
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lysine-based amino-functionalized lipids for gene transfection: 3D phase behaviour and transfection performance†‡
Stephanie
Tassler
 a,
Dorota
Pawlowska
a,
Christopher
Janich
b,
Julia
Giselbrecht
b,
Simon
Drescher
b,
Andreas
Langner
b,
Christian
Wölk
*b and
Gerald
Brezesinski
*a
a,
Dorota
Pawlowska
a,
Christopher
Janich
b,
Julia
Giselbrecht
b,
Simon
Drescher
b,
Andreas
Langner
b,
Christian
Wölk
*b and
Gerald
Brezesinski
*a
aMax Planck Institute of Colloids and Interfaces, Science Park Potsdam-Golm, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: brezesinski@mpikg.mpg.de
bMartin Luther University (MLU) Halle-Wittenberg, Institute of Pharmacy, Wolfgang-Langenbeck-Straße 4, 06120 Halle (Saale), Germany. E-mail: christian.woelk@pharmazie.uni-halle.de
First published on 21st May 2018
Abstract
Based on previous work, the influence of the chain composition on the physical–chemical properties of five new transfection lipids (TH10, TT10, OH10, OT10 and OO10) containing the same lysine-based head group has been investigated in aqueous dispersions. For this purpose, the chain composition has been gradually varied from saturated tetradecyl (T, C14:0) and hexadecyl (H, C16:0) chains to longer but unsaturated oleyl (O, C18:1) chains with double bonds in the cis configuration. In this work, the lipid dispersions have been investigated in the absence and presence of the helper lipid DOPE and calf thymus DNA by small-angle and wide-angle X-ray scattering (SAXS/WAXS) supplemented by differential scanning calorimetry (DSC), attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) and Fourier-transform Raman spectroscopy (FTRS). Lamellar and inverted hexagonal mesophases have been observed in single-component systems. In the binary mixtures, the aggregation behaviour changes with an increasing amount of DOPE from lamellar to cubic. The lipid mixtures with DNA show a panoply of mesophases. Interestingly, TT10 and OT10 form cubic lipoplexes, whereas OO10 complexes the DNA sandwich-like between lipid bilayers in a lamellar lipoplex. Surprisingly, the latter is the most effective lipoplex.
1. Introduction
The concept of gene therapy requires efficient delivery of polynucleotides into biological tissue or cells – the so called gene transfection.1 Three decades after the first reported lipid-mediated transfection by Felgner et al. in 1987, the performance of cationic lipid-based non-viral DNA vehicles is still insufficient.2 Therefore, our research goal is to relate the structure of newly synthesised malonic acid diamide lipids to their physical–chemical properties in 2D and 3D model systems in the absence and presence of DNA and to correlate such data with the biological activity in different cell lines. This work is the continuation of our previous investigations using five lipids containing different alkyl chain patterns but the same small head group type 10 in 2D model systems (Langmuir monolayers).3 TT10, TH10, OT10, OH10 and OO10 are lysine-based amino-functionalized peptide-mimicking lipids, which were synthesised as non-viral carriers for gene transfection (Fig. 1). The hydrophobic lipid domain changes gradually from short and saturated tetradecyl (T = C14:0) to longer saturated hexadecyl (H = C16:0) and finally to unsaturated oleyl (O = C18:1) chains. | ||
| Fig. 1 Chemical structures of N-(2-aminoethyl)-N′-[6-amino-1-oxo-1-[(N-tetradecylamino)hexan-(2S)-2-yl]]-2-tetradecylpropandiamid TT10, N-(2-aminoethyl)-N′-[6-amino-1-oxo-1-(N-tetradecylamino)hexan-(2S)-2-yl]-2-hexadecylpropandiamide TH10, N-(2-aminoethyl)-N′-{6-amino-1-[N-(9Z)-octadec-9-enylamino]-1-oxohexan-(2S)-2-yl}-2-tetradecylpropandiamide OT10, N-(2-aminoethyl)-N′-{6-amino-1-[N-(9Z)-octadec-9-enylamino]-1-oxohexan-(2S)-2-yl}-2-hexadecylpropandiamide OH10 and N-(2-aminoethyl)-N′-{6-amino-1-[N-(9Z)-octadec-9-enylamino]-1-oxohexan-(2S)-2-yl}-2-[(9Z)-octadec-9-enyl]propandiamide OO10. | ||
The oleyl chain was introduced for two reasons. First, because it is known for high transfection efficiency, and second, it ensures a sufficient miscibility with the commonly used helper lipid DOPE with oleyl chains in the cis configuration.4,5
In general, the insertion of one cis double bond decreases the gel to liquid-crystalline phase transition temperature. Further, it can promote the formation of an inverted non-lamellar phase. This effect strongly depends on the position of the cis double bond along the chain. In an oleyl chain, the double bond is in the middle of the chain, where this effect reaches its maximum.6 DOPE, dispersed in water, forms an inverted hexagonal structure at room temperature. The gel to liquid-crystalline phase transition is at around −8 °C, while the Lα to HII transition occurs at around 10 °C.7–9 Adding DOPE to the transfection lipids is believed to promote the formation of inverted hexagonal structures, which rapidly fuse with the endosomal bilayer and allow the release of DNA with high efficiency.7,10,11
Varying the chain composition of these transfection lipids also changes the length and symmetry of the aliphatic chains. The increase in chain length results in stronger van der Waals interactions. Strong asymmetry within the hydrophobic domain of lipids lowers the gel to liquid-crystalline phase transition temperature due to disturbed van der Waals interactions. A sufficient asymmetry in the chain length tends to induce chain interdigitation in bilayers.6,12
The malonic acid/lysine backbone of TT10, TH10, OT10, OH10 and OO10 is biocompatible. Due to this backbone the lipids exhibit a peptide-mimicking character. Head group 10 has a linear ethylene diamine spacer and two primary amine groups (Fig. 1). Going from TH10 to OO10, the pKa value increases from 5 to 5.6, indicating that the readiness to act as proton donors decreases with increasing chain fluidity.13 This can also influence the interaction with DNA (pKa = 9.6 for guanine and pKa = 10.5 for thymine).14
In the present paper, the physical–chemical properties of these lipids (Fig. 1) have been examined in aqueous dispersions regarding phase transitions and structures in the absence and presence of DOPE and DNA. The focus is set on the lipoplex morphology and the resulting transfection performance, in particular the transfection efficiency and cytotoxicity. Consequently, this article gives information on how it is possible to adjust the physical–chemical parameters of lipoplexes by directed variation of the alkyl chain pattern.
