
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNarrowing the gap between experimental and computational determination of methyl group dynamics in proteins†
Falk
Hoffmann
 a,
Mengjun
Xue
b,
Lars V.
Schäfer
*a and
Frans A. A.
Mulder
*b
a,
Mengjun
Xue
b,
Lars V.
Schäfer
*a and
Frans A. A.
Mulder
*b
aTheoretical Chemistry, Ruhr-University Bochum, D-44780 Bochum, Germany. E-mail: lars.schaefer@ruhr-uni-bochum.de; Tel: +49 234 3221582
bInterdisciplinary Nanoscience Center (iNANO) and Department of Chemistry, University of Aarhus, DK-8000 Aarhus, Denmark. E-mail: fmulder@chem.au.dk; Tel: +45 87155889
First published on 18th September 2018
Abstract
Nuclear magnetic resonance (NMR) spin relaxation has become the mainstay technique to sample protein dynamics at atomic resolution, expanding its repertoire from backbone 15N to side-chain 2H probes. At the same time, molecular dynamics (MD) simulations have become increasingly powerful to study protein dynamics due to steady improvements of physical models, algorithms, and computational power. Good agreement between generalized Lipari–Szabo order parameters derived from experiment and MD simulation has been observed for the backbone dynamics of a number of proteins. However, the agreement for the more dynamic side-chains, as probed by methyl group relaxation, was much worse. Here, we use T4 lysozyme (T4L), a protein with moderate tumbling anisotropy, to showcase a number of improvements that reduce this gap by a combined evaluation of NMR relaxation experiments and MD simulations. By applying a protein force field with accurate methyl group rotation barriers in combination with a solvation model that yields correct protein rotational diffusion times, we find that properly accounting for anisotropic protein tumbling is an important factor to improve the match between NMR and MD in terms of methyl axis order parameters, spectral densities, and relaxation rates. The best agreement with the experimentally measured relaxation rates is obtained by a posteriori fitting the appropriate internal time correlation functions, truncated by anisotropic overall tumbling. In addition, MD simulations led us to account for a hitherto unrealized artifact in deuterium relaxation experiments arising from strong coupling for leucine residues in uniformly 13C-enriched proteins. For T4L, the improved analysis reduced the RMSD between MD and NMR derived methyl axis order parameters from 0.19 to 0.11. At the level of the spectral density functions, the improvements allow us to extract the most accurate parameters that describe protein side-chain dynamics. Further improvement is challenging not only due to force field and sampling limitations in MD, but also due to inherent limitations of the Lipari–Szabo model to capture complex dynamics.
1 Introduction
The biological function of proteins is often intimately connected with their dynamical behavior.1,2 NMR spectroscopy is a powerful technique to characterize protein motions on different time scales, both site-specifically and globally,3–7 and combining NMR spectroscopy with molecular dynamics (MD) simulations is particularly useful, as MD simulations can help to interpret NMR relaxation data.8–10 In this paper, several advances in the analysis of protein dynamics from methyl deuterium spin relaxation and MD simulations will be described, which results in an improved agreement between experiments and computations.The motion of a bond vector is described by its time correlation function (TCF)
C(t) = 〈P2[![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) (0)·(t)]〉 (0)·(t)]〉 | (1) |
(t) is the unit vector of the bond, and 〈⋯〉 denotes the average over all time step differences t. The total TCF describes the reorientation motion of the bond in the laboratory frame. In addition to the internal dynamics, a bond also moves together with the full protein. As a consequence, bond orientations become uncorrelated on time scales longer than the overall rotational diffusion (tumbling) correlation time τR of the protein, and the TCF decays to 0 for t ≫ τR. The square of the generalized order parameter S2 describes the internal dynamics of the bond in the molecular frame, and is defined as the long-time limit of the internal TCF,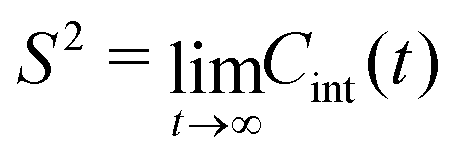 | (2) |
In an NMR spin relaxation experiment, relaxation rates are measured. These relaxation rates determine the spectral densities J(ω) at discrete frequencies that depend on the Larmor frequencies of the involved nuclear spins, as the stochastic internal and overall motions provide the energy for transitions. Fourier transformation of the TCF yields the power spectral density,
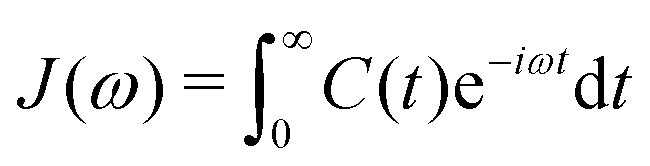 | (3) |
Lipari and Szabo (LS)11,12 described simplified analytical models for interpreting NMR relaxation experiments in terms of generalized order parameters and associated correlation times. The LS formalism, which is related to work by Halle and Wennerström,13 assumes that internal motions and overall tumbling are statistically independent, which is for example the case if they occur on sufficiently different time scales. In this case, the total TCF can be factorized into the internal and overall TCFs, C(t) = Cint(t)CO(t). For a molecule that tumbles isotropically in solution with a global rotational tumbling time τR, the overall motion can be described by a single-exponential decay, CO(t) = e−t/τR. Lipari and Szabo assumed that the internal TCF can also be described by a single-exponential decay on a characteristic time scale of fast internal dynamics, τf ≪ τR. Using the reduced time τred = (τRτf)/(τR + τf), the internal and total TCFs in the original LS model are approximated by
| Cint(t) = S2 + (1 − S2)e−t/τf | (4) |
| C(t) = S2e−t/τR + (1 − S2)e−t/τred | (5) |
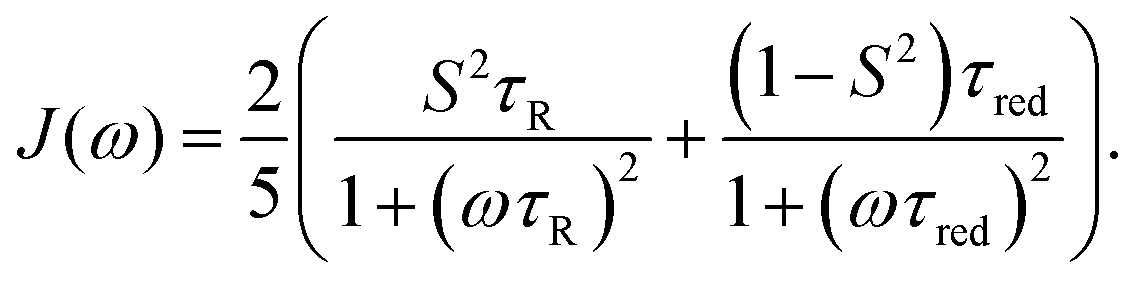 | (6) |
| Cint(t) = S2 + (1 − Sf2)e−t/τf + (Sf2 − S2)e−t/τs | (7) |
| C(t) = S2e−t/τR + (1 − Sf2)e−t/τf,red + (Sf2 − S2)e−t/τs,red | (8) |
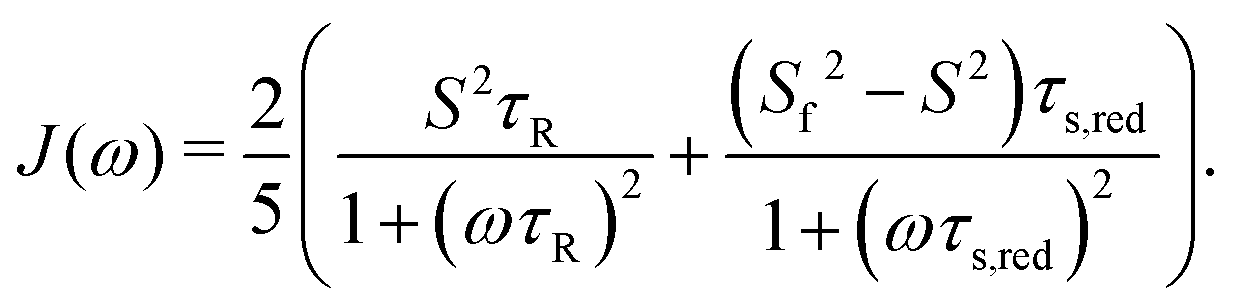 | (9) |
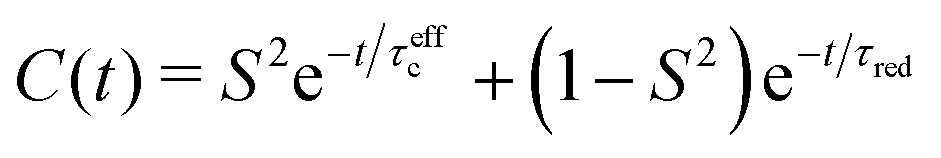 | (10) |
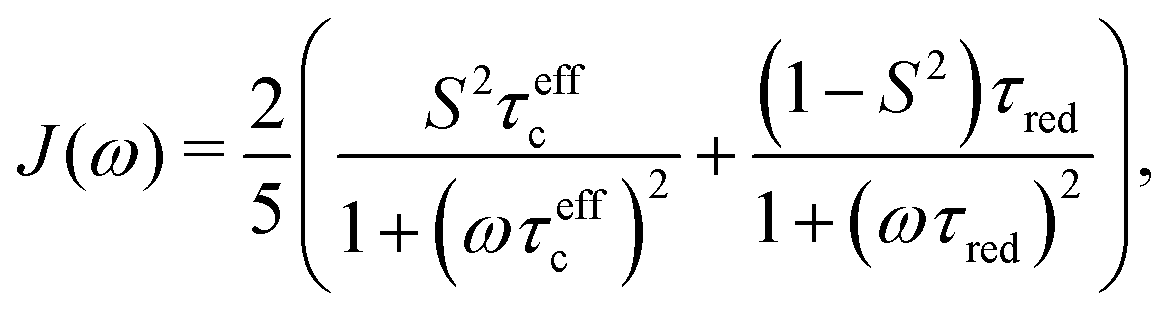 | (11) |
The present work is concerned with the dynamics of methyl groups in protein side-chains. To probe methyl dynamics by deuterium spin relaxation experiments in NMR,16,17 one measures the relaxation rates of fractionally deuterated methyl groups in protein side-chains, which are sensitive to the reorientation motions of 13C–2H bond vectors. For example, the longitudinal, in-phase transverse magnetization, and quadrupolar order rates,
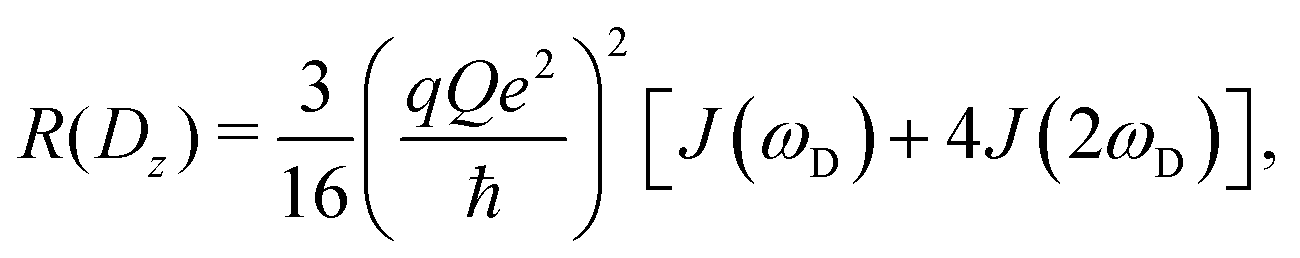 | (12) |
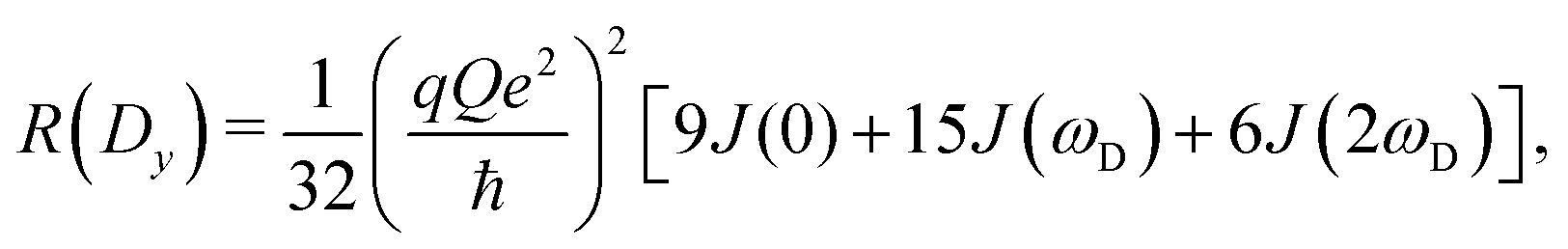 | (13) |
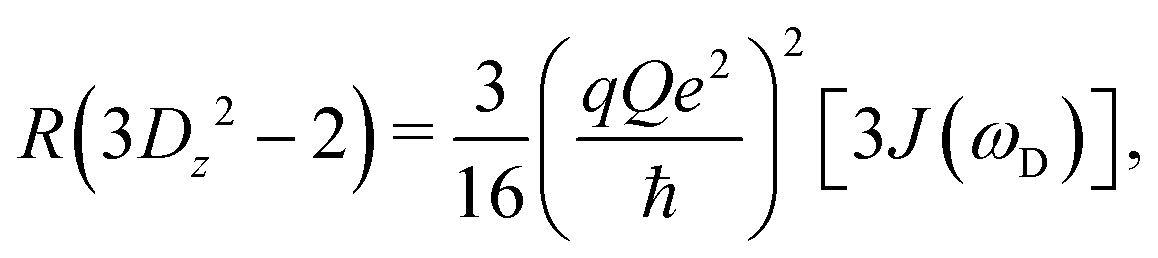 | (14) |
The dynamics of methyl groups comprises the motions of the Cmethyl–2Hmethyl bonds in addition to the dynamics of the C–Cmethyl (or S–Cmethyl, for methionine) bonds that connect the methyl group to the rest of the side-chain. The bonds can undergo different kinds of motions, which are reflected in the corresponding TCFs. Methyl motions include (1) very fast Cmethyl–2Hmethyl librations on the sub-ps time scale; these motions usually decrease the time correlation function to a value of about 0.9 within less than 1 ps.18 (2) Rapid jumps around the 3-fold methyl symmetry axis, typically within less than 100 ps. For ideal tetrahedral methyl geometry this decreases the TCF of the C–2H bond to a value of 1/9C(t = 0). (3) Angular librational motions of the entire side-chain and fast rotamer jumps on the sub-ns time scale, which leads to a further decay of the Cmethyl–2Hmethyl internal TCF to its limiting value of 1/9Saxis2 in the absence of slow internal motions (see point 4); here, Saxis2 is the methyl order parameter of the symmetry axis. (4) Slow internal dynamics of the entire side-chain due to jumps between different rotamer states on a broad range of time scales up to several ns and beyond. Methyl order parameters lower than 0.7 indicate population of more than a single rotamer well.19–22 Internal motions that occur on time scales longer than τR do not affect 2H NMR quadrupolar relaxation, because the TCF has already decayed to zero due to global tumbling. Nevertheless, it is these motions that determine the decay of the internal TCF to its final value according to eqn (2). (5) Overall tumbling of the protein. While isotropic tumbling can be described by a single-exponential decay with a global rotational tumbling time τR, in case of moderate anisotropy one may consider residue-specific τR's (see below).
