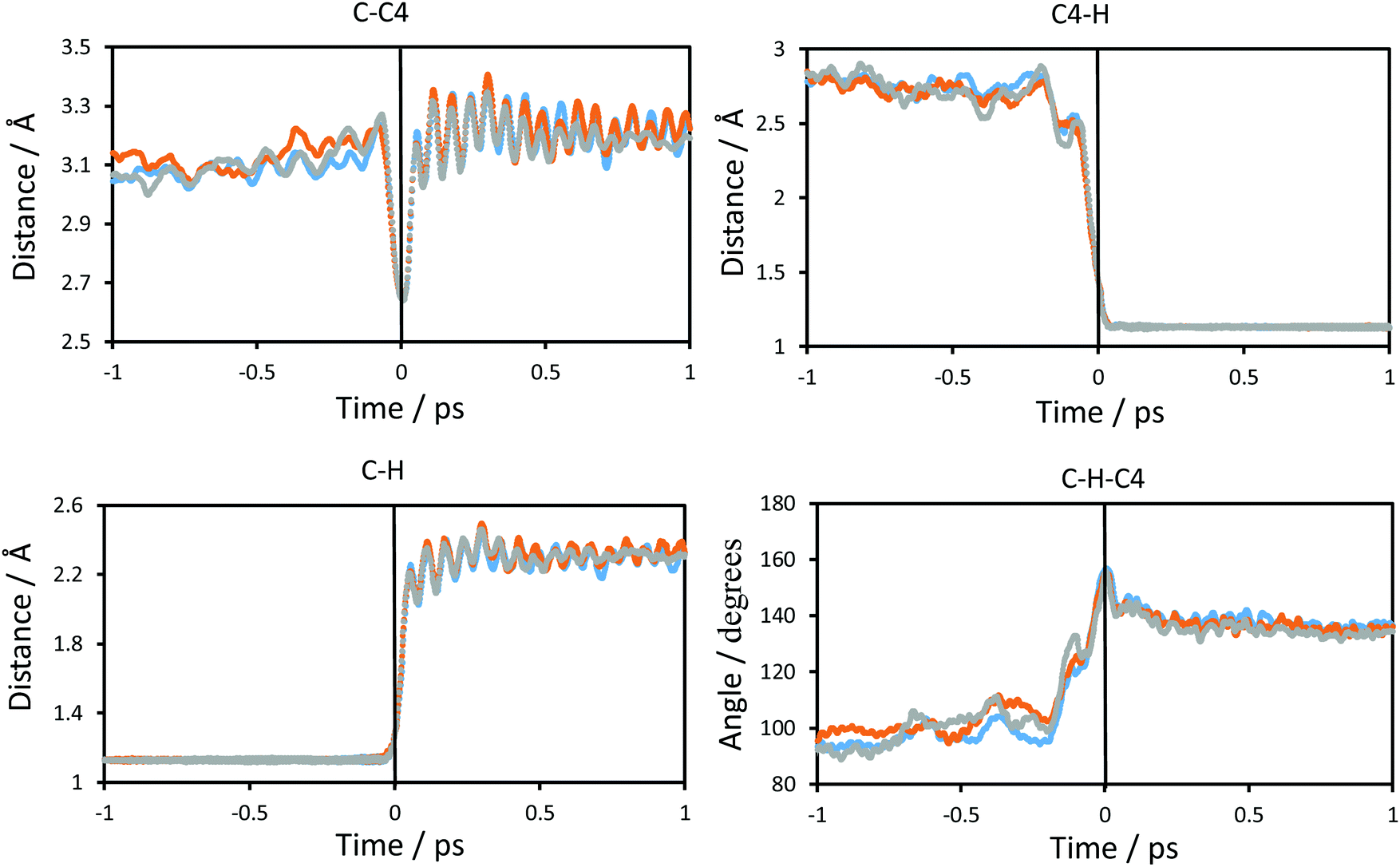 | ||
| Fig. 4 Time evolution of the interatomic distances of the atoms that participate in the definition of the antisymmetric reaction coordinate and of the angle defining the chemical reaction averaged over the reactive trajectories at different temperatures, at 278 K (blue), at 300 K (orange) and at 318 K (grey), for protium as the particle to be transferred. The atoms that define the distances or angle depicted are shown at the top of each graphic. Distances are in Å and angles in degrees. The vertical solid black line at t = 0 shows the position of the TS. | ||
Fig. 5 shows the evolution of the intermolecular distances between the key atoms of the substrate and cofactor and the amino acid residues of the enzyme, averaged over the reactive trajectories at different temperatures and using protium and tritium as the transferred atoms. The first observation that can be deduced is that all the geometrical variations are equivalent at all three temperatures. As observed in Fig. 5(a), the distance between the O8 atom of NAD+ and HE2 atom of His332 decreases before arriving at the TS region. The motion of His332 seems to be activated at around 0.5 ps before crossing the TS and its approach to the cofactor could be polarizing the carbonyl group of the cofactor thus increasing the negative charge on the O8 atom and the positive charge on the C4 atom, as confirmed by the analysis of atomic charges on Fig. 6. If we compare the relative motion of this residue at the three different temperatures, it appears that the distance between the O8 atom of NAD+ and the HE2 atom of His332 fluctuates much more in the reactant region before reaching the TS at higher temperature (318 K) than at lower temperature (278 K) for both protium and tritium. The higher thermal energy at 318 K provokes longer distances between these two atoms compared to the lowest temperature, 278 K. Despite these fluctuations, the behaviour of the active site is quite robust with the reaction progress and is temperature independent.
