
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Morphology control of anatase TiO2 for well-defined surface chemistry†
Gabriel
Jeantelot
 a,
Samy
Ould-Chikh
a,
Julien
Sofack-Kreutzer
a,
Edy
Abou-Hamad
a,
Dalaver H.
Anjum
b,
Sergei
Lopatin
b,
Moussab
Harb
*a,
Luigi
Cavallo
a and
Jean-Marie
Basset
*a
a,
Samy
Ould-Chikh
a,
Julien
Sofack-Kreutzer
a,
Edy
Abou-Hamad
a,
Dalaver H.
Anjum
b,
Sergei
Lopatin
b,
Moussab
Harb
*a,
Luigi
Cavallo
a and
Jean-Marie
Basset
*a
aKaust Catalysis Center (KCC), Physical Science and Engineering Division (PSE), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: samy.ouldchickh@kaust.edu.sa; moussab.harb@kaust.edu.sa
bImaging and Characterization Lab, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
First published on 16th May 2018
Abstract
A specific allotrope of titanium dioxide (anatase) was synthesized both with a standard thermodynamic morphology ({101}-anatase) and with a highly anisotropic morphology ({001}-anatase) dominated by the {001} facet (81%). The surface chemistry of both samples after dehydroxylation was studied by 1H NMR and FT-IR. The influence of surface fluorides on the surface chemistry was also studied by 1H NMR, FT-IR and DFT. Full attribution of the IR spectra of anatase with dominant {001} facets could be provided based on experimental data and further confirmed by DFT. Our results showed that chemisorbed H2O molecules are still present on anatase after dehydroxylation at 350 °C, and that the type of surface hydroxyls present on the {001} facet is dependent on the presence of fluorides. They also provided general insight into the nature of the surface species on both fluorinated and fluorine-free anatase. The use of vanadium oxychloride (VOCl3) allowed the determination of the accessibility of the various OH groups spectroscopically observed.
Introduction
In recent years, a significant amount of interest has been dedicated to the surface chemistry of titanium dioxide1 and its possible role as a support for a large number of catalytic reactions, in particular due to the labile nature of its oxygen atoms.2 These surface oxygen atoms are expected to actively take part in a variety of oxidoreduction processes. Examples of such processes include the selective catalytic reduction of nitrogen oxides,3–6 the liquid and gas phase Wacker process,7,8 conversion of ethane to acetic acid,9,10 conversion of methane to methanol,11,12 steam reforming of methane,13 and alkane oxidative dehydrogenation.14 Furthermore, the semi-conductor properties of TiO2 allow it to actively take part in photocatalytic processes, such as the photocatalytic degradation of organic compounds,15 and photocatalytic water splitting.16 A good understanding and control of the nature of the surface species present on the surface of titanium dioxide would be highly valuable for the application of “catalysis by design” to these processes.17The three most common allotropes of TiO2 are brookite, rutile and anatase.1 The latter will be the focus of this work. Hydrothermally synthesized anatase usually bears {101}, {100} and {001} facets on which water is chemisorbed.1 Several Density Functional Theory (DFT) studies have been conducted on the chemisorption of water on these facets. The general agreement is that water gets chemisorbed non-dissociatively on the {101} facet, leading to weakly bound surface μ1-OH2 groups.18–20 Chemisorbed water on the {100} facet is thought to be the object of an equilibrium between non-dissociated surface μ1-OH2 and μ1-OH and μ2-OH resulting from their dissociation.18,20
Water chemisorption on the {001} surface was shown to proceed in an exclusively dissociative way with the opening of a surface Ti–O bond, leading exclusively to pairs of surface μ1-OH groups linked by a hydrogen bond. Theoretically, a hydroxyl surface coverage of up to 7 OH nm−2 can thus be reached.18–23
A number of studies have investigated the surface hydroxyls of anatase by FT-IR. Multiple isolated O–H stretching peaks are seen in the 3800–3600 cm−1 region. Such studies were however mostly done on anatase samples with uncharacterized morphologies or low faceting,24–29 mixed with other TiO2 phases,30–36 or with physisorbed water still present on the surface,26–28,35–38 thus hampering the understanding of the nature of their surface hydroxyls. Similarly, 1H NMR studies often show the same traits: uncharacterized morphologies,39–43 amorphous surfaces,44–48 mixed phases36 and the presence of a layer of physisorbed water.19,36,41,43,47
In order for a sample to display a simple, uniform and comprehensible surface chemistry, morphology control seems to be required to obtain well-faceted crystallites of a specific allotrope with a high majority of a single facet exposed. In particular, the {001} facet seems ideal for dissociative water chemisorption.
Anatase obtained through hydrothermal synthesis usually exhibits mostly {101} facets, as they have the lowest surface energy among all possible facets upon hydration.18 However, the presence of fluorides drastically lowers the surface energies of the {001} and {100} facets in comparison with the {101} facet.49,50 Experimentally, anatase nanocrystals hydrothermally synthesized at moderate temperatures (<140 °C) in the presence of fluorides have been shown to exhibit mostly {100} and {001} facets, while nanocrystals obtained at higher temperatures (>160 °C) with a similar fluoride concentration show dominant {001} facets with minor {101} facets.50
However, the presence of fluoride ions is known to greatly affect the surface chemistry of titanium dioxide: DFT calculations indicate the substitution of anatase {001} surface fluorides by water molecules (Ti–F + H2O → Ti–OH + HF) to be endothermic.49,51 Consequently, they are expected to prevent dissociative water chemisorption on this facet. These results were corroborated by infrared studies showing a strong decrease in intensity of the ν(OH) bands upon fluorination of P25 samples.33 Furthermore, it could also be shown from XPS data that anatase treatment with dilute HF leads to the replacement of surface HO− and surface-lattice O2− with F−.52
Recently, probing of the Brønsted and Lewis acid sites on the surface of anatase nanoparticles by 31P trimethylphosphine NMR also showed that the nature and the concentration of these sites are strongly dependant on the presence of surface adsorbents such as fluorides.53
Two different methods are known for the removal of surface fluorides from anatase samples: the first one is to perform calcination at 600 °C for 90 minutes to 2 hours. This leads to the desorption of gaseous HF and has been used mostly on micrometer-sized anatase particles49,54–56 or, in a few cases, on nanoparticles with very low anisotropy (no more than 20% exposed {001} facets).56,57
Such thermal treatments have been shown to cause morphology changes on nanometer-sized anatase crystallites, as soon as temperatures above 500 °C are used.53,58,59 Therefore, this approach cannot be applied for removing fluorides from highly anisotropic anatase nanoparticles without significant surface alteration.
The second method for removing fluorides from the surface of titania samples is to wash them with an aqueous solution of sodium hydroxide. This has been shown to fully remove surface fluorides from anatase synthesized with no morphology control,52 as well as from fluorinated Degussa P25 TiO2.33 Other studies have also used this protocol to remove fluorides from the surface of anisotropic anatase with highly exposed {001} facets.57,60–65 However, there is little evidence regarding the full removal of all surface fluorides, considering the small amounts required to constitute a significant surface coverage, and the high affinity of fluorides for the {001} surface of anatase.50,56 Indeed, remaining fluoride was recently reported on nanometer-sized anisotropic anatase that underwent washing with 0.1 M NaOH.53
In this work, the surface chemistry of titanium dioxide was studied as a function of its morphology aiming at obtaining TiO2 with well-defined, mostly identical surface hydroxyl groups.
This was achieved by synthesizing pure anatase samples with either a roughly spherical morphology or “anisotropic” morphology (dominant {001} facets). We then determined the ideal dehydroxylation conditions for the complete removal of physisorbed water and investigated the resulting surface chemistry. The results showed that morphology control using fluorides reduces the variety of surface hydroxyls. The influence of the remaining surface fluorides on the nature of these hydroxyls was also investigated.
Experimental section
The aim of this study is to provide a comparison between the surface chemistry of roughly spherical anatase nanoparticles with low faceting and that of well faceted nanoparticles with highly exposed {001} facets, and to obtain rational insight into the nature of their surface species.TiO2 synthesis
Standard anatase TiO2 crystals without a controlled morphology were synthesized from a 4 M titanium stock solution obtained from the hydrolysis of 20 ml of TiCl4 added dropwise to 25 ml of H2O in an ice bath. This solution was then added to 545 ml of a solution of 8.4 M NH4OH and 0.9 M NH4Cl under vigorous stirring and precipitation occurred immediately. The precipitate was then centrifuged out, and suspended back in 600 ml of pure water, then autoclaved at 180 °C for 24 hours for crystallization to occur. The product was then repeatedly rinsed with Milli-Q water and centrifuged until the conductivity of the supernatant was below 10 μS cm−1. It was then dried at 120 °C in an oven, left to cool down in open air, crushed in an agate mortar, and stored in a glass vial. This sample will be labeled “{101}-anatase”.Anatase TiO2 crystals with highly exposed {001} facets were synthesized following the procedure reported by Han et al.60 Hydrolysis of 130 ml of Ti(OBu)4 was performed with 20 ml of 47 wt% HF in H2O (2H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1Ti) followed by hydrothermal treatment at 180 °C for 24 hours in a Teflon-lined autoclave. The resulting sample was then harvested by centrifugation and rinsed 25 times with 500 ml of pure water (Millipore Milli-Q) to remove any hydrofluoric acid or organic residue from the synthesis, and dried at 120 °C in an oven, left to cool down in open air, crushed in an agate mortar, and stored in a glass vial. This sample is labeled as “{001}-anatase-H2O”.
:1Ti) followed by hydrothermal treatment at 180 °C for 24 hours in a Teflon-lined autoclave. The resulting sample was then harvested by centrifugation and rinsed 25 times with 500 ml of pure water (Millipore Milli-Q) to remove any hydrofluoric acid or organic residue from the synthesis, and dried at 120 °C in an oven, left to cool down in open air, crushed in an agate mortar, and stored in a glass vial. This sample is labeled as “{001}-anatase-H2O”.
