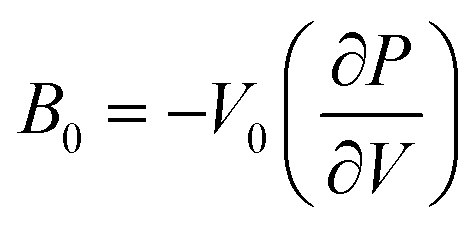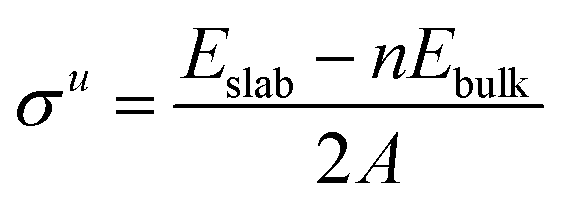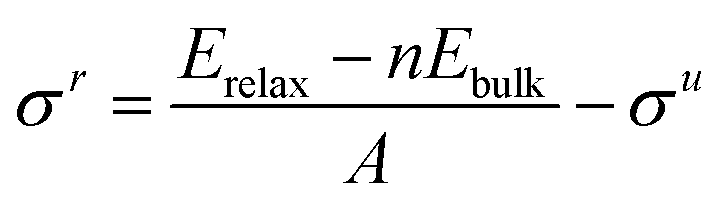Bulk and surface properties of metal carbides: implications for catalysis†
Received
15th September 2017
, Accepted 22nd December 2017
First published on 20th February 2018
Abstract
We present a comprehensive study of the bulk and surface properties of transition metal carbides with rock salt structures and discuss their formation energies and electronic structures. The bonding character of the materials is shown to be dependent on the periodic position of the transition metal as well as the surface termination, which in turn tunes the densities of states and electronic surface properties. Specific focus is given to the possible catalytic implications of the surface properties on CO2 hydrogenation.
Introduction
It has been known since the seminal work of Levy and Boudart in 19731 that the addition of carbon to elemental transition metals can bestow the resulting materials with the properties of precious metals. Subsequent generations of transition metal carbides (TMCs) have demonstrated the ability to catalyse hydrogenation,2 water-gas shift3,4 and CO oxidation reactions.5,6 The focus of this study will be a detailed investigation of the bulk and surface properties of all known transition metal monocarbides. However, we also intend to highlight aspects of these materials that relate to their potential future utilization as catalysts in the reduction of carbon dioxide (CO2). We have chosen this process as an example of a major aim of contemporary chemical science7–9 and because many of the materials that have been used to catalyse such reactions, using CO2 as a feedstock on pathways to fuels and chemicals, are made from expensive metals,10,11 or have low catalytic activity.12 Excitingly, some TMCs have been shown to hydrogenate CO213–16 and such activity hints at the prospect of a possible integrated industrial approach for H2 generation and CO2 conversion.
TMCs combine the physical properties of transition metals, ionic solids and covalent solids.17,18 They have the conductivity of TMs, the structural coordination and high melting point of ionic solids and the hardness of covalent solids. The band structures and therefore electronic properties of each TMC are, to a large degree, driven by the extent of metallic, covalent and ionic bonding character of the metal–metal and metal–carbon bonds.18–20 The physical properties of TMCs also depend on the surface; for instance, the electronic structure of TiC displays more covalent character in the {001} direction and more electron localization (ionic character) in the {111} faces.21 Finally, the position of the TM along the series is also important, with ZrC a considerably more ionic transition metal carbide than δ-MoC.17 Nevertheless, no systematic studies have been reported into the structure and electronic properties of all three low-index surfaces for all available monocarbides. In 2004, Hugosson et al. published a theoretical study into the unrelaxed {001} surface of a range of TMCs;22 this work was then followed by a less extensive study into the properties of the relaxed {001} surfaces of various TMCs.18 However, whilst it is true that studies have pointed to the {001} surface as being the most stable in, for example, TiC23 it is also true that the {111} surfaces of TMCs are generally more active than the corresponding {001} surfaces.17,24 For a good overview of the current literature see ref. 25.