2. Materials
2.1. Experimental details
The synthesis and the analytical data of TT10, TH10, and OT1015 as well as OO1013 and OH103 have already been described. The lipids containing an oleyl chain have been synthesized from oleylamine with technical grade.16 The zwitterionic phospholipid DOPE (C41H78NO8P, 744.03 g mol−1) was purchased from Avanti (Avanti Polar Lipids, Inc., Alabaster, USA) and was used without further purification. Milli-Q Millipore water with a specific resistance of 18.2 MΩ cm was used for all measurements and sample preparation. All chemicals used for the preparation of the buffer were purchased from Sigma Aldrich. The bromide containing buffers had a constant concentration of bromide anions (2 mM) and result from comparison with TRXF experiments using monolayers at the liquid–air interface.13 pH 10 was obtained by using piperazine (pKa1 = 5.7, pKa2 = 9.8). The carbonate buffer (K2CO3) had a constant concentration of 5 mM for SAXS and WAXS and 10 mM for DSC experiments. pH 10 was adjusted with HCl (pKa = −3). The citrate buffer (C8H7NaO7) had a constant concentration of 5 mM, and pH 4 was obtained by adding NaOH (pKb = 0.2). The deoxyribonucleic acid sodium salt from calf thymus (>13 kbp) was purchased from Sigma Aldrich (type 1, CAS: 73049-39-5). A solution of 2 mg mL−1 ct-DNA was freshly prepared in 1 mM NaCl solution by gently stirring at 5 °C overnight. The molar mass of DNA refers to a monomer containing one charge per phosphate moiety with 10% of hydration (M ∼ 370 g mol−1).17To obtain the desired lipid/DOPE mixtures, the lipids and DOPE were dissolved in chloroform/methanol (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v:v) or (8:2 v:v) to a final concentration of 1 mM. Then, the lipids and helper lipids were mixed in the desired ratios. The organic solvent was evaporated under a nitrogen stream, and the film was dried in a desiccator under vacuum for 12 h. The lipids and lipid/DOPE mixtures were dispersed in the aqueous medium to a final concentration of 200 mg mL−1 (20 wt%) (A2 at DESY) or 100 mg mL−1 (10 wt%) (ID02 at ESRF) for the X-ray investigations and 1 mg mL−1 (0.1 wt%) for the DSC experiments. The lipid/DOPE/DNA samples were prepared by adding 2 mg mL−1 calf thymus DNA solution to the lipid mixture in the corresponding N/P ratio (the ratio of primary amines of the cationic lipids to phosphate of the DNA). The lipid dispersions were vortexed, sonicated and heated above the gel to liquid-crystalline phase transition temperature several times. In the case of the lipoplexes, the samples were not heated above 50 °C and only vortexed to avoid the denaturation of DNA. All samples were sealed in glass capillaries for X-ray experiments with an inner diameter of 1.5 mm, a wall thickness of 0.01 mm and a length of 80 mm (GLAS Müller, Berlin, Germany). The SAXS and WAXS samples were stored for 7 to 10 days at 5 °C before the measurements.
:1 v:v) or (8:2 v:v) to a final concentration of 1 mM. Then, the lipids and helper lipids were mixed in the desired ratios. The organic solvent was evaporated under a nitrogen stream, and the film was dried in a desiccator under vacuum for 12 h. The lipids and lipid/DOPE mixtures were dispersed in the aqueous medium to a final concentration of 200 mg mL−1 (20 wt%) (A2 at DESY) or 100 mg mL−1 (10 wt%) (ID02 at ESRF) for the X-ray investigations and 1 mg mL−1 (0.1 wt%) for the DSC experiments. The lipid/DOPE/DNA samples were prepared by adding 2 mg mL−1 calf thymus DNA solution to the lipid mixture in the corresponding N/P ratio (the ratio of primary amines of the cationic lipids to phosphate of the DNA). The lipid dispersions were vortexed, sonicated and heated above the gel to liquid-crystalline phase transition temperature several times. In the case of the lipoplexes, the samples were not heated above 50 °C and only vortexed to avoid the denaturation of DNA. All samples were sealed in glass capillaries for X-ray experiments with an inner diameter of 1.5 mm, a wall thickness of 0.01 mm and a length of 80 mm (GLAS Müller, Berlin, Germany). The SAXS and WAXS samples were stored for 7 to 10 days at 5 °C before the measurements.
pCMV-GFP plasmid DNA (348 bp) was purchased from PlasmidFactory (Bielefeld, Germany). A549 (human lung carcinoma), HeLa (human cervix adenocarcinoma epithelial), and LLC-PK1 (pig kidney epithelial) cells were acquired from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). Cell culture media and fetal bovine serum (FBS) were supplied by Biochrom (Berlin, Germany). AlamarBlue® was purchased from ThermoFisher (Waltham, MA, USA).
3. Methods
3.1. Differential scanning calorimetry (DSC)
The DSC measurements were performed using a VP-DSC (MicroCal Inc. Northampton, Massachusetts, USA) with a heating rate of 60 K h−1 or 20 K h−1 in a temperature range from 2 °C to 95 °C. To ensure reproducibility, several heating and cooling cycles were recorded and the first scan was neglected. The reference cell was filled with buffer. For baseline correction, the buffer-buffer baseline was subtracted from the thermograms of the samples. The DSC scans were evaluated using MicroCal Origin 8.0 software in order to estimate the lipid phase transition temperature Tm and the corresponding change in enthalpy ΔHm. The peak maximum of heat flow curves yields Tm, while the area A under the peak can be correlated to the enthalpy change ΔHm.3.2. Fourier-transform Raman spectroscopy (FTRS)
Raman spectra were recorded using a Bruker Fourier-transform infrared spectrometer RFS 100/S (Bruker Optics GmbH, Ettlingen, Germany). As an excitation source a diode pumped Nd:YAG laser emitting radiation at 1064 nm was used. The spectra were recorded with a power of 500 mV and a resolution of 4 cm−1. 200 scans were taken for each spectrum. 50 mg lipid, hydrated with 500 μL water, was placed in a Norell® glass tube, originally produced for NMR spectroscopy, and spectra were measured in a temperature range between 25 °C and 98 °C. The sample was allowed to equilibrate for 5 minutes. The Bruker software package OPUS was used for data evaluation.3.3. Attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy
Lipid suspensions (70–100 mg mL−1, 7–10 wt% in D2O) were analysed using a Bruker BioATR2 unit with a Bruker Tensor27 FTIR spectrometer (Bruker Optics GmbH, Ettlingen, Germany). Single-channel IR spectra were recorded between 5000 and 900 cm−1 using unpolarized IR light with a resolution of 2 cm−1. The temperature, in the range from 35 to 85 °C (ΔT = 2 K), was adjusted with a Haake Pheonix II thermostat (C25P, Thermo Electron Corporation, Karlsruhe, Germany) and controlled with Delphi-based home-written software. After an equilibration time of 8 min, 64 scans were recorded and accumulated. As a reference, single-channel IR spectra (64 scans), taken from pure D2O at the appropriate temperature, were used and subtracted from the corresponding sample spectra using the Bruker OPUS software.3.4. Small-angle and wide-angle X-ray scattering (SAXS and WAXS)
Small-angle and wide-angle X-ray scattering data were obtained at the Soft Condensed Matter Beamline A218–22 at HASYLAB (DESY, Hamburg, Germany) and the High Brilliance Beamline ID0223 (ESRF, Grenoble, France).The real-space repeating distance d of the lattice planes was calculated from the position of the first diffraction peak by using eqn (1). The Bragg peaks were fitted with the Lorentzian-function.
 | (1) |
3.5. Transfection experiments
000–11000 cells per well 24 h before transfection. Cells were washed once with PBS buffer (pH 7.4) containing Ca2+ and Mg2+. Lipoplexes were added to the cells (0.1 μg DNA per well) and the wells were filled up with DMEM containing 10% FBS to the final volume of 100 μL per well. After 2 h, the cells were washed twice with PBS, and 100 μL PBS was added. The eGFP fluorescence was measured using a POLARstar OMEGA (BMG Labtech, Offenburg, Germany) with a BMG 10 filter (λabs = 485 nm, λem = 520 nm) with a fixed gain. The transfection efficiency is auto-fluorescence corrected and represented in relative fluorescence units (RFU) normalised to the cell count.