These syntheses are summarized in Fig. S1 (ESI†).
Surface treatments
{001}-anatase-steam was obtained by heating 1.5 g of {001}-anatase-H2O at 110 °C on a glass frit in a vertical glass tube, under a combined flow of 100 ml min−1 of 1% oxygen in argon and 200 mg min−1 H2O vapour for 10 h.
{001}-anatase-NaOH was prepared by washing 1 g of {001}-anatase-H2O with 160 ml of 1 M aqueous NaOH 10 times. The solid was then autoclaved in 660 ml of 1 M NaOH at 80 °C for 24 hours, and finally rinsed with milli-Q water until the supernatant conductivity was below 10 μS cm−1. It was then dried at 120 °C in an oven, left to cool to room temperature in open air, crushed in an agate mortar, and stored in a glass vial.
(b) Water removal
Partial dehydroxylations were carried out by heating the samples under a 20 ml min−1 flow of dry 20% O2 in argon (dried with molecular sieves) for 10 h at the desired temperature. The samples were then left to cool down to room temperature, placed under high vacuum (10−5 mbar), transferred to a glovebox under an argon atmosphere, and stored in airtight glass tubes under argon.(c) Vanadium oxychloride grafting
Vanadium oxychloride was grafted on the {001}-anatase-steam sample by the following method:10 ml of vanadium oxychloride was taken in a Schlenk flask under an inert atmosphere and subjected to 5 freeze–pump–thaw cycles at 10−5 mbar. 0.5 g of {001}-anatase-steam dehydroxylated at 200 °C was then transferred to a Schlenk tube. The tube and the VOCl3 flask were connected to a high vacuum line by means of a glass T piece, and a vacuum (10−5 mbar) was pulled over the solid sample. The connection to the vacuum line was then closed, and the connection to the VOCl3 flask was opened, placing the TiO2 support under a vanadium oxychloride atmosphere, while stirred by a small magnetic stir bar. To ensure the complete consumption of the available hydroxyl groups, the support was left to react for 5 hours. To remove any excess VOCl3, it was then placed under dynamic vacuum (10−5 mbar) for 12 hours. The sample was finally stored in a glovebox under an inert atmosphere.
Analytical techniques
The infrared (IR) spectra were acquired using a Thermo Scientific Nicolet 6700 FT-IR spectrometer with a mercury-cadmium telluride (MCT) detector and a Harrick Praying Mantis diffuse reflectance accessory. The optical velocity was set to 0.63, and the aperture was set to 64. The samples were either placed in a Harrick ambient sample chamber with KBr windows, or, for in situ DRIFTS, in a Harrick High Temperature Reaction Chamber with ZnSe windows. The X-ray diffractograms were acquired using a Bruker D8 Advance diffractometer, from 2θ = 10° to 2θ = 90°, 3911 steps, and 2 seconds of integration per step. BET porosimetry was performed on a Micromeritics ASAP 2420 with N2 as the adsorbate and a liquid nitrogen analysis bath. The TEM images were acquired using a Titan 60–300 ST microscope equipped with a CEOS image corrector and a Gatan K2-IS direct electron detector. (Acceleration tension: 300 kV.) STEM imaging was performed at 300 kV using a Titan 60–300 TEM from Thermo Fisher, USA (formerly FEI), equipped with a Cs probe corrector. To obtain Z-contrast images, we used a Fischione high angle annular dark field detector, under the following conditions: 25 mrad probe semi-convergence angle, 100 mm camera length, and 100 pA probe current. Drift corrected frame integration (30 frames with 1 frame per s and post-acquisition alignment) was implemented to minimize scanning noise/distortion and sample drift effects. XPS experiments were performed on a Kratos Axis Ultra DLD spectrometer with a monochromatic Al Kα X-ray source operated at 150 W. Measurements were carried out at ∼10−9 mbar.Computational method
All the DFT calculations were performed using the VASP package,66–69 using the PBE exchange–correlation functional70 and the projected-augmented plane wave (PAW) approach.71 The model includes 2 × 2 × 2.5 unit cells (10 Ti layers) and a 20 Å vacuum region to achieve both good bulk property and a stable interface. Depending on different circumstances, the surface oxygen can be covered by hydrogen, or be replaced by fluorine. Firstly, the models were relaxed with the electronic energy criteria of 10−6 eV, and the Hellmann–Feynman forces criteria of 10−2 eV Å−1. The vibrational frequencies of the key functional groups were obtained from the linear response method using the density functional perturbation theory (DFPT) implemented in VASP as the eigenvalues of the diagonalized dynamical matrix (which are the second derivatives of the energy with respect to the displacements) constructed using the Fourier transform of the Hellmann–Feynman force constant matrix elements.Results
In the following sections, the structure, morphology and the textural properties of the TiO2 anatase samples obtained by the aforementioned methods were characterized by X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET) gas sorption and transmission electron microscopy (TEM/STEM). The resulting surface chemistry was studied by magic angle spinning solid-state nuclear magnetic resonance (MAS SSNMR), diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS), X-ray photolectron spectroscopy (XPS) and elemental analysis.(1) The structural, morphological and textural properties of the synthesized solids
The X-ray diffractograms (Fig. 1) show exclusively anatase diffraction peaks for both samples.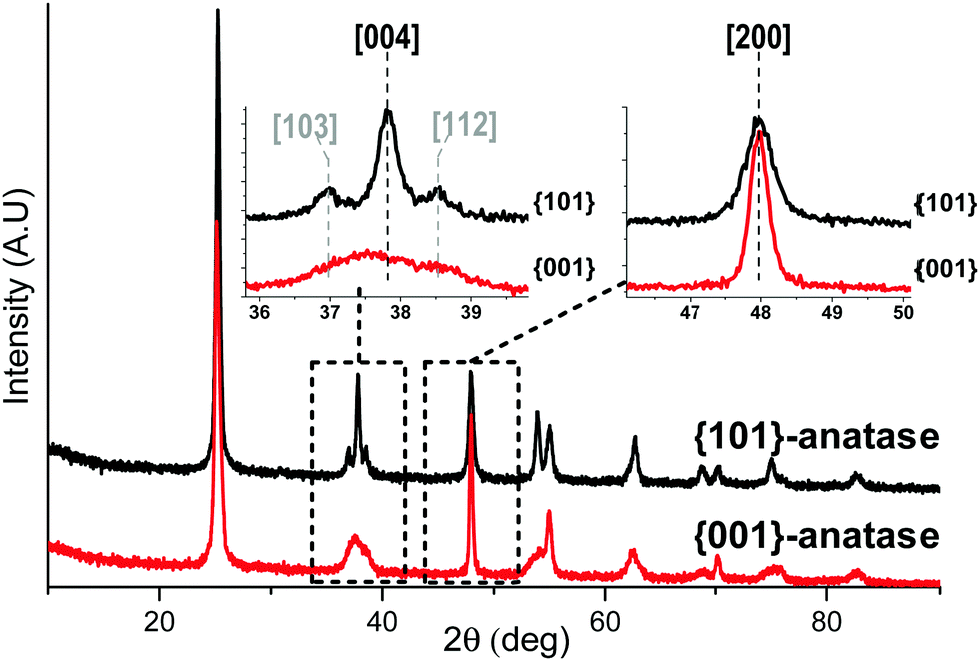 | ||
| Fig. 1 X-ray diffractograms of the {001}-anatase-H2O and {101}-anatase samples. | ||
The coherent domains determined from the peak widths using the Laue–Scherrer law (values in Table 1) are indicative of the high anisotropy of the {001}-anatase sample, which is comparatively thinner than {101}-anatase along the [001] direction and larger along the [101] direction. The TEM (Fig. S5 in the ESI†) and STEM images of the {001}-anatase sample show well faceted platelet-shaped crystallites (seen edge-on in Fig. 2) with dominant {001} faces (81% of the particle surface, detailed calculations are given in the ESI†) and minor {101} facets. The average particle width and thickness measured along the [100] and [001] directions are 43 nm and 4.7 nm, respectively.
| Sample | [001] coherent domain (nm) | [100] coherent domain (nm) | TEM avg. [001] width (nm) | TEM avg. [100] width (nm) | TEM surface area (m2 g−1) | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|---|---|---|---|---|
| {001}-Anatase | 12 | 69 | 4.7 | 43 | 131 | 92 | 0.54 | 40 |
| {101}-Anatase | 35 | 28 | 29 | 20 | 54 | 58 | 0.34 | 25 |
 | ||
| Fig. 2 STEM images of the {001}-anatase (top) and {101}-anatase (bottom) samples. | ||
The difference between the values measured from the TEM images (Fig. S5, ESI†) and the values deduced from X-ray diffraction can be explained by the observed stacking of particles along their {001} facets, which increases the coherent domain as measured by XRD.
The TEM (Fig. S6, ESI†) and STEM images of the {101}-anatase sample show rough truncated octahedral particles with some exposed {101} facets and irregular surfaces, with a considerably lower faceting than that in the previous sample. The average width and thickness measured along the [100] and [001] directions are 29 nm and 20 nm, respectively, in relative agreement with the XRD values. The schematics of the average particle shapes are given in Fig. S2 (ESI†).
The textural properties were investigated by nitrogen physisorption measurements (isotherms in Fig. S3, ESI†). The measured surface area of the {001}-anatase-steam sample is lower than that deduced from the particle dimensions measured by TEM (Table 1). 30% of the surface is therefore inaccessible, due to particle aggregation. The type IV isotherm and H1 hysteresis loop indicate the mesoporous nature of the material, with an average pore diameter of 40 nm, as calculated by the BJH method. The porosity is therefore mainly due to the space in-between the platelet aggregates.
