
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe fate of plasma-generated oxygen atoms in aqueous solutions: non-equilibrium atmospheric pressure plasmas as an efficient source of atomic O(aq)†
J.
Benedikt
 *a,
M.
Mokhtar Hefny
bc,
A.
Shaw
d,
B. R.
Buckley
e,
F.
Iza
d,
S.
Schäkermann
f and
J. E.
Bandow
f
*a,
M.
Mokhtar Hefny
bc,
A.
Shaw
d,
B. R.
Buckley
e,
F.
Iza
d,
S.
Schäkermann
f and
J. E.
Bandow
f
aExperimental and Applied Physics, Faculty of Mathematics and Natural Sciences, Christian-Albrecht-Universität zu Kiel, 24098 Kiel, Germany. E-mail: benedikt@physik.uni-kiel.de
bResearch Department Plasmas with Complex Interactions, Ruhr-Universität Bochum, 44780 Bochum, Germany
cBasic Science Department, Faculty of Engineering and Technology, Future University in Egypt, Cairo, Egypt
dWolfson School of Mechanical, Electrical and Manufacturing Engineering, Loughborough University, UK
eDepartment of Chemistry, Loughborough University, Loughborough, LE11 3TU, UK
fApplied Microbiology, Faculty of Biology and Biotechnology, Ruhr-Universität Bochum, 44780 Bochum, Germany
First published on 19th April 2018
Abstract
Non-equilibrium radio-frequency driven atmospheric-pressure plasma in He/0.6%O2 gas mixture has been used to study the reaction mechanism of plasma-generated oxygen atoms in aqueous solutions. The effluent from the plasma source operated with standard and 18O-labeled O2 gas was used to treat water in the presence of phenol as a chemical probe. Comparing the mass spectrometry and gas chromatography-mass spectrometry data of the solutions treated with plasma under normal and labeled oxygen provides clear evidence that O(aq) originating from the gas phase enters the liquid and reacts directly with phenol, without any intermediate reactions. Additionally, the atmospheric-pressure plasma source demonstrates great potential to be an effective source of O(aq) atoms without the requirement for any precursors in the liquid phase.
Introduction
Reactive oxygen species (ROS) such as ˙OH radical, superoxide (O2˙−), hydrogen peroxide (H2O2), ozone (O3), or singlet oxygen (1O2) play a crucial role in various areas such as regulation of redox-dependent cellular functions in biology, atmospheric chemistry, combustion, and radiation-induced chemistry in water. The importance of these species has led to detailed investigations of their reactivity and reaction mechanisms in the gas phase and especially in aqueous solutions. Surprisingly, very little is known about the reactivity of atomic oxygen, which can be illustrated by the number of records in the NDRL/NIST Solution Kinetics Database, which contains over 2500 records with reactions for ˙OH radicals and hundreds of records for other ROS, but only 8 records for O(aq).1–6This lack of data is due to several reasons; it is difficult to generate O atoms in water or aqueous solutions. For example, O(aq) atoms are believed not to be produced in electron-initiated processes in radiation chemistry,7 where the dominant channel of electron recombination with water clusters such as H3O+ or H+(H2O)2 are H atoms and water molecules. Still, they have been detected in water radiolysis studies,8,9 but without knowing their origin at that time. The triplet ˙OH + ˙OH recombination has been proposed recently10 as the possible source of reactions for O(aq) atoms, where high concentrations of ˙OH radicals are generated. Still it is not clear whether O(aq) can be generated by direct dissociation or in reactions involving water. For the quantitative studies of reactivity, O(aq) has been generated by laser flash photolysis of oxoanions (such as ClO−, ClO3−, or BrOn−n = 1–3),5 4-benzoylpyridine N-oxide (BPyO),11 or from irradiated dibenzothiophene-S-oxide (DBTO) derivatives.12 O(aq) was also suggested to be the precursor of the O2 generated in γ-ray irradiated FeSO4–CuSO4 solutions.13 O atoms cannot be easily generated through dissociation of water and mirrors the case for non-equilibrium (cold) atmospheric plasmas (briefly introduced later), where the humid gas mixture generates preferentially OH radicals and hydrogen peroxide and not O atoms.14,15 Only dry gas mixtures containing oxygen molecules (He/O2 or Ar/O2) leads to the production of O atoms and, subsequently, ozone.16–19 Thus a general method to generate atomic O(aq) without the involvement of chemical precursors would be of significant value.
