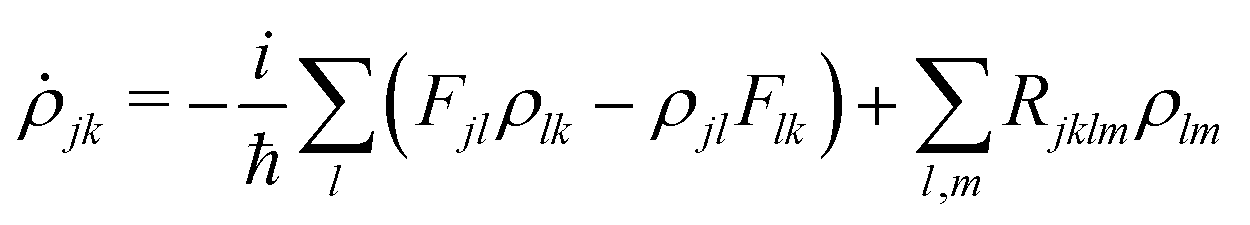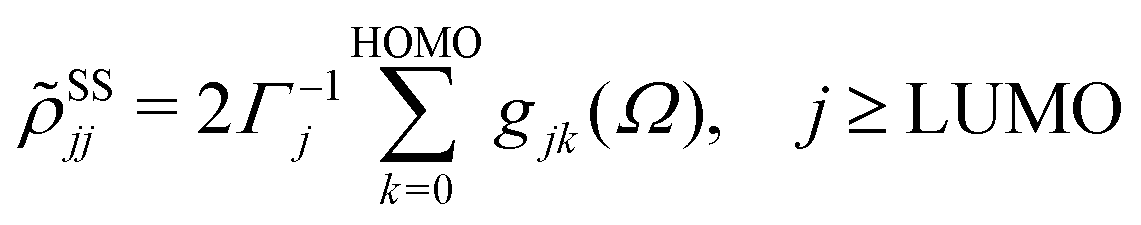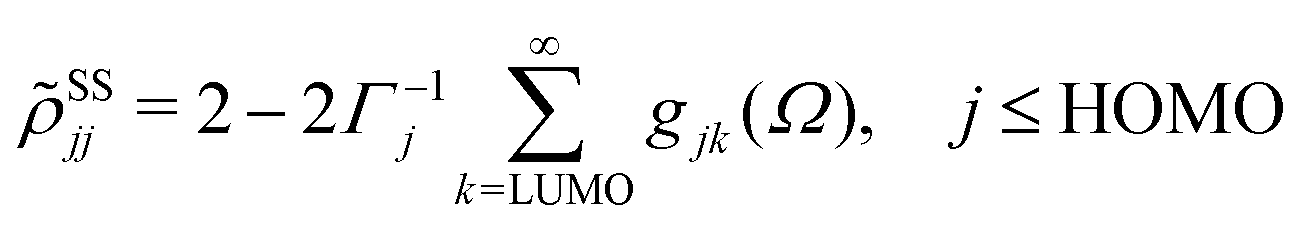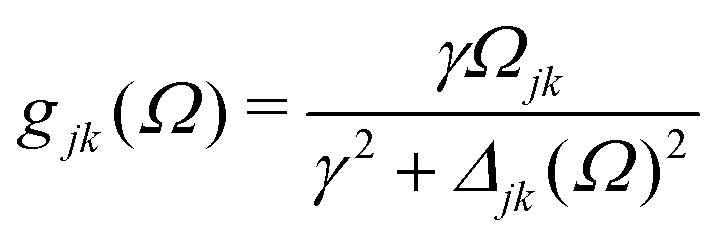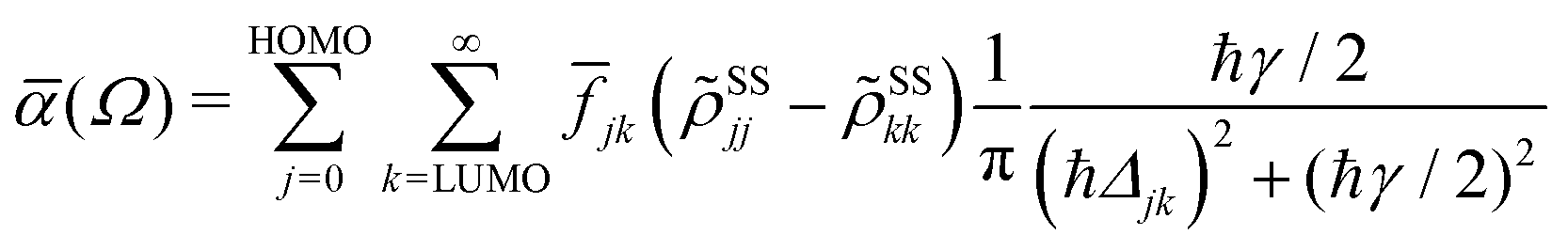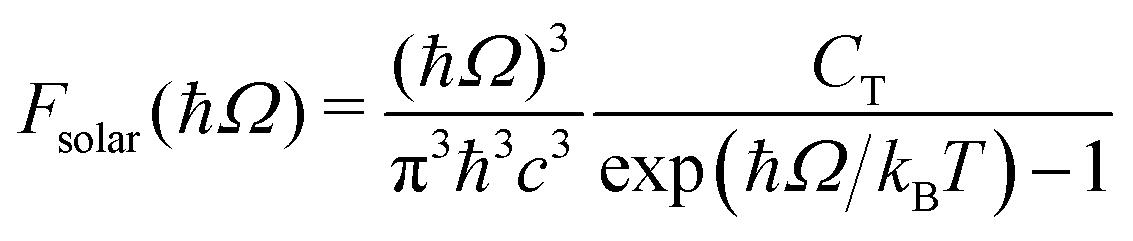Ab initio design of light absorption through silver atomic cluster decoration of TiO2†
Received
4th May 2018
, Accepted 27th June 2018
First published on 27th June 2018
Abstract
A first-principles study of the stability and optical response of subnanometer silver clusters Agn (n ≤ 5) on a TiO2(110) surface is presented. First, the adequacy of the vdW-corrected DFT-D3 approach is assessed using the domain-based pair natural orbital correlation DLPNO-CCSD(T) calculations along with the Symmetry-Adapted Perturbation Theory [SAPT(DFT)] applied to a cluster model. Next, using the DFT-D3 treatment with a periodic slab model, we analyze the interaction energies of the atomic silver clusters with the TiO2(110) surface. Finally, the hybrid HSE06 functional and a reduced density matrix treatment are applied to obtain the projected electronic density of states and photo-absorption spectra of the TiO2(110) surface, with and without adsorbed silver clusters. Our results show the stability of the supported clusters, the enhanced light absorbance intensity of the material upon their deposition, and the appearance of intense secondary broad peaks in the near-infrared and the visible regions of the spectrum, with positions depending on the size and shape of the supported clusters. The secondary peaks arise from the photo-induced transfer of electrons from intra-band valence 5s orbitals of the noble-metal cluster to 3d Ti band states of the supporting material.
1 Introduction
Due to their extraordinary stability and selectivity,1 bare metal clusters of subnanometer size composed of a few atoms have recently emerged as a new generation of catalysts2–4 and photocatalysts.5,6 The synthesis of such molecular-like species has been achieved by kinetic control using an electro-chemical technique without employing surfactants or capping agents.7 Appealing potential applications of these angstrom size objects have been found in important areas such as the development of cancer therapeutic drugs through intercalation of Ag3 clusters,7,8 efficient photocatalytic oxidation,5 carbon monoxide oxidation by deposited Agn and Aun clusters,4,9 and hydrogen photo-production by means of Ag clusters adsorbed on gold nanostructures.6 Realizing these atomic metal clusters as widely used functional materials requires them being stable and accessible through, for example, attachment to solid supports. Moreover, recently developed experimental tools have allowed “soft-landing” conditions in the deposition of metal clusters on solid surfaces to be realized.10–14 In this work, the stability of deposited Agn clusters (with n ≤ 5) on the TiO2(110) rutile surface is analyzed.
Titanium dioxide, TiO2, has wide-ranging and well-known applications in semiconductor photocatalysis (for recent reviews see, e.g., ref. 15 and 16), making it one of the most frequently studied support materials. However, TiO2 can only be activated by UV irradiation, i.e., by about 5–8% of the solar spectrum. The photocatalytic activity of TiO2 surfaces is also much influenced by the dynamics of the photo-generated holes and electrons which need to reach the surface to be catalytically active.17,18 The hole–electron recombination (about 90% of the photo-generated hole–electron pairs) in particular, severely limits the photocatalyst efficiency. As a result, the rate of formation of reaction products divided by the incident photon flow is below 10% in TiO2-based photocatalysts.15,19 A successful alternative is to decorate the surface with adsorbed metal nanoparticles.17,18 They alter the electronic band structure near the surface, increasing the photo-generation of electron–hole pairs very close to the site where the photocatalysis happens. Besides, at the metal–oxide interface a space charge or a depleted region develops that hinders electron–hole recombination. Furthermore, localized surface plasmon resonance (LSPR) at the nanoparticles induces light absorption in the visible region.17
Within this context, stable surface attachment of metal atomic clusters opens a very promising new route to photocalytic enhancement of solid surfaces such as those based on TiO2. Given their atomic scale they are ideally suited for electronic band modification at the very surface, possibly in a profitable way for photocatalytic applications. Indeed, photocatalytic quantum efficiencies increased by three orders of magnitude have already been demonstrated using small Cu clusters.5 Here, once we have analyzed their stability, we explore, using ab initio modelling, such electronic band modification induced by deposited Agn clusters (with n ≤ 5) on TiO2. Furthermore, with this applicability in mind, we have also calculated the corresponding light absorption spectra in search of prominent solar light absorption, an experimentally verifiable fingerprint to identify promising candidates for the development of photocatalytic and photovoltaic applications.
