
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fragmentation of a dioxolanyl radical via nonstatistical reaction dynamics: characterization of the vinyloxy radical by ns time-resolved laser flash photolysis†
Götz
Bucher‡
 *a,
Mukul
Lal
b,
Anup
Rana
b and
Michael
Schmittel
*b
*a,
Mukul
Lal
b,
Anup
Rana
b and
Michael
Schmittel
*b
aLehrstuhl für Organische Chemie II, Ruhr-Universität Bochum, Universitätsstr. 150, 44801 Bochum, Germany
bFB 8 (Chemie-Biologie), Organische Chemie I, Universität Siegen, Adolf-Reichwein-Str. 2, 57068 Siegen, Germany. E-mail: schmittel@chemie.uni-siegen.de
First published on 18th July 2018
Abstract
The photochemistry of two Barton esters, one derived from a dioxolane carboxylic acid and the other from pivalic acid, was investigated by product analysis and nanosecond laser flash photolysis (LFP). As expected, photolysis of the pivalate ester resulted in formation of the pyridine-2-thiyl and the t-butyl radical. Photolysis of the Barton ester of 2,2-dimethyl-1,3-dioxolane-4-carboxylic acid, on the other hand, revealed a complex multi-step fragmentation. In addition to the pyridine-2-thiyl and dioxolanyl radical, we gained evidence for the formation of the vinyloxy radical, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHO˙. The latter was identified in the LFP by its π-complexes with benzene and diphenylether, its rapid quenching by electron-rich arenes and tri-n-butyl tin hydride, and its oxidative power in presence of trifluoroacetic acid as demonstrated by the oxidation of ferrocene to ferrocenium. Formation of CH2CHO˙ can be rationalized via fragmentation of the dioxolanyl radical. As the calculated barriers are too high for the reaction sequence to occur on the LFP time scale, we investigated the fragmentation of the photoexcited Barton ester via Born–Oppenheimer molecular dynamics simulations. In one trajectory, we could observe all reaction steps including ring opening of the dioxolanyl radical, suggesting that the excess energy gained in the ester cleavage and decarboxylation may lead to fragmentation of the hot dioxolanyl radical.
CHO˙. The latter was identified in the LFP by its π-complexes with benzene and diphenylether, its rapid quenching by electron-rich arenes and tri-n-butyl tin hydride, and its oxidative power in presence of trifluoroacetic acid as demonstrated by the oxidation of ferrocene to ferrocenium. Formation of CH2CHO˙ can be rationalized via fragmentation of the dioxolanyl radical. As the calculated barriers are too high for the reaction sequence to occur on the LFP time scale, we investigated the fragmentation of the photoexcited Barton ester via Born–Oppenheimer molecular dynamics simulations. In one trajectory, we could observe all reaction steps including ring opening of the dioxolanyl radical, suggesting that the excess energy gained in the ester cleavage and decarboxylation may lead to fragmentation of the hot dioxolanyl radical.
Introduction
Enol and enol ether radical cations1 are of considerable significance both to chemical2,3 and biological4–7 systems. Due to their relevance, several studies directed at elucidating the reactivity of enol ether radical cations via direct kinetic methods such as laser flash photolysis (LFP) have been published.8–11 The reactivity of radical cations of free enols, on the other hand, remains to be explored, as only few direct kinetic studies are known.12,13 As demonstrated by deuterium kinetic isotope effects, enol radical cations are acidic and thus tend to undergo a facile deprotonation yielding the corresponding vinyloxy radical.13The present work was undertaken with the aim to establish an alternative photochemical route to enol radical cations. It is well known that acetals are acid-sensitive functionalities.14 We therefore reasoned that the acid-catalyzed ring opening of a radical derived from a cyclic acetonide should represent a potential route to obtain the radical cation of a free enol and/or its conjugated base, the vinyloxy radical. Our approach aimed at the generation of ethenol+˙ (1), the radical cation of the simplest enol. Explicitly, the intention was to generate 1 by acid-induced fragmentation of the dioxolanyl radical 3, which in turn should be readily available from decarboxylation of the carbonyloxy radical 4 (Scheme 1). The latter species was intended to be generated by photolysis of the N-hydroxypyridine-2-thione ester (“Barton-ester”) 7. As side products, bis-(2-pyridyl)disulfide (6) and acetone (2) are expected to be formed.
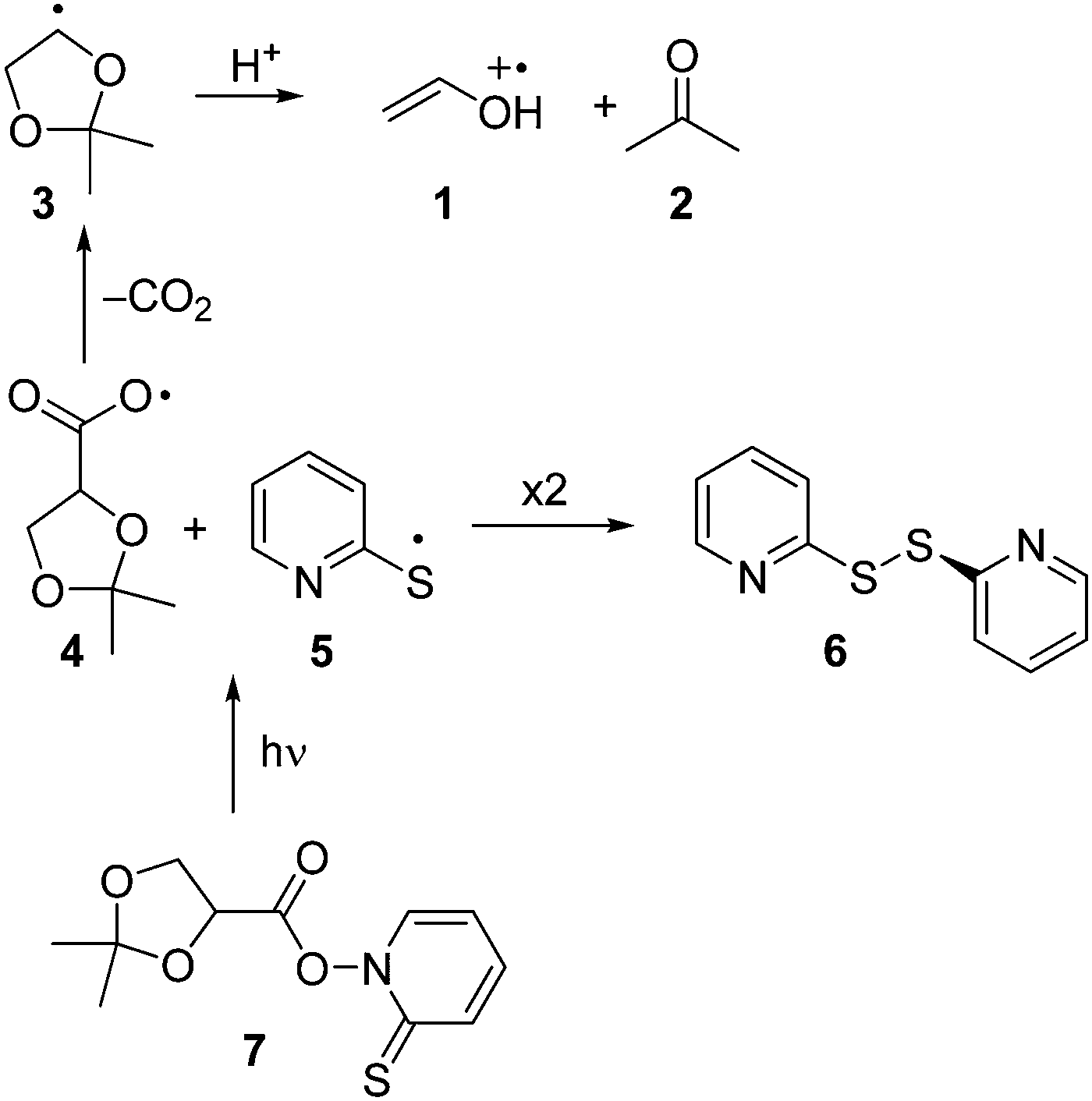 | ||
| Scheme 1 Possible reaction pathways upon photolysis of 7 in acidic medium. | ||
For the present study, we employed product analysis, UV-vis spectroscopic monitoring during steady-state irradiation, and laser flash photolysis (LFP) with UV-vis detection. In addition, we applied density functional theory (DFT) including Born–Oppenheimer molecular dynamic (BOMD) computations to elucidate the reaction pathways occurring in the photolysis. For comparison, we investigated the photochemical decomposition of the t-butyl derivative 8, which should not undergo fragmentation beyond formation of the t-butyl radical (10) (Scheme 2).