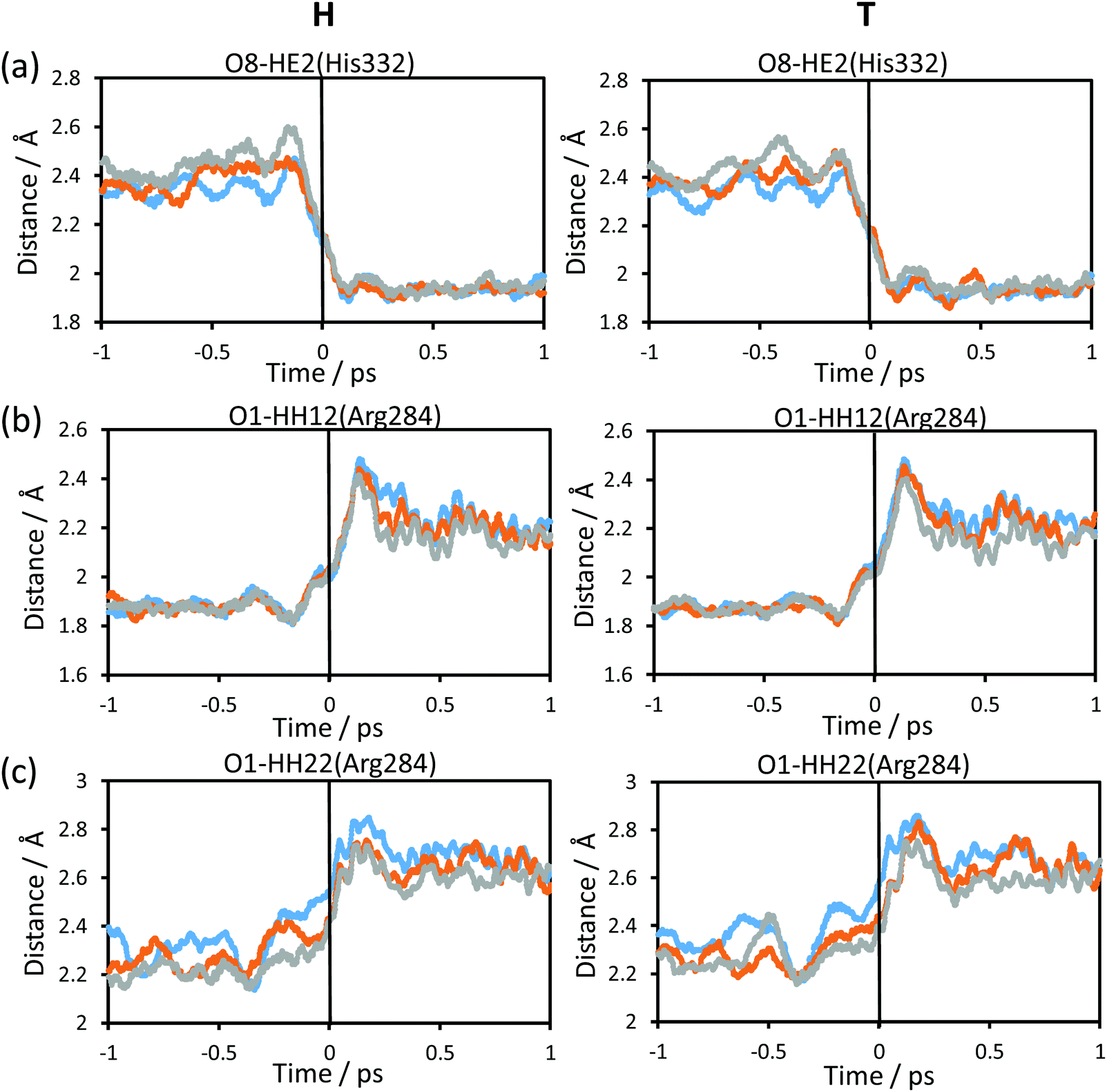 | ||
| Fig. 5 Time evolution of intermolecular distances between key amino acid residues of the enzyme and the formate ion and NAD+ averaged over the reactive trajectories at 278 K (blue), 300 K (orange) and 318 K (grey), for protium (left panels) and tritium (right panels). Distances are in Å. The vertical solid black line at t = 0 shows the position of the TS. | ||
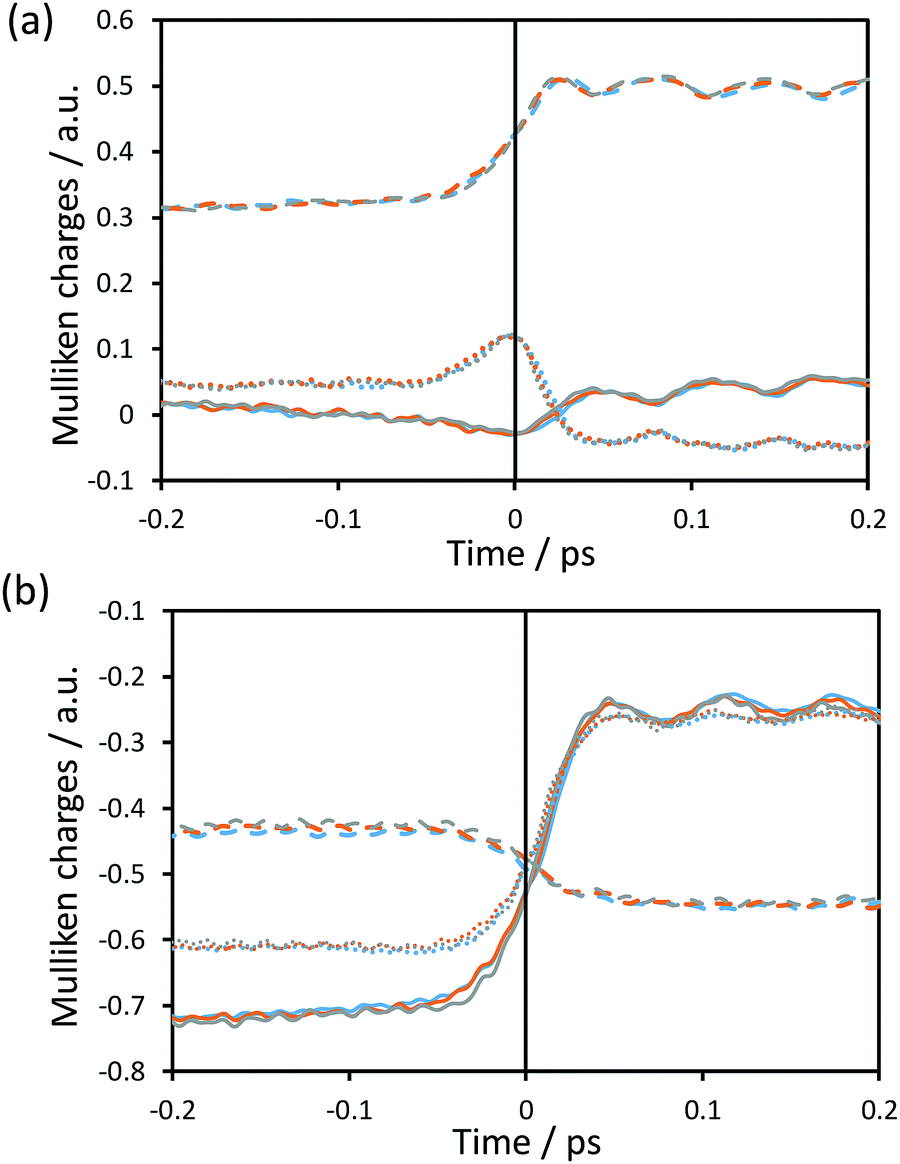 | ||
| Fig. 6 Time evolution of the Mulliken charges on some key atoms averaged over the reactive trajectories for protium as the transferring particle computed at 278 K (blue), 300 K (orange) and 318 K (grey) for (a) C (dashed line), H (solid line) and C4 (dotted line); and (b) O8 (dashed line), O1 (solid line) and O2 (dotted line). The vertical solid black line at t = 0 shows the position of the TS. | ||
The rest of the analysed distances between the amino acid residues and the cofactor or substrate atoms elongate after reaching the TS. Thus, the distances between one of the oxygen atoms of the formate and Arg284 (Fig. 5(b) and (c)), start to increase just before crossing the TS, thus revealing a certain degree of participation in the real reaction coordinate. In Fig. 5(b), the shortest hydrogen bond interaction between the Arg284 amino acid residue and O1 of the formate ion is shown. The evolution of this distance from R to the TS is similar at the three different temperatures, while from TS to P there are more fluctuations leading to a shorter averaged distance at high temperature for both protium and tritium. Fig. 5(c) shows much more fluctuations at lower temperature when going from R to TS, reaching a greater distance at the TS region at lower temperature for both protium and tritium. In this figure, the evolution of the distance from TS to P also displays a shorter averaged distance at high temperature as shown in Fig. 5(b). It is important to point out that Fig. 5(b) and (c) display hydrogen bond interactions between Arg284 and the substrate (formate ion in reactants and carbon dioxide in products), which is small and mobile inside the active site. Thus, there are more fluctuations in these longest distances and more differences between the different temperatures.