In view of the growing importance of TMC materials, we have undertaken a comprehensive theoretical study into the properties of the bulk and three low-index surfaces, i.e. {001} {011} {111}, of all reported TMCs with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry: ScC,26 TiC,27 VC,27 CrC,28 CoC,29 NiC,30 YC,31 ZrC,32 NbC,33 δ-MoC,34 HfC,27 TaC27 and WC.35 Those carbides with groups 3–5 metals form stable bulk crystals,36,37 whereas carbides with 1:1 stoichiometry and post group 5 metals are usually formed from either modifying the surfaces of bulk TMs,38,39 as thin films40,41 or even 2D materials.42,43 Therefore, the future of carbide chemistry will not be limited to those materials that readily anneal into homogeneous bulk crystals and the study of monocarbides with post group five transition metals will also be considered here. Our work employs calculations based on the density functional theory (DFT) with a careful evaluation of three different functionals.
:1 stoichiometry: ScC,26 TiC,27 VC,27 CrC,28 CoC,29 NiC,30 YC,31 ZrC,32 NbC,33 δ-MoC,34 HfC,27 TaC27 and WC.35 Those carbides with groups 3–5 metals form stable bulk crystals,36,37 whereas carbides with 1:1 stoichiometry and post group 5 metals are usually formed from either modifying the surfaces of bulk TMs,38,39 as thin films40,41 or even 2D materials.42,43 Therefore, the future of carbide chemistry will not be limited to those materials that readily anneal into homogeneous bulk crystals and the study of monocarbides with post group five transition metals will also be considered here. Our work employs calculations based on the density functional theory (DFT) with a careful evaluation of three different functionals.
Methodology
All calculations were performed with the Vienna Ab initio Simulation (VASP) package,44–46 which was used to test several types of generalized gradient approximation (GGA) functionals within the framework of periodic density functional theory. Results obtained using the standard version of the Perdew–Burke–Ernzerhof47 exchange functional (PBE) were supplemented first by a version that was developed specifically for solids (PBEsol)48 and finally by a revised version (RPBE).49 We have not considered the computationally much more costly Meta GGA or hybrid functionals, since such methods are most valuable in determining band gaps in semiconducting and insulating materials.50,51 Plane-wave basis sets were applied to the valence electrons, whereas the core electrons were described by the projected augmented wave method (PAW) developed by Blöchl.52 Additionally, long range interactions were modelled using the Grimme dispersion D3 method.53,54 Monkhorst–Pack grids were used to add K points of 11 × 11 × 11 to the bulk calculations and meshes of 11 × 11 × 1 for the surface calculations. All energies converged within a cutoff of 520 eV, although different cutoff values and numbers of K-points were also tested (see ESI†). The threshold for the ionic relaxation was set at 0.01 eV Å−1. The electronic threshold for the self-consistency cycles was set to 10−5 eV and convergence was determined using the tetrahedron method, implementing Blöchl corrected smearing.55 No internal coordinates or symmetry constraints were added to any system and spin polarization was enabled.
Results
Bulk metal carbides
The carbide crystal structures for ScC,26 TiC,27 VC,27 CrC,28 CoC,29 NiC,30 YC,31 ZrC,32 NbC,33 δ-MoC,34 HfC,27 TaC27 and WC35 were taken from the Inorganic Structure Database (ICSD).56 These comprise all the published monocarbide materials with the rock salt structure.56 For every TMC, geometry optimizations were then carried out to relax the atomic coordinates in each unit cell, using all three functionals. This procedure was repeated for a series of different unit cell parameters to produce potential energy curves for each material (see ESI†). From these data sets, lattice constants (a0), unit cell volumes (a03) and bulk moduli (B0) were assigned. Alongside these values, the shortest atomic distances inside each unit cell were calculated and compared to experiment, using a previously described protocol (see Table S14, ESI†).57,58 The lattice parameters are reported in Table 1 and compared to the experimental data. The theoretical results across all functionals are generally in reasonable agreement with experiment and no obvious systematic errors were observed; the exception to this agreement is found for CoC, where a somewhat larger deviation from experiment is observed with all three functionals. The crystal structure for cobalt-carbide was published by Cadeville and Lapierre in 1972 using X-ray crystallographic techniques,29 while the other carbide structures were mostly determined by neutron scattering techniques. As noted above, TMCs with post 5d transition metals such as cobalt do not readily anneal into single crystals with 1:1 stereochemistry. Cadeville and Lapierre29 were able to show that the addition of carbon was able to stabilize the face-centered cubic phase of cobalt-carbide. However, the structure they provide is the only such rock salt crystal phase of cobalt-carbide and the presence of low carbon occupancies throughout the crystal probably explains the discrepancy between experiment and theory. Therefore, it is possible to argue that this crystal structure is more accurately described as cobalt metal stabilized by variable amounts of interstitial carbons, whereas the theoretical models more accurately represent CoC, with a stoichiometry of 1:1.