MD simulations are routinely used to calculate backbone order parameters SNH2 with high accuracy. However, methyl group order parameters are more challenging and less frequently reported in the literature. Kasinath and coworkers23 used an analogous approach to the long-time limit of Cint(t) of the C–Cmethyl bond, eqn (2). They investigated seven proteins (comprising of 73 to 168 residues) in MD simulations of lengths between 112 ns and 260 ns with the CHARMM2724 force field. The reported average Pearson correlation coefficient for methyl order parameters (with respect to experiment) is 0.65 for the seven investigated proteins. Bowman25 simulated ubiquitin and compared three force fields (AMBER ff03,26 AMBER ff99SB-ILDN,27 and CHARMM2724) and three methods (long-time limit approach according to eqn (2), a related truncated-average approximation that employs the average of the internal TCF close to the molecular tumbling time τR, and the Lipari–Szabo model of the internal TCF, eqn (4)). He concluded that microseconds of simulation time are required to obtain statistically reliable methyl order parameters and, furthermore, that the agreement to NMR between the MD-derived methyl order parameters obtained with the long-time limit approach decreases with increasing simulation time, whereas the opposite is the case for the other approaches. Prompers and coworkers28 proposed the isotropic reorientational eigenmode dynamics (iRED) model, in which a principal component analysis of covariance matrices of backbone N–H vectors and their corresponding 2nd rank spherical harmonics from MD trajectories yield reorientational eigenmodes of the protein. The different eigenmodes correspond to correlated dynamics of the bond vectors, and the eigenvalue spectrum shows whether the time scales of internal and overall motions are separable. Genheden29 tested iRED for methyl groups and reported mean unsigned deviations of methyl axis order parameters from experiment of 0.13 and 0.17 for galectin-3 in complex with lactose or a synthetic derivate, respectively.
Most of the proteins studied previously may be described as fairly isotropic. These proteins have the advantage that the tumbling time of all individual methyl groups is close to the global rotational tumbling time τR of the protein. Accounting for anisotropy is more challenging.30–37 Lipari and Szabo already mentioned in their original work12 that anisotropy should be accounted for if the macromolecule has a non-spherical shape. In that case, overall tumbling cannot be captured by an exponential decay with a single global time constant τR, but needs to be described by an anisotropic diffusion tensor. Importantly, the resulting generalized order parameters are defined with respect to the principal axis frame (PAF) of the protein diffusion tensor. To assess rotational diffusion anisotropy in proteins, Ryabov and coworkers38 analyzed a set of 841 protein structures from the Protein Data Bank (PDB), chosen as a representative set of globular single-domain folds. They found that only 11% of 753 prolate diffusion tensors had small anisotropies 1 < A < 1.17 and could be approximated as isotropic for the purposes of NMR studies. Here, A = D‖/D⊥ is the ratio of the diffusion constants along parallel and perpendicular directions of the diffusion tensor. 68% of the tensors had intermediate anisotropies in the range 1.17 < A < 1.6, indicating that, in general, it is imperative to take anisotropic diffusion into account for accurate analysis of NMR relaxation data. As these typical anisotropies are in the applicability range of the quadratic approximation of Brüschweiler and Wright,39 residue-specific correlation times can be calculated based on the overall diffusion tensor and knowledge of the 3D structure. The fitting of diffusion tensors in the analysis of 15N relaxation data is commonplace since several decades,40,41 but is often overlooked in the analysis of 2H relaxation data. However, this approach can also be utilized for side-chain vectors, provided that the direction of the methyl symmetry axis can be accurately determined.
Only a few studies have considered taking anisotropic tumbling into account. Millet and coworkers42 compared the difference in the experimentally derived order parameters for the C–Cmethyl bonds of protein L using an isotropic approach with a single global rotational tumbling time τR for all methyl groups, and an anisotropic approach with residue-specific τR,i values for every methyl group. They found that the effect of anisotropy was small, but can lead to order parameter differences of 0.05 for ALA residues. Skrynnikov and coworkers15 found a qualitatively similar result. Ishima and coworkers43 have shown that for HIV-1 protease, a protein with intermediate anisotropy (D‖/D⊥ = 1.35), the use of the isotropic model led to errors up to 12% in Saxis2. Yet, the effect of anisotropic overall tumbling when analyzing side-chain dynamics is not considered in many studies of methyl relaxation.
Wong and coworkers33 proposed a method to evaluate protein rotational diffusion from MD simulations. The rotational tumbling time τR cannot just be approximated by 1/(6Diso) with the isotropic rotational diffusion constant Diso. In general, diffusion is described by a rank-2 tensor ![[scr D, script letter D]](https://www.rsc.org/images/entities/char_e523.gif) (i.e., a 3 × 3 matrix). Diagonalization yields the principal components of in its principal axis frame (PAF); the trace of this tensor is 3Diso. For a protein that can be approximated as an axially symmetric rotor (i.e., prolate or oblate), the difference from this isotropic value is characterized by the principal components for diffusion of the long (D‖) and short (D⊥) axes.
(i.e., a 3 × 3 matrix). Diagonalization yields the principal components of in its principal axis frame (PAF); the trace of this tensor is 3Diso. For a protein that can be approximated as an axially symmetric rotor (i.e., prolate or oblate), the difference from this isotropic value is characterized by the principal components for diffusion of the long (D‖) and short (D⊥) axes.
A realistic description of the overall rotational diffusion tensor in an MD simulation critically depends on the water model, which has a significant effect on the tumbling times. For example, Maragakis and coworkers44 calculated backbone NH order parameters from a 1.2 µs simulation of ubiquitin using the OPLS-AA force field and the SPC water model by fitting to the Lipari–Szabo TCF (eqn (5)). They reported a rotational tumbling time of τR = 1.98 ns, which is a factor of 2 smaller than the experimental tumbling time. This is in line with the faster self-diffusion of SPC water in comparison to real water. The overall tumbling does not affect the calculation of S2 from eqn (2), but can affect the order parameters obtained from eqn (6). This is especially the case for bond motions that occur on time scales that are comparable to the overall tumbling correlation time of the protein, see below.
In the present work, we use NMR relaxation and MD simulations to derive methyl order parameters Saxis2 as well as relaxation rates for T4 lysozyme (T4L), a prolate protein with intermediate anisotropy (D‖/D⊥ ≈ 1.4). We compare different approaches to obtain Saxis2 from MD simulations. First, we use the long-time limit approach of eqn (2), which has been widely used for methyl order parameters in the literature.22,23,45–50 Second, we use the Lipari–Szabo TCF fitting approach (eqn (5) for LS2, eqn (10) for LS3), which was used for backbone order parameters recently,25,44 but to our knowledge has not been extended to methyls. Third, we compare these methods with a spectral density mapping approach that is similar in nature to the one used to analyze the experimental NMR relaxation data. The generalized order parameters obtained from the MD simulations are compared to those obtained from 2H NMR relaxation data. We show that the discrepancy between NMR and MD can be alleviated by taking the decay of the time correlation functions (and hence spectral densities) due to anisotropic protein tumbling properly into account in the analysis of the MD data. Furthermore, the spectral density approach enables one to extract not only Saxis2 from the MD simulations, but – maybe even more importantly – also relaxation rates that can be compared to the ones directly measured by NMR relaxation experiments, without the need to invoke simplified motional models.
In the following, we describe each of the improvements in detail. First, we show that different spectral densities and order parameter values are obtained in the presence of LEU strong coupling. We identify the origin of this artifact, and describe when to remove faulty data. Second, we discuss how anisotropy affects the interpretation of experimental NMR relaxation data in terms of generalized order parameters of methyl groups. We will illustrate with our experimental data that the use of an isotropic model leads to spurious differences in the order parameters of prochiral methyl groups (e.g., VAL-Cγ,1/Cγ,2 or LEU-Cδ,1/Cδ,2), while an anisotropic model removes these differences. Third, we turn to MD simulations and quantitatively assess the time correlation functions obtained with the Amber ff99SB*-ILDN force field in combination with different water models; in this part we also discuss how anisotropic protein tumbling can be accounted for in the analysis of MD simulations. Then, we compare different ways to calculate generalized methyl order parameters from MD simulations and evaluate their agreement with experimental values. Finally, we show the agreement between the relaxation rates determined by NMR relaxation experiments and MD simulations.
2 Results and discussion
2.1 LEU strong coupling
In an initial analysis of T4L deuterium relaxation data, very different order parameter values were obtained for the prochiral methyl groups of LEU residues (Table S1, ESI†). One can see, that the difference in methyl order parameter between the two prochiral LEU methyl groups can be rather large, e.g., the difference between the methyl order parameter ΔSaxis2 of the two prochiral methyl groups 13Cδ,1 and 13Cδ,2 for LEU79 and LEU118 are 0.23 and 0.28, respectively, which seemingly suggests very different internal dynamics of these methyls. We observed that these differences contrasted the MD simulations, which predicted very similar order parameters for prochiral methyl groups. Using the long-time limit approach (see Methods), a method which is insensitive to protein tumbling, we obtained MD methyl order parameter differences ΔSaxis2 between the two prochiral methyl groups for LEU79 and LEU118 of 0.01 and 0.02, respectively.We therefore hypothesized that the experimental data were flawed, and strong 13C–13C coupling in LEU residues might be responsible. Strong coupling can be expected particularly in the case where the LEU side-chain is rigid. In that case, LEU are typically found in one of the two most favorable side-chain conformations mt or tp51,52 (Table S2, ESI†). This can be recognized from the chemical shift difference of the two prochiral Cδ methyls due to the gamma gauche effect, which reports on the χ2 side-chain angle.53 This leads to the methyl group trans to Cα being shifted down field relative to the methyl group in the gauche position. Therefore, in the case of limited LEU side-chain mobility, one of the methyl groups will have a chemical shift in very close proximity to the 13Cγ chemical shift.