The participation of the environment in the real reaction coordinate can be analysed by the inspection of the transition vector of the optimized TS structures. Thus, from a refined TS structure, a large Hessian matrix that contains the QM subsystem and all the residues that establish hydrogen bond interactions with the QM region such as Ile122, Asn146, Thr282, Arg284, Asp308, Gln313, His332 and Ser334 (160 atoms in total, see Fig. 1) were computed and diagonalised. The coefficients of the negative eigenvalue of the Hessian defining the transition vector (TV) of the reaction on each atom of the Hessian matrix were obtained (see Table S2 of the ESI†). As can be observed, the three key atoms involved in the chemical reaction (donor, acceptor and the transferring hydrogen atom) show the most significant components on the TV, but some atoms of the enzyme (from Ile122, Asn146, Arg284 and His332) show non-negligible components on the TV, revealing a certain participation in the real reaction coordinate (more dramatically Arg284 and His332).
The analysis of the intermolecular distances of the amino acid residues of the enzyme along the reactive trajectories has been complemented by the root mean square fluctuation (RMSF) calculations along 1 ns QM/MM MD simulations restrained at the reactant and TS regions at the three temperatures. In these simulations, a time step of 0.5 fs was used and a force constant of 10000 kJ mol−1 Å−2 was applied to the reaction coordinate. Fig. S2 (see ESI†) shows the RMSF of the amino acid residues along the QM/MM MD simulations restrained at the reactant, the TS and the difference between TS and reactant. Higher RMSF values were obtained at the highest temperature (318 K) for both reactant and TS. When computing the RMSF difference between TS and reactants, displayed in Fig. S2(c) (see ESI†), a negative value for a certain amino acid residue means that this residue has a greater mobility in reactants than in TS. The main difference is observed at residue Thr35, which is outside on the surface of the enzyme and is in contact with the solvent. As in previous geometrical analysis, there is no clear trend of the RMSF difference with the temperature. In fact, larger differences are obtained at the lower temperature in certain cases. The amino acid residues involved in the stabilization of the substrate and cofactor do not show large fluctuations in the differences between reactants and TS, suggesting an important preorganization to stabilize the TS. Nevertheless, there are noticeable RMSF differences between TS and reactants in some residues that are close to the active site, such as Asp125 and Ser147, which establish hydrogen bond interactions with the Arg284 residue and with one oxygen atom of the phosphate groups of the cofactor, respectively. Displaying those hydrogen bond interactions from reactants to TS, there is no significant change, so these fluctuations take place in order to maintain the hydrogen bond interactions of the residues of the active site to stabilize the TS.
3.4. Electrostatic analysis of rare-event trajectories
Fig. 6 shows the time evolution of the Mulliken charges on some key atoms of the cofactor and substrate averaged over the reactive trajectories at the three different temperatures for hydride as the transferring particle. Fig. S3 (see ESI†) displays the time evolution of the Mulliken charges of the same atoms averaged over the reactive trajectories at the three different temperatures for tritium. The trend of the charges is equivalent for protium and tritium transfers, at the three temperatures. In Fig. 6, it is observed that the behaviour of the charges is consistent with the formal description of the reaction involving a charge annihilation in going from the reactant state to the product state and the charge distribution changes take place close to the TS region (between ±0.05 ps around the TS). In Fig. 6(a), it is displayed how the charge on the donor carbon increases while the charge on the acceptor carbon decreases when going from reactants to products, showing a maximum positive value at the position of the TS. The charge of the transferring hydride slightly decreases from reactants to the TS, displaying a minimum negative value at the TS region, and after that gently increases on going to products. Moreover, as observed in Fig. 6(b), the negative charge on the oxygen atoms of the formate decreases, being less negative at the product state, while the charge on the O8 atom of the cofactor becomes more negative as the reaction proceeds.We can analyse the effects of environmental motions during the reaction process by means of the time evolution of an environmental coordinate(s) defined as the difference in the electrostatic potential created by the environment on the acceptor and donor atoms (VC4 and VC, respectively; see Fig. 1) as was done in a previous study.17
