
Sensitivity-enhanced detection of non-labile proton and carbon NMR spectra on water resonances†
Mihajlo
Novakovic
a,
Ricardo P.
Martinho
a,
Gregory L.
Olsen
 a,
Michael S.
Lustig
b and
Lucio
Frydman
*a
a,
Michael S.
Lustig
b and
Lucio
Frydman
*a
aDepartment of Chemical and Biological Physics, Weizmann Institute of Sciences, 76100 Rehovot, Israel. E-mail: lucio.frydman@weizmann.ac.il
bDepartment of Electrical Engineering and Computer Sciences, University of California, Berkeley, California 94720, USA
First published on 14th November 2017
Abstract
Chemical exchange saturation transfer (CEST) experiments enhance the NMR signals of labile protons by continuously transferring these protons’ saturation to an abundant solvent pool like water. The present study expands these principles by fusing into these experiments homonuclear isotropic mixing sequences, enabling the water-enhanced detection of non-exchangeable species. Further opportunities are opened by the addition of coupling-mediated heteronuclear polarization transfers, which then impose on the water resonance a saturation stemming from non-labile heteronuclear species like 13C. To multiplex the ensuing experiments, these relayed approaches are combined with time-domain schemes involving multiple Ramsey-labeling experiments imparting the frequencies of the non-labile sites on the water resonance, via chemical exchange. 13C and 1H NMR spectra were detected in this fashion with about two-fold SNR amplification vis-à-vis conventionally detected spectroscopies. When combined with non-uniform sampling principles, this methodology thus becomes a sensitive alternative to detect non-exchangeable species in biomolecules. Still, multiple parameters including the scalar couplings and solvent exchange rates, will affect the efficiency and consequently the practicality of the overall experiment.
Introduction
Nuclear Magnetic Resonance (NMR) provides unique atomic-level insight into the structure and dynamics of materials, molecules and biological systems. Despite this singular versatility NMR is often hampered by low sensitivity, as a result of weak magnetic interactions leading to both small population differences between the nuclear spin states and to low energies in the emitted signals. NMR's low signal-to-noise ratio (SNR) may be compounded by chemical dilution of the molecules being targeted, and by the low natural abundance of important nuclei such as 13C and 15N. Main strategies to overcome these limitations involve hardware improvements1,2 as well as emerging Dynamic Nuclear Polarization methods transferring polarization to nuclei from the more strongly coupled electrons.3–7 Despite these efforts, NMR has been particularly challenged when applied to characterize metabolites in living tissues, where field strengths and homogeneities have to be tailored to large bodies, coil sizes are dictated by large, lossy and unsteady samples, and observations are complicated by the presence of an overwhelming water signal. A breakthrough in the assessment of metabolic spectral signatures came with the realization by Ward and Balaban that the solvent signal could be exploited for detecting solutes, if the latter species contained exchangeable protons.8,9 A selective radiofrequency (RF) saturation of these labile protons and the subsequent spontaneous transfer of this saturation to the readily detectable water resonance, thus led to the “Chemical Exchange Saturation Transfer” (CEST) experiment for the enhanced detection of labile metabolic resonances.10–12 Such enhancement arises from solute–solvent exchanges that augment the saturation effect on the solvent resonance by factors on the order of kexchTwater1, where kexch is the rate of exchange of the labile protons and Twater1 is the longitudinal relaxation time of the solvent. Compared to a single labile proton response, this product can lead to orders-of-magnitude enhancements, enabling NMR spectroscopy and imaging analyses of previously undetectable species including glucose,13,14 urea,15 creatine,16 lactate17 and endogenous proteins,18,19 as well as applications to map a tissue's pH and temperature.20–23As originally introduced, CEST is a continuous-wave (CW) frequency domain technique. Frequency Labeled Exchange (FLEX)24–26 offers an alternative to CEST, capable of endowing it with time-domain Fourier advantages. In the FLEX experiment the labile protons’ magnetization is amplitude-modulated by applying a pair of selective excitation/storage pulses separated by a delay t1.27 Rather than observing this Ramsey modulation after one exchange period with the solvent, FLEX loops this process multiple times, thereby magnifying its encoding effect on the water resonance. Repeating this as a function of increasing t1 and applying an observation pulse on water enables, after 2D fast Fourier Transform (FFT), the spectral detection of the labile protons with an increased, CEST-like sensitivity. Furthermore, by avoiding the continuous application of a saturating RF, FLEX alleviates some of the resolution limitations associated with CEST. Fig. 1 illustrates this feature by comparing CEST and FLEX spectra recorded on 15N alanine using a relatively low saturation power B1 = 0.25 μT for CEST (equivalent to γB1 = 10.8 Hz), vs. a comparable FLEX acquisition with a spectral resolution of 10 Hz. Notice that whereas both FLEX and CEST easily resolve the 15N–1H J coupling of the amine protons, the latter's intramolecular 1H–1H coupling to the α site is blurred in CEST but clearly resolved in FLEX – a method whose resolving power is akin to that of conventionally-detected acquisitions.
