
Titanium(IV) oxide having a copper co-catalyst: a new type of semihydrogenation photocatalyst working efficiently at an elevated temperature under hydrogen-free and poison-free conditions†
Hiroshi
Kominami
 *a,
Misaki
Shiba
a,
Akimi
Hashimoto
a,
Shota
Imai
a,
Kousuke
Nakanishi
b,
Atsuhiro
Tanaka
a,
Keiji
Hashimoto
a and
Kazuya
Imamura‡
a
*a,
Misaki
Shiba
a,
Akimi
Hashimoto
a,
Shota
Imai
a,
Kousuke
Nakanishi
b,
Atsuhiro
Tanaka
a,
Keiji
Hashimoto
a and
Kazuya
Imamura‡
a
aDepartment of Applied Chemistry, Faculty of Science and Engineering, Kindai University, 3-4-1 Kowakae, Higashiosaka, Osaka 577-8502, Japan. E-mail: hiro@apch.kindai.ac.jp
bMolecular and Material Engineering, Interdisciplinary Graduate School of Science and Engineering, Kindai University, 3-4-1, Kowakae, Higashiosaka, Osaka 577-8502, Japan
First published on 21st May 2018
Abstract
A copper-loaded titanium(IV) oxide photocatalyst exhibited perfect selectivity in hydrogenation of alkynes to alkenes in an alcohol solution at 298 K under hydrogen-free and poison-free conditions. A slight elevation in the reaction temperature to 323 K greatly increased the reaction rate with the selectivity being preserved and the formation of an H2 by-product being suppressed. The apparent activation energy of 4-octyne semihydrogenation was determined to be 54 kJ mol−1, indicating that the rate determining step of this photocatalytic reaction was not an electron production process but a thermocatalytic hydrogenation process under light irradiation.
When titanium(IV) oxide (TiO2) is irradiated by UV light, electrons in the valence band (VB) are excited to the conduction band (CB), leaving positive holes in the VB. The thus-formed electrons and positive holes cause reduction and oxidation, respectively, of compounds adsorbed on the surface of a photocatalyst.1,2 Since positive holes have strong oxidation power, total or partial oxidation of organic compounds has been extensively studied. On the other hand, applications of photocatalytic reduction have been less frequently reported because the reduction potential of many organic compounds is more negative than the reduction potential of the CB. Therefore, target compounds in photocatalytic reduction (or hydrogenation) are basically limited to those having a carbonyl group3 and a nitro group,4 and other photocatalytic reductions of organic compounds have scarcely been reported. Recently, we reported photocatalytic reduction (hydrogenation) of a cyano group (benzonitrile) to an amino group (benzylamine) using palladium-loaded TiO2 (Pd–TiO2) even though the reduction potential of benzonitrile is higher than the potential of the conduction band of TiO2,5 indicating that the applicability of photocatalytic reduction is not limited by the CB position of semiconductor photocatalysts and that a new photocatalytic reduction can be developed if metal co-catalysts are introduced on photocatalysts. Subsequently, we found that difficult reductions can be achieved by combining the TiO2 photocatalyst with silver (Ag) and copper (Cu) co-catalysts. (2,3-Epoxypropyl)benzene and 4-octyne were successfully converted to allylbenzene and cis-4-octene in alcohol suspensions of Ag-loaded TiO2 and Cu-loaded TiO2, respectively.6,7 The excellence of Ag–TiO2 and Cu–TiO2 is that these photocatalysts showed complete chemoselectivity, i.e., no subsequent hydrogenation of alkenes to alkanes occurred over these photocatalysts. In the latter case, photocatalytic hydrogenation consists of two processes: (1) formation of an active hydrogen species over metal particles loaded on TiO2 under light irradiation and (2) reduction (or hydrogenation) of the target compound over metal particles (Scheme 1).
 | ||
| Scheme 1 Semihydrogenation of alkynes to cis-alkenes over a Cu–TiO2 photocatalyst. | ||
In these processes, process (2) is not a photocatalytic process but a catalytic process. If process (1) is the rate determining step, the overall reaction rate can be increased by decreasing electron–hole recombination and increasing carrier trapping, which could be achieved by preparation of photocatalysts having high crystallinity and a large surface area. On the other hand, if process (2) is the rate determining step, acceleration of process (2) would be more effective than acceleration of process (1). In this study, we focused on the hydrogenation of alkynes in an alcohol suspension of a Cu–TiO2 photocatalyst7 because we think that the latter hydrogenation process is the rate determining step. Here we briefly show that the overall reaction rate can be increased by acceleration of the catalytic (thermal) process at an elevated temperature.
