
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of a silver salt on the nanostructure of a Au(111)/ionic liquid interface: an atomic force microscopy study and theoretical concepts†
Viktor
Hoffmann
a,
Giridhar
Pulletikurthi
a,
Timo
Carstens
a,
Abhishek
Lahiri
a,
Andriy
Borodin
a,
Max
Schammer
 bcd,
Birger
Horstmann
bc,
Arnulf
Latz
bcd and
Frank
Endres
*a
bcd,
Birger
Horstmann
bc,
Arnulf
Latz
bcd and
Frank
Endres
*a
aInstitute of Electrochemistry, Clausthal University of Technology, Arnold-Sommerfeld-Strasse 6, 38678 Clausthal-Zellerfeld, Germany. E-mail: frank.endres@tu-clausthal.de
bHelmholtz Institute Ulm, Helmholtzstraße 11, 89081 Ulm, Germany
cGerman Aerospace Center, Pfaffenwaldring 38-40, 70569 Stuttgart, Germany
dUniversität Ulm, Albert-Einstein-Alle 47, 89081 Ulm, Germany
First published on 16th January 2018
Abstract
Ionic liquids (ILs) form a multilayered structure at the solid/electrolyte interface, and the addition of solutes can alter it. For this purpose, we have investigated the influence of the silver bis(trifluoromethylsulfonyl)amide (AgTFSA) concentration in 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)amide ([Py1,4]TFSA) on the layering using in situ atomic force microscopy. AFM investigations revealed that the Au(111)/electrolyte interface indeed depends on the concentration of the salt where a typical “ IL” multilayered structure is retained only at quite low concentrations of the silver salt (e.g. ≤200 μM). However, at 200 μM AgTFSA/[Py1,4]TFSA and above this “IL” multilayered structure is disturbed/varied. A simple double layer structure was observed at 500 μM AgTFSA in [Py1,4]TFSA. Furthermore, the widths of the innermost layers have been found to be dependent on the concentration and on the applied electrode potentials. Our AFM results show that the concentration of solutes strongly influences the structure of the electrode/electrolyte interface and can provide new insights into the electrical double layer structure of the electrode/ionic liquid interface. We also introduce a semi-continuum theory to discuss the double layer structure.
1 Introduction
The electrodeposition of silver on Au(111) is a well-investigated example, both from surface science and from the electrochemical perspective due to quite similar lattice constants and the higher surface energy of gold.1,2 Furthermore, alloying,1,3 surface reconstruction,4,5 surface crystallinity,6 and defects1 can play a crucial role in the initial stages of metal deposition. Moreover, the neat surfaces of gold minimize their surface energy by reconstruction and an electrode potential dependent lifting of Au has been reported.7,8 Specific adsorption of anions can also promote such a lifting process2 and the strong influence of anions on the overlayer structure was observed using atomic force microscopy.2Ionic liquids are interesting solvents for the electrodeposition of reactive metals such as Al and Si, which are not accessible in aqueous media.9,10 These liquids have wide electrochemical and thermal windows, good ionic conductivities, usually low vapor pressures at room temperature and high solubility for a variety of compounds.11,12 However, the practical use of these liquids in applied electrochemistry is hindered, as there is still no thorough understanding of the species in the bulk phase and, especially, at the interface.
Several ILs have been employed as electrolytes to deposit silver, such as tetrafluoroborate,13,14 hexafluorophosphate,15,16 bis(trifluoromethylsulfonyl)amide,13,17 dicyanamide,18 trifluoromethylsulfonate,19,20 acetate,21 and nitrate.21 In most of these studies the focus was on the electrodeposition and the characterization of the deposited films.
In recent years, atomic force microscopy has become a useful tool to explore/probe the electrode/electrolyte interface (EEI) with atomic resolution.22–34 AFM force–distance curves can provide direct insight into the adsorption structure of ions/species in the double layer (or multilayer) on the electrode/IL interface.22
In contrast to aqueous solutions, there are often no simple double layers in ILs, instead the ions adopt a multilayer structure.28 In brief, the interfacial structure can be divided into the innermost layer, the transition zone, and the bulk liquid phase.29,30 The innermost layer is composed of the IL layer adsorbed next to the electrode surface. These layers experience large forces when the AFM tip pushes through them. At the open circuit potential (OCP) and at more negative electrode potentials the innermost layer is usually enriched with cations. At more positive potentials anions are usually dominant at the interface. The forces for penetrating these layers have been shown to be more than one order of magnitude higher than the ones for molecular liquids.25,29,31,35–38 Interfacial properties, including the adsorption/co-adsorption of ions at the interface, are determined by the nature of the electrode and the electrolyte. Even a small variation in the electrolyte composition, e.g. due to impurities39 or dissolved solutes,25,40 can disturb the multilayer arrangement and can also influence electrochemical reactions. The Au(111)/[Py1,4]TFSA interface is rather well described in the literature.35,41 Furthermore, the solvation behaviour of AgTFSA in [EMIm]TFSA has been investigated by spectroscopy and DFT calculations, giving a stable complex like [Ag(TFSA)3]2−.42 We have reported that water has an influence on the Au(111)/ionic liquid interface.43 In the case of [EMIm]TfO, a multilayer structure was predominant up to 30 vol% water and above this concentration the multilayer structure was strongly disturbed. In this context the question came up as to what extent the IL layers are influenced by a solute during electrodeposition. For this purpose AgTFSA, from which Ag is easily deposited, has been taken as an example to study the influence of salt concentration on the electrode/ionic liquid interface. In the present paper we report for the first time on the influence of silver salt concentration on the electrode/electrolyte interface using atomic force microscopy.
The experimental work is accompanied by theoretical considerations. Continuum theories allow the dynamic description of macroscopic systems, e.g., electrochemical cells. The nano-sized surface layers, however, are strongly affected by the particle nature of its constituents. The popular modeling approaches on this scale are density functional theory (DFT)44–47 and molecular dynamics (MD).48 DFT and MD simulations resolve many of the microscopic complexities of individual molecules and intermolecular interactions, but remain limited to small length and time scales. Bazant et al. have proposed a phenomenological continuum theory which captures the multilayer structure of the electrochemical double layer in ionic liquids.28
In this paper, we present a novel semi-continuum model which describes ionic liquids as a continuum fluid of hard spheres. It represents a unified theoretical framework for the description of both scales, bulk phase and surface layers. Our approach is derived from fundamental thermodynamical principles and basic physical assumptions, in contrast to phenomenological continuum theory,28,49,50 Thus, we improve the assignment between material properties and the simulated phenomena. Most importantly, we find that incompressibility leads to crowding and the particle nature of fluids leads to overscreening.
Our consistent continuum model describes both the short-range and long-range behavior of ionic liquids as well as both static and dynamic effects. This generality complements previous modeling efforts. On the one hand, DFT and MD describe electrolytes at smaller length and time scales. Thus, they resolve more detailed ionic properties than our model, e.g., molecular orientation, position, and shape, and make quantitative predictions. The fine resolution, however, comes at the cost of computational complexity. DFT and MD are limited to the nano-scale dimensions of the electrochemical double layer (EDL), whereas our model can calculate properties of ionic liquids from the nano-scale EDL to the micro-scale electrode structure. Most importantly, the simplicity and abstraction of our continuum model brings out the fundamental principles behind the interfacial behavior of ionic liquids clearer than do DFT or MD. Furthermore, our model is time dependent and able to predict the contribution of transport through the EDL structure to the apparent reaction kinetics of ionic liquids.
On the other hand, the phenomenological continuum theory of Bazant et al. is numerically simpler than our model, but remains restricted to binary ionic liquids and does not clearly distinguish between different electrolytes. Our model, instead, applies to more complex electrolytes and clearly connects with material properties because we derive it from non-equilibrium thermodynamics and make explicit the hardcore nature of the molecules. In this paper, for example, we can evaluate the effect of water molecules or silver ions onto the EDL structure of ionic liquids and study the effect of molecular size on electrolyte properties.
2 Methods
2.1 Experimental methods
1-Butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)amide ([Py1,4]TFSA) was purchased from IOLITEC, Germany, in a purity of 99.5%. Silver bis(trifluoromethylsulfonyl)amide (99.5%) was procured from Solvionics, France. The ionic liquid was dried at 100 °C under vacuum for 3 days to water values below 10 ppm. Several concentrations of AgTFSA in [Py1,4]TFSA ranging from 50 μM to 1000 μM were prepared.The electrochemical measurements were carried out using a PARSTAT 2263 potentiostat/galvanostat controlled by PowerCV and PowerStep software. A three electrode cell was used for all electrochemical experiments. Gold on glass was used as the working electrodes (WE) for cyclic voltammetry experiments. The working electrode for the AFM studies was Au(111) on mica purchased from Agilent Technologies. The electrochemical cell was made of Teflon and clamped over a Teflon-covered Viton O-ring onto the working electrode with a geometric surface area of 0.3 cm2. Pt wires were used as the counter and quasi reference electrodes, respectively. The deflection–distance curves were obtained using a Molecular Imaging Pico Plus AFM in contact mode inside an argon-filled glove-box at room temperature and transformed to force–distance curves. A silicon SPM-sensor with a spring constant of 6 N m−1 from Nano World was employed for all AFM measurements.
