
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering L-asparaginase for spontaneous formation of calcium phosphate bioinspired microreactors†
Alexandra
Louka
 ab,
Irina
Matlahov‡
c,
Stefano
Giuntini
ab,
Linda
Cerofolini
a,
Andrea
Cavallo
d,
Serena
Pillozzi
e,
Enrico
Ravera
*ab,
Marco
Fragai
*ab,
Annarosa
Arcangeli
e,
Ayyalusamy
Ramamoorthy
fg,
Gil
Goobes
c and
Claudio
Luchinat
*ab
ab,
Irina
Matlahov‡
c,
Stefano
Giuntini
ab,
Linda
Cerofolini
a,
Andrea
Cavallo
d,
Serena
Pillozzi
e,
Enrico
Ravera
*ab,
Marco
Fragai
*ab,
Annarosa
Arcangeli
e,
Ayyalusamy
Ramamoorthy
fg,
Gil
Goobes
c and
Claudio
Luchinat
*ab
aMagnetic Resonance Center (CERM), University of Florence and Consorzio Interuniversitario Risonanze Magnetiche di Metallo Proteine (CIRMMP), Via L. Sacconi 6, 50019 Sesto Fiorentino, Italy. E-mail: ravera@cerm.unifi.it; fragai@cerm.unifi.it; claudioluchinat@cerm.unifi.it
bDepartment of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 3, 50019 Sesto Fiorentino, Italy
cDepartment of Chemistry, Bar-Ilan University, Ramat Gan 52900, Israel
dCERTEMA, Multidisciplinary Technology Laboratory, S.P. del Cipressino km 10, 58044 Cinigiano, Grosseto, Italy
eDepartment of Experimental and Clinical Medicine, Section of Internal Medicine, University of Florence, Viale G.B. Morgagni 50, 50134 Firenze, Italy
fBiophysics and Department of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA
gInstitute for Advanced Studies, Technical University of Munich, Garching, Munich, Germany
First published on 26th April 2018
Abstract
Active bioinspired materials are appealing biotechnological targets, and their study is gaining momentum. These materials, which comprise of an inorganic matrix and one or more biomolecules, are extremely variable and therefore may result difficult to characterize in their intimate structure. In this work we have prepared a hydroxyapatite–L-asparaginase composite, with the perspective of using it in acute leukemia treatment. We demonstrate that the use of electron microscopy and powder X-ray diffraction, combined with the atomic-resolution information coming from solid-state NMR, allows us to understand the topology of the material and how the different components interplay to obtain an active composite.
Introduction
Biological drugs (biologics1) are the fastest-growing category of approved therapeutics, with most biologics being represented by proteins.2L-Asparaginases, which catalyze the hydrolysis of L-asparagine (Asn) to L-aspartate (Asp), are one of the earliest examples of biologics. The depletion of Asn from circulating blood induces cell death in acute lymphoblastic leukemia (ALL) cells, which lack the Asn-synthetase enzyme.3,4 The most commonly used biologics in therapy are type-II bacterial L-asparaginases (ANSII), with E. coli ANSII being in clinical use since 1967.5 ANSII is indispensable in ALL treatment protocols and is used for remission induction and intensification in all pediatric and most adult regimens.6 However, treatment with ANSII can lead to severe side effects that comprise hepato- and pancreas toxicity, coagulation disorders and, most relevant, allergic reactions. ANSII can be administered both intravenously (IV) or intramuscularly (IM), the latter route being better tolerated and less often resulting in increased hypersensitivity reactions.7 The relevance of antibodies directed against ANSII in children with ALL has been highlighted in the Berlin–Frankfurt–Münster (BFM) studies, and recently reviewed:8 adverse reactions to the enzyme mediated by either IgG or IgE antibodies, or both, have been described.9 The appearance of antibodies increases ANSII clearance and neutralizes the catalytic activity of the enzyme.10 Therefore, it is of capital importance to reduce the interaction of the enzyme with the immune system of the patients. Several strategies have been devised to reduce immunogenicity, the most common being PEGylation.11 However, PEG may cause severe side effects in patients with progressive chronic kidney disease12 and can induce allergic reactions even when administered IM.7A radically different strategy would be to avoid contact with the immune system of the patient altogether. Extracorporeal processing of blood has been proposed almost forty years ago, with limited follow-up in clinical practice, mostly because of the limited compliance of the treatment.13 Nowadays, better-designed biomedical devices that can be used to physically confine active components within a membrane are available. This possibility mitigates the need of obtaining an enzyme that is able to evade immune recognition. On the other hand, the incorporation of an enzyme within a device requires that the formulation has a long lifetime, both on the shelf and in operando, that it is easily handled (weighted, transferred, etc.) and that the active site is accessible. For these reasons, the possibility of extracorporeal processing has triggered a strong interest in the protein immobilization community.14,15
Here, we describe the design of a biomaterial that carries an ANSII chimera attached to a hydroxyapatite (HA) support, because HA-based composites have already been used as new biomedical devices,16–18 drugs and vaccine carriers,19–24 and because immobilization of enzymes often results in stabilization.25
The characterization of bioinspired materials is a complex accomplishment because it needs to rely upon the characterization of both the inorganic matrix and the biomolecular component.26–31 In this context, NMR plays a very important role, as it is the only methodology that can access atomic-level information on both components.32–41