By using the photodeposition method, 0.5 wt% Cu as a co-catalyst was loaded on TiO2. TiO2 powder (P 25 supplied by Nippon Aerosil Co., Ltd) was suspended in 10 cm3 of an aqueous methanol solution (10 vol%) containing copper chloride in a test tube. The test tube was sealed with a rubber septum under argon (Ar) and then photoirradiated for 90 min at λ > 300 nm by a 400 W high-pressure mercury arc (Eiko-sha, Osaka) with magnetic stirring in a water bath continuously kept at 298 K. The color of Cu–TiO2 after the photodeposition process was purple, typically due to surface plasmonic resonance of metallic Cu nanoparticles. The resulting powder was washed repeatedly with distilled water and dried for 1 h in vacuo. The color turned faint blue when Cu–TiO2 was exposed to air.
In a typical run, Cu–TiO2 (50 mg) was suspended in 5 cm3 of methanol containing 50 μmol of 4-octyne in a test tube, which was sealed with a rubber septum and then photoirradiated under Ar with a xenon lamp (Optical Modulex, Ushio, Tokyo) in a water bath. The temperature of the water bath was changed to evaluate the effects of reaction temperature on the rate, product selectivity and chemoselectivity of the photocatalytic reaction. During the photocatalytic reaction, the color changed to gray, that is a typical color of fine metal particles. The amounts of 4-octyne, cis-4-octene, trans-4-octene and octane were determined with an FID-type gas chromatograph (GC-2025, Shimadzu, Kyoto) equipped with a DB-1 column. The amount of H2 as the reduction product of protons (H+) was determined with a TCD-type gas chromatograph (GC-8A, Shimadzu) equipped with an MS-5A column. The color of Cu–TiO2 again changed to faint blue when exposed to air after the reaction.
Fig. 1(a) shows the time course of photocatalytic conversion of 4-octyne in a methanolic suspension of Cu–TiO2 at 298 K. 4-Octyne was consumed along with photoirradiation, while cis-4-octene was produced instead, indicating that 4-octyne was hydrogenated by active hydrogen species that were formed over Cu by reduction of protons by photogenerated electrons. Material balance (MB) was calculated to be almost unity from eqn (1):
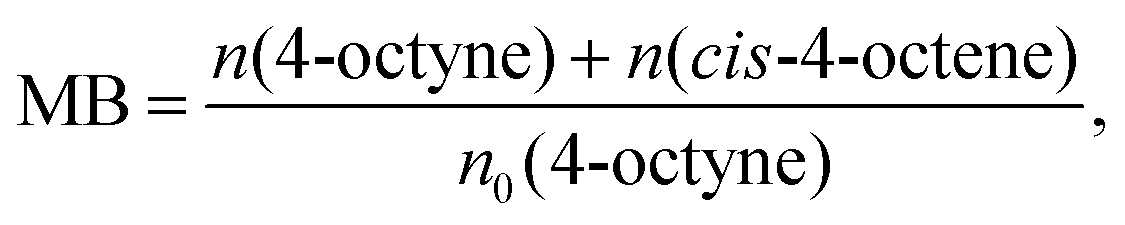 | (1) |
 | ||
| Fig. 1 Time courses of the amounts of 4-octyne, cis-4-octene and H2 and the material balance (MB) in a methanolic suspension of 0.5 wt% Cu–TiO2 photocatalyst under deaerated conditions at (a) 298 K and (b) 323 K. | ||
Fig. 1(b) shows the time course of the same reaction operated at 323 K. The reaction rate for semihydrogenation drastically increased at 323 K, and 4-octyne was almost completely consumed at 1 h and 40 min with chemoselectivity and diastereoselectivity being preserved as they were at 298 K, i.e., 4-octyne was almost quantitatively hydrogenated to cis-4-octene within a short time. We noted that the formation of H2 was completely suppressed during the production of cis-4-octene and that H2 evolved only after consumption of 4-octyne. The results of photocatalytic reaction at 323 K were in contrast to those of the reaction at 298 K shown in Fig. 1(a). After consumption of 4-octyne, no cis-4-octene was consumed, indicating that Cu–TiO2 possessed complete chemoselectivity for semihydrogenation of 4-octyne, i.e., no cis-4-octene was hydrogenated and isomerized to trans-4-octene over Cu–TiO2 even at 323 K.