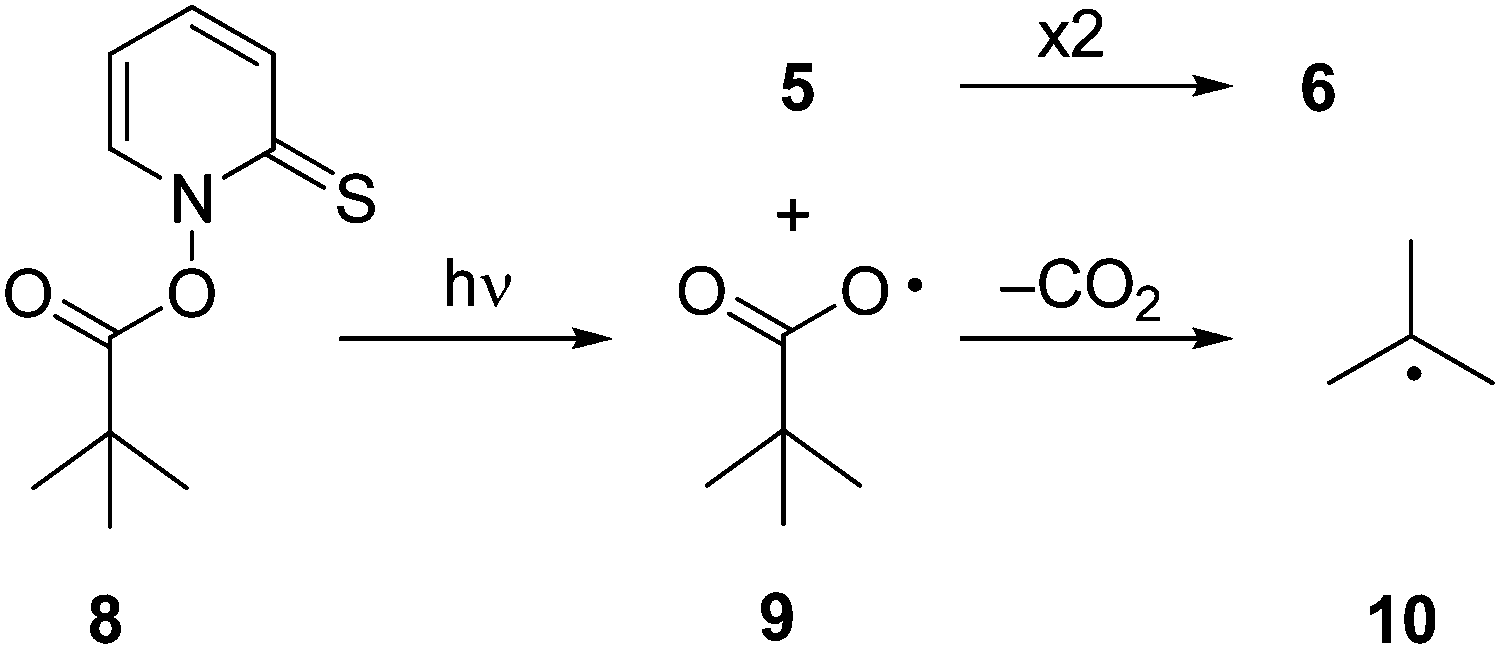 | ||
| Scheme 2 Reactions in the photolytic decomposition of 8. | ||
Results and discussion
Synthesis of the Barton esters
Barton ester 7 was prepared from mannitol 11, which was first protected as bis-acetonide using acetone in the presence of fused zinc chloride. The resulting bis-acetal 12 was then cleaved by sodium metaperiodate furnishing aldehyde 13. The latter was further oxidized with KOH/KMnO4 to afford potassium isopropylideneglycerate (14), which was then converted to acid chloride 15 by treatment with oxalyl chloride in the presence of a catalytic amount of pyridine. Finally, the Barton ester 7 was obtained by reacting 15 with the sodium salt of N-2-thiopyridine oxide (Scheme 3). Similarly, Barton ester 8 was synthesized from pivalyl chloride (16) and the sodium salt of N-2-thiopyridine oxide (for details, see ESI†).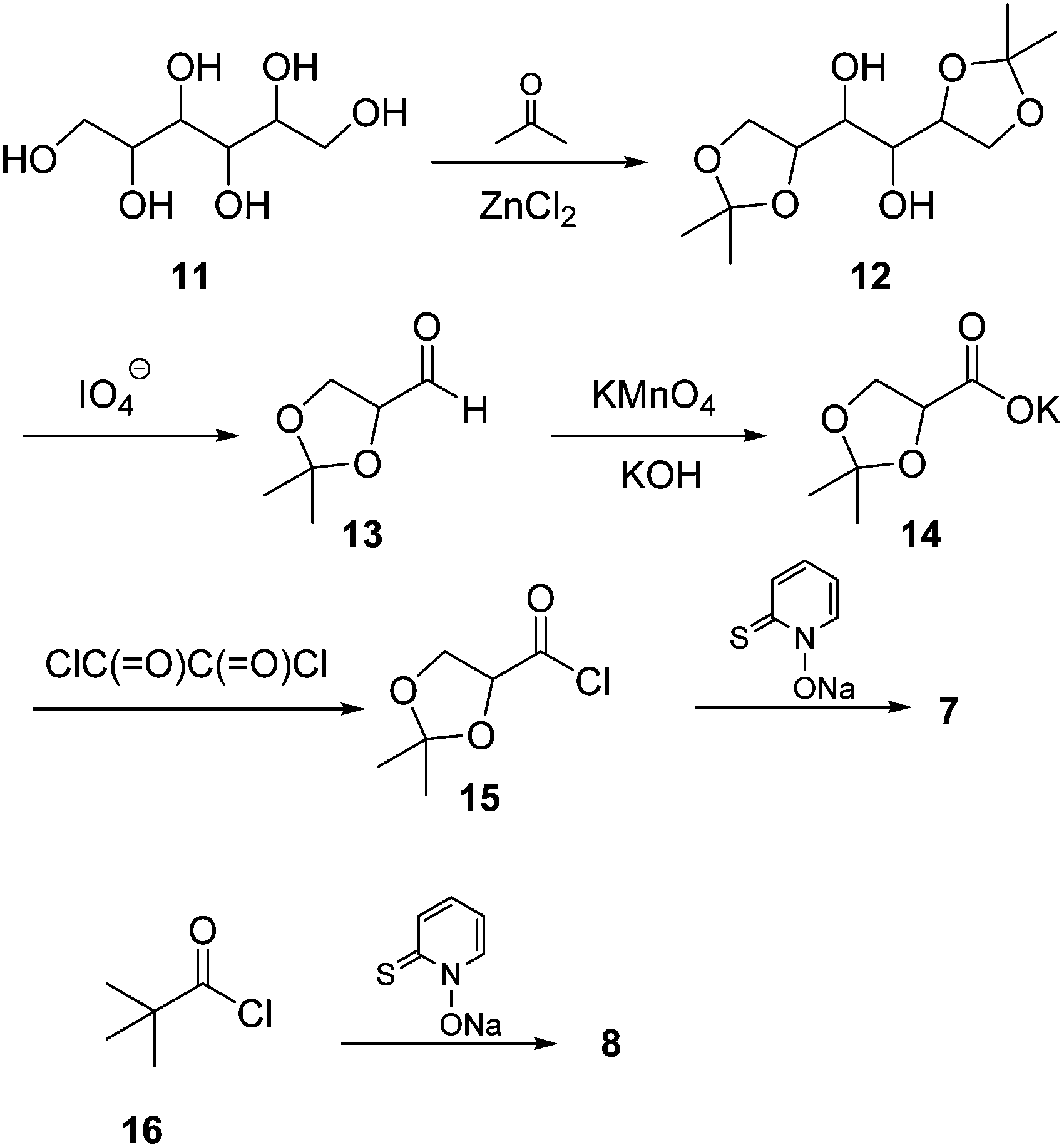 | ||
| Scheme 3 Preparation of Barton esters 7 and 8. | ||
Stability of 7 towards TFA in benzene solution and complex formation with TFA
The acetal unit in 7 is an acid-sensitive functionality that may well be affected by acid, in particular in the presence of trace amounts of water serving as nucleophile.14 We therefore studied the stability of 7 at equimolar ([7] = [TFA] = 0.1 mM) and higher concentrations in dry benzene, which was our choice of solvent for the photolytic experiments (vide infra). After adding an equimolar quantity of TFA to 7, a very small decrease in absorbance was observed at λmax = 360 nm (see ESI†). Further increase in acid concentration not only induced a decrease in the absorbance at λmax = 360 nm but an additional 12 nm hypsochromic shift to λmax = 348 nm. Nevertheless, the original spectrum of the acid-free 7 was retained after addition of a slight excess of pyridine as base (dilution factor taken into account) to the acidified solution clearly suggesting a fully reversible protonation of the chromophore. TFA addition up to 5 equiv. did not show any substantial decay over time (the absorbance at 360 nm was 0.646 after 2 min and it was 0.645 after 4 h).The UV-vis spectral changes in the acid addition experiments indicate that protonation of 7 predominantly occurs at the chromophore of the molecule. At low concentration of TFA, the most likely protonation should afford the cation 17 (Scheme 4).