The evolution of this coordinate takes into account the changes in the enzymatic environment as the reaction proceeds and can provide information on the electrostatic role of the protein. Time evolution of this environmental coordinate along the averaged reactive trajectories is shown in Fig. 7(a). The results in Fig. 7(a) confirm how the protein evolves, though not dramatically, from reactants to products and the difference in the electrostatic potential would favour the transfer of the formal negative charge from the formate to the cofactor.
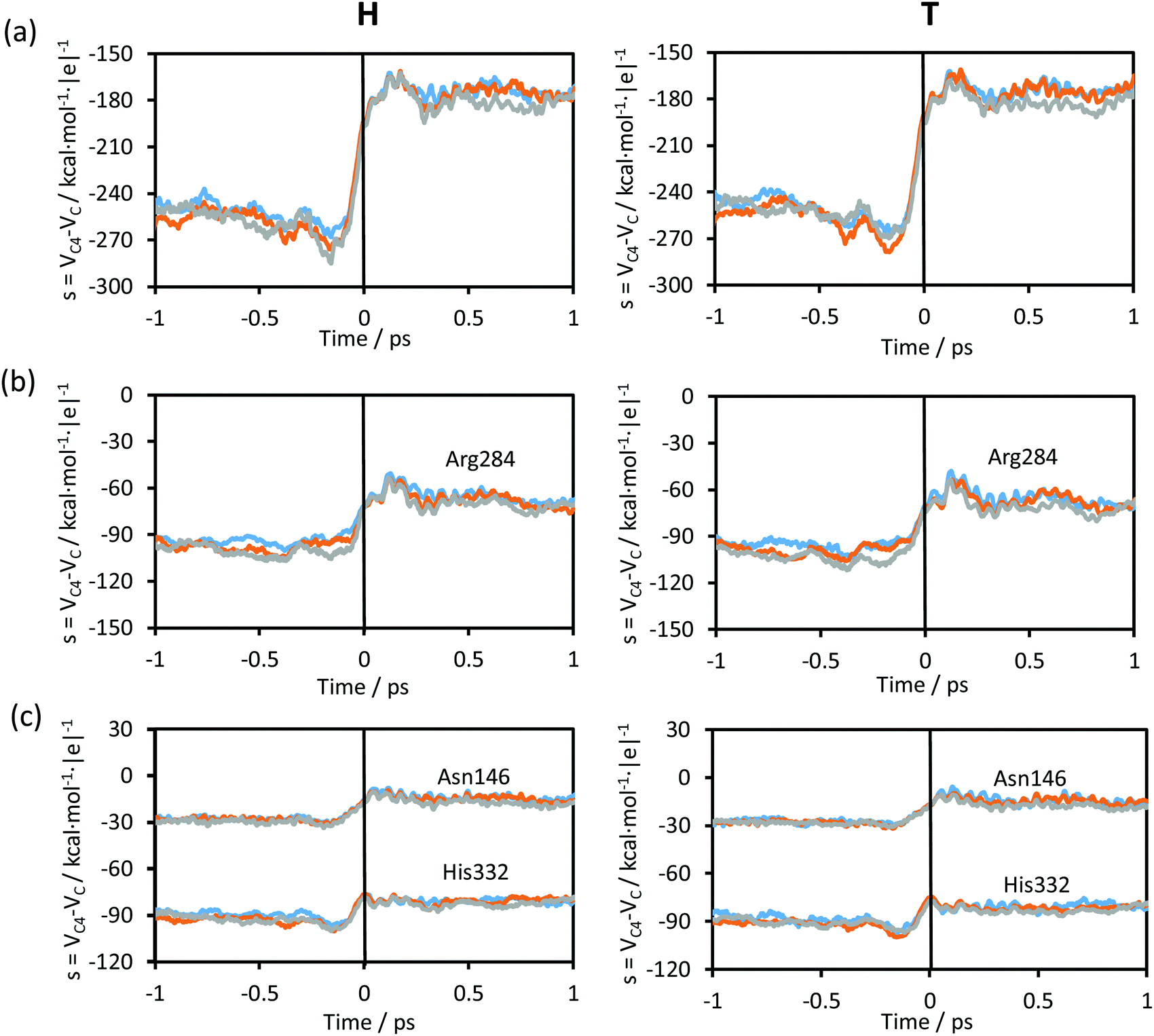 | ||
| Fig. 7 (a) Time evolution of the environmental coordinate defined as the difference in the electrostatic potential created by the environment on the acceptor and donor atoms averaged along the reactive trajectories at 278 K (blue), 300 K (orange) and 318 K (grey), for protium (left panels) and for tritium (right panels), respectively. (b and c) Time evolution of the contribution of some residues to the environmental coordinate averaged along the reactive trajectories. The vertical solid black line at t = 0 shows the position of the TS. | ||
Time evolution of the contributions to the environmental coordinate from some key protein residues, those that establish hydrogen bond interactions with the chemical system, averaged along the reactive trajectories are displayed in Fig. 7(b) and (c). The evolution of the contributions to the environmental coordinates of particular residues (Asn146, Arg284 and His332) follows the same trend as observed in Fig. 7(a) (see Fig. 1 for residue numbering). No differences are observed between the plots obtained at different temperatures and, obviously, the electrostatic variations of the protein do not depend on the mass of the transferring particle. On comparing the plots at the three different temperatures and for protium and tritium as the transferred particles, the evolution was very similar, with the same values of the environmental coordinate for the TS region. While the contribution to the environmental coordinates of Asn146 and His332 slightly changes after crossing the TS (Fig. 7(c)), in the case of the Arg284 (Fig. 7(b)) a fluctuation is shown after crossing the TS as was observed in the plot of the total environmental coordinate, Fig. 7(a).
The change in the environmental coordinate from reactants to products is almost negligible in other amino acid residues that establish hydrogen bond interactions with the chemical system, such as Ile122, Thr282 and Asp308, or residues that establish hydrogen bond interactions with Asp308 and His332 such as Ser334 and Gln313, respectively (see Fig. 1). From these results, one can conclude that Asn146, Arg284 and His332 electrostatically favour the reaction progress, providing a suitable environment to reach the TS and then leading the system toward the formation of products. As obtained in the transition vector analysis, Asn146, Arg284 and His332 are in fact part of the real coordinate definition.
In order to complete the analysis of the contribution of the surroundings to the reaction, the time evolution of the electric field created by the environment, projected along the donor–acceptor (C–C4) vector on the acceptor atom (Fig. 8(a)), transferring atom (Fig. 8(b)) and donor atom (Fig. 8(c)) at the three different temperatures using protium and tritium as transferring particles was computed and depicted in Fig. 8. Small variations in the electric field were observed from reactants to products on the acceptor and the transferring particle (Fig. 8(a) and (b)). Moreover, taking into account the small variations in the charges on these two atoms (see Fig. 6(a)), the electrostatic forces that the protein is generating on these two atoms are not dramatic; however, this is not the case for the donor atom. As observed in Fig. 8(c), there is a dramatic decrease in the electric field from ca. 0.01 a.u. in the reactants to ca. 0 a.u. in the products. This, together with the fact that the charge on the donor atom is always positive, the force created by the environment goes in the direction of the donor–acceptor vector at the reactants and TS, thus facilitating the approach between substrate and cofactor for the hydride transfer to take place. Once the transfer takes place, this force turns into values close to zero, which would facilitate carbon dioxide release.
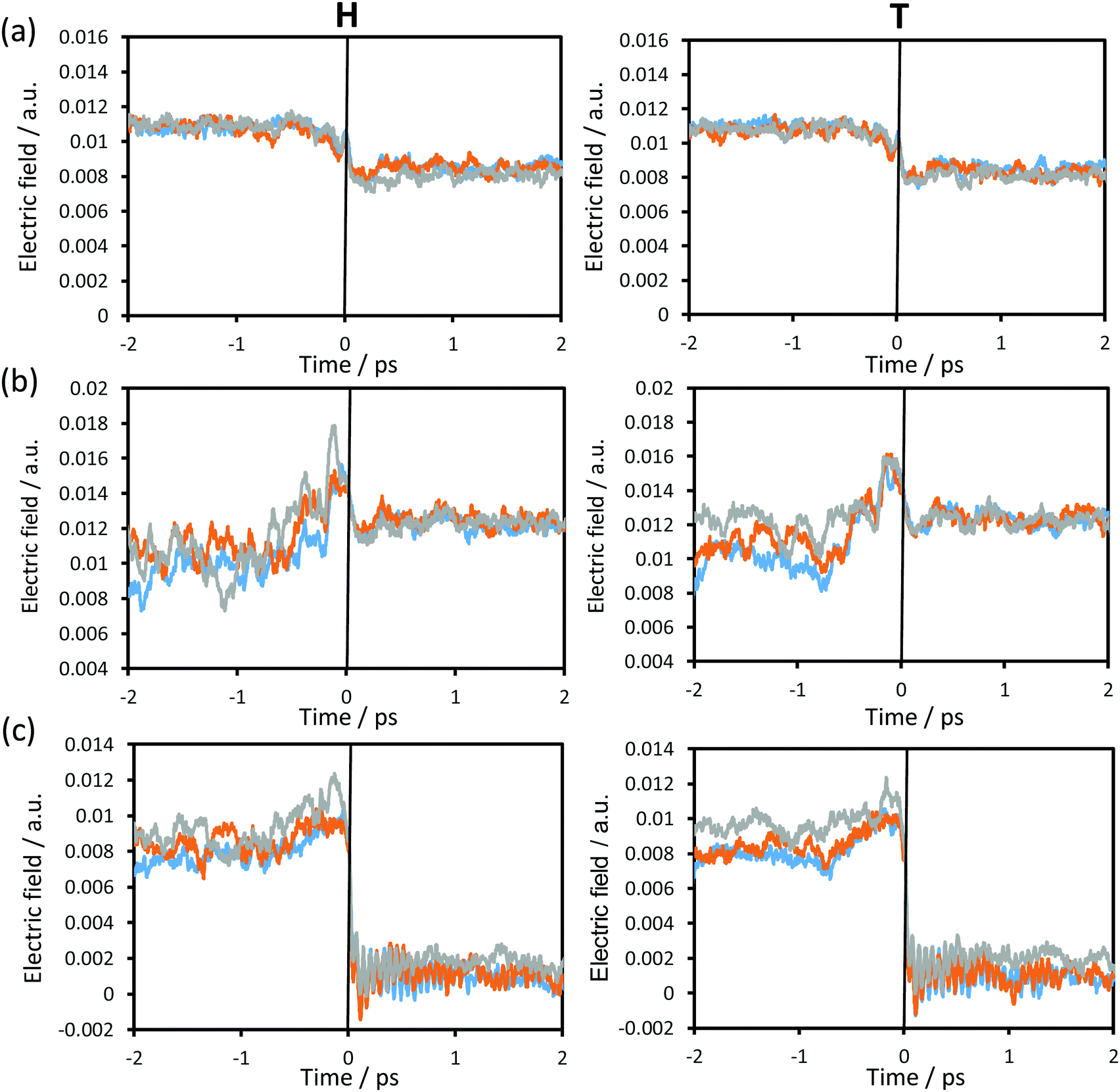 | ||
| Fig. 8 Time evolution of the averaged projection of the electric field created by the environment along the donor–acceptor vector on (a) the acceptor atom, (b) the hydride transferring atom and (c) the donor atom averaged over the reactive trajectories at 278 K (blue), 300 K (orange) and 318 K (grey), for protium (left panels) and for tritium (right panels). | ||
If we compare the results obtained at the three temperatures, the differences are not significant and, obviously, no important differences are observed if the particle to be transferred is protium or tritium.