Another reason for the lack of data for atomic O(aq) reactivity is probably due to the fact that it was believed to react rapidly with water to form two ˙OH radicals, even if this process is thermodynamically unfavorable. This reaction was also observed in classical force-field molecular dynamics (MD) simulations of O radicals in liquid water.20 Using more accurate densityfunctional based tightbinding (DFTB) force potential to examine the interactions of O atoms in bulk water, it was found that the O atoms form oxywater for singlet configuration or they remain stable in solution throughout the simulated time scale of 10 ps for triplet configuration.21 Car–Parrinello MD simulations have demonstrated that O(aq) is stable in liquid with an energy barrier of 10 kcal mole−1 and can even be generated in a triplet ˙OH + ˙OH recombination reaction.10 Other researchers have also suggested O atoms as reaction partners in aqueous solutions. Korang et al. have concluded that photo-induced strand cleavage of pUC19 plasmid DNA (monitored by gel electrophoresis) was at least partially caused by atomic O.12 The plasma generated O atoms and their subsequent reactions in the aqueous environment have been proposed by authors using non-equilibrium atmospheric plasma jets22,23 including our initial work with phenol as a chemical probe.16,24
Non-equilibrium (cold) atmospheric plasmas (CAPs) have attracted much attention from researchers in recent years because of their ability to generate high densities of reactive species at room temperature without the need for expensive vacuum systems.25 This reactivity is achieved by selective heating of electrons by electric fields, leaving the heavy particles (ions, neutral species) cold. The energetic electrons generate high densities of internally excited species (metastables) or radicals in inelastic electron-impact excitation and dissociation collisions, respectively. These reactive species can be used in many applications such as water treatment, generation of nanoparticles, material syntheses, or plasma medicine.26–35 The CAPs exist in many variants, with the most abundant being dielectric barrier discharges (DBD), DBD jets, radio-frequency operated jets or even microwave plasma sources.36–41 Most of these sources are operated in a noble gas with a small admixture of a molecular precursor, but some of them, especially the DBD's can be operated in ambient air without any need for a gas supply.
Herein we report on the fate of oxygen atoms after their generation using plasma in the gas phase and after their transfer into aqueous solution with phenol as chemical probe.42 The main question is whether O atoms can exist as a stable species in water and diffuse towards dissolved (organic or other) molecules to react directly, or whether the O atoms are reacting with water first:
| O(aq) + H2O(aq) → 2˙OH(aq) | (1) |
In the latter scenario the subsequent reactions, such as the hydroxylation of phenol, will involve the newly formed ˙OH radicals. Employing the 18O2 isotope, we can distinguish between these two cases. In the first case, stable O(aq) atoms will have a longer lifetime, will be able to diffuse into the solution and will be available to interact directly with phenol without any intermediate reactions. The subsequent oxidation reaction should result in the incorporation of predominantly 18O atoms when a He/18O2 plasma is used. The latter case would result in equal amounts of 16O and 18O among the newly formed hydroxylated products because half of the reacting ˙OH, in this case, would be derived from water molecules involved in the reaction (1) and therefore will contribute 16O atoms.