Results and discussion
Native, therapeutically active ANSII is a homotetrameric assembly of about 140 kDa with D2 symmetry (a dimer of two intimate dimers)42 with four catalytic sites per tetramer. We have previously provided the NMR characterization of ANSII in its free, PEGylated,43 sugar-conjugated forms44 and bound to gold nanoparticles.45 We fused ANSII with a C-terminal tail encompassing the HA-promoting peptide (HABP) W6p (RWRLEGTDDKEEPESQRRIGRFG; Fig. S1, ESI†).46 Gel-electrophoresis and dynamic light scattering show that ANSII–HABP is smaller (∼80 kDa) than the native enzyme, and likely corresponds to a single intimate dimer (Fig. S2–S4, ESI†). However, it still retains about one-fourth of the catalytic activity per monomer (Fig. S5, ESI†).The HA/ANSII–HABP composite was prepared by self-promoted biomimetic mineralization of HA driven by HABP (see ESI†). After repeated washings to remove the unbound enzyme, the activity of HA/ANSII–HABP was assayed using NMR (Fig. S6 and S7, ESI†), under the same conditions used for metabolomic profiling of serum (Fig. S7, ESI†).47 HA/ANSII–HABP was still active after more than three months of storage at 4 °C. The efficacy of HA/ANSII–HABP was then tested in vitro on human ALL cells (697 cell line), and compared to either the wild-type (WT) enzyme or the commercial product Oncaspar (i.e. PEG-ANSII). All ANSII preparations were added at 0.74 U mL−1 concentration, according to previous reports.48,49 HA/ANSII–HABP was kept separated from leukemic cells via a 3.5 kDa cut-off dialysis membrane. HA/ANSII–HABP reduced the leukemic cell viability to the same extent (after 24 hours of incubation) and even more (at 48 hours of incubation) compared to either the free enzyme or the commercial product (Fig. 1).
 | ||
| Fig. 1 Effects of different ANSII preparations on the proliferation of ALL cells. ALL 697 cells were treated with different ANSII preparations (HA/ANSII–HABP, WT ANSII and Oncaspar) at a concentration of 0.74 U mL−1 for 24 and 48 hours and the number of live cells, determined via the Trypan Blue exclusion test, was measured. HA/ANSII–HABP was confined in a device with walls consisting of a 3.5 kDa cut-off dialysis membrane. Data are mean values ± standard error of the mean of three independent experiments. 48 hours: HA/ANSII–HABP vs. WT ANSII, p = 0.00116; HA/ANSII–HABP vs. Oncaspar, p = 0.00449, as tested by one way ANOVA. | ||
HA/ANSII–HABP was further characterized using powder X-ray diffraction (XRD), scanning electron microscopy and solid-state NMR. The XRD profile (Fig. 2), shows the typical diffraction pattern of HA,50 while the diffraction at low 2θ suggests the simultaneous presence of some octacalcium phosphate (OCP).51 The diffraction peaks are broad, as expected for crystallites of small size. By elemental analysis, the protein content was found to be on the order of 40 ± 1% in the dried material. SEM micrographs show that the material is composed of fused structures, homogeneous in shape and size (Fig. 3), which more closely resemble HA nanosheets,52 rather than OCP spherical particles.
 | ||
| Fig. 2 Top: XRD profile of the HA/ANSII–HABP composite (black line). Bottom: Calculated XRD diffractogram of hydroxyapatite (red) and octacalcium phosphate (blue) shown as stick plots. | ||
 | ||
| Fig. 3 SEM images of the HA/ANSII–HABP composite, taken with (upper panels) and without (lower panels) conductive staining, revealing a highly porous structure, which is most suitable for maintaining the accessibility of the enzyme. The high protein loading at the surface reduces the achievable resolution. | ||
Combustion elemental analysis provided the elemental composition and a quantitative determination of the protein content, found to be of the order of 40% in the dried material. After reacting 1 mL of the mixture, and rinsing it several times, about 28 mg of wet material are obtained. From the content of the reaction mixture (see ESI†), the theoretical yield of the dry material can be calculated: assuming that only HA will form, with a 40% w/w protein content in the material, 4 mg of the composite should precipitate. If, on the contrary, only OCP is formed, about 2 mg of the composite should precipitate. Thus, in the wet material, we can estimate that 6.5–13% will consist of HA/ANSII–HABP.
HA/ANSII–HABP was further characterized in terms of their elemental composition using energy dispersive X-ray spectroscopy (EDS). Inductively coupled plasma (ICP) emission was additionally used for elemental analysis of the total Ca and P content in the sample (Table 1). The ICP measurement shows a Ca/P ratio of 1.78, higher than the theoretical HA value of 1.67 (Tables 1 and 2). Such a phosphate depletion has been also observed53 for HA prepared in the presence of a mineral binding peptide, and is possibly due to a disordered phase in addition to the ordered HA phase.53
| Element | Average wt% | Std% | Average atom count | Std |
|---|---|---|---|---|
| C | 23 | 3 | 1.9 | 0.2 |
| N | 10 | 3 | 0.7 | 0.2 |
| Ca | 26 | 4 | 0.7 | 0.1 |
| P | 14 | 2 | 0.44 | 0.06 |
| Ratio | Measured – EDS | Measured – ICP | Expected for HA | Expected for OCP |
|---|---|---|---|---|
| Ca/P | 1.6 ± 0.3 | 1.78 | 1.67 | 1.33 |
| Ratio | Measured | Expected for current protein chimeric construct |
|---|---|---|
| C/N | 2.8 ± 0.9 | 3.57 |
| Protein% | 47 ± 7 |
In order to fully understand the activity of the material, it is necessary to link the bulk observations to the molecular scale, answering the following questions:
(a) are the ordered and the disordered layers in contact with each other?