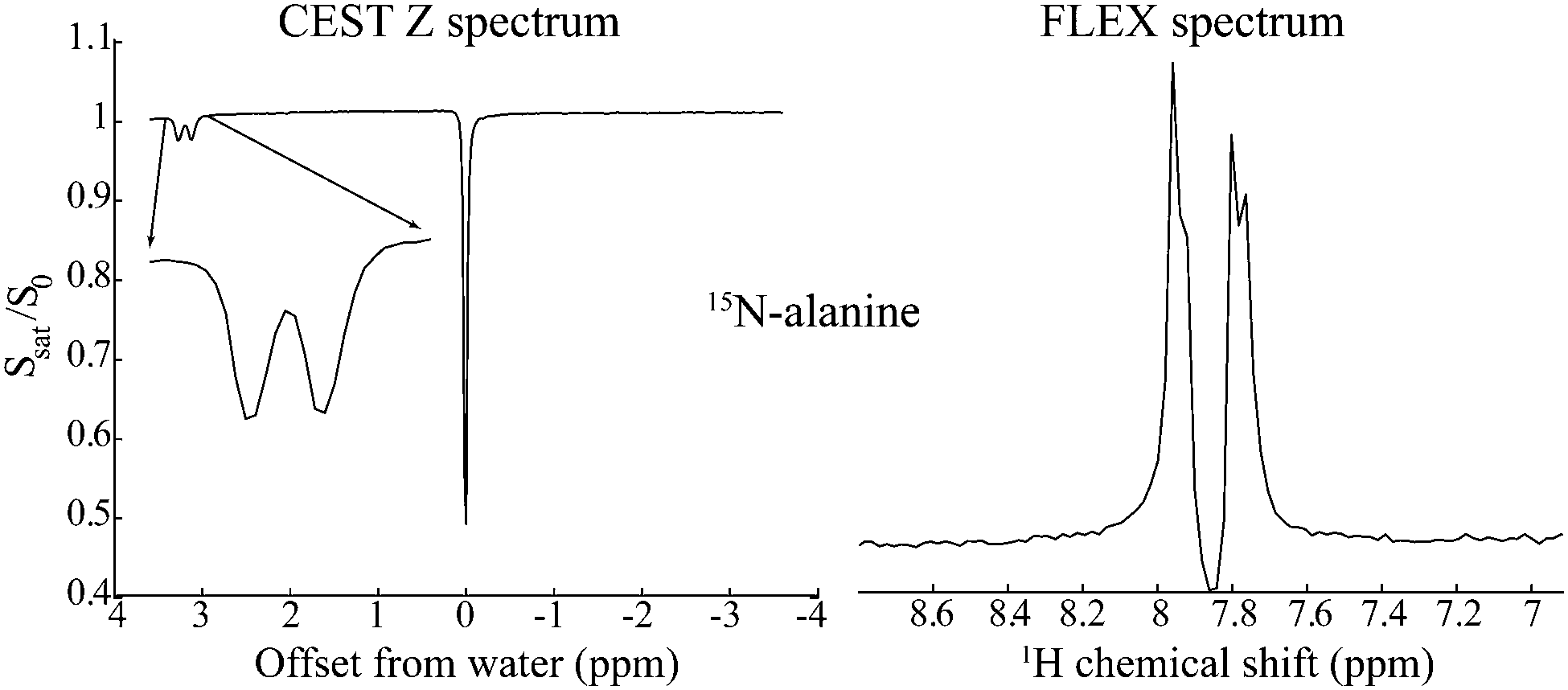 | ||
| Fig. 1 Comparison between CEST and FLEX spectra detecting on the water resonance the labile amine protons of 15N-labeled alanine at 2 °C. Whereas the latter experiment reveals both the larger 15N–1H and the smaller 1H–1H couplings, CEST's “Z spectrum” fails to resolve the smaller 1H–1H coupling due to the additional broadening brought about by the application of the CW RF. | ||
Both CEST and FLEX experiments have traditionally focused on detecting the exchangeable protons. In principle, however, any perturbation of these labile hydrogens could transfer to – and become observable on – the water pool. CEST effects often appear in vivo upfield from the water peak, for instance, as a result of perturbations on the non-labile protons associated to proteins and membranes in tissues that get transmitted to labile protons via cross relaxation, and from there onwards to the water resonance.28,29 Likewise, the use of an 15N-selective pulse was recently proposed to invert the magnetization of 15N bonded urea protons, thereby enabling the acquisition of 15N-contrasted water images.30 Related ideas have led to the introduction of HetFLEX,31 an experiment that combines the FLEX principles with Heteronuclear Multiple-Quantum Correlation (HMQC) experiments. HetFLEX thereby allows one to observe on the water resonance either 1D 15N NMR spectra from sites directly connected to labile protons, or 2D correlation spectra between these 15N nuclei and their labile 1H partners – always while incorporating the sensitivity enhancement that characterizes CEST. In this study we discuss a variant of these principles and methodologies, which allows one to record the NMR spectra of non-labile hydrogens or even heteronuclear sites like 13C, via the Fourier analysis of the strong modulations that these nuclei will impart on the water 1H peak. To make these experiments possible the FLEX-based detection of labile protons via water's modulation is combined with homonuclear isotropic mixing sequences,32,33 capable of transferring information from non-labile to labile sites; to make the second of these options feasible, an additional heteronuclear coherence transfer step is added prior to the isotropic mixing. These additions extend the sensitivity benefits arising from water-based FLEX experiments to non-labile species; the potential and limitations found for these RElayed FLEX (REFLEX) experiments, are briefly discussed.
Materials and methods
NMR experiments
15N-Alanine and D-glucose (U–13C6, 24–25%) were purchased from Cambridge Isotope Laboratories. 13C sucrose spectra were recorded on household sugar; dimethyl sulfoxide-d6 (d6-DMSO) was obtained from Merck. Alanine samples were prepared at 100 mM and pH = 2.2 (adjusted with HCl) in H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O (25%:75%). A U–13C6-glucose sample was prepared in H2O:d6-DMSO (20%:80%) at 100 mM concentration, whereas natural abundance sucrose was dissolved in the same solvent at 400 mM. All NMR experiments were conducted using an 11.7 T Magnex magnet (Abingdon, UK) run by a Varian iNova console (Palo Alto, USA) and equipped with a triple resonance HCN Varian 5 mm probe possessing x, y, z-gradients. Temperature was regulated with an FTS TC-84 temperature controller (Stone Ridge, USA). REFLEX spectra were recorded with 128 hypercomplex complex points in t134 and a spectral width of 3000 Hz for the homonuclear and of 8000 Hz for the heteronuclear experiments. For each complex increment two transients were collected using a 0/180° phase cycle. 90° pulse widths were ≈7 μs for 1H and 31 μs for 13C; DIPSI-133 was used for the isotropic mixing with a total duration of between 40 and 64 ms, tuned to yield 360° total rotations.
:D2O (25%:75%). A U–13C6-glucose sample was prepared in H2O:d6-DMSO (20%:80%) at 100 mM concentration, whereas natural abundance sucrose was dissolved in the same solvent at 400 mM. All NMR experiments were conducted using an 11.7 T Magnex magnet (Abingdon, UK) run by a Varian iNova console (Palo Alto, USA) and equipped with a triple resonance HCN Varian 5 mm probe possessing x, y, z-gradients. Temperature was regulated with an FTS TC-84 temperature controller (Stone Ridge, USA). REFLEX spectra were recorded with 128 hypercomplex complex points in t134 and a spectral width of 3000 Hz for the homonuclear and of 8000 Hz for the heteronuclear experiments. For each complex increment two transients were collected using a 0/180° phase cycle. 90° pulse widths were ≈7 μs for 1H and 31 μs for 13C; DIPSI-133 was used for the isotropic mixing with a total duration of between 40 and 64 ms, tuned to yield 360° total rotations.