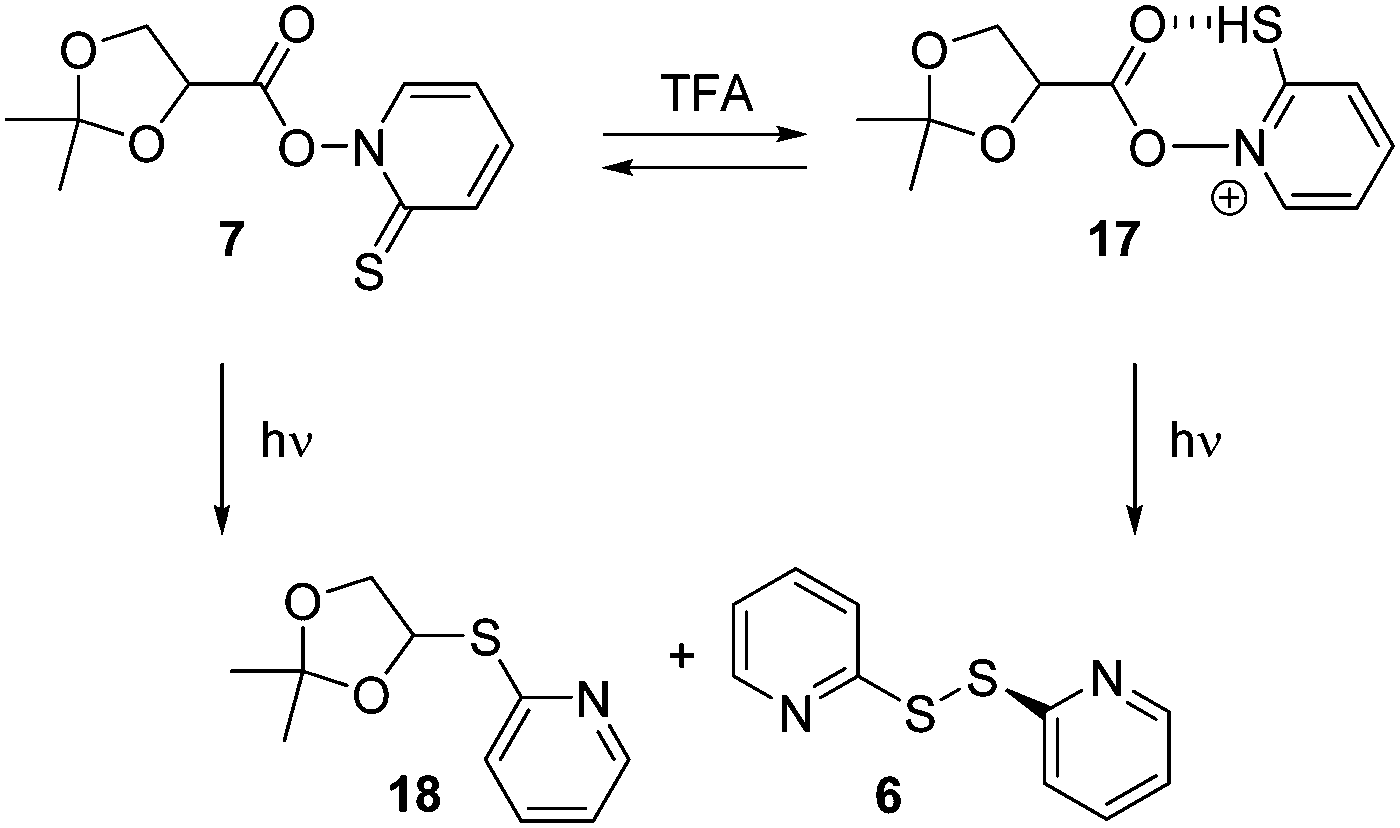 | ||
| Scheme 4 Protonation and photolysis of 7. | ||
Photolysis in absence and presence of TFA
The photolysis of 7 in benzene (c = 0.2 mM) in the absence of acid using a 300 W UV lamp was followed by both UV-vis and NMR spectroscopy. The band at 360 nm, characteristic for Barton ester 7, disappeared completely in 60 s. Photolysis of 7 (0.49 mM) in the presence of 1 equiv. of trifluoroacetic acid (TFA) equally showed a rapid decrease of the absorbance of 7 at 360 nm. Importantly, no absorbing species was detectable in the spectrum up to 800 nm (Fig. 1). | ||
| Fig. 1 UV-vis changes observed upon photolysis of Barton ester 7 (490 μM), (a) in dry benzene containing TFA (490 μM) and (b) in presence of both TFA and ferrocene (both 490 μM) in dry benzene (25 ml). The UV-vis spectrum is an overlay of spectra measured every second over a period of 90 s. | ||
For preparative purposes the photolyses were conducted at higher concentrations of 7 (1.57 mM). Product formation was studied by IR, 1H NMR, 13C NMR and HPLC (for quantification). Notably formation of product 6 was increased, most likely because at higher concentrations it arises from the reaction of radical 5 with 7.15 HPLC quantification showed that the overall conversion of 7 → 18 in the absence of TFA was 89% (by 1H NMR no further product was detectable) and 59% in the presence of acid (see ESI†). Formation of 6 was the same for both acid-free and acidic conditions.
Photolysis of 7 in presence of ferrocene.
Photolysis of 7 (0.49 mM, dry benzene) in the presence of both TFA and ferrocene (equimolar concentrations) afforded a broad band at ∼620 nm that was assigned to the ferrocenium cation. The photolysis was monitored by UV-vis spectroscopy over a period of 90 s. By adding a strong acid, such as TFA, the photolysis of 7 was slowed down slightly suggesting that protonation of the chromophore stabilizes 7 somewhat against photo-degradation.A control experiment was conducted with Barton ester 8. Photolysis of 8 (568 μM) in the presence of an equimolar concentration of ferrocene and TFA showed a decrease of the absorbance at 370 nm and the formation of 6 and 19. Again, no formation of any long absorbing species above 400 nm was detectable. Photolysis of 8 (568 μM) in the presence of an equimolar concentration of ferrocene and TFA showed a decrease in the 370 nm band but no formation of any long absorbing species above 400 nm. Even with 9 equiv. of TFA (5.19 mM) no formation of any long absorbing species was observed (see ESI†).
The above experiments with 8 in presence of TFA and ferrocene suggest that the putative oxidants 6+˙ or protonated 5·H+ are either not formed during the photolytic conditions or have very short transient lifetimes insufficient for intermolecular electron transfer with ferrocene. Laser flash photolysis of 6 has been carried out by Osamu Ito et al. in various solvents,16 but without any evidence for 6+˙ or 5·H+. Thus, the finding of ferrocenium formation with 7, TFA, ferrocene and light suggests that another, yet unknown strong oxidant is formed in the photolysis that is a result of the dioxolane's follow-up reactions.
As Barton esters are known to be prone to decomposition in radical chain reactions, a plausible mechanism for the formation of 18 (or 19) appears to be the attack of radical 3 (or 10) onto the sulfur atom in 7 (or 8), which would eventually release another equivalent of radical 3 (or 10) to continue the radical chain process (vide infra). Disulfide 6 in turn could be formed by dimerization of radical 5 or by radical-induced decomposition of 7 (or 8),15 both mechanisms explaining increased formation of 6 at higher concentrations of 7 (or 8).
In summary, the observations indicate that in the presence of TFA and light an oxidizing species is formed in the solution from 7 that is derived from the dioxolane unit. While the data would be consistent with formation of ethenol radical cation (1) as hypothesized in Scheme 1, we note that the oxidation of ferrocene could also be due to reactions of other short-lived oxidants.
Laser flash photolysis
Experiments with 7 and 8 employing LFP with UV-vis detection were performed employing the third harmonic of a Nd-YAG laser (λexc = 355 nm) as excitation light source in combination with benzene as solvent.LFP of pivalate 8 (c = 0.1 mM) in benzene solution gave the transient spectrum shown in Fig. 2. We observed a single transient with λmax ∼ 480 and ca. 570 nm (second order decay, half-life in the μs range), which we assign to the pyridine 2-thiyl radical (5), complexed to a benzene molecule. Our spectrum agrees well with the spectrum reported by Aveline, Kochevar and Redmond,15 using benzene as solvent, whereas in THF solution, radical 5 has been reported to show an absorption maximum with λmax = 490 nm.16 π-Complex formation with benzene is predicted (TD-B3LYP/6-311++G(d,p)(pcm,benzene)//M06-2X/cc-pVTZ)(pcm,benzene) to result in a broadening of the spectrum with two absorption maxima at λmax = 425 and 556 nm, in reasonable agreement with the experimental observation (for the calculated absorption spectrum of a complex of 5 and benzene, see the ESI†). Adding 0.5 mM TFA did not change the transient spectrum. In agreement with the absorption maxima of the precursor, there is a bleach at λ = 295 and 375 nm. Interestingly, at both wavelengths we observed an apparent recovery of the signal, which occurred with a first-order rate constant k = 7.4 × 104 s−1, corresponding to τ = 13.5 μs. Usually, bleach and recovery like this is observed, if a long-lived triplet excited state returns to the ground state and the precursor concentration thus is replenished. This certainly cannot be the case here. We therefore suggest that the bleach and recovery reflects the reaction of the t-butyl radical (which we do not observe directly) with the precursor 8, yielding a species that absorbs at a similar place as 8, yet stronger.