{101}-Anatase shows a surface area of 58 m2 g−1, with no measurable surface loss due to the possible aggregation. Again, the type IV isotherm and H1 hysteresis loop are apparent, resulting from the mesoporous nature of the material. The pores are on average 30 nm in diameter.
(2) Surface chemistry
Our objective is to study the surface chemistry of the previously described samples, with a focus on the surface hydroxyl species. However, under ambient conditions, TiO2 nanoparticles are known to be covered with several layers of physisorbed water, linked by hydrogen bonds.72 As a result, under these conditions, the IR absorption bands and NMR signals from the surface hydroxyl species are drowned in the physisorbed water peaks.Therefore, in order to study the surface hydroxyls of our samples, dehydroxylation treatments have to be applied to remove physisorbed water by heating the materials under vacuum, leaving only the more strongly bound chemisorbed surface species.
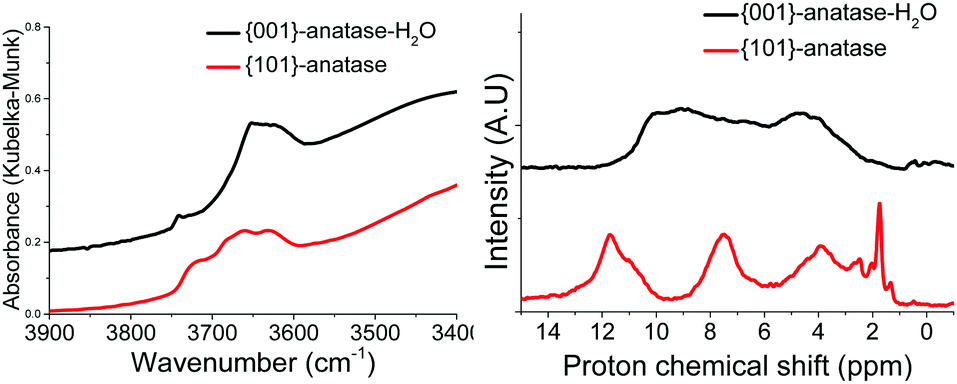 | ||
| Fig. 3 DRIFTS (left) and 1H MAS SSNMR (right) spectra of the {001}-anatase and {101}-anatase samples after dehydroxylation at 300 °C. | ||
The spectra show the existence of a large variety of surface hydroxyl species on the {101}-anatase sample. This diversity is thought to arise mostly from the low faceting of this sample, leading to an increased number of defect sites on the surface of the crystallites. Comparatively, the {001}-anatase-steam sample exhibits a much simpler and understandable surface chemistry.
The {001}-anatase-H2O sample was dehydroxylated at 300 °C under a flow of 20% oxygen in argon dried over 4 Å molecular sieves. An IR spectrum of the sample was acquired in situ by DRIFTS. Then, the temperature was lowered to 150 °C, and a flow of O2/Ar was bubbled in a 50% D2O–50% H2O mix and passed over the sample for 24 hours. In this way, all labile surface protons can be exchanged with deuterium carried by the gas flow. The sample was dehydroxylated again at 300 °C under dry O2/Ar, and a second in situ IR spectrum was acquired. This was then repeated with the gas being bubbled in pure D2O, and the third spectrum was acquired. All spectra are shown in Fig. 4.
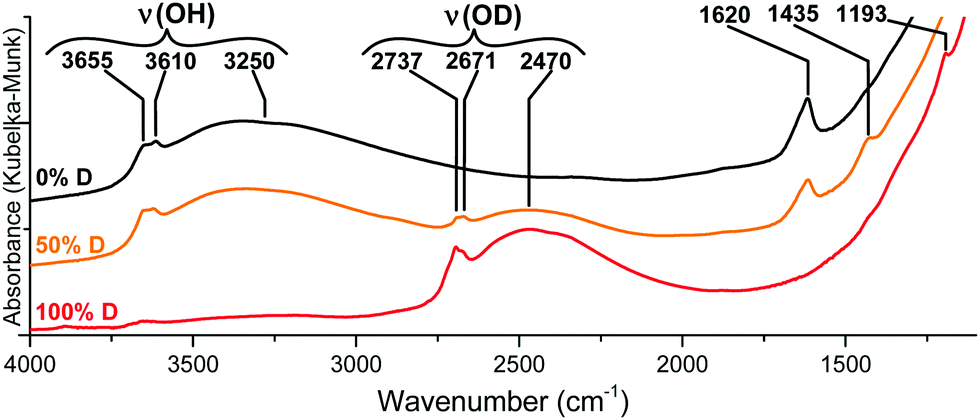 | ||
| Fig. 4 IR spectra of the {001}-anatase-H2O sample dehydroxylated at 300 °C under flowing O2/Ar before (top) and after (bottom) isotopic exchange with D2O. | ||
The replacement of hydrogen by deuterium in an IR-active moiety is expected to result in a lower corresponding wavenumber due to the isotopic shift that results from the important mass difference between both atoms.
As shown in Fig. 4, all ν(OH) vibrations are entirely shifted towards lower wavenumbers, indicating a complete substitution of the hydrogens by deuterium. Therefore, all the OH groups of {001}-anatase-H2O are accessible to D2O, and should be located on the TiO2 surface.
We therefore conclude that the surface fluorine atoms on our samples have identical chemical environments that remain unaffected by dehydroxylation treatments.
{001}-Anatase-steam was placed in an in situ DRIFTS cell under a 20 ml min−1 flow of 20% oxygen in argon. The temperature was raised to 100 °C for 2 h, and an initial IR spectrum was acquired. The temperature was then raised up to 360 °C by 20 °C increments. For each increment, a spectrum was collected after a 30 minute equilibration period (Fig. 5).
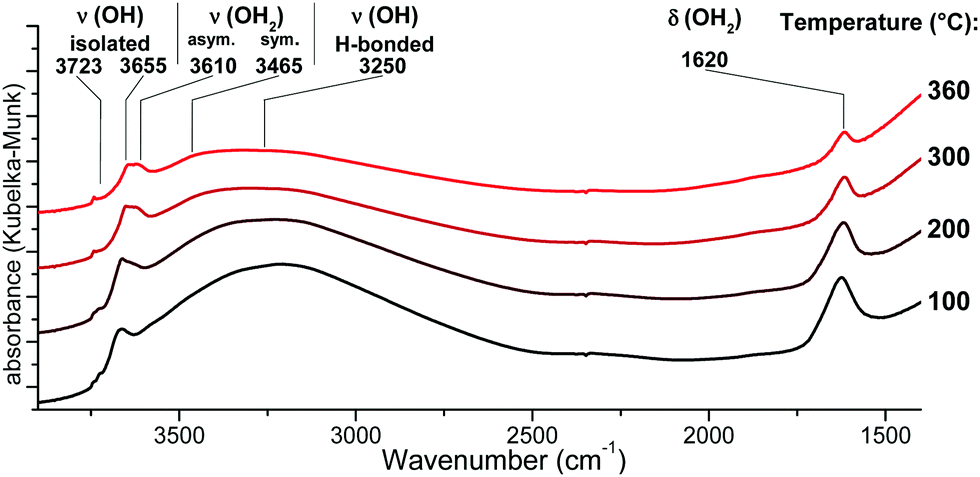 | ||
| Fig. 5 DRIFTS infrared spectra of the {001}-anatase-steam sample after dehydroxylation at different temperatures. (Full data are available in the ESI,† Fig. S9.) | ||
An absorption peak is observed at 1620 cm−1, a value compatible with the scissors-bending motion of H2O.25,74 Furthermore, this peak does shift to 1193 cm−1 when the sample is fully deuterated, as shown in Fig. 4. When the sample is partly deuterated, the third intermediate peak appears at 1435 cm−1, indicating the existence of a semi-deuterated species.
The 1620 cm−1 peak therefore appeared due to a surface species bearing two labile protons, confirming that it arises from surface H2O. It is present up to and including 360 °C, at which point no physisorbed water is expected to remain. It is therefore attributed to the surface H2O species chemisorbed on our sample.
As the hydroxyl stretching peaks in the 4000–2500 cm−1 region show a significant overlap, the acquired spectra had to be fitted with individual Gaussian curves to study their positions and intensities in detail. An example of such a fitting is shown in Fig. 6.
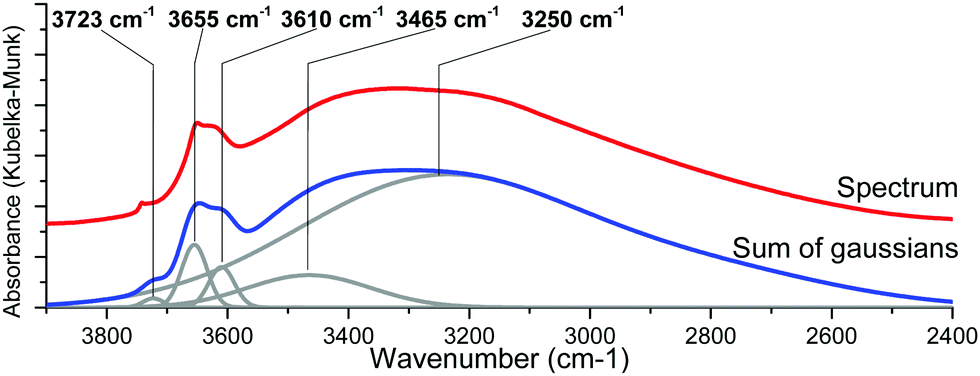 | ||
| Fig. 6 In situ DRIFTS spectra of the {001}-anatase-steam sample at 300 °C (top) and its Gaussian fitting (bottom). | ||
Non-dissociatively chemisorbed surface water is expected to display two stretching modes (antisymmetric and symmetric) in the O–H stretching area. The two peaks at 3610 and 3465 cm−1 display an identical behaviour relative to the temperature (Fig. 7), as the ratio of their intensities remains constant (the inset of Fig. 7). They therefore appear to belong to the same surface species, and are here attributed to the antisymmetric and symmetric stretching of chemisorbed surface H2O, respectively. Two other O–H stretching peaks are present at 3655 cm−1 and 3723 cm−1. These high wavenumbers and thin widths are indicative of the isolated OH stretching peaks.
