
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the possibility of an Eley–Rideal mechanism for ammonia synthesis on Mn6N5+x (x = 1)-(111) surfaces†
Constantinos D.
Zeinalipour-Yazdi
 ab
ab
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: c.zeinalipour-yazdi@ucl.ac.uk
bUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Labs, Harwell Campus, OX11 0FA, UK
First published on 25th June 2018
Abstract
Recently we reported an Eley–Rideal/Mars–van Krevelen mechanism for ammonia synthesis on cobalt molybdenum nitride (Co3Mo3N). In this mechanism hydrogenation of activated dinitrogen occurs directly from the gas phase in a low barrier step forming a hydrazinylidene intermediate ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NNH2. In this paper we study whether such a mechanism of ammonia synthesis could occur on the (111) surface of another metal nitride, Mn6N5+x (x = 1), as this would explain the low-T ammonia synthesis activity of Co3Mo3N. We find that although N2 adsorbs more strongly than H2 on the (111) surface, having also examined the (110) and the (100) surface, N2 is not significantly activated when adsorbed in an end-on configuration. The hydrogenation reactions via an Eley–Rideal mechanism are all high barrier processes (>182 kJ mol−1) and therefore an Eley–Rideal mechanism for ammonia synthesis is predicted to not occur on this material unless there are high temperatures. Our study indicates that the fact that an Eley–Rideal/Mars–van Krevelen mechanism occurs on Co3Mo3N is a result of the stronger activation of dinitrogen at nitrogen vacancies when dinitrogen is adsorbed in an end-on configuration.
NNH2. In this paper we study whether such a mechanism of ammonia synthesis could occur on the (111) surface of another metal nitride, Mn6N5+x (x = 1), as this would explain the low-T ammonia synthesis activity of Co3Mo3N. We find that although N2 adsorbs more strongly than H2 on the (111) surface, having also examined the (110) and the (100) surface, N2 is not significantly activated when adsorbed in an end-on configuration. The hydrogenation reactions via an Eley–Rideal mechanism are all high barrier processes (>182 kJ mol−1) and therefore an Eley–Rideal mechanism for ammonia synthesis is predicted to not occur on this material unless there are high temperatures. Our study indicates that the fact that an Eley–Rideal/Mars–van Krevelen mechanism occurs on Co3Mo3N is a result of the stronger activation of dinitrogen at nitrogen vacancies when dinitrogen is adsorbed in an end-on configuration.
1. Introduction
Metal nitrides, which are also called interstitial nitrides, have varying amounts of lattice nitrogen that occupies octahedral coordination sites, with the general formula MNy, with M = 3d metal and y < 1.1 The structure that has received most attention is η-Mn3N22 which was found to be anti-ferromagnetic.3 ε-Mn4N4 has the lowest percent nitrogen content, in which the atoms occupy a face-centered cubic (fcc) arrangement with interstitial nitrogen. The following in nitrogen content is ζ-Mn2N0.865 (occasionally labelled as Mn2N6 and Mn5N27) which has a hexagonal close packed (hcp) structure. The manganese nitride with the largest nitrogen content is θ-MnN8 that has a slightly tetragonally distorted rocksalt phase (occasionally labelled as θ-Mn6N5+x2). The manganese nitride phase studied here is θ-Mn6N5+x (for x = 1) which however did not contain any intrinsic nitrogen vacancies in the 3 × 1 × 1 unit cell and had therefore a 3 times smaller unit cell as shown in Fig. 1. This manganese nitride is refered to in this paper as θ-Mn6N5. This manganese nitride has the advantage of having well defined low Miller index surfaces, which are depicted in Fig. 2.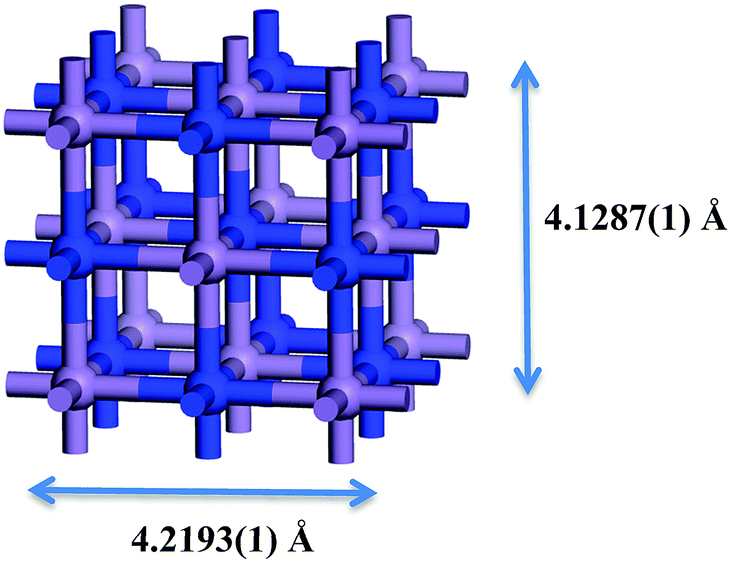 | ||