The impact of artifacts arising from strong 13C–13C coupling on methyl group order parameters can best be demonstrated with the data of chicken α-spectrin SH3 from Agarwal54 (Table S3, ESI†). The two methyl order parameter of 6 of the 7 LEU side-chains have been measured for samples produced from 3-13C-pyruvate and uniformly 13C-labeled glucose. In the sample produced from 13C6-glucose LEU methyl groups are 13C-enriched, as well as the attached carbons in the side-chain, which is avoided by use of 3-13C-pyruvate55 such that strong 13C–13C spin coupling cannot occur. The 13Cδ chemical shift of one of the two methyl groups for all of these side-chains was higher than 24 ppm, indicating that these methyl groups could experience strong coupling. In 4 of these 6 cases, the methyl order parameter difference between glucose- and pyruvate-derived samples is larger than 0.1, while it is always lower than 0.1 for the LEU methyl groups with 13Cδ chemical shifts lower than 24 ppm. For example, for LEU8, Saxis2 order parameters for the two methyl groups are 0.67 and 0.97 when derived for a uniformly 13C-labeled sample. When producing the protein from 3-13C-pyruvate, these values are 0.61 and 0.71, respectively, clearly showing the impact of the strong coupling artifact. In the absence of complete chemical shift assignments for the side-chain 13C shifts, therefore, signals appearing within approximately 2–3 standard deviations from the 13Cγ shift (26 ± 1.1 ppm in BMRB) should be eliminated or treated with great caution. Thus, in practice, 2H relaxation data for LEU with 13Cδ chemical shifts larger than 24 ppm may be compromised. Although this was mentioned in a footnote by Xue,56 an exploration of the effect, to our knowledge, has hitherto not been published.
We measured 13C chemical shifts of all methyl groups of T4L and identified 9 LEU methyl groups with 13Cδ chemical shifts that are close to their respective 13Cγ chemical shifts. All LEU methyl groups that could be affected by LEU strong coupling (highlighted in red in Table S1, ESI†) were eliminated from further analysis.
2.2 NMR relaxation data show anisotropy of T4L
The three experimental quadrupolar relaxation rates R(Dz), R(Dy), and R(3Dz2 − 2) at 950 MHz were measured for the Cys-free wild-type of T4 lysozyme and analyzed with the spectral density mapping approach, where the relaxation rates are converted into spectral density values at the frequencies 0, ωD and 2ωD. The LS2 and LS3 models were used to fit the spectral density points. The results for the measured relaxation rates and the fitted model parameters can be found in Table S4 and Fig. S1, ESI.†Table 1 compares the methyl order parameter of specific methyl groups using an isotropic Lipari–Szabo model with a global rotational tumbling time τR = 10.70 ns, and an anisotropic model with methyl group-specific rotational tumbling times. The chosen residues fit well to the LS2 model in both cases.
| Methyl group | τ R,iso [ns] | S iso 2 | τ R,aniso [ns] | S aniso 2 |
|---|---|---|---|---|
| LEU39-Cδ,1 | 10.70 | 0.63 | 11.00 | 0.62 |
| LEU39-Cδ,2 | 10.70 | 0.54 | 10.14 | 0.57 |
| ALA97-Cβ | 10.70 | 0.79 | 9.69 | 0.88 |
| ALA98-Cβ | 10.70 | 0.94 | 11.57 | 0.88 |
| ILE100-Cγ,2 | 10.70 | 0.97 | 11.89 | 0.89 |
| ILE100-Cδ,1 | 10.70 | 0.63 | 9.77 | 0.68 |
| VAL149-Cγ,1 | 10.70 | 0.76 | 9.82 | 0.83 |
| VAL149-Cγ,2 | 10.70 | 0.90 | 11.62 | 0.84 |
Table 1 shows that the order parameters of the prochiral methyl groups of LEU39 and of VAL149, respectively, are very different in the isotropic model (ΔSaxis2 = 0.09 and 0.14 for LEU39-Cδ,1/δ,2 and VAL149-Cγ,1/γ,2, respectively). Likewise, the order parameters of ALA97-Cβ and ALA98-Cβ, both of which are part of the same α-helix, differ by 0.15 in the isotropic model. If one employs an anisotropic model (prolate), derived from backbone 15N relaxation data, and takes the orientation of the methyl symmetry axis into account, the artificial differences between these order parameters are diminished. This indicates that the methyl groups in the two consecutive ALA residues in the same α-helix as well as the prochiral methyl groups in the same side-chain (LEU39, VAL149) have similar internal dynamics, but tumble differently in the reference frame due to their different orientations with respect to the PAF of the protein diffusion tensor. For example, the Cα–Cβ vector of ALA98 is oriented along the direction of the slowly tumbling long axis of the protein, while the same bond in ALA97 points almost perpendicular to it. Furthermore, artifactually high order parameters such as for ILE100-Cγ,2 can also result from neglecting anisotropic protein tumbling (Table 1).
A proper interpretation of order parameters from NMR relaxation data for proteins of even moderate anisotropy clearly seems to require adequate consideration of overall tumbling.
2.3 Effect of the water model on protein dynamics in MD simulations
In principle, MD simulations can provide the spectral density at all frequencies. In practice, this requires accurate time correlation functions, which describe the internal motions and the overall tumbling of the bond in the laboratory frame. Here, we compare two commonly used water models, TIP3P57 and TIP4P/2005,58 in terms of the internal backbone dynamics and the overall tumbling of the protein. For all simulations, the Amber ff99SB*-ILDN force field27,59,60 was used for the protein, including recent adjustments of the methyl rotation barriers.61 In each case, ten MD simulations were carried out; the simulations with TIP4P/2005 water were 300 ns each, and the simulations with TIP3P were 100 ns each. Thus, the accumulated simulation times are 3 µs and 1 µs for TIP4P/2005 and TIP3P water, respectively, and each individual MD trajectory exceeds τR by about a factor 30.Since the Amber force fields were originally parametrized together with the TIP3P model, we first tested the influence of the water model on the backbone order parameters and rotational tumbling times. We calculated the TCFs of all backbone N–H bonds viaeqn (1) and extracted SNH2 and corresponding τR,i values by fitting to eqn (10). The rotational tumbling times from eqn (10), which are defined in the laboratory frame, were used together with the initial structure of the MD simulations to calculate the diffusion tensor of the protein using the program QUADRIC (see Methods). Fig. 1 shows the backbone NH order parameters, and the rotational tumbling times are shown in Fig. S2, ESI.†
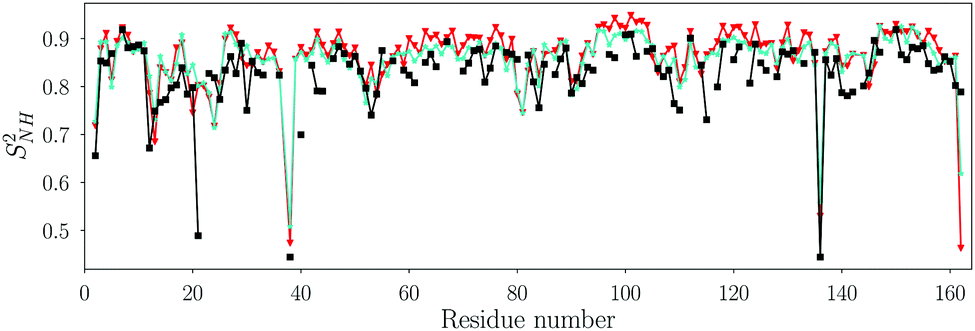 | ||
| Fig. 1 Order parameter SNH2 of backbone N–H vectors fitted with the Lipari–Szabo TCF model (eqn (10)) for the MD simulations with the TIP3P (cyan stars) and the TIP4P/2005 (red triangles) water models. The Pearson and Spearman correlation coefficients between the TIP3P and TIP4P/2005 SNH2 are RP = 0.95 and RS = 0.89, respectively. The order parameters from NMR relaxation experiments are shown as black squares. The RMSD between NMR and MD (TIP4P/2005 water) is 0.05. | ||
Fig. 1 shows that the backbone order parameters extracted from the two water models are very similar, and both agree with the experimental order parameters for most N–H vectors (see also Table S5, ESI†). The order parameters from MD are in general a little bit higher than those from NMR. The difference in SNH2 between both water models is less than 0.05 for all non-terminal residues except for ASP40, where it is 0.09. These results show that the water model does not influence the internal dynamics of the backbone, in line with previous findings.29,33,62,63
The τR,i values of individual backbone N–H vectors are highly correlated between the two water models, but the time scales differ substantially (Fig. S2, ESI†). The residue-averaged tumbling times are 4.3 ± 0.3 ns and 10.9 ± 0.5 ns for TIP3P and TIP4P/2005, respectively. This difference by a factor of ca. 2.5 coincides with the ratio of the self-diffusion constants of the two water models, which are 6.05 × 10−9 m2 s−1 and 2.49 × 10−9 m2 s−1 for TIP3P and TIP4P/2005, respectively.64,65 Although the overall rotational tumbling times in TIP4P/2005 and TIP3P water differ substantially, tumbling is much slower than internal dynamics of the N–H vectors for both water models, explaining the similar backbone amide SNH2 order parameters in TIP4P/2005 and TIP3P water extracted from the LS model.
This work aims at a quantitative comparison of side-chain dynamics from MD and NMR, which demands accurate spectral densities from MD and hence correct global tumbling time scales. The NMR relaxation experiments yield a global tumbling time for T4L of τiso = 10.70 ns (see above), which is very close to τiso = 10.78 ns obtained from the MD simulations in TIP4P/2005 water.‡ Therefore, in the following the data from the TIP4P/2005 simulations are used.
2.4 Comparison of methyl order parameters from MD and NMR
After having established that the overall rotational diffusion is realistically described in the MD simulations with the TIP4P/2005 water model, we next turn to the analysis of simulations in terms of the internal dynamics of methyl groups. Several methods have been proposed for calculating order parameters from MD simulations. In addition to the widespread long-time limit approach (eqn (2)), we used the method proposed by Maragakis and coworkers44 for calculating backbone NH order parameters by directly fitting the total TCFs with a Lipari–Szabo model. Here, we applied both these approaches to the C–Cmethyl vectors. The results are compared to the spectral density mapping approach, which we applied to Cmethyl–Hmethyl vectors. As detailed in Methods, the total TCFs of the Cmethyl–Hmethyl bond vector reorientation motions were factorized into TCFs for internal motions and overall tumbling; the latter is described by an anisotropic diffusion tensor. This tensor is (again) obtained from MD simulations, and our approach does thus not draw on any experimental NMR information§ or system-specific adjustable parameters. The spectral densities from MD, obtained by Fourier transformation of the TCF, were fitted to the LS2 and LS3 models. The MD-based spectral densities and TCFs are shown in Fig. S1 and S3, ESI,† respectively. We compare the methyl order parameters obtained with all three methods to the values obtained from our NMR relaxation experiments analyzed with the anisotropic (prolate) model.Fig. 2 shows the correlation between MD and NMR; the correlation coefficients and RMSD values are also listed in Table 2. The spectral density mapping approach yields the best agreement with the NMR relaxation experiments, with an RMSD of 0.11 and correlation coefficients of about 0.75 (Fig. 2C). We attribute this result to the fact that the MD-based spectral density mapping approach is very similar in nature to the way the experimental NMR relaxation data is analyzed, and, most importantly, that it properly takes the anisotropic protein diffusion tensor into account. Direct fitting of the raw TCF to a LS model yields comparably accurate results as spectral density mapping (Fig. 2B and Table 2), as this approach also accounts for the decay of the TCF due to anisotropic tumbling. However, the raw TCFs from the MD simulations suffer from statistical noise at long lag-times, which is avoided in the spectral density approach due to the smoothening of both Cint(t) and CO(t) by the 6- and single-exponential fits, respectively (see Methods; the validity of these assumed functional forms was previously demonstrated for ubiquitin61). Interestingly, Fig. 2B shows that the direct TCF fits tend to yield too high methyl order parameters. To further investigate this effect, an additional analysis was carried out in which we discriminated between methyl groups described by the LS2 and LS3 models (as determined from the NMR experiments). To that end, for the LS2 methyls, τR was not used as a fitting parameter but set to the fixed value obtained from the MD-based diffusion tensor (see Methods). In principle, this approach is the real time space correspondence of the spectral density mapping in Fourier time space. However, as shown in Fig. S4, ESI,† the results are very similar to those shown in Fig. 2B. The RMSD in Saxis2 is 0.12, RP = 0.70, RS = 0.67, and the MD order parameters are on average still higher than the NMR ones. This result demonstrates the robustness of the approach. A possible explanation why the direct TCF fitting approach tends to overestimate Saxis2 is provided in Section 2.5 below.