As far as the stability of the adsorbed atomic clusters is concerned, we have chosen the DFT-D3 method20,21 among the vdW-corrected DFT treatments after validation using both the domain-based pair natural orbital correlation approach DLPNO-CCSD(T)22 and the Symmetry-Adapted Perturbation Theory [SAPT(DFT)] method.23,24 Regarding the electronic band structure calculations, we have relied on the hybrid HSE06 functional25,26 since it has demonstrated renowned accuracy for TiO2 band gap calculations. Finally, to calculate the absorption spectra, we use the reduced density matrix (RDM) theory within the Redfield approximation27 combined with HSE06 calculations. This RDM treatment has been successfully applied to Agn clusters adsorbed on semiconductor silicon surfaces.28–30 In the next section, we briefly present the methods used in this study. Our results are reported in Section 3. Finally, the main conclusions are provided in Section 4.
2 Method
2.1 Structural models
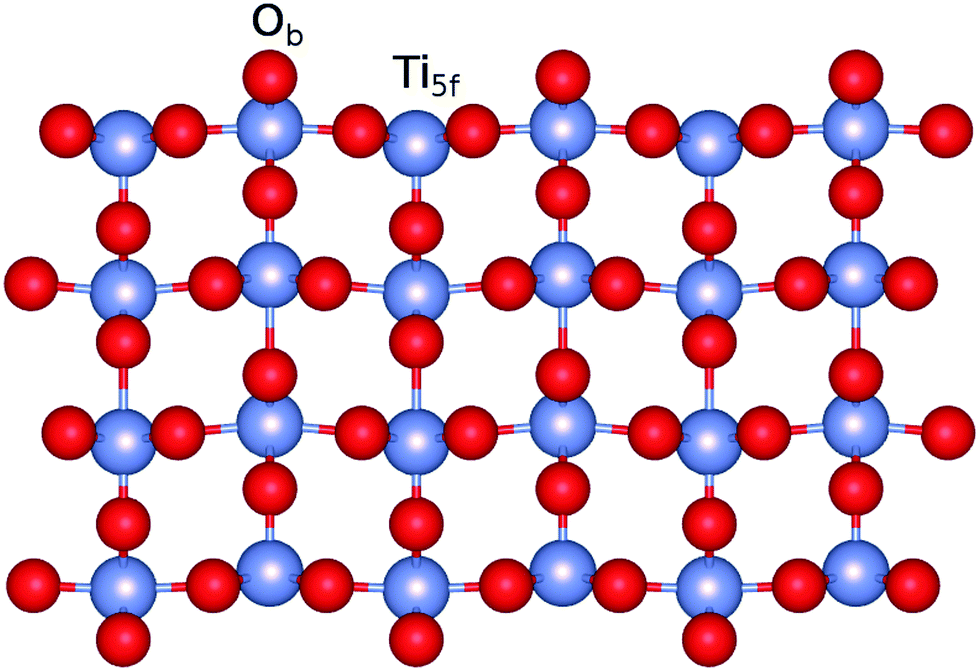
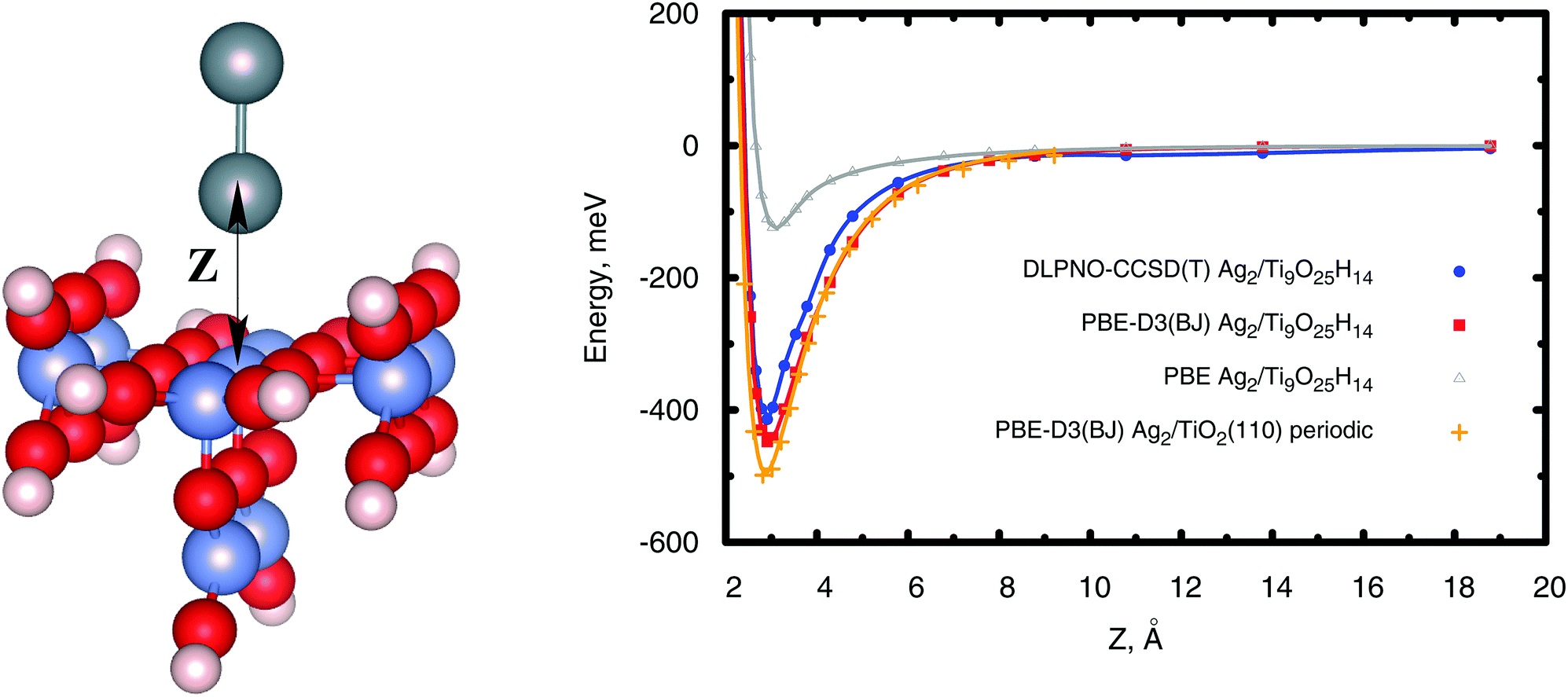
The calculations on the periodic slabs were carried out using the 4 × 2 supercell (four TiO2 trilayers giving a ca. 13 Å width slab) model shown in Fig. 1 and reported in ref. 31. The adsorption of the silver clusters has been modeled on one side of the slab and the vacuum region above the slab was 38 Å thick. This large vacuum region allowed the description of long-range tails of the cluster–surface interaction potentials while avoiding unphysical overlaps of electronic densities. Periodic calculations were carried out by freezing the positions of the O and Ti atoms to those determined by Busayaporn et al.32 using X-Ray Diffraction (XRD). Following ref. 33, the hydrogen-saturated cluster of stoichiometry Ti9O25H14 (see Fig. 2) was used in our finite cluster calculations. The geometry of the model cluster has also been frozen to the experimental geometry of the TiO2(110)-(1 × 1) surface.32 Both periodic and cluster structural models have been previously used to study the adsorption of noble gases on the same surface,34 showing a good agreement for calculated adsorption energies with experimental measurements.35
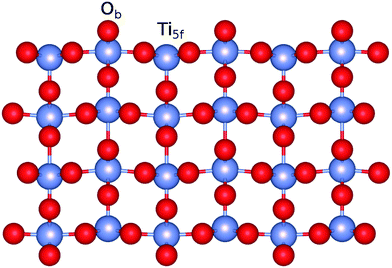 |
| | Fig. 1 Figure illustrating a side view of the slab model of the TiO2(110) surface used in this work. The positions of five-fold (Ti5f) and bridging oxygen (Ob) atoms are also indicated. | |
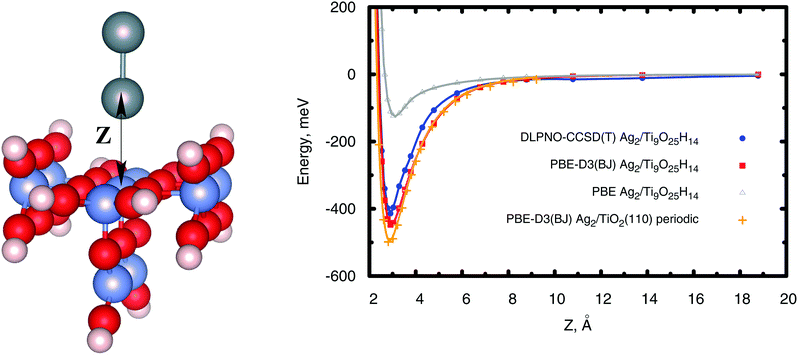 |
| | Fig. 2 Left-hand panel: Figure illustrating the hydrogen-saturated Ti9O25H14 cluster modelling the Ag2/TiO2(110) system. Gray spheres represent silver atoms while red and blue spheres stand for oxygen and titanium atoms. Hydrogen atoms saturating dangling bonds are also shown with small white spheres. Right-hand panel: Scan of the interaction energies between the Ag2 dimer and the TiO2(110) surface using different methods and both cluster (Ti9O25H14) and periodic models of the surface. | |
In periodic and cluster calculations, the Ag2 internuclear distance was fixed to the experimentally determined value36 (2.531 Å). Complementary results on the optimization of supported and unsupported Ag2 structures are presented in the ESI.†
2.2 Theoretical approaches using surface cluster models
Using the cluster shown in Fig. 2 to model the Ag2/TiO2(110) interaction, the DLPNO-CCSD(T) approach22 and the SAPT(DFT) method have been applied to assess the performance of the chosen vdW-corrected DFT treatment. The interaction energies were calculated as a function of the cluster–surface distance and included the counterpoise correction37 to account for the basis set superposition error. The frozen-core treatment was used for 140 chemical core electrons. The SAPT(DFT) method was applied adopting the Perdew–Burke–Ernzerhof (PBE) density functional.38 This method23,24,39,40 has allowed the interaction energy to be decomposed as a sum of first- and second-order interaction terms, namely first-order electrostatic Eelec and exchange Eexch, and second-order induction Eind and dispersion Edisp terms, along with their respective exchange corrections (Eexch–ind and Eexch–disp). The δ(HF) estimate41,42 of the higher-order induction plus exchange–induction contributions was added to the SAPT(DFT) interaction energies. Specifically, the δ(HF) correction is defined as,41| | | δ(HF) = Eint(HF) − Eelec(HF) − Eexch(HF) − Eind(HF) | (1) |
where Eint is the supermolecular HF interaction energy and the other terms are calculated using the HF density matrices. The dispersion and exchange–dispersion SAPT(DFT) contributions were summed to provide the total exchange–repulsion part, as were the induction, exchange–induction and δ(HF) contributions into the total induction term.
2.3 Theoretical approaches using periodic models
The periodic vdW-corrected PBE-D3 treatment has been applied to calculate interaction energies and optimized geometries after assessing its performance with the DLPNO-CCSD(T) and SAPT(DFT) approaches. Due to the underestimation of the band gap from the spurious electron self-interaction in the semilocal PBE treatment, the projected density of states and photo-adsorption spectra were instead calculated with the hybrid HSE06 screened density functional.25,26 This functional has been chosen because of its capability to provide accurate band gaps of dielectric and semiconductor compounds.43 It has also been the preferred approach in previous studies of optical and other electronic properties of TiO2.44–46 In this work, the HSE06 treatment has provided a direct band gap of 3.26 eV for the TiO2(110) surface, agreeing very well with experimental measurements for bulk rutile (3.3 ± 0.5 eV).47