Results and discussion
The plasma source used in this work is an earlier version of the COST reference jet41 (see Fig. 1 and ESI† for more details). It is a capacitively coupled plasma source with two stainless steel electrodes at 1 mm distance in between two quartz glass plates. One of the two electrodes is powered by a 13.56 MHz sinusoidal voltage. As feed gas, helium with the flow of 1.4 standard liters per minute (slm) is used and admixed with 0.6% of molecular oxygen (16O2) or molecular oxygen isotope (18O2, 99 atomic percent 18O).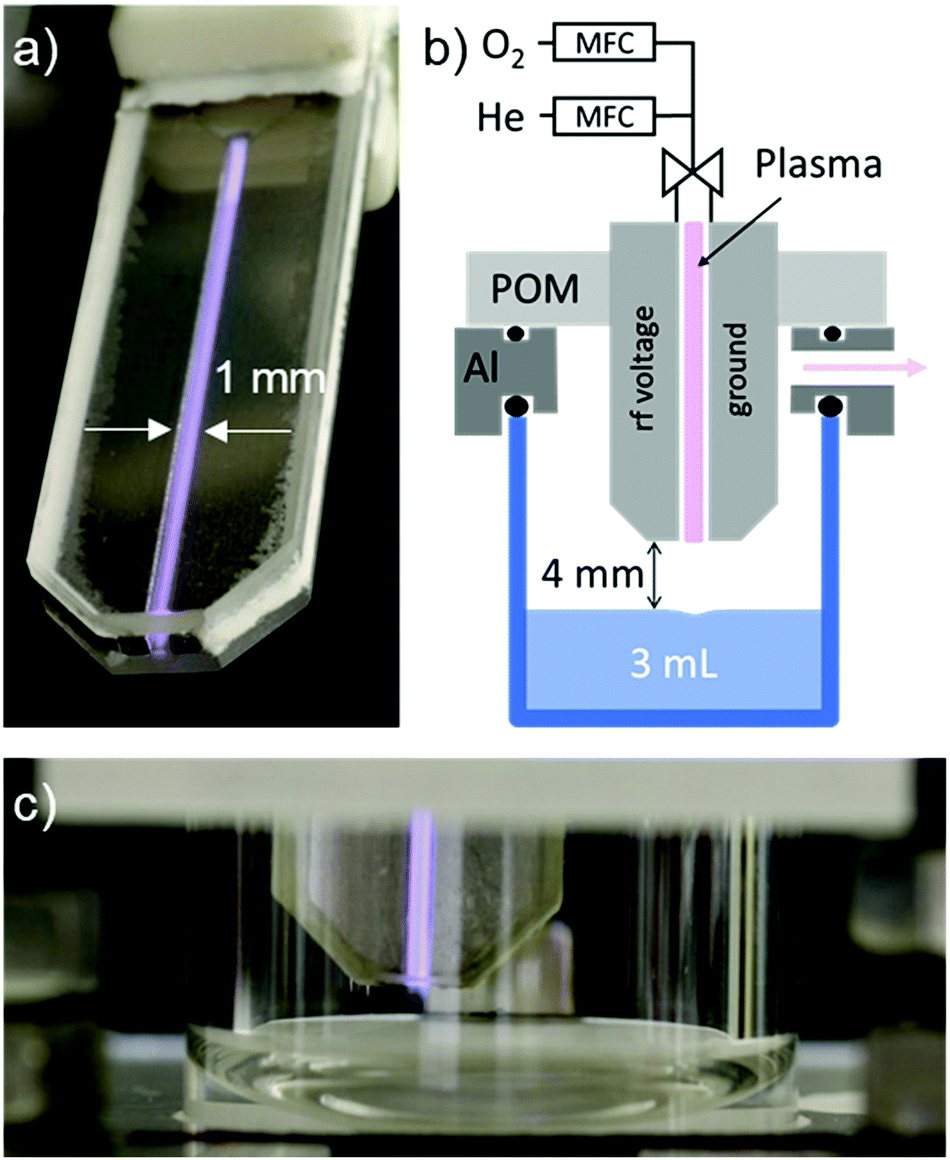 | ||
| Fig. 1 (a) Plasma jet with He gas in operation. (b) Scheme of the closed treatment chamber for plasma liquid treatment under atmospheric pressure. (c) Plasma jet in the chamber during solution treatment. | ||
The O2 admixture of 0.6% was previously shown to yield the maximum O density of 8 × 1014 cm−3 when measured 4 mm away from the jet nozzle by molecular beam mass spectrometry.43 Simulation of the gas flow, diffusion/convection transport of O atoms, and the gas phase recombination reactions have shown that more than 50% of O atoms leaving the plasma source reach the surface of the liquid and the integrated flux of O atoms to the liquid surface is around 9 × 1015 s−1.16 It should be noted that the generated plasma is strongly non-thermal. Only electrons are heated by electric fields, and the gas temperature stays below 40 °C.41
Plasma treatments of aqueous solutions were performed in a small glass chamber with a closed atmosphere, without contact to the ambient air. The shear forces between the He/O2 flow and liquid surface result in the movement of the liquid. The short-lived reactive species can react, therefore, with the whole volume of the liquid in a matter of minutes.16
Two different phenol concentrations, 0.5 mM and 5 mM, were treated for four and eight minutes, respectively, by the plasma feed gas containing 16O2 or labeled 18O2 (referred to unlabeled and labeled treatments respectively).
When 0.5 mM phenol solution is treated with the exhaust gas of the He/O2 plasma, it starts to degrade at a rate of around 5 × 1015 s−1 into the following reaction products: catechol (CC), resorcinol (RS), hydroquinone (HQ), and hydroxy-1,4-benzoquinone (HBQ).16 The maximum concentrations of these products are reached after four minutes of treatment. We have argued that the observed chemistry in the liquid phase is due to the plasma-generated oxygen atoms, which enter the liquid and react with phenol.
To distinguish among the oxidized products with different O isotopes, high-resolution mass spectrometry (MS) and gas chromatography-mass spectrometry (GC-MS) were employed to determine the composition of treated solutions. Details of the treatment procedure and both diagnostics are provided in ESI.†
The mass spectra of untreated samples and samples treated with He/16O2 plasma are shown in Fig. 2(a) and (b), respectively. The treatment results in appearance of new peaks. The molecular ion peak of m/z = 109.03 corresponds to CC, RS, or HQ (diols, all mass 110). Its relative abundance compared to the phenol peak is about 148%. An additional molecular ion peak at m/z = 123.01 corresponds to HBQ (mass 124 amu) with 45% abundance. The molecular ion peak of m/z = 125.02, which corresponds to phloroglucinol (C6H6O3, mass 126 amu), has about 71% abundance. Fig. 2(c) shows the mass spectrum of 0.5 mM phenol solution treated with He/18O2 plasma. Depending on whether one or two heavy O atoms were incorporated, the compounds are two or four mass units heavier than those treated with He/16O2 plasma in Fig. 2(b).