(b) is the protein in contact with the ordered phase, with the disordered one or with both?
(c) is the protein exposed to the solvent, thus accessible to the substrate?
We here demonstrate how solid-state NMR is the methodology of choice to answer these questions.
A Bloch decay (BD), a homonuclear decoupled 1H spectrum of HA/ANSII–HABP and a BD spectrum of HA are shown in Fig. 4. The presence of the HA phase is confirmed by the BD spectrum (red), where HA/ANSII–HABP has the same OH− resonance at 0.2 ppm as synthetic HA (green). The BD spectrum shows a broad intense water resonance at 5.3 ppm (consistent with the high water content in HA/ANSII–HABP); however, as will be shown, these water molecules are not necessarily residing at the enzyme–mineral interface. The peak at 1.3 ppm is associated with the aliphatic side-chain protons of the protein chimera but may include some contribution from HA as can be seen by comparing with the mineral spectrum. In the homonuclear decoupled wPMLG spectrum (violet) of HA/ANSII–HABP, the water line is extensively attenuated, exposing three clear bands at 1.3 ppm, 4.5 ppm and 7.2 ppm from ANSII–HABP protons.
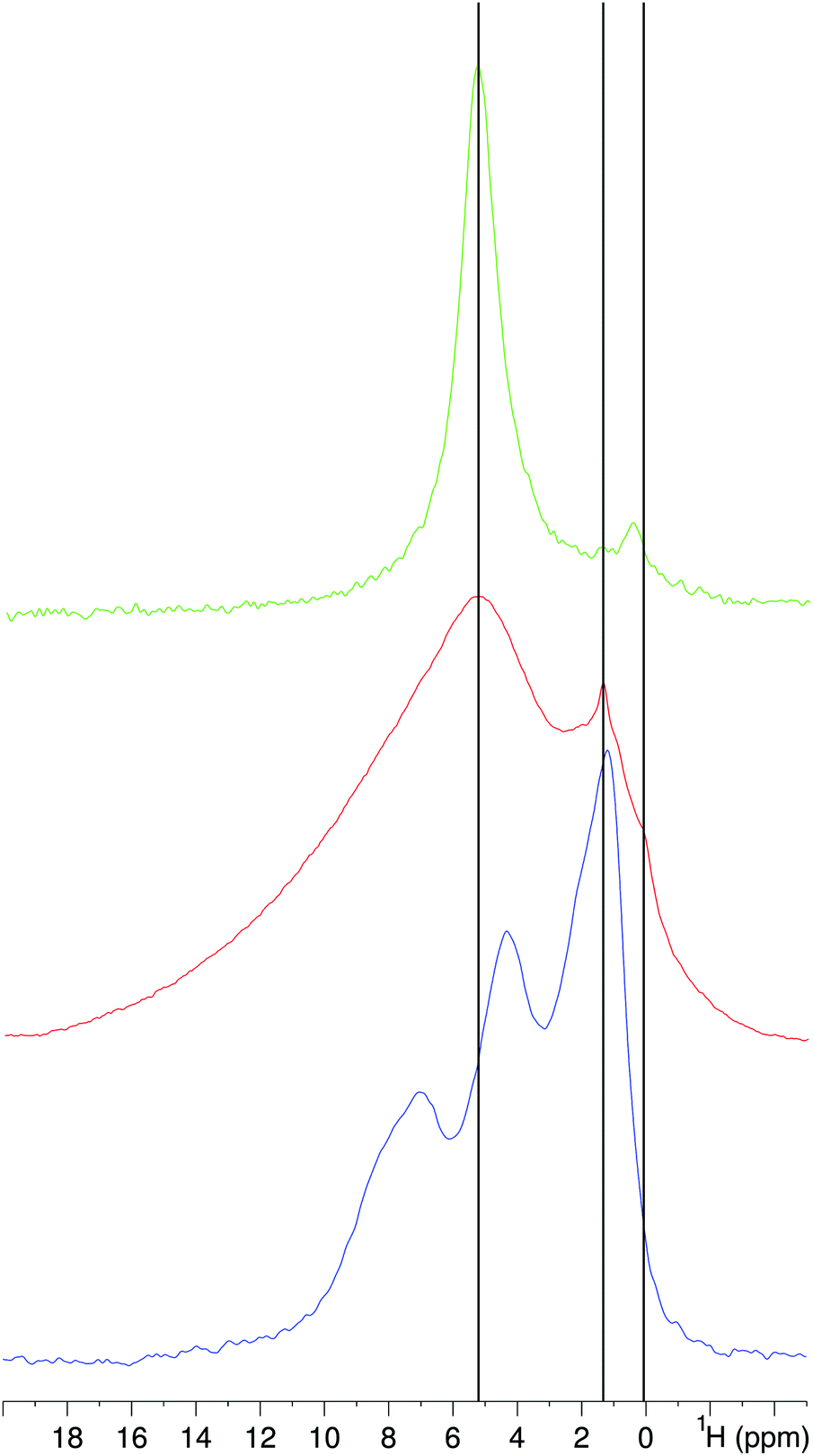 | ||
| Fig. 4 1H NMR spectra of ANSII–HABP acquired using wPMLG for homonuclear-decoupling (violet) and a simple 90°-acquisition (i.e. a Bloch decay) (red). A 1H Bloch decay spectrum (green) of synthetic hydroxyapatite prepared without calcination is also shown for comparison. | ||
Although the wPMLG experiment is intended to enhance resolution by 1H–1H decoupling, in this biomaterial sample we find that it improves the capability to observe the protein proton lines via a simple solvent suppression mechanism. Exposing the protons to a strong effective field tilted at the magic angle causes exclusive broadening of the water signal beyond detection due to coincidental matching of the water motional time scale with the field's decoupling cycle period (15 μs). The motions experienced by ANSII–HABP are quite different and therefore are not affected similarly by the homonuclear decoupling. The widths of the ANSII–HABP proton lines are 3–6 ppm and they result from immobile protein molecules: these lines are collectively associated with ANSII–HABP protons belonging to side-chains, Hα and amide protons. The peak at 1.3 ppm may mask a smaller contribution from HA (see the spectrum of synthetic HA).