2.4 Reduced density matrix treatment
2.4.1 Steady state density matrix.
In order to calculate light absorption in the system, we model the involved relaxation processes by means of the reduced density matrix (RDM) approach in the Redfield approximation27 and using an electronic basis set of orbitals generated from DFT calculations.48,49 When a monochromatic electromagnetic field of frequency Ω is applied, the evolution equation for the RDM, ρ, in the Schrödinger picture reads,| | 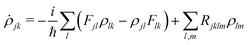 | (2) |
| | ![[scr E, script letter E]](https://www.rsc.org/images/entities/char_e140.gif) (t) = 0(eiΩt + e−iΩt) (t) = 0(eiΩt + e−iΩt) | (4) |
where ![[F with combining circumflex]](https://www.rsc.org/images/entities/i_char_0046_0302.gif) KS is the effective Kohn–Sham Hamiltonian, the index refers to the Kohn–Sham basis set,
KS is the effective Kohn–Sham Hamiltonian, the index refers to the Kohn–Sham basis set, ![[D with combining circumflex]](https://www.rsc.org/images/entities/b_char_0044_0302.gif) is the electric dipole moment operator and Rjklm stand for Kohn–Sham components of the relaxation tensor (Redfield coefficients). The Redfield coefficients are defined as in ref. 27 and are implemented as described in ref. 48. This approximation is valid for medium phenomena relaxing over times that are long compared with transient energy exchange between the adsorbate and its medium.
is the electric dipole moment operator and Rjklm stand for Kohn–Sham components of the relaxation tensor (Redfield coefficients). The Redfield coefficients are defined as in ref. 27 and are implemented as described in ref. 48. This approximation is valid for medium phenomena relaxing over times that are long compared with transient energy exchange between the adsorbate and its medium.
Within the Redfield approximation used here, the relaxation tensor not only describes the effect of the fast electronic dissipation due to electronic fluctuations in the medium at long times but also the relatively slow relaxation owing to the atomic lattice vibrations. It is convenient to change to a rotating frame accounting for the electromagnetic field oscillation,
| | ![[small rho, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e4.gif) ij(t) = ρij(t) ij(t) = ρij(t)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(iΩt), εi > εj exp(iΩt), εi > εj | (5) |
| | | ij(t) = ρij(t)exp(−iΩt), εi < εj | (6) |
| | | ii(t) = ρii(t) | (7) |
where
εi is the energy of the
ith Kohn–Sham orbital. After averaging out over time the fast terms in the equation of motion for the RDM, its stationary-state solution for the diagonal elements reads,
48,50| | 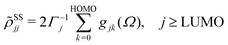 | (8) |
| | 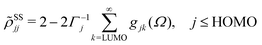 | (9) |
where LUMO stands for the lowest unoccupied molecular orbital and HOMO denotes the highest occupied one.
Γj is a depopulation rate and
| | 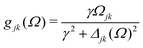 | (10) |
Here
γ is a decoherence rate,
Ωjk are the Rabi frequencies given by
Ωjk = −
Djk·
0/
ħ and
Δjk(
Ω) =
Ω − (
εj −
εk) are the detunings. The diagonal elements provide the populations of the KS orbitals. The population relaxation rate
ħΓ and the decoherence rate
ħγ were not computed but fixed to values of 0.15 meV and 150 meV (27 ps and 27 fs) and have been chosen to be of the order of rates for semiconductors following decay of phonon and electronic density excitations (see,
e.g.,
ref. 51 for values from measurements of adsorbates on TiO
2).
As far as the populations are concerned, an averaging over many light periods, as happens to be the case in the usual measurements, is equivalent to take the stationary solutions in the rotating frame.48
2.4.2 Absorption spectrum.
In terms of the stationary populations the absorbance is given by,29,30,52| | 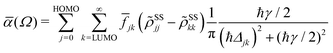 | (11) |
where ![[f with combining macron]](https://www.rsc.org/images/entities/i_char_0066_0304.gif) jk is an oscillator strength per active electron. This is the purely dissipative contribution to the absorbance. We are assuming a thin slab neglecting dispersive effects, i.e., assuming refraction index ca. 1. The solar flux absorption spectrum is then given by,
jk is an oscillator strength per active electron. This is the purely dissipative contribution to the absorbance. We are assuming a thin slab neglecting dispersive effects, i.e., assuming refraction index ca. 1. The solar flux absorption spectrum is then given by,| | F(ħΩ) = ![[small alpha, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c2.gif) (Ω)Fsolar(ħΩ)ħΩ (Ω)Fsolar(ħΩ)ħΩ | (12) |
where the solar flux is approximated by the black body flux distribution, normalized to an incident photon flux of 1 kW m−2,| | 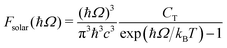 | (13) |
with CT the flux normalization constant and the temperature T set to 5800 K.
2.5 Computational details
2.5.1 Surface cluster calculations.
Surface cluster calculations were carried out employing correlation-consistent basis sets such as the (augmented) polarized correlation-consistent triple-ζ (aug-cc-pVTZ) basis of Woon and Dunning, Jr.,53 as reported in ref. 33 for oxygen and titanium atoms, while the aug-cc-pVTZ-PP basis was used for silver,54,55 including a small-core (19-valence-electron) relativistic pseudopotential.56 When applying the DLPNO-CCSD(T) and PBE-D3 treatments, the ORCA package of programs57 was employed (version 4.0.1.2).