To understand the semihydrogenation of 4-octyne under light irradiation, we carried out additional experiments under different conditions: dark reactions at 323 K under argon (Ar, 1 atm) and H2 (1 atm, 1.3 mmol). Since the Cu species was partly oxidized as shown in Fig. 1(a), Cu–TiO2 was used for the dark reactions after reduction of the Cu species in methanol under light irradiation in the absence of 4-octyne. No reactions occurred in the dark under Ar and H2 at 323 K. The result under Ar indicates that no thermal reaction occurred over Cu metal at 323 K and that light irradiation is indispensable for inducing hydrogenation of 4-octyne. The result under H2 is an interesting feature of this system, i.e., an excess of H2 (1.3 mmol) in the gas phase did not contribute to the hydrogenation of 4-octyne over Cu–TiO2 at 323 K, although semihydrogenation of alkynes with H2 over Cu supported on silica (Cu/SiO2) has been reported in both liquid-batch and gas-flow systems.8
There are some possible reasons for the differences between the present and previously reported results. First, the reaction temperature (323 K) is still low for dissociative adsorption of H2 on Cu metal in a methanolic suspension. Second, H2 pressure (1 atm) is insufficient for H2 to be dissolved in methanol or to be adsorbed on Cu–TiO2 suspended in methanol. Third, the hydrogen active species formed by dissociative adsorption of H2 and that formed photocatalytically are somehow different.
Since the reaction temperature showed a great effect on the reaction rate of photocatalytic semihydrogenation of 4-octyne to cis-4-octene, photocatalytic reactions were carried out at various temperatures. Fig. 2(a) shows the amounts of 4-octyne and cis-4-octene and the material balance calculated by eqn (1) after 1 h irradiation. The reaction rate for semihydrogenation of 4-octyne to cis-4-octene increased with elevation of the reaction temperature. The results were used for estimation of the apparent activation energy (Ea) for this reaction over Cu–TiO2, and an Arrhenius plot is shown in Fig. 2(b). A linear correlation was observed in the Arrhenius plot between 298 K and 323 K. From the slope of the plot, Ea in this reaction system was estimated to be 54 kJ mol−1.
 | ||
| Fig. 2 (a) Effect of reaction temperature on semihydrogenation of 4-octyne to cis-4-octene in a methanolic suspension of 0.5 wt% Cu–TiO2 photocatalyst for 1 h. (b) Arrhenius plot (logarithm of k vs. reciprocal of T). | ||
For comparison, photocatalytic reactions in a methanolic suspension of Cu–TiO2 in the absence of 4-octyne were examined at various temperatures (Fig. 3(a)). In this case, H2 was solely evolved as the reduced product because there is no other electron acceptor. The results were used for estimation of Ea for H2 evolution over Cu–TiO2, and an Arrhenius plot is shown in Fig. 3(b). A linear correlation was observed in the Arrhenius plot between 298 K and 323 K, and Ea of H2 evolution under this condition was estimated from the slope of the plot to be 12 kJ mol−1. The value of Ea was close to values of photocatalytic evolution of H2 from aqueous solutions of ethylene glycol over gold-loaded TiO2 (15 kJ mol−1)9 and 2-propanol over platinum-loaded TiO2 (20 kJ mol−1).10 These results indicate that semihydrogenation of 4-octyne on Cu metal should overcome the relatively large Ea. Due to the large Ea of semihydrogenation, the reaction rate was small at temperatures around room temperature, resulting in a large yield of H2. The reaction rate of semihydrogenation became large with elevation of the reaction temperature and finally exceeded the reaction rate of H2 evolution at 323 K. Switching of the predominant reaction from H2 formation to semihydrogenation at 323 K over Cu–TiO2 in this system is attributed to the large frequency factor of the Arrhenius equation in semihydrogenation. These results indicate that the rate determining step is not a photocatalytic process but a thermocatalytic process. If the rate determining step is not a photocatalytic process, the reaction rate is independent of the light intensity. Photocatalytic reactions of 4-octyne in methanol suspensions of Cu–TiO2 under photoirradiation of different intensities were examined (Table 1). Almost constant rates show that the light intensity is not a decisive factor in this reaction and that a photocatalytic process is not the rate determining step.