| Fig. 1 Bulk unit cell of θ-Mn6N5+x (x = 1). Blue atoms correspond to nitrogen atoms and light blue to manganese atoms. Structure taken from ref. 9. | ||
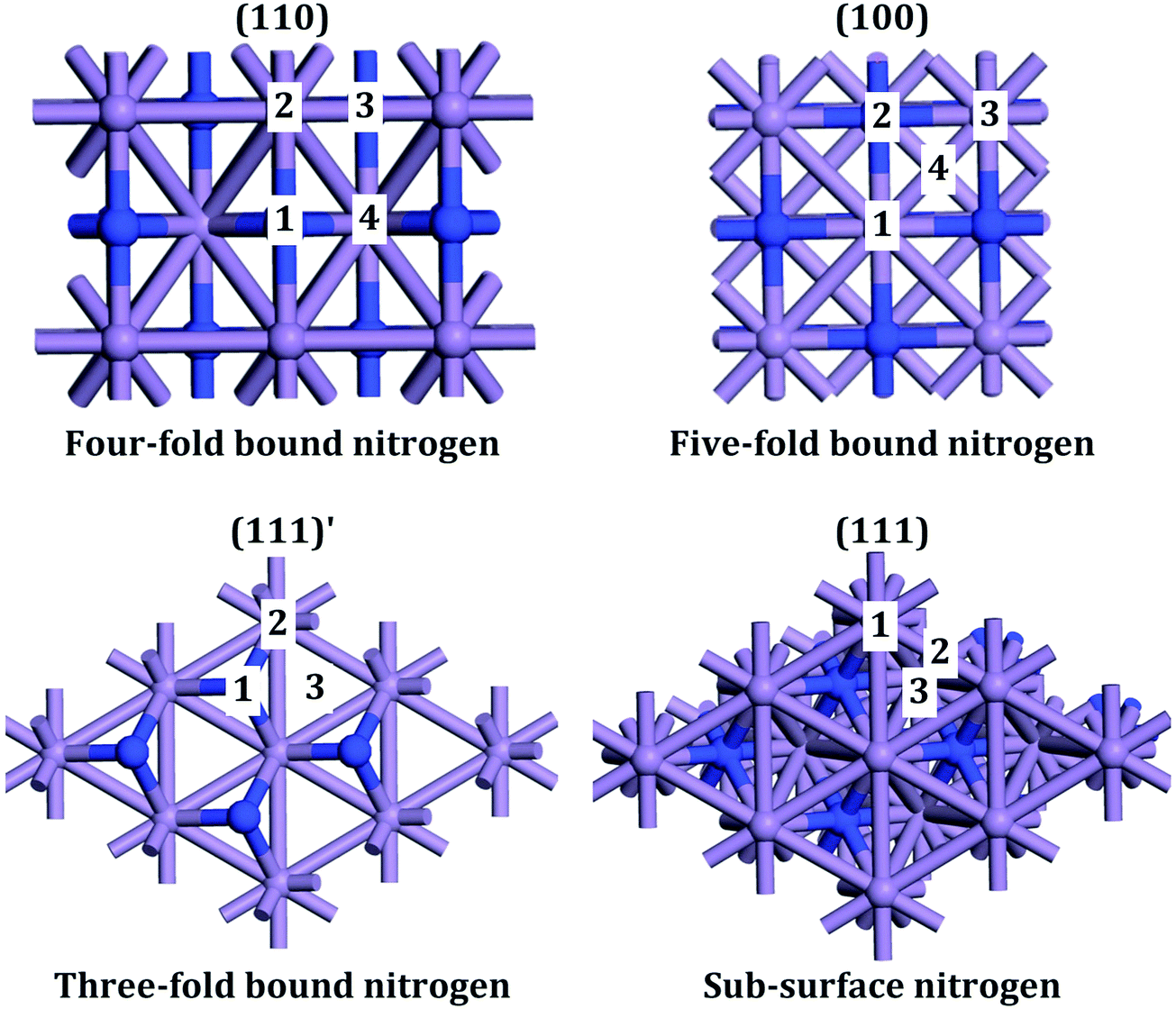 | ||
| Fig. 2 Low Miller index surfaces of θ-Mn6N5. (111)′ indicates that the surface is parallel to the (111) surface. Blue and light-blue atoms are nitrogens and manganese, respectively. | ||
Manganese nitrides have also been studied as a looping catalyst in ammonia synthesis10,11 and for the reverse reaction, ammonia decomposition, by thermogravimetric analysis and neutron diffraction.12 Mn6N5+x and lithium imide (Li2NH) when combined result in a larger rate of ammonia decomposition than for either of the separate parts.13 Additionally, CaNH can exert a strong synergistic effect on Mn6N5+x leading to greatly enhanced catalytic activity for ammonia decomposition.14 Manganese nitrides were also found to be an active and durable electrocatalyst in the oxygen evolution reaction (OER) from water under alkaline conditions.15
Manganese nitrides can be synthesized by nitriding the metal with nitrogen or ammonia.16 The rate of manganese nitride formation is higher at lower temperatures when ammonia instead of nitrogen is used as the source of lattice nitrogen,17 which is a result of ammonia having a lower dissociation barrier than dinitrogen. Most of the manganese nitrides have been studied for their magnetic properties (e.g. ε-Mn4N is ferromagnetic) however little is known about their activity as ammonia synthesis catalysts or nitrogen transfer reagents. There are only a few previous reports that mention a manganese nitride with the chemical formula θ-Mn6N5. There is an electron diffraction study of the phase transition of this material from tetragonal to cubic at temperatures that exceed 440 ± 20 °C. The phase transition is attributed to magnetic reordering of the material rather than changes of the position of the atoms.18 Another report which addresses the magnetic transition in more detail is a neutron diffraction study of both η-Mn3N2 and θ-Mn6N5.9
The potential use of metal nitrides in ammonia synthesis as catalysts or as reagents has attracted considerable recent interest.19 Directly relevant to this is the use of Mg3N2 as a nitrogen transfer reagent in the Paal–Knorr pyrrole reaction.20 We have recently reported this material to be an efficient nitrogen transfer reagent especially when doped with lithium.11 The discovery of catalysts or reagents that can synthesize ammonia at lower temperature for small-scale production is highly desirable for in situ and “on demand” production of ammonia.11 However to use a catalyst in a decentralised plant, exact knowledge of the reaction mechanism and kinetic models is necessary. Currently the mechanism of ammonia synthesis is not known on manganese nitrides, whether it follows a conventional Langmuir–Hinshelwood mechanism such as the one observed on the iron or ruthenium21 ammonia synthesis catalysts or an Eley–Rideal/Mars–van Krevelen mechanism such as the one we recently found on cobalt molybdenum nitride (Co3Mo3N).22,23 Furthermore, the details of the adsorption and activation of molecular nitrogen and hydrogen are not known and whether hydrogen poisons the surface of the catalyst as the adsorption energy of H2 usually exceeds that of N2, as we have recently calculated on other metal nitrides.24,25
In this DFT study we have calculated the adsorption and activation of N2 and H2 on various low Miller index surfaces of θ-Mn6N5. We found the surface of this material that will adsorb N2 favourably instead of H2. We then calculated the complete pathway for ammonia synthesis via an Eley–Rideal mechanism. The results are then discussed in reference to another metal nitride, Co3Mo3N, that has previously exhibited the possibility of an Eley–Rideal/Mars–van Krevelen mechanism for ammonia synthesis.