The SAPT(PBE) calculations were performed with the MOLPRO package58 using the density-fitting technique39 and adopting the same computational setup as ref. 33. The exchange–correlation PBE potential was asymptotically corrected59 with the ionization potential value reported in the NIST Chemistry Web Book for the silver dimer (7.66 eV from ref. 60). For the cluster model of the TiO2(110) surface, an ionization potential value of 5.0 eV was chosen on the basis of work function values characterizing TiO2(110) surfaces61 (in between 4.9 and 5.5 eV).
2.5.2 Periodic calculations.
To perform the periodic calculations, the computational setup reported in a previous study of the He/TiO2(110) interaction31 was applied. For this purpose, we used the Vienna Ab initio Simulation Package (VASP 5.4.4).62,63 To describe the electron–ion interaction, the projector augmented-wave method was applied,63,64 using the PAW-PBE pseudopotentials supplied with the same code. The electrons from O(2s, 2p), Ti(3s, 4s, 3p, 3d) and Ag(4d, 5s) orbitals were treated explicitly as valence electrons. As in ref. 31, we used plane wave basis sets with a kinetic energy cutoff of 700 eV and a Gaussian smearing of 0.05 eV to account for partial occupancies, with the Brillouin zone sampled at the Γ point only. Test calculations showed that interaction energies at the potential minimum using a 5 × 5 × 1 Monkhorst–Pack65k-point mesh are within ca. 0.005 eV of those calculated at the Γ point (see ESI†).
All interaction energies were determined using the supermolecular approach as follows,
| | | Etotint = EAgn/TiO2(110) − EAgn − ETiO2(110) | (14) |
where
EAgn/TiO2(110) is the total energy of the system,
ETiO2(110) stands for the energy of the substrate, and
EAgn denotes the energy of the bare silver cluster.
3 Results and discussion
3.1 Assessment of the PBE-D3(BJ) approach at DLPNO-CCSD(T) and SAPT(DFT) levels
Cost-efficient calculations accounting for the extended nature of cluster–surface systems use vdW-corrected DFT methods. When applying a vdW-corrected DFT approach, benchmarking against accurate reference values is crucial. The coupled-cluster single, doubles and non-iterative triples [CCSD(T)] approach is ideally suited for this purpose. When applied to clusters modelling the TiO2(110) surface, the incremental method66 has allowed the CCSD(T) approach to be applied to adsorbate-TiO2(110) systems (see e.g., ref. 12 and 33), with the employment of localized orbitals making it very efficient. Using the domain-based local pair natural orbital approximation, DLPNO-CCSD(T)22 is another ab initio option that, when applied to molecules adsorbed on TiO2(110), has provided excellent (within 1 kcal mol−1) agreement with experimental data.67 In addition, previous work on the He–TiO2(110) system using cluster models has shown that the SAPT(DFT) approach provides interaction energies very close to those obtained at the CCSD(T) level.12,33 The incremental approach has been also applied to describe the Ag2/graphene interaction at the CCSD(T) level,68 with results showing overall good performance with the PBE-D3 scheme including the Becke-Johnson (BJ) damping of the D3 dispersion correction.20,21 The PBE-D3 approach has thus been chosen for our periodic calculations while the DLPNO-CCSD(T) and SAPT(DFT) methods are used to validate its adequacy.
From Fig. 2 and Table 1 it can be observed that the PBE-D3(BJ) approach closely reproduces the DLPNO-CCSD(T) and SAPT(DFT) results, with the well-depth values agreeing to within 10%. As can be also seen in Fig. 2, the PBE-D3(BJ) potential energy curves obtained using periodic and cluster models are rather close to each other, confirming the adequacy of both the cluster model and the electronic basis set to provide reference DLPNO-CCSD(T) values.
Table 1 Decomposition of the Ag2/(Ti9O25H14) interaction energy using the SAPT(PBE) method. Ze stands for the equilibrium distance between the Ag atom closest to the surface and the five-fold coordinated Ti5f atom (see the left-hand panel of Fig. 2). Well-depths and Ze values of DLPNO-CCSD(T) and PBE-D3(BJ) potential energy curves are also shown (see the right-hand panel of Fig. 2)
|
|
SAPT(PBE) |
DLPNO-CCSD(T) |
PBE-D3(BJ) |
|
Z
e, Å |
2.86 |
2.90 |
2.92 |
|
E
tot, meV |
−429 |
−414 |
−447 |
|
E
elec, meV |
−358 |
— |
— |
|
E
exch–rep, meV |
1141 |
— |
— |
|
E
ind, meV |
−574 |
— |
− |
|
E
disp, meV |
−648 |
— |
— |
Table 1 also shows a SAPT-based decomposition of the total energy to the electrostatic Eelec, exchange–repulsion Eexch–rep, induction Eind, and dispersion Edisp contributions. For the perpendicular orientation of the silver dimer, the attractive interaction is dominated by both dispersion and induction contributions, as can be expected from the pronounced polarization of the silver electronic density towards the titania surface. The importance of including dispersion corrections is also clear when comparing PBE and PBE-D3(BJ) interaction potentials, with the well-depth of the former being much smaller (−447 vs. −124 meV). The crucial role of the dispersion contribution has been also found for the interaction of silver atomic clusters with a TiO2 (anatase) surface.69
3.2 Interaction potentials and optimized geometries of Agn clusters on the TiO2(110) surface
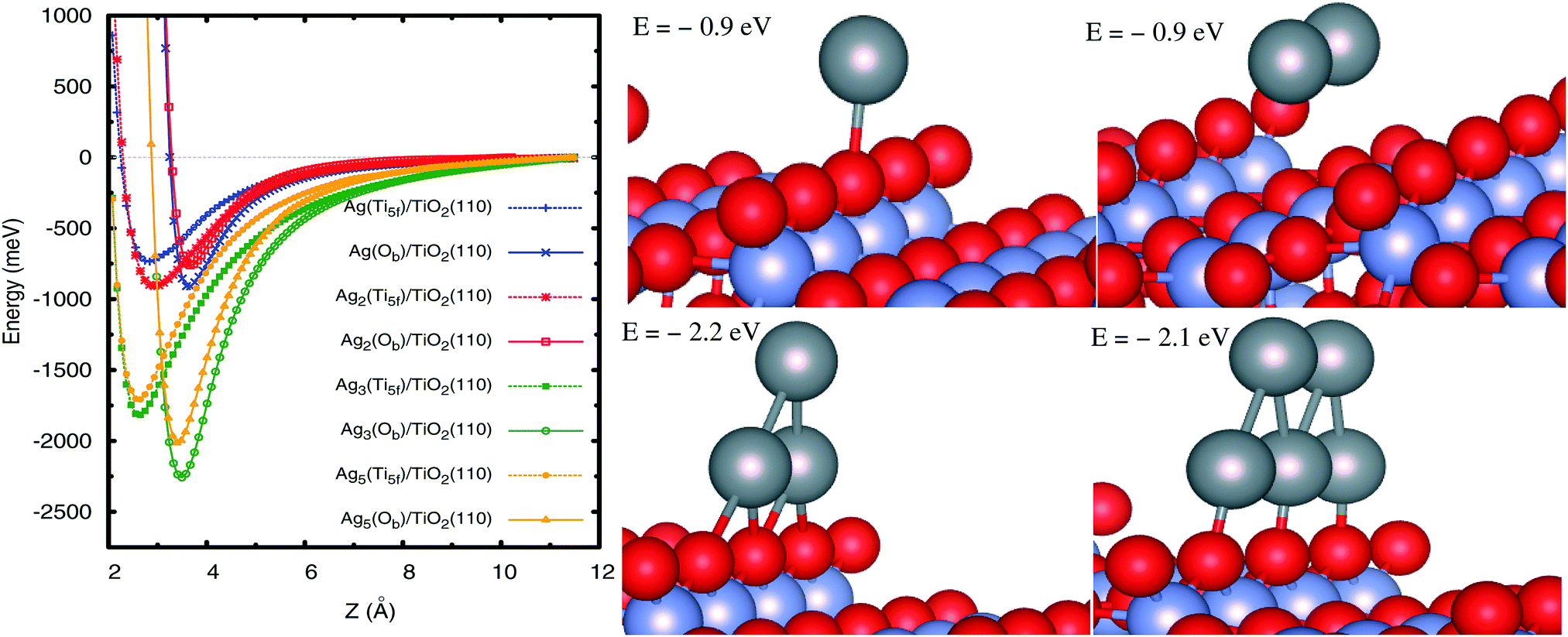
Once the PBE-D3(BJ) approach has been validated, we analyze the Agn–TiO2(110) interaction by focusing on the supported, not yet explored, Ag3 and Ag5 clusters. First, the geometries of the free-standing clusters were optimized at the second-order Möller–Plesset perturbation theory (MP2) level, resulting in isosceles planar triangular (Ag3) and trapezoidal (Ag5) structures (see ESI†). These configurations were found as the most stable in previous studies using coupled-cluster approaches.70,71 A planar trapezoidal structure for Ag5 was also revealed from experimental observations.72 Keeping these geometries fixed, Fig. 3 shows scans of the interaction potentials as a function of the adsorbate–surface distance. We have considered two representative adsorption sites in which the cluster center-of-mass is located on top of either the fivefold coordination Ti atom (Ti5f site from now on) or the bridging O atom (Ob site). The energies at the potential minima are presented in Table 2. As can be seen in Fig. 3 (right-hand panel), we have chosen configurations in which the planar structures are oriented perpendicular to the surface plane while the internuclear Ag–Ag (in Ag2/Ag3) and Ag–Ag–Ag (in Ag5) axes lie parallel to the row of Ti5f atoms and Ob atoms. These configurations are found to be more stable for Ag3 and one Ag5 isomers (see below). We have also explored configurations for supported Ag3 clusters which are parallel to the surface plane, providing smaller adsorption energies (by 0.6 eV) when considering the adsorption Ob site. However, the internuclear Ag–Ag axis for the most stable Ag2 isomer is oriented perpendicular to the Ti5f row, and not shown here. For brevity, complementary results on this structure have been moved to ESI.†