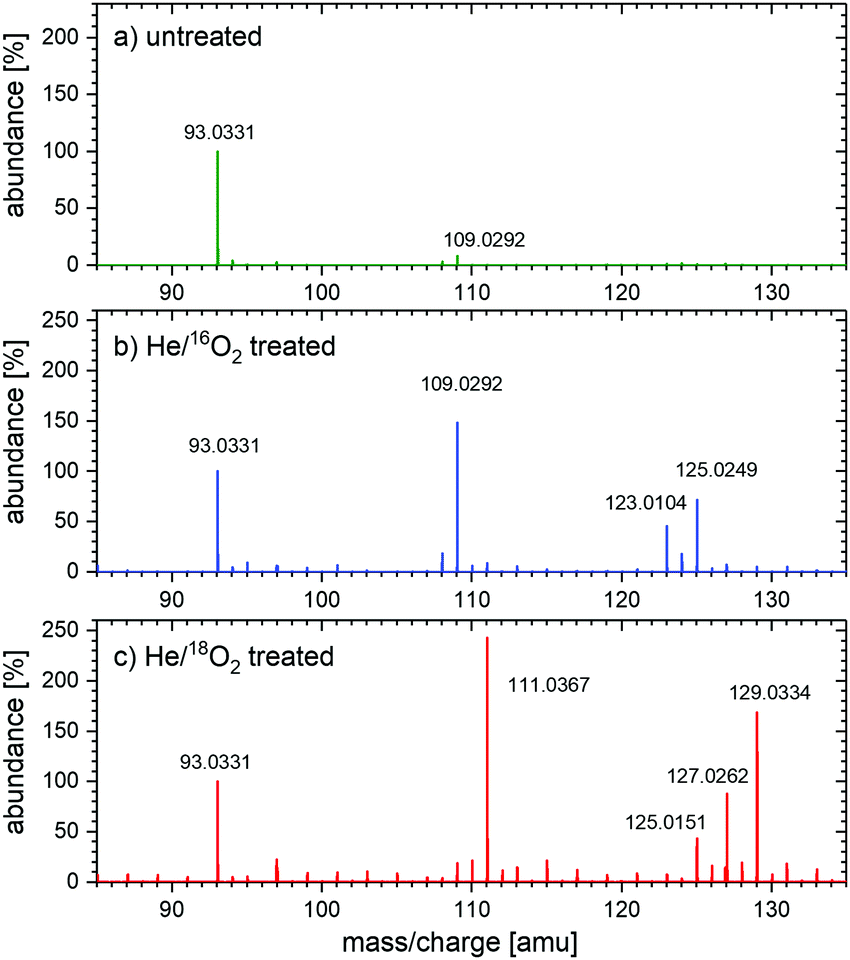 | ||
| Fig. 2 Abundance relative to the phenol peak as measured by mass spectrometry in negative ionization mode for (a) untreated sample of 0.5 mM phenol solution, (b) 0.5 mM phenol solution treated with He/16O2 plasma, and (c) 0.5 mM phenol solution treated with He/18O2 plasma. The spectra represents an average of two or three individual spectra recorded for the repetitions. | ||
The treatment of 0.5 mM phenol solutions with two different types of plasmas, He/16O2 plasma and He/18O2 plasma and study of the products of the treatment using MS led to the following observations (see also Table S2 in the ESI†):
(a) The ratio of labeled diols (HQ, CC, or RS, m/z = 111) to unlabeled diols (m/z = 109) changes from 0.005 for He/16O2 treatment to 11.2 for He/18O2. The former ratio is close to the natural concentration of the 18O isotope (0.002), and the latter one shows that 92% of diols are formed with 18O isotope in the He/18O2 treatment. Considering the fact that the purity of the oxygen isotope was around 99% and that any oxygen-containing impurity in the gas phase (O2, H2O vapor) will lead to the formation of the unlabeled O or OH groups and also unlabeled diols, we can conclude that O atoms are stable in the pure water and can be, therefore, directly involved in the formation of diols in the aqueous phenol solutions.
(b) The ratio HBQ labeled with two 18O atoms (m/z = 127) to normal HBQ (m/z = 123) is around 0.01 for He/16O2 treatment and increases to 10.86 for He/18O2 treatment. These ratios are very close to the ratios for diols and fully corroborate the conclusion about the stability of the O atoms in an aqueous environment.
(c) The ratio of triols labeled with two isotopes to normal triols changes from 0.01 to 3 when normal treatment changes to the treatment with 18O2. Again, a clear trend is observed. The ratio 3 is lower than in the case of diols or HBQ because m/z = 125 mass used to estimate the number of unlabeled triols can also contain HBQ molecules labeled just with one 18O (again due to finite 99% purity of the gas and due to impurities), reducing the ratio compared to the diol case. Considering this effect, the measurement of triols corroborates our conclusion as well.
These MS results provide clear evidence that atomic O can survive in the aqueous solution and directly react with the phenol, without reaction (1) being involved.
Ozone is another plasma chemistry product generated in large quantities in the gas phase by the used He/O2 plasma. However, as we have discussed previously,16 its contribution to the aqueous chemistry is negligible due to its very low Henry constant resulting in concentrations in the solution below 0.6 μM. The absence of ozone was corroborated in this work by the absence of cis,cis-muconic acid, a known product of the reaction of ozone with phenol, in the high precision liquid chromatography (HPLC) measurements (detection limit <0.25 μM).
The samples with 5.0 mM phenol concentration treated for 8 minutes were analyzed by GC-MS. The higher phenol concentration and longer treatment is necessary because the sensitivity of the GC-MS is lower than that of the MS. Fig. 3(a) shows the time-resolved signal intensities at m/z = 94, 110, and 112 amu of 16O2 plasma treated samples. Mass 110 represents diols and mass 112 the same species labeled with 18O in the OH group.