4. Results and discussion
4.1. Phase behaviour of the synthesised cationic lipids
 | ||
| Fig. 2 DSC curves of lipid dispersions (1 mg mL−1, scanning rate was 20 K h−1) in water or carbonate buffer (10 mM, pH 10). The curves were shifted vertically for clarity. | ||
| Lipid | T m [°C] heating | ΔH [kJ mol−1] | T m [°C] cooling | Hysteresis [K] | Additional peaks/shoulder in heating scan [°C] |
|---|---|---|---|---|---|
| In water (pH ∼ 5.8) | |||||
| TH10 | 89.5 | 27.8 | 70.8 | 18.7 | 92.2 |
| TT10 | 86.6 | 29.1 | 67.6 | 19 | — |
| OH10 | 66.3 | 9.8 | 49.1 | 17.2 | 78.9/85.3 |
| OT10 | 65.2 | 11.2 | 49.6 | 15.6 | 77.2 |
| In carbonate buffer (pH 10) | |||||
| TH10 | >95 | n.d. | 93 | — | |
| TT10 | 92.1 | n.d. | 88.4 | 3.7 | 34.8 |
| OH10 | 81.8 | n.d. | 69.5 | 12.3 | |
| OT10 | 81.6 | n.d. | 72.1 | 9.5 | |
A clear dependence of the main transition temperature from the alkyl chain pattern was observed. TH10 has the highest phase transition temperature at 89.5 °C. The replacement of the hexadecyl chain by a tetradecyl chain (TH10 → TT10) results in a decrease of Tm by 2.9 K to 86.6 °C. The enthalpy values of the main transition of both lipids and the observed hysteresis are comparable (see Table 1). If the lysine bound tetradecyl chain was exchanged by an oleyl chain (TH10 → OH10, TT10 → OT10) the main transition was shifted to lower temperatures by either 23.2 K or 21.4 K. Furthermore, the transition becomes broader and the enthalpy decreases by ∼18 kJ mol−1 (see Table 1). Additional transitions with lower enthalpy can be seen above the main transition (see Fig. 2 and Table 1). This is most probably an effect of the saturated components in the oleyl chain of technical grade.16 Very noticeable is the pronounced hysteresis (up to 19 K) between Tm in the heating and cooling scans. Such pronounced hysteresis effects can be found in lipid bilayers stabilized by hydrogen bond networks.26,27 Hysteresis between heating and cooling the sample indicates a kinetically hindered formation of the ordered gel phase which could be connected with the electrostatic repulsion between the protonated head groups and the formation of a highly ordered hydrogen bond network.
DSC experiments were also performed in carbonate buffer pH 10 (Fig. 2). The evaluation of these curves is more difficult because the samples tend to form cloudy precipitates. Therefore, the transition enthalpies have not been estimated because of the precipitation of lipid aggregates leading to some loss of material (sticking to the glass wall etc.). The precipitate formation is a result of the uncharged head groups (deprotonation of ammonium ions to primary amines) due to the high pH. Another effect of the pH induced shift of the ammonium/amine equilibrium to the uncharged amine form is the increase of the phase transition temperature compared to the measurements in water (Table 1). A second effect is the decrease of the hysteresis between the heating and cooling scans (Table 1). This means the formation of the gel state is less kinetically hindered due to the disappearance of electrostatic repulsions between the lipids. Furthermore, an additional weak transition occurs at ∼35 °C for TT10 in carbonate buffer, whose origin is yet unknown (see Fig. 2).
The symmetric methylene stretching mode gives information about the alkyl chain conformation. Fig. 3A demonstrates a slight increase of the maximum of the νs(CH2) band with increasing temperature starting from 2846.5 cm−1 at 25 °C to 2848 cm−1 at 80 °C. By a further increase of the temperature, a pronounced increase of the wavenumber by 4 cm−1 occurs between 85 °C and 95 °C. This sudden jump results from a drastic increase of the amount of gauche conformers in the alkyl chains28 and thereby indicates a gel to liquid-crystalline phase transition which perfectly fits with the phase transition observed in the DSC experiment (Table 1). Another possibility to determine phase transitions from ordered alkyl chains to disordered phases is the determination of intensity ratios in the C–H stretching range.29,30Fig. 3A demonstrates that the sudden increase of the I(2953 cm−1)/I(2882 cm−1) ratio, which is indicative for lattice disorder and intra-chain conformational disorder, matches perfectly with the increase in wavenumber of the νs(CH2) stretching mode. The corresponding intensities are determined from the spectra presented in Fig. 3B. The amide I band (see Fig. 3C), which is mainly composed of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes of the amides, is sensitive to hydration and hydrogen bonding.31 The formation of hydrogen bonds is connected with a red shift (towards smaller wavenumbers). It was observed that with increasing temperature the peak maximum slightly shifts to higher wavenumbers and the shoulder at 1650 cm−1 slightly decreases in intensity, which is a hint of weakening the hydrogen bond network (see Fig. 3D and E). With the phase transition the amide I band shows a strong broadening and a pronounced decrease in intensity, a behaviour which can be explained by the strong increase of the hydration and the breakdown of the hydrogen bond network.
O stretching modes of the amides, is sensitive to hydration and hydrogen bonding.31 The formation of hydrogen bonds is connected with a red shift (towards smaller wavenumbers). It was observed that with increasing temperature the peak maximum slightly shifts to higher wavenumbers and the shoulder at 1650 cm−1 slightly decreases in intensity, which is a hint of weakening the hydrogen bond network (see Fig. 3D and E). With the phase transition the amide I band shows a strong broadening and a pronounced decrease in intensity, a behaviour which can be explained by the strong increase of the hydration and the breakdown of the hydrogen bond network.
 | ||
| Fig. 3 Raman experiments of TH10 dispersed in water. (A) Wavenumber of the symmetric CH2 stretching mode and relative intensity I(2953 cm−1)/I(2882 cm−1) vs. temperature. (B) Temperature dependence of the C–H stretching mode region. The curves are shifted vertically for clarity. (C) Temperature dependence of the amide I band region. The curves are shifted vertically for clarity. (D and E) Deconvolution of the amide I band at 83 °C (D) and 25 °C (E) by three Gaussian functions. The three bands are assigned to amides in different hydrogen bonded states. The red arrows indicate the changes induced by the temperature increase from 25 °C to 83 °C. The maximum of the band at around 1665 cm−1 shifts to higher wavenumbers, and the band at around 1650 cm−1 decreases in intensity. | ||
The ATR-FTIR measurements give comparable information. The pronounced increase of the position of the band maximum of the symmetric stretching vibrational mode (Fig. 4, upper panel) indicates a gel to liquid-crystalline phase transition32,33 connected with changes in the packing density.34 TT10 exhibits a pronounced increase above 80 °C, which fits with the phase transition temperature determined by DSC (Table 1). OH10 shows an extended transition region from the gel to the liquid-crystalline state between 60 °C and 70 °C, which again perfectly matches with the DSC results showing a broad peak in this temperature range (Fig. 2). A second increase in wavenumbers can be seen at around 80 °C, indicating a second transition, which is also supported by a second weak transition in the corresponding DSC experiments (compare Fig. 2 and 4). Additionally and concordant with the DSC, a hysteresis between 45 °C and 60 °C has been observed between heating and cooling curves. However, the transition occurs at higher temperatures on cooling the sample. Lastly, OT10 shows a sudden increase in the νs(CH2) wavenumbers between 70 °C and 80 °C, 5 K higher than the Tm determined by DSC, but in the range of the second transition in the calorimetric experiment (compare Fig. 2 and 4). The hysteresis of 15 K is comparable to the DSC experiments.