The deconvolution54 of 1H–31P cross-polarization (CP)53 spectra of HA/ANSII–HABP obtained under MAS (Fig. 5a and Fig. S8, ESI†) reveals two phosphate species, as synthetic HA,53 that can be attributed to two different layers. The peak at 2.9 ppm (FWHM 1.2 ppm) is typical of the phosphate resonance in HA crystals (denoted hereafter as PHA) while the peak at 2.6 ppm (FWHM 3 ppm) is attributed to hydrogen phosphate ions (HPO42−) (denoted hereafter as PHP) which are part of the disordered calcium phosphate phase that resides at the HA/ANSII–HABP interface55 (this peak cannot be attributed to beta-glycerophosphate because it exhibits a higher chemical shift56). Mono-acid phosphates were shown to adopt a large range of chemical shifts, depending on the surroundings ions. In HA/ANSII–HABP this is reflected in the significant width, 7 ppm (FWHM), of the PHP peak.55
 | ||
| Fig. 5 (a) An example of the 31P CP spectrum of HA/ANSII–HABP obtained using 3.6 ms contact time deconvoluted using the DMFIT program. The experimental spectrum (blue) is shown with the sum of simulated peaks (red) and individual lines at 2.6 ppm (green) and 2.9 ppm (purple). (b) 31P intensity buildup with increasing CP contact time of PHA phosphates (black) and PHP phosphates (red). | ||
To characterize the proximity of interfacial and bulk protons and phosphates, 31P CP build-up curves were analyzed.5331P CP build-up measurements were carried out by varying the contact time from 100 μs to 10 ms, and the intensities of the PHA (black) and PS (red) peaks were plotted against the contact time (see Fig. 5b). Recently, similar magnetization build-up curves were used to isolate magnetization transfers between interfacial protons and phosphates and bulk protons and phosphates in apatite samples prepared with a bone protein binding peptide which displays similar properties of having a crystalline phase of HA and a disordered phase containing calcium phosphate ions and the biomolecule.53 Here, we follow a similar strategy.
These intensity buildup curves show the rate of magnetization transfer from adjacent protons to the two phosphate species. The relative line intensity in Table 3 is derived from the fit to the experimental phosphate line, in Fig. 5 as described below. The integral (integrated intensity) of the experimental phosphate peak for each contact time point was calculated and the values were normalized to the highest integral to obtain the buildup normalized to 1. Each spectrum was then deconvolved into the two phosphate contributions and the calculated area under each deconvolved peak was taken as a fraction of one. Therefore, the intensity per phosphate peak and per contact point was overall normalized to the fractional intensity of that phosphate species within each contact time spectrum and to the intensity of the highest signal in the CP buildup curve. The curve of PHA was fitted using eqn (1) and that of PHP was fitted using the extended eqn (2) which accounts for relaxation processes which further influence transfer.
 | (1) |
 | (2) |
| Peak position (ppm) | Assignment | Width (ppm) | τ cp (ms) | T 1ρ (ms) | Normalized intensity |
|---|---|---|---|---|---|
| 2.9 | PHA | 1.4 | 3.1 | — | 0.27 |
| 2.6 | PHP | 5.0 | 0.5 | 13.1 | 0.73 |
In these equations, I0 is the overall intensity of phosphate magnetization, τcp is the typical CP time and (TH1ρ) is the longitudinal rotating frame relaxation time of the protons.
The kinetic parameters, summarized in Table 1, were deduced from best-fitted curves calculated by minimizing the deviations of signal intensity I0, the magnetization transfer time constant (τcp) and the 1H rotating frame relaxation time (TH1ρ) in calculations and in experiments.