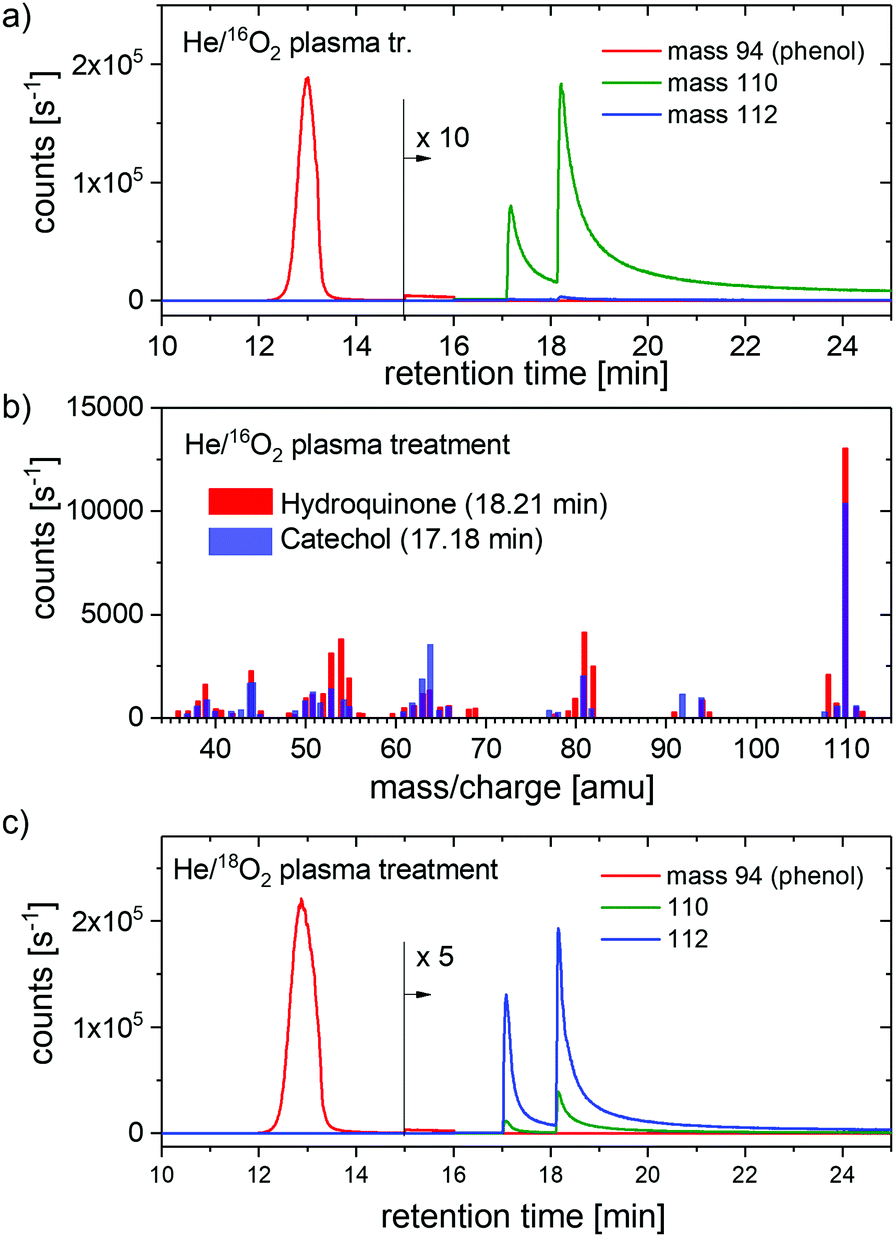 | ||
| Fig. 3 (a) The time-resolved signal intensity at m/z of 94, 110, and 112 amu of 5.0 mM phenol solution treated with He/16O2 plasma. (b) The mass spectra of CC and HQ as measured at indicated times. (c) The time-resolved signal intensity at m/z of 94, 110, and 112 of 5.0 mM phenol solution treated with He/18O2 plasma. | ||
Two signal maxima observed at 110 mass units have been identified as CC (at 17.2 min) and HQ (at 18.2 min). Fig. 3(b) shows the mass spectra of CC and HQ, respectively, as measured at the retention times mentioned above and with the most important peaks labeled. The signal intensity at mass 112 is negligibly small.
Fig. 3(c) shows the time-resolved signal intensities of the same masses measured for He/18O2 plasma treated samples. Additionally, mass 96 was recorded to ensure that there is no O exchange in the phenol molecule (not shown), and the mass m/z = 114 for double labeled diols have been checked as well for the same reason. Mass 112 is now a dominant mass of the products with much smaller signal intensity at mass 110. The signal intensity at masses 96 and 114 are negligibly small (not shown). Moreover, no other products could be observed in the analysis.
The treatment of 5 mM with two different types of plasmas, He/16O2 plasma and He/18O2 plasma and study of the products of the treatment using GC-MS leads to the following observations (see also Table S3 in the ESI†):
(a) The temporal resolution of the gas chromatography allows distinguishing among diol isomers CC, HQ, or RS, where only CC and HQ were detectable. It should be, however, noted that RS had been identified in our previous work as one of the products after treatment of 0.5 mM phenol solution. Its absence here is very probably due to higher phenol concentration (5 mM) and treatment time that have not been optimized to reach maximum concentration of the products. The RS concentration stays, therefore, very probably under the detection limit of this diagnostics.
(b) The ratio of labeled CC (mass 112) compared to unlabeled (mass 110) changes from 0.01 for normal treatment to 16.7 for treatment with He/18O2 plasma. Again, most of the CC (∼95%) is formed labeled in the latter treatment in agreement with MS measurements. The higher observed ratio here, compared to MS results, after the labeled treatment can be very probably attributed to the uncertainty in the subtraction of the signal component due to the ion fragment at the mass 110. Additionally, the small change of the impurity concentration in the feed gas can result in the observed higher ratio.