2.2 Mathematical model
Our mathematical model of ionic liquids is based on the methodology of rational thermodynamics and describes a thermodynamically consistent transport theory. The modeling methodology has been applied to electrolytes with a dominant neutral solvent before, as outlined by Latz and Zausch.51 However, ionic liquids possess unique physical properties which must be captured by fundamental principles and which differ from those of uncharged solvent electrolytes. Therefore, transport theories for such media are not applicable to ionic liquids. This motivates the formulation of a novel transport theory for ionic liquids.Furthermore, our definition of incompressibility is based on a volumetric constraint52,53 and differs from the one used in ref. 51. Here we show how to supplement the continuum theory with non-local interactions, modeling the particle nature of the constituents. Therefore, our approach can be used to examine both the bulk behavior, and the quasi-crystalline double-layer behavior of ionic liquids.
We divide the free energy
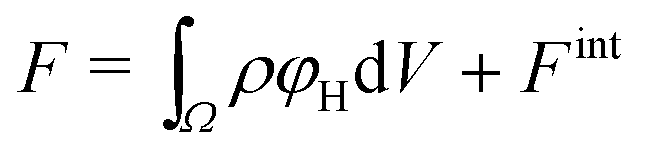 | (1) |
The electrolyte equation of state is formulated as a constraint on the concentrations cα and the partial molar volumes να, such that 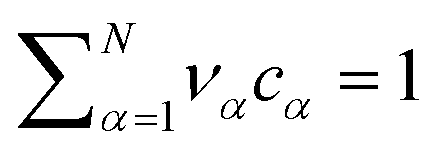 .52–54 If the partial molar volumes do not depend on pressure, this describes incompressibility. As consequence, we derive the convective equation of state
.52–54 If the partial molar volumes do not depend on pressure, this describes incompressibility. As consequence, we derive the convective equation of state 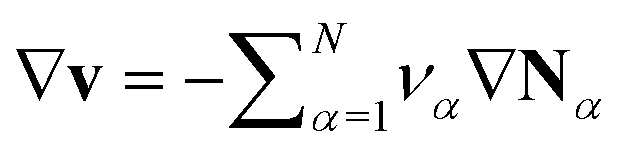 , where v is the convective bulk velocity and the ionic fluxes Nα measure transport in the center-of-mass frame.
, where v is the convective bulk velocity and the ionic fluxes Nα measure transport in the center-of-mass frame.
The single-particle free energy density is chosen as
 | (2) |
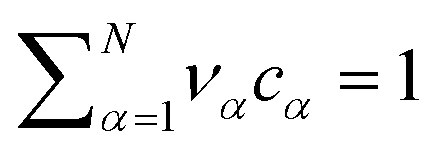 because we let the bulk modulus diverge
because we let the bulk modulus diverge ![[scr K, script letter K]](https://www.rsc.org/images/entities/char_e148.gif) →∞. The elastic contribution determines the force density via the Navier–Stokes equation
→∞. The elastic contribution determines the force density via the Navier–Stokes equation | (3) |
![[v with combining dot above]](https://www.rsc.org/images/entities/b_char_0076_0307.gif) = 0 in the highly viscous ionic liquids. Thus, we can express elastic forces in terms of electrostatic forces in our transport equations below. The remnant of the single-particle free energy density is analogous to the one used in ref. 51.
= 0 in the highly viscous ionic liquids. Thus, we can express elastic forces in terms of electrostatic forces in our transport equations below. The remnant of the single-particle free energy density is analogous to the one used in ref. 51.
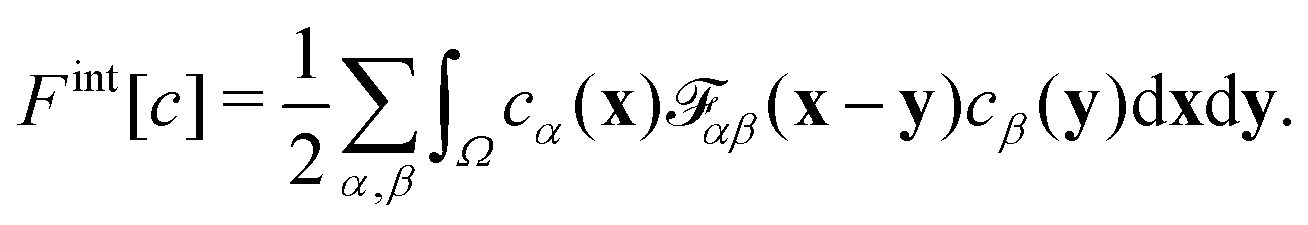 | (4) |
![[scr F, script letter F]](https://www.rsc.org/images/entities/char_e141.gif) αβ(x − y) models the impenetrable hard spheres, i.e., the repulsive interaction is non-zero in a small volume Ω determined by the particle-radii. The mathematical form of αβ is given in the ESI.† The modification of the chemical potentials due to this inter-particle repulsion is derived with functional derivatives