 | ||
| Fig. 2 Transient absorption spectra, monitored after different time intervals following LFP (λexc = 355 nm, laser attenuated to ca. 20 mJ per pulse) of a 0.1 mM solution of 8 in benzene, purged with argon. | ||
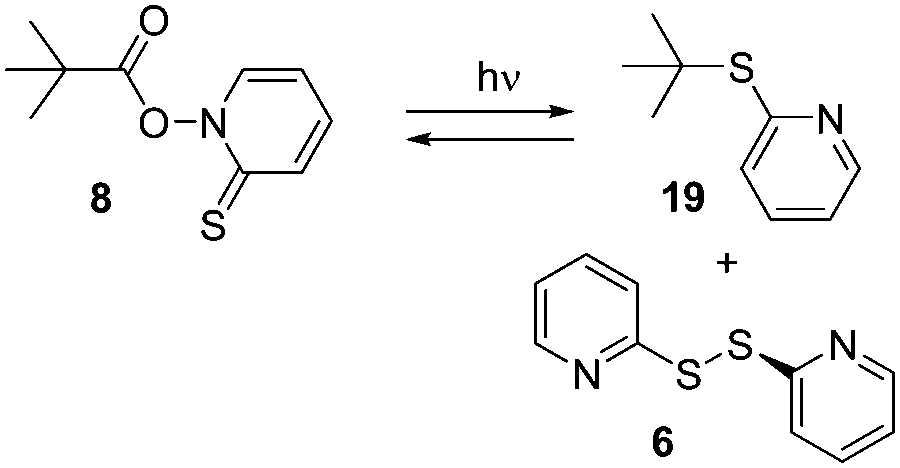
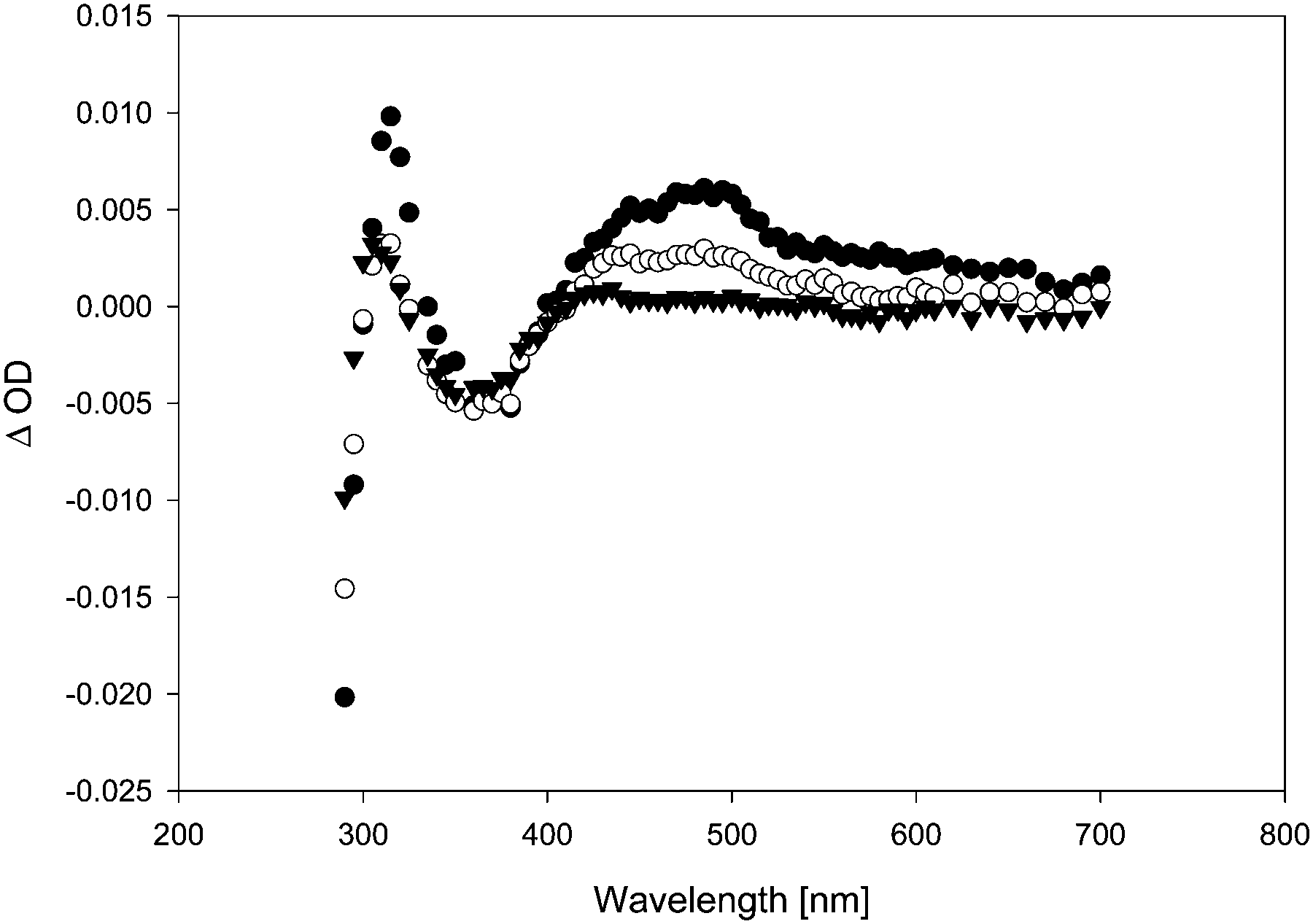
LFP of acetonide 7 gave significantly weaker transient signals. A larger number of different transient species observed conveyed a much more complex mechanistic picture. Fig. 3 shows the transient spectrum observed upon LFP of a 0.1 mM solution of 7 in benzene, purged with argon. We observed a transient A decaying according to 2nd order kinetics (λmax = 445 and 485 nm) that is again assigned to a complex of thiyl radical 5 with benzene.16 Additionally, a transient B with λmax = 315 nm and a much shorter lifetime (1st order: τ = 170 ns) was observed (Fig. 3). If the laser power was reduced to ca. 20 mJ per pulse, we again observed growth or recovery at the absorption maxima of the precursor. This reaction took place with a lifetime τ = 170 ns. On a yet shorter time-scale, we detected another transient C, showing a λmax = 500 nm that appears to be formed in a monophotonic process (the plot of transient intensity vs. laser power is linear, see Fig. S6, ESI†). Its lifetime is extremely short (τ = 34 ns) (Fig. 4).
 | ||
| Fig. 3 Transient spectra, observed upon LFP (λexc = 355 nm, 120 mJ per pulse) of 7 (0.1 mM) in benzene, purged with argon. Black circles: 400 ns after LFP. White circles: 3.4 μs after LFP. Black triangles: 15 μs after LFP. | ||
 | ||
| Fig. 4 Transient spectra, observed upon LFP (λexc = 355 nm, 120 mJ per pulse) of 7 (0.1 mM) in benzene, purged with argon. Black circles: 14 ns after LFP. White circles: 34 ns after LFP. Black triangles: 150 ns after LFP. | ||
Addition of 0.03% TFA to the solution did not affect the transient spectra of A, B or C. The lifetimes of the three transients also did not change. We could, however, observe certain changes in the reactivity of transient C. This species turned out to be quenched by electron-rich aromatic quenchers such as anisole at a rate that was close to or at the diffusion-controlled limit (Fig. 5). Adding TFA further increased the reactivity slightly. Table 1 shows quenching rate constants measured both in the presence and in the absence of TFA.
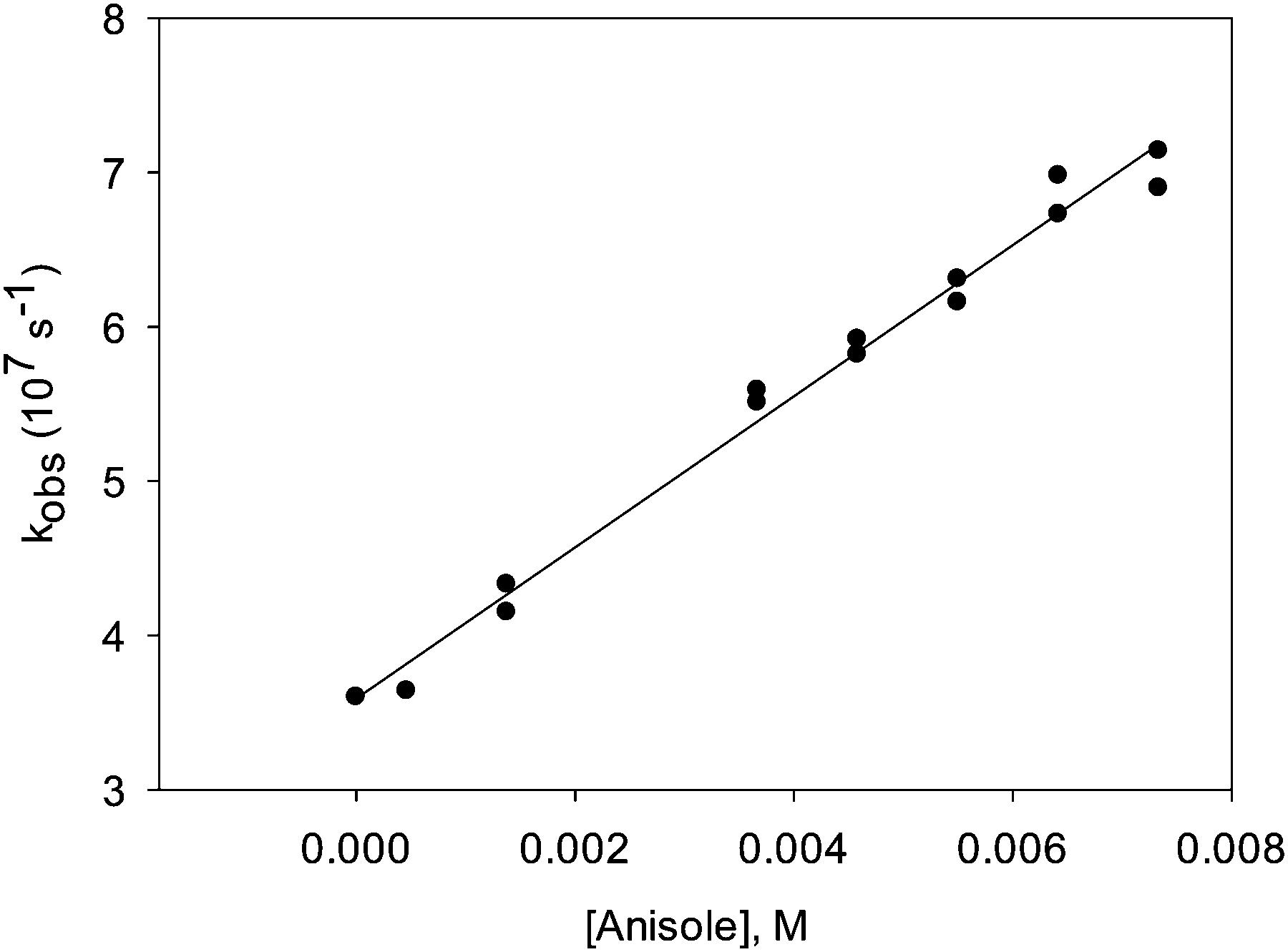 | ||
| Fig. 5 Plot of the transient decay rate constant, measured at λ = 500 nm after LFP (λexc = 355 nm) of 7 in benzene, purged with argon, versus the concentration of anisole. The straight line represents a linear fit to the data. | ||
| Quencher | Rate constant in benzene | Rate constant in benzene + 0.03% TFA |