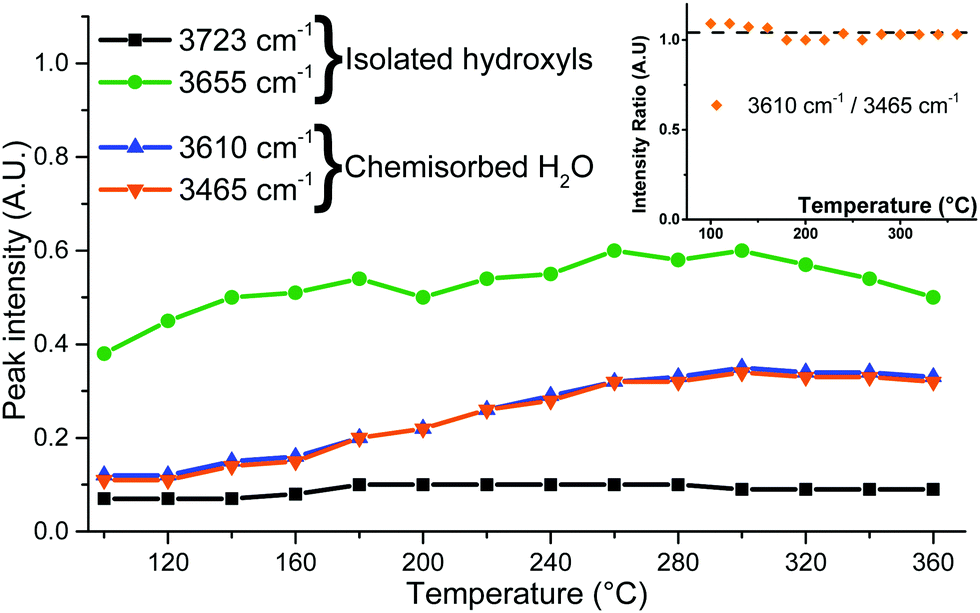 | ||
| Fig. 7 Peak intensities determined from IR spectra fitting and the intensity ratio of the 3610 and 3465 cm−1 peaks (inset). | ||
Finally, a broad peak at around 3250 cm−1 can be attributed to the ν(O–H) bands of the surface hydroxyls involved in the hydrogen bonding. Its great width and slight asymmetry seem to indicate that several species contribute to it.
The infrared spectra of the {001}-anatase-H2O and {001}-anatase-NaOH samples were also acquired after dehydroxylation at 300 °C (Fig. 8).
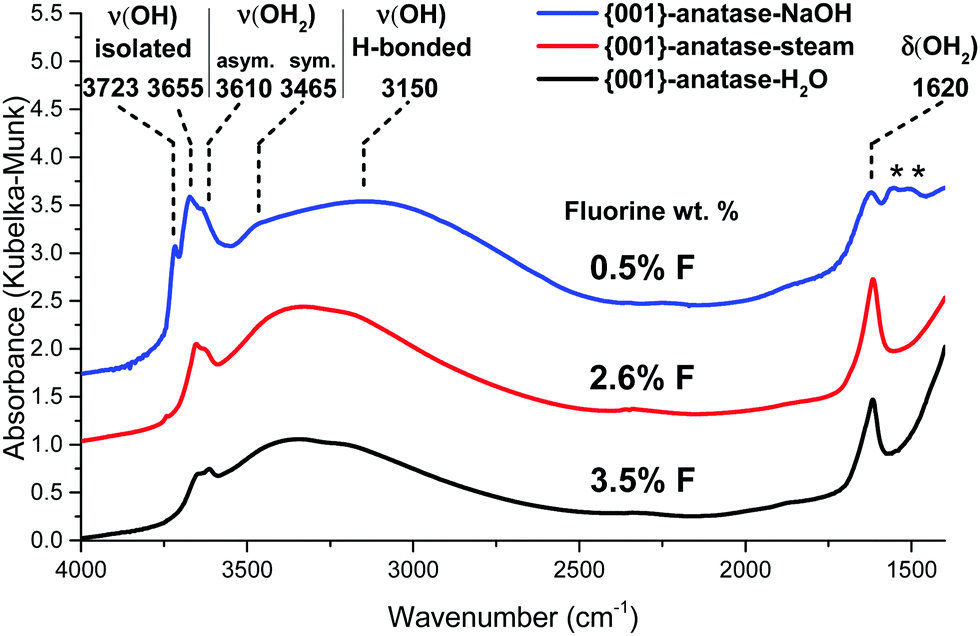 | ||
| Fig. 8 DRIFTS infrared spectra of the {001} anatase-NaOH, {001} anatase-steam and {001}-anatase-H2O samples after dehydroxylation at 300 °C. (*: carbonates contamination from NaOH.) | ||
They show an increase in the amount of free hydroxyl groups on the surface with decreasing fluoride content: the 3655 cm−1 peak increases first when the fluorine content decreases from 3.5 wt% to 2.6 wt%. The 3723 cm−1 peak appears at 2.6 wt% fluorine, and its intensity drastically increases with decreasing fluorine content. It must therefore belong to the hydroxyl groups from the non-fluorinated {001} surface of anatase. Based on the previous DFT studies, this surface is expected to bear pairs of μ1-OH groups, with a hydrogen bond between the proton from one group and the oxygen from the other.18–23 The value of 3723 cm−1 is compatible with the isolated μ1-OH groups.33 Here, this vibration is attributed to the isolated μ1-OH of the predicted OH pairs of the non-fluorinated {001} surface.
The increased intensity and the change in the shape of the broad 3250 cm−1 peak at 0.42% F indicates additional contribution from the OH species involved in a hydrogen bond, which is here attributed to the other, hydrogen-bonded, μ1-OH groups predicted on the non-fluorinated {001} surface.
The wavenumber of the 3655 cm−1 peak appears compatible with the O–H stretching of the μ2-OH groups. As it is present even at a high fluorine content, it is attributed here to {001} μ2-OH groups stabilized by the presence of fluorine.
(3) DFT study
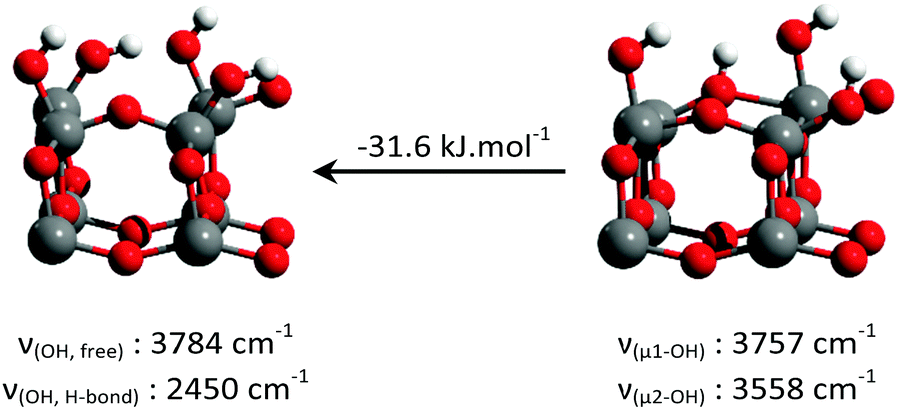
DFT calculations were performed in order to determine the nature of the surface hydroxyls on the {001} surface of anatase at different degrees of fluorination, as well as the O–H stretching frequencies of all the species described above.Six different structures (Table 2) were constructed for the {001} anatase surface at three different fluorination levels. The optimized structures, and their relative enthalpies and O–H stretching wavenumbers are presented in Table 2.
| F:Ti | μ1-OH | ΔH (kJ mol−1) | μ2-OH |
|---|---|---|---|
| 0 |
|
||
| 0.5 |
|
||
| 1 |
|
||
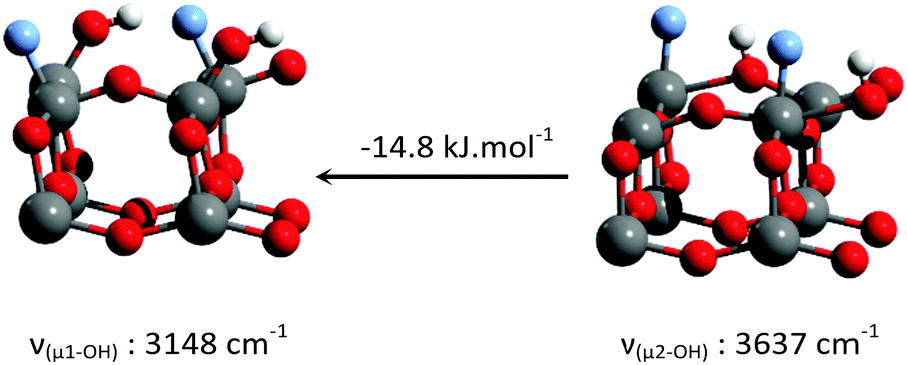
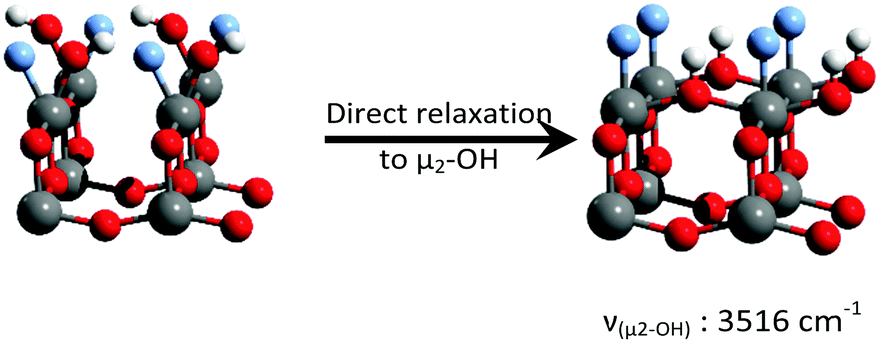
Consistent with previous reports,18–23 it can be seen that μ1-OH pairs are the most stable species on the non-fluorinated {001} surface. This species becomes less stable with increased surface fluorination level. Using this simplified model, μ2-OH becomes the dominant species at an F to surface Ti ratio of 1:1. This result is consistent with our experimental attribution to the IR spectra discussed above.