αβ(x − y) models the impenetrable hard spheres, i.e., the repulsive interaction is non-zero in a small volume Ω determined by the particle-radii. The mathematical form of αβ is given in the ESI.† The modification of the chemical potentials due to this inter-particle repulsion is derived with functional derivatives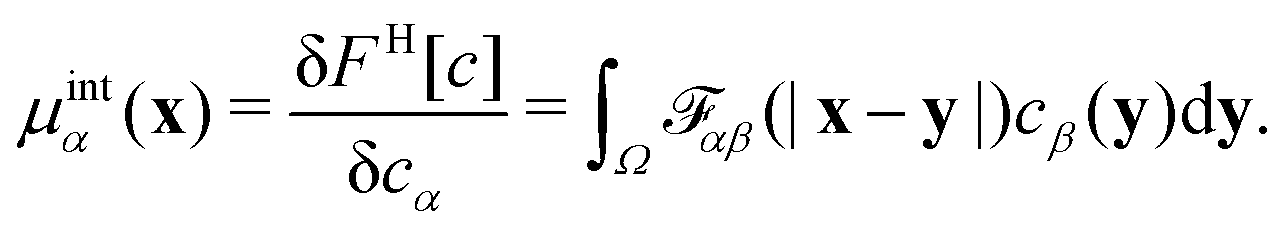 | (5) |
| ∂tρF = −∇J − ∇(ρFv), | (6) |
| ∂tc3 = −∇N3 − ∇(c3v), | (7) |
| ρF = −εRεΔΦ, | (8) |
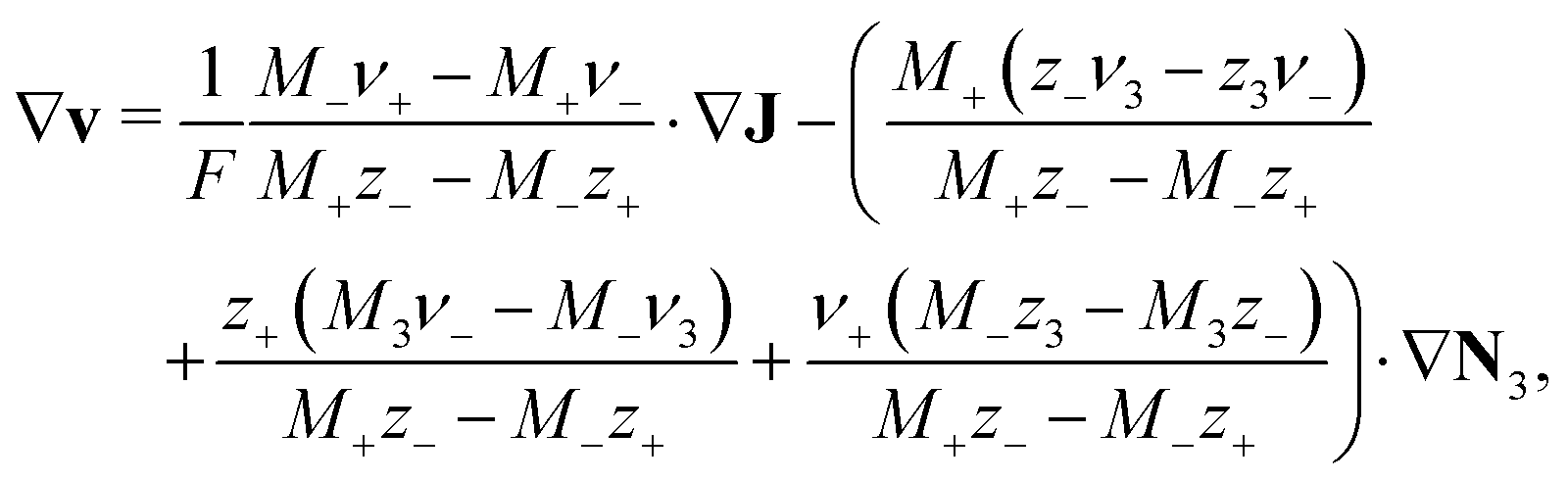 | (9) |
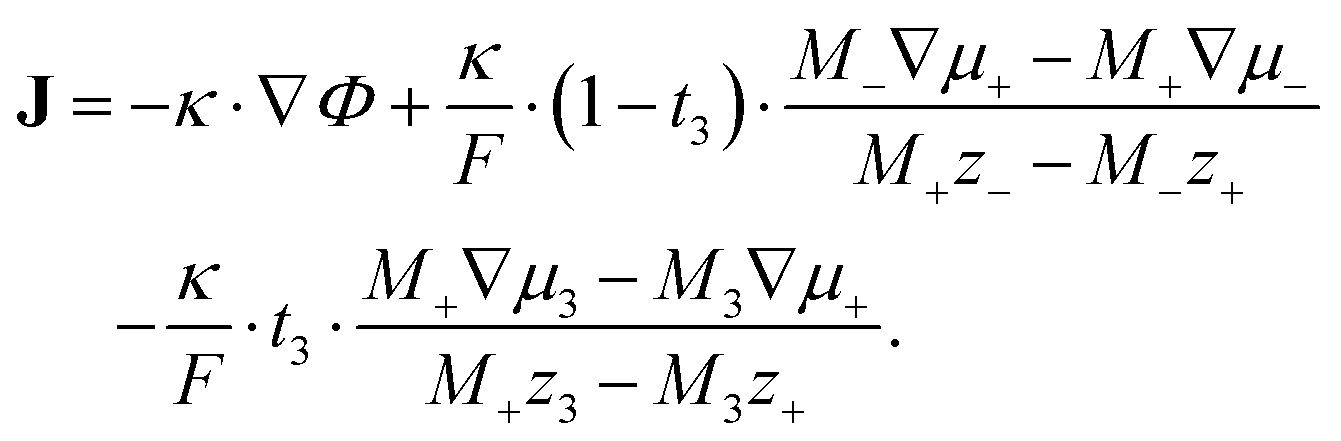 | (10) |
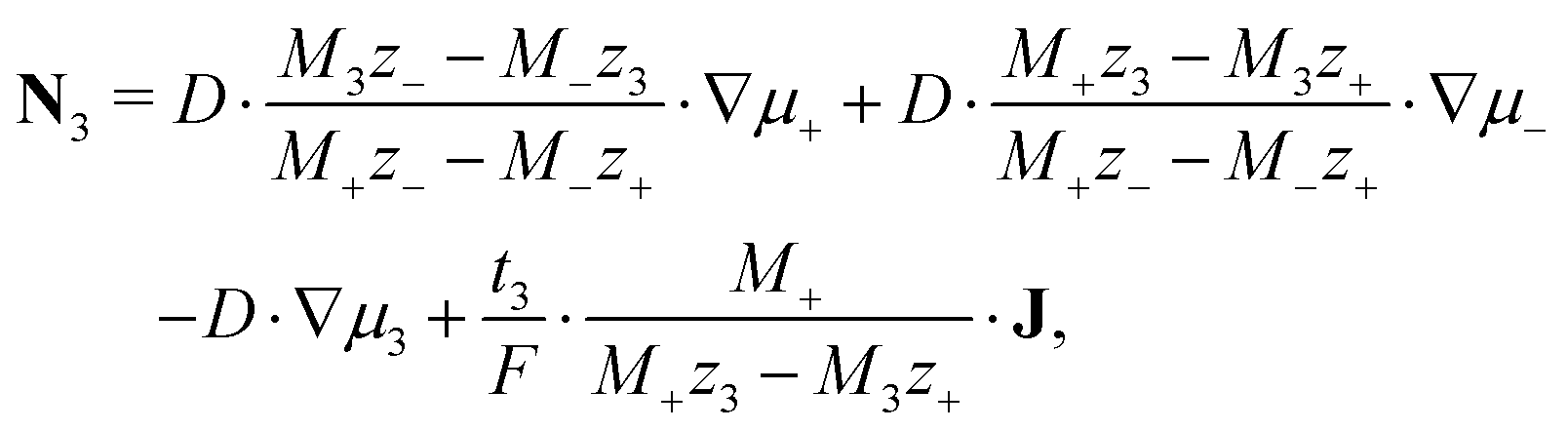 | (11) |
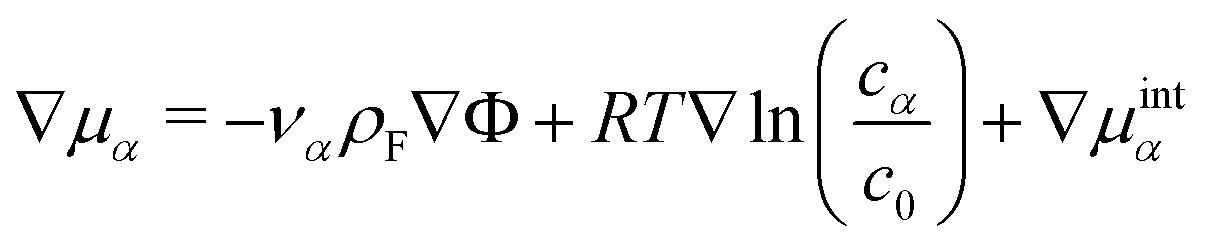 | (12) |
 | (13) |
The formalism outlined above can be generalized to electrolytes with a neutral solvent and an arbitrary number of solutes. In future work, we will extend the transport theory in this regard and include viscous media and heat transport. It is the strength of our consistent approach that the contributions of the solvent are not hidden as in standard concentrated solution theory.57
3 Results and discussions – electrochemical and interfacial investigations of the electrolytes
3.1 Experimental results
Cyclic voltammetry has been employed to investigate the electrochemical behaviour of the electrolytes at varying concentrations of AgTFSA in [Py1,4]TFSA on gold at RT. Multiple-cycles have been recorded for AgTFSA/[Py1,4]TFSA electrolytes to investigate changes in the interfacial processes/redox processes. All cyclic voltammograms have been recorded first in the negative direction from the Open Circuit Potential (OCP) to the chosen switching potentials at a scan rate of 10 mV s−1. For the neat liquid, a rise in reduction current has been observed at ∼−0.7 V, which can be due to the adsorption of IL on Au (Fig. 1a).35 Quite a weak oxidation peak has been observed at −0.9 V in the backward scan of the CV. Upon addition of 50 μM AgTFSA to [Py1,4]TFSA, three reduction processes have been noticed in the forward scan of the CV (Fig. 1b). The first two reduction processes C1 and C2 could be due to the adsorption processes of the electrolyte species and to the underpotential deposition of Ag. The reduction process C3 can be clearly correlated with the bulk deposition of silver.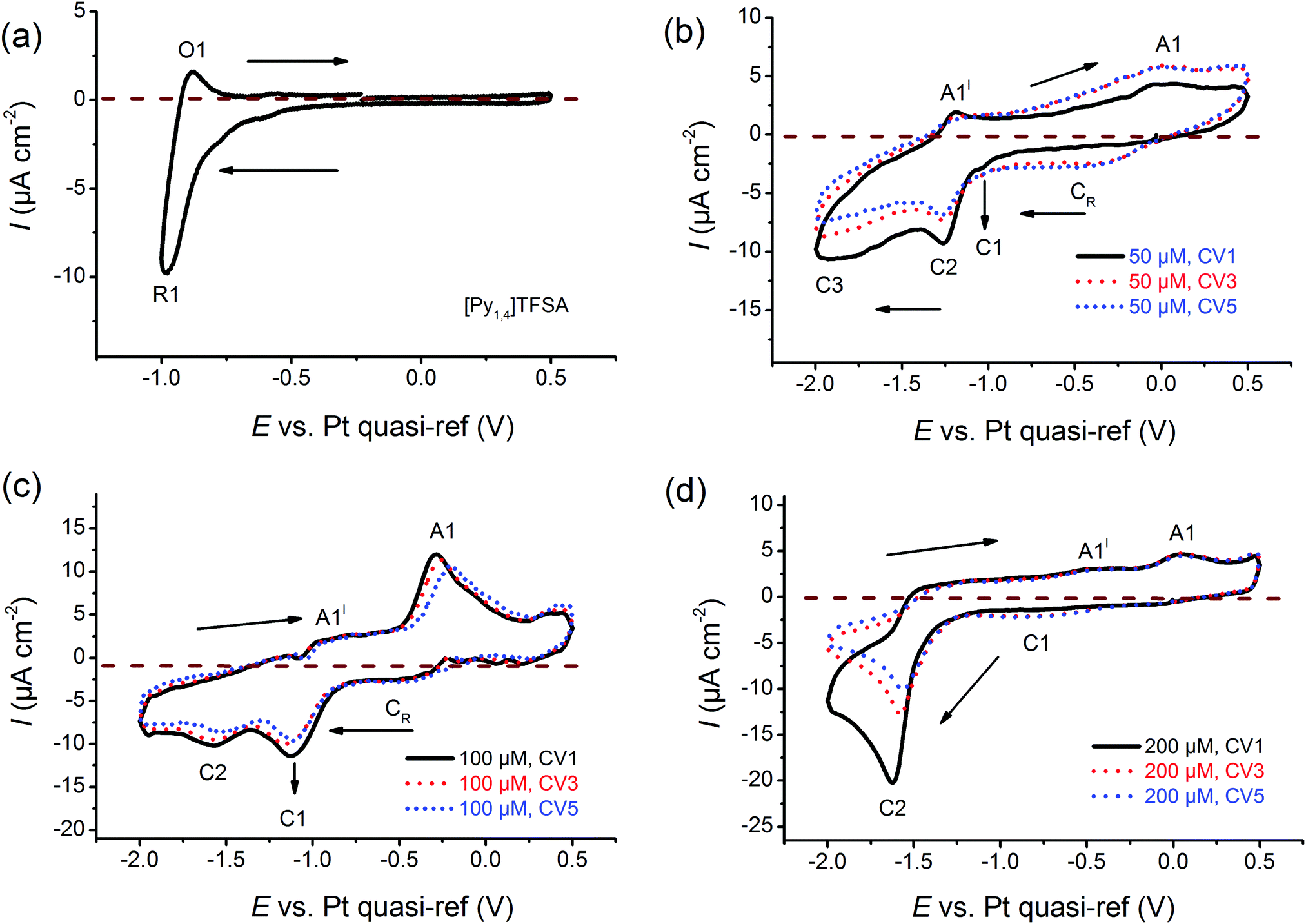 | ||
| Fig. 1 Comparison of cyclic voltammograms of (a) neat [Py1,4]TFSA, (b) 50 μM AgTFSA/[Py1,4]TFSA, (c) 100 μM AgTFSA[Py1,4]TFSA, and (d) 200 μM AgTFSA/[Py1,4]TFSA. Scan rate = 10 mV s−1. | ||
For 50 μM AgTFSA/[Py1,4]TFSA, two oxidation processes have been observed in the backward scan of the CV (Fig. 1c). The oxidation process A1′ is related to the reduction process C3 and the oxidation process A1 is associated with the reduction process C2. A broad reduction (CR) wave has been noticed after the first scan, which can be related to the surface reduction process due to a change in the electrode surface. The third and fifth CVs also exhibit similar features to that of the first scan except for a broad reduction process CR. For the CV of 100 μM AgTFSA/[Py1,4]TFSA, a reduction wave is seen at C1 followed by a reduction step at C2. Here, the reduction step C1 could be due to an adsorption of the electrolyte, and C2 is due to the deposition of silver, respectively. The processes A1′ and A1 are correlated with the desorption of adsorbed layers of the electrolyte from the electrode surface and with the dissolution of the deposited silver films, respectively.