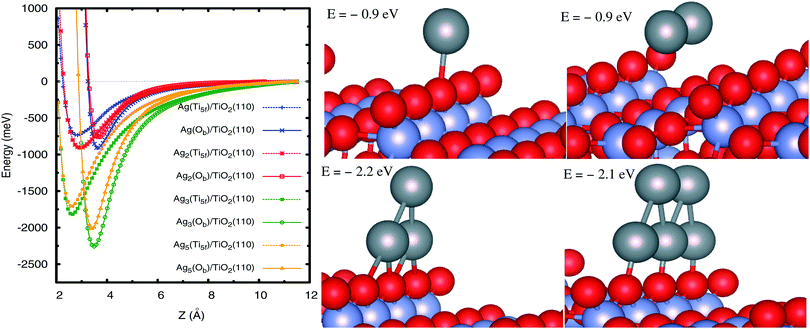 |
| | Fig. 3 Left-hand panel: Radial scan of the interaction energies between atomic silver clusters Agn and the TiO2(110) surface, as calculated with the PBE-D3(BJ) method. The cluster center-of-mass is located on top of either the five-fold titanium atom Ti5f (Ti5f site, dashed lines) or the bridging oxygen atom Ob (Ob site, full lines). Z stands for the distance between the silver atom closest to the surface and the Ti5f atom along the surface normal direction. Right-hand panel: Geometries of Agn clusters on the Ob site. For both adsorption sites, the internuclear Ag–Ag (in Ag2/Ag3) and Ag–Ag–Ag (in Ag5) axes lie parallel to the direction of the row of Ti5f and Ob atoms. Gray spheres represent silver atoms while red and blue spheres stand for oxygen and titanium atoms. | |
Table 2 Energies (in meV) at the potential minima for the Agn–TiO2(110) interaction, considering Ti5f and Ob adsorption sites (see Fig. 3). Values in parenthesis stand for the vertical distance (in Å) from the Ag atom closest to the plane where the Ti5f atoms are located
| Site |
Ag |
Ag2 |
Ag3 |
Ag5 |
| Ti5f |
−733 (2.83) |
−894 (2.94) |
−1811 (2.59) |
−1694 (2.61) |
| Ob |
−939 (3.64) |
−756 (3.68) |
−2251 (3.45) |
−2016 (3.42) |
From Fig. 3 it can be observed that the Ob site is clearly favored over the Ti5f position for all Agn clusters composed of an odd number of atoms n, with energies at the potential minima differing by up to ca. 0.4 eV (see Table 2). The same preferred adsorption site is found at the PBE level, with the D3(BJ) dispersion correction making the energy differences smaller. For instance, for the Ag3 cluster, the energies of the Ob and Ti5f isomers are about −0.8 and −1.5 eV at the PBE level, with an energy difference of 0.7 eV, reduced to ca. 0.4 eV upon inclusion of the D3(BJ) dispersion (see Table 2).
As can be seen in Table 2, the order of stabilities is inverse for the closed-shell Ag2 case, as also found for the closed-shell Xe adatom (see ESI†). As discussed in previous work on noble gases on TiO212,33,34,73 and Ag2 on graphene,68 the Ag2–TiO2 preferential adsorption is also significantly influenced by a subtle balance between short-range exchange–repulsion and long-range dispersion contributions: the exchange–repulsion is smaller on top of the Ti5f site, allowing the Ag atoms to benefit from the attractive dispersion interaction with Ob atoms at both sides of the Ti5f atom. The analysis of the projected density of states (PDOS) for the silver dimer (see Fig. 5) indicates that there is chemical mixing between Ag(4d) orbitals and O(2p) states of the support but no net charge-transfer. This finding is consistent with previous work on the Ag4–TiO2 (anatase) interaction.69
Obviously, the outer electrons of open-shell adsorbates are more prone to participate in chemical bonding with the support either by sharing or transferring them. Atomic charges were estimated with the Bader decomposition scheme.74 The Bader charge on open-shell silver clusters was positive, having a value of about +0.8|e|, which is twice as large as found for the closed-shell Ag2 cluster (+0.4|e| only). The TiO2 surface thus attracts more electronic charge from open-shell silver clusters.
In going from a single Ag adatom to Ag3 and Ag5 clusters, it can be observed that the well-depth values increase steeply (see Fig. 3 and Table 2), with the preferential adsorption along the row of Ob atoms being also very clear. From the PDOS of the supported open-shell species (see Fig. 5), the mixing between Ag(4d) and O(2p) orbitals (valence band) and between Ag(5s) and Ti(3d) orbitals (conduction band) can be observed, indicating the partial covalent nature of the adsorbate–surface interaction. However, the non-covalent vdW-type dispersion interaction is essential, contributing to the increase of the binding energy by 40–45% for Ag3 and Ag5 clusters.
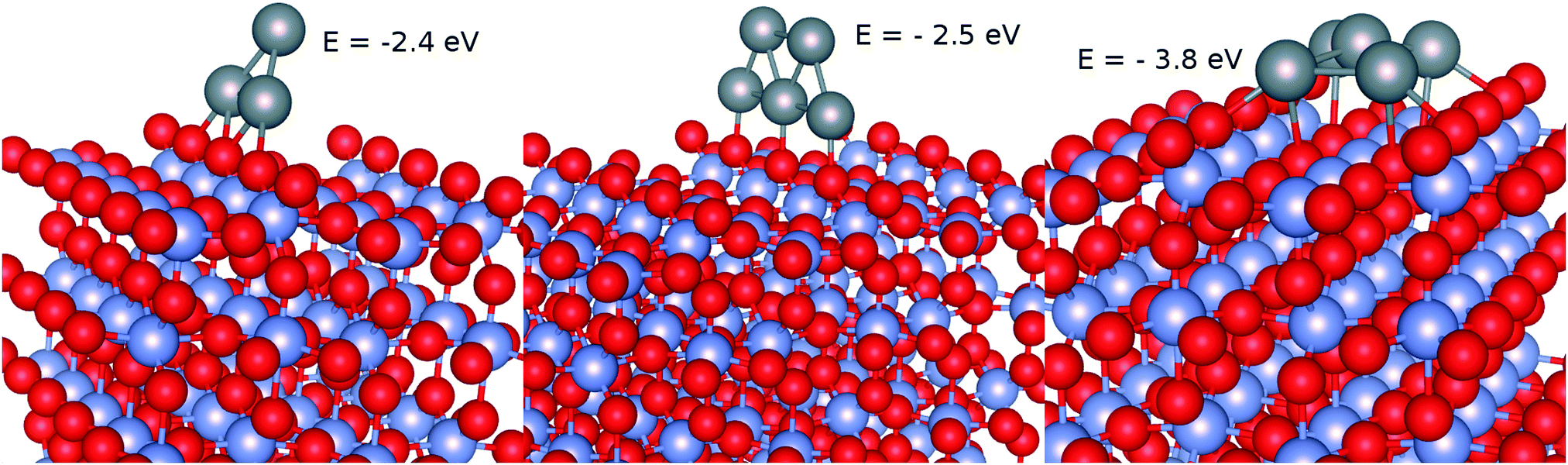
As a second step, the geometries of supported Ag3 and Ag5 clusters were relaxed starting with those optimized for the free clusters with the PBE-D3(BJ) approach. As can can be seen in Fig. 4, both the (triangular) structure and the interaction energy (−2.5 eV) of the most stable Ag3 isomer remain rather similar to those corresponding to the isolated cluster. Upon adsorption, the triangular Ag3 structure becomes just a little bit tilted towards the Ti5f site, signaling stability of the supported cluster. Also, the structure of the trapezoidal Ag5 isomer is only slightly modified from that of the isolated cluster (see the middle panel of Fig. 4). The most pronounced change is in the stretching of the Ag–Ag bonds so that three Ag atoms now lie almost aligned with the Ob atoms along the surface normal direction. As found for the supported Ag3 cluster, the diffusion of the Ag5 cluster along the Ob row is rather constrained due to high energy barriers. For instance, when the cluster moves such that its center-of-mass lies in between two Ob atoms, the energy penalty is above 0.8 eV, indicating constrained diffusion and subsequent aggregation. Interestingly, we also found a more stable Ag5 isomer (by ca. 1.3 eV) for which the planar trapezoid isomer transforms into a (slightly) pyramidal structure with four Ag atoms lying at the corners of a rectangle centered by one Ag atom atop a Ti5f atom (see the right-hand panel of Fig. 4). Future work will consider the minimum-energy pathway from the trapezoidal to the pyramidal isomer.