 | ||
| Fig. 3 (a) Time courses of H2 evolution from a methanolic suspension of 0.5 wt% Cu–TiO2 photocatalyst at various temperatures. (b) Arrhenius plot (logarithm of k vs. reciprocal of T). | ||
| Light sourceb | UV intensityc/mW cm−2 | Temp./K | Rated/μmol h−1 |
|---|---|---|---|
| a 4-octyne: 50 μmol, methanol: 5.0 cm3, 0.5 wt% Cu–TiO2: 50 mg. b Xenon lamp: Optical Modulex (Ushio, Tokyo), UV LED: PJ-1505-2CA (CCS, Kyoto). c Determined using a spectroradiometer USR-45D (Ushio, Tokyo). d Rate of cis-4-octene formation. | |||
| Xenon lamp | 31.7 | 298 | 11.5 |
| UV LED | 4.7 | 296 | 11.0 |
| UV LED | 2.3 | 296 | 11.1 |
Since a slight elevation in the reaction temperature drastically increased the reaction rate and improved selectivity in electron utilization in diastereoselective semihydrogenation of 4-octyne, this effect was evaluated in another system, i.e., hydrogenation of 5-hexynenitrile having another functional group (Scheme 2).
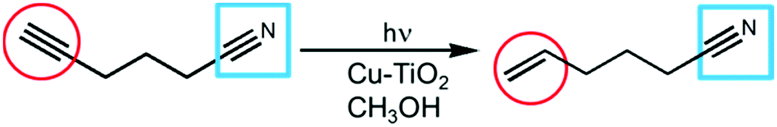 | ||
| Scheme 2 Photocatalytic semihydrogenation of 5-hexynenitrile. | ||
A previous study revealed that only the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond was hydrogenated to the C
C triple bond was hydrogenated to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond over a Cu–TiO2 photocatalyst with the nitrile group being preserved.7Table 2 shows results of photocatalytic conversion of 5-hexynenitrile in a methanol suspension of Cu–TiO2 at 298 K and 323 K. At 298 K, 5-hexynenitrile was almost completely consumed and 5-hexenenitrile was obtained in 93% yield after 7 h. When the reaction was carried out at 323 K, 5-hexynenitrile was almost quantitatively converted to 5-hexenenitrile only after 1.5 h. These results clarify that a slight elevation in the reaction temperature over a Cu–TiO2 photocatalyst is effective for drastically increasing a reaction rate without the change in the chemoselectivity.
C double bond over a Cu–TiO2 photocatalyst with the nitrile group being preserved.7Table 2 shows results of photocatalytic conversion of 5-hexynenitrile in a methanol suspension of Cu–TiO2 at 298 K and 323 K. At 298 K, 5-hexynenitrile was almost completely consumed and 5-hexenenitrile was obtained in 93% yield after 7 h. When the reaction was carried out at 323 K, 5-hexynenitrile was almost quantitatively converted to 5-hexenenitrile only after 1.5 h. These results clarify that a slight elevation in the reaction temperature over a Cu–TiO2 photocatalyst is effective for drastically increasing a reaction rate without the change in the chemoselectivity.
In summary, a slight increase in the reaction temperature greatly increased the reaction rate for photocatalytic semihydrogenation of alkynes to alkenes in a methanolic suspension of Cu–TiO2. A large amount of H2 simultaneously evolved at 298 K, while only semihydrogenation of 4-octyne occurred at 323 K. These results indicate that the rate determining step is not a photocatalytic process but a thermocatalytic process. Switching of the predominant reaction from H2 formation to semihydrogenation at 323 K over Cu–TiO2 in this system is attributed to a large Ea and a large frequency factor for semihydrogenation and a small Ea for H2 formation over a Cu co-catalyst. The results obtained in this study show that photocatalytic reactions can be thermally accelerated if a thermocatalytic process is the rate determining step in the photocatalytic reaction and Ea of the process is relatively large.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by JSPS KAKENHI Grant Numbers 17H03462, 15J11412, 17H04967, and 16K18292. This work was also supported by MEXT-Supported Program for the Strategic Research Foundation at Private Universities 2014-2018, subsidy from MEXT and Kindai University. K. N. is grateful to the Japan Society for the Promotion of Science (JSPS) for a Research Fellowship for young scientists. A. T. is grateful for financial support from the Faculty of Science and Engineering, Kindai University.References
- J. Colmenares and Y.-J. Xu, Heterogeneous Photocatalysis: From Fundamentals to Green Applications, Springer-Verlag, Berlin, Heidelberg, 2016 Search PubMed.