The CV of 200 μM AgTFSA/[Py1,4]TFSA (Fig. 1d) consists of a clear reduction peak at C2 along with a broad reduction wave at C1 in the forward scan and three oxidation steps in the backward scan. The reduction process C2 with a peak at −1.6 V is due to the bulk deposition of Ag and the process C1 is likely to be correlated with the underpotential deposition of Ag. The oxidation process A1 can be related to the reduction process C2. Moreover, it has been observed that silver nanoparticles can form chemically under light irradiance. The third and fifth CVs exhibit similar features to those of the first scan. Furthermore, the adsorption process that was observed in the neat IL and at low concentrations of AgTFSA/[Py1,4]TFSA (50 and 100 μM) has not been observed for 200 μM AgTFSA/[Py1,4]TFSA, obviously an effect of the increase in concentration of AgTFSA. In the case of the zinc ions in ILs, it has been proposed that such interfacial processes can be varied by the arrangement of IL anions and cations adjacent to the electrode in the compact layer (IHP, Inner Helmholtz Plane), which can dictate the kinetics of the redox processes.58 Based on these electrochemical results the question arises as to how the interfacial processes of ILs vary in the presence of a solute and how are they dependent on its concentration.
In order to describe the electrode/electrolyte interface (or the interfacial structure) of ILs and solutions with a varying concentration of silver salts, we have performed in situ Atomic Force Microscopy (AFM) investigations. To the best of the authors' knowledge, such measurements, where a systematic variation of salt concentration has been performed, have not yet been reported so far.
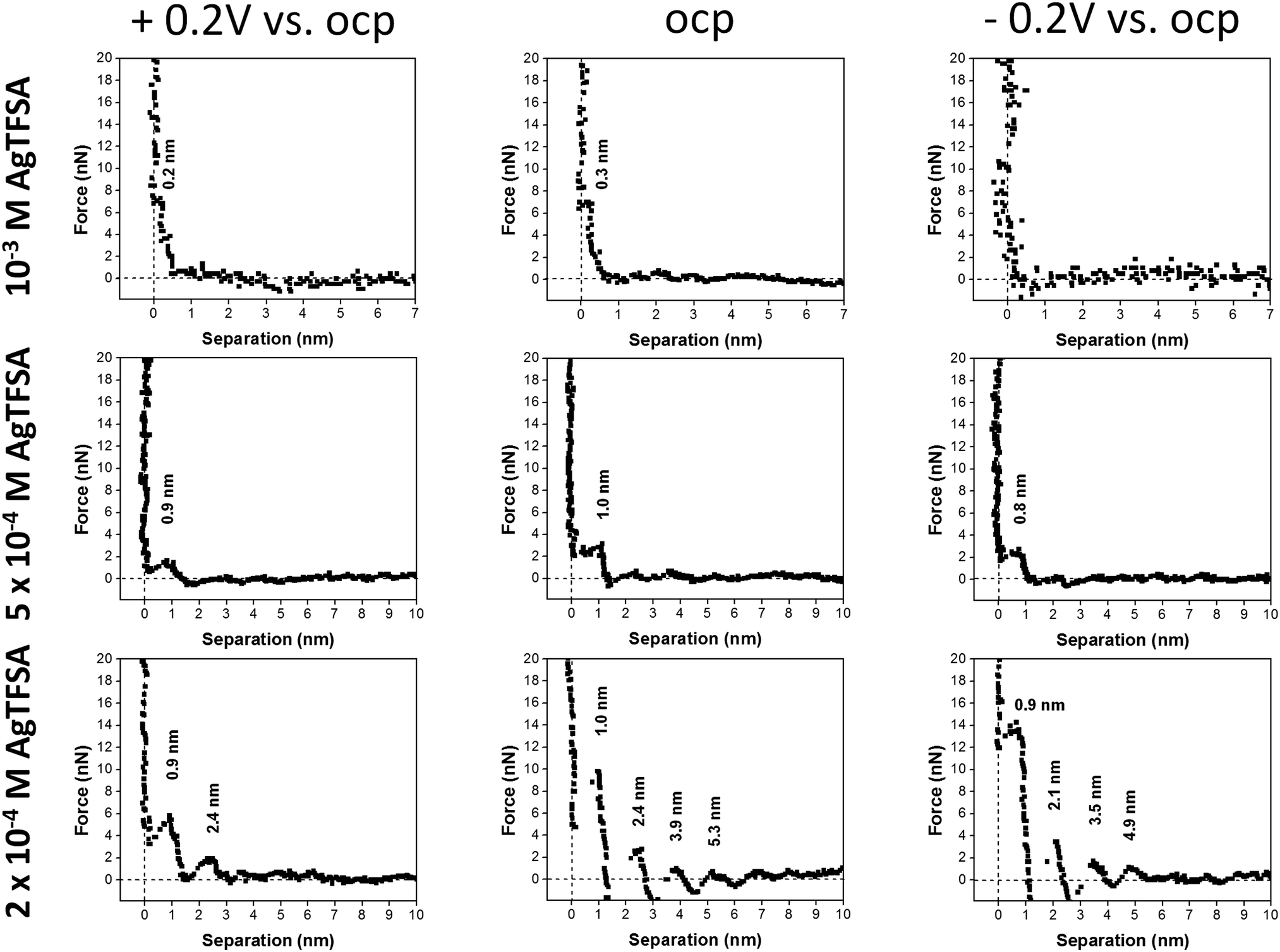
Fig. 2 shows the force vs. separation curves for an AFM tip approaching the Au(111) surface in the neat IL and in mixtures with two different concentrations of AgTFSA and at three different electrode potentials (OCP, +0.2 V and −0.2 V). The appearance of the AFM force curves differ with varying concentrations of AgTFSA (addition of solute) from the force curves of the Au(111)/IL interface.25,40,41 For the neat IL, the interfacial structure consists of at least four discrete steps at the measured electrode potentials, which is in agreement with the literature. This behavior reveals a strong ion–ion interaction between the solvation layers, as in this case by displacing the innermost solvation layers, the AFM tip pulls the other layers as well. For the Au(111) surface the first layer is observed at ∼0.6 nm followed by the 1.5, 2.4, and 3.3 nm steps, which correspond to widths of ∼0.6 and 0.9 nm, respectively. The steps correspond to the rupturing of successive adjacent surface layers as the AFM tip approaches the gold surface. Furthermore, the force required to rupture decreases for the layers farther away from the electrode surface to the bulk, indicating a weaker ordering of the layered structure away from the electrode surface. Upon changing the electrode potentials to either +0.2 V or to −0.2 V, a similar multi-layered structure with a variation in the separation of the layers can be detected (Fig. 2). For the neat IL, the force required to rupture the layer adjacent to the electrode was found to vary with the applied electrode potential. The rupturing forces were found to be 10 nN, 6 nN, and ∼3 nN for the neat IL at −0.2 V, at OCP, and at +0.2 V, respectively, revealing that a relatively strong near surface structure is present at −0.2 V. This indicates an increase in the strength of adsorption at more negative electrode potentials. For the neat IL at −0.2 V, the widths of the innermost and successive layers were found to be 0.5 and ∼0.9 nm, revealing a strong near surface interaction of the ions due to the applied electrode potential. This observation is in agreement with literature data.
 | ||
| Fig. 2 A comparison of the typical force–separation curves of neat [Py1,4]TFSA and of two different concentrations of AgTFSA in [Py1,4]TFSA. The curves have been obtained at +0.2 V vs. OCP (left column), at OCP (middle column), and at −0.2 V vs. OCP (right column). [AgTFSA] = 50 μM and 100 μM. | ||
For 50 μM AgTFSA/[Py1,4]TFSA, a few changes have been recorded in the force–distance curves at all measured electrode potentials. At OCP, the width of the innermost layer is ∼0.8 nm, which can either be due to a rearrangement of the ions or to a change in the composition of the electrolyte (or species like AgTFSA complexes are present). The widths of the subsequent layers have been slightly increased for 50 μM AgTFSA in comparison to the neat IL, with an average width of 1 nm at all electrode potentials. However, the innermost layer widths were found to be 0.7 nm at −0.2 V and at +0.2 V. The increase in the width of the layers for 50 μM AgTFSA can be attributed either to the presence of AgTFSA complexes or to their changes in orientation (or conformers) of the species. Furthermore, there is a marginal change in the rupturing force in the force–distance curves. The rupturing forces were found to be 3 nN for all electrode potentials, indicating a rather weak near surface interaction of the ions with the electrode.