|---|---|---|
| 1,2,3-Trimethoxybenzene | (8.2 ± 0.8) × 109 l mol−1 s−1 | (1.1 ± 0.1) × 1010 l mol−1 s−1 |
| Anisole | (4.9 ± 0.5) × 109 l mol−1 s−1 | (8.9 ± 0.9) × 109 l mol−1 s−1 |
| Diphenylether | Lifetime increases | (1.4 ± 0.1) × 1010 l mol−1 s−1 |
| Toluene | No quenching observed | No quenching observed |
| Tri-n-butylstannane | (1.6 ± 0.2) × 109 l mol−1 s−1 | Not measured |
| Cyclohexane | No quenching observed | No quenching observed |
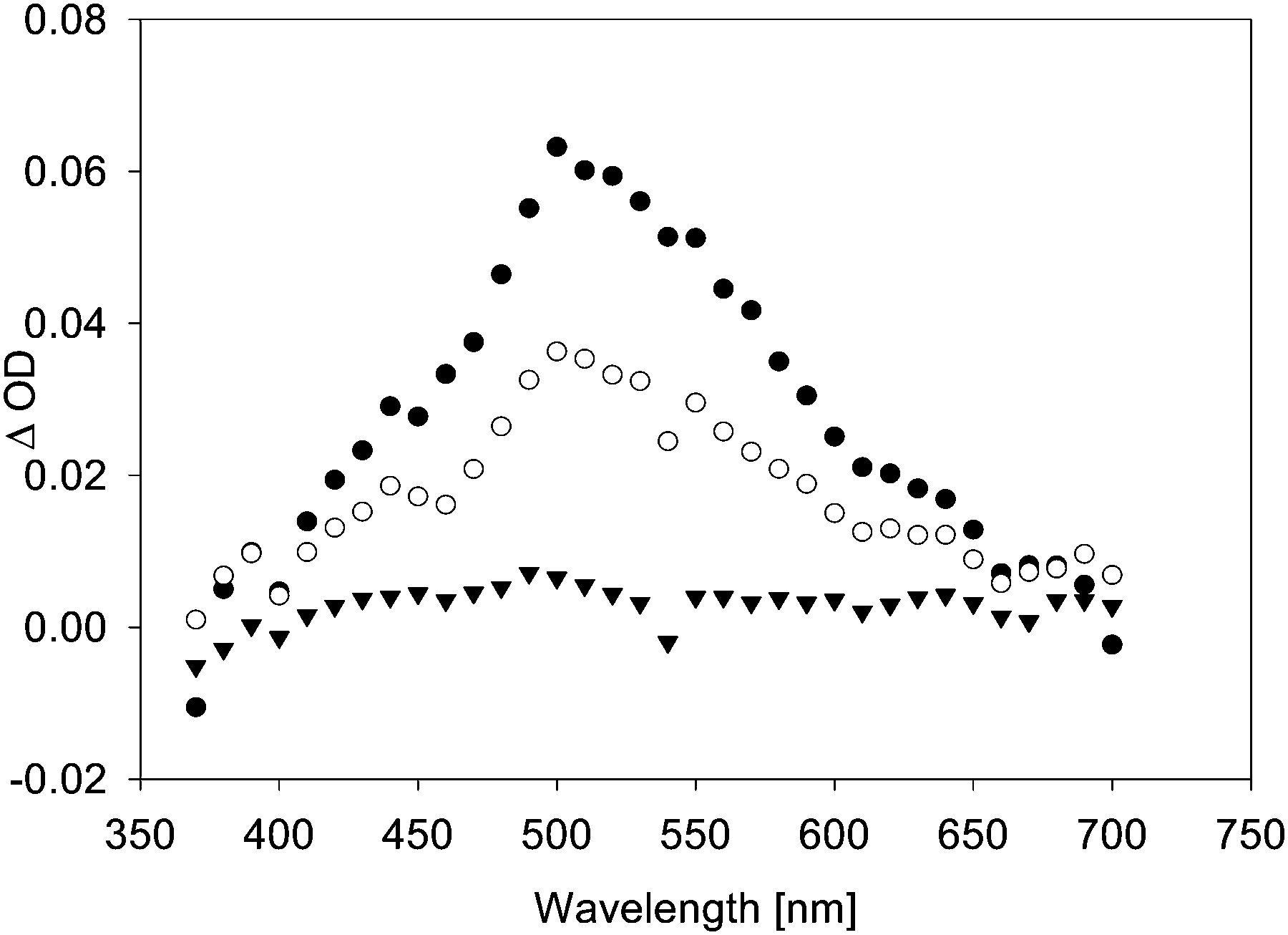
In the presence of TFA, the quenching rate constants turned out to be consistently slightly higher than in the absence of TFA. This effect was particularly obvious in the case of anisole as quencher. When diphenylether (DPE) was added, the transient lifetime measured at λ = 500 nm increased in the absence of TFA (τ = 90 ns in the presence of 0.08% diphenylether), and decreased in the presence of TFA. In the absence of TFA, we also observed a bathochromic shift in λmax (from λmax = 500 nm in the absence of DPE to a double maximum at ca. 500 and 630 nm in the presence of DPE), and the transient absorption spectrum became more intense and much broader (Fig. 6). Whether the growth (τ = 7 ns) that is seemingly resolved during the initial stages of the transient trace (Fig. 7, upper trace) is real or not is uncertain, as this growth lifetime is very close to the rise time of our LFP detector.17
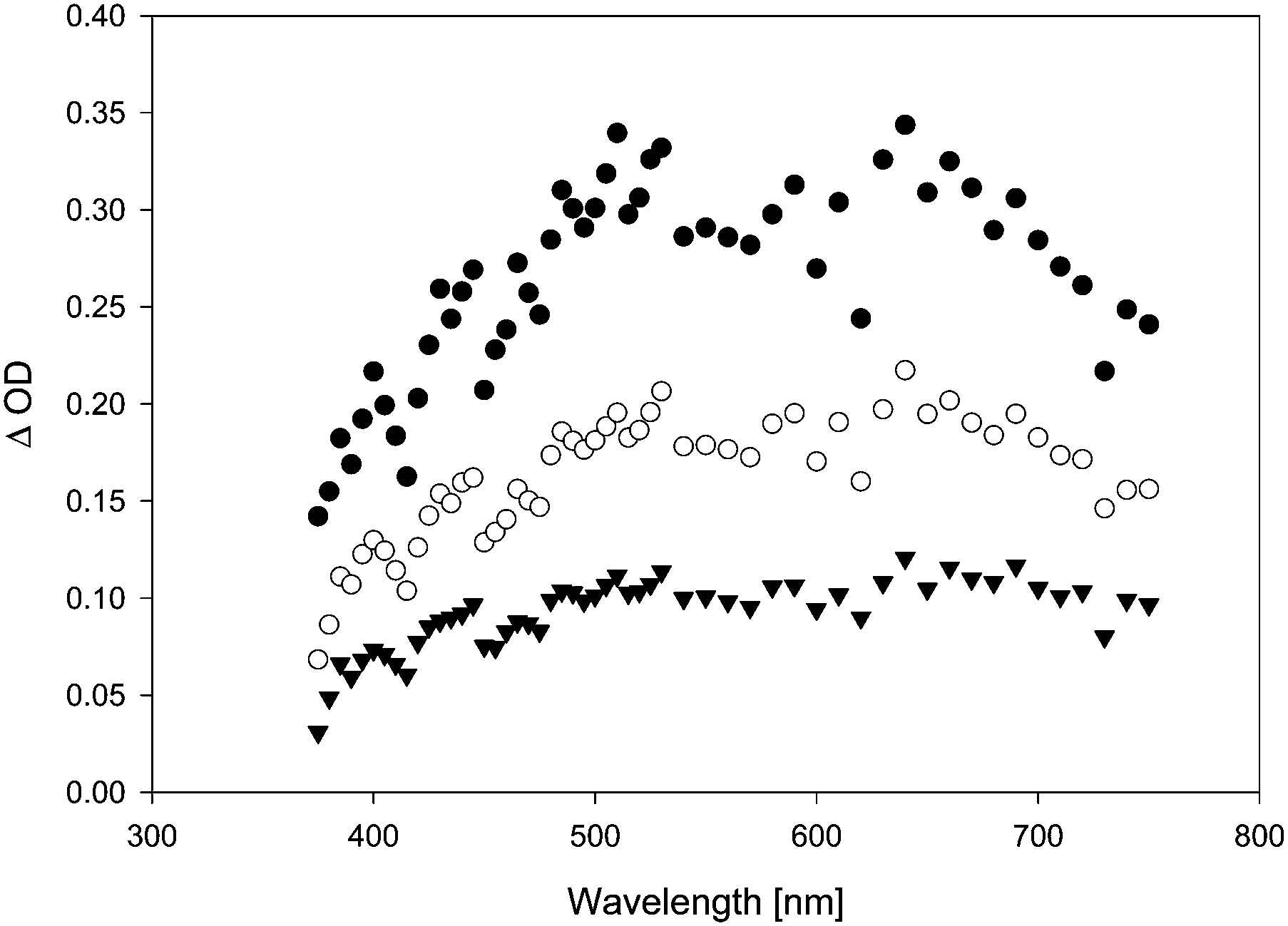 | ||
| Fig. 6 Transient spectra, observed upon LFP (λexc = 355 nm, 120 mJ per pulse) of 7 (0.1 mM) in benzene containing 0.08% diphenylether, purged with argon. Black circles: 25 ns after LFP. White circles: 80 ns after LFP. Black triangles: 165 ns after LFP. | ||
 | ||
| Fig. 7 Transient traces, monitored at λ = 500 nm upon LFP (355 nm) of 7 in benzene. Bottom trace: Without diphenylether. Top trace: In the presence of 0.08% diphenylether. | ||
Considering the three different transient species observed, the assignment of A to thiyl radical 5 is straightforward based on a comparison with literature data,16 as well as a comparison with a calculated spectrum of the complex 5·benzene. The identity of transients B (λmax = 315 nm, τ = 170 ns) and C (λmax = 500 nm, τ = 34 ns), however, still needs to be discussed. In the experiments with pivalate 8, we observed a growth or recovery in the absorption bands of the precursor that occurred with a lifetime τ = 13.5 μs. As there is no transient observed decaying with this lifetime, it appears reasonable to attribute this growth to a reaction of the t-butyl radical, which is not expected to be detectable in our experiments owing to the lack of a good chromophore absorbing at λ > 300 nm.