2. Computational methods
We have generated a slab model of the various surfaces of θ-Mn6N5 based on the structure reported in a neutron diffraction study.9 The surfaces were modelled via slabs about 1 nm in width that had a 20 Å vacuum gap, for which the surface structure is shown in Fig. 2. All DFT calculations were periodic Γ-point26 spin-polarised performed with the VASP 5.4.1 code.27,28 Exchange and correlation (XC) effects were evaluated within the generalized gradient approximation (GGA) using the revised Perdew–Burke–Ernzerhof (revPBE) XC-functional.29 We used the projector augmented-wave (PAW) method30,31 to represent core states, 1s for N and H and 1s to 3p for Mn. The cut-off energy for the plane wave expansion was set to 600 eV for periodic bulk and slab calculations. Geometry optimizations were performed with a residual force threshold of 0.01 eV Å−1 using the conjugate-gradient algorithm whereas the convergence criterion for electronic relaxation was set to 10−4 eV.In a previous computational study we have tested the bond dissociation enthalpy (BDE) of N2 with the use of various XC functionals, the % error for revPBE was found to be the lowest among various GGA and hybrid GGA functionals. In particular, we found that the % error was for revPBE = 0.2 < B3LYP = 1.7 < PBE0 = 3.5 < HSE06 = 4.1 < PBE = 4.9 < PW91 = 7.0.23 Therefore, we have combined this XC functional with pairwise additive correction for van der Waals (VdW) interactions via the D3 method,32 denoted as revPBE-D3, with which all subsequent calculations have been performed to yield more accurate barrier heights according to the calculated BDE of the N–N bond and the inclusion of VdW interactions.
For the bulk unit cell, convergence of the energetic and magnetic properties was observed for a 5 × 5 × 5 Monkhorst–Pack grid (MP-grid). The convergence plots are given as supporting information Fig. S1 and S2 (ESI†). A similar or slightly larger grid density (so that the grid obtains numbers to the nearest integer) was used for the slab calculations, for consistency. A Γ-point centred MP k-point grid was used with a grid density of 3 × 3 × 1, 2 × 2 × 1, 2 × 2 × 1 and 3 × 2 × 1 for the (100), (111), (111)′ and (110) surface of θ-Mn6N5, respectively, shown in Fig. 2.
The initial charge density was obtained by superposition of atomic charges. Initial adsorption configurations were such that the distance between the adsorbate and the nearest surface site was set to 2 Å in an end-on configuration for N2 and H2. This adsorption configuration was found to also result in end-on and tilt adsorbed adsorbates. The various adsorption sites were every symmetry unique surface site shown in Fig. 2. This resulted in 4, 4, 3 and 3 symmetry unique adsorption sites for the (110), (100), (111)′ and (111) surface, respectively. The adsorption energy was taken as the total difference between the energy of the fully relaxed bound state of the surface-adsorbate complex and that of the fully relaxed surface slab and the isolated molecules, given by:
| ΔEads,D3 = Eslab-X2 − Eslab − EX2, | (1) |
The effective magnetic moment per Mn atom in the optimised bulk structure of θ-Mn6N5 was found to be 3.95 μB, which was in good agreement with the experimentally measured value which ranges between 3.3 and 3.8 μB.9 Furthermore, the optimised structure of the bulk unit cell was found to converge to the lattice parameters determined by neutron diffraction (a = b = 4.2193(1) Å, c = 4.1287(1) Å, and α = β = γ = 90°),9 when the initial structure was the neutron diffraction crystal structure, for cutoff energies of the plane wave expansion greater than 500 eV. When the plane wave expansion had a cutoff smaller than 500 eV, these lattice parameters were severly underestimated.
We have studied an Eley–Rideal mechanism for ammonia synthesis on θ-Mn6N5 but without the missing N in the 3 × 1 × 1 unit cell. This reduces the size of the unit cell considerably to the 1 × 1 × 1 unit cell shown in Fig. 1, which is a face-centred cubic arrangement of Mn atoms with N atoms octahedrally coordinated. This unit cell has a chemical formula Mn4N4 (or MnN) in which lower Miller index surfaces are shown in Fig. 2. Although our calculations for simplicity were done on Mn4N4, we refer to it as θ-Mn6N5.