For 100 μM AgTFSA/[Py1,4]TFSA, a further increase in the widths of the layers has been observed at all measured electrode potentials. Typically, the width of the innermost layer was found to be 0.9 ± 0.1 nm at +0.2 V, OCP and at −0.2 V. The average widths of the successive layers were found to be 1.4 nm at OCP and at −0.2 V. The force required to rupture the layers increased to 8 nN at all the measured potentials. However, a clear change was noticed at the 200 μM concentration wherein the number of layers varied upon varying the electrode potential. Four clear and distinct layers with step widths of about 1.0, 1.4, 1.5, and 1.4 nm have been observed at OCP and 0.9, 1.2, 1.4, 1.4 nm have been noticed at −0.2 V. However, only two steps with widths of about 0.9 and 1.5 nm have been recorded at +0.2 V. The force required to rupture the innermost layers is found to be large at OCP (10 nN) and at −0.2 V (14 nN) compared to the one at +0.2 V, which indicates a difference in the strength of the adsorption of the innermost layer with the substrate. Furthermore, the number of steps has been found to be the same at −0.2 V while a decrease in the step width has been noticed (ca. 0.9 nm). Quite an interesting result has been obtained at and above the 500 μM AgTFSA concentration, wherein only a single step was noticed at all measured electrode potentials. Furthermore, a change in the widths of the layers by varying the electrode potentials has been found for 500 μM AgTFSA (Fig. 3). Here in this case, one clear step with a layer width of 1 nm has been observed at OCP, whereas the step widths were found to decrease upon varying the electrode potential (e.g. 0.9 nm at +0.2 V and 0.8 nm at −0.2 V). Moreover, the forces required to rupture these layers have been nearly the same (ca. 2–3 nN). However, no clear and discrete steps have been observed at the OCP and at 0.2 V for 1 mM AgTFSA in the force–distance profiles, rather gradients have been noticed. These gradient widths were found to be ∼0.3 and ∼0.2 nm at the OCP and at +0.2 V, respectively. The forces needed to break the layers were around 6–7 nN in both cases. Furthermore, no clear signal was seen at −0.2 V, which indicates that the applied electrode potential affects the interfacial structure. The analysis of force–separation profiles at various concentrations of AgTFSA in [Py1,4]TFSA has shown that the number of solvation layers decreases from four layers to a single layer with an increase in concentration from 50 to 200 μM AgTFSA in [Py1,4]TFSA. A significant change in the widths of the innermost layers can also be observed at 1 mM AgTFSA.
 | ||
| Fig. 3 A comparison of the typical force–separation curves of three different concentrations of AgTFSA in [Py1,4]TFSA. The curves have been obtained at +0.2 V vs. OCP (left column), at OCP (middle column), and at −0.2 V vs. OCP (right column). [AgTFSA] = 200 μM, 500 μM, and 1 mM. | ||
In order to interpret the present findings of the Au(111)/[Py1,4]TFSA interfacial nanostructure at various concentrations of AgTFSA, we compare the results with the reported ones in the literature at varying concentrations of alkali metal salts in ILs41,59 (e.g. LiTFSA/[Py1,4]TFSA41 and NaFSA/[Py1,4]TFSA59). At OCP, five solvation layers were reported for 0.1 M LiTFSA/[Py1,4]TFSA on Au(111). The innermost layer width was found to be 0.16 nm for 0.1 M LiTFSA/[Py1,4]TFSA while the innermost layer width was found to be 0.65 nm for [Py1,4]TFSA. In the case of the 0.1 M LiTFSA/[Py1,4]TFSA, the widths of the successive layers were found to be same (∼0.7 nm) and these steps could be related to the presence of the ion-pairs of [Py1,4]TFSA. A similar behaviour was also seen for [Py1,4]TFSA, where a multilayered structure can be observed. The widths of the innermost layers rose by increasing the concentration from 0.1 M to ≥0.5 M LiTFSA/[Py1,4]TFSA. A decrease in the number of layers can also be observed with an increase in the concentration of LiTFSA. Moreover, the widths of the successive layers (e.g. 2nd, 3rd and 4th layers for 0.5 M LiTFSA in [Py1,4]TFSA and 2nd and 3rd layers for 1 M LiTFSA/[Py1,4]TFSA) were found to be between ∼0.85 and ∼0.95 nm. This indicates that the presence of [Py1,4]TFSA ion-pairs in the successive layers was not influenced by the presence of Li+ at the interface for varying concentrations of Li+ between 0.1 M and 0.5 M. However, the interfacial nanostructure was disturbed at 1 M LiTFSA/[Py1,4]TFSA upon varying the electrode potential, which was attributed to the formation of upd layers on the electrode. This study clearly showed that the addition of Li+ can significantly change the interfacial structure where [Py1,4]+ ions are replaced by Li+, which was observed as a decrease in the innermost layer widths on Au(111). Furthermore, the widths of the innermost layers were increased on increasing the concentration of LiTFSA, which was attributed to the change in configuration between Li+ and TFSA−.41
For NaFSA in [Py1,4]FSA, the interfacial nanostructure did not vary upon the addition of 0.05 M NaFSA to [Py1,4]FSA59 in which the innermost layer width was found to be nearly the same (∼0.65 nm) as that of the innermost layer width for [Py1,4]TFSA. However, the interfacial nanostructure was changed upon increasing the concentration of NaFSA from 0.05 to ≥0.25 M in [Py1,4]FSA, wherein the width of the innermost layer was found to be ∼0.35 nm for 0.25 M and 0.5 M NaFSA in [Py1,4]FSA. In the case of NaFSA/[Py1,4]FSA at OCP, the widths of the innermost layers were found to be invariant upon increasing the concentration of NaFSA. Based on these two studies we can conclude that the structure of the Au(111)/IL interface at a higher concentrations of metal salts (e.g. 1 M LiTFSA [Py1,4]TFSA or 0.5 M NaFSA/[Py1,4]FSA) differed with that of the interfacial structure at lower concentrations (e.g. 0.1 M LiTFSA in [Py1,4]TFSA or 0.05 M NaFSA in [Py1,4]FSA). However, in contrast to the present studies with AgTFSA rather high concentrations of LiTFSA and NaFSA were needed to disturb the interfacial layers.
The following salient features can be drawn from the above discussion. An increase in the width of the innermost layer has been noticed upon increasing the concentration of LiTFSA from 0.1 to 0.5 M in [Py1,4]TFSA (0.16 nm for 0.1 M vs. 0.25 nm for 0.5 M), which was attributed to changes in the complexation (i.e. a change in the coordination number, C.N.) and to configurational changes (i.e. cis/trans and/or monodentate/bidentate).41 For the nanostructure of the Au(111)/NaFSA-[Py1,4]FSA interface, the authors have observed no significant increase in the push-through forces for 0.05 M NaFSA in [Py1,4]FSA at various electrode potentials. These push-through forces were almost comparable with those of [Py1,4]FSA under similar conditions. A significant change in the nanostructure of Au(111)/NaFSA-[Py1,4]FSA has been observed for 0.25 M NaFSA in [Py1,4]FSA, where the innermost layer width significantly decreased (0.37 nm for 0.25 M FSA/[Py1,4]FSA vs. 0.68 nm for 0.05 M NaFSA/[Py1,4]FSA). A stable interfacial structure has been found upon further increasing the concentration from 0.25 M to 0.5 M NaFSA.59 Here, in the aforementioned two cases, the concentrations of metal salts (LiTFSA/NaFSA) have been varied in the regime of mM to M. However, in the case of AgTFSA reported in the present paper, the interfacial structure was studied at quite low concentrations of the salt from 50 μM to 1 mM of AgTFSA. Moreover, we observe a similar solvation structure for salt concentrations between 50 and 200 μM AgTFSA. The solvation layers formed due to the presence of AgTFSA complexes from 50 to 200 μM should be the same. Therefore, the widths of the innermost layers do not differ largely in this regime of concentration. We indeed observed a similar solvation structure at the interface of the Au(111)/electrolyte in this regime of AgTFSA concentrations, where the innermost layer widths were between 0.8 and 1 nm. However, not only the orientation (cis/trans configurations) of the solvated species on the electrode surface change but also the solvation layers change upon further increasing the concentration to 500 μM and above. The above in situ AFM results reveal that the Au(111)/AgTFSA/[Py1,4]TFSA interfacial structure is rather complex and further thorough investigations are needed to comprehensively describe the near surface ionic arrangement at the electrode/electrolyte interface.
In order to investigate the dynamics of the interfacial processes that occur on Au(111), we have recorded the force–distance curves of 50 μM AgTFSA/[Py1,4]TFSA by varying the electrode potentials to more negative values like −0.8 V vs. OCP and then back to a more positive value, e.g. −0.3 V. The corresponding force–distance curves are shown in Fig. 4. Four distinct steps have been observed in the force–distance profiles upon varying the electrode potentials from OCP to −0.8 V. The average widths of the innermost and the next successive layers were found to be 0.7 and 1 nm, respectively. However, by applying −0.8 V for two hours under the conditions of Ag deposition the force–distance profiles show no steps indicating that the interfacial structure is totally disturbed during the deposition of silver on gold. The deposition process consumes silver ions that are present at the interface resulting in a depletion of silver complexes at the interface. A multilayered structure is resumed after changing the potential back to −0.3 V where the deposited silver is slowly oxidized. The reappearance of this multilayer is typical for quite a low concentration of AgTFSA/[Py1,4]TFSA and for the neat IL, which is due to the presence of either AgTFSA complexes or to the ion-pair of the IL at the interface.
