
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-covalent interactions in electrochemical reactions and implications in clean energy applications†
Botao
Huang
 *ab,
Sokseiha
Muy
ac,
Shuting
Feng
ad,
Yu
Katayama
ae,
Yi-Chun
Lu
f,
Gang
Chen
g and
Yang
Shao-Horn
*abcg
*ab,
Sokseiha
Muy
ac,
Shuting
Feng
ad,
Yu
Katayama
ae,
Yi-Chun
Lu
f,
Gang
Chen
g and
Yang
Shao-Horn
*abcg
aElectrochemical Energy Laboratory, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: huang73@mit.edu; shaohorn@mit.edu
bResearch Laboratory of Electronics, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
cDepartment of Material Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
dDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
eDepartment of Applied Chemistry, Graduate School of Sciences and Technology for Innovation, Yamaguchi University, Ube 755-8611, Japan
fElectrochemical Energy and Interfaces Laboratory, Department of Mechanical and Automation Engineering, The Chinese University of Hong Kong, Shatin, N.T. 999077, Hong Kong SAR, China
gDepartment of Mechanical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
First published on 8th May 2018
Abstract
Understanding and controlling non-covalent interactions associated with solvent molecules and redox-inactive ions provide new opportunities to enhance the reaction entropy changes and reaction kinetics of metal redox centers, which can increase the thermodynamic efficiency of energy conversion and storage devices. Here, we report systematic changes in the redox entropy of one-electron transfer reactions including [Fe(CN)6]3−/4−, [Fe(H2O)6]3+/2+ and [Ag(H2O)4]+/0 induced by the addition of redox inactive ions, where approximately twenty different known structure making/breaking ions were employed. The measured reaction entropy changes of these redox couples were found to increase linearly with higher concentration and greater structural entropy (having greater structure breaking tendency) for inactive ions with opposite charge to the redox centers. The trend could be attributed to the altered solvation shells of oxidized and reduced redox active species due to non-covalent interactions among redox centers, inactive ions and water molecules, which was supported by Raman spectroscopy. Not only were these non-covalent interactions shown to increase reaction entropy, but they were also found to systematically alter the redox kinetics, where increasing redox reaction energy changes associated with the presence of water structure breaking cations were correlated linearly with the greater exchange current density of [Fe(CN)6]3−/4−.
Introduction
Understanding and controlling non-covalent interactions (<85 kJ mol−1, including hydrogen bonding, cation–water bonding, cation–OH bonding and electrostatic interactions)1 associated with solvent molecules and redox-inactive ions provide new opportunities to enhance the reaction entropy changes of metal redox centers, which can increase the thermodynamic efficiency of thermogalvanic (TG) cells2,3 and thermally regenerative electrochemical cycles (TRECs).4–6 In addition, these interactions can greatly influence the reaction kinetics of not only facile one-electron transfer reactions7–9 but also multi-electron reactions such as hydrogen evolution reaction (HER) in electrolytic devices for making sustainable fuels10,11 and oxygen reduction reaction (ORR) in fuel cells.1 The entropy change of a redox or an electron transfer reaction (half-cell reaction) is the difference between the entropy of the reduced and oxidized species of a redox couple such as Mn+1/Mn. Generally speaking, the electron transfer reaction entropy changes of transition metal redox couples can be described by the Born model:12–14where T is temperature, e is elementary charge and NA is the Avogadro constant. ε0 is the permittivity of free space; εr is the relative dielectric constant of the solvent; z and r are the charge and ionic radii of ions. This model assumes that each redox active center can be treated as a sphere, having the first solvation layer dielectrically saturated. Unfortunately, this assumption