Data processing
All NMR data were processed using customized Matlab® software (The Mathworks Inc., Natick, USA) based on the procedures described by Martinho et al.,31 as well as Mestre Nova® software (version 11.0.4, Mestrelabs Research, Spain). In some of the examples below (Fig. 2 and Fig. S2, ESI†) this included applying on the 2D REFLEX data a conventional 2D FFT; in other cases a 1D FFT was applied along the direct dimension, the water peak was integrated for each t1 increment to create a complex time-domain free induction decay (FID), this S(t1) FID was apodized with an exponential window function and zero filled, and then processed with either a FFT or a non-uniform Fourier transform to yield the indirect dimension spectrum (additional details on the processing of compressed sensing data are given in the main text). Conventional NMR spectra were processed using standard Mestre Nova parameters. Pulse sequences and processing scripts are available upon request.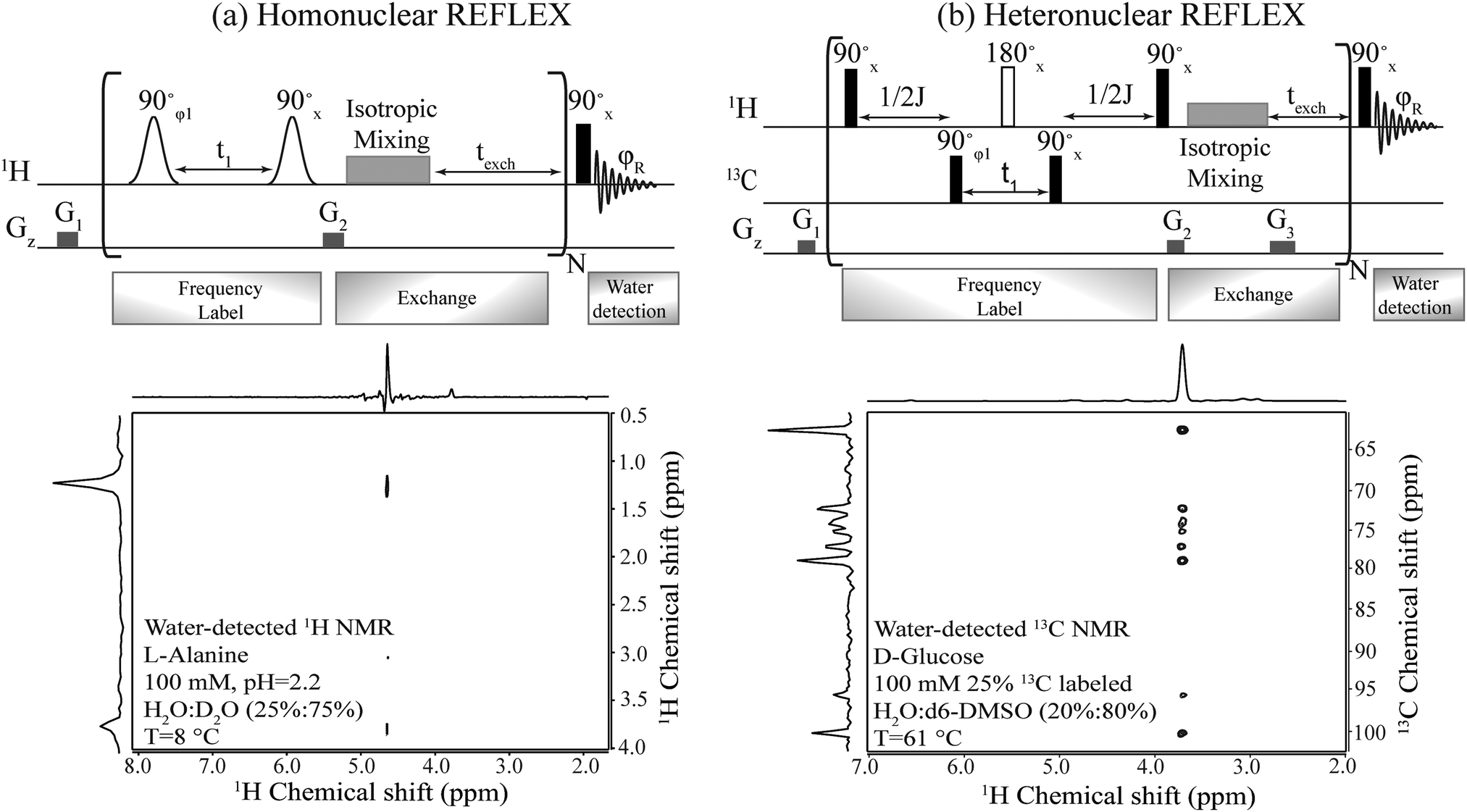 | ||
| Fig. 2 Top: Pulse sequences proposed for the homo- and hetero-nuclear versions of the REFLEX experiment. In all cases t1 is the 1H or 13C encoding delay, which is followed by an isotropic mixing plus an exchange period texch and repeated multiple (N) times, before a single hard pulse is applied to detect the water FID. Open shapes correspond to frequency selective (e.g., sinc) 90° pulses; filled black rectangles are hard 90° pulses; open rectangles are broadband 180° pulses; G1, G2 and G3 are purging gradients. The isotropic mixing was implemented using DIPSI-1; φ1 was incremented by 90° to achieve quadrature detection and phase-cycled as ±x together with φR to achieve a cancellation of unmodulated water. Bottom: Homo- and hetero-REFLEX results processed as 2D experiments, showing the correlation of non-labile protons in alanine and of carbons in D-glucose, with the water resonance. Shown on top and side of each 2D spectrum is the projection along the respective axis. | ||
Results
The REFLEX sequence
Fig. 2 summarizes the principles proposed for recording NMR spectra of non-exchangeable 1Hs or heteronuclei using a coherent, relayed transfer of their evolution to the water resonance. Like the original FLEX scheme these experiments involve variable t1 evolution periods during which the targeted coherences evolve under the action of their offset frequencies, before being stored as Mz magnetizations. These excite/evolve/store blocks are repeated numerous times N and are followed in each instance by an exchange period texch during which the encoded magnetizations exchange with the large, untouched water pool; the experiment thereby will benefit from the full sensitivity enhancement that characterizes CEST. The novel ingredient of the REFLEX sequences is that, unlike CEST or FLEX, their encoding is now done onto non-labile sites. To pass the t1 modulation of these non-labile protons to the water resonance (Homo REFLEX, Fig. 2a) each storage pulse is followed by an isotropic mixing block, whereby the encoded protons pass their chemical shift evolution onto hitherto untouched labile sites. Repeating this procedure multiple times up to a duration dictated by the water's longitudinal relaxation T1, eventually yields a water modulation pattern that reflects, for each t1 increment, a measure of the non-labile protons’ evolution. In order to detect solely this desired modulation and eliminate signals from non-exchanged water molecules, a two-step phase cycling is implemented where the phase of the initial excitation pulse and of the receiver are shifted by 180°. Quadrature components are also generated by using orthogonal pulse phases for the excitation and storage pulses according to the states procedure,34 enabling a sign discrimination of the FLEX resonances. A 2D spectrum of L-alanine detected in this fashion and showing both the α and the methyl group hydrogens correlated with water resonance, is shown in Fig. 2a.Fig. 2b shows an adaptation of these principles to heteronuclear-oriented