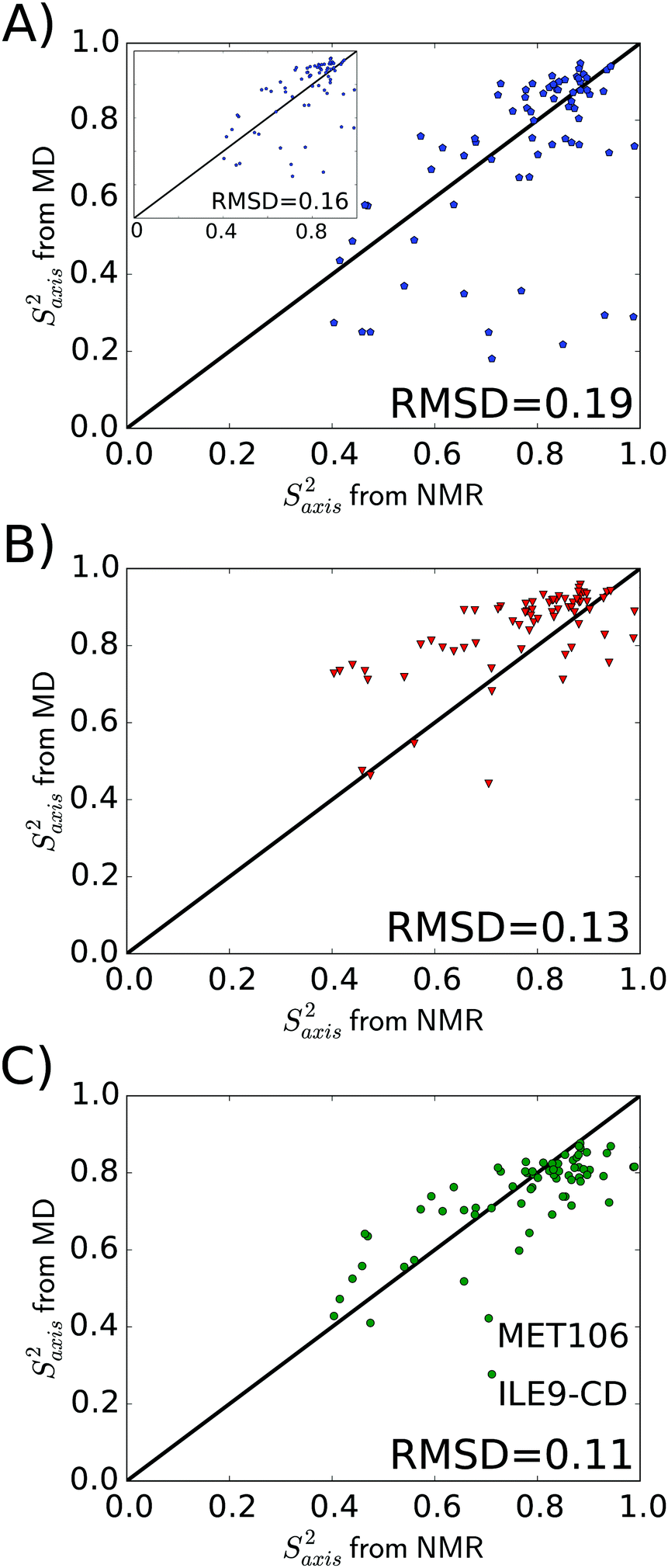 | ||
| Fig. 2 Correlation of methyl order parameters of T4 lysozyme from NMR relaxation and MD simulations using (A) the long-time limit approach, (B) direct fitting of the Lipari–Szabo TCF, and (C) spectral density mapping. The inset in (A) shows the order parameters obtained from the value of Cint(t) around τR instead of in the long-time limit; these Saxis2 were obtained by averaging over Cint(t) for lag-times between 9 and 11 ns. | ||
| Method | R P | R S | RMSD |
|---|---|---|---|
| Long-time limit | 0.56 | 0.59 | 0.19 |
| Lipari–Szabo TCF fit | 0.65 | 0.65 | 0.13 |
| Spectral density mapping | 0.74 | 0.76 | 0.11 |
Compared to the previous two methods that take anisotropy into account, taking the long-time limit of Cint yields considerably worse results (Fig. 2A and Table 2). There are several methyl groups with high NMR order parameter (Saxis2 > 0.7) that have a too low MD order parameter with the long-time limit method. These methyl groups undergo motions on slow time scales τ ≥ τR, which contribute to Cint in the long-time limit but are not detectable by NMR spin relaxation, because the TCF has already decayed to zero beyond τR. This cannot be remedied by simply taking the value of Cint at the overall tumbling time τR instead of in the long-time limit (Fig. 2A, inset). This procedure does only slightly decrease the RMSD between MD and NMR (from 0.19 to 0.16) by eliminating some of the too low MD order parameters, as it still neglects the smooth decay of the TCF due to anisotropic protein tumbling.
To judge the above results, the findings reported here for T4L can be compared to previous MD simulation studies of ubiquitin, in which methyl order parameters from MD and NMR were compared.23,25,50,67 The reported RMSD in Saxis2 between MD and NMR range between 0.10423 and ca. 0.15;25 some of these studies23,50 used the long-time limit approach. These results are comparable to our own ubiquitin simulations,61 in which we obtained an RMSD of 0.13 using the spectral density mapping approach to analyze the MD simulations. Hence, for the small isotropic protein ubiquitin, the simple long-time limit and the spectral density mapping approaches seem to yield comparable methyl order parameters. Interestingly, employing the long-time limit approach, Kasinath and coworkers23 reported a considerably worse agreement between MD and NMR for larger proteins, such as hen egg white lysozyme, calmodulin, and cytochrome c2. Our present work shows that by taking the decay of the TCF due to anisotropic protein tumbling into account in the analysis of the MD simulations, the same level of agreement with experimental NMR relaxation data can be achieved for a protein as large and complex as T4L as was possible before for small isotropic proteins such as ubiquitin.
In addition, to test the quality of the predictions, it is illustrative to compare the MD-derived methyl order parameters for T4L to a “null model” that simply assigns, to each type of methyl group, the average Saxis2 determined from NMR relaxation for a range of proteins,21,68i.e., averaging out all site-to-site variations between methyl groups of the same type. To that end, Saxis2 values of 12 different proteins were extracted from the literature and compared to our NMR values for T4L (Fig. S5, ESI†). The null model yields an overall rather poor prediction of the actual order parameters of T4L (RMSD = 0.21, RP = 0.47, RS = 0.51). Given the large computational effort involved in running the MD simulations, it is somewhat disappointing to notice that the long-time limit approach is only slightly better than the “null model”. In contrast, the MD-based spectral density mapping and direct TCF fitting methods perform substantially better, and are able to successfully predict the site-to-site variability between methyl groups. This is an encouraging result, because one is often interested in the difference ΔSaxis2 of a (set of) methyl group(s), e.g., upon mutation, binding of a ligand, changes of external conditions, etc. However, at the same time, the agreement between MD and NMR is still worse for side-chain methyl groups than for the backbone (the RMSD in Saxis2 is 0.11, as compared to 0.05 for SNH2, see Fig. 1), showing that there is still need for future improvements.
2.5 Can Lipari–Szabo order parameters represent methyl group dynamics adequately?
In the previous section it was observed that the best agreement between NMR and MD was 0.11 (RMSD) for the methyl axis order parameter. Although lower than for most other comparisons to date, the discrepancy between computation and experiment is about twice that observed for backbone dynamics (cf.Fig. 1), and we examine below possible reasons for this discrepancy.In folded proteins, side-chain dynamics are much richer than those of the backbone, both in time scale and amplitude of the motions: methyl groups are positioned at the ends of side-chains of varying number of bonds relative to the main chain, and may undergo multiple rotamer transitions as well as experience librational motion in each rotamer well along the chain. Thus, the internal TCF decays by the simultaneous action of all these motions, which for the more dynamic side-chains may range from sub-ps to several ns and beyond. To investigate this quantitatively the following analysis is based solely on analyses of the MD trajectories: a comparison was made of order parameters obtained from (i) fitting the internal TCF by a multiple-exponential decay (eqn (16)) and (ii) the internal TCF computed by Lipari–Szabo analysis. In the latter case we first computed the spectral density points from the MD simulation including tumbling, and then fitted Saxis2, τf (LS2) or Saxis2, τf, τeffc (LS3). Saxis2 and τf were used to calculate Cint(t) with eqn (4) in both instances.