fails to capture the non-covalent interactions,15 which can alter the solvation shells of oxidized and reduced species via interactions between redox-inactive ions and solvent molecules, and those among solvent molecules. For instance, charged redox centers can attract ions with opposite charge by an electrostatic interaction,16 thereby affecting the solvation structures and entropy changes of redox couples (ESI1,†). This is probably the physical origin of the deviation of experimental data (Fig. 1) from Born model prediction. [Fe(CN)6]3−/4− redox centers can attract cations such as Li+ and repulse anions such as Cl− by electrostatic interactions, which can alter the solvation shell of the [Fe(CN)6]3−/4− redox center.7 These interactions can greatly influence the reaction kinetics. For example, the kinetics of ferri/ferrocyanide,7 ORR,1,17,18 HER17 and methanol oxidation reaction1 are cation-dependent in the order of Li+ > Na+ > K+ > Cs+. Unfortunately, the role of non-covalent interactions in the entropy changes of redox reactions and reaction kinetics is not understood due to the lack of description or measurement of the ternary interactions of redox centers, counter ions and solvent molecules.1
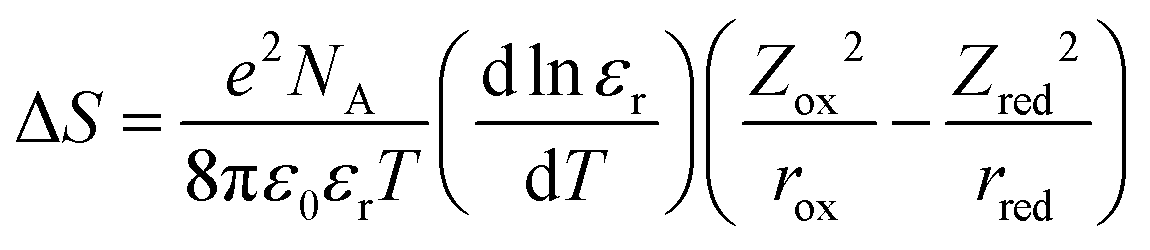
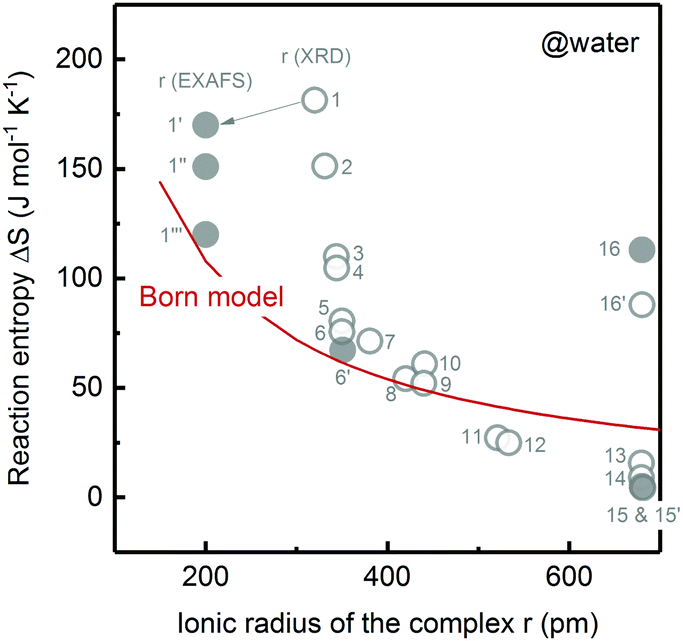 | ||
| Fig. 1 Comparison of the measured redox reaction entropy in aqueous electrolyte (open circle: data from ref. 12 and solid circle: measured in this work) and prediction from the Born model (red curve). 1: [Fe(H2O)6]2+/3+; 1′: [Fe(H2O)6]2+/3+ with 0.6 M LiClO4; 1′′: [Fe(H2O)6]2+/3+ only; 1′′′: [Fe(H2O)6]2+/3+ with 0.6 M LiCl; 2: [Ru(H2O)6]2+/3+; 3: [Ru(NH3)4(H2O)2]2+/3+; 4: [Ru(NH3)5(H2O)]2+/3+; 5: [Os(NH3)6]2+/3+; 6 and 6′: [Ru(NH3)6]2+/3+; 7: [Ru(NH3)5(py)]2+/3+; 8: [Ru(en)3]2+/3+; 9: [Ru(NH3)4(bpy)]2+/3+; 10: [Ru(NH3)4(phen)]2+/3+; 11: [Ru(NH3)2(bpy)2]2+/3+; 12: [Ru(H2O)2(bpy)2]2+/3+; 13: [Cr(bpy)3]2+/3+; 14: [Fe(bpy)3]2+/3+; 15 and 15′: [Ru(bpy)3]2+/3+; 16 and 16′: [Co(bpy)3]2+/3+. en: ethylenediamine, py: pyridine, bpy: 2,2′-bipyridine, phen: 1,10-phenanthroline. | ||
In this study, we employ structure making/breaking ions19–21 that are redox inactive to perturb the solvation structure of redox metal centers in aqueous electrolytes, where changes are examined by redox reaction entropy measurements. Previous studies19 described that structure making ions (Fig. 2a) can attract water molecules and form a large solvation/hydration shell, where water molecules are more structured than those in pure bulk water.19 On the other hand, structure breaking ions (Fig. 2b) interact with water molecules weakly and water molecules adjacent to water structure breakers are less structured than in pure bulk water. Generally speaking, small highly charged ions such as Li+ and F− are structure makers while large ions with low charge density such as Cs+ and I− are structure breakers.19 In addition, the ordering tendency of water molecules increases with decreasing ion size or increasing charge density (Li+ > Na+ > K+ > Cs+).22,23 Here, we examined the role of a number of cations and anions with varying tendency for structure making or breaking, as described by the structural entropy of ions (<50 J mol−1 K−1 for structure makers; >50 J mol−1 K−1 for structure breakers),19,20 in the reaction entropy changes and reaction kinetics of a few metal center redox couples. Reaction entropy changes of [Fe(H2O)6]3+/2+, [Fe(CN)6]3−/4− and [Ag(H2O)4]+/0 were found to increase with the addition of opposite charged ions with greater structure breaking tendency. The trend can be attributed to the altered solvation shells of oxidized and reduced metal centers due to non-covalent interactions induced by inactive ions in the electrolyte. This hypothesis is supported by Raman spectroscopy showing altered interactions between water molecules and the redox center due to different ions in the electrolytes. Moreover, these non-covalent interactions were found to considerably influence the redox kinetics of [Fe(CN)6]3−/4−, where the presence of water structure breaking cations increased the exchange current density.