(c) The ratio of labeled HQ (mass 112) compared to unlabeled (mass 110) changes from 0.01 for normal treatment to 19.4 for treatment with He/18O2 plasma. Again, the most of HQ (96%) is formed labeled in the latter treatment, in agreement with other results. The uncertainties in the subtraction of the baseline and signal due to fragment ion and the possible change of the impurity level are probably responsible for the higher ratio of the labeled products.
The analysis of the aqueous phenol solutions by using two different diagnostics shows clearly that the mass spectra of the solution treated with He/18O2 plasma regarding phenol byproducts is dominant for products attached with 18O atoms. This result imply that the plasma-generated O atoms are transferred from the gas phase into the phenol solutions and react directly with phenol generating CC, RS & HQ products. These observations are fully consistent with our conclusion from the previous work, which has been drawn based on indirect evidence16 and agrees with theoretical predictions,10 where the authors concluded that the equilibrium clearly favors the left-hand side of reaction (1), implying a slow isotope exchange. We should note here that the hydrogen atom on the new OH group can either originate from the aromatic ring or from water and that our methods were not able to distinguish between these two pathways.
We expect that oxidizing reactions of O(aq) atoms are at least as fast as reactions of ˙OH radicals due to its increased reactivity as a diradical.44 For example, the reactions of ˙OH with phenol, CC, and HQ have reaction rate constants of 6.6 × 109 M−1 s−1, 1.1 × 1010 M−1 s−1, and 1.0 × 1010 M−1 s−1, respectively.
Assuming the same reaction rate constants for O(aq) atoms as for ˙OH radicals the reaction lifetime of O in phenol solution with 0.5 mM phenol concentration will be around 53 ns. This time is much shorter than the reaction lifetime due to recombination reaction:
| O(aq) + O2(aq) → O3(aq), | (2) |
Fig. 4 summarizes the reaction pathways of O atoms in the gas phase and the aqueous solution with phenol. Resorcinol RS is also shown here as it was identified as a product in our previous work.19
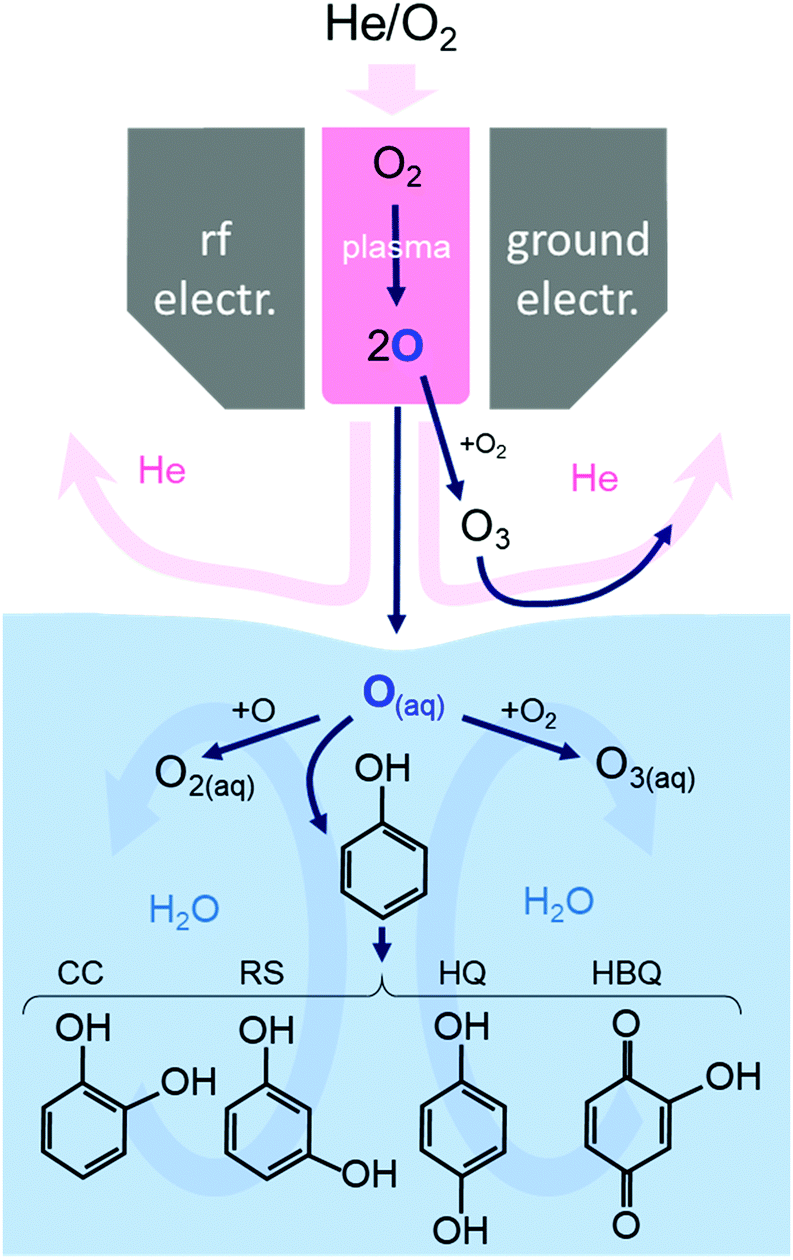 | ||
| Fig. 4 The reaction pathways of O atoms in the gas phase and the phenol solution. The solid arrows summarize the main reaction pathways, and the broad arrows in the background indicate the gas and liquid movement. | ||
Conclusions
In summary, we have employed non-equilibrium radio-frequency driven atmospheric-pressure plasma in He/0.6%O2 gas mixture to study the reaction mechanism of plasma-generated oxygen atoms in aqueous solutions. The gas mixture with O2 as the only reactive component, without humidity and influence of ambient atmosphere, leads preferentially to the production of O(gas) atoms in the plasma. The plasma-generated O(gas) atoms enter the aqueous solution and initiate there chemical reactions. The experiments with 18O2 isotope demonstrate that the O(aq) atoms react with phenol molecules directly, without dissociating water molecules in intermediate reactions. Additionally, the used atmospheric-pressure plasma source demonstrates its potential as an effective source of O(aq) atoms without the need for chemical precursors in the liquid phase or photolytic activation. Atmospheric