One additional structure was also generated for the {101} anatase surface, with non-dissociated chemisorbed H2O, in agreement with the previous DFT studies.18–20 The optimized structure can be seen in Fig. 9.
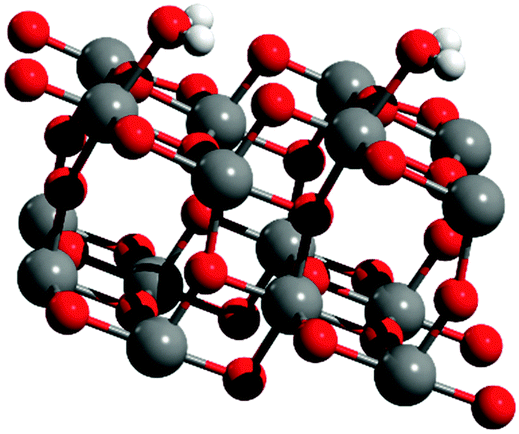 | ||
| Fig. 9 DFT-optimized structure of the {101} anatase surface with non-dissociated chemisorbed H2O. | ||
Two O–H stretching vibrations are observed for this structure, at 3573 and 3515 cm−1, corresponding to the the anti-symmetrical and symmetrical stretching of H2O, respectively. Another vibration mode was found at 1599 cm−1, which corresponds to the H–O–H “scissors” bending motion.
All the calculated O–H stretching wavenumbers for the species on both the {001} and {101} surfaces appear to be in the same order as the experimentally attributed values above.
(4) Solid-state nuclear magnetic resonance study
1H MAS SSNMR spectra were acquired for the samples after dehydroxylation.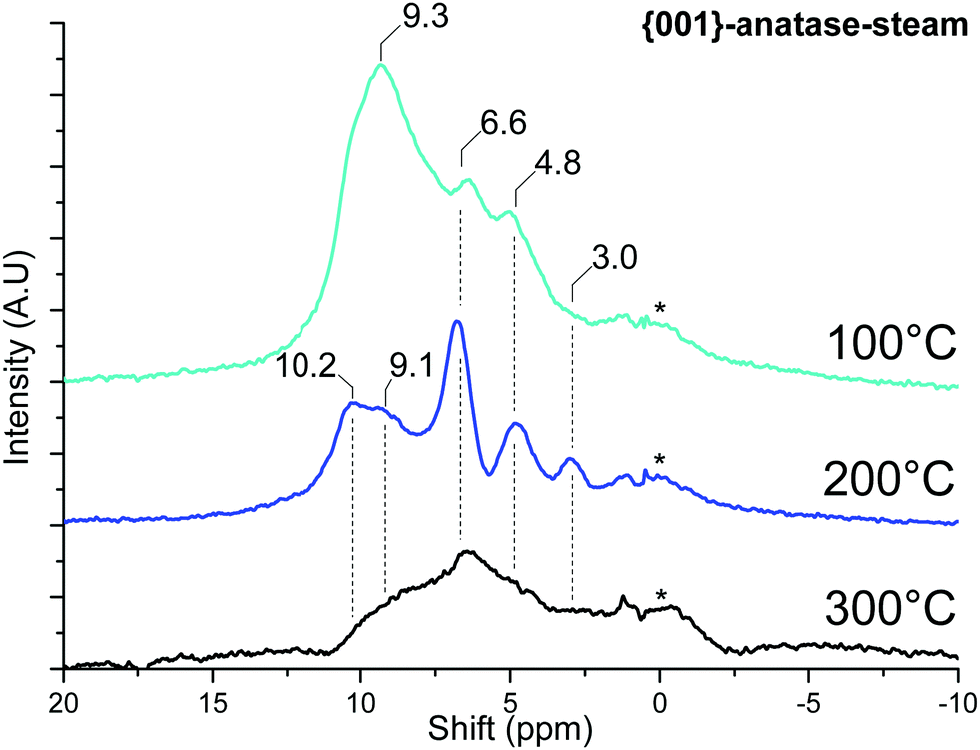 | ||
| Fig. 10 MAS SSNMR spectra of {001}-anatase-steam at various dehydroxylating temperatures. (A.U.: arbitrary units; *: NMR bore contamination). | ||
Once the physisorbed water is removed, the hydroxyl peaks can be seen over a relatively wide range of shifts, from 10.2 ppm to 3.0 ppm.
The effects of surface fluoride content on the proton NMR can be seen in Fig. 11: the 10.2 and 9.1 ppm resonances are relatively unaffected by fluorination levels up to 3.5% F. These high shifts, similar to the 9.3 ppm resonance of physisorbed water on this sample, are indicative of very electron-poor hydrogens. Similar shifts can be seen for the hydrogen bond donor protons on “trivalent” oxygen, as previously seen for H3O+ solvated in SO278 or hydrogen-bonded Si–OH2 in HY–zeolite.79 Inter-platelet hydrogen-bonded H2O or Ti–OH2 species therefore seem to be good candidates for these resonances.
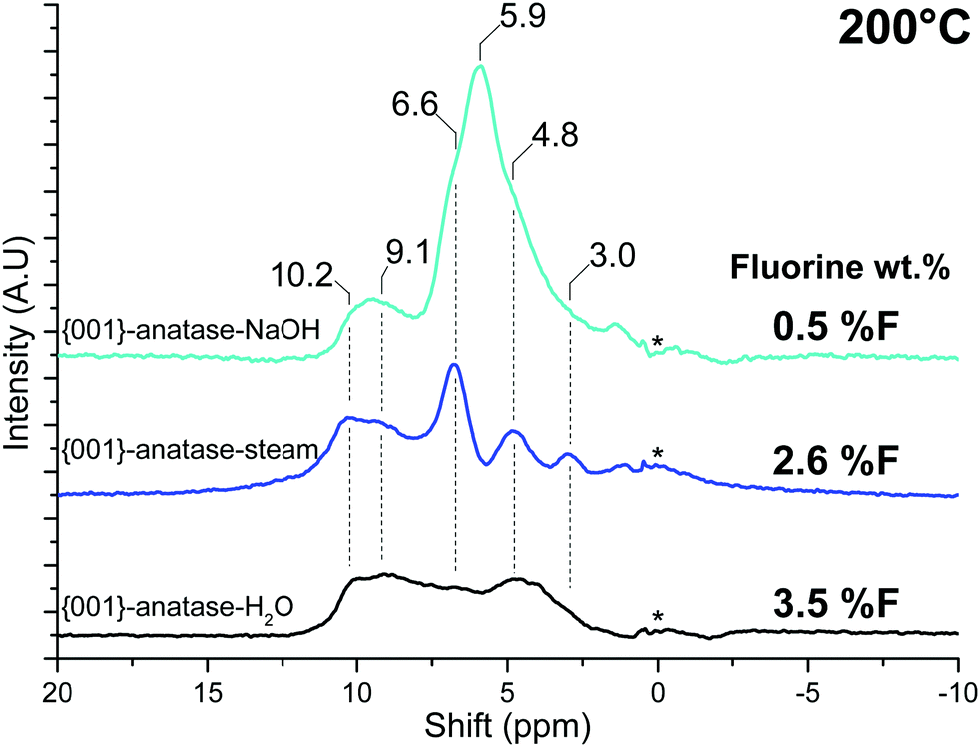 | ||
| Fig. 11 NMR spectra of all the {001}-anatase samples at 200 °C. (A.U.: arbitrary units; *: NMR bore contamination.) | ||
A strong resonance at 5.9 ppm is seen exclusively for {001}-anatase-NaOH, matching the expected presence of the μ1-OH hydroxyl pairs at the lowest fluorination level (Tables 2 and 3). The presence of a single peak for these species including both the hydrogen bond donor and hydrogen bond acceptor OH can be explained by fast exchange between both positions by a simple rotation of the Ti–O bond, yielding an average of both exchanging states on the NMR spectrum.80
| Facet | Species | Representation | Associated IR absorption bands | Exp. value (cm−1) | DFT value (cm−1) |
|---|---|---|---|---|---|
| {101} | {101} μ1-OH2 |
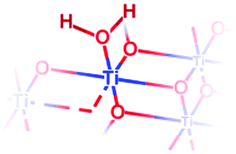
|
O–H stretching (asym.): | 3610 | 3573 |
| O–H stretching (sym.): | 3465 | 3515 | |||
| H–O–H bending: | 1620 | 1599 | |||
| {001} | {001} μ1-OH “pairs” |
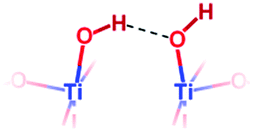
|
Free O–H stretching: | 3723 | 3784 |
| H-Bonded O–H stretching: | ∼3250 | 2450 | |||
| F-{001} μ1-OH |
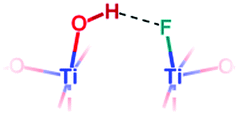
|
H-Bonded O–H stretching: | ∼3250 | 3148 | |
| F-{001} μ2-OH |
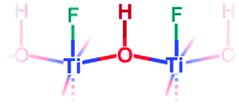
|
O–H stretching: | 3666 | 3639–3520 | |
| Inter-platelet hydroxyls and H2O |
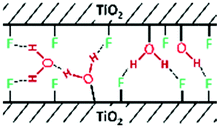
|
H-Bonded O–H stretching: | ∼3250 | ||
| H–O–H bending: | 1620 |
| M–OH(s) + VOCl3(g) → M–O–VOCl2(s) + HCl(g) |
Since the VOCl3 molecule is relatively large (a V–Cl distance of 2.14 Å (ref. 82)), it should not be able to access the inter-platelet hydroxyls of the samples studied here. By determining the reactivity of hydroxyl groups towards it, it should be possible to gain insight into their physical location on the sample.