3.5. Quantum effects
The contribution of quantum mechanical tunnelling, κ(T), to the rate constant is shown in Table 2. The comparison of the tunnelling transmission coefficients between protium and tritium shows how, as expected, the coefficient is larger for the lighter particle at all tested temperatures. This result means that the rate constant computed using eqn (1)–(3), for the lighter particle (protium) will be larger than for the heavier particle (tritium) at the three temperatures. On the other hand, the analysis of the temperature dependency of the tunnelling coefficients shows how it decreases with the temperature, as observed in our previous studies of the temperature-dependence of hydride transfer reactions catalysed by TSase or DHFR.43,70,71,110,112The quantum vibrational corrections (ΔW‡vib(T,ξ)) to the free energy barrier and the total value of the quasi-classical free energy barrier (ΔGQCact(T,ξ)) obtained from the 2D-PMFs with their corresponding standard deviations are shown in Table 2. The vibrational corrections to the activation free energy barrier are smaller for the heavy particle (tritium), so the quasi-classical free energy barriers are lower for the light particle. In Table 2 the effective free energy activation (ΔGeff) is shown as well, in which the effects of tunnelling and recrossing are included. Like the quasi-classical free energy barriers, the effective free energy activation is lower for the lighter particle.
3.6. Kinetic isotope effects
The KIEs, obtained using eqn (4) are shown in Table 2, while a graphical representation of these values, and all their contributions to the KIEs, are displayed in Fig. 9. Theoretical standard deviations are reported as bars on each point. The first conclusion derived from the results is that the KIEs are slightly temperature dependent, decreasing with the temperature. The recrossing and tunnelling contributions are temperature independent, within the uncertainty associated with the standard deviations. Thus, the larger contribution of the possible temperature effect to the total KIE comes exclusively from the quasi-classical vibrational contribution.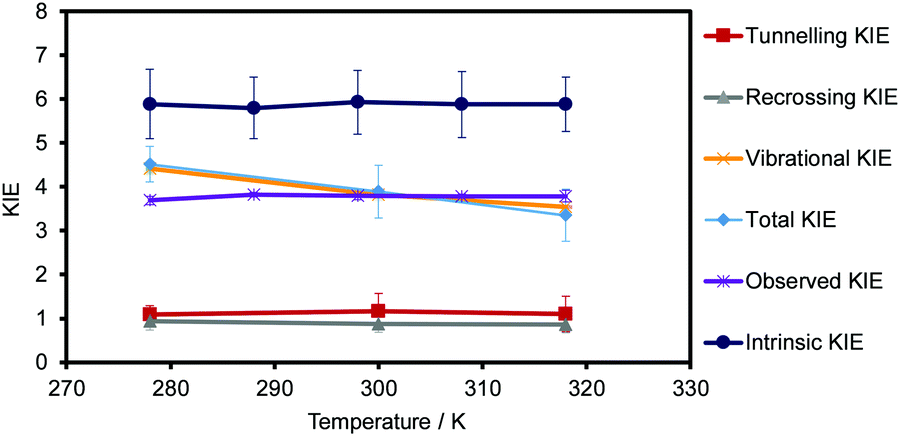 | ||
| Fig. 9 Temperature dependence of total KIEs (in light blue), the recrossing contribution (in grey), the tunnelling contribution (in red), the quantum vibrational contribution to the quasi-classical free energy barrier (in orange), the experimentally observed KIEs (in purple) and the experimentally intrinsic KIEs (in dark blue). Standard deviations are displayed as error bars for each value. The standard deviation of the quantum vibrational contribution and the error bars of the observed KIEs are also displayed but they appear as indistinguishable compared with the standard deviations of the other terms. | ||
The experimentally observed KIEs and the intrinsic KIEs obtained by Kohen and co-workers for the related CbFDH44 are shown in Table S3 of the ESI† and are also displayed in Fig. 9. The comparison of the theoretical predictions with the experimental data obtained by Kohen and co-workers44 renders interesting conclusions. The theoretical values obtained (see Table 2 and light blue data in Fig. 9) are similar to the experimentally observed KIEs (see Table S3 of the ESI† and purple data in Fig. 9) but quite different from the intrinsic KIEs calculated by Kohen and co-workers44 (using Northrop's method see Table S3 of the ESI† and dark blue data in Fig. 9). In addition, the experimental KIEs are temperature-independent, while the theoretical results obtained are slightly temperature-dependent. As observed in Fig. 9, this small dependence is due to the quantum vibrational corrections from −2.83 to −2.72 kcal mol−1 for the transfer of protium at 278 and 318 K, respectively. This trend was also observed by Gao, Truhlar and co-workers in a theoretical study of the temperature dependence of KIEs in the hydride transfer on DHFR89 and in our previous study on the thymidylate synthase.71 The novelty in the present study is that variations of the other terms of the rate constant are not cancelling each other and, in turn, it provokes the slight dependency. Major and co-workers also computed the primary KIE by means of nuclear classical mechanical and quantum mechanical potentials of mean force, and using the values of the activation barriers at 310 K for protium and tritium (13.2 and 14.0 kcal mol−1, respectively) obtained a value of the KIE of 3.66.86 This value agrees with our KIEs obtained at 300 and 318 K (3.90 and 3.4, respectively, see Table 2) within the uncertainty associated with the standard deviations.