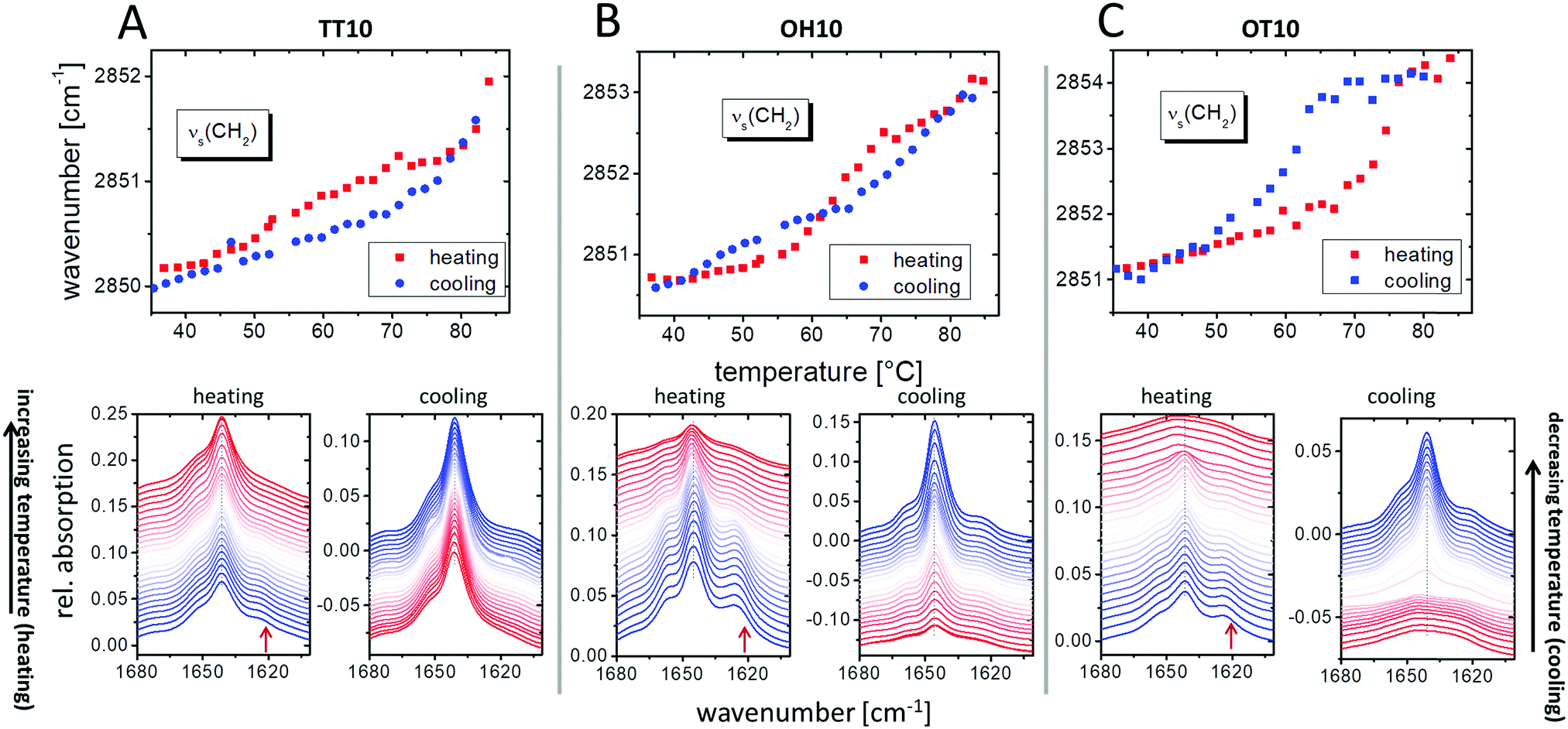 | ||
| Fig. 4 ATR-FTIR measurements of TT10 (A), OH10 (B), and OT10 (C) dispersed in D2O. The upper panel shows the position of the maximum of the symmetric methylene stretching vibrational mode during the heating and cooling scans as a function of the temperature. The lower panel presents the amide I region of the infrared spectra during heating (left) and cooling (right) scans. Blue indicates the lower (starting with 35 °C) and red the higher (starting with 85 °C) temperatures. The curves are shifted vertically for clarity. | ||
Further, the amide I band (see Fig. 4, lower panel) was examined. This band is quite complex because different amide groups are involved. Comparable to the Raman experiments, the amide I band gives information about hydrogen bond formation.34 The shoulder at 1620 cm−1 (the red arrow in the heating scans in Fig. 4) indicates that there are amide functions which belong to a hydrogen bond network, while the signals at higher wavenumbers (the signal maximum and the second shoulder at higher wavenumbers) are less involved in hydrogen bonds.35 The intensity of the shoulder at 1620 cm−1 decreases with increasing temperature indicating a loss of hydrogen bonds. For OH10 and OT10, a decrease and broadening of the amide I signal after the phase transition can be seen, which is comparable to the observation made in the Raman experiments for TH10. This seems to be an effect of the disorder in lipid packing and the result of highly increased head group solvation.
:2 ratio indicating a multi-lamellar structure (see Fig. 5A). In agreement with the DSC, the SAXS pattern of OO10 is different. Here, the Bragg peaks are in the ratio 1:√3:2:√7, characteristic of a hexagonal phase (see Fig. 5A). The parameters, obtained at 25 °C, are summarized in Table 2. The characteristic repeating distance d of lamellar phases, describing the thickness of a lipid bilayer with an additional water layer, is obtained by  . The theoretical length of the single C14:0 chain (both chains of TT10) is 13 × 1.26 Å (all-trans CH2) + 1.46 Å (–CH3 terminus) = 17.8 Å,36,37 consequently a double layer of non-tilted chains should be ∼36 Å thick. Two hexadecyl chains give ∼41 Å. The fluid oleyl chain is shorter than C14:0 or C16:0 due to the cis-double bond in the middle. This calculation shows that only 17 Å (TH10) and 20 Å (TT10) remain for two head groups and the water layer between the head groups (dpolar (calc.), see Table 2). Possible reasons for such small values could be chain interdigitation or a high chain tilt, both leading to a smaller thickness of the hydrophobic part of the bilayer and therefore to larger values of dpolar (calc.). The d-values of OH10 and OT10 are much larger indicating a larger water layer between the lipid bilayers.
. The theoretical length of the single C14:0 chain (both chains of TT10) is 13 × 1.26 Å (all-trans CH2) + 1.46 Å (–CH3 terminus) = 17.8 Å,36,37 consequently a double layer of non-tilted chains should be ∼36 Å thick. Two hexadecyl chains give ∼41 Å. The fluid oleyl chain is shorter than C14:0 or C16:0 due to the cis-double bond in the middle. This calculation shows that only 17 Å (TH10) and 20 Å (TT10) remain for two head groups and the water layer between the head groups (dpolar (calc.), see Table 2). Possible reasons for such small values could be chain interdigitation or a high chain tilt, both leading to a smaller thickness of the hydrophobic part of the bilayer and therefore to larger values of dpolar (calc.). The d-values of OH10 and OT10 are much larger indicating a larger water layer between the lipid bilayers.