VOCl3 was grafted on the {001}-anatase-steam sample dehydroxylated at 200 °C. The results are shown in Fig. 12.
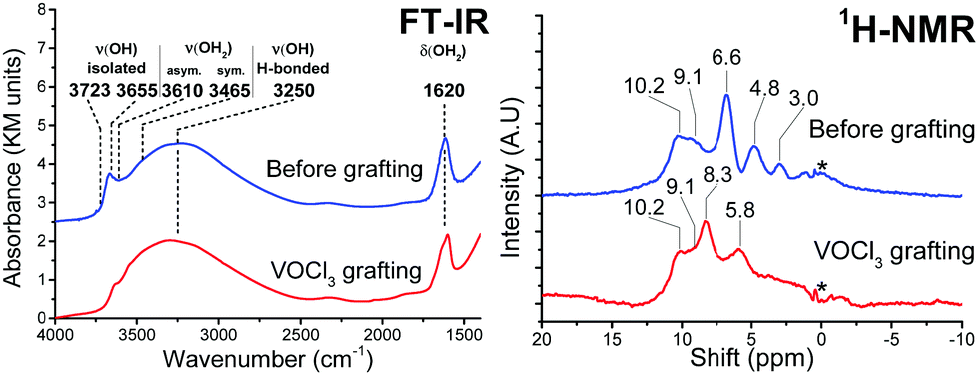 | ||
| Fig. 12 FT-IR (left) and 1H MAS SSNMR spectra of the {001}-anatase-steam sample dehydroxylated at 200 °C, before and after VOCl3 grafting. | ||
FT-IR shows a complete consumption of the isolated hydroxyls in the 3723–3465 cm−1 region, indicating their accessibility towards VOCl3.
The broad, 3250 cm−1 hydrogen-bonded hydroxyl region remains largely unchanged, pointing the inter-platelet hydroxyls as a main species contributing to it.
The 1620 cm−1 H–O–H bending vibration shows only partial consumption from VOCl3 grafting, therefore also pointing at the presence of the hydrogen bond donor H2O species in-between the platelets, their O–H stretching vibrations contributing to the broad vibration of the hydrogen-bonded hydroxyls.
Two new resonances are visible in 1H NMR, at 8.3 and 5.8 ppm. The adsorption of gaseous HCl on this sample (Fig. S11, ESI†) also leads to the apparition of a 5.8 ppm resonance. It is therefore attributed to HCl adsorbed on TiO2, while the 8.3 ppm resonance is attributed HCl adsorbed on the grafted vanadium surface species.
The protons in the 10.2 and 9.1 ppm resonance appear unreactive towards VOCl3. This corroborates their previous attribution to the hydrogen-bonded inter-platelet H2O species.
Complete consumption of the protons from the 6.6, 4.8 and 3.0 ppm resonances can be seen, which must therefore be accessible to VOCl3.
The high shift of the 6.6 ppm resonance seems indicative of a hydrogen bond donor species. Of all the species proposed so far and expected to be present on this sample, {001} μ1-OH hydrogen-bonded to a surface fluoride fits this description (Table 3). This resonance is also seen to disappear at high fluorination levels (Fig. 11), in accordance with the predicted stabilizing effect of the surface fluorides for μ2-OH against μ1-OH (Table 2).
The 4.8 ppm resonance, however, does appear to remain at the highest fluorination level, suggesting that it corresponds to the expected μ2-OH groups. In addition to this, its shift is in the middle of the 3.6–5.3 ppm range within which protons from similar μ2-OH groups have consistently been observed in zeolites.83–88
Finally, the 3.0 ppm resonance falls close to the 3.2 ppm of gas-phase water.89 This appears compatible with the protons from the predicted {101}-OH2 groups, as, unlike inter-platelet H2O or physisorbed H2O, they are not involved in hydrogen bonding (Fig. 9).
As shown in Table 4, a correlation can be seen between the O–H bond length and the 1H NMR shift in our current attribution, with shorter bond lengths corresponding to smaller shifts. Such a correlation is in agreement with the literature data for other systems.77
| Facet | Species | Representation | VOCl3 accessible | O–H bond length (DFT) (Å) | Associated NMR resonance (ppm) |
|---|---|---|---|---|---|
| {101} | {101} μ1-OH2 |
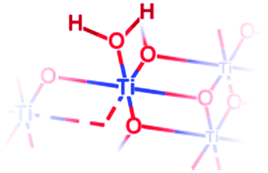
|
Yes | 0.985 | 3.0 |
| {001} | F-{001} μ2-OH |
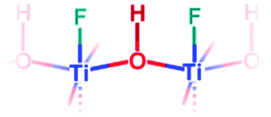
|
Yes | 0.987 | 4.8 |
| {001} μ1-OH “pairs” |
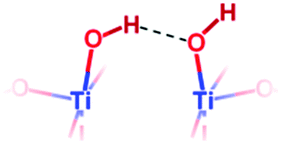
|
— | 0.971 (free OH) | 5.9 | |
| 1.038 (H-Bonded) | |||||
| F-{001} μ1-OH |
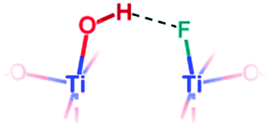
|
Yes | 1.001 | 6.6 | |
| Inter-platelet hydroxyls and H2O |
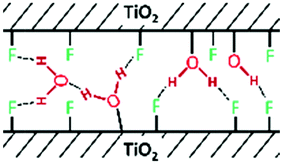
|
No | — | 10.2, 9.1 | |
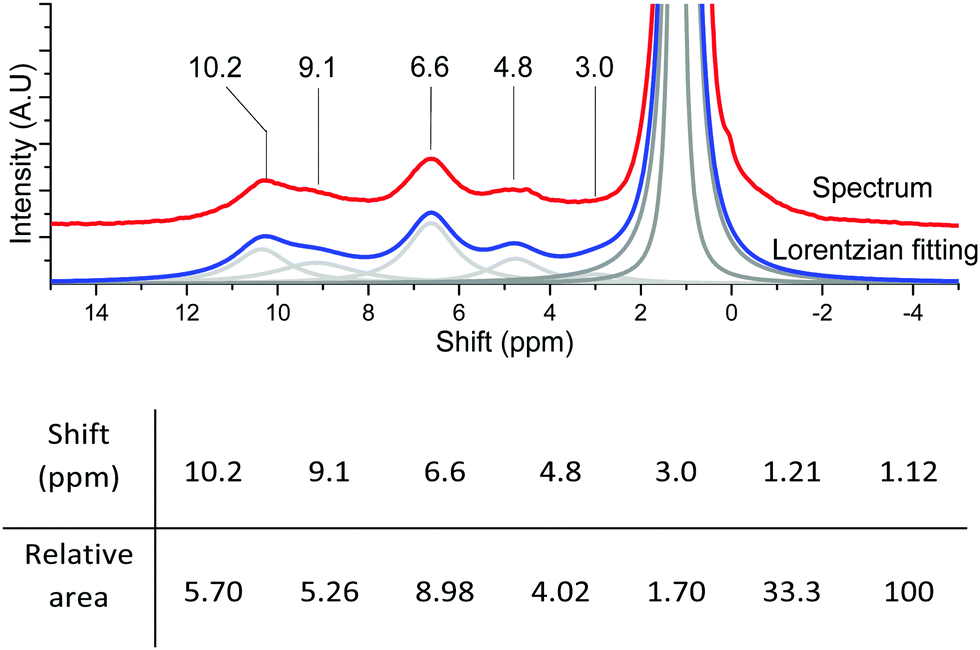 | ||
| Fig. 13 Lorentzian fitting and relative areas of the fitted peaks of {001}-anatase-steam dehydroxylated at 200 °C with 1.23% adamantane. | ||
The surface coverage of a given OH species can be determined by applying the following formula:
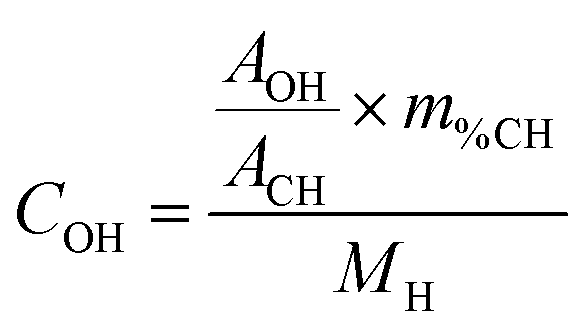
where COH is the concentration of the species in the sample (mol g−1), AOH is the area of its corresponding NMR peak, ACH is the combined area of both adamantane CH peaks, m%CH is the mass fraction of hydrogen from adamantane in the sample (m%adamantane × 0.1175), NA is Avogadro's number, SOH is the surface area of the facet bearing the OH species in question (nm2 g−1), and θOH is the corresponding hydroxyl coverage of this surface (OH per nm2).