The relatively low normalized PHA intensity, 0.27, is indicative of the low content of crystalline HA in ANSII–HABP. This line typically has an intensity >0.6 in other co-precipitation procedures of proteins with HA.53 The extracted magnetization transfer time of PHA, 3.1 ms (Table 3), closely resembles the value for synthetic HA (see Fig. 6),53 and is somewhat intermediate between hydroxyl-to-phosphate transfer time in apatite crystals grown with an osteonectin binding peptide and interfacial transfer time from protons outside the crystals to crystalline phosphates in that same sample.53
 | ||
| Fig. 6 31P CP buildup curves of PHA (black) and of the HA phosphate line in hydroxyapatite prepared via a titration method at 310 K (green53). It shows that the rate of magnetization buildup of PHA in ANSII–HABP is similar to that of HA, suggesting that this line represents a 31P nucleus in a crystalline environment. | ||
This finding suggests that either water or protein molecules are located close to the faces of the HA crystal and transfer magnetization to ordered (crystalline) phosphate ions in exposed crystal surfaces. The transfer rate of the PHP line, 0.5 ms, is similar to the magnetization transfer observed for interfacial phosphates in the aforementioned apatite grown with an osteonectin binding peptide.
To further probe the local environment sensed by phosphate groups and identify their distribution within the sample, we recorded two-dimensional 1H–31P HETCOR MAS NMR spectra (Fig. 7).
 | ||
| Fig. 7 1H–31P heteronuclear correlation (HETCOR) spectra of HA/ANSII–HABP using contact times of 0.5 ms. | ||
During the 1H magnetization evolution under the chemical shift in t1, the PMLG decoupling sequence was used to suppress 1H–1H dipolar couplings. Fig. 7 shows an intense cross peak between HA hydroxyl protons at 0.2 ppm and phosphates at 2.9 ppm (i.e. PHA). It also shows a group of narrower cross peaks correlating 1H at 1.3 ppm, 4.0–6.5 and 7.0–8.5 ppm along F1 with PHP phosphates. These resonances are attributed to the protein protons that transfer magnetization to proximal PHP phosphates in the mineral. The homonuclear decoupling employed in t1 effectively removes water proton excitation, and therefore no correlations are observed between water and phosphate species. This is quite different from other preparations of apatite minerals,53,57 whereby a strong correlation of water protons with phosphates in a disordered calcium phosphate layer on HA crystallites was observed. The absence of the water resonance in the 2D HETCOR spectra is further demonstrated in the 1H projections (taken along the phosphate maximal intensity) of these measurements as compared to similar measurements in synthetic HA where the relative intensity is much different (see Fig. S9, ESI†). Similar 1H–31P 2D HETCOR measurements without PMLG decoupling did not show any water cross peaks with phosphates, so it is apparent that water molecules are distantly located from phosphates.
In summary, these spectra show that the protein resonances correlate with PHP in the disordered layer, which in turn is connected with the PHA phase. The latter crystalline phase is mostly excluded from water, suggesting also that this bulk mineral phase is overall distant from the biomolecule. From these data, we can conclude that ANSII–HABP is interacting with a layer of interfacial phosphates (PHP), probably of the mono-acid type, and has no direct interactions with HA crystallites. Finally, no correlations are here observed between water and phosphate species: this means that the water molecules are either located far from the phosphates or are too dynamic to be detected in dipolar-based magnetization transfer experiments.
The 1H–13C CP58,59 MAS spectrum with ramped 1H amplitude (ramp-CP60) has low intensity and resolution (Fig. 8, middle trace) that are both increased when ramp-CP is hybridized with the nuclear Overhauser effect (NOE, Fig. 8, top trace):61 this sequence results in the appearance of a larger number of signals. Most of these additional peaks appear in the aliphatic region (0–75 ppm) and some peaks from aromatic side chains also appear (110–140 ppm). Moreover, some new peaks can be observed also in the carbonyl region (150–180 ppm). The observed peaks around 153 ppm belong to Cζ of the arginine side-chains, while those around 178 ppm relate to Cγ or Cδ of the aspartate and asparagine or glutamate and glutamine side-chains, respectively (Fig. S10, ESI†). These observations suggest that the additional peaks, observed via NOE, originate from those chemical groups or amino acid residues that undergo significant motion on the milliseconds (or faster) timescale, and are therefore not detectable by the dipolar-coupling based CP. One needs to consider the hypothesis of a substantial water–protein magnetization transfer (either through intermolecular NOE, which is prevailing at high spinning rates and low temperature, or through chemical exchange followed by spin diffusion, which is most probably dominant under the present experimental conditions62) at the time of the NOE mixing, plus the contribution due to direct 13C excitations which is intrinsic for the pulse sequence applied.61 Either of the two mechanisms does not detract from the idea, but rather corroborates it, that a large share of the protein is sizably exposed to water.
 | ||
| Fig. 8 13C NMR spectra of HA/ANSII–HABP (black) and PEGylated wtANSII43 (red) obtained under MAS using CP only (middle and bottom spectra) or CP–NOE (topmost). The spinning sidebands are marked as ‘*’. NOE mixing was set to 1.3 s and the CP contact time was 1.2 ms. | ||
The topology of HA/ANSII–HABP (Fig. 9) that can be derived from our integrated analysis is summarized below.
 | ||
| Fig. 9 Scheme of the arrangement of the components of HA/ANSII–HABP. | ||
(a) The microscopic characterization demonstrates that the material is composed of small lamellar structures that closely resemble hydroxyapatite, and PXRD confirms this conclusion.