- D. Friedmann, A. Hakki, H. Kim, W. Choi and D. Bahnemann, Green Chem., 2016, 18, 5391 RSC.
- (a) C. Joyce-Pruden, J. K. Pross and Y. Li, J. Org. Chem., 1992, 57, 5087 CrossRef; (b) J. W. Park, M. J. Hong and K. K. Park, Bull. Korean Chem. Soc., 2001, 22, 1213 Search PubMed; (c) Y. Matsushita, S. Kumada, K. Wakabayashi, K. Sakeda and T. Ichimura, Chem. Lett., 2006, 35, 410 CrossRef; (d) S. Kohtani, E. Yoshioka, K. Saito, A. Kudo and H. Miyabe, Catal. Commun., 2010, 11, 1049 CrossRef.
- (a) F. Mahdavi, T. C. Bruton and Y. Li, J. Org. Chem., 1993, 58, 744 CrossRef; (b) J. L. Ferry and W. H. Glaze, Langmuir, 1998, 14, 3551 CrossRef; (c) O. V. Makarova, T. Rajh, M. C. Thurnauer, A. Martin, P. A. Kemme and D. Cropek, Environ. Sci. Technol., 2000, 34, 4797 CrossRef; (d) V. Brezová, P. Tarábek, D. Dvoranová, A. Staško and S. Biskupič, J. Photochem. Photobiol., A, 2003, 155, 179 CrossRef; (e) H. Tada, T. Ishida, A. Takao and S. Ito, Langmuir, 2004, 20, 7898 CrossRef PubMed; (f) T. Zhang, L. You and Y. Zhang, Dyes Pigm., 2006, 68, 95 CrossRef; (g) S. O. Flores, O. R. Bernij, M. A. Valenzuela, I. Córdova, R. Gómez and R. Gutiérrez, Top. Catal., 2007, 44, 507 CrossRef; (h) H. Kominami, S. Iwasaki, T. Maeda, K. Imamura, K. Hashimoto, Y. Kera and B. Ohtani, Chem. Lett., 2009, 15, 410 CrossRef; (i) K. Imamura, S. Iwasaki, T. Maeda, K. Hashimoto, B. Ohtani and H. Kominami, Phys. Chem. Chem. Phys., 2011, 13, 5114 RSC; (j) S. Chen, H. Zhang, X. Yu and W. Liu, Chin. J. Chem., 2010, 28, 21 CrossRef; (k) K. Imamura, K. Hashimoto and H. Kominami, Chem. Commun., 2012, 48, 4356 RSC; (l) K. Imamura, T. Yoshikawa, K. Hashimoto and H. Kominami, Appl. Catal., B, 2013, 134-135, 193 CrossRef.
- K. Imamura, T. Yoshikawa, K. Nakanishi, K. Hashimoto and H. Kominami, Chem. Commun., 2013, 49, 10911 RSC.
- H. Kominami, S. Yamamoto, K. Imamura, A. Tanaka and K. Hashimoto, Chem. Commun., 2014, 50, 4558 RSC.
- H. Kominami, M. Higa, T. Nojima, T. Ito, K. Nakanishi, K. Hashimoto and K. Imamura, ChemCatChem, 2016, 8, 2019 CrossRef.
- (a) J. T. Wehrli, D. J. Thomas, M. S. Wainwright, D. L. Trimm and N. W. Cant, Appl. Catal., 1991, 70, 253 CrossRef; (b) A. Fedorov, H.-J. Liu, H.-K. Lo and C. Copéret, J. Am. Chem. Soc., 2016, 138, 16502 CrossRef PubMed.
- G. R. Bamwenda, S. Tsubota, T. Kobayashi and M. Haruta, J. Photochem. Photobiol., A, 1994, 77, 59 CrossRef.
- F. H. Hussein and R. Rudham, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 2817 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2. See DOI: 10.1039/c8cp02316f |
| ‡ Present address: Research Laboratory of Hydrothermal Chemistry, Faculty of Science and Technology, Kochi University, Akebono-cho 2-5-1, Kochi City, Kochi 780-8520, Japan. |
| This journal is © the Owner Societies 2018 |