 | ||
| Fig. 5 (A) Examples of SAXS patterns of lamellar (TH10) and hexagonal (OO10) mesophases of lipids (indicated) dispersed in water (20 wt%) at 25 °C. (B) Temperature dependence of the lamellar repeat distance d and the lattice parameter a, respectively, for the lipids dispersed in water (20 wt%). TH10, TT10, OH10, and OT10 form lamellar phases while OO10 and DOPE form hexagonal lyotropic liquid crystals. SAXS (C) and the corresponding WAXS (D) patterns of TH10 (black), OH10 (red) and OO10 (blue) at 25 °C as 10 wt% dispersions in bromide containing buffer (pH 10). The corresponding Miller indices are given as numbers in brackets at the peaks. | ||
| Lipid | Mesophase | Repeat distance d [Å] | d chain (calc.) [Å] | d polar (calc.) [Å] | Lattice parameter a [Å] |
|---|---|---|---|---|---|
| TH10 | Lamellar | 57.8 | 41 | 17 | — |
| TT10 | 55.8 | 36 | 20 | — | |
| OH10 | 74.2 | 41 | 33 | — | |
| OT10 | 61.2 | 36 | 25 | — | |
| OO10 | Hexagonal | — | — | 56.5 | |
| DOPE | — | — | 72 |
Upon heating (see Fig. 5B), the repeating distance of TH10 did not change significantly in the investigated temperature range from 25–95 °C. Since the phase transition is at around 90 °C (DSC and Raman), either the temperature in the X-ray experiments was not high enough or during the gel to liquid-crystalline transition the decrease of the hydrocarbon chain layer thickness (transition from the trans to gauche conformation) is compensated for by an increase of the thickness of the hydrophilic layer (water + head groups). For TT10, a sudden increase of the d-value occurs between 90 °C and 95 °C in agreement with the phase transition around 87 °C observed by DSC. The dramatic increase in the d-value can only be explained by a drastic increase of the water layer after the alkyl chain melting. OT10 exhibits also a pronounced increase in the d-value between 80 °C and 85 °C. This increase in the d-value is not accompanied by the main but the smaller second phase transition at 77 °C observed in DSC (compare Fig. 5B with Table 1). OH10 shows a continuous increase of the repeating distance between 25 °C and 75 °C which can be explained by an increasing interfacial water layer or a change in the molecule tilt. Between 75 °C and 80 °C, a pronounced increase in d by 63 Å occurs followed by a strong decrease (Fig. 5B). This behaviour can only be explained by changes in the interfacial water layer.
The hexagonal mesophase forming lipids show also temperature dependent changes of the lattice parameter a (Fig. 5B). Since the alkyl chains are in the fluid state, these changes can result from changes in the hydration and therefore the interfacial water layer. For OO10, the lattice parameter a slightly increases with increasing temperature, while in the case of DOPE a decreases continuously.
Grazing incidence X-ray diffraction experiments using monolayers of the investigated lipids indicate a head group lattice based on hydrogen bonds.3 SAXS and WAXS measurements of OO10, OH10 and TH10 in bromide containing buffer at pH 10 were successfully performed (see Fig. 5C and D). The SAXS experiments show a comparable mesophase structure to the experiments in water with only slight to moderate variations in the d and a values (compare Tables 2 and 3). The WAXS pattern indicates a hydrogen bond network. In the WAXS pattern of TH10 one broad signal composed of three Bragg peaks (Fig. 5D) can be seen. The peak qH = 1.32 Å−1 can be correlated to –N–H⋯OC– hydrogen-bonds, since the real spacing distance of 4.76 Å is a typical value for the strand separation in a peptide β-sheet structure due to hydrogen bonds.38–40 The other two Bragg peaks, namely q11 = 1.43 Å−1 and q02 = 1.5 Å−1, describe an orthorhombic chain lattice. The cross-sectional area A0 of the chains is equal to ∼21 Å2. Therefore, TH10 forms lamellar aggregates with chains in the all-trans conformation (gel state). In the case of OH10, the WAXS pattern is similar. The peak at qH = 1.29 Å−1 can be associated with hydrogen-bonds, while the other two Bragg peaks q11 = 1.47 Å−1 and q02 = 1.39 Å−1 arise from the orthorhombic chain lattice. In the orthorhombic unit cell the chains have a large cross-sectional area A0 of ∼22 Å2. Hence, the exchange of a tetradecyl chain by an oleyl chain increases the unit cell area.
| Phase state | q(001) [Å−1] | d [Å] | q 11 [Å−1] | q 02 [Å−1] | A 0 [Å2] | q H-bond [Å−1] | q Cα–Cα [Å−1] | |
|---|---|---|---|---|---|---|---|---|
| TH10 | Lβ′ | 0.103 | 61.1 | 1.43 | 1.5 | 21.6 | 1.32 | Shoulder ∼1.6 |
| OH10 | Lβ′ | 0.116 | 54 | 1.47 | 1.39 | 21.9 | 1.29 | Shoulder ∼1.6 |
| Phase state | q(010) [Å−1] | a [Å] | q 11 [Å−1] | q 02 [Å−1] | A 0 [Å2] | q H-bond [Å−1] | q Cα–Cα [Å−1] | |
|---|---|---|---|---|---|---|---|---|
| OO10 | Hα | 0.125 | 57.9 | 1.37 | 1.52 | 22.8 | 1.28 | Shoulder ∼1.6 |
| Halo → molten chains | ||||||||
Due to that fact that oleyl chains generally do not order at ambient temperature, they should only cause diffuse scattering (a broad halo in the WAXS pattern) indicating molten chains in the gauche conformation. Surprisingly, OO10 exhibits distinct signals in the WAXS region. The peak at qH = 1.28 Å−1 can be correlated to –N–H⋯OC– hydrogen-bonds between the head groups. It is possible that an ordered structure in the head group and/or backbone region is formed by an H-bond lattice. The additional peaks at q11 = 1.37 Å and q01 = 1.52 Å−1 could be connected with a chain lattice. Such a lattice might be caused by traces of saturated alkyl chains present in the oleylamine with a technical grade of purity.16 The latter hypothesis is supported by the DSC thermogram of OO10 showing a weak transition around the Tm of OT10 and OH10.
Besides the distinct peaks, TH10, OH10 and OO10 show a shoulder at q ≈ 1.6 Å−1 in the WAXS pattern, which can be associated with the Cα–Cα spacing (3.79 Å) comparable to β-sheet structures in proteins.41 For TH10 and OH10, the peptide-mimicking character has been already found by grazing incidence X-ray diffraction (GIXD) at the air–water interface.3
Furthermore, hydrogen bonds and the Cα–Cα spacing have been found in WAXS for the very similar lipid TH4.42 The only structural difference between TH10 and TH4 is the spacer group.
4.2. Phase behaviour of lipid/DOPE mixtures
In order to enhance the transfection rates, the often used helper lipid DOPE was added to the liposomes. In general, zwitterionic helper lipids dilute the charge density of cationic liposomes. An optimum charge density is high enough to avoid endosomal entrapment and low enough to efficiently release their DNA.43 Furthermore, the complex structure can be adjusted by the choice of helper lipids. DOPE forms an inverted hexagonal structure due to its conical molecule shape. Therefore, adding DOPE to the transfection lipids is believed to promote the formation of inverted hexagonal structures, which rapidly fuse with the endosomal bilayer and allow the release of DNA with high efficiency.7,10,11The 2:1 and 1:1 mixtures of TT10, TH10 and OT10 with DOPE are multi-lamellar. In this case, the added co-lipid did not change the structure of the cationic lipid (see Table 2). The structure only changes from lamellar to non-lamellar for the lipid/DOPE mixtures in the ratio 1:4 (Fig. 6A–C).
 | ||
| Fig. 6 (A–C) SAXS pattern of TT10/DOPE (A), TH10/DOPE (B), and OT10/DOPE (C) mixtures (20 wt% dispersion in water) at 25 °C. (D) WAXS patterns of TH10/DOPE mixtures (20 wt% dispersion in water) at 25 °C. The corresponding SAXS patterns are presented in (B). | ||
TT10/DOPE (1:4) and TH10/DOPE (1:4) form a cubic lattice with Im3m symmetry (Fig. 7A and B and Table S1, ESI‡). The characteristic cubic lattice parameter a was determined as the slope of the linear function  passing through the origin (0,0). Here, h, k and l are Miller indices of the cubic lattice and the slope is equal to 1/a.44 The Im3m lattice parameters a for TT10/DOPE (1:4) and TH10/DOPE (1:4) amount to 195 Å and 138 Å, respectively. The structure of OT10/DOPE (1:4) at 25 °C remains unidentified (data not shown) due to the small number of peaks, which are additionally not well resolved.