4. Conclusions
Hybrid QM/MM MD simulations have been carried out in the present study to gain deep insight into the hydride transfer step in formate dehydrogenase. Our calculations, carried out at different temperatures for the transfer of protium and tritium, have allowed us to explore the temperature dependence of not only the free energy barriers but the possible dependence on the different contributions to the rate constant (recrossing effects, quantum tunnelling effects and quantum vibrational corrections) by applying the VTST. In addition, the ratios between the computed rate constants with protium and tritium have provided estimated values of KIEs that can be compared with previous experimental data.44The first conclusion that can be derived from our calculations is that the classical mechanical free energy of activation does not depend on the temperature in the range between 278 and 318 K. Structural analysis of averaged geometrical parameters on going from reactants to products reveals that the active site of the protein is well prepared to accommodate the substrate and cofactor and the variations observed along the reaction are temperature-invariant. It is interesting to note that a rigid model of the protein would not render realistic results since the relative position of some residues is evolving as the reaction proceeds, such as Ile122, Asn146 and Arg284, which are anchoring the substrate that is transforming from the formate ion to carbon dioxide, or the Thr282, Asp308 and the dyad Gln313–His332 that interact with the amide group of the nicotinamide ring of the cofactor. Nevertheless, the analysis of the Hessian matrix on the located TS structures confirms that some of these residues have a non-negligible component on the transition vector, thus indicating that they are part of the real reaction coordinate. This conclusion is confirmed by the dynamic analysis of the evolution of the chemical system and the protein along the reaction progress. Thus, collective coordinates such as the electrostatic potential or the electric field created by the protein on the key atoms reveal that although none of them show dramatic changes from reactants to products, confirming that the protein is preorganised to accommodate the reaction in the active site, significant variations are detected. It is worth pointing out that by combining the evolution of charges on the key atoms and the electric field created by the protein and projected on the donor–acceptor atoms, the protein is exerting an electric force on the formate that would favour the approach to the acceptor cofactor. This force disappears on crossing the TS, thus facilitating the release of the carbon dioxide moiety.
The recrossing trajectories appear to be clearly independent of the temperature, as well as the classical mechanical free energy barrier, as indicated above. This result would be in agreement with the proposal of a well-prepared machinery to favour the reaction to proceed. Nevertheless, the quantum vibrational corrections and the tunnelling effect slightly depend on the temperature and the mass of the transferring particle, the former provoking a slight temperature-dependence of the primary KIEs. This dependence, taking into account the standard deviations of our results, can be considered as negligible. These values are in very good agreement with the experimentally observed KIEs of Kohen and co-workers.44 The absence of temperature dependence suggests a well-organized active site capable of adjusting to the variations in temperature explored in the present study.
This study shows how the contribution assigned to the dynamics by some authors must be revisited. The contribution of the movements of the protein (“dynamic effects”) to the catalysis should be considered as an ill definition of the reaction coordinate. The real reaction coordinate is not exclusively defined by coordinates of the substrate and cofactor (donor atom, acceptor atom and the transferring hydride) but part of the coordinates of the protein should be taken into account to properly define this conflictive term. The protein cannot be considered as a passive spectator of the chemical system but as part of the chemical reaction. The inclusion of all the involved degrees of freedom into the reaction coordinate, in the framework of the VTST, can provide results in perfect agreement with experiments and shed light on their interpretation.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Spanish Ministerio de Economía y Competitividad and FEDER funds through CTQ2015-66223-C2, CTQ2015-65207-P and CTQ2015-74523-JIN(AEI/FEDER) projects, Universitat de València UV-INV-AE11-40931 project and by Universitat Jaume I UJI-B2016-28 and UJI-B2017-31 projects. M. R. thanks Ministerio de Economía y Competitividad for a ‘Ramón y Cajal’ contract (RYC-2014-16592). The authors acknowledge the computational facilities of Universitat Jaume I and Universitat de València (Tirant Supercomputer).References
- A. Warshel and M. Levitt, J. Mol. Biol., 1976, 103, 227 CrossRef CAS PubMed.
- A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 5250 CrossRef CAS.
- A. Warshel, Computer Modeling of Chemical Reactions in Enzymes and Solutions, John Wiley & Sons, New York, 1991 Search PubMed.
- J. Villa and A. Warshel, J. Phys. Chem. B, 2001, 105, 7887 CrossRef CAS.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210 CrossRef CAS PubMed.
- M. Roca, S. Martí, J. Andrés, V. Moliner, M. Tuñón, J. Bertran and A. H. Williams, J. Am. Chem. Soc., 2003, 125, 7726 CrossRef CAS PubMed.
- S. Martí, M. Roca, J. Andrés, V. Moliner, E. Silla, I. Tuñón and J. Bertran, Chem. Soc. Rev., 2004, 33, 98 RSC.
- S. Martí, J. Andrés, V. Moliner, E. Silla, I. Tuñón and J. Bertran, J. Am. Chem. Soc., 2004, 126, 311 CrossRef PubMed.
- M. Roca, J. Andrés, V. Moliner, I. Tuñón and J. Bertran, J. Am. Chem. Soc., 2005, 127, 10648 CrossRef CAS PubMed.
- M. Roca, V. Moliner, I. Tuñón and J. T. Hynes, J. Am. Chem. Soc., 2006, 128, 6186 CrossRef CAS PubMed.
- B. Szefczyk, F. Claeyssens, A. J. Mulholland and W. A. Sokalski, Int. J. Quantum Chem., 2007, 107(12), 2274 CrossRef CAS.
- J. J. Ruiz-Pernía, I. Tuñón, V. Moliner, J. T. Hynes and M. Roca, J. Am. Chem. Soc., 2008, 130, 7477 CrossRef PubMed.
- R. Castillo, M. Oliva, S. Martí and V. Moliner, J. Phys. Chem. B, 2008, 112, 10012 CrossRef CAS PubMed.
- A. J. Adamczyk, J. Cao, S. C. L. Kamerlin and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14115 CrossRef CAS PubMed.
- G. Jindal, B. Ramachandran, R. P. Bora and A. Warshel, ACS Catal., 2017, 7, 3301 CrossRef CAS PubMed.
- A. Krzeminska, V. Moliner and K. Świderek, J. Am. Chem. Soc., 2016, 138, 16283 CrossRef CAS PubMed.
- M. Roca, V. Moliner and I. Tuñón, ACS Catal., 2018, 8, 815 CrossRef CAS.
- M. García-Viloca, J. Gao, M. Karplus and D. G. Truhlar, Science, 2004, 303, 186 CrossRef PubMed.
- E. J. Loveridge, E. M. Behiry, J. Guo and R. K. Allemann, Nat. Chem., 2012, 4, 292 CrossRef CAS PubMed.
- D. R. Glowacki, J. N. Harvey and A. J. Mulholland, Nat. Chem., 2012, 4, 169 CrossRef CAS PubMed.
- D. Antoniou, S. Caratzoulas, C. Kalyanaraman, J. S. Mincer and S. D. Schwartz, Eur. J. Biochem., 2002, 269, 3103 CrossRef CAS PubMed.
- E. Z. Eisenmesser, D. A. Bosco, M. Akke and D. Kern, Science, 2002, 295, 1520 CrossRef CAS PubMed.
- E. Z. Eisenmesser, O. Millet, W. Labeikovsky, D. M. Korzhnev, M. Wolf-Watz, D. A. Bosco, J. J. Skalicky, L. E. Kay and D. Kern, Nature, 2005, 438, 117 CrossRef CAS PubMed.
- D. Kern, E. Z. Eisenmesser and M. Wolf-Watz, in Nuclear Magnetic Resonance of Biological Macromolecules, Part C, ed. T. L. James, 2005, vol. 394, p. 507 Search PubMed.