(b) The secondary electron analysis shows that there is a phosphate depletion, consistent with previous reports on HA/peptide composites.
(c) Solid-state NMR investigation of the inorganic part shows that the ordered HA phase is not in contact with the protein and the disordered layer is in intimate contact with the protein. This further shows that the two inorganic phases are in contact with each other, and water is closer to the protein and largely excluded from the mineral phases.
(d) Protein solid-state NMR indicates that the enzyme is not completely immobile in the material, but undergoes extensive motions (Fig. S10, ESI†), suggesting that it is mainly sitting at the surface of the particles, rather than being entrapped within the material (Fig. 9). Despite the preserved mobility, the observed chimeric ANSII–HABP is stably linked to the mineral particles, as discussed above.
(e) The tests against the cells and those in vitro confirm that the protein is stable in the construct for several months and demonstrate that the material is active.
Nowadays ANSII is still indispensable6 in the treatment of ALL patients and new ANSII preparations are needed to overcome the limitations of the current ANSII therapy, especially immunogenicity. The results obtained in an in vitro assay on human leukemic cells with HA/ANSII–HABP, and the protocols for its characterization, provide new perspectives for the development of microreactors to be used as possible innovative biomedical devices able (i) to increase the stability of ANSII, (ii) to extend Asn depletion in the serum and at the same time (iii) virtually to reduce immunogenicity. Collectively, our results show that enzymes processing small substrates can be efficiently and profitably bound to the surface of a HA matrix by engineering the wild-type construct with HA promoting peptides.
Author contributions
AL and MF designed the chimeric construct; AL and SG expressed and purified the protein and prepared the composite; IM, LC, ER, MF, GG, AR and CL performed and interpreted the NMR experiments; IM, AC and GG recorded and interpreted the EM data; SG, SP and AA performed and interpreted the in-cell analysis; ER, MF, AR, GG and CL designed the research; and all authors wrote the manuscript.Abbreviations
| SSNMR | Solid-state nuclear magnetic resonance |
| HA | Hydroxyapatite |
| ANSII | Type-II bacterial asparaginase |
| HABP | Hydroxyapatite binding peptide W6p RWRLEGTDDKEEPESQRRIGRFG |
| HA/ANSII–HABP | HA-bound ANSII–HABP chimera composite |
| OCP | Octacalcium phosphate |
| CP | Cross-polarization |
| HETCOR | Heteronuclear correlation |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Fondazione CR Firenze, MIUR Contract MEDINTECH CTN01_001177_962865, EC Contracts pNMR No. 317127 and iNEXT No. 653706, EU ESFRI Instruct-ERIC (Core Centre CERM Italy), Regione Toscana (CERM-TT, CERTEMA), University of Florence (PIA), FIRC (17941), and IAS “Hans Fischer Senior fellowship” (COFUND). XRD measurements are courtesy of Samuele Ciattini and Laura Chelazzi, CRIST, University of Florence. Elementary analysis is courtesy of Stefania Pucci, Laboratorio di Microanalisi, University of Florence. Shani Hazan (Bar-Ilan University) is acknowledged for help with the analysis and fitting of the 31P CP buildup curves. FEI-Magellan images were collected at the Bar-Ilan University Nanotechnology Center. Gigliola Lusvardi (Università degli Studi di Modena e Reggio Emilia) and Guido Pintacuda (École normale supérieure de Lyon) are acknowledged for fruitful discussions.References
- R. A. Rader, Nat. Biotechnol., 2008, 26, 743–751 CrossRef CAS PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- J. H. Parmentier, M. Maggi, E. Tarasco, C. Scotti, V. I. Avramis and S. D. Mittelman, Leuk. Res., 2015, 39, 757–762 CrossRef CAS PubMed.
- A. Emadi, N. A. Bade, B. Stevenson and Z. Singh, Pharmaceuticals, 2016, 9, 12 CrossRef PubMed.
- J. M. Hill, J. Roberts, E. Loeb, A. Khan, A. MacLellan and R. W. Hill, JAMA, 1967, 202, 882–888 CrossRef CAS PubMed.
- R. A. Egler, S. P. Ahuja and Y. Matloub, J. Pharmacol. Pharmacother., 2016, 7, 62–71 CrossRef CAS PubMed.
- T. MacDonald, K. Kulkarni, M. Bernstein and C. V. Fernandez, J. Pediatr. Hematol./Oncol., 2016, 38, 341–344 CrossRef PubMed.
- I. M. van der Sluis, L. M. Vrooman, R. Pieters, A. Baruchel, G. Escherich, N. Goulden, V. Mondelaers, J. S. de Toledo, C. Rizzari, L. B. Silverman and J. A. Whitlock, Haematologica, 2016, 101, 279–285 CrossRef CAS PubMed.
- R. Pieters, S. P. Hunger, J. Boos, C. Rizzari, L. Silverman, A. Baruchel, N. Goekbuget, M. Schrappe and C.-H. Pui, Cancer, 2011, 117, 238–249 CrossRef CAS PubMed.
- B. Zalewska-Szewczyk, W. Andrzejewski, W. Młynarski, K. Jedrychowska-Dańska, H. Witas and J. Bodalski, Leuk. Lymphoma, 2007, 48, 931–936 CrossRef CAS PubMed.
- M. L. Graham, Adv. Drug Delivery Rev., 2003, 55, 1293–1302 CrossRef CAS PubMed.