passing through the origin (0,0). Here, h, k and l are Miller indices of the cubic lattice and the slope is equal to 1/a.44 The Im3m lattice parameters a for TT10/DOPE (1:4) and TH10/DOPE (1:4) amount to 195 Å and 138 Å, respectively. The structure of OT10/DOPE (1:4) at 25 °C remains unidentified (data not shown) due to the small number of peaks, which are additionally not well resolved.
 | ||
| Fig. 7 SAXS pattern of TT10/DOPE (1:4) (A), TH10/DOPE (1:4) (B), OH10/DOPE (1:4) (C), and OO10/DOPE (1:1) (D) mixtures (20 wt% dispersion in water) at different temperatures (indicated). (A and B) The indexed peaks belong to the cubic Im3m lattice, and the indexed reflexes of the Im3m lattice are plotted vs. their peak positions in the insets. For TT10/DOPE (1:4) the reflex √10 is missing. Red dashed lines are linear fits. (C and D) The indexed peaks belong to the cubic Pm3n lattice, and the indexed reflexes of the Pm3n lattice have been plotted vs. their peak positions in the insets. Red dashed lines are linear fits. | ||
For OH10/DOPE (1:4), high temperatures are needed to form a well-defined structure with DOPE. At 80 °C, OH10/DOPE (1:4) forms a micellar cubic lattice with Pm3n symmetry (Fig. 7C and Table S2, ESI‡). There are three additional peaks in the SAXS pattern belonging to a hexagonal phase (q-ratio 1:√3:2). The obtained Pm3n lattice parameter is 203 Å. The cylinder parameter a of the hexagonal phase is 164.5 Å.
OO10 has the highest transfection efficiency with DOPE in the ratio 1:1 (Fig. 8). A coexistence of two inverted hexagonal (HII1 and HII2) and one cubic phase has been obtained (Fig. 7D). The peak positions could be indexed with a cubic Pm3n lattice. The determined Pm3n lattice parameter equals 563 Å. The cylinder parameters a of HII1 and HII2 are 60.6 Å and 52.6 Å, respectively, which implies phase-separated OO10 with different amounts of DOPE. The addition of DOPE induces the formation of non-lamellar phases (see also Table 4). Additionally, DOPE disturbs the chain packing and a liquid-crystalline state occurs in the mixture TH10/DOPE (1:4) (Fig. 6D and Table 4). This increased chain fluidity can possibly increase the interaction with other lipid structures like cellular membranes, e.g. endosomal membranes, because a lipid mixing is more likely in the liquid-crystalline state compared to the gel state.45
 | ||
| Fig. 8 GFP expression of cationic lipid/DOPE 1:4 (n/n) complexed with pCMV-GFP plasmid DNA (0.1 μg per well) at a N/P ratio of 4 in three different cell lines (A549, HeLa, and LLC-PK1) after 24 h (n = 12). The corresponding cell viability examined by the alamarBlue assay is presented by the blue squares. The corresponding cationic lipid of the mixtures is mentioned at the x-axis. The right graph shows the results of OO10/DOPE mixtures in different molar ratios complexed with pCMV-GFP plasmid DNA (0.1 μg per well) at a constant N/P ratio of 4. Note that the experiments have been performed at different days with different cell passages compared to the left graph. The term ‘relative transfection efficiency’ indicates the transfection efficiency relative to the most effective (100%) in each diagram. | ||
| Phase state | d/a [Å] | q 11 [Å−1] | q 02 [Å−1] | A 0 [Å2] | |
|---|---|---|---|---|---|
| TH10 | Lβ′ | 57.8 | 1.54 | 1.35 | 20.7 |
| TH10/DOPE 2:1 |
Lβ′ | 81.6 | Chains in gel state | ||
| TH10/DOPE 1:1 |
Lβ′ | 80.2 | Chains in gel state | ||
| TH10/DOPE 1:4 |
Qα229 | 138 | Halo → molten chains | ||
| DOPE | Hα | 72.9 | Halo → molten chains | ||
4.3. Transfection experiments
The transfection efficiency of the cationic lipid/DOPE mixtures in a molar ratio of 1:4 was examined after complexing a plasmid DNA, encoding for the reporter gene GFP, at a N/P ratio of 4 (Fig. 8). The OO10/DOPE 1:4 mixture was the most effective one, but also the most toxic formulation. Decreasing the amount of the co-lipid in OO10/DOPE mixtures from 1:4 to 1:2 decreases the efficiency in A549 cells, while the 1:1 mixture has the highest. At the moment, we have no secured explanation for this behaviour, but de-mixing and therefore patches with different charge densities could be the reason. The other cationic lipids in mixtures with DOPE show a continuous decrease in the efficiency if the amount of co-lipid decreases in the mixture.
4.4. Phase behaviour of lipid/DOPE/DNA mixtures
Formulations with the best transfection rates have been examined. The results for the lipoplexes with TT10, TH10, OT10 and OH10 as well as OO10 are shown in Fig. 9. In general, the observed liquid-crystalline phases change by adding calf thymus DNA. Due to the co-existence of different phases, the lipoplex structure determination is rather complex. | ||
| Fig. 9 (A) SAXS pattern of lipoplexes at N/P = 4 composed of TH10/DOPE (1:4)/DNA (black), TT10/DOPE (1:4)/DNA (green), OT10/DOPE (1:4)/DNA (violet) and OH10/DOPE (1:4)/DNA (red) as 20 wt% dispersions in water at 25 °C. (B) SAXS of OO10/DOPE (1:1) (red) and of OO10/DOPE (1:1)/DNA at N/P = 4 (black) at 25 °C as 10 wt% dispersion in water. (C) Schematic representation of the structures found in OO10/DOPE (1:1)/DNA lipoplexes (see B). | ||
The SAXS patterns of the mixtures TT10/DOPE (1:4) and OT10/DOPE (1:4) with DNA indicate a coexistence of three different phases. Besides the bicontinuous cubic Im3m phase, additionally two inverted hexagonal phases appear. Most probably these phases consist of phase-separated DOPE. Pure DOPE as a 20 wt% dispersion in water has a cylinder lattice parameter a of 72 Å. For HII1 and HII2 the rod diameter a equals 76 Å and 68 Å, respectively. The small parameter a might indicate that ct-DNA is not incorporated into the hexagonal phases. More likely, ct-DNA is part of the cubic phase with Im3m symmetry indexed as QDNA in Fig. 9A. The cubic phase is already formed in the corresponding binary mixture in the absence of DNA (see Fig. 7). In a general phase diagram, the cubic phase is in between the lamellar phase (TT10) and the hexagonal mesophase (DOPE). The cationic lipid provides the positive charge suitable for the complexation of DNA. The characteristic Im3m lattice parameter a of TT10/DOPE (1:4) is equal to 195 Å without DNA. After ct-DNA is added to the lipid mixture, a decreases to 162 Å. Obviously, the cubic mesophase shrinks due to condensation caused by electrostatic interactions between cationic lipids and negatively charged ct-DNA. Since the samples have been measured in water, there are no counter ions to screen the charges. OT10/DOPE (1:4)/DNA lipoplexes with N/P = 4 show similar Bragg peaks as found for TT10/DOPE (1:4)/DNA (N/P 4). In contrast to the Bragg peaks, which correspond to a cubic Im3m structure, the Bragg peaks indicating two hexagonal phases are not well-resolved. The lattice parameter a of the Im3m structure is equal to 165 Å and therefore slightly larger for the OT10-containing lipoplex than for the TT10-containing cubic lipoplex. Since the replacement of the shorter and saturated tetradecyl chain in TT10 by the longer and unsaturated oleyl chain OT10 results in higher fluidity, the sponge-like bicontinuous system can bind more water. Due to this swelling and the increase in chain length from C14:0 to C18:1, the increased lattice parameter is not surprising. The cylinder lattice parameters of the two hexagonal mesophases of OT10/DOPE (1:4)/DNA (N/P 4) are too small to host ct-DNA, which leads to the conclusion that the calf thymus DNA is mostly complexed in the cubic phase. Both mixtures, TT10/DOPE (1:4) and OT10/DOPE (1:4), incorporate calf thymus DNA in cubic lipoplexes. Cubic lipoplexes containing DNA or siRNA have already been described in the literature.46,47
In contrast, the lipoplexes of the hexadecyl-containing derivatives, namely TH10/DOPE (1:4)/DNA (N/P 4) and OH10/DOPE (1:4)/DNA (N/P 4), are slightly different. Due to the small number of Bragg peaks, the structure of the TH10/DOPE (1:4)/DNA (N/P 4) lipoplex remains unresolved. For the OH10/DOPE (1:4)/DNA (N/P 4) mixture it seems that the ct-DNA is incorporated in a lamellar lipoplex with a repeating distance d equal to 64 Å (indicated as LDNA in Fig. 9A). Pure OH10 has a d-values of 54 Å. In this case, ct-DNA might partially penetrate into the lipid bilayer due to electrostatic interactions because the layer thickness increases only by about 10 Å instead of 20 Å (the double helix diameter is 20 Å).48,49 Additional peaks occur which can be the result of an additional repeat pattern (indicated as L2 in Fig. 9A). This could be either a lamellar phase without DNA resulting in a d of 50 Å or ct-DNA aligned in a 1D lattice between the lipid bilayers with an in-plane distance of 50 Å. A hexagonal phase with a cylinder lattice parameter a of 67 Å is additionally present in the SAXS pattern. Since the rod diameter is similar to DOPE, it might be phase-separated DOPE.