Fig. 3 shows example dynamics for two ILE side-chains that display markedly different mobility. Fig. 3A displays data for the Cδ,1 methyl group of ILE150, which displays three-fold rotation of the methyl group. The ILE150 side-chain is otherwise largely immobile, with dynamics confined to single rotamer wells. Trajectory analysis shows only exceedingly rare excursions to alternative χ1 or χ2 angles during the simulation (Fig. S6 in ESI†). The internal TCF of the methyl C–H vector was fitted by eqn (16), and is shown in red. Fits by the Lipari–Szabo functions LS2 and LS3 are shown in cyan and magenta, respectively, and are virtually superimposable. This good agreement indicates that the simple LS2 model is sufficient to capture the rather limited dynamics of ILE150-Cδ,1, and can be adequate for extracting the time constant for methyl spinning (about 5 ps) as well as the order parameter (see inset). In contrast, the Cδ,1 methyl group of ILE27 (Fig. 3B) undergoes much more intricate dynamics. As can be seen from the red curve in Fig. 3 and from trajectory analysis (Fig. S6 in ESI†), the methyl group rotates on a 5 ps time scale about the methyl axis, but it also undergoes frequent two-site jumps about the χ2 angle on a time scale of about 1–2 ns. The presence of this additional dynamic mode causes the internal TCF to decay further over multiple nanoseconds. The result of this complex motion is that neither the LS2 nor the LS3 model fit the internal TCF correctly over the entire range. Surprisingly, LS2 fitting is able to provide a good estimate for Saxis2, but at the expense of a large overestimate for the time scale of fast dynamics. LS3 fitting, on the other hand, leads to a strong overestimation of the order parameter, while also still overestimating τf. To gauge how well the LS functions fit the internal TCFs for all methyl groups, we compared the agreement of the two curves by computing the following root mean square relative error (RMSRE),
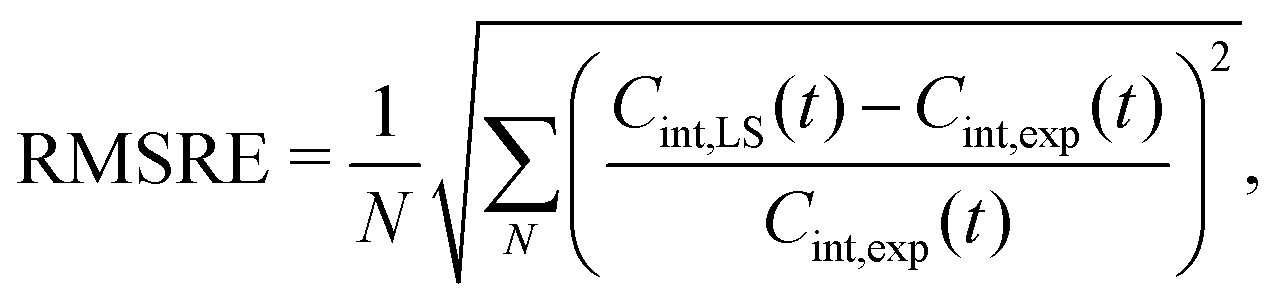 | (15) |
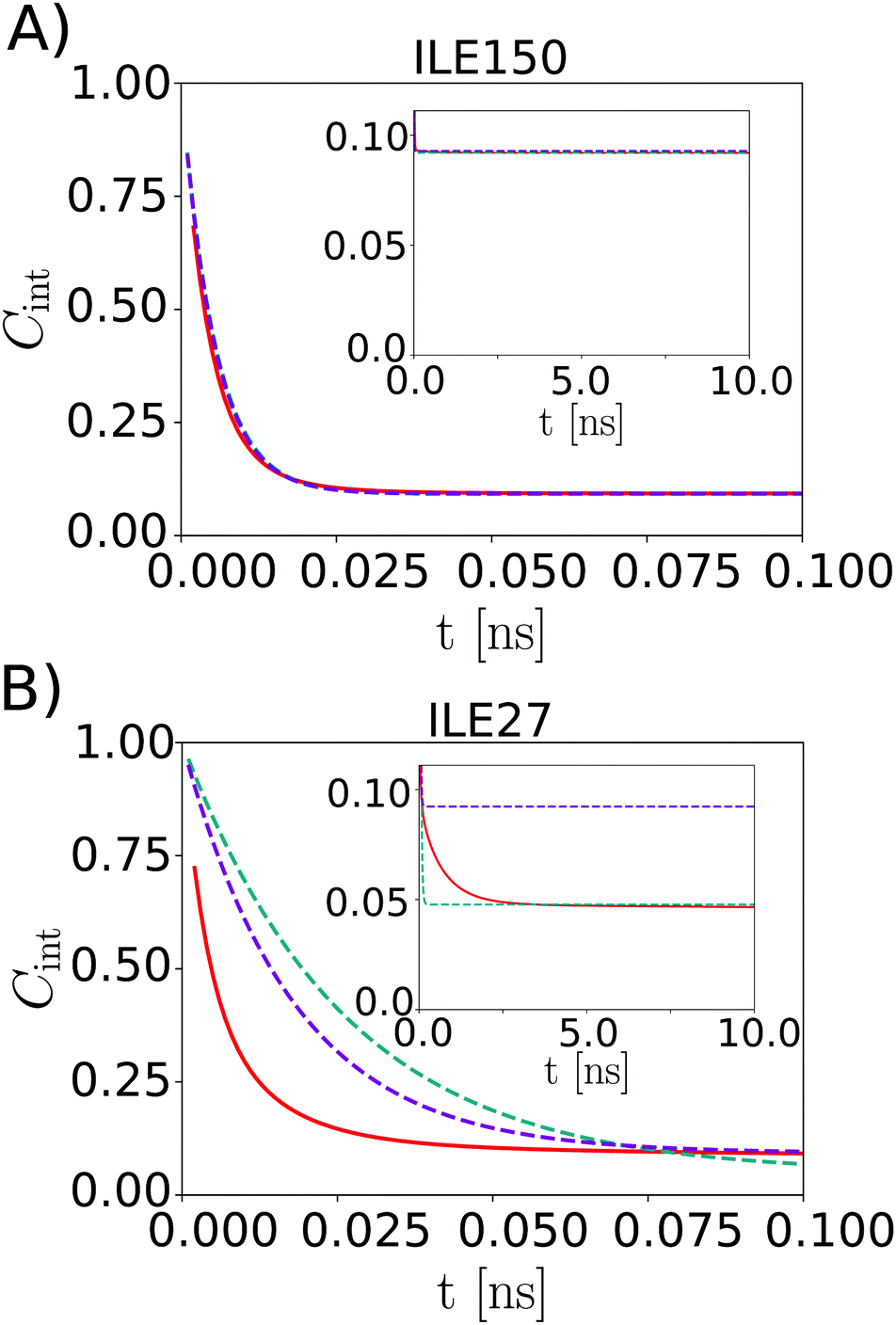 | ||
| Fig. 3 Internal TCF Cint,exp from eqn (16) (red) and from the LS2 (cyan) and LS3 model (magenta) for ILE150-Cδ,1 (A) and ILE27-Cδ,1 (B). | ||
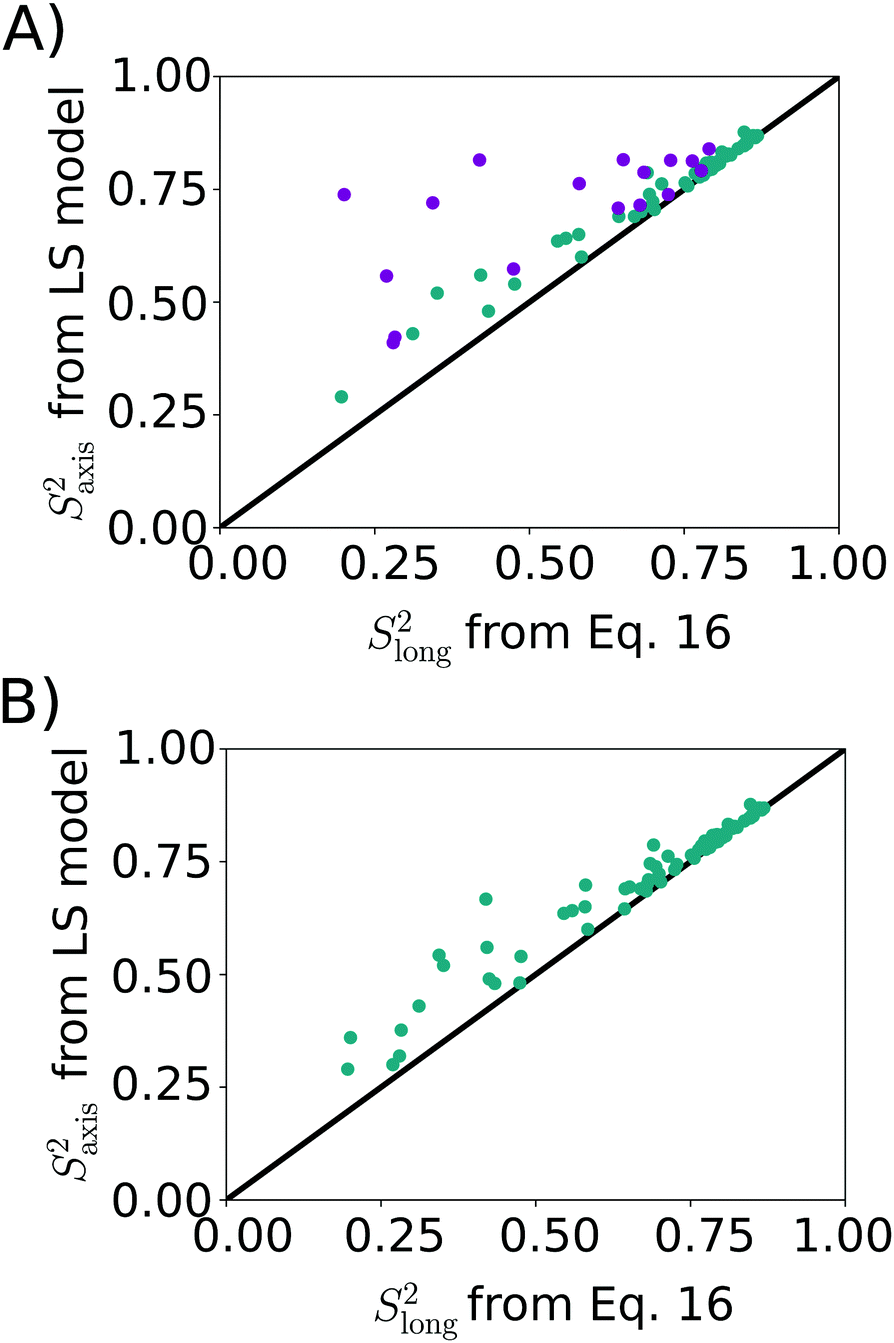 | ||
| Fig. 4 Correlation between methyl axis order parameter from LS model (LS2: cyan, LS3: magenta) and Slong2 (eqn (16)). (A) LS2/LS3 model picked via AIC. The RMSD between the two data sets is 0.13, RP = 0.77, RS = 0.83. (B) LS2 model used for all methyl groups. RMSD = 0.06, RP = 0.97, RS = 0.98. | ||
As a final analysis we considered what happens if LS2 is enforced on fitting the MD data. The result, shown in Fig. 4B, indicates that a rather good agreement is obtained, with an RMSD of 0.06 between the two datasets. Thus, somewhat counterintuitively, LS2 fitting approximates the amount of order surprisingly well, despite yielding poorer fits of the experimental data. This outcome underlines that the estimation of the amount of order from NMR spin relaxation data is challenging, as simple functional forms for the TCF are not able to fully capture the convoluted dynamics of mobile methyl-containing side-chains.