- A. Attar, M. Lémann, A. Ferguson, M. Halphen, M. C. Boutron, B. Flourié, E. Alix, M. Salmeron, F. Guillemot, S. Chaussade, A. M. Ménard, J. Moreau, G. Naudin and M. Barthet, Gut, 1999, 44, 226–230 CrossRef CAS PubMed.
- J. A. Jackson, H. R. Halvorson, J. W. Furlong, K. D. Lucast and J. D. Shore, J. Pharmacol. Exp. Ther., 1979, 209, 271–274 CAS.
- V. M. Balcão, C. Mateo, R. Fernández-Lafuente, F. X. Malcata and J. M. Guisán, Enzyme Microb. Technol., 2001, 28, 696–704 CrossRef.
- A. Ulu and B. Ates, Bioconjugate Chem., 2017, 28, 1598–1610 CrossRef CAS PubMed.
- E. Boanini, P. Torricelli, M. C. Cassani, G. A. Gentilomi, B. Ballarin, K. Rubini, F. Bonvicini and A. Bigi, RSC Adv., 2013, 4, 645–652 RSC.
- R. Gelli, B. Del, P. Tempesti, M. Bonini, F. Ridi and P. Baglioni, J. Colloid Interface Sci., 2018, 511, 145–154 CrossRef CAS PubMed.
- P. Tempesti, G. S. Nicotera, M. Bonini, E. Fratini and P. Baglioni, J. Colloid Interface Sci., 2018, 509, 123–131 CrossRef CAS PubMed.
- X. Wang, H. C. Schröder, M. Wiens, H. Ushijima and W. E. Müller, Curr. Opin. Biotechnol., 2012, 23, 570–578 CrossRef CAS PubMed.
- Y. Mizushima, T. Ikoma, J. Tanaka, K. Hoshi, T. Ishihara, Y. Ogawa and A. Ueno, J. Controlled Release, 2006, 110, 260–265 CrossRef CAS PubMed.
- B. Palazzo, M. Iafisco, M. Laforgia, N. Margiotta, G. Natile, C. L. Bianchi, D. Walsh, S. Mann and N. Roveri, Adv. Funct. Mater., 2007, 17, 2180–2188 CrossRef CAS.
- G. Wang, X. Li, L. Mo, Z. Song, W. Chen, Y. Deng, H. Zhao, E. Qin, C. Qin and R. Tang, Angew. Chem., Int. Ed., 2012, 51, 10576–10579 CrossRef CAS PubMed.
- K. Tomoda, H. Ariizumi, T. Nakaji and K. Makino, Colloids Surf., B, 2010, 76, 226–235 CrossRef CAS PubMed.
- C. Kojima and K. Watanabe, J. Drug Delivery, 2012, 1–4 Search PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- J. M. Bolivar, I. Eisl and B. Nidetzky, Catal. Today, 2016, 259, 66–80 CrossRef CAS.
- K. Yang, S. Bai and Y. Sun, Chem. Eng. Sci., 2008, 63, 4045–4054 CrossRef CAS.
- J. Hubbuch and M. R. Kula, Bioprocess Biosyst. Eng., 2008, 31, 241–259 CrossRef CAS PubMed.
- P. Torres-Salas, A. del Monte-Martinez, B. Cutiño-Avila, B. Rodriguez-Colinas, M. Alcalde, A. O. Ballesteros and F. J. Plou, Adv. Mater., 2011, 23, 5275–5282 CrossRef CAS PubMed.
- J. M. Bolivar, A. Hidalgo, L. Sánchez-Ruiloba, J. Berenguer, J. M. Guisán and F. López-Gallego, J. Biotechnol., 2011, 155, 412–420 CrossRef CAS PubMed.
- P. Batalla, J. M. Bolívar, F. Lopez-Gallego and J. M. Guisan, J. Chromatogr. A, 2012, 1262, 56–63 CrossRef CAS PubMed.
- C. Gröger, K. Lutz and E. Brunner, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 54, 54–68 CrossRef.
- A. Roehrich, J. Ash, A. Zane, D. L. Masica, J. J. Gray, G. Goobes and G. Drobny, Proteins at Interfaces III State of the Art, American Chemical Society, 2012, vol. 1120, pp. 77–96 Search PubMed.
- M. Fragai, C. Luchinat, T. Martelli, E. Ravera, I. Sagi, I. Solomonov and Y. Udi, Chem. Commun., 2013, 50, 421–423 RSC.
- G. Goobes, Isr. J. Chem., 2014, 54, 113–124 CrossRef CAS.
- E. Ravera, V. K. Michaelis, T.-C. Ong, E. G. Keeler, T. Martelli, M. Fragai, R. G. Griffin and C. Luchinat, ChemPhysChem, 2015, 16, 2751–2754 CrossRef CAS PubMed.
- Y. Geiger, H. E. Gottlieb, Ü. Akbey, H. Oschkinat and G. Goobes, J. Am. Chem. Soc., 2016, 138, 5561–5567 CrossRef CAS PubMed.
- T. Martelli, E. Ravera, A. Louka, L. Cerofolini, M. Hafner, M. Fragai, C. F. W. Becker and C. Luchinat, Chem. – Eur. J., 2016, 4, 425–432 CrossRef PubMed.