3. Results and discussion
3.1. Adsorption and activation of N2 and H2
The adsorption of N2 was investigated on every atop, bridge and hollow adsorption site of the (111), (111)′, (110) and (100) surfaces of θ-Mn6N5 as depicted in Fig. 2. The dispersion-corrected adsorption energy and the percent activation of the N–N bond are shown in Fig. 3a. The adsorption energy was found to be between −5 kJ mol−1 and −43 kJ mol−1, which is weak, indicating physisorption except for the (111) surface where a value of −120 kJ mol−1 was calculated. This adsorption energy is unusually large for dinitrogen and it clearly suggests chemisorption. Although this adsorption energy is very large, the percent activation of the N–N bond is less than 4%. This is much lower than the activation of end-on adsorbed N2 on Co3Mo3N at nitrogen vacancies where 11% activation was found. However, it is comparable to the activation of N2 found on another metal nitride, Ta3N5, which was less than 3%.24 Therefore, the adsorption and activation of N2 on this metal nitride is molecular in the absence of nitrogen vacancies. It is intriguing that among the various metal nitrides studied computationally thus far, the (111) surface of θ-Mn6N5 is the only surface where the adsorption energy of N2 exceeds that of H2; we will return to this point later. | ||
| Fig. 3 (a) Nitrogen and (b) hydrogen adsorption energy and percent activation as a function of the Miller index of the surface of θ-Mn6N5. Percent N2 activation is defined as [r(N2,ads) − 1.098] × 100/1.09. Percent H2 activation is defined as [r(H2,ads) − 0.74] × 100/0.74. | ||
We have studied the adsorption of H2 on all low Miller index surfaces of θ-Mn6N5. The adsorption of H2 was found to be molecular with an adsorption energy that ranged between −79 kJ mol−1 and −87 kJ mol−1. This adsorption energy is rather large for molecular hydrogen and identical in strength with the value calculated on other metal nitrides such as Ta3N5 where it was found to be between −81 kJ mol−1 and −87 kJ mol−1. In contrast to what was observed on Co3Mo3N surfaces, we do not observe dissociative adsorption of hydrogen when nitrogen vacancies are not present and the adsorption of H2 is molecular on every symmetry unique adsorption site of the (111), (111)′, (110) and (100) surfaces of θ-Mn6N5. This indicates that there is a barrier for the dissociative chemisorption of H2 on θ-Mn6N5, however the dissociative chemisorption of H2 at N-vacancies was not considered. In total 15 adsorption sites were investigated and these corresponded to every atop, bridge and hollow site on the three low Miller index surfaces of θ-Mn6N5. The adsorption energy of H2 and the percent activation of the H–H bond are plotted in Fig. 3b. The activation of the H–H bond is also very constant for these adsorption configurations, which was found to be between 1.3% and 1.7%. The most prevalent adsorption configuration of H2 was the end-on adsorption although a tilt was also frequent and one side-on configuration was also found.
If we address the question of competitive adsorption of N2 and H2 on the various low Miller index surfaces of θ-Mn6N5, certain conclusions can be drawn based on these adsorption data. These conclusions are relevant to surfaces of this material that completely lack intrinsic nitrogen vacancies. (i) All surfaces apart from the (111) surface would have mostly hydrogen adsorbed to them, (ii) only the (111) surface would activate the N–N bond significantly, (iii) no activation of the H–H bond is observed when H2 is adsorbed molecularly, and (iv) there is a barrier for the dissociative adsorption of H2. For the above four reasons we have modelled the ammonia synthesis mechanism on the (111) surface and only considered hydrogenation reactions via an Eley–Rideal type mechanism. We were primarily interested if an Eley–Rideal mechanism is possible on this manganese nitride, as we have seen on another metal nitride, Co3Mo3N.22 We note that a potential Langmuir–Hinshelwood mechanism and the participation of intrinsic nitrogen vacancies in the mechanism are not considered in this study and will be the topic of a subsequent study.
3.2. Mechanism of ammonia synthesis on θ-Mn6N5
There are only a few studies of the ammonia synthesis mechanism via DFT on metal surfaces, nanoclusters and in organometallic complexes. Most of the early computational studies are focused on understanding the adsorption, activation and dissociation of N2 on the Ru(0001) surface where it was found to occur primarily at steps rather than planar terraces with an activation barrier of 0.5 eV.34 These findings were in agreement with earlier experimental studies by Ertl and co-workers who found through variable-temperature temperature programmed desorption (TPD) measurements the dissociation activation barriers of 0.3 eV35 and 0.6 eV36 on Ru/MgO. The effect of promoters such as Na and Cs was attributed to an electrostatic interaction that lowers the dissociation activation barrier on Ru(0001).37 A partial study of the ammonia synthesis mechanism by Hu et al.38 revealed that the hydrogenation steps of nitrogen can be as high in barrier as the N–N dissociation steps. Logadóttir and Nørskov21 studied the ammonia synthesis mechanism on Ru(0001) surfaces at steps and at terraces. The influence of an electric field on ΔE for the dissociation of N2 and the formation of NNH was studied on Ru surfaces (i.e. (0001) and (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1)) and found to increase and decrease, respectively.39 Moreover, the barrier for NNH formation was found to decrease with the application of an electric field. There are few theoretical studies of electrochemical ammonia synthesis on metal nanoclusters40 and surfaces10,41,42 at ambient temperature and pressure. Abghoui and Skúlasson43 studied a Mars–van Krevelen mechanism for electrochemical ammonia synthesis on the (100) facet of VN and the (110) facet of RuN. The same authors showed that H-assisted N2 activation is significantly more facile than direct N2 dissociation on transition metal nitrides (TMNs).44 Michalsky and Steinfeld45 explored the possibility of a solar-driven ammonia synthesis cycle where the Fe doping of manganese nitrides