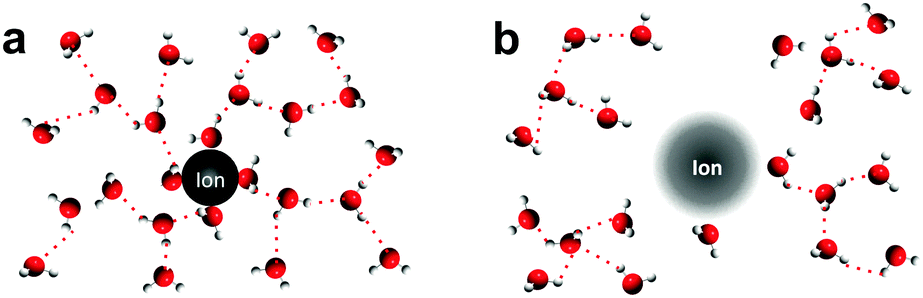 | ||
| Fig. 2 Schematics of water molecule arrangement within the solvation shell of structure making and breaking ions. (a) Structure making ions enhance the proton network in liquid water; (b) structure breaking ions weaken the proton network in bulk water. | ||
Experimental section
Chemicals and electrolyte preparation
The [Fe(CN)6]3−/4− electrolyte was prepared from deionized water (Millipore, >18.2 MΩ cm), deuterium oxide (99.9% atom% D, Aldrich), potassium hexacyanoferrate(III) (>99%, Alfa Aesar) and potassium hexacyanoferrate(II) trihydrate (99.5%, Sigma). Iron(III) chloride hexahydrate (>98%, Sigma-Aldrich) and iron(II) chloride tetrahydrate (98%, Sigma-Aldrich) were used for the preparation of the [Fe(H2O)6]3+/2+ electrolyte. For cation dependent measurements, tetrabutylammonium chloride (TBACl, >98%, Sigma-Aldrich), tetraethylammonium chloride (TEACl >98%, Sigma-Aldrich), tetramethylammonium chloride (TMACl >98%, Sigma-Aldrich), lithium chloride (>98%, Alfa Aesar), sodium chloride (99.99%, Alfa Aesar), potassium chloride (99.995%, Alfa Aesar), rubidium chloride (99.8%, Alfa Aesar), cesium chloride (99.9%, Alfa Aesar), lithium perchlorate (>95%, Sigma-Aldrich), potassium perchlorate, sodium perchlorate (>99%, Alfa Aesar), lithium nitrate (99%, Alfa Aesar), potassium nitrate (99%, Alfa Aesar), rubidium nitrate (99.7%, Sigma-Aldrich) and cesium nitrate (99.99%, Sigma-Aldrich) were used. Furthermore, both [Fe(CN)6]3−/4− and [Fe(H2O)6]3+/2+ redox couples were stable in the presence of perchlorate salts, as shown in the cyclic voltammograms (ESI2,†).Electrochemical measurements
All electrochemical measurements were conducted using a Biologic SP-300 potentiostat in an isothermal three-electrode electrochemical system (ESI3,†) under an Ar atmosphere, where the temperature was controlled by a thermal bath circulator (Thermo Neslab RTE 7). The working electrode was a platinum (Pt) rotating disc electrode (RDE) (Pine instrument). Potentials were referenced to a saturated calomel electrode (SCE).The entropy change of the half-cell reaction of ferro/ferricyanide ([Fe(CN)6]3− + e− ↔ [Fe(CN)6]4−) and aquo iron ([Fe(H2O)6]3+ + e− ↔ [Fe(H2O)6]2+), defined as ΔS = Sred − Sox, was determined by the temperature-dependence of the potential of the redox couples24 (ESI4,†). The formal potential of the redox couples (2 mM) was measured by using the open circuit voltage (OCV) between the Pt working electrode and the reference electrode (ESI5,†) in an electrolyte. The temperature of the electrochemical cell was increased from 20 to 60 °C by an increment of 10 °C. The temperature-dependent potential of the SCE was calibrated in a non-isothermal set-up (ESI6,†). The standard deviation of the reaction entropy measurements was estimated to be 2% based on ten independently repeated measurements (ESI7,†). The influence of structure breaking ions (Cs+, Rb+, NO3− and ClO4−), making ions (Na+, Li+ and TBA+) and border ions (K+ and Cl−) on the entropy changes of half-cell [Fe(CN)6]3−/4− and [Fe(H2O)6]3+/2+ reactions was studied using 2 mM equimolar [Fe(CN)6]3−/4− or [Fe(H2O)6]3+/2+ mixed with ions at a concentration of 0.2 M, 0.4 M or 0.6 M in the electrolyte.
The effect of cations in the supporting electrolyte on the kinetics of ferro/ferricyanide [Fe(CN)6]3−/4− redox was examined by cyclic voltammogram at a scan rate of 10 mV s−1, from which the exchange current density was extracted using the Butler–Volmer equation (ESI8,†). In these electrochemical measurements, the electrolytes consisted of 2 mM equimolar [Fe(CN)6]3−/4− and 0.6 M chloride salts of Li+, Na+, K+, Rb+, Cs+ or TBA+.