- E. Ravera, L. Cerofolini, T. Martelli, A. Louka, M. Fragai and C. Luchinat, Sci. Rep., 2016, 6, 27851 CrossRef PubMed.
- E. Ravera, T. Martelli, Y. Geiger, M. Fragai, G. Goobes and C. Luchinat, Coord. Chem. Rev., 2016, 327, 110–122 CrossRef.
- N. Adiram-Filiba, A. Schremer, E. Ohaion, M. Nadav-Tsubery, T. Lublin-Tennenbaum, K. Keinan-Adamsky and G. Goobes, Biointerphases, 2017, 12, 02D414 CrossRef PubMed.
- A. L. Swain, M. Jaskólski, D. Housset, J. K. Rao and A. Wlodawer, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 1474–1478 CrossRef CAS.
- E. Ravera, S. Ciambellotti, L. Cerofolini, T. Martelli, T. Kozyreva, C. Bernacchioni, S. Giuntini, M. Fragai, P. Turano and C. Luchinat, Angew. Chem., Int. Ed., 2016, 55, 2446–2449 CrossRef CAS PubMed.
- S. Giuntini, E. Balducci, L. Cerofolini, E. Ravera, M. Fragai, F. Berti and C. Luchinat, Angew. Chem., Int. Ed., 2017, 56, 14997–15001 CrossRef CAS PubMed.
- S. Giuntini, L. Cerofolini, E. Ravera, M. Fragai and C. Luchinat, Sci. Rep., 2017, 7, 17934 CrossRef PubMed.
- G. Wang, R.-Y. Cao, R. Chen, L. Mo, J.-F. Han, X. Wang, X. Xu, T. Jiang, Y.-Q. Deng, K. Lyu, S.-Y. Zhu, E.-D. Qin, R. Tang and C.-F. Qin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7619–7624 CrossRef CAS PubMed.
- P. Bernini, I. Bertini, C. Luchinat, P. Nincheri, S. Staderini and P. Turano, J. Biomol. NMR, 2011, 49, 231–243 CrossRef CAS PubMed.
- H. Takahashi, J. Inoue, K. Sakaguchi, M. Takagi, S. Mizutani and J. Inazawa, Oncogene, 2017, 36, 4267–4276 CrossRef CAS PubMed.
- S. Pillozzi, M. Masselli, E. D. Lorenzo, B. Accordi, E. Cilia, O. Crociani, A. Amedei, M. Veltroni, M. D’Amico, G. Basso, A. Becchetti, D. Campana and A. Arcangeli, Blood, 2011, 117, 902–914 CrossRef CAS PubMed.
- J. D. Hanawalt, H. W. Rinn and L. K. Frevel, Ind. Eng. Chem., Anal. Ed., 1938, 10, 457–512 CrossRef CAS.
- Y.-H. Tseng, C.-Y. Mou and J. C. C. Chan, J. Am. Chem. Soc., 2006, 128, 6909–6918 CrossRef CAS PubMed.
- X.-Y. Zhao, Y.-J. Zhu, F. Chen, B.-Q. Lu and J. Wu, CrystEngComm, 2013, 15, 206–212 RSC.
- T. Iline-Vul, I. Matlahov, J. Grinblat, K. Keinan-Adamsky and G. Goobes, Biomacromolecules, 2015, 16, 2656–2663 CrossRef CAS PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- P. Conte, D. ŠMejkalová, A. Piccolo and R. Spaccini, Eur. J. Soil Sci., 2008, 59, 584–591 CrossRef CAS.
- L. C. Gatiboni, G. Brunetto, D. dos, S. Rheinheimer, J. Kaminski, C. M. Pandolfo, M. Veiga, A. F. C. Flores, M. A. S. Lima, E. Girotto and A. C. C. Copetti, Rev. Bras. Cienc. Solo, 2013, 37, 640–648 CrossRef CAS.
- I. Matlahov, T. Iline-Vul, M. Abayev, E. M. Y. Lee, M. Nadav-Tsubery, K. Keinan-Adamsky, J. J. Gray and G. Goobes, Chem. Mater., 2015, 27, 5562–5569 CrossRef CAS.
- A. Pines, M. G. Gibby and J. S. Waugh, J. Chem. Phys., 1973, 59, 569–590 CrossRef CAS.
- D. Marks and S. Vega, J. Magn. Reson., Ser. A, 1996, 118, 157–172 CrossRef CAS.
- G. Metz, X. L. Wu and S. O. Smith, J. Magn. Reson., Ser. A, 1994, 110, 219–227 CrossRef CAS.
- R. Zhang, K. H. Mroue and A. Ramamoorthy, J. Magn. Reson. San Diego Calif 1997, 2016, 266, 59–66 CAS.
- A. Lesage, C. Gardinnet, A. Loquet, R. Verel, G. Pintacuda, L. Emsley, B. H. Meier and A. Bockmann, Angew. Chem., Int. Ed. Engl., 2008, 47, 5851–5854 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Protein cloning, expression and purification; SDS-PAGE data; SEC-LS data; biomineralization details; cell viability assay and analysis; enzyme kinetics by NMR; experimental details of EM, PXRD and solid-state NMR. See DOI: 10.1039/c8cp00419f |
| ‡ Present address: Department of Structural Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15260, USA. |
| This journal is © the Owner Societies 2018 |