While we can only speculate about the nature of this reaction, our possible candidate for transient B is the dioxolanyl radical 3. TD-DFT calculations (TD-B3LYP/6-31 + G*//B3LYP/6-31G*) reveal that this radical in the gas phase should have a longest-wavelength transition at λ = 302 nm, which should not be particularly intense (f = 0.0016). Thus there is qualitative agreement between the calculated absorption maximum of 3 and the absorption maximum of B. Given the fact, that formation of thioether 18 as a major product is strongly indicative of the intermediacy of 3, the assignment appears reasonable. Its rather short lifetime (τ = 170 ns) likely stems from a rapid reaction with the precursor. Given a diffusion-controlled reaction between 3 and 7, a sub-μs lifetime would be expected for 3 in the sub-millimolar concentration range employed for precursor 7.
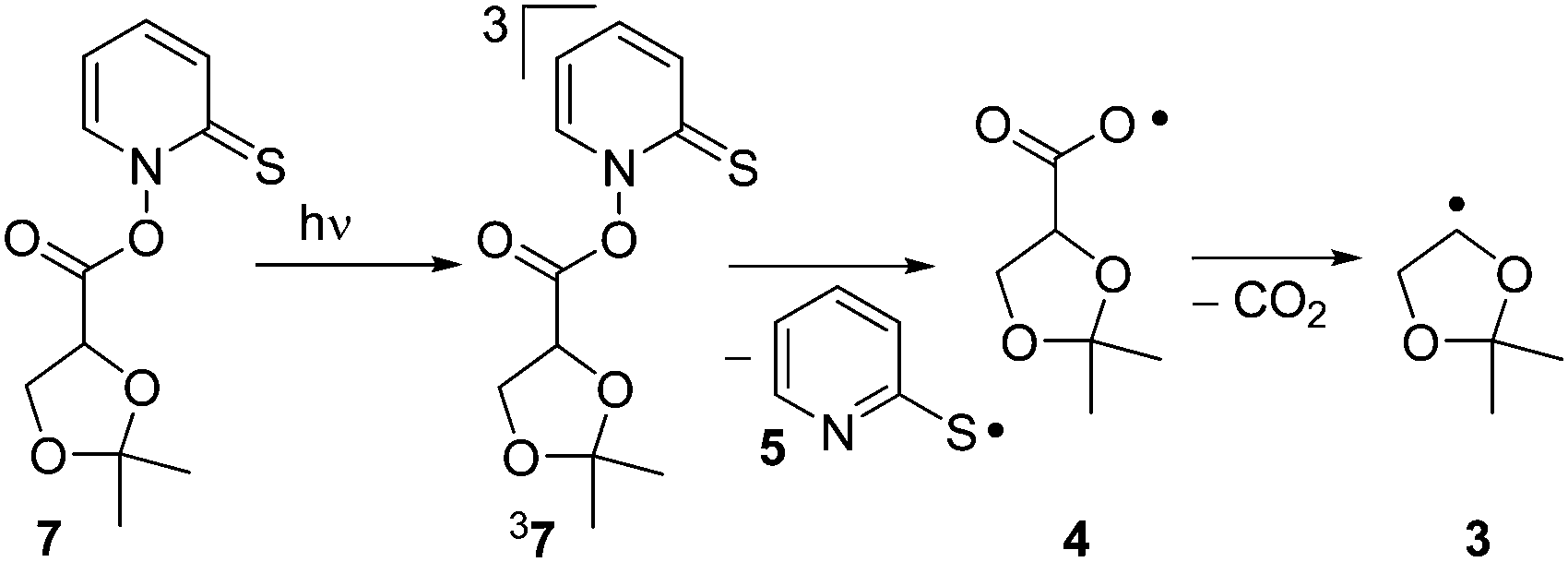
Assigning the short-lived transient C is less straightforward. Possible candidates include a triplet excited state of precursor 7, carbonyloxy radical 4, the ring-opened radicals 20 or 21, and the vinyloxy radical 22 (Scheme 5 and Fig. 8).
 | ||
| Scheme 5 Possible intermediates in the photochemical fragmentation of Barton ester 7 in the absence of TFA. | ||
 | ||
| Fig. 8 Calculated (M06-2X/cc-pVQZ or CCSD(T)/cc-pVTZ//M06-2X/cc-pVQZ) energies (in kcal mol−1) for stationary points in the fragmentation of 3, relative to 3 = 0.0 kcal mol−1. Normal font: M06-2X free energies, gas phase; in brackets: M06-2X electronic energies, gas phase. Italics: M06-2X free energies, benzene solution (scrf = pcm). Underlined: CCSD(T) electronic energies, gas phase. | ||
As far as the triplet-excited Barton ester 37 or carbonyloxy radical 4 are concerned, the barriers for fragmentation are expected to be very small. In fact, in both cases DFT optimizations resulted in either spontaneous N–O bond cleavage (37, M06-2X/cc-pVDZ) or in a very small barrier to decarboxylation (4, M06-2X/cc-pVQZ: ΔU‡ = 0.8 kcal mol−1). Born–Oppenheimer molecular dynamics simulations (M06-2X/cc-pVDZ) also in both cases resulted in C–C cleavage (4) or N–O cleavage followed by C–C cleavage (37), within a few femtoseconds each. Hence, an assignment of transient C to either species does not appear to represent a viable option. Among the remaining radical species 20–22, the vinyloxy radical (or formylmethyl radical) 22 represents the most likely assignment, as DFT calculations indicate that the barrier for decay of both 20 and 21 is smaller than the barrier for their formation, meaning that 20 or 21 should normally only be present in quasistationary concentration (Fig. 8). We note that the results obtained using the M06-2X/cc-pVQZ method are in excellent agreement with the results from the CCSD(T) single point energy calculations. Over all, the fragmentation of 3 is predicted to be endothermic, but exergonic (at T = 298 K), hence entropy-driven.
In the gas phase, the longest-wavelength absorption maximum of 22 is calculated (TD-B3LYP/6-31+G*) at λmax = 271 nm. However, if π-complexation to benzene or DPE is taken into account, additional weak long-wavelength CT bands appear at λmax = 468 nm (π-complex with benzene) or λmax = 464 and 689 nm (π-complex with DPE) (all values TD-UB3LYP/aug-cc-pVTZ//UM062X/cc-pVQZ). Hence, with these CT bands, the calculated UV-vis spectra of the vinyloxy/arene complexes qualitatively match the experimental transient spectra shown in Fig. 4 and 6, including the double maximum observed for the DPE complex. Fig. 9 shows a juxtaposition of the calculated vs. the experimental spectrum of 22·DPE, recorded in the presence of 0.08% DPE.
 | ||
| Fig. 9 Bottom: Calculated UV/vis spectrum of complex 22·DPE (TD-UB3LYP/aug-cc-pVTZ//UM062X/cc-pVQZ). Inset: Experimental transient spectrum, observed after LFP (355 nm) of 7 in benzene (1 atm. Ar) in the presence of 0.08% DPE. Black circles: 25 ns after LFP. White circles: 80 ns after LFP. Black triangles: 165 ns after LFP. | ||
As far as the quenching reactions of the short-lived transient C are concerned, we note that both the high reactivity towards electron-rich arenes and the increase in reactivity upon addition of TFA would also be consistent with an assignment to the vinyloxy radical (22). A hydrogen-bridged complex of 22 with TFA would be very similar to a free ethenol radical cation in both structure and likely also oxidative power. Such complex would explain the pronounced reactivity of 22 in acidic medium, in particular its capability to rapidly react with DPE or Fc, whereas no reaction (or an increase in lifetime by complex formation) was observed in the absence of TFA.