was found to increase the number of nitrogen vacancies. Michalsky and Peterson10 also studied the possibility of low-pressure ammonia synthesis for energy storage through chemical looping of metal nitrides. There are also a few DFT studies of the enzymatic ammonia synthesis mechanism. Rod and Nørskov46 studied the ammonia synthesis mechanism on the FeMo cofactor in nitrogenases and found that the reaction is an associative mechanism in which end-on nitrogen adsorbed to the Fe of the cluster undergoes stepwise hydrogenation, forming first hydrazine, which after further hydrogenation forms ammonia. An associative mechanism for ammonia synthesis was studied on mono- and dinuclear molybdenum complexes and cubane-type metal–sulfido clusters.47 More recently an associative ammonia synthesis mechanism has been studied on singly dispersed bimetallic catalyst Rh1Co3/CoO(011)48 similar to how the biological fixation of N2 occurs by nitrogenases in plants.49 The basic characteristic of these ammonia synthesis mechanisms is that N2 is hydrogenated by H+ which causes gradual weakening and dissociation of the N
1)) and found to increase and decrease, respectively.39 Moreover, the barrier for NNH formation was found to decrease with the application of an electric field. There are few theoretical studies of electrochemical ammonia synthesis on metal nanoclusters40 and surfaces10,41,42 at ambient temperature and pressure. Abghoui and Skúlasson43 studied a Mars–van Krevelen mechanism for electrochemical ammonia synthesis on the (100) facet of VN and the (110) facet of RuN. The same authors showed that H-assisted N2 activation is significantly more facile than direct N2 dissociation on transition metal nitrides (TMNs).44 Michalsky and Steinfeld45 explored the possibility of a solar-driven ammonia synthesis cycle where the Fe doping of manganese nitrides was found to increase the number of nitrogen vacancies. Michalsky and Peterson10 also studied the possibility of low-pressure ammonia synthesis for energy storage through chemical looping of metal nitrides. There are also a few DFT studies of the enzymatic ammonia synthesis mechanism. Rod and Nørskov46 studied the ammonia synthesis mechanism on the FeMo cofactor in nitrogenases and found that the reaction is an associative mechanism in which end-on nitrogen adsorbed to the Fe of the cluster undergoes stepwise hydrogenation, forming first hydrazine, which after further hydrogenation forms ammonia. An associative mechanism for ammonia synthesis was studied on mono- and dinuclear molybdenum complexes and cubane-type metal–sulfido clusters.47 More recently an associative ammonia synthesis mechanism has been studied on singly dispersed bimetallic catalyst Rh1Co3/CoO(011)48 similar to how the biological fixation of N2 occurs by nitrogenases in plants.49 The basic characteristic of these ammonia synthesis mechanisms is that N2 is hydrogenated by H+ which causes gradual weakening and dissociation of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond.50,51
N bond.50,51
There is only our previous study of an Eley–Rideal/Mars–van Krevelen mechanism on Co3Mo3N which we have shown to proceed efficiently by nitrogen activation in an end-on configuration at nitrogen vacancies.22 The question that arises is whether an Eley–Rideal mechanism also exist on other metal nitrides? We have therefore modelled a complete pathway for ammonia synthesis via an Eley–Rideal mechanism. The reaction mechanism was modelled on the (111) surface of θ-Mn6N5, which we found adsorbs nitrogen in an end-on configuration. The elementary reaction steps of the mechanism are shown in Scheme 1. The mechanism is an associative mechanism where the hydrogenation of N2 causes gradual weakening of the NN bond and the formation of ammonia. The reaction mechanism is depicted in Fig. 4 for which a brief description is given next. Two N2 adsorb end-on on two manganese atoms that are separated by a nearest neighbour manganese atom (steps B and C). H2 was then fixed by one of its atoms at a 3 Å separation from the terminal nitrogen of adsorbed N2 (step E). The reason for this is that H2 has an exothermic interaction with adsorbed N2, and therefore this increases the reaction barrier for its reaction forming diazanylidene, NNH2. Here a clear distinction has to be made with respect to an Eley–Rideal mechanism occurring at high pressure and at ambient pressure. For the first the adsorption step of H2 could be ignored as the H2 coming from the gas phase has high enough velocity that it would not allow any structural perturbation of the adsorbate at the adsorption site, before the bimolecular collision and reaction. So at high pressure this phenomenon would decrease the barrier for hydrogenation by an amount equal to the exothermicity of the adsorption step. Based on this rational higher pressures would act favourably on the barriers of the hydrogenation steps via an Eley–Rideal mechanism. In step F H2 adsorbs again at a 3 Å distance from the terminal nitrogen of the second adsorbed N2. A second diazanylidene is formed upon hydrogenation (step G). Two diazanylidene adsobates then react with an incoming molecular hydrogen (step H), forming two diazanes, -NNH3 (step I). This elementary reaction step although it appears to be trimolecular is in fact bimolecular, as the two diazanylidene molecules are bound to the same substrate. In steps J and K the diazanes decompose forming two ammonia molecules and leaving nitride groups on the surface. H2 adsorbs to the first nitride group (step L) forming an azanylium, >NH2 (step M). H2 then adsorbs to the second nitride group (step N) and a second azanylium group forms (step O). The two azanylium groups are aligned in such a way that they can stereochemically interact favourably with a H2 coming from the gas phase (step P). There are other alignments of the azanylium group that were higher in energy and therefore not considered in the mechanism. This reaction forms two surface adsorbed ammonia groups at positions that are separated by a Mn atom (step Q). The surface adsorbed ammonia desorbs from the manganese nitride surface (step R) and an adsorbate free surface is recovered so that a new catalytic cycle can be initiated.