plasma-based sources have, therefore, a very large potential for being used in future studies of O(aq) reactivity in solutions and in future applications based on reactions of O(aq) radicals for applications such as waste remediation or therapeutic treatments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. P. Kusalik and Dr P. Lukeš for fruitful discussions and the COST Action TD1208 for financial support; JB acknowledges funding from the German Research Foundation (BE 4349/5-1); JEB acknowledges funding from the German Research Foundation (BA 4193/7-1) and the German federal state of North-Rhine Westphalia for the financial support for the mass spectrometer; AHS, FI, BRB acknowledge the support of the Loughborough University Centre for doctoral training on Gas Plasma Interactions with Organic Liquids and Robert Bentham for his technical assistance with the GCMS.Notes and references
- W. G. Brown and E. J. Hart, J. Phys. Chem., 1978, 82, 2539 Search PubMed.
- E. J. Hart and W. G. Brown, J. Phys. Chem., 1980, 84, 2237 Search PubMed.
- E. J. Hart and W. G. Brown, Radiat. Phys. Chem., 1980, 15, 163 CrossRef CAS.
- M. C. Sauer, W. G. Brown and E. J. Hart, J. Phys. Chem., 1984, 88, 1398 CrossRef CAS.
- U. K. Kläning, K. Sehested and T. Wolff, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 2969 RSC.
- NDRL/NIST Solution Kinetics Database, NIST Standard Reference Database 40 (Web Version), A compilation of kinetics data on solution-phase reactions, 2002.
- B. C. Garrett, D. A. Dixon, D. M. Camaioni, D. M. Chipman, M. A. Johnson, C. D. Jonah, G. A. Kimmel, J. H. Miller, T. N. Rescigno, P. J. Rossky, S. S. Xantheas, S. D. Colson, A. H. Laufer, D. Ray, P. F. Barbara, D. M. Bartels, K. H. Becker, K. H. Bowen, S. E. Bradforth, I. Carmichael, J. V. Coe, L. R. Corrales, J. P. Cowin, M. Dupuis, K. B. Eisenthal, J. A. Franz, M. S. Gutowski, K. D. Jordan, B. D. Kay, J. A. Laverne, S. V. Lymar, T. E. Madey, C. W. McCurdy, D. Meisel, S. Mukamel, A. R. Nilsson, T. M. Orlando, N. G. Petrik, S. M. Pimblott, J. R. Rustad, G. K. Schenter, S. J. Singer, A. Tokmakoff, L.-S. Wang, C. Wettig and T. S. Zwier, Chem. Rev., 2005, 105, 355 CrossRef CAS PubMed.
- W. G. Brown and E. J. Hart, Radiat. Res., 1972, 51, 249 CrossRef CAS PubMed.
- W. G. Brown, E. J. Hart and M. C. Sauer, Radiat. Res., 1978, 76, 533 CrossRef CAS.
- E. Codorniu-Hernández, K. W. Hall, A. D. Boese, D. Ziemianowicz, S. Carpendale and P. G. Kusalik, J. Chem. Theory Comput., 2015, 11, 4740 CrossRef PubMed.
- J. M. Carraher and A. Bakac, Phys. Chem. Chem. Phys., 2014, 16, 19429 RSC.
- J. Korang, I. Emahi, W. R. Grither, S. M. Baumann, D. A. Baum and R. D. McCulla, RSC Adv., 2013, 3, 12390 RSC.
- E. Bjergbakke and E. J. Hart, Radiat. Res., 1971, 45, 261 CrossRef CAS PubMed.
- C. A. Vasko, D. X. Liu, E. M. van Veldhuizen, F. Iza and P. J. Bruggeman, Plasma Chem. Plasma Process., 2014, 34, 1081 CrossRef CAS.
- G. Willems, J. Benedikt and A. v. Keudell, J. Phys. D: Appl. Phys., 2017, 50, 335204 CrossRef.
- M. M. Hefny, C. Pattyn, P. Lukes and J. Benedikt, J. Phys. D: Appl. Phys., 2016, 49, 404002 CrossRef.
- D. Ellerweg, A. von Keudell and J. Benedikt, Plasma Sources Sci. Technol., 2012, 21, 34019 CrossRef.
- S. Zhang, W. van Gaens, B. van Gessel, S. Hofmann, E. van Veldhuizen, A. Bogaerts and P. Bruggeman, J. Phys. D: Appl. Phys., 2013, 46, 205202 CrossRef.