Table 1 Calculated (a0) and experimental (aexp0) lattice parameters. All distances are given in Å and the relative errors (%a0) are given in percent
| Carbide |
a
exp0
|
a
0
|
a
0
|
a
0
|
%a0a |
%a0b |
%a0c |
|
PBE values.
PBEsol values.
RPBE values.
|
| ScC |
4.51026 |
4.651 |
4.614 |
4.681 |
−3.12 |
−2.32 |
−3.79 |
| TiC |
4.32827 |
4.308 |
4.276 |
4.319 |
0.46 |
1.20 |
0.21 |
| VC |
4.16527 |
4.132 |
4.098 |
4.138 |
0.80 |
1.61 |
0.66 |
| CrC |
4.03028 |
4.043 |
4.010 |
4.057 |
−0.33 |
0.49 |
−0.68 |
| CoC |
3.55129 |
3.977 |
3.932 |
3.982 |
−12.00 |
−10.72 |
−12.13 |
| NiC |
4.07730 |
4.047 |
4.009 |
4.070 |
0.73 |
1.66 |
0.18 |
| YC |
5.08631 |
5.060 |
5.017 |
5.090 |
0.51 |
1.35 |
−0.08 |
| ZrC |
4.69832 |
4.682 |
4.650 |
4.694 |
0.34 |
1.01 |
0.09 |
| NbC |
4.46933 |
4.458 |
4.430 |
4.458 |
0.25 |
0.87 |
0.25 |
| δ-MoC |
4.27334 |
4.351 |
4.317 |
4.353 |
−1.82 |
−1.04 |
−1.87 |
| HfC |
4.63127 |
4.662 |
4.588 |
4.633 |
−0.66 |
0.93 |
−0.05 |
| TaC |
4.45327 |
4.456 |
4.276 |
4.459 |
−0.08 |
3.97 |
−0.14 |
| WC |
4.37435 |
4.362 |
4.328 |
4.360 |
0.28 |
1.04 |
0.31 |
By utilizing eqn (1), it was subsequently possible to use the previous geometric calculations to compute bulk moduli;
| | 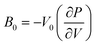 | (1) |
The bulk moduli (
B0), calculated for each functional, are given in
Table 2 alongside a range of experimentally determined values for some of the TMCs. It should be noted that there is a lack of experimental data on the
B0 of 3d carbides beyond group 5 (CrC-NiC), since, as mentioned earlier, only monocarbides with groups 3–5 metals form stable bulk crystals, with rock salt structures.
36,37 It is also important to keep in mind that variations in the numbers of vacancies as well as the precise physical conditions of the materials can lead to a broad range of experimental values.
18,59,60 Therefore, it can be problematic to make direct comparisons between calculated and experimental bulk moduli. However, in many cases our computed values are reasonably close to the experimentally determined ones.
Table 2 Calculated (B0) and experimental (Bexp0) bulk modulus. All values are given in GPa
| Carbide |
B
exp0
|
B
0
|
B
0
|
B
0
|
|
PBE values.
PBEsol values.
RPBE values.