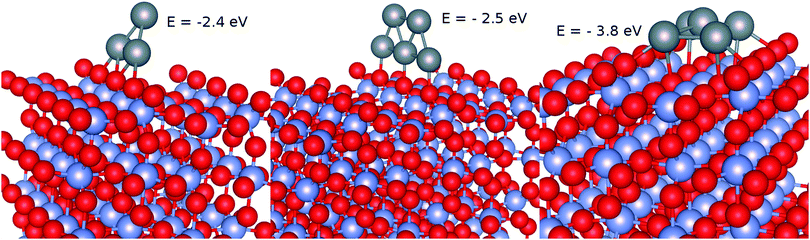 |
| | Fig. 4 Optimized geometries and binding energies of Agn clusters (n ≤ 5) upon adsorption on the TiO2(110) surface. Gray spheres represent silver atoms while red and blue spheres stand for oxygen and titanium atoms. | |
3.3 Projected electronic density of states and absorption spectra
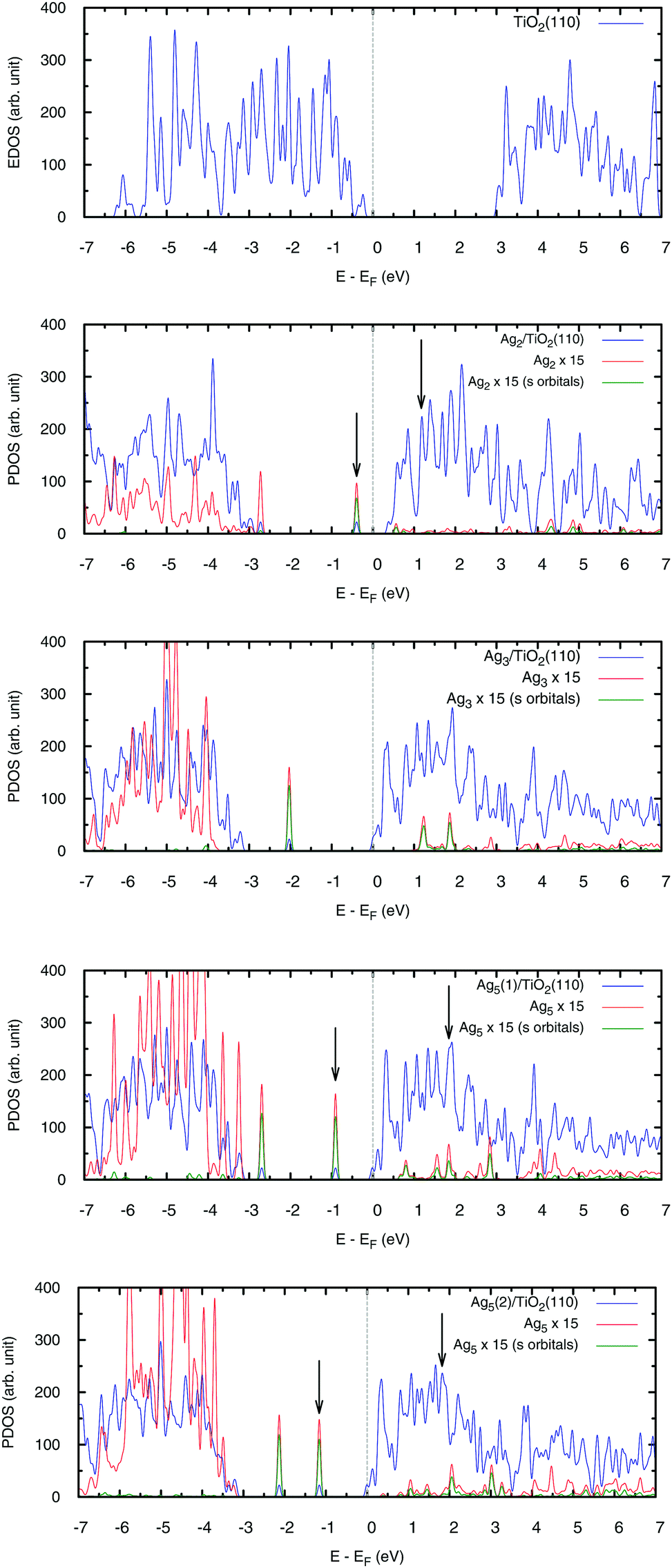
Using the cluster structures optimized at the PBE-D3(BJ) level, Fig. 5 shows the electronic density of states (EDOS) of the bare TiO2(110) surface and the PDOS for the Agn/TiO2(110) systems, as calculated with the hybrid HSE06 density functional, while the absorption spectra are presented in Fig. 6. As can be observed in Fig. 5 (upper panel), our computational set-up provides an estimation of the band gap (3.3 eV) that agrees well with the experimental value (3.3 ± 0.5 eV from ref. 47). From Fig. 5, it can be seen that the supported Agn clusters add one or two sharp peaks in the band-gap region of TiO2. These sharp peaks correspond to localized states in the Ag atomic clusters, as attested by the PDOS onto the Ag orbitals shown in Fig. 5. More specifically, these peaks arise from frontier orbitals mainly made of Ag(5s) orbitals but involving also hybridization with Ag(4p) orbitals. Notice also, that the Agn atomic clusters add intra-gap states closer to the bottom of the TiO2 conduction band with increasing n. In fact, for n = 3 and 5, the conduction band begins to be populated giving an incipient metallic character to the system.
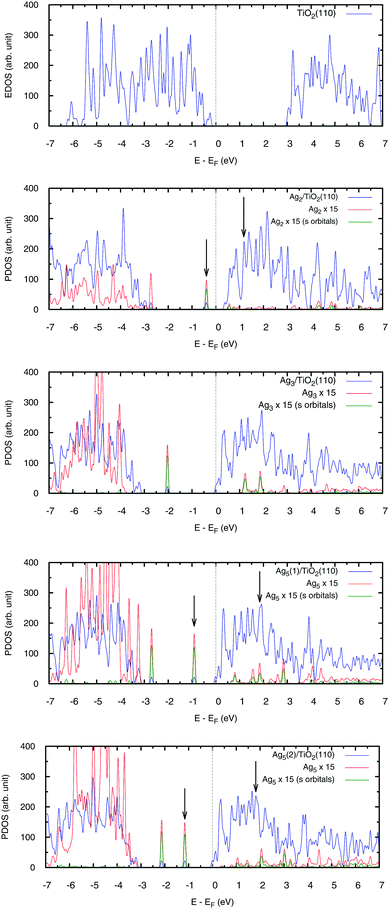 |
| | Fig. 5 EDOS of the TiO2(110) surface and PDOS of the most stable Agn–TiO2(110) systems obtained with the hybrid HSE06 functional, with Ag5(1) and Ag5(2) standing for the trapezoidal and pyramidal isomers (see Fig. 4). The zero of energy corresponds to the Fermi level, defined here as the highest occupied level. The arrows indicate the energies of the most responsible orbitals for the intense secondary peaks in the photo-absorption spectra (see Fig. 6). | |
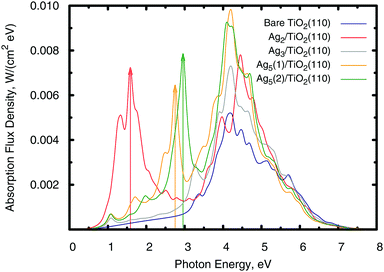 |
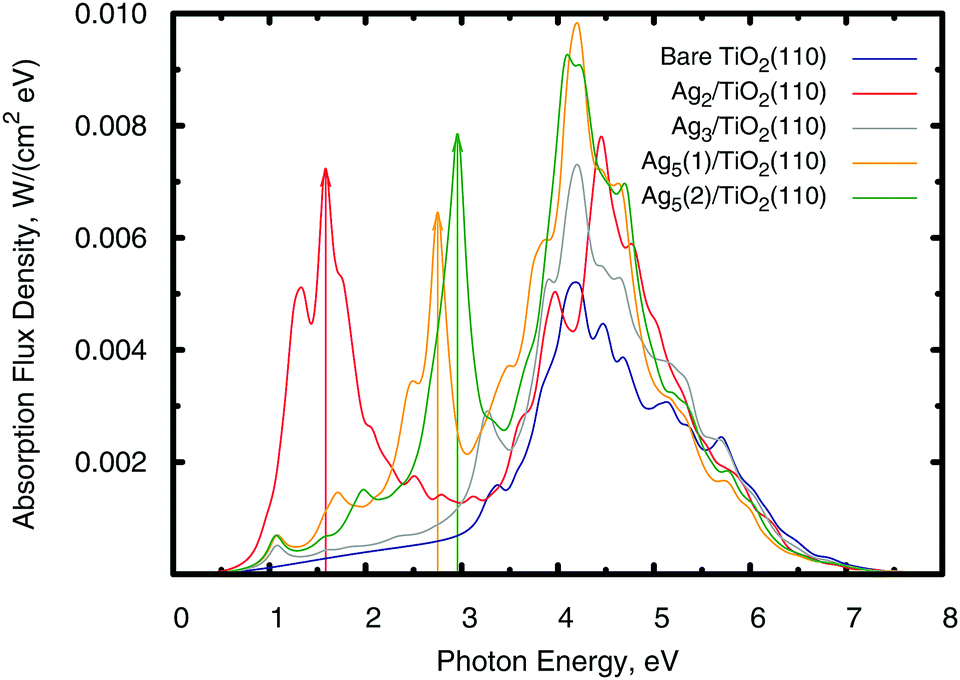
| | Fig. 6 Photo-absorption spectra of the TiO2(110) surface, without adsorbates (shown in blue color) and with adsorbed Agn clusters. The most stable supported Agn clusters were considered, with Ag5(1) and Ag5(2) standing for the trapezoidal and pyramidal isomers (see Fig. 4). The arrows indicate the positions of the most intense secondary peaks across the near-infrared and visible regions of the solar spectrum. | |
The intra-gap localized states have deep consequences for the light absorption behaviour. As shown in Fig. 6, the photo-absorption spectra develop broad absorption peaks in the visible (n = 5) and near-infrared (n = 2) spectra owing to photo-induced electronic transitions from the highest-energy localized orbitals to the conduction Ti(3d) states. The energy position of the frontier and the Ti(3d) orbitals involved in the transition with the largest oscillation strength are indicated by arrows in Fig. 5. In the case of the Ag3 cluster, its only intra-gap state is far enough from the conduction band edge to contribute mainly to the absorption in the UV region. In this respect, notice also that the Ag2 and Ag5 clusters also enhance UV absorption in part owing to the contribution coming from the second lower energy intra-gap states present in these clusters. However, the observed enhancement of UV absorption comes not only because of the intra-gap peaks but also from the the energy band changes that the atomic clusters produce in the system.