In the LFP of 7 and 8, at low laser powers, an apparent recovery was observed at the wavelength of the precursor absorption band. The kinetics of this recovery in case of 7 matched the kinetics of the decay of radical 3, monitored at λ = 315 nm. The fact that the recovery in case of the LFP of 7 was only observed when the laser power was reduced would be consistent with an assignment of this transient process to a reaction of the precursor Barton esters 7 and 8, as use of high laser power would result in complete bleaching of the precursor, so that its subsequent radical chain reactions would be suppressed. It cannot be the growth of thioethers 18 or 19 that is resolved here (Scheme 6), as the UV-vis spectrum of the very similar 2-methylthiopyridine, which may serve as a model system, has neither been reported18a nor is expected to show absorption around λmax = 390 nm. Likewise, the UV-vis spectrum of di-2-pyridyldisulfide (6) has not been reported to show absorption at this wavelength.18b This hypothesis would also be consistent with the observation that the lifetime of the t-butyl radical (10) in the presence of 0.1 mmol L−1 of 7 is τ = 13.5 μs, whereas the lifetime of 3 under analogous conditions ([8] = 0.1 mmol L−1) is only τ = 170 ns. The rate constant of the reaction of 8 with the sterically hindered radical 10 can be expected to be smaller than the rate constant of the analogous reaction of 7 with the more accessible radical 3. Reactions of free radicals like 3 or 10 with Barton esters are expected to proceed via addition to the sulfur atom of the reactive CS bond, followed by cleavage of the rather weak N–O bond in adducts such as 23 (Scheme 6).
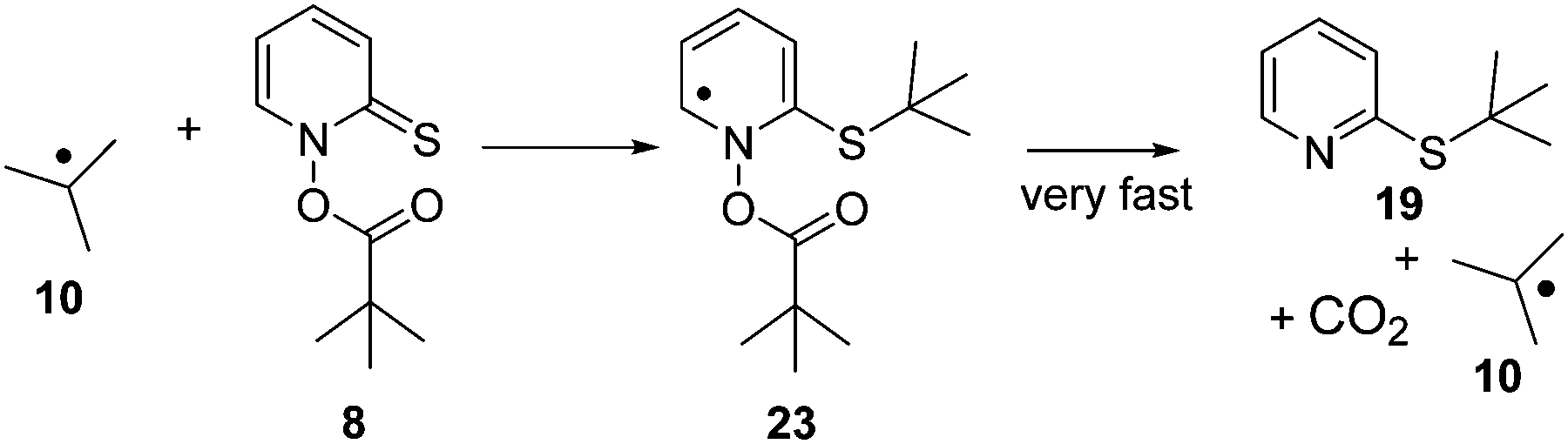 | ||
| Scheme 6 Reaction pathway in the reaction of Barton ester 7 with radical 10. | ||
It is noted that the barrier for cleavage of the radical adduct 23 is expected to be very small. At the M06-2X/6-311++G(2df,p) (pcm,benzene) level of theory, a relaxed surface scan systematically stretching the N–O bond in 23 resulted in an increase in energy of only 0.8 kcal mol−1 required to reach a point where fragmentation into 19, 10 and CO2 occurred. Hence, any free radical intermediate formed along the reaction coordinate linking 8/10 with 10/19 will be extremely short-lived. We will therefore leave the assignment of the species responsible for the growth at λ = 390 nm open at present, merely noting that kinetic data indicate that this species results from reaction of radicals 3/10 with Barton esters 7/8. The results of an in-depth computational study on reactions of Barton esters with free radicals will be published in due course.
Notably, we have been able to detect radical 3 in the photolysis of 7 in benzene by time resolved EPR (see ESI†). Radical 3, generated by hydrogen abstraction or photoionization, had been detected earlier by EPR spectroscopy and had not demonstrated any fragmentation at ambient temperature.19,20 The barriers for its fragmentation are significant – even the more favourable pathway via radical 20 bears a 21.7 kcal mol−1 free energy of activation in the initial step (see Fig. 8), which would normally make a fragmentation on the ps timescale completely impossible. Our observation of the vinyloxy radical (22), however, necessitates such a rapid fragmentation. The BDE of the N–O bond in Barton esters is small, estimates range between 16 and 28 kcal mol−1.21,22 Given the fact that a 355 nm photon carries an energy of 82 kcal mol−1, and that the decarboxylation of 4 is exothermic by 35 kcal mol−1 (M06-2X/cc-pVQZ), a sizeable excess energy of at least 90 kcal mol−1 remains for the complete photoinduced fragmentation of 7 into thiyl radical 5, dioxolanyl radical 3, and CO2. Even if this excess energy is evenly spread over all vibrational degrees of freedom in all three fragments, sufficient energy (ca. 66 kcal mol−1) will remain in 3 to potentially result in rapid fragmentation. In order to gain further evidence for potential nonstatistical reaction dynamics23 in the fragmentation of 3, we performed Born–Oppenheimer molecular dynamic (BOMD) simulations at the UB3LYP-D3(BJ)/6-31G* level. Four possible conformers of 7 (Fig. 10) were considered with 7c being the most stable one. Quasiclassical trajectories were initialized from normal mode sampling of 7c in the T1 state to approximate a quantum mechanical Boltzmann distribution of the vibrational levels at 298 K. The overlay of the sampled geometries is shown in Fig. 11. A total of 98 trajectories were initiated from the sampled structures.
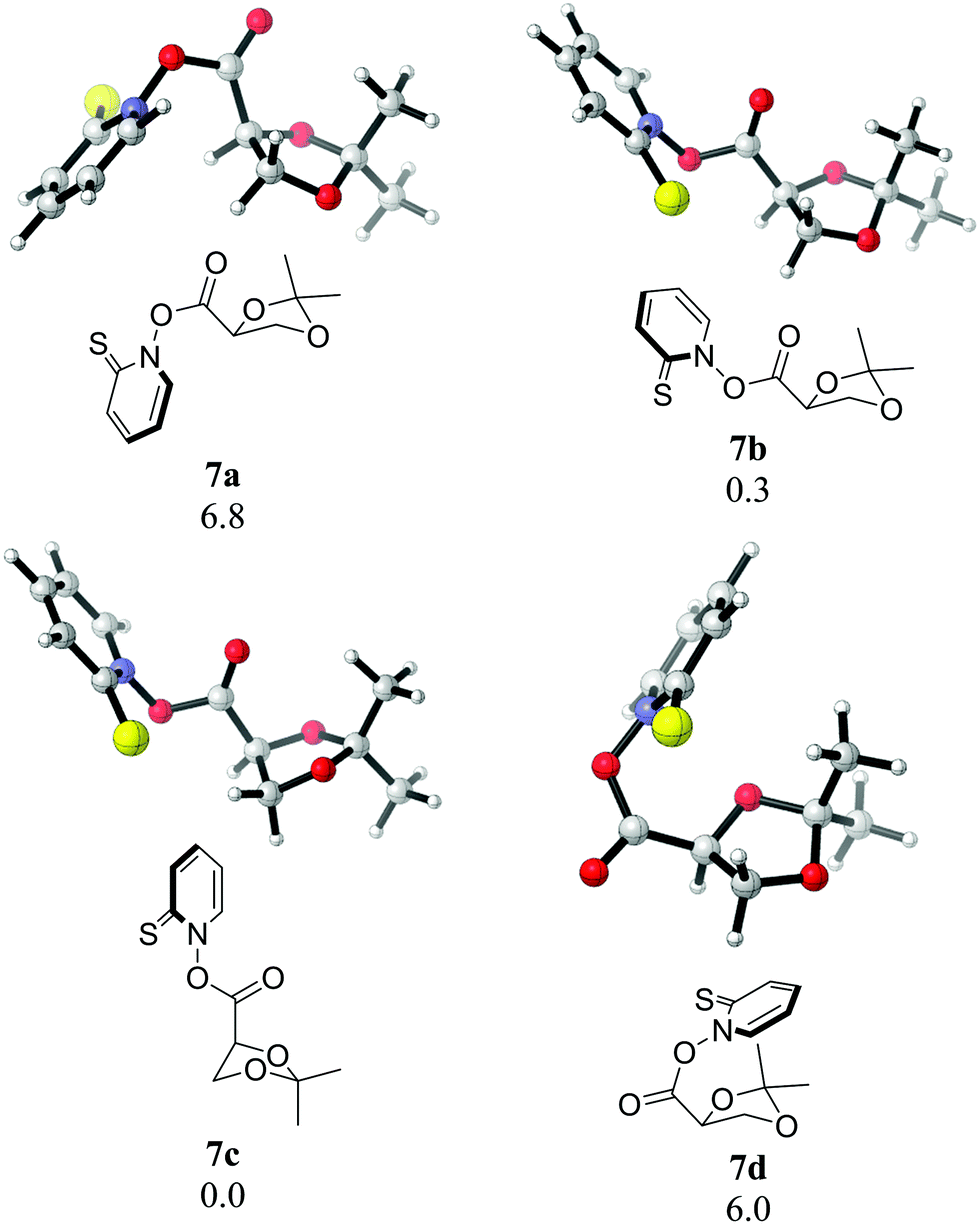 | ||
| Fig. 10 Conformers of 7. Numerical values are relative free energies (in kcal mol−1) at 25 °C (B3LYP-D3(BJ)/6-31G* level). | ||
 | ||
| Fig. 11 Overlay of the sampled structures of 7c in the T1 state. | ||
For all the trajectories, N1–O2 and C3–C4 (Fig. 12) bonds cleave within the fs time scale. The average time to break the N1–O2 bond, as defined by lengthening of the same beyond 1.60 Å, is 196 fs whereas the C3–C4 bond cleaves at 276 fs, considering a bond distance larger than 1.90 Å. Accordingly, the average time lag between the two bond cleavage events is 80 fs. The minimum time required to cleave the N1–O2 bond is 23 fs while the maximum time is 787 fs. Interestingly, the sluggish trajectory took only 45 fs to cleave the second bond. Although all the trajectories ended up eliminating CO2 within 1121 fs, only one went further to cleave the third bond (O8–C7) dynamically. It took a total 630 fs to disrupt all three bonds. Thereafter, the dihedral angle C4–C5–O6–C7 started to rotate but the C5–O6 bond did not break within the simulation time limit of 1352 fs. Thus, the finding indicates that the C5–O6 bond breaking is the rate determining step, in contrast to the result obtained via the calculated barrier heights (cf.Fig. 8).