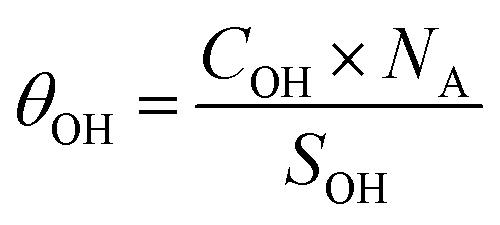
From the data given in Table 1, and assuming that aggregation happens exclusively on the {001} facet, we determined the SOH of the accessible {001} and {101} surfaces to be 26 and 64 m2 g−1, respectively (2.6 × 1019 and 6.4 × 1019 nm2 g−1).
Using these values and those given in Fig. 13 for the 6.6 and 4.8 ppm peaks, we found an accessible {001} surface OH coverage of θ{001}-OH = 1.32 OH per nm2, and a {101} surface H2O coverage of θ{101}-OH2 = 0.21 H2O per nm2.
These values are lower than the theoretical maximum coverages of 7.0 OH per nm2 for the {001} surface and 5.2 OH2 per nm2 for the {101} surface. This can be explained by the high fluorine content leading to the formation of Ti–F–Ti bridges on the {001} surface,50 as well as the easy removal of non-dissociated H2O from the {101} surface by dehydroxylation treatments.18
Discussion
Previous DFT studies are in global agreement that chemisorbed water on the {101} facet of anatase exists as non-dissociated surface H2O.18–20 We believe that these surface H2O groups correspond to the 3610 cm−1 and 3465 cm−1 IR absorption bands. As they are not involved in hydrogen bonding, we expect their NMR shift to be similar to that of water in the gas phase, and attribute the 3.0 ppm shift to them.The strong increase in the intensity of the 3723 cm−1 IR peak with decreasing fluorine content indicates that it belongs to species of the non-fluorinated {001} facet. As already shown by several DFT studies,18,19,21–23 and further confirmed here, water chemisorbs dissociatively on this facet, leading to pairs of hydroxyl groups, linked by a hydrogen bond. Therefore, the hydrogen bonded hydroxyls should contribute to the broad 3250 cm−1 peak, explaining its increase in intensity at 0.42% F, while the free hydroxyl would correspond to the 3723 cm−1 infrared peak. Similarly, a 5.9 ppm resonance appears in NMR at a low fluorine content. This resonance is therefore attributed to those hydroxyl pairs, averaged by fast exchange.
Few studies are available regarding the fully fluorinated {001} surface, and none on the partly fluorinated surface. It was earlier suggested by DFT that the fully fluorinated surface bears bridging μ2-OH,50 which was further confirmed here. In addition, a previous study of trimethylphosphine adsorption on anatase by 31P NMR showed Brønsted acid sites from bridging hydroxyls to be associated with the fluorinated {001} facet.53 These would fit the 3655 cm−1 IR peak, as bridging hydroxyl peaks are expected within the 3750–3723 cm−1 range of free hydroxyls.33 The 4.8 ppm resonance observed for our samples is within the expected range of such bridging hydroxyls. Their replacement with bridging surface fluorides at high fluorination levels50 provides an explanation for the slight decrease in intensity of their IR and NMR peaks at 3.5% F.
The broad peak centered at 3250 cm−1 shows a non-Gaussian shape (a sum of two broad Gaussian curves had to be used for proper fitting), indicating contributions from several species.
Both fluorine-free {001} μ1-OH pairs linked by a hydrogen bond and partly fluorinated {001} μ1-OH in a hydrogen bond with a nearby fluorine are expected to contribute to this broad peak. A 6.6 ppm NMR resonance matches with the latter.
Another major contribution to the broad hydrogen-bonded OH stretching peak is that of the surface hydroxyls in-between the particles stacked along their {001} facets (cf.Fig. 2): groups from the surface of one crystallite interact with the nearby surface of the other crystallite, causing the observed aggregation.58 Their inaccessibility to vanadium oxychloride indicates that the protons from the 10.2 and 9.1 ppm resonance are part of such inter-platelet hydroxyls.
Conclusions
A specific allotrope of titanium dioxide (anatase) was synthesized, both with a standard thermodynamic morphology ({101}-anatase) and with a highly anisotropic morphology ({001}-anatase) dominated by the {001} facet (81%).The surface chemistry of both samples after dehydroxylation was studied by 1H NMR and FT-IR. It could thus be determined that morphology control provides us with a simpler surface chemistry. The influence of surface fluorides on the surface chemistry was also studied by 1H NMR, FT-IR and DFT.
As a result, a full attribution of the IR spectra of anatase with dominant {001} facets could be provided, based on experimental data and further confirmed by DFT. Our results showed that chemisorbed H2O is still present on anatase after dehydroxylation at 350 °C, and that the type of surface hydroxyls present on the {001} facet is dependent on the presence of fluorides. They also provided general insight into the nature of surface species on both fluorinated and fluorine-free anatase.
The use of vanadium oxychloride (VOCl3) allowed the determination of the accessibility of the various OH groups spectroscopically observed. This, along with the previously gained insights into their nature from FT-IR and DFT, allowed a full attribution of the 1H-NMR spectra of the {001}-anatase samples.
Future work will be focused on using these surface hydroxyl groups to perform the grafting of metal complexes via Surface Organometallic Chemistry (SOMC) techniques for photocatalysis and heterogeneous catalysis processes.
Abbreviations
| DFT | Density functional theory |
| XPS | X-ray photoelectron spectroscopy |
| FT-IR | Fourier-transform infrared spectroscopy |
| DRIFTS | Diffuse reflectance Fourier-transform infrared spectroscopy |
| BET | Brunauer–Emmett–Teller porosimetry |
| TEM | Transmission electron microscopy |
| STEM | Scanning transmission electron microscopy |
| NMR | Nuclear magnetic resonance |
| MAS SSNMR | Magic angle spinning solid state nuclear magnetic resonance |
| SOMC | Surface organometallic chemistry. |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the King Abdullah University of Science and Technology (KAUST). We warmly acknowledge the help of the KAUST Core labs, and the Supercomputing Laboratory at KAUST for the CPU time attributed to this research work. The authors thank Zhen Cao (KCC-KAUST) for the fruitful discussion.Notes and references
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef.
- P. C. Gravelle, F. Juillet, P. Meriaudeau and S. J. Teichner, Discuss. Faraday Soc., 1971, 52, 140 RSC.
- D. a. Peña, B. S. Uphade and P. G. Smirniotis, J. Catal., 2004, 221, 421–431 CrossRef.
- B. Shen, T. Liu, N. Zhao, X. Yang and L. Deng, J. Environ. Sci., 2010, 22, 1447–1454 CrossRef.
- L. Chen, J. Li and M. Ge, J. Phys. Chem. C, 2009, 113, 21177–21184 Search PubMed.
- R. Jin, Y. Liu, Z. Wu, H. Wang and T. Gu, Chemosphere, 2010, 78, 1160–1166 CrossRef PubMed.
- K.-M. Choi, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Lett., 2003, 32, 180–181 CrossRef.
- A. W. Stobbe-Kreemers, M. Makkee and J. J. F. Scholten, Appl. Catal., A, 1997, 156, 219–238 CrossRef.
- M. Sopa, A. Wącław-Held, M. Grossy, J. Pijanka and K. Nowińska, Appl. Catal., A, 2005, 285, 119–125 CrossRef.
- M. Roy, M. Gubelmann-Bonneau, H. Ponceblanc and J.-C. Volta, Catal. Lett., 1996, 42, 93–97 CrossRef.
- M. H. Ab Rahim, M. M. Forde, C. Hammond, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock and G. J. Hutchings, Top. Catal., 2013, 56, 1843–1857 CrossRef.
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock, D. M. Murphy, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2013, 52, 1280–1284 CrossRef PubMed.
- V. M. Shinde and G. Madras, RSC Adv., 2014, 4, 4817 RSC.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4, 3357–3380 CrossRef.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef.
- J. D. A. Pelletier and J.-M. Basset, Acc. Chem. Res., 2016, 49, 664–677 CrossRef PubMed.
- C. Arrouvel, M. Digne, M. Breysse, H. Toulhoat and P. Raybaud, J. Catal., 2004, 222, 152–166 CrossRef.
- M. Sumita, C. Hu and Y. Tateyama, J. Phys. Chem. C, 2010, 114, 18529–18537 Search PubMed.
- C. Sun, L.-M. Liu, A. Selloni, G. Q. (Max) Lu and S. C. Smith, J. Mater. Chem., 2010, 20, 10319 RSC.
- R. Erdogan and I. Onal, Int. J. Quantum Chem., 2011, 111, 2149–2159 CrossRef.
- R. Erdogan, O. Ozbek and I. Onal, Surf. Sci., 2010, 604, 1029–1033 CrossRef.
- A. Hussain, J. Gracia, B. E. Nieuwenhuys and J. W. H. Niemantsverdriet, ChemPhysChem, 2010, 11, 2375–2382 CrossRef PubMed.
- S. Dzwigaj, C. Arrouvel, M. Breysse, C. Geantet, S. Inoue, H. Toulhoat and P. Raybaud, J. Catal., 2005, 236, 245–250 CrossRef.
- K. S. Finnie, D. J. Cassidy, J. R. Bartlett and J. L. Woolfrey, Langmuir, 2001, 17, 816–820 CrossRef.
- P. C. Shih, C. H. Huang, T. H. Chen, L. W. Lai, Y. S. Lu and D. S. Liu, Mater. Res. Bull., 2014, 52, 177–182 CrossRef.
- M. Umadevi, M. Sangari, R. Parimaladevi, A. Sivanantham and J. Mayandi, J. Fluorine Chem., 2013, 156, 209–213 CrossRef.
- M. K. Lee and Y. C. Park, Thin Solid Films, 2017, 638, 9–16 CrossRef.