Raman spectroscopy
Raman spectroscopy was performed on a LabRAM HR800 microscope (Horiba Jobin Yvon) using an external 20 mW He:Ne 532 nm laser (Horiba, Jobin Yvon), focused with a 50× long working distance objective and a 10−0.3 neutral density filter. A silicon substrate was used to calibrate the Raman shift. Raman spectra were recorded in the range of 100–4000 cm−1 with laser power at 10% and an acquisition time of 15 s and 10 accumulations per run. Glass vials (2 ml volume) were used as sample holders. A higher concentration than that used in the entropy and exchange current density measurements, 0.1 M (K3Fe(CN)6 or K4Fe(CN)6), was used in the Raman measurements to obtain reliable CN stretching signals. 2 M chloride salts of CsCl, RbCl, KCl, NaCl, LiCl or TBACl were added in order to have a reasonable cation/redox ratio, allowing the observation of the appropriate cation effect.DFT calculations
To investigate the effects of cation type (cation = Li+, Na+, K+, Rb+ and Cs+) on the aqueous solvation energy, ab-initio molecular dynamics (AIMD) simulations were performed on the system containing 93 water molecules and one ferricyanide [Fe(CN)6]3− anion as well as 3 cations to make the overall system electrically neutral. AIMD simulations were carried out using the PBE functional25 as implemented in the Vienna Ab-Initio Simulation Package (VASP).26 The Projector Augmented Wave (PAW)27 method was used to treat the core-electrons with a plane wave kinetic energy cutoff of 550 eV (750 eV for systems containing Na+). All calculations were performed using a reciprocal space containing only the Γ point. We used the NVT ensemble with the temperature fixed to 300 K. The size of the unit cell for each system (ESI9,† and Table S1) was decided based on the density of the aqueous solution and the size of the cations.19,28 The time step was chosen to be 0.5 fs and the Grimme DFT-D2 van der Waals correction29 was added to better treat the interaction between water molecules. Each simulation was run for 10 ps. The initial geometries of systems containing Na+, K+, Rb+ and Cs+ were generated using the optimized geometries obtained for smaller ions with the replacement of smaller ions with larger ions. For example, the initial geometry of the system containing Na+ was obtained from replacing the Li+ ions in the optimized Li+-containing system. For these calculations, 1 × 10−6 and −0.04 eV Å−1 were used as stopping criteria for the electronic and ionic relaxation loops, respectively. The reaction enthalpy ΔE of the ferri/ferrocyanide [Fe(CN)6]3−/4− redox is estimated by| ΔE = E(Fe(CN)64−) − E(Fe(CN)63−) |
Results and discussion
Anion- and cation-dependent entropy of redox reactions
We show that the redox reaction entropy of [Fe(H2O)6]3+/2+ and [Fe(CN)6]3−/4− couples can be strongly anion- or cation-dependent. The reaction entropy of 2 mM equimolar [Fe(H2O)6]3+/2+ in 0.1 M HCl (Fig. 3a and Fig. S10a, ESI†) was found to be positive (150 ± 2 J mol−1 K−1), suggesting greater entropy for [Fe(H2O)6]2+ than [Fe(H2O)6]3+, which can be attributed to more disorder in the solvation shell of the less charged [Fe(H2O)6]2+ redox center. On the other hand, the redox entropy of 2 mM equimolar [Fe(CN)6]3−/4− (Fig. 3b and Fig. S10b, ESI†) was negative (−179 ± 3 J mol−1 K−1), indicating greater entropy for the less charged [Fe(CN)6]3− than [Fe(CN)6]4− redox center. These values are in agreement with previous findings.3,24 Increasing the concentration of anions such as ClO4− and NO3− previously known as water structure breakers19,20 in the electrolyte led to greater redox entropy for [Fe(H2O)6]3+/2+ while adding Cl−, known as less water structure breaking, significantly reduced entropy, as shown in Fig. 3a. In contrast, changing cations for a given anion did not cause significant changes in the redox entropy of [Fe(H2O)6]3+/2+. A similar anion dependence was observed for [Ag(H2O)4]+/0 (ESI11,†). The lower redox entropy for [Fe(H2O)6]3+/2+ in the presence of Cl− ions suggests that the reduction in the entropy of less charged [Fe(H2O)6]2+ associated with less disordered water in the solvation shell is greater than more charged [Fe(H2O)6]3+. This hypothesis is supported by the pronounced cation-dependent entropy changes of negatively charged [Fe(CN)6]3−/4−, where the redox entropy was found to become less negative with increasing concentration of structure making, monovalent cations, as shown in Fig. 3b. At a given cation concentration, the increase of reaction entropy was in the sequence of tetrabutylammonium+ (TBA+) > Li+ > K+ > Rb+ > Cs+, which scaled with decreasing structural entropy of these cations reported previously.19,20 Despite their large sizes, quaternary ammonium cations such as tetramethylammonium (TMA+), tetraethylammonium (TEA+) and tetrabutylammonium (TBA+) are water structure makers due to their longer alkyl hydrophobic chains, which could weaken the quaternary ammonium cation/water interaction and promote water/water hydrogen bond networking.33,34 Similar trends were found for water-making divalent cations such as Ni2+, Ca2+ and Mg2+ (Table S2, ESI†). The less negative redox entropy for [Fe(CN)6]3−/4− in the presence of cations suggests that the reduction in the entropy of less charged [Fe(CN)6]3− associated with less disordered water in the solvation shell is greater than more charged [Fe(CN)6]4−.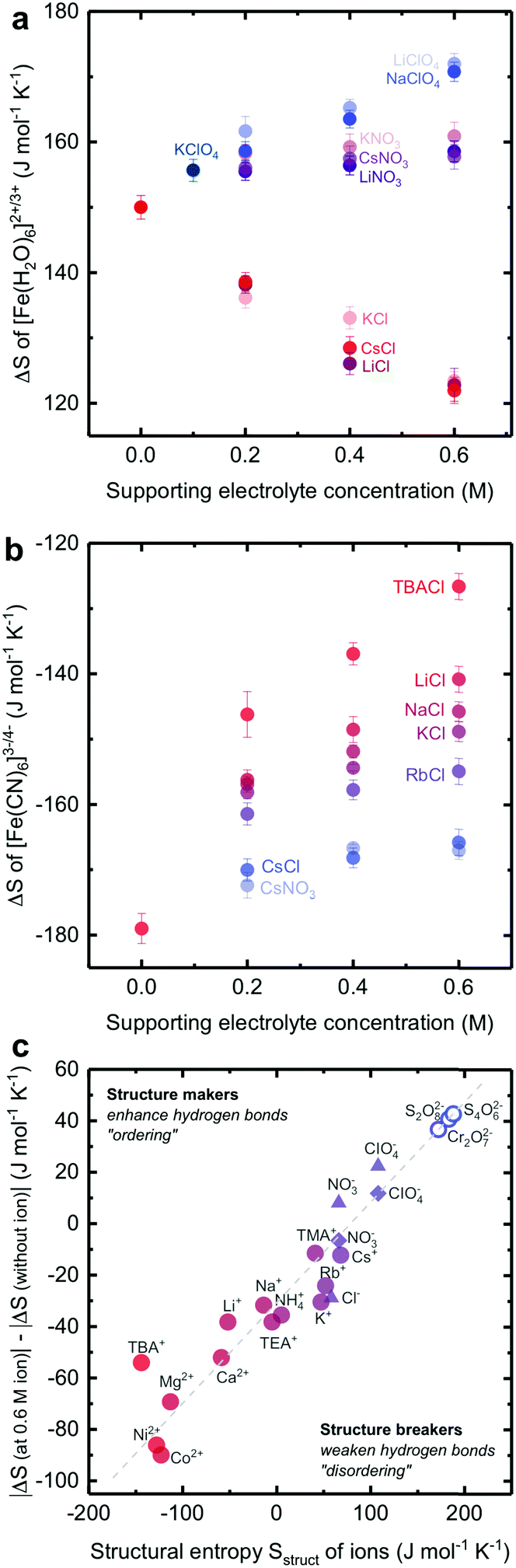 | ||