2.6 Relaxation rates
As the above section shows, Lipari–Szabo analysis may lead to systematic errors for side-chain dynamic parameters, and hence it is desirable to sidestep this kind of analysis, as the spectral density function can be extracted directly from computation. A first test would be if the MD simulations are able to accurately predict the nuclear spin relaxation rates. This has previously been complicated by unrealistic protein tumbling times in commonly used water force fields, but Section 2.3 indicates that this problem is solved with improved water models and longer simulations.Fig. 5 compares the relaxation rates R(Dz), R(3Dz2 − 2) and R(Dy) from the computed spectral densities to the relaxation rates that were directly measured by NMR relaxation, which does not require any (simplified) motional models. All rates are listed in Table S4, ESI.†
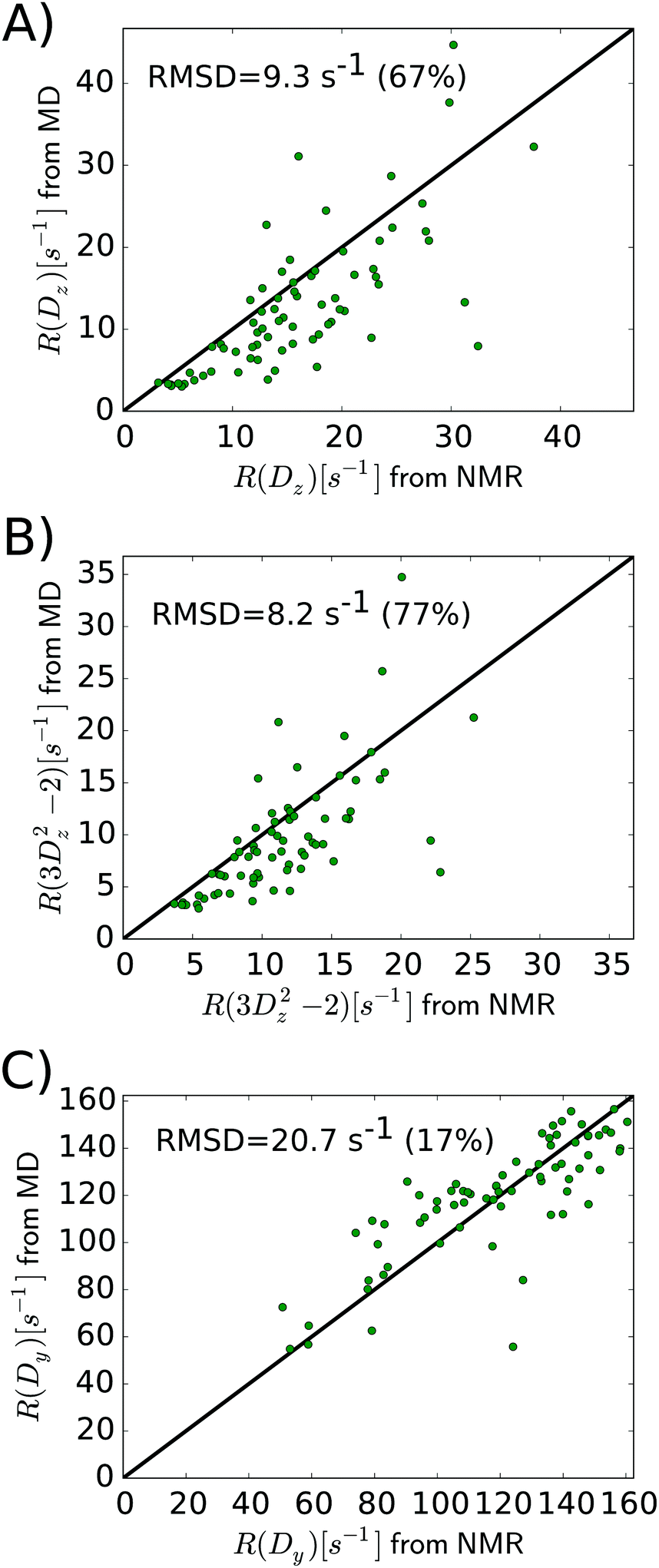 | ||
| Fig. 5 Relaxation rates R(Dz) (A), R(3Dz2 − 2) (B), and R(Dy) (C) from MD simulations are compared to the experimental values. The data point for ALA146 is outside of the plotted range (but included in the calculation of correlation coefficients and RMSD). | ||
In general, the relaxation rates obtained from the MD-based spectral density mapping approach match the experimental relaxation rates. For all three measured relaxation rates, the correlation coefficients between calculated rates from MD and NMR are higher than 0.7 (see Table 3). The relative RMSD of the relaxation rates R(Dz), R(3Dz2 − 2), and R(Dy) between MD and NMR are 0.67, 0.77 and 0.17, respectively. This shows that R(Dy), which contributes most to the spectral density at zero frequency, J(0), and therefore to the methyl order parameter, can be obtained with very high accuracy, whereas the rates R(Dz) and R(3Dz2 − 2), which contribute to the spectral densities at higher frequencies, deviate more strongly from experiment. This result might also influence the interpretation of the methyl motions. The slow motions, represented by J(0), contribute to the agreement between MD and NMR in Saxis2. Fast motions, e.g., on the time scale of τf, determine the spectral density at higher frequencies, which is dominated by R(Dz) and R(3Dz2 − 2). Fig. 6 shows that despite the higher relative RMSD in these two rates, the agreement between MD and NMR in τf is decent, too. However, the spread in the τf values in Fig. 6 is larger than that in Saxis2 (Fig. 2, bottom panel), suggesting that the MD simulations better recapitulate the amplitude of methyl dynamics than their precise time scales. This conclusion is in line with earlier work.45,56,61
| Relaxation rate | R P | R S | RMSD [s−1] | Relative RMSD |
|---|---|---|---|---|
| R(Dz) | 0.72 | 0.78 | 9.3 | 0.67 |
| R(3Dz2 − 2) | 0.73 | 0.77 | 8.2 | 0.77 |
| R(Dy) | 0.77 | 0.82 | 20.7 | 0.17 |
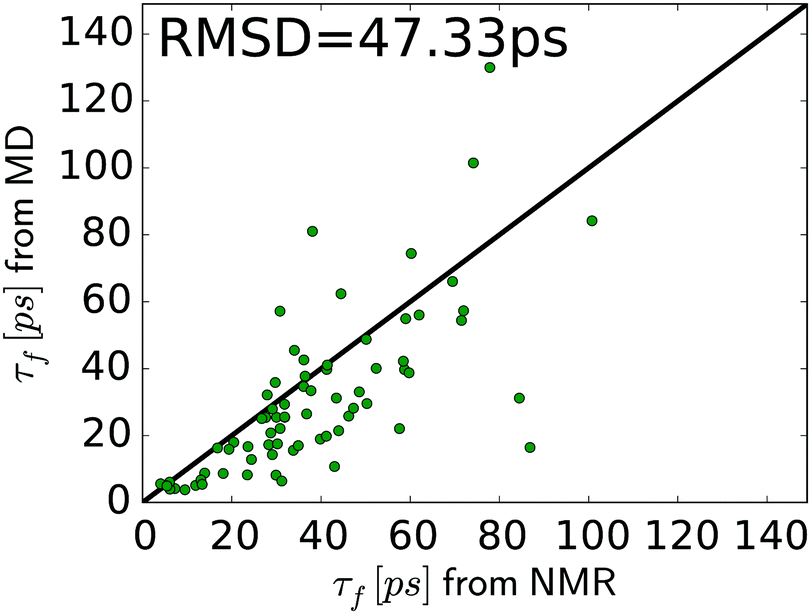 | ||
| Fig. 6 Correlation of τf for T4L methyl groups from NMR and MD. The RMSD is 47.3 ps, and the Pearson and Spearman correlation coefficients are RP = 0.67 and RS = 0.78, respectively. The value of τf for ALA146 is not shown (491 ps in MD, 129 ps in NMR). | ||
Notably, the relaxation rates of one particular methyl group, ALA146-Cβ, are much higher in MD than in NMR (by a factor of 2–3, see Table S4, ESI†). Similar deviations have been observed previously in MD simulations of staphylococcal nuclease,45 which yielded a broader range of methyl rotation correlation times than observed in NMR. Close inspection of our simulations revealed that the rotation of the ALA146 methyl group around its 3-fold symmetry axis is strongly slowed down. The ALA146 methyl group is in a sterically confined local microenvironment, being packed between the indole ring of TRP138 and the hydroxyphenyl ring of TYR139. Whether and if so, how exactly, contacts with these π-systems affect the dynamics of the ALA146 methyl group and to which extent the force field can capture these interactions are intriguing questions but beyond the scope of this work.
3 Conclusions
The present work combines NMR relaxation experiments and MD simulations of T4 lysozyme to investigate the side-chain dynamics, as encoded in the deuterium relaxation of methyl groups. It is shown that to obtain accurate results, it is imperative to take protein anisotropy into account in the analysis of both the experimental NMR relaxation data and the MD simulations. Near-quantitative agreement between MD and NMR for generalized order parameters of methyl groups Saxis2 and associated correlation times τf was achieved by properly accounting for the decay of the time correlation functions due to anisotropic protein tumbling in solution, which is adequately captured by the TIP4P/2005 water model. The employed MD-based spectral density mapping approach closely mimics the way the experimental NMR data is analyzed; directly fitting the raw TCFs, as obtained from the MD simulations, to an extended Lipari–Szabo model yielded almost comparably accurate results, with somewhat too high Saxis2. Both these approaches do not draw on any experimental NMR information or adjustable parameters that are specific to a particular system and hence enable true predictions from the MD simulations, with the only reservation that the statistical noise in the MD simulations render it challenging to pick the same Lipari–Szabo model (LS2 or LS3) as in NMR. In addition to Saxis2 and τf, which are derived from the data using Lipari–Szabo motional models, good agreement between experiments and simulations is also seen for the spectral densities and relaxation rates that are directly accessible to NMR deuterium relaxation without the need to invoke simplified motional models, which might be problematic for complex side-chain dynamics.Furthermore, we describe and critically discuss the LEU strong coupling effect, which can compromise deuterium NMR relaxation data for uniformly 13C-enriched protein samples if the Cδ and Cγ chemical shifts are close to each other. Here, MD simulations helped to identify experimental artifacts for many LEU residues in T4 lysozyme, e.g., spurious large differences between the Saxis2 order parameters of LEU prochiral methyl groups. Taking all these aspects into account results in overall very good agreement between MD simulations and NMR relaxation.
This work shows that MD simulations can now provide equally accurate predictions for the side-chain dynamics of rather large and anisotropic proteins such as T4 lysozyme as was previously possible for small isotropic proteins such as ubiquitin. This opens new ways for scrutinizing protein force fields against NMR relaxation data, fueling ongoing efforts to improve the accuracy of the potential energy functions used in the simulations. The improvements described in this study will aid MD simulations in guiding the interpretation of NMR relaxation experiments, as shown here for side-chain methyl dynamics. These developments could also be helpful for deriving improved analytical models that are able to more accurately capture the complex dynamics of side-chains than the highly simplified motional models that have been originally derived for backbone dynamics but are widely used today also for side-chains. Finally, given that close links have been established between NMR order parameters and conformational entropy,23,69–74 improvements in the accuracy of the parameters obtained from MD simulations might enable to revisit this close connection, e.g., to gauge and – if necessary – recalibrate an empirical entropy meter.
4 Methods
4.1 NMR sample preparation
Uniformly 15N-labeled T4 lysozyme (WT*, C54T/C97A) was produced in M9 media with 15NH4Cl as the sole nitrogen source. 50% 2H-enriched 15N/13C/2H-labeled samples were prepared from M9 media using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio of H2O/D2O with 15NH4Cl and 13C6-glucose as sole nitrogen and carbon sources, respectively. Purified protein samples were dialyzed into buffer containing 50 mM sodium phosphate and 25 mM NaCl at pH 5.5. NMR samples were prepared in 50 mM phosphate buffer with 25 mM NaCl, pH 5.5, 93% H2O/7% D2O (v/v).75
:1 volume ratio of H2O/D2O with 15NH4Cl and 13C6-glucose as sole nitrogen and carbon sources, respectively. Purified protein samples were dialyzed into buffer containing 50 mM sodium phosphate and 25 mM NaCl at pH 5.5. NMR samples were prepared in 50 mM phosphate buffer with 25 mM NaCl, pH 5.5, 93% H2O/7% D2O (v/v).75
4.2 NMR spectroscopy
All NMR spectra were collected on Bruker 500, 700, and 950 MHz spectrometers. Temperature was calibrated to 25 °C using a methanol d4 standard. Backbone 15N, 1H and side-chain methyl 13C, 1H resonance assignments of 15N/13C/2H-labeled WT* T4L were obtained primarily through analysis of 3D HNCACB, CC(CO)NH, and H(CCO)NH. Backbone dynamics were studied by measuring amide 15N T1, T2, and {1H}-15N NOE values at 500 and 700 MHz for WT* T4L (about 1.0 mM) through standard experiments.76 Side-chain 2H R(Dz), R(Dy) and R(3Dz2 − 2) relaxation rates were obtained at 950 MHz for 15N/13C/2H-labeled WT* TL4 (about 1.0 mM), as described previously.16,174.3 Analysis of backbone and side-chain dynamics from NMR relaxation
Backbone and side-chain relaxation rates were obtained by fitting the peak intensity decay as a function of the relaxation delay in the NMR relaxation experiments to a single exponential. The global rotational diffusion parameters were estimated with the programs QUADRIC (http://www.palmer.hs.columbia.edu) and ROTDIF.77–80 The WT* T4L X-ray structure (PDB 1L63) was used in QUADRIC, after centering with PDBINERTIA (http://www.palmer.hs.columbia.edu). The rigid residues were selected by excluding residues that are subject to fast internal motions and residues with conformational exchange. The former applies if the {1H}-15N NOE < 0.65, and the latter applies if [(T1,av − T1,i)/T1,av + (T2,av − T2,i)/T2,av] > standard deviation, where T1,av and T2,av are the average values, and T1,i and T2,i are the relaxation times for each individual residue i. These conditions identify the rigid residues with relaxation parameter values corresponding to the global rotational dynamics. The local correlation times were obtained through the R2 and R1 values of the selected rigid residues using R2R1_TM (http://www.palmer.hs.columbia.edu). The results indicate that WT* T4L is best approximated by a prolate (axially symmetric) diffusion tensor with a ratio D‖/D⊥ = 1.45. The program DYNAMICS was used to calculate SNH2 from the relaxation data through the model free approach, using a prolate diffusion tensor and a fixed chemical shift anisotropy of −172 ppm.81,82Saxis2, τf, and τeffc of methyl side-chains were obtained from the relaxation rates of 2H nuclei in CH22H isotopomers by nonlinear least-squares optimization.15 Methyl-specific τR,i values were calculated using the orientation of the C–Cmethyl vector with respect to the diffusion frame from τR,i = 1/6(Diso − P2(cosθ)(D‖ − D⊥)/3) with (Diso, D‖, D⊥) = (1.6, 2.0, 1.4) × 107 s−1, and the PDB file produced by QUADRIC where the molecular structure is aligned with the PAF of the diffusion tensor. Deuterium relaxation rates for each methyl group were used as input in model free approaches LS2 and LS3 using a quadrupolar coupling constant of 167 kHz. The selection of the LS2 (with fit parameters Saxis2, τf) or the LS3 model (Saxis2, τf, τeffc)15 was done by the Akaike information criterion (AIC) test.