- K. A. Henzler-Wildman, V. Thai, M. Lei, M. Ott, M. Wolf-Watz, T. Fenn, E. Pozharski, M. A. Wilson, G. A. Petsko, M. Karplus, C. G. Huebner and D. Kern, Nature, 2007, 450, 838 CrossRef CAS PubMed.
- Z. D. Nagel and J. P. Klinman, Nat. Chem. Biol., 2009, 5, 543 CrossRef CAS PubMed.
- S. J. Benkovic and S. Hammes-Schiffer, Science, 2003, 301, 1196 CrossRef CAS PubMed.
- S. Hay and N. S. Scrutton, Nat. Chem., 2012, 4, 161 CrossRef CAS PubMed.
- H.-H. Limbach, K. B. Schowen and R. L. Schowen, J. Phys. Org. Chem., 2010, 23, 586 CrossRef CAS.
- J. P. Klinman, Nat. Chem., 2010, 2, 907 CrossRef CAS PubMed.
- V. L. Schramm, in Ann. Rev. Biochem., ed. R. D. Kornberg, C. R. H. Raetz, J. E. Rothman and J. W. Thorner, 2011, vol. 80, p. 703 Search PubMed.
- G. Bhabha, J. Lee, D. C. Ekiert, J. Gam, I. A. Wilson, H. J. Dyson, S. J. Benkovic and P. E. Wright, Science, 2011, 332, 234 CrossRef CAS PubMed.
- P. Singh, K. Francis and A. Kohen, ACS Catal., 2015, 5, 3067 CrossRef CAS PubMed.
- M. H. M. Olsson, W. W. Parson and A. Warshel, Chem. Rev., 2006, 106, 1737 CrossRef CAS PubMed.
- S. C. L. Kamerlin and A. Warshel, Proteins, 2010, 78, 1339 CAS.
- K. Świderek, J. J. Ruiz-Pernía, V. Moliner and I. Tuñón, Curr. Opin. Chem. Biol., 2014, 21, 11 CrossRef PubMed.
- J. P. Klinman, Acc. Chem. Res., 2015, 48, 449 CrossRef CAS PubMed.
- A. Kohen, Acc. Chem. Res., 2015, 48, 466 CrossRef CAS PubMed.
- I. Tuñón, D. Laage and J. T. Hynes, Arch. Biochem. Biophys., 2015, 582, 42 CrossRef PubMed.
- A. Warshel and R. P. Bora, J. Chem. Phys., 2016, 144 Search PubMed.
- L. Y. P. Luk, J. J. Ruiz-Pernía, W. M. Dawson, M. Roca, E. Joel Loveridge, D. R. Glowacki, J. N. Harvey, A. J. Mulholland, I. Tuñón, V. Moliner and R. K. Allemann, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16344 CrossRef CAS PubMed.
- J. J. Ruiz-Pernía, L. Y. Luk, R. García-Meseguer, S. Martí, E. J. Loveridge, I. Tuñón, V. Moliner and R. K. Allemann, J. Am. Chem. Soc., 2013, 135, 18689 CrossRef PubMed.
- J. J. Ruiz-Pernía, E. Behiry, L. Y. P. Luk, E. J. Loveridge, I. Tuñón, V. Moliner and R. K. Allemann, Chem. Sci., 2016, 7, 3248 RSC.
- J. N. Bandaria, C. M. Cheatum and A. Kohen, J. Am. Chem. Soc., 2009, 131, 10151 CrossRef CAS PubMed.
- J. P. Klinman and A. Kohen, in Ann. Rev. Biochem., ed. R. D. Kornberg, 2013, vol. 82, p. 471 Search PubMed.
- P. Pagano, Q. Guo, A. Kohen and C. M. Cheatum, J. Phys. Chem. Lett., 2016, 7, 2507 CrossRef CAS PubMed.
- D. R. Kipp, R. G. Silva and V. L. Schramm, J. Am. Chem. Soc., 2011, 133, 19358 CrossRef CAS PubMed.
- R. G. Silva, A. S. Murkin and V. L. Schramm, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18661 CrossRef CAS PubMed.
- J. E. Masterson and S. D. Schwartz, J. Phys. Chem. A, 2013, 117, 7107 CrossRef CAS PubMed.
- C. R. Pudney, A. Guerriero, N. J. Baxter, L. O. Johannissen, J. P. Waltho, S. Hay and N. S. Scrutton, J. Am. Chem. Soc., 2013, 135, 2512 CrossRef CAS PubMed.
- M. D. Toney, J. N. Castro and T. A. Addington, J. Am. Chem. Soc., 2013, 135, 2509 CrossRef CAS PubMed.
- Z. Wang, D. Antoniou, S. D. Schwartz and V. L. Schramm, Biochemistry, 2016, 55, 157 CrossRef CAS PubMed.
- Z. Wang, E. P. Chang and V. L. Schramm, J. Am. Chem. Soc., 2016, 138, 15004 CrossRef CAS PubMed.
- D. Glasstone, K. J. Laidler and H. Eyring, The theory of rate processes: the kinetics of chemical reactions, viscosity, diffusion and electrochemical phenomena, McGraw-Hill, New York, 1941 Search PubMed.
- J. Keck, Adv. Chem. Phys., 1967, 13, 85 Search PubMed.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, J. Phys. Chem., 1996, 100, 12771 CrossRef CAS.
- C. Alhambra, J. Corchado, M. L. Sánchez, M. García-Viloca, J. Gao and D. G. Truhlar, J. Phys. Chem. B, 2001, 105, 11326 CrossRef CAS.
- D. G. Truhlar, J. L. Gao, C. Alhambra, M. García-Viloca, J. Corchado, M. L. Sánchez and J. Villa, Acc. Chem. Res., 2002, 35, 341 CrossRef CAS PubMed.
- D. G. Truhlar, J. Gao, M. García-Viloca, C. Alhambra, J. Corchado, M. Luz Sánchez and T. D. Poulsen, Int. J. Quantum Chem., 2004, 100, 1136 CrossRef CAS.
- J. P. Bergsma, B. J. Gertner, K. R. Wilson and J. T. Hynes, J. Chem. Phys., 1987, 86, 1356 CrossRef CAS.
- R. F. Grote and J. T. Hynes, J. Chem. Phys., 1980, 73, 2715 CrossRef CAS.
- J. T. Hynes and M. Baer, The Theory of Chemical Reaction Dynamics, Boca Raton, FL, 1985 Search PubMed.
- B. J. Gertner, K. R. Wilson and J. T. Hynes, J. Chem. Phys., 1989, 90, 3537 CrossRef CAS.
- R. Castillo, M. Roca, A. Soriano, V. Moliner and I. Tuñón, J. Phys. Chem. B, 2008, 112 Search PubMed.
- K. Świderek, I. Tuñón, I. H. Williams and V. Moliner, J. Am. Chem. Soc., 2018, 140, 4327 CrossRef PubMed.
- N. Kanaan, M. Roca, I. Tuñón, S. Martí and V. Moliner, Phys. Chem. Chem. Phys., 2010, 12, 11657 RSC.
- M. Roca, M. Oliva, R. Castillo, V. Moliner and I. Tuñón, Chem. – Eur. J., 2010, 16, 11399 CrossRef CAS PubMed.