|
| ScC |
148 |
222 |
243 |
247 |
| TiC |
232–390 |
286 |
270 |
276 |
| VC |
308–390 |
268 |
273 |
291 |
| CrC |
— |
348 |
369 |
351 |
| CoC |
— |
681 |
505 |
432 |
| NiC |
— |
207 |
174 |
198 |
| YC |
— |
131 |
125 |
129 |
| ZrC |
159–224 |
241 |
232 |
235 |
| NbC |
300–330 |
331 |
368 |
331 |
| δ-MoC |
319 |
508 |
461 |
464 |
| HfC |
241 |
276 |
256 |
266 |
| TaC |
214–404 |
373 |
359 |
369 |
| WC |
329–587 |
401 |
354 |
350 |
Heats of formation
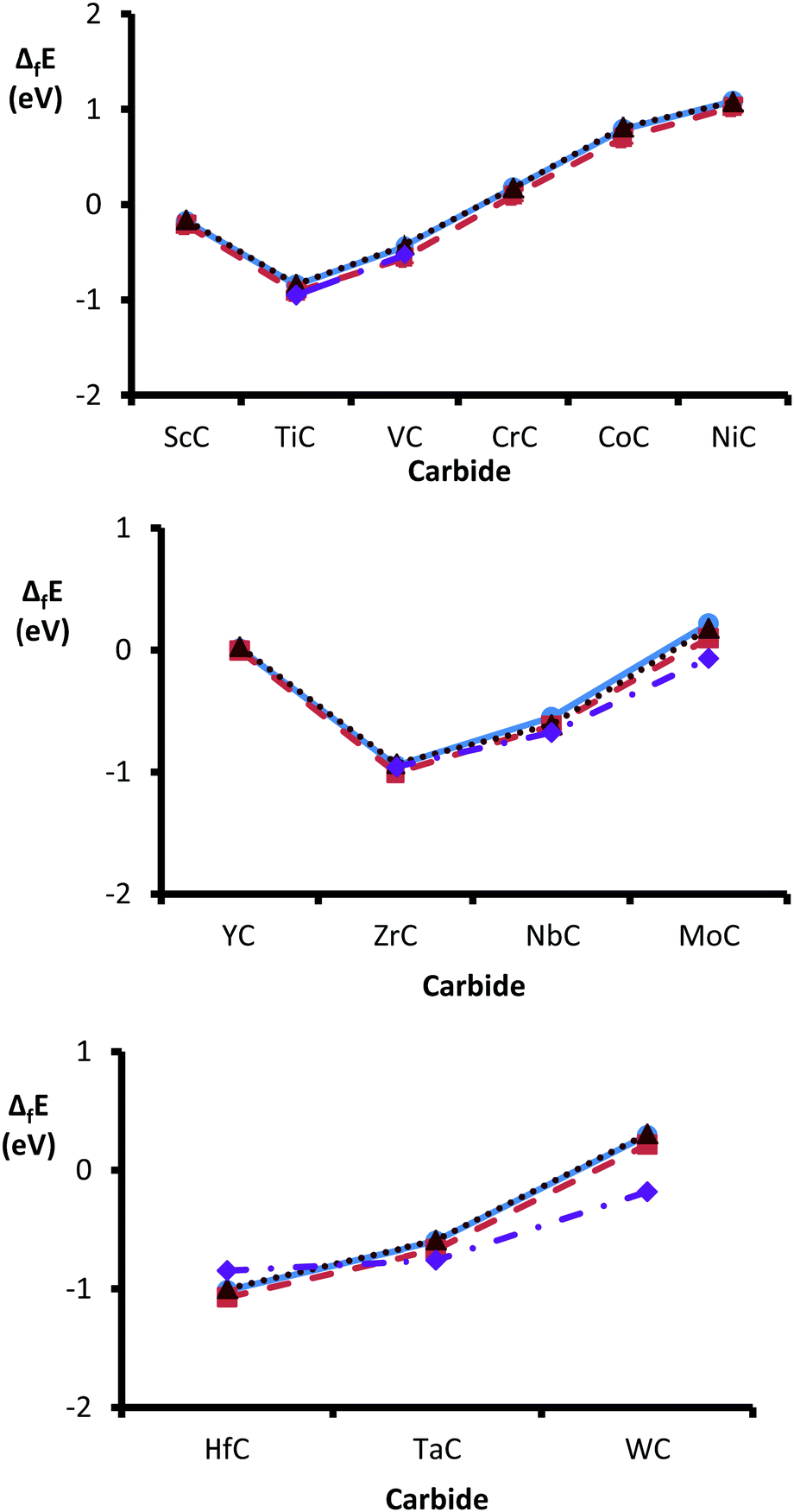
Heats of formation were calculated by subtracting the sum (per atom values) of bulk metal and graphite from the energy (per atom) of the bulk carbide. The heats of formation (ΔfE) were found to show a clear trend across each series, with an early minimum in ΔfE around group 4d (see Fig. 2). As previously noted, carbides that contain TMs early in the periodic series readily anneal into monocarbides, whilst metals with increasingly filled d-bands form more TM-rich carbides.17,61,62 Inserting interstitial carbons into a transition metal lattice causes the weakening of TM–TM bonds and replaces them with TM–C bonds. Therefore, a carbide's heat of formation reflects the competition between these two effects. The nobleness of the parent metal increases from left to right (along each period) causing a weakening in the TM–C bonds.62,63 Our data are in excellent agreement with the available experimental values: TiC,64 VC,64 ZrC,64 NbC,64 δ-MoC,64 HfC,65 TaC64 & WC.65 No experimental data are available for the monocarbides, containing 3d transition metals past group five. However, formation enthalpies (per atom) are available for some of the more transition metal-rich stoichiometries: (Cr3C2) −0.21 eV,66 (Co2C) 0.06 eV64 and (Ni3C) 0.09 eV.64