4.4 MD simulations
All MD simulations were carried out with Gromacs version 5.0.6.83 The X-ray structure of the cysteine-free T4L SER44GLY mutant (PDB 107L)84 was used as starting structure for our simulations, after changing GLY at position 44 back to SER. Crystallographic water molecules were kept. The protein was centered in a periodic truncated dodecahedron box with a minimum distance between protein and the box boundary of 1.2 nm. The system was solvated with 12221 TIP3P57 or TIP4P/200558 water molecules. Sodium and chloride ions were added at a concentration of 0.15 mol l−1 to yield an overall neutral simulation system. Prior to MD simulation, the system was energy-minimized (5000 steps steepest descent) and equilibrated in the NPT ensemble for 200 ps with harmonic position restraints on all protein heavy atoms (with force constants of 1000 kJ mol−1 nm−2). The Amber ff99SB*-ILDN protein force field27,59,60 was used, including our recent adjustments of the methyl rotation barriers.61 To keep the temperature constant at 300 K, the velocity rescaling thermostat of Bussi and coworkers85 was applied, with coupling time constants of τT = 0.1 ps. Isotropic Parrinello–Rahman pressure coupling was used to maintain constant 1 bar pressure, with a 2 ps coupling time constant and a compressibility of 4.5 × 10−5 bar−1. The SETTLE86 and LINCS87 algorithms were applied to constrain the internal degrees of freedom of water molecules and the bonds in the protein, respectively, allowing for integrating the equations of motion with 2 fs time steps. Lennard-Jones (6,12) interactions were smoothly shifted to zero at a cut-off distance of 1.0 nm; this cut-off distance was also used for the short-range Coulomb interactions. Analytical dispersion corrections were added to energy and pressure to correct for the truncation of the Lennard-Jones interactions. Long-range Coulomb interactions were treated with the particle mesh Ewald (PME) method88 with a 0.12 nm grid spacing and cubic spline interpolation. Finally, ten 300 ns production MD simulations were carried out in the NPT ensemble using different random seeds for generating initial velocities from a Maxwell–Boltzmann distribution at 300 K. Coordinates were saved to disk every 1 ps.4.5 Calculation of order parameters, tumbling times, and relaxation rates from MD
Time correlation functions for the C–Cmethyl and for the three Cmethyl–Himethyl (i = 1, 2, 3) vector orientations for all methyl groups of all 10 trajectories are calculated up to a maximum lag-time of 150 ns. The TCFs of the three C–H vectors of the same methyl group were averaged. The total TCFs were calculated directly from the trajectory, i.e., without removing overall rotation. Internal TCFs were calculated after aligning all trajectory frames to the initial structure. The TCFs of the 10 individual simulations were averaged.The methyl order parameters according to the long-time limit approach of eqn (2) were calculated as the average value between 75 ns and 150 ns of the internal TCF of the C–Cmethyl bond vector orientation. For the Lipari–Szabo TCF fitting,44 the methyl order parameters and rotational tumbling times were determined by directly fitting the total TCFs of the N–H and C–Cmethyl vectors to eqn (10) up to a lag-time of 30 ns. We used QUADRIC with the initial structure of the MD simulation to convert the rotational tumbling from the laboratory frame to the diffusion frame of the protein using a prolate diffusion tensor. The tumbling times of the C–Cmethyl vectors were converted to the PAF of the diffusion tensor, obtained from the backbone N–H vectors, as described by Lee and Palmer.89 Based on the initial structure of our MD simulation, we calculated the second order Legendre polynomial P2(cosθ) of the cosine of the orientation of each C–Cmethyl vector with respect to the principal axis of the diffusion tensor. The diffusion tensor obtained from the N–H analysis was diagonalized and its principal values Dxx, Dyy, and Dzz were used to calculate the rotational diffusion constant in the isotropic model from the trace, Diso = Tr/3, and the methyl-specific individual diffusion constants Di = Diso − P2(cosθ)(Dzz − Dyy)/3. From this, we calculated the methyl-specific tumbling times τR,i = 1/(6Di). A prolate model was used for T4L with Dzz = D‖ and Dxx = Dyy = D⊥ as the principal components of the long and short axes of the diffusion tensor, respectively. We obtained D‖/D⊥ = 1.36 using the initial structure of the simulations (i.e., the energy-minimized X-ray structure); calculating this ratio using 3000 snapshots from the MD simulations yielded values in the range [1.2–1.6], with the same average of 1.36. As an additional check, we repeated our analyses using a snapshot after 300 ns of MD simulation (instead of the initial structure, i.e., the energy-minimized X-ray structure) for projecting the C–Cmethyl vectors onto the PAF of the diffusion tensor. In this snapshot, ca. 15 methyl groups adopted a different rotamer state than in the X-ray structure. However, this did not affect the methyl order parameters, with only one single methyl group showing differences larger than 0.015 (Saxis2 of LEU66-Cδ,2 changes from 1.04 to 0.98).
In the spectral density approach, the internal TCFs of the Cmethyl–Hmethyl vectors (i.e., after removing overall tumbling) were fitted with six exponentials plus an offset,90
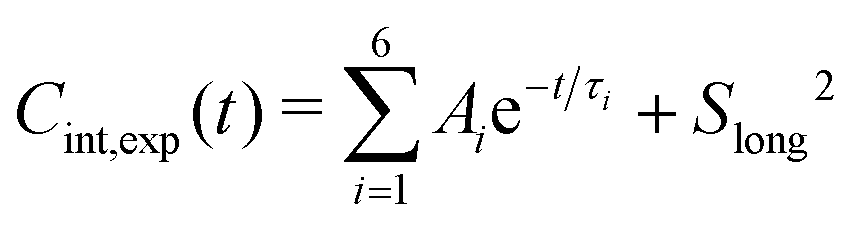 | (16) |
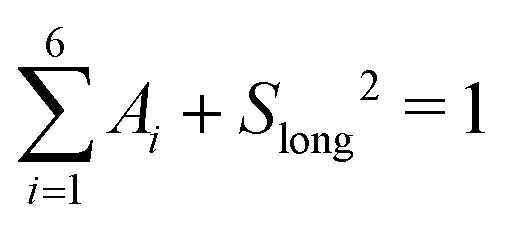 , 0 ≤ Ai ≤ 1, 0 ≤ Slong2 ≤ 1, 0 ≤ τ1 ≤ 10 ps, 0 ≤ τ2 ≤ 50 ps, 0 ≤ τ3 ≤ 200 ps, 0 ≤ τ4 ≤ 500 ps, 0 ≤ τ5 ≤ 1000 ps and τ6 ≥ 0.
, 0 ≤ Ai ≤ 1, 0 ≤ Slong2 ≤ 1, 0 ≤ τ1 ≤ 10 ps, 0 ≤ τ2 ≤ 50 ps, 0 ≤ τ3 ≤ 200 ps, 0 ≤ τ4 ≤ 500 ps, 0 ≤ τ5 ≤ 1000 ps and τ6 ≥ 0.
The resulting internal TCF was multiplied by a single-exponential15 using the anisotropic tumbling time τR,i of the corresponding methyl group
| C(t) = Cint,exp(t)e−t/τR,i | (17) |
The methyl group-specific tumbling times were extracted based on the diffusion tensor calculated from the rotational tumbling times of the backbone N–H vectors, as obtained by the Lipari–Szabo TCF fitting approach, and the orientation of the C–Cmethyl vector with respect to the PAF of this diffusion tensor, as described above. These τR,i values were also used for the direct TCF fits with the LS2 model (eqn (5)). The total TCF was converted into a spectral density
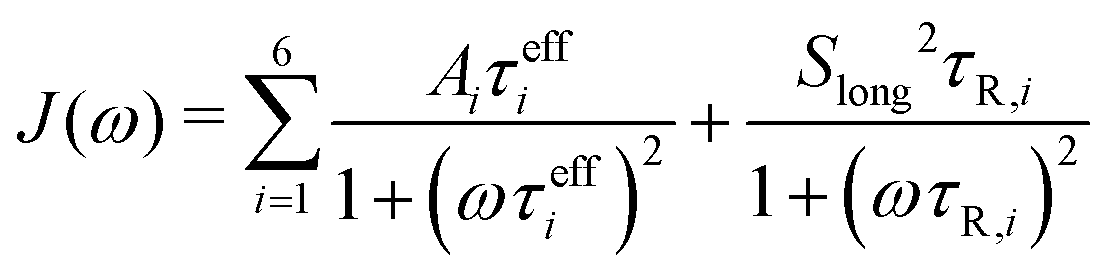 | (18) |
(1) LS2 grid search within Saxis;LS22 ∈ [0, 0.01, …, 2Slong2] and τf;LS2 ∈ [0 ps, 1 ps, …, 2τefffit]. Slong2 and 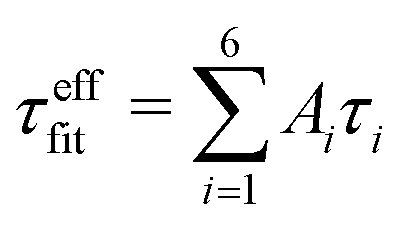 were used as initial values for the fitting parameters.
were used as initial values for the fitting parameters.
(2) The results from the grid search were used as starting points for a direct, i.e., gradient-based fit of Saxis;LS22 and τf;LS2 to yield the final LS2 parameter pair Saxis;LS22; τf;LS2.
(3) LS3 grid search within Saxis;LS32 ∈ [0, 0.01, …, 2Saxis;LS22], τf;LS3 ∈ [τf;LS2 − 50 ps, τf;LS2 − 49 ps, …, τf;LS2 + 50 ps] and τeffc ∈ [τR,i − 5 ns, τR,i − 4.9 ns, …, τR,i + 5 ns]. The LS2 results (Saxis;LS22; τf;LS2) were used to initiate Sf;LS32 and τf;LS3. Furthermore, the methyl-specific τR,i's from eqn (17) were used as starting points for fitting τeffc.
(4) Direct fit of Saxis;LS32, τf;LS3, and τeffc using the results from the previous grid search as starting points, yielding the final LS3 parameter triple (Saxis;LS32; τf;LS3; τeffc).
At every step of the grid search or gradient-based minimization, Jmodel(0), Jmodel(ωD), and Jmodel(2ωD) were determined from eqn (6) or eqn (11), and Rmodel(Dz), Rmodel(3Dz2 − 2), Rmodel(Dy), and χ2 were calculated from
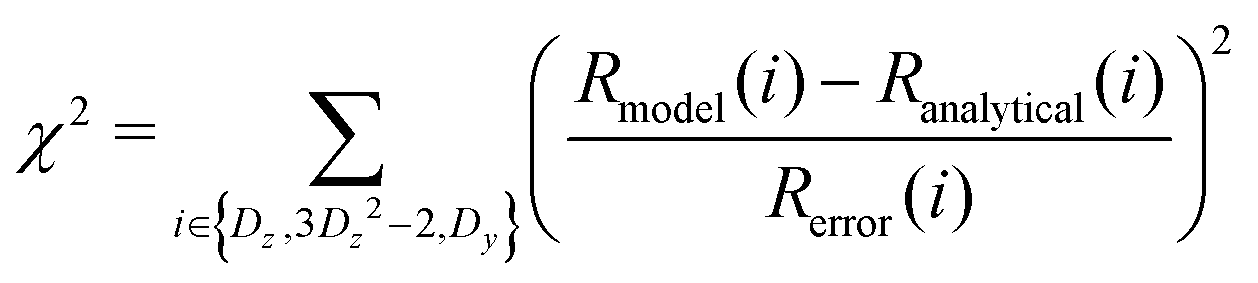 | (19) |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge insightful discussions with Kresten Lindorff-Larsen (University of Copenhagen) and Nikolai Skrynnikov (Purdue University). This work was performed using the NMR spectrometers at the Danish Center for Ultra-High Field NMR Spectroscopy. The Deutsche Forschungsgemeinschaft (DFG) supported this work through Cluster of Excellence RESOLV (EXC 1069) and Emmy-Noether grant SCHA 1574/3-1 to L. S. The Steinbuch Centre for Computing (SCC), Karlsruhe/Germany, is acknowledged for providing computational resources.References
- K. Henzler-Wildman and D. Kern, Nature, 2007, 450, 964–972 CrossRef PubMed.