experiments. This sequence encodes the heteronuclear non-labile shifts with an HMQC block, during which multiple quantum coherences (e.g. H±C±) are allowed to evolve under chemical shifts over a time t1. The resulting modulation is thereby imprinted on the non-labile protons of the 13C–1H pair but, rather than detecting the resulting transverse 1H coherences as in an HMQC experiment, these are passed via a homonuclear isotropic mixing block to nearby labile sites (in the present study this mixing was preceded, at no expense in SNR, by a storage pulse and purging gradient acting as z-filters,35cf.Fig. 2b). As a result of all this the 13C chemical shift modulation is transferred from directly 13C-bound protons to remote labile protons, and onwards to the water magnetization due to the latter's chemical exchange. To facilitate this further, a texch exchange time can be added after the mixing sequence, and the overall procedure repeated N times to build up the imprint of the 13C modulation on the water. Each encoding t1 increment thus results in a 13C-imposed amplitude modulation of the aqueous 1H resonance, which can be readily isolated by a suitable phase cycling that removes the pedestal arising from the non-modulated water signal. Full correlations of the 13C resonances in D-glucose with water obtained in this manner are shown in Fig. 2b.
Enhancement-defining parameters
Since REFLEX depends on multiple transfers from the solute nuclei that are being targeted to the water, a number of independent and interconnected parameters control its efficiency and hence its potential SNR gain. These include fixed parameters like the J-couplings between the non-labile and labile sites, and the ratio between labile and non-labile nuclei. There are also tunable variables like the mixing time tmix, the exchange time texch and the number of exchange loops N; in the heteronuclear case there are additional HMQC-derived timings to be optimized. Also amenable to manipulation are the T1 of the water as well as the exchange rate kexch between the labile protons and water, which can be controlled by adjusting pH or temperature, or by adding relaxation agents or a non-protonated solvent – for instance in this study kexch was controlled by adding to the water certain amounts of DMSO. All of these parameters are important, as they define the efficiency of both the chemical exchange process and the isotropic mixing. Fig. 3a stresses kexch's influence, by comparing the efficiencies of homonuclear REFLEX and of regular CEST experiments executed on sucrose, as a function of temperature. While the efficiency of a CEST experiment focusing on directly saturating sucrose's labile protons increases monotonically in this temperature range thanks to the continuous rise in kexch, the dependence of a REFLEX experiment focusing on the non-labile sucrose protons is non-monotonic. At lower temperatures the REFLEX-driven modulation increases as in CEST thanks to the faster exchange rates, but it eventually plateaus and begins to decay at higher temperatures. This decay tracks the behavior illustrated in Fig. 3b, which presents experimental results measuring the isotropic mixing transfer efficiency with respect to chemical exchange rate. The exponential decay that is empirically observed is relatively independent of the mixing scheme assayed (in the present study DIPSI-1 appeared as the most robust scheme and hence was adopted), and it reflects the averaging of the J-couplings between the non-labile and labile sites with increasing kexch. This decay places a fundamental constraint to the potential sensitivity gains that this approach can deliver on a per-scan basis. To further explore this feature a model based on the Bloch-McConnell equations36,37 was developed, which successfully reproduced REFLEX's experimental behavior and which allowed us to explore REFLEX's potential over a range of conditions (ESI,† Fig. S1). According to these simulations the exchange rates that maximize the experiment's SNR are on the same