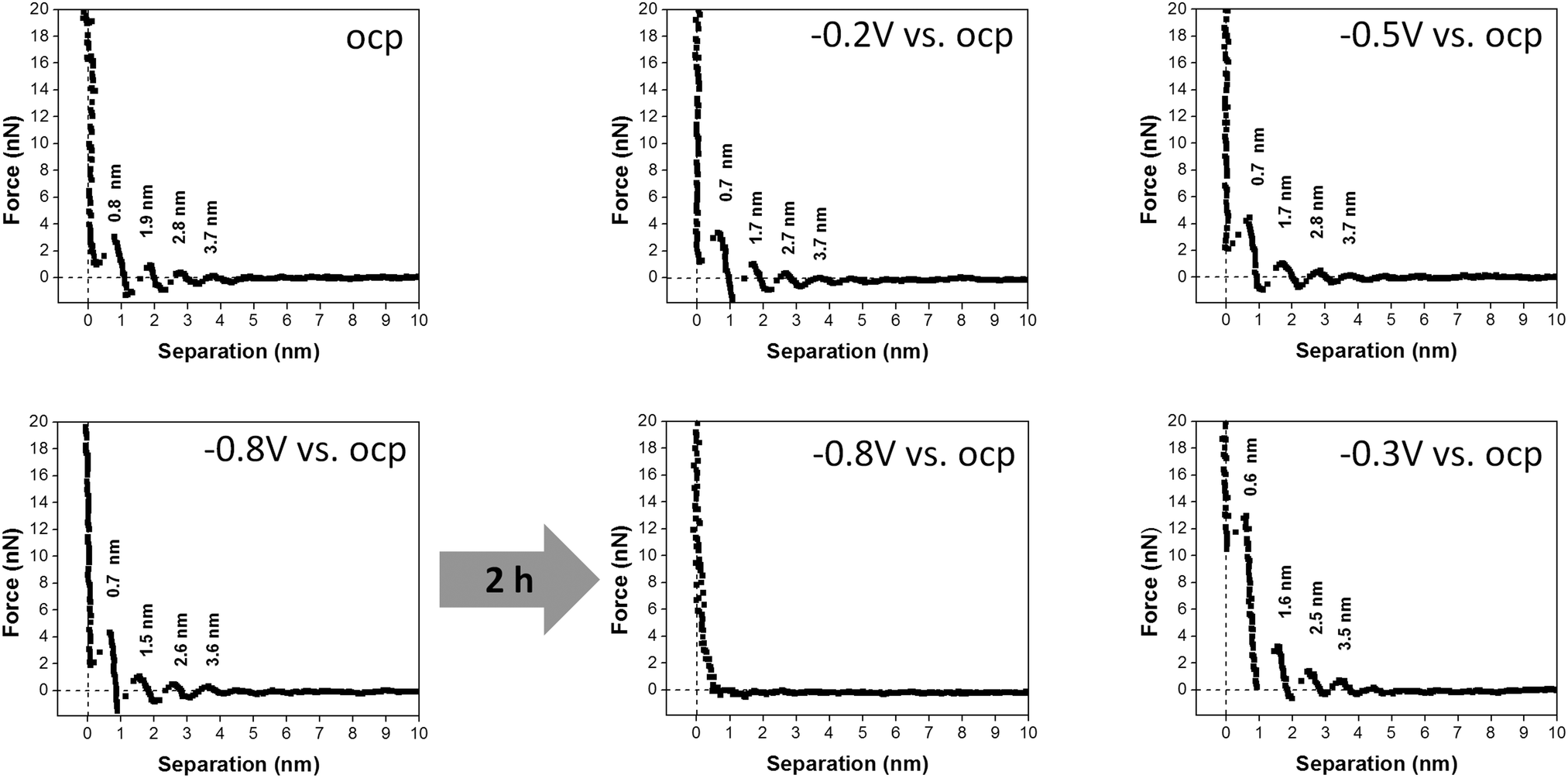 | ||
| Fig. 4 A comparison of typical force–separation profiles of 50 μM AgTFSA in [Py1,4]TFSA. The curves have been obtained at various electrode potentials from OCP to −0.8 V, the potential was held at −1 V for 2 h, and switched back to −0.3 V. | ||
In summary, the force–distance profiles obtained for [Py1,4]TFSA and for AgTFSA/[Py1,4]TFSA are different. The results obtained in this investigation indicate that the multilayered structure is retained in IL solutions with quite low concentrations of the silver salt, and slight changes have been observed for 50 and 100 μM AgTFSA at all measured electrode potentials. The innermost layer thickness for the neat IL varies upon changing the electrode potentials (e.g. ±0.2 V vs. OCP). There is a slight increase in the width of the innermost layer for AgTFSA/[Py1,4]TFSA at the measured potentials, indicating an increase in the dimensions of the ions, which could be due to the presence of the ion-pairs of the IL in a different orientation along with the Ag+ complexes in the interface. Furthermore, the presence of quite low concentrations of AgTFSA complexes may not influence the interfacial structure strongly and the innermost layer is still dominated by IL ion pairs. A transition in the interfacial structure has been observed at 200 μM AgTFSA in [Py1,4]TFSA, wherein the step width is increased with a further increase in the average widths of subsequent layers (ca. 1.5 nm). The increase in width of the innermost layer and the layers extending to the bulk (i.e. either in the transition zone or in the diffusion zone) could be due to the presence of AgTFSA complexes as these species are large in their sizes compared to the ion-pair sizes of [Py1,4]TFSA. A comparably large force is required to rupture the innermost layer at this concentration compared to the one for the neat IL and for lower concentrations of AgTFSA/[Py1,4]TFSA, which is due to an increased strength of adsorption of the AgTFSA complexes on the electrode surface.41 Furthermore, the number of steps has been found to decrease to 2 at +0.2 V. The decrease in the number of layers indicates a change in the ordering of the layers (layers in the diffusion zone/transition zone) away from the electrode surface, which is also dependent on the electrode potential due to changes in the electrostatic attraction.60,61
Rather a simple double layer structure is observed at 500 μM AgTFSA, where only a single step is observed in the force–distance profiles at all electrode potentials. Furthermore, the tip experiences quite a low rupturing force of 2 nN indicating that the adsorbed layer can be ruptured easily with minimum force. Such behaviour indicates that the adsorbed layer weakly interacts with the surface. An increase in the concentration of AgTFSA to 1 mM changes the interfacial structure completely as we could not observe any steps in the force–distance measurements. This indicates that the cantilever cannot find any adsorbed layers until it reaches the electrode surface. The AFM results signify that the concentration has a considerable influence on the interfacial structure for Au(111)/AgTFSA in [Py1,4]TFSA. This behaviour is rather complex and it is obviously different for aqueous electrolytes, where a classical EDL structure can be observed at very dilute solutions of aqueous electrolytes.
3.2 Simulation results
For comparison with experiments, we simulate the static equilibrium EDL at 200 mV overpotential. First, we simulate the behavior of the binary ionic liquid [Py1,4]TFSA at a metallic surface. Second, we discuss the effect of neutral or charged additives. The details of our computational method can be found in the ESI.†In Fig. 5, we neglect the hardcore constraint to reach a better understanding of the model (Fint = 0). The simulations show the crowding of cations at the negative electrode (see Fig. 5(a)). The shape of the ionic layers depends on the applied potential difference. In particular, at small electrode potentials (−0.02 V in Fig. 5(a)), the concentration of cations exhibits a distribution of the Boltzmann-type. The cation concentration at the surface increases with electrode potential until, at sufficiently large polarization (−0.05 V in Fig. 5(a)), it becomes saturated due to incompressibility because limited space is available for the ions. This implies that the concentration of cations at the electrode surface is bound from above. Thereby, the concentration profile changes to the Fermi–Dirac-type. During further increase of electrode polarization (up to −0.2 V in Fig. 5(a)), the cation layer broadens to screen the electric field of the electrode. The decay length of the ionic layers corresponds to the Debye-length expected for a dilute electrolyte, 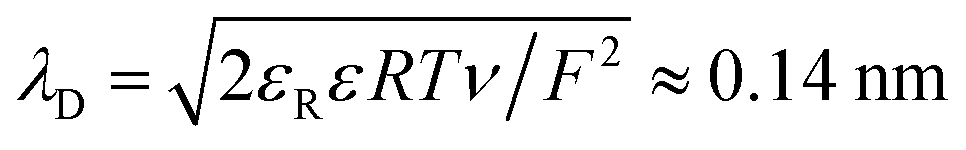 .
.