The same electron transfer mechanisms found here for visible/near-infrared absorption have been proposed as being responsible for the photocatalytic hydrogen production enhancement observed after deposition of gold clusters onto TiO2,75 but instead of photo-transfer from localized orbitals of the adsorbate, the gold clusters are large enough to develop their own conduction band as corroborated by the observation of a localized surface plasmon resonance band in the reflectivity. Notice also, that the localized nature of the Agn intra-gap states probably implies long lived hole excitations. Long lived photo-excitations are relevant to extract charge from photovoltaic devices. In this respect, the reduced density matrix treatment provides a pathway to calculate the hole lifetime, as previously shown for the Ag3 cluster deposited on a Si(111) surface.28 By the same token, the atomic clusters lack a properly developed populated conduction band so that they cannot sustain localized surface plasmons by themselves. However, since the Agn clusters give an incipient metal character to the system, probably already at room temperature, properly nanostructured Agn–TiO2 may sustain localized surface plasmons, which have a well demonstrated beneficial effect in photovoltaic devices.76
Before concluding this section, we note that an interesting future direction of research would be to study the influence of the dispersion on single energy levels and the EDOS. For this purpose, it would be necessary to apply dispersion corrections depending on the electronic density such as the non-local vdW-DF77 and vdW-DF278 approaches. Work along this line is currently in progress.
4 Conclusions
Motivated by the extraordinary properties of sub-nanometer silver clusters, we have presented a first-principles study of the stability and light absorption response of Agn clusters (n ≤ 5) upon deposition on a TiO2 surface. The vdW-corrected DFT-D3 scheme applied to analyze their stability has been first validated using a local version of the CCSD(T) method as well as the SAPT(DFT) approach, demonstrating excellent agreement (to within 10%). The DFT-D3 results show that the silver clusters are strongly bound to the support, with binding energies ranging from −0.9 eV for a single adatom to −3.8 eV for one Ag5 isomer. The adsorbate–surface interaction is of polar-covalent nature, with the support attracting electronic charge from the cluster. The vdW dispersion also plays a crucial role, increasing very significantly the binding energies. The optimized structures of supported clusters differ slightly from those of their bare counterparts with the exception of one Ag5 isomer. High energy penalties are found for the diffusion of the supported clusters so that high temperatures are predicted for their aggregation.
Using hybrid DFT calculations we have studied the influence of the Ag atomic clusters on the electronic density of states. One or two (depending on the number of Ag atoms) localized intra-gap states develop while at the same time the highest occupied level (at zero temperature) strongly shifts towards the system conduction band so that for n = 3 and 5 the initial conduction band state becomes populated.
Applying a scheme combining the hybrid DFT calculations with a reduced density matrix treatment, we have analyzed the light absorption response of the Agn–TiO2 system. Besides increasing the intensity of the TiO2 absorption spectra in the UV region, intense broad absorption peaks are also found in the infrared (the Ag2–TiO2 system) and the visible (the Ag5–TiO2 system) regions: an electron “jumps” from the Agn atomic cluster to the system conduction band leaving a hole in the cluster, when a photon is absorbed. The Ag5-induced separation of photo-generated electrons and holes is expected to have a pronounced influence on the visible light photocatalytic activity of the modified TiO2 material since hole–electron recombination is a key factor limiting the photocatalyst efficiency of unmodified TiO2. Other surface locations for the atomic silver clusters are possible, with similar binding energies, and can be studied with the same methods. It is expected that the general features of light absorbance described here will remain the same for related structures. Also, as a “reporter” of the photocatalytic activity of the modified TiO2 material, the photoreactivity of molecular oxygen adsorbed on the supported clusters will be addressed in future studies, following pioneering first-principles studies of O2 photodesorption from bare TiO2 surfaces,79,80 including cluster-based embedding techniques.81
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work has been partly supported by the Spanish Agencia Estatal de Investigación (AEI) and the Fondo Europeo de Desarrollo Regional (FEDER, UE) under Grant No. MAT2016-75354-P and COST Action CM1405 “Molecules in Motion” (MOLIM). The CESGA super-computer center (Spain) is acknowledged for having provided the computational resources used in this work. Work by D. A. M. has been partly supported by the National Science Foundation of the USA under grant. no. CHE-1445825 and by the HPC Computing Center of the University of Florida. MPdLC is greatly thankful to Arturo M. López-Quintela for very helpful suggestions and discussions.
References
-
Y. Piñeiro, D. Buceta, J. Rivas and A. M. López-Quintela, in Metal Nanoparticles and Clusters, ed. F. Deepak, Springer, 2018, pp. 1–30 Search PubMed.
- N. Vilar-Vidal, J. Rivas and M. A. López-Quintela, ACS Catal., 2012, 2, 1693–1697 CrossRef.
- E. C. Tyo and S. Vajda, Nat. Nanotechnol., 2015, 10, 577–588 CrossRef PubMed.
- P.-T. Chen, E. C. Tyo, M. Hayashi, M. J. Pellin, O. Safonova, M. Nachtegaal, J. A. van Bokhoven, S. Vajda and P. Zapol, J. Phys. Chem. C, 2017, 121, 6614–6625 CrossRef.
- N. Vilar-Vidal, J. R. Rey and M. A. López Quintela, Small, 2014, 10, 3632–3636 CrossRef PubMed.
- Y. A. Attia, D. Buceta, C. Blanco-Varela, M. B. Mohamed, G. Barone and M. A. López-Quintela, J. Am. Chem. Soc., 2014, 136, 1182–1185 CrossRef PubMed.
- D. Buceta, N. Busto, G. Barone, J. M. Leal, F. Domínguez, L. J. Giovanetti, F. G. Requejo, B. García and M. A. López-Quintela, Angew. Chem., Int. Ed., 2015, 54, 7612–7616 CrossRef PubMed.
- J. Neissa, C. Pérez-Arnaiz, V. Porto, N. Busto, E. Borrajo, J. M. Leal, M. A. López-Quintela, B. García and F. Domínguez, Chem. Sci., 2015, 6, 6717–6724 RSC.
- S. Lee, C. Fan, T. Wu and S. L. Anderson, J. Am. Chem. Soc., 2004, 126, 5682–5683 CrossRef PubMed.
- E. Loginov, L. F. Gómez and A. F. Vilesov, J. Phys. Chem. A, 2011, 115, 7199–7204 CrossRef PubMed.