order as the J-coupling between non-labile and labile sites. Under these conditions simulations predict that isotropic mixings times ≈0.5 J−1 will provide a maximum transfer, and that no additional texch is needed since sufficient chemical exchange already occurs during this mixing period (even if hardware duty cycle limits may demand making texch ≠ 0). An increase in the water's T1 will then always benefit sensitivity, since on a per-scan basis additional loops can then be applied without suffering from relaxation penalties. These predictions (ESI,† Fig. S1) match well the experimental results, which revealed maximum per-scan sensitivity enhancements of ≈50% for the homonuclear cases. Somewhat better results were obtained in REFLEX's heteronuclear variant, where the 1D projection arising from a 25% 13C-labeled 100 mM glucose sample gave a ca. two-fold better SNR per scan then the 1D 13C trace available by projecting its HMQC counterpart (Fig. 4). An interesting feature of this heteronuclear variant is associated to the fact that in it, multiple 1Hs are correlated by virtue of the isotropic mixing (ESI,† Fig. S2). As a result of this heteronuclear REFLEX shows, in addition to the correlations to the water resonance arising via the chemical exchange, correlations between multiple 13C and 1H sites characteristic of HMQC-TOCSY experiments. These correlations (ESI,† Fig. S2) also contribute to the overall sensitivity upon computing F1 traces of the kind shown in Fig. 4.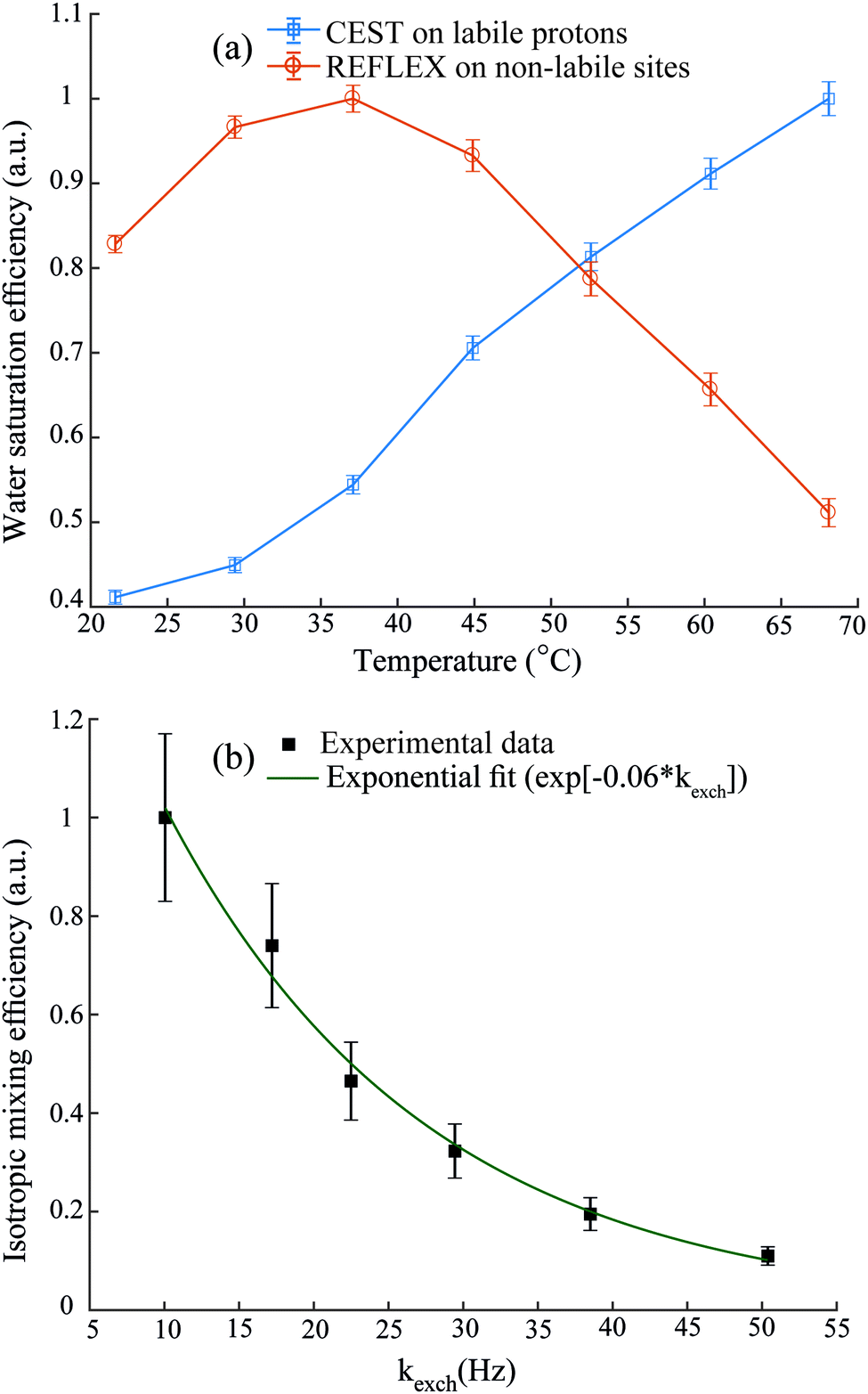 | ||
| Fig. 3 (a) Relative responses of labile protons in CEST and of non-labile protons in homonuclear REFLEX, as monitored on the water 1H NMR signal of a solution of sucrose in H2O/DMSO (2/8), with increasing temperature. (b) Dependence of the intensity observed in a 2D TOCSY experiment between the cross peaks of the non-labile alpha protons and the labile amino resonances of L-alanine, plotted against the latter's exchange rate with water at pH = 2.2. Chemical exchange rates in (b) were estimated from the line width and eventual coalescence of the amine proton peak at higher temperatures, and extrapolated down to the reported temperatures based on an Arrhenius behavior. Also shown is the best fit of this isotropic mixing efficiency by an exponential decay. All y-axes are normalized with respect to their highest experimental values. | ||
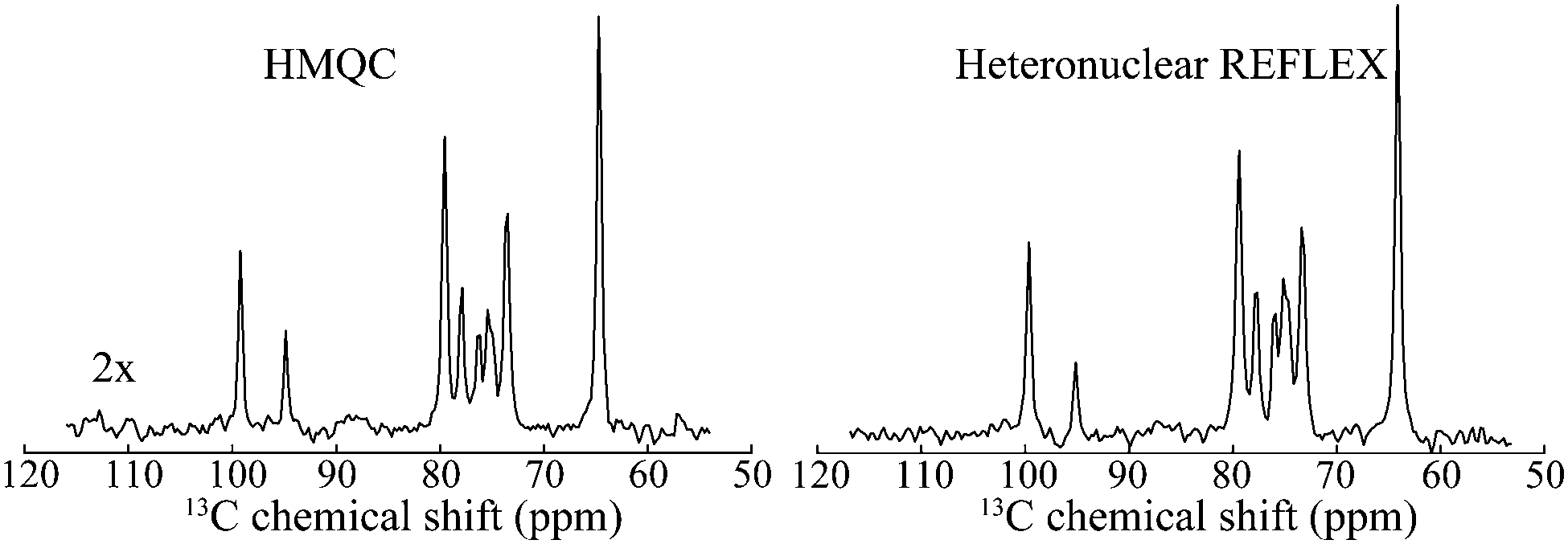 | ||
| Fig. 4 Comparison between the F1 13C NMR 1D projections arising in HMQC and in heteronuclear relayed FLEX for D-glucose. Shown next to the HMQC spectrum is the vertical scale amplification used to bring the spectra to the same intensities. Notice the similar resolution of both traces, reflecting the behavior shown in Fig. 1. Spectra were acquired under same conditions as in Fig. 2b. | ||
Compressed sensing
A disadvantage of FLEX experiments, both in their relayed as well as in their original acquisition formats, is their targeting of 1D spectra via the t1 dimension of a 2D acquisition. This implies that samples with short longitudinal relaxation times and/or narrow lines, may lead to spectra with worse SNR per unit acquisition time than if performing a conventional 1D direct detection. To alleviate this limitation we explored the use of non-uniform sampling (NUS) schemes,38,39 whereby only a subset of t1 increments is acquired and spectra are reconstructed by non-FFT principles. Furthermore, such compressed sensing also allows one to alleviate the demands of hypercomplex acquisitions,34 and still succeed in determining the absolute frequency sign of the t1 precession from sampling randomly alternated in- or quadrature-phase components. This is an interesting feature, which as discussed by Schuyler et al. could find additional use in other kinds of nD NMR acquisitions.43Fig. 5 illustrates this potential to collect FLEX/REFLEX spectra using a fraction of the conventionally sampled time-domain data, by comparing the 13C glucose spectrum arising from a full sampling of 512 complex t1 REFLEX increments (1024 phase-cycled experiments acquired in total), against a projection reconstructed by randomly extracting 8.79% of either real or imaginary data points from this original t1 FID. The spectrum reconstructed from this data subset yields the same resolution and an even better SNR than the FFT of the original set (Fig. 5b). This SNR improvement reflects the nature of the algorithm employed, which finds the singular values of a Hankel matrix deriving from the complex time-domain signal, attributes the M main singular values to the actual spectrum that it reconstructs, and neglects smaller singular values that end up being related to noise. Similarly, an experimentally acquired array of 90 randomly distributed points (including once again a random sampling of either real or imaginary components) also results in a fully resolved spectrum (Fig. 5c). A conventional 1D 13C NMR spectrum collected in the same number of scans is presented in Fig. 5d, evidencing slightly better resolution but poorer per-scan SNR than its water-detected counterparts. This reduced resolution can be traced to limitations of the HMQC segment of the 2D technique for this particular sample.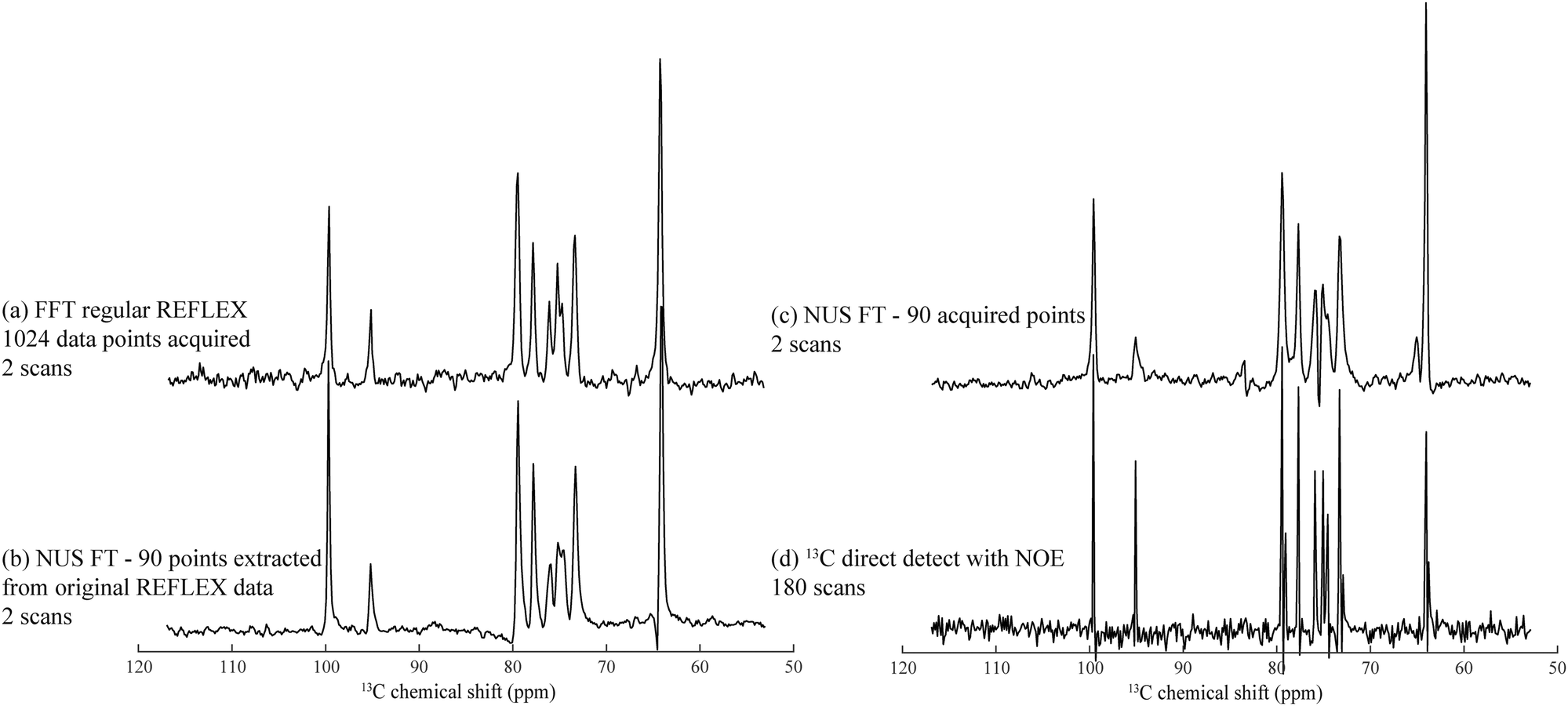 | ||
| Fig. 5 Heteronuclear 13C REFLEX spectra of 25% 13C-labeled D-glucose collected under the same conditions as in Fig. 2 after: (a) FFT of a fully sampled t1-domain FID containing 512 complex points. (b) Reconstruction involving the extraction of 90 random data points from the real or imaginary t1 components in the original FID, and subject the array to a non-uniform FT by application of the Cadzow recovery procedure.40–42 (c) Spectrum resulting from NUS FT processing of another 90 acquired points. (d) Conventional 1H-decoupled 1D 13C NMR spectrum acquired with the same number of scans as in (c). Spectral width in all spectra was 8000 Hz with a digital resolution of 15.6 Hz per point. | ||
Conclusions
The FLEX concept endows CEST experiments with multiplexing capabilities, bringing to the latter the advantages of Fourier spectroscopy as well as an enhanced resolution at no apparent cost in sensitivity. This makes it an attractive alternative for the detection of labile protons, with a large gain in sensitivity. This work, together with recent counterparts,30,31 explores routes that extend these sensitivity enhancements to species other than labile protons while keeping the advantages of FFT NMR. REFLEX achieves this by introducing relayed J-driven transfers from non-labile protons and heteronuclei to labile species; this passes the chemical shift information of the non-labile species through bonds to labile protons, and then onward to the water via chemical exchange. Repeating this modulation transfer to the abundant water peak numerous times carries significant signal amplification promises, yet these are dampened by the opposing demands of the relayed vs. the exchange transfers. Indeed, whereas rapid exchange rates can amplify the signals of labile protons by several orders of magnitude, they will also lead to a self-decoupling between these labile protons and their non-labile neighbors. This will in turn decrease the efficiency of J-driven transfers, on which both HetFLEX and REFLEX experiments rely. Simulations and experiments indicate that the ensuing enhancements are then limited to factors ≤10×, depending on the molecule in question. In order to reap the maximum sensitivity, relayed FLEX experiments benefit from multiple exchanging sites, from strong 1H–1H or 13C–1H couplings to the exchanging hydrogens, as well as from exchange rates on the order of the J-couplings. For small metabolites, including sugars and amino acids, these optimal conditions can be adjusted by manipulating the temperature and pH, while for amides in structured proteins those conditions will often arise naturally. The applicability of these principles to larger biomolecules is thus also promising, and it is currently under investigation. Also under investigation is the implementation of strategies known to