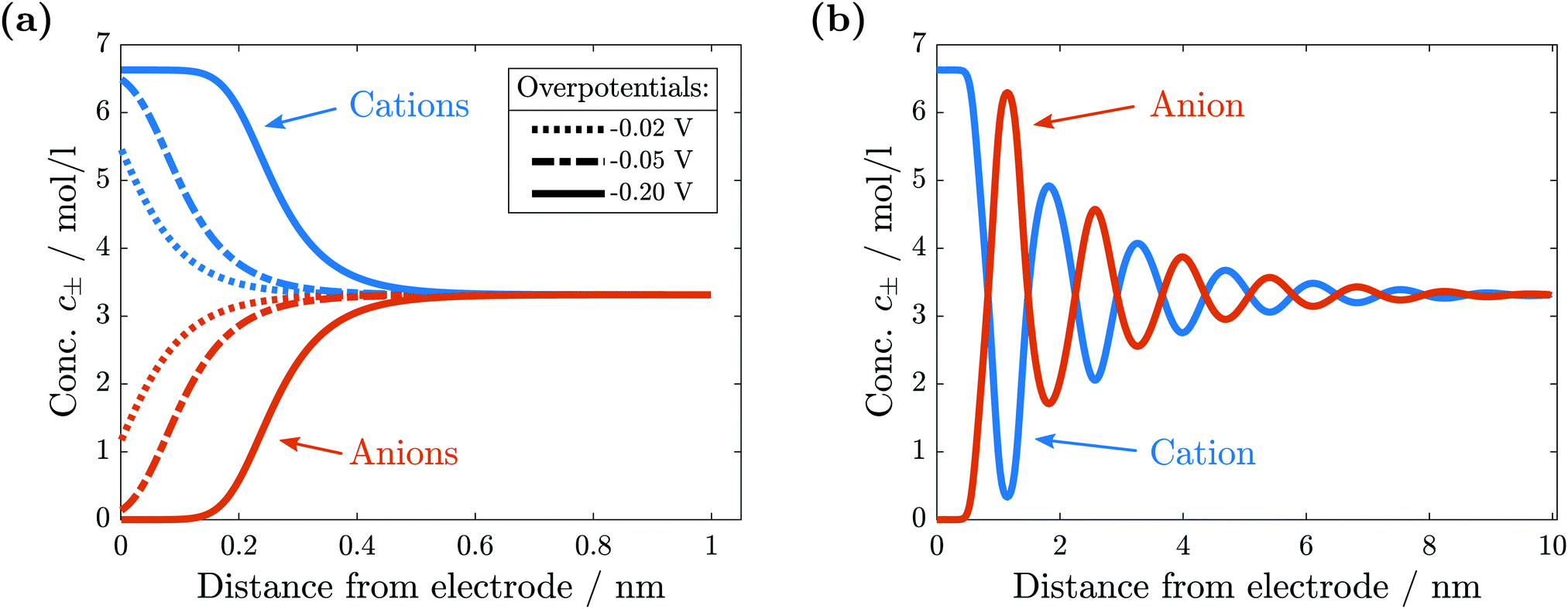 | ||
| Fig. 5 Comparison of ionic concentrations in the vicinity of a negatively charged electrode surface. (a) Without hardcore interactions we observe the depletions of anions and the crowding of cations. (b) With hardcore interactions a quasi-crystalline structure of alternating layers forms. | ||
In conclusion, without the hardcore constraint we observe the standard behavior for electrolytes at electrode interfaces. The electric potential jump is screened by the accumulation of ionic charges.
The simulation results change significantly if the hardcore constraint is taken into account (see Fig. 5(b)). We observe the formation of various alternating layers extending over 5 nm. The first layer adjacent to the negatively charged electrode consists entirely of cations, whereas the anions are depleted completely. The successive layers alternate in the dominating species and decay in amplitude, i.e., the total depletion of the minor species does not appear any more. Finally, the concentrations of the two species converge in the bulk electrolyte. The electrolyte potential is in accordance with the damped oscillations of the ionic concentrations (see Fig. 6). These oscillations show that the ionic diameter is incommensurate with the charge required to screen the electric potential jump.
 | ||
| Fig. 6 Electric potential in the electrolyte in the vicinity of a negatively charged electrode surface. | ||
We calculate the mechanical force as outlined in Section 2.2. The simulated force (see Fig. 7) exhibits the characteristic peaks observed in the AFM experiments. The spacing between the positive peaks agrees with the molecular radius of 0.67 nm (see the ESI†) within numerical accuracy. As shown in Fig. 5(b), the innermost layers are dominated by a single species, in alternating sequence. As these layer widths are qualitatively in good agreement with the experimentally measured layer widths for the pure IL, our model supports the interpretation of AFM measurements to correlate the force peaks with molecular mono-layers. The deviations between the modeled and measured layer widths are not surprising, as our modeling approach neglects microscopic properties, e.g., non-spherical molecular shapes and orientation-dependent interactions.
 | ||
| Fig. 7 Simulated force on AFM tip in the vicinity of a negatively charged electrode surface. | ||
The deviation between the simulated and experimental forces lies between one to two orders of magnitude. This agreement is very good if we take into account the differing set-ups. The atomic force microscope measures the penetration into a many-particle crystalline structure, including the replacement of particles. In contrast, we simulate elastic perturbations of an equilibrium state.
We discuss the impact of neutral and charged additives, e.g., water H2O and ionic silver Ag+ onto the EDL. The dynamics of this third species in the electrolyte is determined by eqn (11). The small concentration of the additive species allows us to neglect the corresponding contributions to convection and to the electric current, setting M3 ≈ 0, ν3 ≪ ν±, and t3 ≪ 1 in this principal discussion of the EDL structure. In the stationary case (J = 0 and N3 = 0) we find from eqn (10) and (11)
 | (14) |
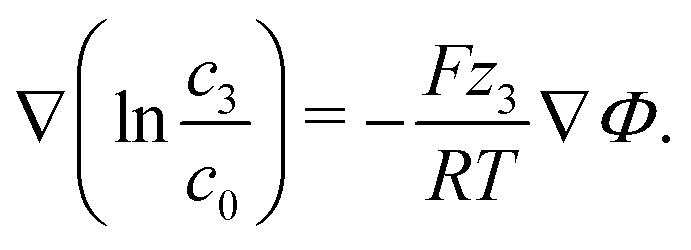 | (15) |
For the neutral additive water, our theoretical analysis in eqn (15) predicts a uniform distribution throughout the EDL, c3 = cH2O = const. This is a consequence of the entropy forcing neutral additives into the EDL. Therefore, large bulk concentrations of neutral additives are required to reach significant concentrations in the EDL and to affect its multilayer structure. Indeed, previous experiments43 show that the addition of water perturbs the EDL and destroys the multilayer structure only if it makes up more than half of the solution volume.
Electric forces are to be discussed for charged additives. If we consider silver (zAg = 1) as a third species, eqn (15) shows that its concentration obeys a Boltzmann distribution,
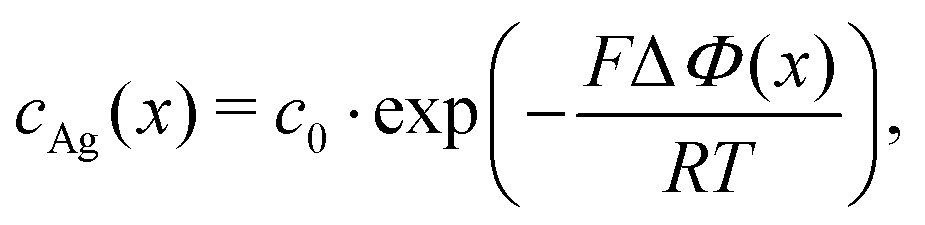 | (16) |
The screening of charges and electric fields cause the electrochemical surface layer. The quasi-crystalline structure emerges because the screening length does not fit the molecular size of the ionic liquids. Metal ions in the surface layer can efficiently shield electric forces and introduce an additional very small molecular length scale. Therefore, we expect that the silver ions very effectively destroy the EDL multi-layer structure.
This argument, however, is complicated by the presence of chemical interactions between the molecules. The chemical interactions could, for example, lead to strong pairing forces. In this case metal ions would mainly be present as a neutral complex and might behave as neutral solutes. Alternatively, neutral solutes could have preferred and strongly polarized bonds with solvent molecules that destroy the quasi-crystalline structure at low concentrations. It remains for further studies to evaluate and classify the effect of the chemical interactions on the EDL.
4 Conclusions
The electrode/electrolyte interface of Au(111)/[Py1,4]TFSA and of Au(111)/AgTFSA/[Py1,4]TFSA has been investigated using atomic force microscopy. The interfacial structure (or the arrangement of ions) remarkably varies with the concentration of AgTFSA until the layer structure is completely disrupted. Furthermore, changing the electrode potential has some influence on the interfacial structure. The force–separation profiles indicate that a multi-layer structure is found for the neat IL and this multi-layer structure is retained at quite low concentrations of the silver salt (50 to 100 μM AgTFSA/[Py1,4]TFSA). However, a clear change in the electrical double layer structure has been noticed at a 200 μM concentration of AgTFSA in [Py1,4]TFSA wherein the interfacial layers were found to be different on changing the applied electrode potentials. Furthermore, the widths of the corresponding innermost layers at 200 μM concentration are different from those ones observed at lower concentrations of AgTFSA. A simple double layer structure has been observed for 500 μM AgTFSA, which does not vary with the applied electrode potential. A remarkable difference is observed at this concentration and above, in which no layer structure has been noticed at 1 mM concentration. The observed changes in the widths of the innermost layer from AFM measurements could be attributed to the orientation of the Ag species (or to the conformational changes of the anions that are coordinated to the Ag+).Furthermore, we present a model analysis supporting these measurements of the EDL structure. We simulate force profiles that show the multilayer structure and agree with the experimental observations. The distances between force peaks correspond to ionic monolayers. We can show that in contrast to neutral additives ionic additives like AgTFSA are attracted by an electrified interface. Therefore, we predict in good agreement with experiments that roughly 400 μM concentrations are sufficient to reach large concentrations in the EDL and disrupt its structure. By neglecting the microscopic details of chemistry, our model highlights the fundamental relations behind the multilayer EDL structure of ionic liquids.
The AFM results on the dynamics of the interfacial processes reveal that the multilayer structure is disrupted completely during the deposition of silver upon applying −0.8 V for 2 h and the multilayer structure is retained after changing the electrode potential back to −0.3 V. This study signifies that the EDL structure of Au(111)/IL is indeed influenced by the presence of varying concentrations of an added salt to the IL. The general question arises to what extent different solutes alter IL interfacial layers and whether the dynamic processes alter the deposit morphology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the German Ministry of Education and Research (BMBF) (project LUZI, BMBF: 03SF0499A, 03SF0499E) and by the German Research Foundation (DFG) (DFG: EN 370/26-1) is gratefully acknowledged.References
- V. Rooryck, F. Reniers, C. Buess-Herman, G. Attard and X. Yang, J. Electroanal. Chem., 2000, 482, 93–101 CrossRef CAS.