- W. El-Alami, D. G. Sousa, C. F. Rodríguez, O. G. Díaz, J. M. D. Rodríguez, M. El Azzouzi and J. Araña, J. Photochem. Photobiol., A, 2017, 348, 139–149 CrossRef.
- D. A. Panayotov and J. T. Yates, Chem. Phys. Lett., 2005, 410, 11–17 CrossRef.
- M. Takeuchi, G. Martra, S. Coluccia and M. Anpo, J. Phys. Chem. B, 2005, 109, 7387–7391 CrossRef PubMed.
- G. Martra, Appl. Catal., A, 2000, 200, 275–285 CrossRef.
- M. Minella, M. G. Faga, V. Maurino, C. Minero, E. Pelizzetti, S. Coluccia and G. Martra, Langmuir, 2010, 26, 2521–2527 CrossRef PubMed.
- M. M. Kumar, S. Badrinarayanan and M. Sastry, Thin Solid Films, 2000, 358, 122–130 CrossRef.
- H. Jensen, A. Soloviev, Z. Li and E. G. Søgaard, Appl. Surf. Sci., 2005, 246, 239–249 CrossRef.
- S. Kaewgun, C. A. Nolph, B. I. Lee and L. Q. Wang, Mater. Chem. Phys., 2009, 114, 439–445 CrossRef.
- J. J. Murcia, M. C. Hidalgo, J. A. Navío, J. Araña and J. M. Doña-Rodríguez, Appl. Catal., B, 2013, 142–143, 205–213 CrossRef.
- M. Wang, F. Zhang, X. Zhu, Z. Qi, B. Hong, J. Ding, J. Bao, S. Sun and C. Gao, Langmuir, 2015, 31, 1730–1736 CrossRef PubMed.
- C. Dorémieux-Morin, M. A. Enriquez, J. Sanz and J. Fraissard, J. Colloid Interface Sci., 1983, 95, 502–512 CrossRef.
- M. A. Enríquez, C. Dorémieux-Morin and J. Fraissard, Appl. Surf. Sci., 1981, 40, 233–240 Search PubMed.
- V. M. Gun’ko, J. P. Blitz, V. I. Zarko, V. V. Turov, E. M. Pakhlov, O. I. Oranska, E. V. Goncharuk, Y. I. Gornikov, V. S. Sergeev, T. V. Kulik, B. B. Palyanytsya and R. K. Samala, J. Colloid Interface Sci., 2009, 330, 125–137 CrossRef PubMed.
- V. M. Mastikhin and A. V. Nosov, React. Kinet. Catal. Lett., 1992, 46, 123–130 CrossRef.
- P. Conte, V. Loddo, C. De Pasquale, V. Marsala, G. Alonzo and L. Palmisano, J. Phys. Chem. C, 2013, 117, 5269–5273 Search PubMed.
- S. Yurdakal, V. Augugliaro, J. Sanz, J. Soria, I. Sobrados and M. J. Torralvo, J. Catal., 2014, 309, 97–104 CrossRef.
- X. Sun, M. Dyballa, J. Yan, L. Li, N. Guan and M. Hunger, Chem. Phys. Lett., 2014, 594, 34–40 CrossRef.
- J. Sanz, I. Sobrados, J. Soria, S. Yurdakal and V. Augugliaro, Catal. Today, 2017, 281, 198–204 CrossRef.
- J. Soria, J. Sanz, M. J. Torralvo, I. Sobrados, C. Garlisi, G. Palmisano, S. Çetinkaya, S. Yurdakal and V. Augugliaro, Appl. Catal., B, 2017, 210, 306–319 CrossRef.
- M. J. Torralvo, J. Sanz, I. Sobrados, J. Soria, C. Garlisi, G. Palmisano, S. Çetinkaya, S. Yurdakal and V. Augugliaro, Appl. Catal., B, 2018, 221, 140–151 CrossRef.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef PubMed.
- Z. Lai, F. Peng, Y. Wang, H. Wang, H. Yu, P. Liu and H. Zhao, J. Mater. Chem., 2012, 22, 23906–23912 RSC.
- C. Sun, A. Selloni, A. Du and S. C. Smith, J. Phys. Chem. C, 2011, 115, 17092–17096 Search PubMed.
- Q. Wang, C. Chen, D. Zhao, W. Ma and J. Zhao, Langmuir, 2008, 24, 7338–7345 CrossRef PubMed.
- Y. K. Peng, Y. Hu, H. L. Chou, Y. Fu, I. F. Teixeira, L. Zhang, H. He and S. C. E. Tsang, Nat. Commun., 2017, 8, 1–13 CrossRef PubMed.
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. M. Lu, J. Am. Chem. Soc., 2009, 131, 4078–4083 CrossRef PubMed.
- M. Liu, L. Piao, L. Zhao, S. Ju, Z. Yan, T. He, C. Zhou and W. Wang, Chem. Commun., 2010, 46, 1664 RSC.
- G. Liu, C. Sun, H. G. Yang, S. C. Smith, L. Wang, G. Q. M. Lu and H.-M. Cheng, Chem. Commun., 2010, 46, 755–757 RSC.
- S. Liu, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2010, 132, 11914–11916 CrossRef PubMed.
- K. Lv, Q. Xiang and J. Yu, Appl. Catal., B, 2011, 104, 275–281 CrossRef.
- Z. Wei, E. Kowalska and B. Ohtani, Molecules, 2014, 19, 19573–19587 CrossRef PubMed.
- X. Han, Q. Kuang, M. Jin, Z. Xie and L. Zheng, J. Am. Chem. Soc., 2009, 131, 3152–3153 CrossRef PubMed.
- T. R. Gordon, M. Cargnello, T. Paik, F. Mangolini, R. T. Weber, P. Fornasiero and C. B. Murray, J. Am. Chem. Soc., 2012, 134, 6751–6761 CrossRef PubMed.
- M.-V. Sofianou, M. Tassi, V. Psycharis, N. Boukos, S. Thanos, T. Vaimakis, J. Yu and C. Trapalis, Appl. Catal., B, 2015, 162, 27–33 CrossRef.
- J. Yu, L. Qi and M. Jaroniec, J. Phys. Chem. C, 2010, 114, 13118–13125 Search PubMed.
- Q. Xiang, K. Lv and J. Yu, Appl. Catal., B, 2010, 96, 557–564 CrossRef.
- X. Yu, B. Jeon and Y. K. Kim, ACS Catal., 2015, 5, 3316–3322 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef.
- G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 50, 17953–17979 CrossRef.
- A. Y. Nosaka, J. Nishino, T. Fujiwara, T. Ikegami, H. Yagi, H. Akutsu and Y. Nosaka, J. Phys. Chem. B, 2006, 110, 8380–8385 CrossRef PubMed.
- I. E. Grey and N. C. Wilson, J. Solid State Chem., 2007, 180, 670–678 CrossRef.
- D. F. Hornig, H. F. White and F. P. Reding, Spectrochim. Acta, 1958, 12, 338–349 CrossRef.
- L. Brammer, A. Bruton and P. Sherwood, New J. Chem., 1999, 23, 965–968 RSC.
- J. Emsley, Chem. Soc. Rev., 1980, 9, 91 RSC.
- C. Chizallet, G. Costentin, H. Lauron-Pernot, M. Che, C. Bonhomme, J. Maquet, F. Delbecq and P. Sautet, J. Phys. Chem. C, 2007, 111, 18279–18287 Search PubMed.
- V. Gold, J. L. Grant and K. P. Morris, J. Chem. Soc., Chem. Commun., 1976, 397–398 RSC.
- S. Li, A. Zheng, Y. Su, H. Fang, W. Shen, Z. Yu, L. Chen and F. Deng, Phys. Chem. Chem. Phys., 2010, 12, 3895 RSC.
- V. B. Kazalsky and V. Y. Borovkov, Colloids Surf., 1984, 12, 429–442 CrossRef.
- H. Zhu, S. Ould-Chikh, H. Dong, I. Llorens, Y. Saih, D. H. Anjum, J.-L. Hazemann and J.-M. Basset, ChemCatChem, 2015, 7, 3332–3339 CrossRef.
- R. Bjornsson, H. Früchtl and M. Bühl, Phys. Chem. Chem. Phys., 2011, 13, 619–627 RSC.
- V. B. Kazansky, A. I. Serykh, V. Semmer-Herledan and J. Fraissard, Phys. Chem. Chem. Phys., 2003, 5, 966–969 RSC.
- M. Dyballa, U. Obenaus, S. Lang, B. Gehring, Y. Traa, H. Koller and M. Hunger, Microporous Mesoporous Mater., 2015, 212, 110–116 CrossRef.
- H. Huo, L. Peng, Z. Gan and C. P. Grey, J. Am. Chem. Soc., 2012, 134, 9708–9720 CrossRef PubMed.
- R. R. Pinto, P. Borges, M. A. N. D. A. Lemos, F. Lemos, J. C. Védrine, E. G. Derouane and F. R. Ribeiro, Appl. Catal., A, 2005, 284, 39–46 CrossRef.
- M. Hunger, Catal. Rev., 1997, 39, 345–393 Search PubMed.
- J. Kanellopoulos, C. Gottert, D. Schneider, B. Knorr, D. Prager, H. Ernst and D. Freude, J. Catal., 2008, 255, 68–78 CrossRef.
- D. Mammoli, E. Canet, R. Buratto, P. Miéville, L. Helm and G. Bodenhausen, Sci. Rep., 2016, 6, 38492 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Equations used, BET isotherms, XRD and BET determination of stability under dehydroxylation conditions, TEM micrographs, XPS spectra, in situ DRIFTS at all temperatures, and 19F NMR and 1H NMR after HCl adsorption. See DOI: 10.1039/c8cp01983e |
| This journal is © the Owner Societies 2018 |