The OO10/DOPE (1:1)/DNA (N/P 4) lipoplex at 25 °C is multi-lamellar, which means the ct-DNA is sandwich-like complexed between the bilayers (LCα).40,50 The ordered 1D alignment of the ct-DNA causes a broad Bragg peak (dDNA) in the SAXS pattern (Fig. 9B). The in-plane distance of 37.8 Å indicates that DNA is not densely packed (Fig. 9C). Furthermore, a fraction of phase-separated OO10 can be seen in the SAXS pattern, which implies that not all OO10 molecules are involved in the lipoplex formation.
5. Conclusions
In this work, the influence of the chain composition on the physical–chemical properties of five lipids, designed for non-viral gene transfection, were investigated in 3D model systems. The focus was set on the phase behaviour and structures of the pure lipids, their mixtures with the commonly used helper-lipid DOPE, and the selected transfection mixtures with DNA. It was shown that the replacement of saturated tetradecyl (C14:0) and hexadecyl (C16:0) chains by unsaturated oleyl (C18:1) chains decreases the gel to liquid-crystalline phase transition temperature. The exchange of a short tetradecyl (C14:0) chain by a longer hexadecyl (C16:0) chain enhances the van der Waals interactions and leads to an increase of the gel to liquid-crystalline phase transition temperature. The fluidity increases with shorter chains and double bonds: TH10 < TT10 < OH10 < OT10 < OO10, a behaviour also described in monolayer experiments.3 Additionally, a hydrogen bond network was found for the pure cationic lipids. The increasing volume of the chain domain changed the molecular shape such that TT10, TH10, OT10 and OH10 are cylinder-like while OO10 has an inverted cone-like molecular shape. Therefore, the first four lipids form lamellar mesophases and the last one an inverted hexagonal phase. The addition of DOPE has several effects. It destroys the hydrogen bond network, reduces the charge density in the lipid mixtures and disturbs the alkyl chain packing resulting in an increased fluidity of the mixtures. As a result, the aggregation behaviour of the lipid mixtures changes with an increasing amount of DOPE from lamellar to cubic. The lipid mixtures with DNA show a panoply of mesophases. Interestingly, TT10 and OT10 form cubic lipoplexes, while OO10 complexes the DNA sandwich-like between lipid bilayers in a lamellar lipoplex. Surprisingly, the latter is the most effective lipoplex.Against all expectations, it emerges that the miscibility with DOPE decreases with an increasing amount of oleyl chains. Maybe the liposomes are too instable due to a lack of rigid chains, which eventually results in phase separation.
Abbreviation
| ATR-FTIR | Attenuated total reflection Fourier-transform infrared spectroscopy |
| arb. units | Arbitrary units |
| ct-DNA | Calf thymus deoxyribonucleic acid |
| DOPE | 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine |
| FTRS | Fourier-transform Raman spectroscopy |
| GIXD | Grazing incidence X-ray diffraction |
| Hα | Hexagonal phase state with molten chains |
| Lβ′ | Lamellar phase state with tilted chains in all-trans conformation |
| OH10 | N-(2-Aminoethyl)-N′-{6-amino-1-[N-(9Z)-octadec-9-enylamino]-1-oxohexan-(2S)-2-yl}-2-hexadecylpropandiamide |
| OO10 | N-(2-Aminoethyl)-N′-{6-amino-1-[N-(9Z)-octadec-9-enylamino]-1-oxohexan-(2S)-2-yl}-2-[(9Z)-octadec-9-enyl]propandiamide |
| OT10 | N-(2-Aminoethyl)-N′-{6-amino-1-[N-(9Z)-octadec-9-enylamino]-1-oxohexan-(2S)-2-yl}-2-tetradecylpropandiamide |
| SAXS | Small-angle X-ray scattering |
| TH10 | N-(2-Aminoethyl)-N′-[6-amino-1-oxo-1-(N-tetradecylamino)hexan-(2S)-2-yl]-2-hexadecylpropandiamide |
| T m | Gel to liquid-crystalline phase transition temperature |
| TT10 | N-(2-Aminoethyl)-N′-[6-amino-1-oxo-1-[(N-tetradecylamino)hexan-(2S)-2-yl]]-2-tetradecylpropandiamid |
| WAXS | Wide-angle X-ray scattering |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Max Planck Society. We thank DESY and ESRF for beamtime and support at the beam line by Dr Sérgio S. Funari (A2, DESY) and Dr Gudrun Lotze (ID02, ESRF). Open Access funding provided by the Max Planck Society. We thank Prof. Hinderberger (Institute of Chemistry, MLU Halle/Wittenberg) for the access to the DSC and ATR-FTIR. S. D. was financially supported by a grant from the Deutsche Forschungsgemeinschaft (DFG) project DR 1024/1-1.Notes and references
- B. L. Davidson and P. B. McCray Jr, Current prospects for RNA interference-based therapies, Nat. Rev. Genet., 2011, 12, 329–340, DOI:10.1038/nrg2968.
- P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7413–7417 CrossRef.
- S. Tassler, D. Pawlowska, C. Janich, B. Dobner, C. Woelk and G. Brezesinski, Phys. Chem. Chem. Phys., 2018, 20, 6936–6944 RSC.
- Z. Hyvönen, M. Ruponen, S. Rönkkö, P. Suhonen and A. Urtti, Eur. J. Pharm. Sci., 2002, 15, 449–460 CrossRef.