- H. Frauenfelder, S. Sligar and P. Wolynes, Science, 1991, 254, 1598–1603 CrossRef PubMed.
- J. W. Peng, J. Phys. Chem. Lett., 2012, 3, 1039–1051 CrossRef PubMed.
- P. J. Sapienza and A. L. Lee, Curr. Opin. Pharmacol., 2010, 10, 723–730 CrossRef PubMed.
- A. Mittermaier and L. E. Kay, Science, 2006, 312, 224–228 CrossRef PubMed.
- A. G. Palmer, Chem. Rev., 2004, 104, 3623–3640 CrossRef PubMed.
- R. Brüschweiler, Curr. Opin. Struct. Biol., 2003, 13, 175–183 CrossRef.
- D. A. Case, Acc. Chem. Res., 2002, 35, 325–331 CrossRef PubMed.
- K. Lindorff-Larsen, R. B. Best, M. A. DePristo, C. M. Dobson and M. Vendruscolo, Nature, 2005, 433, 128–132 CrossRef PubMed.
- N. Salvi, A. Abyzov and M. Blackledge, J. Phys. Chem. Lett., 2016, 7, 2483–2489 CrossRef PubMed.
- G. Lipari and A. Szabo, J. Am. Chem. Soc., 1982, 104, 4546–4559 CrossRef.
- G. Lipari and A. Szabo, J. Am. Chem. Soc., 1982, 104, 4559–4570 CrossRef.
- B. Halle and H. Wennerström, J. Chem. Phys., 1981, 75, 1928–1943 CrossRef.
- G. M. Clore, A. Szabo, A. Bax, L. E. Kay, P. C. Driscoll and A. M. Gronenborn, J. Am. Chem. Soc., 1990, 112, 4989–4991 CrossRef.
- N. R. Skrynnikov, O. Millet and L. E. Kay, J. Am. Chem. Soc., 2002, 124, 6449–6460 CrossRef PubMed.
- D. R. Muhandiram, T. Yamazaki, B. D. Sykes and L. E. Kay, J. Am. Chem. Soc., 1995, 117, 11536–11544 CrossRef.
- O. Millet, D. R. Muhandiram, N. R. Skrynnikov and L. E. Kay, J. Am. Chem. Soc., 2002, 124, 6439–6448 CrossRef PubMed.
- D. C. Chatfield and S. E. Wong, J. Phys. Chem. B, 2000, 104, 11342–11348 CrossRef.
- D. Yang, A. Mittermaier, Y. K. Mok and L. E. Kay, J. Mol. Biol., 1998, 276, 939–954 CrossRef PubMed.
- J. J. Chou, D. A. Case and A. Bax, J. Am. Chem. Soc., 2003, 125, 8959–8966 CrossRef PubMed.
- R. B. Best, J. Clarke and M. Karplus, J. Am. Chem. Soc., 2004, 126, 7734–7735 CrossRef PubMed.
- H. Hu, J. Hermans and A. L. Lee, J. Biomol. NMR, 2005, 32, 151–162 CrossRef PubMed.
- V. Kasinath, K. A. Sharp and A. J. Wand, J. Am. Chem. Soc., 2013, 135, 15092–15100 CrossRef PubMed.
- N. Foloppe and A. D. MacKerell, J. Comput. Chem., 2000, 21, 86–104 CrossRef.
- G. R. Bowman, J. Comput. Chem., 2016, 37, 558–566 CrossRef PubMed.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang and P. Kollman, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 Search PubMed.
- J. J. Prompers and R. Brüschweiler, J. Am. Chem. Soc., 2002, 124, 4522–4534 CrossRef PubMed.
- S. Genheden, J. Mol. Graphics, 2017, 71, 80–87 CrossRef PubMed.
- D. E. Woessner, J. Chem. Phys., 1962, 37, 647–654 CrossRef.
- H. Shimizu, J. Chem. Phys., 1962, 37, 765–778 CrossRef.
- P. Luginbühl, K. V. Pervushin, H. Iwai and K. Wüthrich, Biochemistry, 1997, 36, 7305–7312 CrossRef PubMed.
- V. Wong and D. A. Case, J. Phys. Chem. B, 2008, 112, 6013–6024 CrossRef PubMed.
- J. S. Anderson, G. Hernández and D. M. LeMaster, J. Chem. Theory Comput., 2017, 13, 3276–3289 CrossRef PubMed.
- P.-c. Chen, M. Hologne and O. Walker, J. Phys. Chem. B, 2017, 121, 1812–1823 CrossRef PubMed.
- M. Linke, J. Köfinger and G. Hummer, J. Phys. Chem. B, 2018, 122, 5630–5639 CrossRef PubMed.
- O. H. S. Ollila, H. A. Heikkinen and H. Iwai, J. Phys. Chem. B, 2018, 122, 6559–6569 CrossRef PubMed.
- Y. E. Ryabov, C. Geraghty, A. Varshney and D. Fushman, J. Am. Chem. Soc., 2006, 128, 15432–15444 CrossRef PubMed.
- R. Brüschweiler, X. Liao and P. E. Wright, Science, 1995, 268, 886–889 CrossRef.
- G. Barbato, M. Ikura, L. E. Kay, R. W. Pastor and A. Bax, Biochemistry, 1992, 31, 5269–5278 CrossRef PubMed.
- J. M. Schurr, H. P. Babcock and B. S. Fujimoto, J. Magn. Reson., 1994, 105, 211–224 CrossRef PubMed.
- O. Millet, A. Mittermaier, D. Baker and L. E. Kay, J. Mol. Biol., 2003, 329, 551–563 CrossRef PubMed.
- R. Ishima, A. P. Petkova, J. M. Louis and D. A. Torchia, J. Am. Chem. Soc., 2001, 123, 6164–6171 CrossRef PubMed.
- P. Maragakis, K. Lindorff-Larsen, M. P. Eastwood, R. O. Dror, J. L. Klepeis, I. T. Arkin, M. Ø. Jensen, H. Xu, N. Trbovic, R. A. Friesner, A. G. Palmer and D. E. Shaw, J. Phys. Chem. B, 2008, 112, 6155–6158 CrossRef PubMed.
- D. C. Chatfield, A. Szabo and B. R. Brooks, J. Am. Chem. Soc., 1998, 120, 5301–5311 CrossRef.
- R. B. Best, J. Clarke and M. Karplus, J. Mol. Biol., 2005, 349, 185–203 CrossRef PubMed.
- J. Huang and A. D. MacKerell, J. Comput. Chem., 2013, 34, 2135–2145 CrossRef PubMed.
- T. Rajitha Rajeshwar, J. C. Smith and M. Krishnan, J. Am. Chem. Soc., 2014, 136, 8590–8605 CrossRef PubMed.
- K. A. Sharp, E. OBrien, V. Kasinath and A. J. Wand, Proteins, 2015, 83, 922–930 CrossRef PubMed.
- E. S. OBrien, A. J. Wand and K. A. Sharp, Protein Sci., 2016, 25, 1156–1160 CrossRef PubMed.
- F. A. A. Mulder, B. Hon, A. Mittermaier, F. W. Dahlquist and L. E. Kay, J. Am. Chem. Soc., 2002, 124, 1443–1451 CrossRef PubMed.
- R. L. Dunbrack and F. E. Cohen, Protein Sci., 1997, 6, 1661–1681 CrossRef PubMed.
- F. A. A. Mulder, ChemBioChem, 2009, 10, 1477–1479 CrossRef PubMed.
- V. Agarwal, A. Diehl, N. R. Skrynnikov and B. Reif, J. Am. Chem. Soc., 2006, 128, 12620–12621 CrossRef PubMed.
- A. L. Lee, J. L. Urbauer and J. A. Wand, J. Biomol. NMR, 1997, 9, 437–440 CrossRef PubMed.
- Y. Xue, M. S. Pavlova, Y. E. Ryabov, B. Reif and N. R. Skrynnikov, J. Am. Chem. Soc., 2007, 129, 6827–6838 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef.
- J. L. F. Abascal and C. Vega, J. Chem. Phys., 2005, 123, 234505 CrossRef PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef PubMed.
- R. B. Best and G. Hummer, J. Phys. Chem. B, 2009, 113, 9004–9015 CrossRef PubMed.
- F. Hoffmann, F. A. A. Mulder and L. V. Schäfer, J. Phys. Chem. B, 2018, 122, 5038–5048 CrossRef PubMed.
- T. Zeiske, K. A. Stafford, R. A. Friesner and A. G. Palmer, Proteins, 2013, 81, 499–509 CrossRef PubMed.
- J. S. Anderson and D. M. LeMaster, Biophys. Chem., 2012, 168–169, 28–39 CrossRef PubMed.
- I.-C. Yeh and G. Hummer, J. Phys. Chem. B, 2004, 108, 15873–15879 CrossRef.
- S. Tazi, A. Botan, M. Salanne, V. Marry, P. Turq and B. Rotenberg, J. Phys.: Condens. Matter, 2012, 24, 284117 CrossRef PubMed.
- M. Linke, J. Köfinger and G. Hummer, J. Phys. Chem. Lett., 2018, 9, 2874–2878 CrossRef PubMed.
- D. Long, D.-W. Li, K. F. A. Walter, C. Griesinger and R. Brüschweiler, Biophys. J., 2011, 101, 910–915 CrossRef PubMed.
- A. Mittermaier, L. E. Kay and J. D. Forman-Kay, J. Biomol. NMR, 1999, 13, 181–185 CrossRef PubMed.
- M. Akke, R. Brüschweiler and A. G. Palmer, J. Am. Chem. Soc., 1993, 115, 9832–9833 CrossRef.
- D. Yang and L. E. Kay, J. Mol. Biol., 1996, 263, 369–382 CrossRef PubMed.
- Z. Li, S. Raychaudhuri and A. J. Wand, Protein Sci., 1996, 5, 2647–2650 CrossRef PubMed.
- D. M. LeMaster, J. Am. Chem. Soc., 1999, 121, 1726–1742 CrossRef.
- K. K. Frederick, M. S. Marlow, K. G. Valentine and A. J. Wand, Nature, 2007, 448, 325–329 CrossRef PubMed.
- C. M. Petit, J. Zhang, P. J. Sapienza, E. J. Fuentes and A. L. Lee, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18249–18254 CrossRef PubMed.
- F. A. A. Mulder, B. Hon, D. R. Muhandiram, F. W. Dahlquist and L. E. Kay, Biochemistry, 2000, 39, 12614–12622 CrossRef PubMed.
- N. A. Farrow, R. Muhandiram, A. U. Singer, S. M. Pascal, C. M. Kay, G. Gish, S. E. Shoelson, T. Pawson, J. D. Forman-Kay and L. E. Kay, Biochemistry, 1994, 33, 5984–6003 CrossRef PubMed.
- D. Fushman, R. Xu and D. Cowburn, Biochemistry, 1999, 38, 10225–10230 CrossRef PubMed.
- R. Varadan, O. Walker, C. Pickart and D. Fushman, J. Mol. Biol., 2002, 324, 637–647 CrossRef PubMed.
- O. Walker, R. Varadan and D. Fushman, J. Magn. Reson., 2004, 168, 336–345 CrossRef PubMed.
- D. Fushman, R. Varadan, M. Assfalg and O. Walker, Prog. Nucl. Magn. Reson. Spectrosc., 2004, 44, 189–214 CrossRef.
- D. Fushman, S. Cahill and D. Cowburn, J. Mol. Biol., 1997, 266, 173–194 CrossRef PubMed.
- J. B. Hall and D. Fushman, J. Biomol. NMR, 2003, 27, 261–275 CrossRef PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- M. Blaber, X. J. Zhang and B. W. Matthews, Science, 1993, 260, 1637–1640 CrossRef PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef.
- L. K. Lee, M. Rance, W. J. Chazin and A. G. Palmer, J. Biomol. NMR, 1997, 9, 287–298 CrossRef PubMed.
- T. Bremi, R. Brüschweiler and R. R. Ernst, J. Am. Chem. Soc., 1997, 119, 4272–4284 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp03915a |
| ‡ Including a hydrodynamic correction to account for box-size dependence66 yields 9.30 ns. |
| § Except from the choice of the LS2 or LS3 model, see below. |
| This journal is © the Owner Societies 2018 |