enhance water-oriented experiments, including the reliance on Carr-Purcell Meiboom-Gill44 and on steady-state free precession45 methods capable of melding into one the encoding, the exchange and the observation periods,46 and thereby further the sensitivity per unit time of this kind of experiments.Among the inherent drawbacks of the approach here discussed is its reliance on a slow, indirect-domain-based detection method for obtaining a 1D trace. It is likely that in a number of instances this can be overcome using compressed sensing methods, which in the preliminary tests shown in Fig. 5 and in Fig. S3 (ESI†), evidence the possibility to accelerate these 2D-based detections of 1D spectra on both enriched and natural abundance compounds. In fact, joint non-quadrature NUS of the t1 and hypercomplex acquisition parameters allowed us to speed up these acquisitions by close to an order of magnitude (Fig. S3, ESI†). Yet another intrinsic complication of these experiments stems from their reliance on monitoring small percentage differences in the amplitude of a water resonance that, by contrast to what usually happens in CEST MRI experiments, gives in NMR an extremely strong response. Instrumental fluctuations and radiation damping effects complicate the monitoring of such strong resonance with ideal stability and consistency, leading to the presence of t1 noise.47 These effects compromise the SNR of the weaker solute signals being targeted by REFLEX, a complication that we here alleviated by diluting protonated water with deuterated solvents. We are looking for alternative roads to alleviate both of these complications, including the use of deconvolution methods48,49 and of ultrafast 2D NMR techniques.50
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Koby Zibzener for technical assistance. This work was supported by the Kimmel Institute for Magnetic Resonance (Weizmann Institute), Israel Science Foundation Grant 795/13, the Program of the Planning and Budgeting Committee from the Israel Science Foundation (iCORE) Project 1775/12, the EU Horizon 2020 programme (Maria Sklodowska-Curie Grant 642773), and the generosity of the Perlman Family Foundation.Notes and references
- J. H. Ardenkjaer-Larsen, G. S. Boebinger, A. Comment, S. Duckett, A. S. Edison, F. Engelke, C. Griesinger, R. G. Griffin, C. Hilty, H. Maeda, G. Parigi, T. Prisner, E. Ravera, J. Van Bentum, S. Vega, A. Webb, C. Luchinat, H. Schwalbe and L. Frydman, Angew. Chem., Int. Ed., 2015, 54, 9162–9185 CrossRef CAS PubMed.
- K. Hashi, S. Ohki, S. Matsumoto, G. Nishijima, A. Goto, K. Deguchi, K. Yamada, T. Noguchi, S. Sakai, M. Takahashi, Y. Yanagisawa, S. Iguchi, T. Yamazaki, H. Maeda, R. Tanaka, T. Nemoto, H. Suematsu, T. Miki, K. Saito and T. Shimizu, J. Magn. Reson., 2015, 256, 30–33 CrossRef CAS PubMed.
- F. Shi, Q. Zhang, P. Wang, H. Sun, J. Wang, X. Rong, M. Chen, C. Ju, F. Reinhard, H. Chen, J. Wrachtrup, J. Wang and J. Du, Science, 2015, 347, 1135–1138 CrossRef CAS PubMed.
- J. H. Ardenkjaer-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef CAS PubMed.
- T. Harris, G. Eliyahu, L. Frydman and H. Degani, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18131–18136 CrossRef CAS PubMed.
- A. Abragam and M. Goldman, Reports Prog. Phys., 1978, 41, 395–467 CrossRef CAS.
- J. Kemsley, Chem. Eng. News, 2008, 86, 12–15 Search PubMed.
- V. Guivel-Scharen, T. Sinnwell, S. D. Wolff and R. S. Balaban, J. Magn. Reson., 1998, 133, 36–45 CrossRef CAS PubMed.
- K. M. Ward, A. H. Aletras and R. S. Balaban, J. Magn. Reson., 2000, 143, 79–87 CrossRef CAS PubMed.
- P. C. M. Van Zijl and N. N. Yadav, Magn. Reson. Med., 2011, 65, 927–948 CrossRef CAS PubMed.
- F. Kogan, H. Hariharan and R. Reddy, Curr. Radiol. Rep., 2013, 1, 102–114 CrossRef PubMed.
- E. Vinogradov, A. D. Sherry and R. E. Lenkinski, J. Magn. Reson., 2013, 229, 155–172 CrossRef CAS PubMed.
- K. W. Y. Chan, M. T. McMahon, Y. Kato, G. Liu, J. W. M. Bulte, Z. M. Bhujwalla, D. Artemov and P. C. M. Van Zijl, Magn. Reson. Med., 2012, 68, 1764–1773 CrossRef CAS PubMed.