- A. Ahmad, H. M. Evans, K. Ewert, C. X. George, C. E. Samuel and C. R. Safinya, J. Gene Med., 2005, 7, 739–748 CrossRef PubMed.
- J. M. Seddon and R. H. Templer, Handbook of Biological Physics, 1995, ch. 3, vol. 1, pp. 99–149 Search PubMed.
- I. Koltover, T. Salditt, J. O. Rädler and C. R. Safinya, Science, 1998, 281, 78–81 CrossRef PubMed.
- P. W. M. V. Dijck, Biochim. Biophys. Acta, Biomembr., 1979, 555, 89–101 CrossRef.
- P. R. Cullis, P. W. M. V. Dijck, B. D. Kruijff and J. D. Gier, Biochim. Biophys. Acta, Biomembr., 1978, 513, 21–30 CrossRef.
- I. S. Zuhorn, U. Bakowsky, E. Polushkin, W. H. Visser, M. C. A. Stuart, J. B. F. N. Engberts and D. Hoekstra, Mol. Ther., 2005, 11, 801–810 CrossRef PubMed.
- R. Koynova, Lipid Insights, 2008, 2, 41–59 CrossRef.
- A. Zumbuehl, B. Dobner and G. Brezesinski, Curr. Opin. Colloid Interface Sci., 2014, 19, 17–24 CrossRef.
- S. Tassler, C. Wölk, C. Janich, B. Dobner and G. Brezesinski, Phys. Chem. Chem. Phys., 2017, 19, 20271–20280 RSC.
- V. Verdolino, R. Cammi, B. H. Munk and H. B. Schlegel, J. Phys. Chem. B, 2008, 112, 16860–16873 CrossRef PubMed.
- C. Wölk, S. Drescher, A. Meister, A. Blume, A. Langner and B. Dobner, Chem. – Eur. J., 2013, 19, 12824–12838 CrossRef PubMed.
- M. Dittrich, M. Böttcher, J. S. L. Oliveira, B. Dobner, H. Möhwald and G. Brezesinski, Soft Matter, 2011, 7, 10162–10173 RSC.
- T. Schindler and E. Nordmeier, Macromol. Chem. Phys., 1997, 198, 1943–1972 CrossRef.
- S. S. Funari, F. Barceló and P. V. Escribá, J. Lipid Res., 2003, 44, 567–675 CrossRef PubMed.
- C. Boulin, R. Kempf, M. H. J. Koch and S. M. McLaughlin, Nucl. Instrum. Methods Phys. Res., Sect. A, 1986, 249, 399–407 CrossRef.
- C. J. Boulin, R. Kempf, A. Gabriel and M. H. J. Koch, Nucl. Instrum. Methods Phys. Res., Sect. A, 1988, 269, 312–320 CrossRef.
- M. H. J. Koch and J. Bordas, Nucl. Instrum. Methods, 1983, 208, 461–469 CrossRef.
- G. Rapp, A. Gabriel, M. Dosiére and M. H. J. Koch, Nucl. Instrum. Methods Phys. Res., 1995, 357, 178–182 CrossRef.
- T. Narayanan, O. Diat and P. Bösecke, Nucl. Instrum. Methods Phys. Res., Sect. A, 2001, 467–468, 1005–1009 CrossRef.
- T. C. Huang, H. Toraya, T. N. Blanton and Y. Wu, J. Appl. Crystallogr., 1993, 26, 180–184 CrossRef.
- O. Kenji, K. Setsuo and H. Masao, Bull. Chem. Soc. Jpn., 1972, 45, 2651–2652 CrossRef.
- P. Garidel, Phys. Chem. Chem. Phys., 2002, 4, 1934–1942 RSC.
- P. Garidel, Phys. Chem. Chem. Phys., 2006, 8, 2265–2275 RSC.
- S. Raudenkolb, S. Wartewig, G. Brezesinski, S. S. Funari and R. H. Neubert, Chem. Phys. Lipids, 2005, 136, 13–22 CrossRef PubMed.
- W. H. Kirchhoff and I. W. Levin, J. Res. Natl. Bur. Stand., 1987, 92, 113–128 CrossRef.
- I. W. Levin, T. E. Thompson, Y. Barenholz and C. Huang, Biochemistry, 1985, 24, 6282–6286 CrossRef PubMed.
- K. Shirota, K. Yagi, T. Inaba, P.-C. Li, M. Murata, Y. Sugita and T. Kobayashi, Biophys. J., 2016, 111, 999–1007 CrossRef PubMed.
- A. Blume, Curr. Opin. Colloid Interface Sci., 1996, 1, 64–77 CrossRef.
- F. Severcan and D.-O. Dorohoi, J. Mol. Struct., 2008, 887, 117–121 CrossRef.
- R. Mendelsohn, C. R. Flach and D. J. Moore, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 923–933 CrossRef PubMed.
- P. Garidel, B. Fölting, I. Schaller and A. Kerth, Biophys. Chem., 2010, 150, 144–156 CrossRef PubMed.
- J. N. Israelachvili, Intermolecular and Surface Forces, Saunders College Publishing, 1985 Search PubMed.
- C. Tanford, The Hydrophobic Effect, Wiley, New York, N.Y., 1973 Search PubMed.
- T. Yu, O. S. Lee and G. C. Schatz, J. Phys. Chem. A, 2013, 117, 7453–7460 CrossRef PubMed.
- M. O. Guler, R. C. Claussen and S. I. Stupp, J. Mater. Chem., 2005, 15, 4507–4512 RSC.
- R. Sabaté and J. Estelrich, J. Phys. Chem. B, 2005, 109, 11027–11032 CrossRef PubMed.
- I. W. Hamley, A. Dehsorkhi, V. Castelletto, M. N. M. Walter, C. J. Connon, M. Reza and J. Ruokolainen, Langmuir, 2015, 31, 4490–4495 CrossRef PubMed.
- C. Janich, S. Taßler, A. Meister, G. Hause, J. Schäfer, U. Bakowsky, G. Brezesinski and C. Wölk, Soft Matter, 2016, 12, 5854–5866 RSC.
- K. K. Ewert, A. Zidovska, A. Ahmad, N. F. Bouxsein, H. M. Evans, C. S. McAllister, C. E. Samuel and C. R. Safinya, in Nucleic Acid Transfection, ed. W. Bielke and C. Erbacher, Springer Verlag, Berlin, Germany, 2010, pp. 191–226 Search PubMed.
- P. Alexandridis, U. Olsson and B. Lindman, Langmuir, 1998, 14, 2627–2638 CrossRef.
- A. Blume and P. Garidel, Handbook of Thermal Analysis and Calorimetry, Elsevier Science, 1st edn, 1999, pp. 109–173 Search PubMed.
- A. Bilalov, U. Olsson and B. Lindman, Soft Matter, 2009, 5, 3827–3830 RSC.
- C. Leal, K. K. Ewert, R. S. Shirazi, N. F. Bouxsein and C. R. Safinya, Langmuir, 2011, 27, 7691–7697 CrossRef PubMed.
- R. Podgornik, D. C. Rau and V. A. Parsegian, Macromolecules, 1989, 22, 1780–1786 CrossRef.
- C. Symietz, M. Schneider, G. Brezesinski and H. Möhwald, Macromolecules, 2004, 37, 3865–3873 CrossRef.
- G. Caracciolo, D. Pozzi, R. Caminiti, G. Mancini, P. Luciani and H. Amenitsch, J. Am. Chem. Soc., 2007, 129, 10092–10093 CrossRef PubMed.
Footnotes |
| † This article is dedicated to the memory of the late Professor Helmuth Möhwald. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp01922c |
| This journal is © the Owner Societies 2018 |