- A. Volk, P. Thaler, M. Koch, E. Fisslthaler, W. Grogger and W. E. Ernst, J. Chem. Phys., 2013, 138, 214312 CrossRef PubMed.
- M. P. de Lara-Castells, N. F. Aguirre, H. Stoll, A. O. Mitrushchenkov, D. Mateo and M. Pi, J. Chem. Phys., 2015, 142, 131101 CrossRef PubMed.
- Q. Wu, C. J. Ridge, S. Zhao, D. Zakharov, J. Cen, X. Tong, E. Connors, D. Su, E. A. Stach, C. M. Lindsay and A. Orlov, J. Phys. Chem. Lett., 2016, 7, 2910–2914 CrossRef PubMed.
- R. Fernández-Perea, Luis F. Gómez, C. Cabrillo, M. Pi, A. O. Mitrushchenkov, A. F. Vilesov and M. P. de Lara-Castells, J. Phys. Chem. C, 2017, 121, 22248–22257 CrossRef.
- Q. Guo, C. Zhou, Z. Ma, Z. Ren, H. Fan and X. Yang, Chem. Soc. Rev., 2016, 45, 3701–3730 RSC.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef PubMed.
- Z. Zhang and J. T. Yates Jr., Chem. Rev., 2012, 112, 5520–5551 CrossRef PubMed.
- M. A. Henderson, Surf. Sci. Rep., 2011, 66, 185–197 CrossRef.
- P. Salvador and C. Gutierrez, J. Phys. Chem., 1984, 88, 3696–3698 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed.
- A. J. Misquitta, B. Jeziorski and K. Szalewicz, Phys. Rev. Lett., 2003, 91, 033201 CrossRef PubMed.
- A. Heßelmann and G. Jansen, Chem. Phys. Lett., 2003, 367, 778–784 CrossRef.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
-
V. May and O. Kühn, Charge and Energy Transfer Dynamics in Molecular Systems, Wiley-VCH, 2011 Search PubMed.
- D. S. Kilin and D. A. Micha, J. Phys. Chem. Lett., 2010, 1, 1073–1077 CrossRef.
- T. Vazhappilly, D. S. Kilin and D. A. Micha, J. Phys. Chem. C, 2012, 116, 25525–25536 CrossRef.
- R. H. Hembree, T. Vazhappilly and D. A. Micha, J. Chem. Phys., 2017, 147, 224703 CrossRef PubMed.
- N. F. Aguirre, D. Mateo, A. O. Mitrushchenkov, M. Pi and M. P. de Lara-Castells, J. Chem. Phys., 2012, 136, 124703 CrossRef PubMed.
- W. Busayaporn, X. Torrelles, A. Wander, S. Tomić, A. Ernst, B. Montanari, N. M. Harrison, O. Bikondoa, I. Jourmard and J. Zegenhagen,
et al.
, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 153404 CrossRef.
- M. P. de Lara-Castells, H. Stoll and A. O. Mitrushchenkov, J. Phys. Chem. A, 2014, 118, 6367–6384 CrossRef PubMed.
- A. A. Tamijani, A. Salam and M. P. de Lara-Castells, J. Phys. Chem. C, 2016, 120, 18126–18139 CrossRef.
- N. G. Petrik and G. A. Kimmel, Phys. Chem. Chem. Phys., 2014, 16, 2338–2346 RSC.
- B. Simard, P. A. Hackett, A. M. James and P. R. R. Langridge-Smith, Chem. Phys. Lett., 1991, 186, 415 CrossRef.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- A. Heßelmann, G. Jansen and M. Schütz, J. Chem. Phys., 2005, 122, 014103 CrossRef PubMed.
- A. J. Misquitta, R. Podeszwa, B. Jeziorski and K. Szalewicz, J. Chem. Phys., 2005, 123, 214103 CrossRef PubMed.
- K. Patkowski, K. Szalewicz and B. Jeziorski, J. Chem. Phys., 2006, 125, 154107 CrossRef PubMed.
-
B. Jeziorski, R. Moszynski, A. Ratkiewicz, S. Rybak, K. Szalewicz and H. L. Williams, in Mehods and Techniques in Computational Chemistry; METECC94, ed. E. Clementi and D. Reidel, Publishing Company, STEF, Cagliari, 1993, vol. B, p. 79 Search PubMed.
- P. Deák, B. Aradi and T. Frauenheim, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155207 CrossRef.
- J. Anderson, V. de Walle and G. Chris, Phys. Status Solidi B, 2010, 248, 799–804 Search PubMed.
- A. Janotti, J. B. Varley, P. Rinke, N. Umezawa, G. Kresse and C. G. Van de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085212 CrossRef.
- V. Francesc, L. Oriol, C. K. Kyoung, Y. L. Jin and I. Francesc, J. Comput. Chem., 2017, 38, 781–789 CrossRef PubMed.
- Y. Tezuka, S. Shin, T. Ishii, T. Ejima, S. Suzuki and S. Sato, J. Phys. Soc. Jpn., 1994, 63, 347–357 CrossRef.
- D. S. Kilin and D. A. Micha, J. Phys. Chem. C, 2009, 113, 3530–3542 CrossRef.
- D. A. Micha, Adv. Quantum Chem., 2015, 71, 195–220 CrossRef.
- D. A. Micha, Adv. Quantum Chem., 2017, 14, 107–127 CrossRef.
- K. Ozawa, S. Yamamoto, R. Yukawa, R.-Y. Liu, N. Terashima, Y. Natsui, H. Kato, K. Mase and I. Matsuda, J. Phys. Chem. C, 2018, 122, 9562–9569 CrossRef.
- T. Vazhappilly and D. A. Micha, J. Phys. Chem. C, 2014, 118, 4429–4436 CrossRef.
- D. E. Woon and T. H. Dunning, Jr., J. Chem. Phys., 1994, 100, 2975–2988 CrossRef.
- K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283 Search PubMed.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113 CrossRef.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
-
H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. O. Mitrushchenkov, G. Rauhut, et al., MOLPRO, version2012.1, a package of ab initio programs, see http://www.molpro.net Search PubMed.
- M. Grüning, O. V. Gritsenko, S. V. A. van Gisbergen and E. J. Baerends, J. Chem. Phys., 2001, 114, 652–660 CrossRef.
-
S. Lias, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. Linstrom and W. Mallard, National Institute of Standards and Technology, Gaithersburg, 2005, ch. Ionization Energy Evaluation Search PubMed.
- A. Borodin and M. Reichling, Phys. Chem. Chem. Phys., 2011, 13, 15442–15447 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef.
- P. E. Blöch, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- H. Stoll, J. Chem. Phys., 1992, 97, 8449–8454 CrossRef.
- A. Kubas, D. Berger, H. Oberhofer, D. Maganas, K. Reuter and F. Neese, J. Phys. Chem. Lett., 2016, 7, 4207–4212 CrossRef PubMed.
- M. P. de Lara-Castells, A. O. Mitrushchenkov and H. Stoll, J. Chem. Phys., 2015, 143, 102804 CrossRef PubMed.
- A. R. Puigdollers, P. Schlexer and G. Pacchioni, J. Phys. Chem. C, 2015, 119, 15381–15389 CrossRef.
- M. Chen, J. E. Dyer, K. Li and D. A. Dixon, J. Phys. Chem. A, 2013, 117, 8298–8313 CrossRef PubMed.
- V. Bonacicić-Koutecky, V. Veyret and R. Mitrić, J. Chem. Phys., 2001, 115, 10450–10460 CrossRef.
- T. L. Haslett, K. A. Bosnick and M. Moskovits, J. Chem. Phys., 1998, 108, 3453–3457 CrossRef.
- M. P. de Lara-Castells, N. F. Aguirre and A. O. Mitrushchenkov, Chem. Phys., 2012, 399, 272–280 CrossRef.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef.
- C. Gomes Silva, R. Juárez, T. Marino, R. Molinari and H. García, J. Am. Chem. Soc., 2011, 133, 595–602 CrossRef PubMed.
- K. Ueno, T. Oshikiri, Q. Sun, X. Shi and H. Misawa, Chem. Rev., 2018, 118, 2955–2993 CrossRef PubMed.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef PubMed.
- K. Lee, E. Eamonn, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081101(R) CrossRef.
- M. P. de Lara-Castells and J. L. Krause, J. Chem. Phys., 2003, 118, 5098–5105 CrossRef.
- M. P. de Lara-Castells, A. O. Mitrushchenkov, O. Roncero and J. L. Krause, Isr. J. Chem., 2005, 45, 59–76 CrossRef.
- M. P. de Lara-Castells and A. O. Mitrushchenkov, J. Phys. Chem. C, 2011, 115, 17540–17557 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Complementary results on optimized structures of bare Ag3 and Ag5 clusters, Xe- and Ag2–TiO2(110) interaction potentials, and the Ag2–TiO2(110) interaction. See DOI: 10.1039/c8cp02853b |
|
| This journal is © the Owner Societies 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Carlos
Cabrillo
b,
David A.
Micha
*a,
Carlos
Cabrillo
b,
David A.
Micha