- S. Zhang, R. Trokowski and A. D. Sherry, J. Am. Chem. Soc., 2003, 15288–15289 CrossRef CAS PubMed.
- A. P. Dagher, A. Aletras, P. Choyke and R. S. Balaban, J. Magn. Reson. Imaging, 2000, 12, 745–748 CrossRef CAS PubMed.
- F. Kogan, M. Haris, A. Singh, K. Cai, C. Debrosse, R. P. R. Nanga, H. Hariharan and R. Reddy, Magn. Reson. Med., 2014, 71, 164–172 CrossRef PubMed.
- S. Aime, D. Delli Castelli, F. Fedeli and E. Terreno, J. Am. Chem. Soc., 2002, 124, 9364–9365 CrossRef CAS PubMed.
- J. Zhou, J.-F. Payen, D. Wilson, R. Traystman and P. C. M. van Zijl, Nat. Med., 2003, 9, 1085–1090 CrossRef CAS PubMed.
- P. C. van Zijl, C. K. Jones, J. Ren, C. R. Malloy and A. D. Sherry, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4359–4364 CrossRef CAS PubMed.
- D. L. Longo, W. Dastrù, G. Digilio, J. Keupp, S. Langereis, S. Lanzardo, S. Prestigio, O. Steinbach, E. Terreno, F. Uggeri and S. Aime, Magn. Reson. Med., 2011, 65, 202–211 CrossRef CAS PubMed.
- D. Dellicastelli, E. Terreno and S. Aime, Angew. Chem., Int. Ed., 2011, 50, 1798–1800 CrossRef CAS PubMed.
- L. Q. Chen, C. M. Howison, J. J. Jeffery, I. F. Robey, P. H. Kuo and M. D. Pagel, Magn. Reson. Med., 2014, 72, 1408–1417 CrossRef PubMed.
- S. Aime, A. Barge, D. D. Castelli, F. Fedeli, A. Mortillaro, F. U. Nielsen and E. Terreno, Magn. Reson. Med., 2002, 47, 639–648 CrossRef CAS PubMed.
- S. Garcia, L. Chavez, T. J. Lowery, S. I. Han, D. E. Wemmer and A. Pines, J. Magn. Reson., 2007, 184, 72–77 CrossRef CAS PubMed.
- J. I. Friedman, M. T. McMahon, J. T. Stivers and P. C. M. van Zijl, J. Am. Chem. Soc., 2010, 132, 1813–1815 CrossRef CAS PubMed.
- N. N. Yadav, C. K. Jones, J. Xu, A. Bar-Shir, A. A. Gilad, M. T. McMahon and P. C. M. van Zijl, Magn. Reson. Med., 2012, 68, 1048–1055 CrossRef CAS PubMed.
- N. F. Ramsey, Phys. Rev., 1950, 78, 695–699 CrossRef CAS.
- W. Ling, R. R. Regatte, G. Navon and A. Jerschow, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2266–2270 CrossRef CAS PubMed.
- P. C. M. Van Zijl, J. Zhou, N. Mori, J. F. Payen, D. Wilson and S. Mori, Magn. Reson. Med., 2003, 49, 440–449 CrossRef CAS PubMed.
- H. Zeng, J. Xu, N. N. Yadav, M. T. McMahon, B. Harden, D. Frueh and P. C. M. van Zijl, J. Am. Chem. Soc., 2016, 138, 11136–11139 CrossRef CAS PubMed.
- R. P. Martinho, M. Novakovic, G. L. Olsen and L. Frydman, Angew. Chem., Int. Ed., 2017, 3521–3525 CrossRef CAS PubMed.
- L. Braunschweiler and R. R. Ernst, J. Magn. Reson., 1983, 53, 521–528 CAS.
- S. Rucker and A. J. Shaka, Mol. Phys., 1989, 68, 509–517 CrossRef CAS.
- D. J. States, R. A. Haberkorn and D. J. Ruben, J. Magn. Reson., 1982, 48, 286–292 CAS.
- O. W. Sørensen, M. Rance and R. R. Ernst, J. Magn. Reson., 1984, 56, 527–534 Search PubMed.
- H. M. McConnell, J. Chem. Phys., 1958, 28, 430–431 CrossRef CAS.
- M. Helgstrand, T. Hard and P. Allard, J. Biomol. NMR, 2000, 18, 49–63 CrossRef CAS PubMed.
- M. Mobli and J. C. Hoch, Prog. Nucl. Magn. Reson. Spectrosc., 2014, 83, 21–41 CrossRef CAS PubMed.
- K. Kazimierczuk and V. Orekhov, Magn. Reson. Chem., 2015, 53, 921–926 CrossRef CAS PubMed.
- J. A. Cadzow, IEEE Trans. Acoust., Speech, Signal Process., 1988, 36, 49–62 CrossRef.
- C. Brissac, T. E. Malliavin and M. A. Delsuc, J. Biomol. NMR, 1995, 6, 361–365 CrossRef CAS PubMed.
- P. J. Shin, P. E. Z. Larson, M. A. Ohliger, M. Elad, J. M. Pauly, D. B. Vigneron and M. Lustig, Magn. Reson. Med., 2014, 72, 959–970 CrossRef PubMed.
- H. Monajemi, D. L. Donoho, J. C. Hoch and A. D. Schuyler, 2017, arXiv: 1702.01830.
- H. Y. Carr and E. M. Purcell, Phys. Rev., 1954, 94, 630–638 CrossRef CAS.
- K. Scheffler and S. Lehnhardt, Eur. J. Radiol., 2003, 13, 2409–2418 CrossRef PubMed.
- S. Zhang, Z. Liu, A. Grant, J. Keupp, R. E. Lenkinski and E. Vinogradov, J. Magn. Reson., 2017, 275, 55–67 CrossRef CAS PubMed.
- A. F. Mehlkopf, D. Korbee, T. A. Tiggelman and R. Freeman, J. Magn. Reson., 1984, 58, 315–323 CAS.
- G. A. Morris, H. Barjat and T. J. Home, Prog. Nucl. Magn. Reson. Spectrosc., 1997, 31, 197–257 CrossRef CAS.
- A. Gibbs, G. Morris, A. Swanson and D. Cowburn, J. Magn. Reson., Ser. A, 1993, 101, 351–356 CrossRef CAS.
- A. Tal and L. Frydman, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 241–292 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp07046b |
| This journal is © the Owner Societies 2018 |