- S. Garcia, D. Salinas, C. Mayer, E. Schmidt, G. Staikov and W. J. Lorenz, Electrochim. Acta, 1998, 43, 3007–3019 CrossRef CAS.
- A. Morgante, K. Prince, G. Paolucci and E. Tosatti, Surf. Sci., 1987, 189-190, 620–627 CrossRef CAS.
- K. Ogaki and K. Itaya, Electrochim. Acta, 1995, 40, 1249–1257 CrossRef CAS.
- J. Wang, B. M. Ocko, A. J. Davenport and H. S. Isaacs, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 10321–10338 CrossRef CAS.
- G. A. Attard and D. A. King, Surf. Sci., 1987, 188, 589–598 CrossRef CAS.
- D. Kolb, G. Lehmpfuhl and M. Zei, J. Electroanal. Chem. Interfacial Electrochem., 1984, 179, 289–295 CrossRef CAS.
- D. Kolb and J. Schneider, Surf. Sci., 1985, 162, 764–775 CrossRef CAS.
- G. Pulletikurthi, A. Lahiri, T. Carstens, N. Borisenko, S. Zein El Abedin and F. Endres, J. Solid State Electrochem., 2013, 17, 2823–2832 CrossRef CAS.
- S. Zein El Abedin, E. M. Moustafa, R. Hempelmann, H. Natter and F. Endres, ChemPhysChem, 2006, 7, 1535–1543 CrossRef PubMed.
- M. Galiński, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef.
- S. Zhang, N. Sun, X. He, X. Lu and X. Zhang, J. Phys. Chem. Ref. Data, 2006, 35, 1475–1517 CrossRef CAS.
- A. Basile, A. I. Bhatt, A. P. O'Mullane and S. K. Bhargava, Electrochim. Acta, 2011, 56, 2895–2905 CrossRef CAS.
- Y. Katayama, S. Dan, T. Miura and T. Kishi, J. Electrochem. Soc., 2001, 148, C102 CrossRef CAS.
- C. Fu, H. Zhou, W. Peng, J. Chen and Y. Kuang, Electrochem. Commun., 2008, 10, 806–809 CrossRef CAS.
- P. He, H. Liu, Z. Li, Y. Liu, X. Xu and J. Li, Langmuir, 2004, 20, 10260–10267 CrossRef CAS PubMed.
- J. M. Reyna-González, J. C. Reyes-López and M. Aguilar-Martínez, Electrochim. Acta, 2013, 94, 344–352 CrossRef.
- M.-C. Tsai, D.-X. Zhuang and P.-Y. Chen, Electrochim. Acta, 2010, 55, 1019–1027 CrossRef CAS.
- S. Zein El Abedin and F. Endres, Electrochim. Acta, 2009, 54, 5673–5677 CrossRef.
- A. Ispas, M. Pölleth, K. H. T. Ba, A. Bund and J. Janek, Electrochim. Acta, 2011, 56, 10332–10339 CrossRef CAS.
- B. H. R. Suryanto, C. A. Gunawan, X. Lu and C. Zhao, Electrochim. Acta, 2012, 81, 98–105 CrossRef CAS.
- I. Siretanu, D. Ebeling, M. P. Andersson, S. L. S. Stipp, A. Philipse, M. C. Stuart, D. van den Ende and F. Mugele, Sci. Rep., 2015, 4, 4956 CrossRef PubMed.
- J. R. MacDonald, J. Electroanal. Chem. Interfacial Electrochem., 1987, 223, 1–23 CrossRef CAS.
- J. Wang and A. J. Bard, J. Phys. Chem. B, 2001, 105, 5217–5222 CrossRef CAS.
- F. Endres, N. Borisenko, S. Z. El Abedin, R. Hayes and R. Atkin, Faraday Discuss., 2012, 154, 221–233 RSC.
- A. Elbourne, S. McDonald, K. Voichovsky, F. Endres, G. G. Warr and R. Atkin, ACS Nano, 2015, 9, 7608–7620 CrossRef CAS PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- M. Z. Bazant, B. D. Storey and A. A. Kornyshev, Phys. Rev. Lett., 2011, 106, 046102 CrossRef PubMed.
- R. Atkin, N. Borisenko, M. Drüschler, S. Z. El Abedin, F. Endres, R. Hayes, B. Huber and B. Roling, Phys. Chem. Chem. Phys., 2011, 13, 6849 RSC.
- R. Hayes, D. Wakeham and R. Atkin, Ionic Liq. UnCOILed, 2012, 51–85 Search PubMed.
- R. Atkin and G. G. Warr, J. Phys. Chem. C, 2007, 111, 5162–5168 CAS.
- R. Hayes, S. Z. El Abedin and R. Atkin, J. Phys. Chem. B, 2009, 113, 7049–7052 CrossRef CAS PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2010, 12, 1709–1723 RSC.
- R. Hayes, N. Borisenko, M. K. Tam, P. C. Howlett, F. Endres and R. Atkin, J. Phys. Chem. C, 2011, 115, 6855–6863 CAS.
- R. Atkin, S. Z. El Abedin, R. Hayes, L. H. Gasparotto, N. Borisenko and F. Endres, J. Phys. Chem. C, 2009, 113, 13266–13272 CAS.
- X. Zhang, Y.-X. Zhong, J.-W. Yan, Y.-Z. Su, M. Zhang and B.-W. Mao, Chem. Commun., 2012, 48, 582–584 RSC.
- A. Labuda and P. Grütter, Langmuir, 2012, 28, 5319–5322 CrossRef CAS PubMed.
- J. M. Black, D. Walters, A. Labuda, G. Feng, P. C. Hillesheim, S. Dai, P. T. Cummings, S. V. Kalinin, R. Proksch and N. Balke, Nano Lett., 2013, 13, 5954–5960 CrossRef CAS PubMed.
- F. Endres, E. Abedin, S. Zein and N. Borissenko, Z. Phys. Chem., 2006, 220, 1377–1394 CrossRef CAS.
- R. Hayes, N. Borisenko, B. Corr, G. B. Webber, F. Endres and R. Atkin, Chem. Commun., 2012, 48, 10246–10248 RSC.
- A. Lahiri, T. Carstens, R. Atkin, N. Borisenko and F. Endres, J. Phys. Chem. C, 2015, 119, 16734–16742 CAS.
- T. Liu, Y. Danten, J. Grondin and R. Vilar, J. Raman Spectrosc., 2016, 47, 449–456 CrossRef CAS.
- T. Cui, A. Lahiri, T. Carstens, N. Borisenko, G. Pulletikurthi, C. Kuhl and F. Endres, J. Phys. Chem. C, 2016, 120, 9341–9349 CAS.
- J. Wu, T. Jiang, D.-e. Jiang, Z. Jin and D. Henderson, Soft Matter, 2011, 7, 11222 RSC.
- D. Henderson and J. Wu, J. Phys. Chem. B, 2012, 116, 2520–2525 CrossRef CAS PubMed.
- D.-e. Jiang and J. Wu, J. Phys. Chem. Lett., 2013, 4, 1260–1267 CrossRef CAS PubMed.
- J. Jiang, D. Cao, D.-e. Jiang and J. Wu, J. Phys.: Condens. Matter, 2014, 26, 284102 CrossRef PubMed.
- M. H. Ghatee and Y. Ansari, J. Chem. Phys., 2007, 126, 154502 CrossRef PubMed.
- Z. A. Goodwin, G. Feng and A. A. Kornyshev, Electrochim. Acta, 2017, 225, 190–197 CrossRef CAS.
- R. Blossey, A. C. Maggs and R. Podgornik, Phys. Rev. E, 2017, 95, 060602 CrossRef CAS PubMed.
- A. Latz and J. Zausch, Beilstein J. Nanotechnol., 2015, 6, 987–1007 CrossRef CAS PubMed.
- J. Stamm, A. Varzi, A. Latz and B. Horstmann, J. Power Sources, 2017, 360, 136–149 CrossRef CAS.
- F. Single, B. Horstmann and A. Latz, J. Electrochem. Soc., 2017, 164, E3132–E3145 CrossRef CAS.
- S. Clark, A. Latz and B. Horstmann, ChemSusChem, 2017, 10, 4735–4747 CrossRef CAS PubMed.
- IoLiTec Ionic Liquids Technologies Gmbh, Technical Data Sheet IL-0035, 2012.
- M. Huang, Y. Jiang, P. Sasisanker, G. W. Driver and H. Weingärtner, J. Chem. Eng., 2011, 1494–1499 CAS.
- J. S. Newman and K. E. Thomas-Alyea, Electrochemical Systems, John Wiley & Sons, 2004, ISBN: 978-0-471-47756-3 Search PubMed.
- T. J. Simons, P. C. Howlett, A. A. J. Torriero, D. R. MacFarlane and M. Forsyth, J. Phys. Chem. C, 2013, 117, 2662–2669 CAS.
- T. Carstens, A. Lahiri, N. Borisenko and F. Endres, J. Phys. Chem. C, 2016, 120, 14736–14741 CAS.
- S. Millefiorini, A. H. Tkaczyk, R. Sedev, J. Efthimiadis and J. Ralston, J. Am. Chem. Soc., 2006, 128, 3098–3101 CrossRef CAS PubMed.
- J. Restolho, J. L. Mata and B. Saramago, J. Phys. Chem. C, 2009, 113, 9321–9327 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp08243f |
| This journal is © the Owner Societies 2018 |