| Fig. 3 The dependence of the reaction entropy of (a) [Fe(H2O)6]3+/2+ at a 2 mM equimolar concentration and pH = 1 and (b) [Fe(CN)6]3−/4− at a 2 mM equimolar concentration. (c) The changes in the redox reaction entropy of [Fe(H2O)6]3+/2+ (triangle), [Fe(CN)6]3−/4− (circle), and [Ag(H2O)4]+/0 (diamond) between the electrolyte with and without 0.6 M supporting electrolyte plotted against the structural entropy Sstruct of non-redox active ions from previous work.20 For TEA+ and TMA+, the data are presented in the ESI10.† The projected values of reaction entropy gain with Cr2O72−, S2O82− and S4O62− ions in aqueous electrolyte (open circle). Divalent cations (Ni2+, Co2+, Mg2+ and Ca2+) have also been studied in the [Fe(CN)6]3−/4− electrolyte (data shown in the ESI10,†). Error bars were obtained from the standard deviation of 3 independent measurements. | ||
Considering approximately twenty redox-inactive ions in the electrolyte in this study, we report a universal trend for the absolute change in the redox entropy of [Fe(H2O)6]3+/2+, [Fe(CN)6]3−/4− and [Ag(H2O)4]+/0 measured at a concentration of 0.6 M (where the ionic strength, activity coefficient of redox and the dielectric constant35–37 of electrolytes are approximately the same) with the structural entropy of redox-inactive cations or anions reported previously,20 as shown in Fig. 3c. The greater the structural entropy, the larger the change in the redox reaction entropy. Remarkably, a large reduction of up to ∼80 J mol−1 K−1 for the redox entropy of [Fe(CN)6 ]3−/4− can be induced by adding water structure making cations in the electrolytes. Following this trend, one can potentially increase redox reaction entropy by adding ions with high structural entropy such as I−, Cr2O72−, S2O82−, and S4O62−.20 Unfortunately, these ions are incompatible with [Fe(H2O)6]3+/2+ and [Fe(CN)6]3−/4− redox couples in the aqueous electrolytes due to (1) the strong oxidizing power of Cr2O72− (E0 of Cr2O72−/Cr3+ = 1.36 VNHE) and S2O82− (E0 of S2O82−/SO42− = 2.01 VNHE), (2) the strong oxidizing power of I− (E0 (I3−/I−) = 0.45 VNHE), and (3) the low solubility of S4O62− (the solubility of Na2S4O6 < 0.1 M). This trend can be attributed to the altered solvation or hydration shell structure of redox metal centers by the addition of inert structure breaking or making ions with opposite charge via electrostatic interactions, which will be discussed in detail.
Tuning non-covalent interactions within the solvation shell of redox species
The change of non-covalent interaction in the solvation shell of [Fe(CN)6]3− and [Fe(CN)6]4− redox centers was probed by Raman spectroscopy, where we examined the changes in the CN stretching frequency in the presence of different water structure making cations. Note that Raman spectroscopy was chosen because of its high sensitivity towards the CN stretching vibration compared to Fourier transform infrared (FT-IR) spectroscopy (see the ESI12,† for FT-IR results). CN stretching of ferrocyanide [Fe(CN)6]4− has two separated peaks at around 2090 cm−1 and 2060 cm−1, corresponding to two vibration modes, A1g and Eg, respectively. CN stretching peaks of ferricyanide [Fe(CN)6]3− are closer than those of ferrocyanide [Fe(CN)6]4−. These observations and assignments are consistent with previous work.38 In this work, we found that the CN stretching frequency was sensitive to the non-covalent interaction with surrounding water molecules (Fig. 4a). Although the frequency of the CN stretching vibration is mostly determined by the characteristics of the covalent bond between CN and the redox metal center, it can also reflect the strength of non-covalent interaction, such as the CN–water interaction (see the ESI12,† for detail). For example, heavy water (D2O) shifted the CN stretching frequency of [Fe(CN)6]4− (0.2 M) to lower values than normal water (H2O), as shown in Fig. 4b. This negative shift in the CN vibration frequency is due to either higher molecular mass39 or stronger H/D-bonds in D2O compared to H2O.40 More interestingly, the CN stretching frequencies of 0.1 M [Fe(CN)6]4− (Fig. 4c) and [Fe(CN)6]3− (Fig. 4d) were found to shift negatively in the order of Cs+ > Rb+ > K+ > Na+ > Li+ > TBA+. It is noteworthy that the trend in the wavenumber shift cannot be explained by the molecular mass of the cation (TBA+ > Cs+ > Rb+ > K+ > Na+ > Li+) as well as the strength in direct electrostatic interaction between the cation and the CN ligand governed by charge density of the cations (TBA+ > Cs+ > Rb+ > K+ > Na+ > Li+). We therefore believe that this shift is due to the change in the non-covalent water–CN interaction induced by the cations. By electrostatic interactions, the negatively charged [Fe(CN)6]3− and [Fe(CN)6]4− redox centers attract these cations, which could affect the non-covalent interaction between CN and water in the vicinity of the complexes due to their different structure making/breaking ability. The relatively large negative shift observed for strong water structure maker ions such as Li+ and TBA+ can be explained by the weakening of the CN bond due to the existence of a strong noncovalent interaction with water molecules. A similar cation dependent trend was observed by FT-IR spectroscopy, as shown in the ESI12,† and Fig. S12. | ||
| Fig. 4 Raman spectra of CN stretching for probing interaction between ferro-/ferricyanide redox species and surrounding water molecules in the solvation shell. (a) Schematic showing possible interaction that can affect the CN ligand, including the interaction of CN–Fe redox center, CN–inactive ions, and CN–water; (b) 0.2 M K4Fe(CN)6 in water and heavy water; (c) 0.1 M K4Fe(CN)6 and 2 M chloride salts (CsCl, RbCl, KCl, NaCl, LiCl and TBACl) in water; (d) 0.1 M K3Fe(CN)6 and 2 M chloride salts (CsCl, RbCl, KCl, NaCl, LiCl and TBACl) in water. | ||
The role of water structure in reaction kinetics
Here, we show the cation-dependent redox reaction kinetics of [Fe(CN)6]3−/4− (Fig. 5) and discuss its physical origin in the context of entropy changes associated with the reorganization of the solvation shells of [Fe(CN)6]3− and [Fe(CN)6]4− during the electron transfer process.41 To examine the role of water structure in the solvation shell in the kinetics of ferro/ferricyanide electron transfer reaction, cyclic voltammetry was performed in the electrolytes containing a 2 mM equimolar mixture of [Fe(CN)6]3−/4− redox reaction and 0.6 M chloride salt, as shown in Fig. 5a. The limiting current density in the cyclic voltammogram (Fig. 5a) was found to decrease in the order: Cs+ > Rb+ > K+ > Na+ > Li+ > TBA+, which can be attributed to the increased viscosity19–21 of electrolytes in the presence of these inert ions and the reduced diffusivity of the redox species.42 Cyclic voltammograms for overpotentials ranging between −0.03 VNHE and 0.03 VNHE have been fitted to the Butler–Volmer relation to extract exchange current density, j0. The Tafel plots of the cyclic voltammograms showed that the current density is higher for electrolytes containing 0.6 M Cs+ than that with 0.6 M TBA+ (Fig. 5b), indicating that cations in the electrolyte could affect the ferri/ferrocyanide redox reaction rate.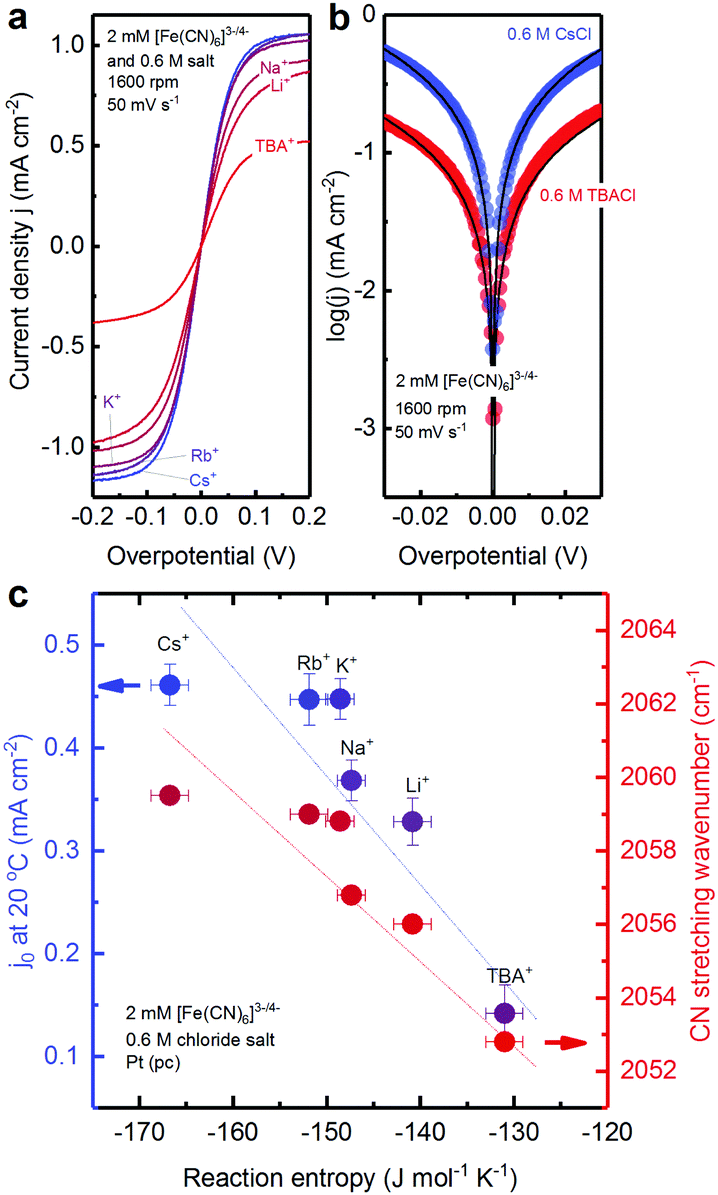 | ||
| Fig. 5 Cation dependent exchange current density j0 of 2 mM equimolar [Fe(CN)6]3−/4− measured in an aqueous solution containing 0.6 M chloride salts of TBA+, Li+, Na+, K+, Rb+ and Cs+. (a) CV measured at 50 mV s−1 and 1600 rpm in Ar-saturated electrolytes at 20 °C on the Pt RDE electrode. (b) Tafel plots of CVs for electrolytes containing 0.6 M CsCl and 0.6 M TBACl. (c) CN stretching frequency in Raman spectra (red points) and exchange current density (blue points) extracted from the Butler–Volmer equation with charge transfer coefficient α = 0.5 plotted against the experimental reaction entropy change of [Fe(CN)6]3−/4− redox. Error bars were obtained from the standard deviation of 4–6 independent measurements. Lines were added to guide the eye. (See ESI8,† for additional data.) | ||
The exchange current density of [Fe(CN)6]3−/4− redox was found to increase in the order of TBA+ < Li+ < Na+ < K+ < Rb+ < Cs+ (Fig. 5c). The exchange current density decreases almost linearly with increasing reaction entropy change of the ferri/ferrocyanide redox. The presence of structure making ions, such as Li+ and TBA+, decreases the exchange current density significantly. This decrease in exchange current density is correlated with the Raman shift of CN stretching values (Fig. 5c) and thus associated with the change of the water structure in the solvation shell of the redox center. Structure making cations bind strongly with water molecules and the water molecule arrangement is difficult to reorganize during electrochemical processes, thus lowering the electron transfer kinetics.41 On the other hand, the structure breaking cations interact weakly with water where the water structure is easy to reorganize, thus there is a higher kinetics for charge transfer.41