 | ||
| Fig. 12 Effective trajectory yielding 20. | ||
Conclusions
Photolysis of dioxolane-carboxylic acid Barton ester 7 results in formation of the vinyloxy radical 22, which was detected via ns LFP as its benzene or diphenylether π-complexes. Its formation on a ps timescale can only be rationalized by invoking contribution of dynamic effects, for which evidence could also be gained by performing Born–Oppenheimer molecular dynamics simulations. The vinyloxy radical 22 is a potent oxidant, as shown by very rapid quenching by electron-rich arenes. Addition of trifluoroacetic acid further increases its oxidative power, and the complex of CH2CHO with CF3COOH essentially behaves like the radical cation of ethenol.
Experimental part and computational methodologies
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank both the University of Glasgow (G. B.) and Universität Siegen (A. R.) for CPU time on the local clusters (Glasgow High Performance Cluster, HorUS Cluster). We are indebted to Dr Dirk Grote (Ruhr-Universität Bochum) for measuring the EPR spectrum.References
- M. Schmittel, M. Lal, R. Lal, M. Röck, A. Langels, Z. Rappoport, A. Basheer, J. Schlirf, H.-J. Deiseroth, U. Flörke and G. Gescheidt, A comprehensive picture of the one-electron oxidation chemistry of enols, enolates and α-carbonyl radicals: Oxidation potentials and characterization of radical intermediates, Tetrahedron, 2009, 65, 10842 CrossRef.
- Y. Yamaguchi, Y. Okada and K. Chiba, Understanding the reactivity of enol ether radical cations: investigation of anodic four-membered carbon ring formation, J. Org. Chem., 2013, 78, 2626 CrossRef PubMed.
- E. Taxil, L. Bagnol, J. H. Horner and M. Newcomb, Efficient production of enol ether radical cations by heterolytic cleavage of beta-mesylate radicals, Org. Lett., 2003, 5, 827 CrossRef PubMed.
- R. Paul and M. M. Greenberg, Mechanistic Studies on RNA Strand Scission from a C2′-Radical, J. Org. Chem., 2016, 81, 9199 CrossRef PubMed.
- M. J. E. Resendiz, V. Pottiboyina, M. D. Sevilla and M. M. Greenberg, Direct strand scission in double stranded RNA via a C5-pyrimidine radical, J. Am. Chem. Soc., 2012, 134, 3917 CrossRef PubMed.
- J. Stubbe and W. A. van der Donk, Protein Radicals in Enzyme Catalysis, Chem. Rev., 1998, 98, 705 CrossRef PubMed.
- A. Abend, V. Bandarian, G. H. Reed and P. A. Frey, Identification of cis-ethanesemidione as the organic radical derived from glycolaldehyde in the suicide inactivation of dioldehydrase and of ethanolamine ammonia-lyase, Biochemistry, 2000, 39, 6250 CrossRef PubMed.
- E. Taxil, L. Bagnol, J. H. Horner and M. Newcomb, Efficient production of enol ether radical cations by heterolytic cleavage of beta-mesylate radicals, Org. Lett., 2003, 5, 827 CrossRef PubMed.
- J. H. Horner, E. Taxil and M. Newcomb, Laser Flash Photolysis Kinetic Studies of Enol Ether Radical Cations. Rate Constants for Heterolysis of α-Methoxy-β-phosphatoxyalkyl Radicals and for Cyclizations of Enol Ether Radical Cations, J. Am. Chem. Soc., 2002, 124, 5402 CrossRef PubMed.
- J. H. Horner and M. Newcomb, Enol Ether Radical Cation Reaction Kinetics. Laser Flash Photolysis Calibration of Radical Cation Clocks, J. Am. Chem. Soc., 2001, 123, 4364 CrossRef PubMed.
- M. Newcomb, N. Miranda, M. Sannigrahi, X. Huang and D. Crich, Direct Measurement of Enol Ether Radical Cation Reaction Kinetics, J. Am. Chem. Soc., 2001, 123, 6445 CrossRef PubMed.
- N. P. Schepp, Generation and absolute reactivity of an aryl enol radical cation in solution, J. Org. Chem., 2004, 69, 4931 CrossRef PubMed.
- M. Schmittel, G. Gescheidt and M. Röck, The First Spectroscopic Identification of an Enol Radical Cation in Solution: The Anisyl-dimesitylethenol Radical Cation, Angew. Chem., Int. Ed. Engl., 1994, 33, 1961 CrossRef.
- P. J. Kocienski, Protecting Groups, Georg Thieme, Stuttgart, 2005, ch. 2, p. 2 Search PubMed.
- B. M. Aveline, I. E. Kochevar and R. W. Redmond, Photochemistry of N-hydroxypyridine-2-thione derivatives: involvement of the 2-pyridylthiyl radical in the radical chain reaction mechanism, J. Am. Chem. Soc., 1995, 117, 9699 CrossRef.
- M. M. Alam, A. Watanabe and O. Ito, Laser flash photolysis of dithio-2,2′-dipyridine; Structure and reactivity of pyridyl-2-thio radical, J. Org. Chem., 1995, 60, 3440 CrossRef.
- G. Bucher, Laser Flash Photolysis Study on N,N-Diethyl-2-azidobenzylamine: The Reactivity of Iminoquinonemethides in Solution, Eur. J. Org. Chem., 2001, 2463 CrossRef.
- (a) While 2-methylthiopyridine has been reported to show absorption slightly below 400 nm (see A. Albert, G. B. Barlin, Ionization Constants of Heterocyclic Substances. Part III. Mercapto-Derivatives of Pyridine, Quinoline, and Isoquinoline. J. Chem. Soc., 1959, 2384), this report likely is erroneous. Other publications do not mention this band (see, e.g., S. J. Dunne, L. A. Summers, E. I. von Nagy-Felsobuki, Conformational and Photoelectron Analysis of the Methylchalcogenopyridines, Phosph., Sulf. Silicon Rel. Elements, 1992, 72, 103); (b) S. Stoyanov, I. Petkov, L. Antonov, T. Stoyanova, P. Karagiannidis and P. Aslanidis, Thione-thiol tautomerism and stability of 2- and 4-mercaptopyridines and 2-mercaptopyrimidines, Can. J. Chem., 1990, 68, 1482 CrossRef.
- G. Trampe, J. Mattay and S. Steenken, Ionisation of 1,3-dioxoles in aqueous solution by 248 nm laser flash photolysis characterisation of the radical cations, J. Phys. Chem., 1989, 93, 7157 CrossRef.
- K. H. Lee, K.-L. Chin and S. Brumby, Electron spin resonance spectra of free radicals. Part 3. 2,2-Disubstituted 1,3-dioxolan-4-yl radicals, J. Chem. Soc., Perkin Trans. 2, 1985, 161 RSC.
- D. G. Harman and S. J. Blanksby, Investigation of the gas phase reactivity of the 1-adamantyl radical using a distonic radical anion approach, Org. Biomol. Chem., 2007, 5, 3495 RSC.
- C. Dietlin, X. Allonas, F. Morlet-Savary, J. P. Fouassier, M. Visconti, G. Norcini and S. Romagnano, Investigation of Barton Esters a Radical Photoinitiators, J. Appl. Polym. Sci., 2008, 109, 825 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648 CrossRef.
- R. Ditchfield, W. J. Hehre and J. A. Pople, Self-Consistent Molecular Orbital Methods. 9. Extended Gaussian-type basis for molecular-orbital studies of organic molecules, J. Chem. Phys., 1971, 54, 724 CrossRef.
- A. D. McLean and G. S. Chandler, Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z = 11-18, J. Chem. Phys., 1980, 72, 5639 CrossRef.
- T. H. Dunning Jr., Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999 CrossRef PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilization of ab-initio molecular potentials for the prevision of solvent effects, Chem. Phys., 1981, 55, 117 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, characterization data, photolysis, EPR, computational data. See DOI: 10.1039/c8cp03311k |
| ‡ Current address: WestCHEM, School of Chemistry, University of Glasgow, Joseph-Black-Building, University Avenue, Glasgow G12 8QQ, UK. E-mail: goetz.bucher@glasgow.ac.uk |
| This journal is © the Owner Societies 2018 |