 | ||
| Scheme 1 Elementary reaction mechanism steps of ammonia synthesis via an Eley–Rideal mechanism on θ-Mn6N5-(111). The letters t-, b-, h- and a- are to indicate the adsorption position which is top, bridge, 3-fold hollow and adsorbate, respectively. | ||
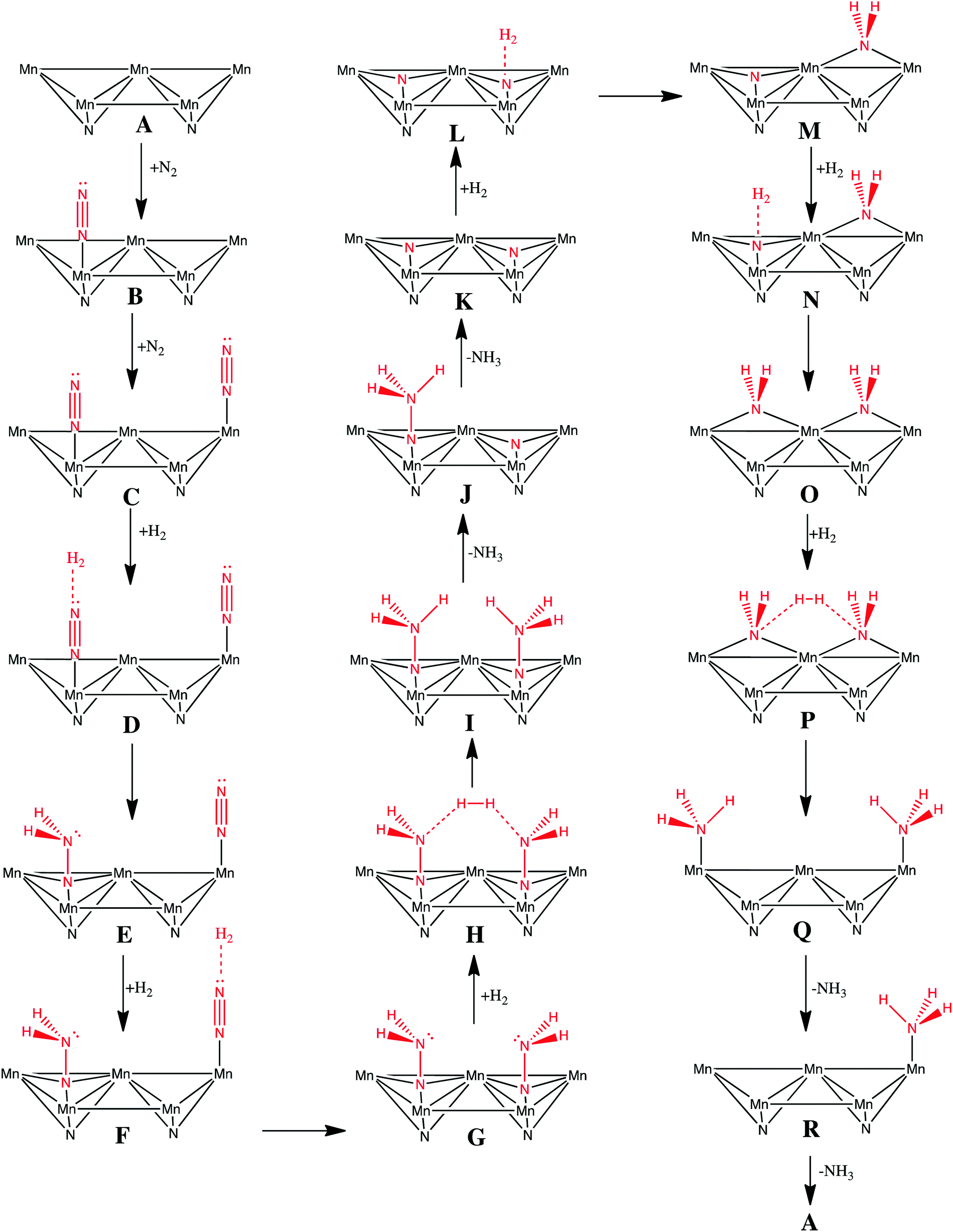 | ||
| Fig. 4 Reaction mechanism for ammonia synthesis via an Eley–Rideal mechanism on θ-Mn6N5-(111). | ||
3.3. Potential energy surface of reaction
In Fig. 5 we present the potential energy curve for the Eley–Rideal mechanism on θ-Mn6N5-(111). The values of the activation energy barriers (Eact) and the reaction energies (ΔEreact) are tabulated in Table 1. We observe that the adsorption of N2 to the (111) surface is exothermic by −122 kJ mol−1 (step B and C). This is followed by an exothermic adsorption of molecular H2 of −76 kJ mol−1 (step D). The previous adsorption energies indicate that the surface will be entirely covered with end-on N2 before the reaction of H2 starts to occur. H2 then reacts with the adsorbed N2 in a high barrier process with an activation energy of 327 kJ mol−1 (step D to E). The adsorption of a second H2 is only slightly exothermic by −17 kJ mol−1. The next hydrogenation barrier is 478 kJ mol−1 high which is considerably higher than the first hydrogenation barrier which could be due to stereochemical repulsion between the diazanylidene and the incoming H2. Here the adsorption of H2 to the two diazanylidenes is slightly exothermic by −20 kJ mol−1. The third hydrogenation barrier of this H2 that forms two diazanes is much smaller (182 kJ mol−1) compared to the barrier of N2 hydrogenation to NNH2. The decomposition of the diazane into ammonia and a nitride is slightly endothermic by 11 kJ mol−1 for the first ammonia (step J) and 67 kJ mol−1 for the second ammonia (step K). The adsorption of the fourth H2 to the nitride was found to be again exothermic by −78 kJ mol−1 (step L) with a high hydrogenation barrier of 265 kJ mol−1 (step L to M) in order to form azanylium. This was followed by the adsorption of the fifth H2, which was again found to be exothermic by −81 kJ mol−1 (step N).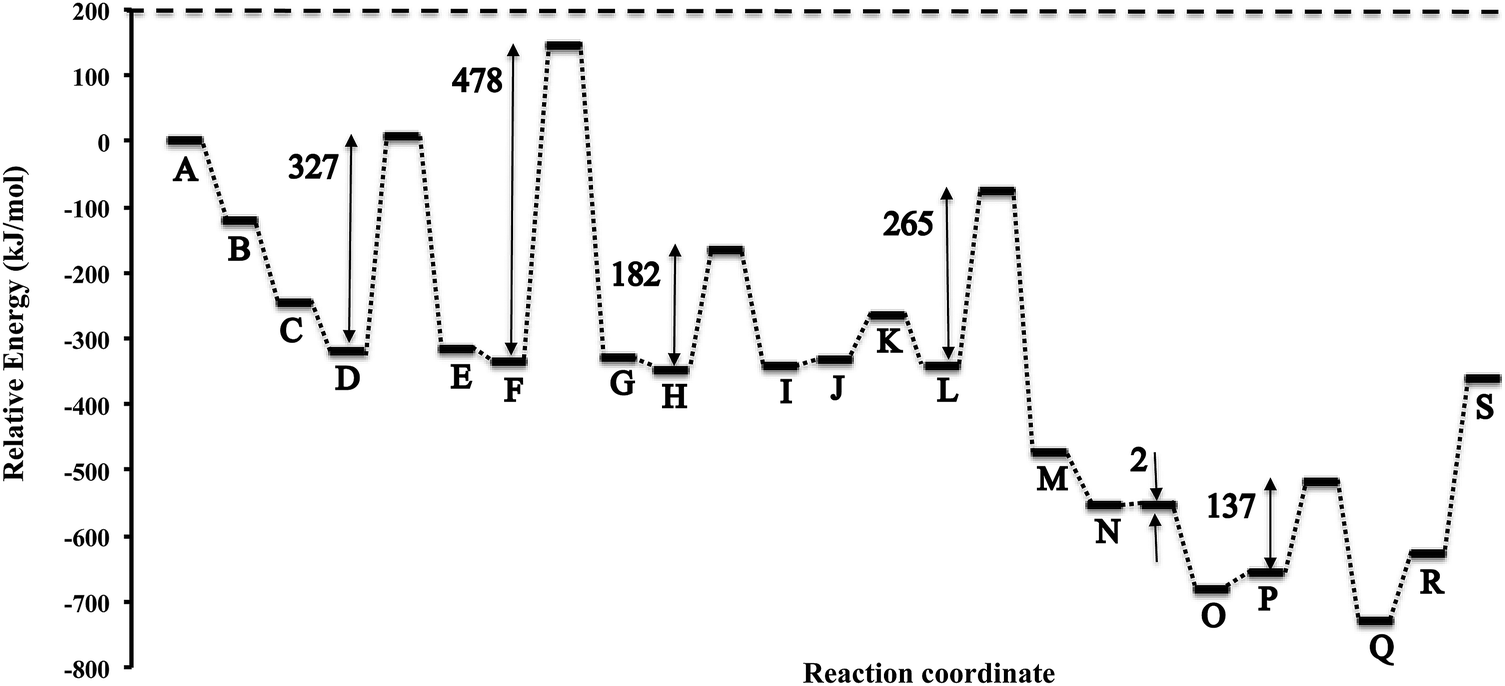 | ||
| Fig. 5 Potential energy diagram of the ammonia synthesis reaction via an Eley–Rideal mechanism on the θ-Mn6N5-(111) slab without surface nitrogen vacancies. | ||
| Label | ΔEreact (kJ mol−1) | Label | E act (kJ mol−1) |
|---|---|---|---|
| ΔE (AB) | −122 | ||
| ΔE (BC) | −122 | ||
| ΔE (CD) | −76 | ||
| ΔE (DE) | 3 | E act (DE) | 327 |
| ΔE (EF) | −17 | E act (FG) | 478 |
| ΔE (FG) | 6 | ||
| ΔE (GH) | −20 | ||
| ΔE (HI) | 5 | E act (HI) | 182 |
| ΔE (IJ) | 11 | ||
| ΔE (JK) | 67 | ||
| ΔE (KL) | −78 | ||
| ΔE (LM) | −131 | E act (LM) | 265 |
| ΔE (MN) | −81 | ||
| ΔE (NO) | −126 | E act (NO) | 2 |
| ΔE (OP) | 26 | ||
| ΔE (PQ) | −76 | E act (PQ) | 137 |
| ΔE (QR) | 103 | ||
| ΔE (RS) | 267 |