Both experimental and theoretical techniques were used to estimate the energy of redox reactions. The formal potentials of the ferri-/ferrocyanide redox couple measured at the 2 mM [Fe(CN)6]3−/4− redox couple with 0.6 M chloride salts (e.g. CsCl, RbCl, KCl, NaCl and LiCl) slightly decreased with cations with increasing structure making tendency (Fig. 6). The difference in the formal potential found among inactive cations was found to be comparable to that for reaction entropy induced by non-covalent interactions induced by inactive cations added, indicating that the reaction enthalpy of redox centers was not changed significantly by these non-covalent interactions. This argument is supported by DFT calculations, where the changes in the energy of [Fe(CN)6]3−/4− induced by the presence of cations (e.g. Cs+, Rb+, K+, Na+ and Li+) were found to be comparable to the energy contribution from entropy (ESI9,† and Fig. S9), considering computational uncertainty. Therefore, it is proposed that the faster kinetics in the presence of water structure breaking ions such as Cs+ compared to TBA+ by ∼five times can be attributed to the smaller reorganization energy of the [Fe(CN)6]3−/4− redox as suggested by large entropy changes of this redox reaction (Fig. 5c). Therefore, tuning the water structure in the redox solvation shell by adding inactive ions with opposite charge could alter the reaction kinetics, which highlights a new path to control electrocatalytic activity by exploiting the rich chemical physics of liquid electrolytes.
 | ||
| Fig. 6 Formal potential of 2 mM equimolar [Fe(CN)6]3−/4− measured in aqueous electrolyte containing 0.6 M chloride salts of Li+, Na+, K+, Rb+ and Cs+ by OCV measured at 50 mV s−1 and 1600 rpm in Ar-saturated electrolytes at 20 °C on the Pt RDE electrode. | ||
Conclusions
The contribution of non-covalent interactions associated with redox centers, inactive ions and water molecules within the solvation shell to the reaction entropy changes and the electrocatalytic activity of one-electron transfer reactions was probed. Around twenty redox inactive structure making/breaking ions19–21 were employed to perturb the solvation structure of redox metal centers in aqueous electrolytes. A systematic trend for redox entropy change of one-electron transfer reactions, such as [Fe(CN)6]3−/4−, [Fe(H2O)6]3+/2+ and [Ag(H2O)4]+/0, associated with the disordering within the redox solvation shell, was reported. Reaction entropy changes of these redox couples varied linearly with the concentration of inactive ions with opposite charge. It was also found to increase with the addition of inactive ions with greater structure breaking tendency. The trend can be attributed to the altered solvation shells of oxidized and reduced metal centers due to non-covalent interactions induced by inactive ions in the electrolyte. This hypothesis is supported by Raman spectroscopy, showing altered interactions between water molecules and the redox center due to different ions in the electrolytes. Moreover, these non-covalent interactions were found to considerably influence the redox kinetics of [Fe(CN)6]3−/4−, where the presence of water structure breaking cations increased the exchange current density. These findings suggest that tailoring a redox solvation shell by choosing an appropriate supporting electrolyte (inactive ion) with opposite charge is a new strategy for tuning reaction entropy change and electrocatalytic activity, which opens a new path to enhance the efficiency of energy-storage and energy-conversion devices.Author contributions
B. H. and Y. S.-H. designed the experiments. B. H. carried out reaction entropy, Raman and electrochemical experiments, B. H. prepared the initial manuscript, S. F. and S. M. performed DFT calculations, and Y. K. conducted FTIR measurements. All authors contributed to the discussion and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by “Solid State Solar-Thermal Energy Conversion Center (S3TEC)”, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, BES under Award Number DE-SC0001299/DE-FG02-09ER46577 and by a grant from the Innovation and Technology Commission of the Hong Kong Special Administrative Region, China (Project No. ITS/020/16FP). We thank Livia Giordano for the helpful discussion. This study used resources of the National Energy Research Scientific Computing Center, a DOE office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-5CH11231.Notes and references
- D. Strmcnik, K. Kodama, D. van der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, Nat. Chem., 2009, 1, 466–472 CrossRef PubMed.