- S. Reuter, J. Winter, A. Schmidt-Bleker, D. Schroeder, H. Lange, N. Knake, V. Schulz-von der Gathen and K.-D. Weltmann, Plasma Sources Sci. Technol., 2012, 21, 24005 CrossRef.
- M. Yusupov, E. C. Neyts, P. Simon, G. Berdiyorov, R. Snoeckx, A. C. T. van Duin and A. Bogaerts, J. Phys. D: Appl. Phys., 2013, 47, 25205 CrossRef.
- C. C. W. Verlackt, E. C. Neyts and A. Bogaerts, J. Phys. D: Appl. Phys., 2017, 50, 11LT01 CrossRef.
- Y. Gorbanev, D. O'Connell and V. Chechik, Chemistry, 2016, 22, 3496 CrossRef CAS PubMed.
- K. Wende, P. Williams, J. Dalluge, W. van Gaens, H. Aboubakr, J. Bischof, T. von Woedtke, S. M. Goyal, K.-D. Weltmann, A. Bogaerts, K. Masur and P. J. Bruggeman, Biointerphases, 2015, 10, 29518 CrossRef PubMed.
- S. Bekeschus, K. Wende, M. M. Hefny, K. Rödder, H. Jablonowski, A. Schmidt, T. von Woedtke, K.-D. Weltmann and J. Benedikt, Sci. Rep., 2017, 7, 2791 CrossRef PubMed.
- T. Yokoyama, M. Kogoma, S. Kanazawa, T. Moriwaki and S. Okazaki, J. Phys. D: Appl. Phys., 1990, 23, 374 CrossRef CAS.
- K. Oehmigen, M. Hähnel, R. Brandenburg, C. Wilke, K.-D. Weltmann and T. von Woedtke, Plasma Processes Polym., 2010, 7, 250 CrossRef CAS.
- A. M. Hirst, M. S. Simms, V. M. Mann, N. J. Maitland, D. O'Connell and F. M. Frame, Br. J. Cancer, 2015, 112, 1536 CrossRef CAS PubMed.
- V. Arora, V. Nikhil, N. K. Suri and P. Arora, Dentistry, 2014, 4, 189 Search PubMed.
- J. Benedikt, A. Hecimovic, D. Ellerweg and A. von Keudell, J. Phys. D: Appl. Phys., 2012, 45, 403001 CrossRef.
- H. A. Aboubakr, P. Williams, U. Gangal, M. M. Youssef, S. A. A. El-Sohaimy, P. J. Bruggeman and S. M. Goyal, Appl. Environ. Microbiol., 2015, 81, 3612 CrossRef CAS PubMed.
- J. Pawłat, Eur. Phys. J.: Appl. Phys., 2013, 61, 24323 CrossRef.
- J. Benedikt, V. Raballand, A. Yanguas-Gil, K. Focke and A. von Keudell, Plasma Phys. Controlled Fusion, 2007, 49, B419 CrossRef CAS.
- J. Patel, L. Nemcova, P. Maguire, W. G. Graham and D. Mariotti, Nanotechnology, 2013, 24, 245604 CrossRef CAS PubMed.
- G. Fridman, G. Friedman, A. Gutsol, A. B. Shekhter, V. N. Vasilets and A. Fridman, Plasma Processes Polym., 2008, 5, 503 CrossRef CAS.
- E. Edengeiser, J.-W. Lackmann, E. Bründermann, S. Schneider, J. Benedikt, J. E. Bandow and M. Havenith, J. Biophotonics, 2015, 8, 918 CrossRef CAS PubMed.
- L. Schaper, S. Reuter, J. Waskoenig, K. Niemi, V. S.-v. d. Gathen and T. Gans, J. Phys.: Conf. Ser., 2008, 162, 012013 CrossRef.
- X. Li, Y. Li, P. Zhang, P. Jia and L. Dong, Sci. Rep., 2016, 6, 35653 CrossRef CAS PubMed.
- R. Brandenburg, Plasma Sources Sci. Technol., 2017, 26, 53001 CrossRef.
- A. A. Fridman, Plasma medicine, John Wiley et Sons, Chichester, West Sussex, UK, 2013 Search PubMed.
- L. Bárdos and H. Baránková, Thin Solid Films, 2010, 518, 6705 CrossRef.
- J. Golda, J. Held, B. Redeker, M. Konkowski, P. Beijer, A. Sobota, G. Kroesen, N. S. J. Braithwaite, S. Reuter, M. M. Turner, T. Gans, D. O’Connell and V. Schulz-von der Gathen, J. Phys. D: Appl. Phys., 2016, 49, 84003 CrossRef.
- P. Lukes, E. Dolezalova, I. Sisrova and M. Clupek, Plasma Sources Sci. Technol., 2014, 23, 15019 CrossRef.
- D. Ellerweg, J. Benedikt, A. von Keudell, N. Knake and V. Schulz-von der Gathen, New J. Phys., 2010, 12, 13021 CrossRef.
- E. Codorniu-Hernández, K. W. Hall, D. Ziemianowicz, S. Carpendale and P. G. Kusalik, Phys. Chem. Chem. Phys., 2014, 16, 26094 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp00197a |
| This journal is © the Owner Societies 2018 |