- D. H. Lu, T. N. Truong, V. S. Melissas, G. C. Lynch, Y. P. Liu, B. C. Garrett, R. Steckler, A. D. Isaacson, S. N. Rai, G. C. Hancock, J. G. Lauderdale, T. Joseph and D. G. Truhlar, Comput. Phys. Commun., 1992, 71, 235 CrossRef CAS.
- Y. P. Liu, G. C. Lynch, T. N. Truong, D. H. Lu, D. G. Truhlar and B. C. Garrett, J. Am. Chem. Soc., 1993, 115, 2408 CrossRef CAS.
- N. Kanaan, S. Ferrer, S. Martí, M. García-Viloca, A. Kohen and V. Moliner, J. Am. Chem. Soc., 2011, 133, 6692 CrossRef CAS PubMed.
- K. Świderek, K. Arafet, A. Kohen and V. Moliner, J. Chem. Theory Comput., 2017, 13, 1375 CrossRef PubMed.
- M. Delgado, S. Gorlich, J. E. Longbotham, N. S. Scrutton, S. Hay, V. Moliner and I. Tuñón, ACS Catal., 2017, 7, 3190 CrossRef CAS.
- V. I. Tishkov and V. O. Popov, Biomol. Eng., 2006, 23, 89 CrossRef CAS PubMed.
- R. Wichmann, C. Wandrey, A. F. Buckmann and M. R. Kula, Biotechnol. Bioeng., 2000, 67, 791 CrossRef CAS PubMed.
- G. Krix, A. S. Bommarius, K. Drauz, M. Kottenhahn, M. Schwarm and M. R. Kula, J. Biotechnol., 1997, 53, 29 CrossRef CAS.
- A. S. Bommarius, M. Schwarm, K. Stingl, M. Kottenhahn, K. Huthmacher and K. Drauz, Tetrahedron: Asymmetry, 1995, 6, 2851 CrossRef CAS.
- A. M. Rojkova, A. G. Galkin, L. B. Kulakova, A. E. Serov, P. A. Savitsky, V. V. Fedorchuk and V. I. Tishkov, FEBS Lett., 1999, 445, 183 CrossRef CAS PubMed.
- V. S. Lamzin, Z. Dauter, V. O. Popov, E. H. Harutyunyan and K. S. Wilson, J. Mol. Biol., 1994, 236, 759 CrossRef CAS PubMed.
- I. G. Shabalin, E. V. Filippova, K. M. Polyakov, E. G. Sadykhov, T. N. Safonova, T. V. Tikhonova, V. I. Tishkov and V. O. Popov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 1315 CrossRef CAS PubMed.
- Q. Guo, L. Gakhar, K. Wickersham, K. Francis, A. Vardi-Kilshtain, D. T. Major, C. M. Cheatum and A. Kohen, Biochemistry, 2016, 55, 2760 CrossRef CAS PubMed.
- V. I. Tishkov, A. G. Galkin and A. M. Yegorov, Dokl. Akad. Nauk SSSR, 1991, 317, 745 CAS.
- M. Iida, K. Kitamura-Kimura, H. Maeda and S. Mineki, Biosci., Biotechnol., Biochem., 1992, 56, 1966 CrossRef CAS.
- V. I. Tishkov and V. O. Popov, Biochemistry, 2004, 69, 1252 CAS.
- B. Schiott, Y. J. Zheng and T. C. Bruice, J. Am. Chem. Soc., 1998, 120, 7192 CrossRef CAS.
- J. N. Bandaria, S. Dutta, S. E. Hill, A. Kohen and C. M. Cheatum, J. Am. Chem. Soc., 2008, 130, 22 CrossRef CAS PubMed.
- A. Vardi-Kilshtain, D. T. Major, A. Kohen, H. Engel and D. Doron, J. Chem. Theory Comput., 2012, 8, 4786 CrossRef CAS PubMed.
- D. T. Major and J. Gao, J. Chem. Theory Comput., 2007, 3, 949 CrossRef CAS PubMed.
- J. N. Bandaria, S. Dutta, M. W. Nydegger, W. Rock, A. Kohen and C. M. Cheatum, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17974 CrossRef CAS PubMed.
- J. Pu, S. Ma, J. Gao and D. G. Truhlar, J. Phys. Chem. B, 2005, 109, 8551 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Acc. Chem. Res., 2006, 39, 93 CrossRef CAS PubMed.
- D. Roston, C. M. Cheatum and A. Kohen, Biochemistry, 2012, 51, 6860 CrossRef CAS PubMed.
- H. Li, A. D. Robertson and J. H. Jensen, Proteins, 2005, 61, 704 CrossRef CAS PubMed.
- D. C. Bas, D. M. Rogers and J. H. Jensen, Proteins, 2008, 73, 765 CrossRef CAS PubMed.
- C. R. Sondergaard, M. H. M. Olsson, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 2284 CrossRef CAS PubMed.
- M. H. M. Olsson, C. R. Sondergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525 CrossRef CAS PubMed.
- M. J. Field, A practical introduction to the simulation of molecular systems, Cambridge University Press, Cambridge, 1999 Search PubMed.
- M. J. Field, M. Albe, C. Bret, F. Proust-De Martín and A. Thomas, J. Comput. Chem., 2000, 21, 1088 CrossRef CAS.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902 CrossRef CAS.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1986, 7, 718 CrossRef CAS.
- M. J. Field, P. A. Bash and M. Karplus, J. Comput. Chem., 1990, 11, 700 CrossRef CAS.
- W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 1988, 110, 1657 CrossRef CAS PubMed.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187 CrossRef.
- S. Kumar, D. Bouzida, R. H. Swendsen, P. A. Kollman and J. M. Rosenberg, J. Comput. Chem., 1992, 13, 1011 CrossRef CAS.
- K. Nam, X. Prat-Resina, M. García-Viloca, L. S. Devi-Kesavan and J. L. Gao, J. Am. Chem. Soc., 2004, 126, 1369 CrossRef CAS PubMed.
- A. D. Isaacson, D. G. Truhlar, S. N. Rai, R. Steckler, G. C. Hancock, B. C. Garrett and M. J. Redmon, Comput. Phys. Commun., 1987, 47, 91 CrossRef CAS.
- K. Świderek, I. Tuñón, S. Martí and V. Moliner, ACS Catal., 2015, 5, 1172 CrossRef PubMed.
- L. Y. P. Luk, J. J. Ruiz-Pernía, W. M. Dawson, E. J. Loveridge, I. Tuñón, V. Moliner and R. K. Allemann, J. Am. Chem. Soc., 2014, 136, 17317 CrossRef CAS PubMed.
- L. Y. P. Luk, J. J. Ruiz-Pernía, A. S. Adesina, E. J. Loveridge, I. Tuñón, V. Moliner and R. K. Allemann, Angew. Chem., Int. Ed., 2015, 54, 9016 CrossRef CAS PubMed.
- J. Pu, J. Gao and D. G. Truhlar, Chem. Rev., 2006, 106, 3140 CrossRef CAS PubMed.
- L. Y. Luk, J. J. Ruiz-Pernía, W. M. Dawson, E. J. Loveridge, I. Tuñón, V. Moliner and R. K. Allemann, J. Am. Chem. Soc., 2014, 136, 17317 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S3 and Fig. S1–S3. See DOI: 10.1039/c8cp04244f |
| This journal is © the Owner Societies 2018 |