- B. T. Huang, M. Roger, M. Bonetti, T. J. Salez, C. Wiertel-Gasquet, E. Dubois, R. Cabreira Gomes, G. Demouchy, G. Mériguet, V. Peyre, M. Kouyaté, C. L. Filomeno, J. Depeyrot, F. A. Tourinho, R. Perzynski and S. Nakamae, J. Chem. Phys., 2015, 143, 054902 CrossRef PubMed.
- A. Gunawan, C.-H. Lin, D. A. Buttry, V. Mujica, R. A. Taylor, R. S. Prasher and P. E. Phelan, Nanoscale Microscale Thermophys. Eng., 2013, 17, 304–323 CrossRef.
- S. W. Lee, Y. Yang, H.-W. Lee, H. Ghasemi, D. Kraemer, G. Chen and Y. Cui, Nat. Commun., 2014, 5, 3942 CrossRef PubMed.
- Y. Yang, S. W. Lee, H. Ghasemi, J. Loomis, X. Li, D. Kraemer, G. Zheng, Y. Cui and G. Chen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17011–17016 CrossRef PubMed.
- Y. Yang, J. Loomis, H. Ghasemi, S. W. Lee, Y. J. Wang, Y. Cui and G. Chen, Nano Lett., 2014, 14, 6578–6583 CrossRef PubMed.
- L. M. Peter, W. Dürr, P. Bindra and H. Gerischer, J. Electroanal. Chem. Interfacial Electrochem., 1976, 71, 31–50 CrossRef.
- F. C. Anson, Anal. Chem., 1961, 33, 939–942 CrossRef.
- W. M. Taama, R. E. Plimley and K. Scott, Electrochim. Acta, 1996, 41, 549–551 CrossRef.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256 CrossRef PubMed.
- J. Staszak-Jirkovsky, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K.-C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197–203 CrossRef PubMed.
- J. T. Hupp and M. J. Weaver, Inorg. Chem., 1984, 23, 3639–3644 CrossRef.
- S. Sahami and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1981, 122, 155–170 CrossRef.
- S. Sahami and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1981, 122, 171–181 CrossRef.
- K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143–168 CrossRef.
- Y. Yamato, Y. Katayama and T. Miura, J. Electrochem. Soc., 2013, 160, H309–H314 CrossRef.
- J. Tymoczko, V. Colic, A. Ganassin, W. Schuhmann and A. S. Bandarenka, Catal. Today, 2015, 244, 96–102 CrossRef.
- J. Suntivich, E. E. Perry, H. A. Gasteiger and Y. Shao-Horn, Electrocatalysis, 2013, 4, 49–55 CrossRef.
- Y. Marcus, Chem. Rev., 2009, 109, 1346–1370 CrossRef PubMed.
- Y. Marcus, J. Solution Chem., 1994, 23, 831–848 CrossRef.
- H. D. B. Jenkins and Y. Marcus, Chem. Rev., 1995, 95, 2695–2724 CrossRef.
- D. J. Miller and J. M. Lisy, J. Am. Chem. Soc., 2008, 130, 15381–15392 CrossRef PubMed.
- D. J. Miller and J. M. Lisy, J. Am. Chem. Soc., 2008, 130, 15393–15404 CrossRef PubMed.
- E. L. Yee, R. J. Cave, K. L. Guyer, P. D. Tyma and M. J. Weaver, J. Am. Chem. Soc., 1979, 101, 1131–1137 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz and A. F. Izmaylov, Gaussian, Inc., Wallingford CT, 2016.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- K. Udachin, S. Alavi and J. A. Ripmeester, J. Chem. Phys., 2011, 134, 121104 CrossRef PubMed.
- S. Alavi and J. A. Ripmeester, J. Chem. Phys., 2012, 137, 054712 CrossRef PubMed.
- J. B. Hasted, D. M. Ritson and C. H. Collie, J. Chem. Phys., 1948, 16, 1–21 CrossRef.
- K. Nörtemann, J. Hilland and U. Kaatze, J. Phys. Chem. A, 1997, 101, 6864–6869 CrossRef.
- T. Chen, G. Hefter and R. Buchner, J. Phys. Chem. A, 2003, 107, 4025–4031 CrossRef.
- B. I. Swanson and J. J. Rafalko, Inorg. Chem., 1976, 15, 249–253 CrossRef.
- R. E. Weston, Spectrochim. Acta, 1962, 18, 1257–1277 CrossRef.
- Y. Harada, T. Tokushima, Y. Horikawa, O. Takahashi, H. Niwa, M. Kobayashi, M. Oshima, Y. Senba, H. Ohashi, K. T. Wikfeldt, A. Nilsson, L. G. M. Pettersson and S. Shin, Phys. Rev. Lett., 2013, 111, 193001 CrossRef PubMed.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 17031 CrossRef.
- A. Voronel, E. Veliyulin, V. S. Machavariani, A. Kisliuk and D. Quitmann, Phys. Rev. Lett., 1998, 80, 2630–2633 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Proposed solvation model, DFT method and experimental details, additional electrochemical and reaction entropy measurements and FITR spectroscopy data. See DOI: 10.1039/c8cp02512f |
| This journal is © the Owner Societies 2018 |