Interestingly, the hydrogenation of the nitride into an azanylium that had an adjacent azanylium (step N to O) had a barrier of only 2 kJ mol−1. We note that this elementary reaction step may be a low energy pathway for the hydrogenation of surface nitrides in metal nitrides that have 3-fold bound nitrogen which is relevant to the formation of nitrogen vacancies on manganese nitrides on surfaces such as (111)′ shown in Fig. 2.
The adsorption of the sixth H2 becomes endothermic by 26 kJ mol−1 (step P). The subsequent barrier for hydrogenation is 137 kJ mol−1 which forms two ammonia molecules adsorbed to the surface. The desorption of these ammonia from the surface is highly endothermic, with ΔE of 103 kJ mol−1 (step R) and 267 kJ mol−1, respectively, for the desorption of the first and second ammonia. From the above potential energy surface of the reaction it is apparent that the mechanism does not proceed at low temperatures via Eley–Rideal chemistry although it is possible at high T. The hydrogenation barriers, apart from the 5th hydrogenation barrier, are high and therefore other mechanisms should be examined, such as Langmuir–Hinshelwood mechanisms or other pathways that occur at nitrogen vacancies. It is intriguing though that an Eley–Rideal mechanism for the hydrogenation of surface activated nitrogen exists on Co3Mo3N, which could be the underlying reason for the known enhanced activity of this tertiary metal nitride.
Conclusions
The adsorption and activation of N2 and H2 was studied on every low Miller index surface of Mn6N5+x (x = 1). The adsorption of N2 was molecular in the absence of nitrogen vacancies with adsorption energies of −5 kJ mol−1 to −43 kJ mol−1. Only the (111) surface was found to activate N2, 4%, with a large exothermic adsorption energy (−120 kJ mol−1). This surface would inhibit the poisoning of the surface by H2 which was found to bind strongly (−79 kJ mol−1 to −87 kJ mol−1) in a molecular form in the absence of nitrogen vacancies to all surfaces of Mn6N5+x (x = 1). The complete reaction Eley–Rideal reaction pathway was modelled on the (111) surface of Mn6N5+x (x = 1). It shows that an Eley–Rideal mechanism, in contrast with what was found on Co3Mo3N, would only proceed at high temperatures. This suggests that other mechanisms such as Langmuir–Hinshelwood or mechanisms that invoke nitrogen vacancies should be the focus of future studies of mechanistic pathways.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by EPSRC funding (EP/L026317/1, EP/K014714/1). Via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202/1), this work used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk). The authors acknowledge the use of the Grace High Performance Computing Facility (Grace@UCL), and associated support services, in the completion of this work.References
- W. R. L. Lambrecht, M. Prokhodko and M. S. Miao, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 174411 CrossRef.
- A. Leineweber, R. Niewa, H. Jacobs and W. Kockelmann, J. Mater. Chem., 2000, 10, 2827–2834 RSC.
- G. Kreiner and H. Jacobs, J. Alloys Compd., 1992, 183, 345–362 CrossRef.
- G. W. Wiener and J. A. Berger, Trans. Am. Inst. Min., Metall. Pet. Eng., 1955, 203, 360–368 Search PubMed.
- M. N. Eddine and E. F. Bertaut, Solid State Commun., 1977, 23, 147–150 CrossRef.
- M. Mekata, J. Haruna and H. Takaki, J. Phys. Soc. Jpn., 1968, 25, 234–238 CrossRef.
- F. Lihl, P. Ettmayer and A. Kutzelnigg, Z. Metallkd., 1962, 53, 715–719 Search PubMed.
- K. Suzuki, T. Kaneko, H. Yoshida, Y. Obi, H. Fujimori and H. Morita, J. Alloys Compd., 2000, 306, 66–71 CrossRef.
- A. Leineweber, R. Niewa, H. Jacobs and W. Kockelmann, J. Mater. Chem., 2000, 10, 2827–2834 RSC.
- R. Michalsky, A. M. Avram, B. A. Peterson, P. H. Pfromm and A. A. Peterson, Chem. Sci., 2015, 6, 3965–3974 RSC.
- S. Laassiri, C. D. Zeinalipour-Yazdi, C. R. A. Catlow and J. S. J. Hargreaves, Appl. Catal., B, 2018, 223, 60–66 CrossRef.
- T. J. Wood, J. W. Makepeace and W. I. F. David, Phys. Chem. Chem. Phys., 2018, 20, 8547–8553 RSC.
- J. Guo, P. Wang, G. Wu, A. Wu, D. Hu, Z. Xiong, J. Wang, P. Yu, F. Chang, Z. Chen and P. Chen, Angew. Chem., Int. Ed., 2015, 54, 2950–2954 CrossRef PubMed.
- P. Yu, J. Guo, L. Liu, P. Wang, G. Wu, F. Chang and P. Chen, ChemSusChem, 2016, 9, 364–369 CrossRef PubMed.
- C. Walter, W. Menezes Prashanth, S. Orthmann, J. Schuch, P. Connor, B. Kaiser, M. Lerch and M. Driess, Angew. Chem., Int. Ed., 2017, 57, 698–702 CrossRef PubMed.
- U. Zwicker, Z. Metallkd., 1951, 42, 274–276 Search PubMed.
- M. D. Lyutaya and A. B. Goncharuk, Powder Metall. Met. Ceram., 1977, 16, 208–212 CrossRef.
- N. Otsuka, Y. Hanawa and S. Nagakura, Phys. Status Solidi, 1977, 43, K127–K129 CrossRef.
- J. S. J. Hargreaves, Appl. Petrochem. Res., 2014, 4, 3–10 CrossRef.
- G. E. Veitch, K. L. Bridgwood and S. V. Ley, Org. Lett., 2008, 10, 3623–3625 CrossRef PubMed.
- Á. Logadóttir and J. K. Nørskov, J. Catal., 2003, 220, 273–279 CrossRef.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2018, 122, 6078–6082 CrossRef.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2015, 119, 28368–28376 CrossRef.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves, S. Laassiri and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2017, 19, 11968–11974 RSC.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2016, 120, 21390–21398 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef PubMed.
- G. Kresse and D. Jouber, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Mills, H. Jónsson and G. K. Schenter, Surf. Sci., 1995, 324, 305–337 CrossRef.
- S. Dahl, A. Logadottir, R. C. Egeberg, J. H. Larsen, I. Chorkendorff, E. Törnqvist and J. K. Nørskov, Phys. Rev. Lett., 1999, 83, 1814–1817 CrossRef.
- F. Rosowski, O. Hinrichsen, M. Muhler and G. Ertl, Catal. Lett., 1996, 36, 229–235 CrossRef.
- O. Hinrichsen, F. Rosowski, A. Hornung, M. Muhler and G. Ertl, J. Catal., 1997, 165, 33–44 CrossRef.
- J. J. Mortensen, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 80, 4333–4336 CrossRef.
- C. J. Zhang, M. Lynch and P. Hu, Surf. Sci., 2002, 496, 221–230 CrossRef.
- R. Manabe, H. Nakatsubo, A. Gondo, K. Murakami, S. Ogo, H. Tsuneki, M. Ikeda, A. Ishikawa, H. Nakai and Y. Sekine, Chem. Sci., 2017, 8, 5434–5439 RSC.
- J. G. Howalt, T. Bligaard, J. Rossmeisl and T. Vegge, Phys. Chem. Chem. Phys., 2013, 15, 7785–7795 RSC.
- E. Skulason, T. Bligaard, S. Gudmundsdottir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jonsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- Y. Abghoui, A. L. Garden, V. F. Hlynsson, S. Bjorgvinsdottir, H. Olafsdottir and E. Skulason, Phys. Chem. Chem. Phys., 2015, 17, 4909–4918 RSC.
- Y. Abghoui and E. Skúlasson, Proc. Comp. Sci., 2015, 51, 1897–1906 CrossRef.
- Y. Abghoui and E. Skúlason, Catal. Today, 2017, 286, 69–77 CrossRef.
- R. Michalsky, P. H. Pfromm and A. Steinfeld, Interface Focus, 2015, 5, 1–9 CrossRef PubMed.
- T. H. Rod and J. K. Nørskov, J. Am. Chem. Soc., 2000, 122, 12751–12763 CrossRef.
- H. Tanaka, Y. Nishibayashi and K. Yoshizawa, Acc. Chem. Res., 2016, 49, 987–995 CrossRef PubMed.
- X.-L. Ma, J.-C. Liu, H. Xiao and J. Li, J. Am. Chem. Soc., 2018, 140, 46–49 CrossRef PubMed.
- S. Kuriyama, K. Arashiba, K. Nakajima, Y. Matsuo, H. Tanaka, K. Ishii, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2016, 7, 12181 CrossRef PubMed.
- I. Coric, B. Q. Mercado, E. Bill, D. J. Vinyard and P. L. Holland, Nature, 2015, 526, 96 CrossRef PubMed.
- B. M. Hoffman, D. Lukoyanov, Z. Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp02381f |
| This journal is © the Owner Societies 2018 |