These energies also support our theoretical findings, since even the more transition metal-rich carbides (containing post group six TMs) show endothermic formation energies. Therefore, the insertion of even relatively low amounts of interstitial carbon into the transition metal lattices of post group six metals is not thermodynamically favoured. The trends reported here are also consistent with a previous study into the relative formation energies of groups IV to VIII transition metal carbides, published in 2016 by Wang et al.62 The authors reported highly exothermic formation energies per atom, of −0.81 eV (TiC), −0.82 eV (ZrC) and −0.94 eV (HfC) for group four carbides, followed by less exothermic ΔfE for group five monocarbides: −0.42 eV (VC), −0.53 eV (NbC) and −0.59 eV (TaC). Apart from δ-MoC (−0.09 eV) and WC (−0.13), all other TMCs analysed had positive ΔfE, which the authors suggest as an explanation for the lack of stable group seven and eight monocarbides with 1:1 stereochemistry.62
Evaluation of the different functionals
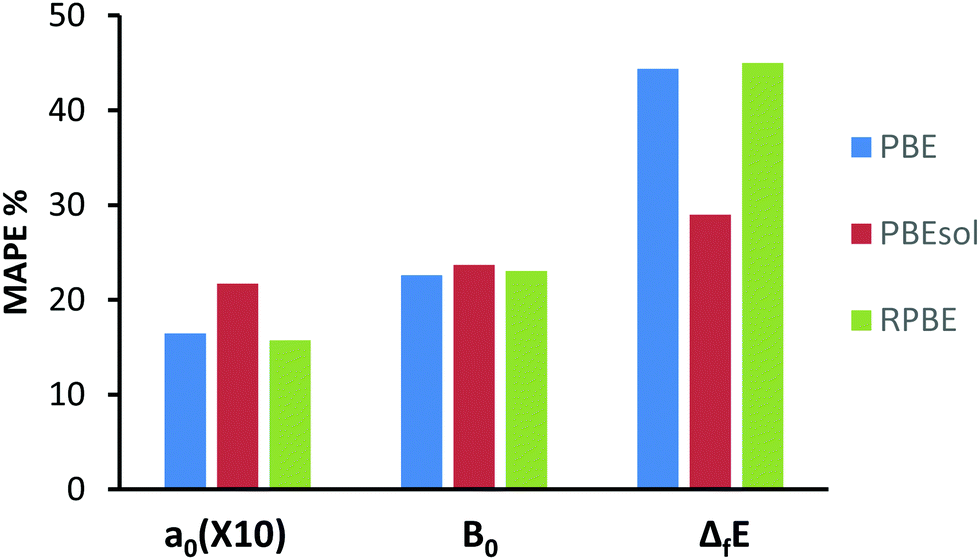
In order to better describe the performance of each functional, we have performed a statistical analysis of their performance versus the experimentally determined values, by summarizing the mean absolute errors of the a0 and B0 as previously described,57,58 presenting our findings in Chart 1. Here we see, that although some functionals do perform better in certain types of analysis, no one functional can be said to clearly outperform the others and all functionals show reasonable overall performance.
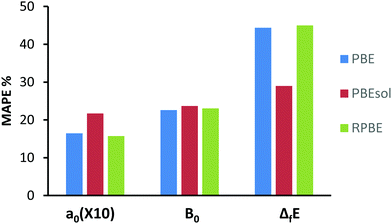 |
| | Chart 1 Mean Absolute Percentage Error (MAPE) for the lattice parameters (a0), energies of formation ΔfE and bulk moduli (B0). MAPE of a0 has been multiplied by a factor of 10 for a better presentation. Data for functions; PBE, RPEsol and RPBE are shown. | |
Low-index surfaces of metal carbides
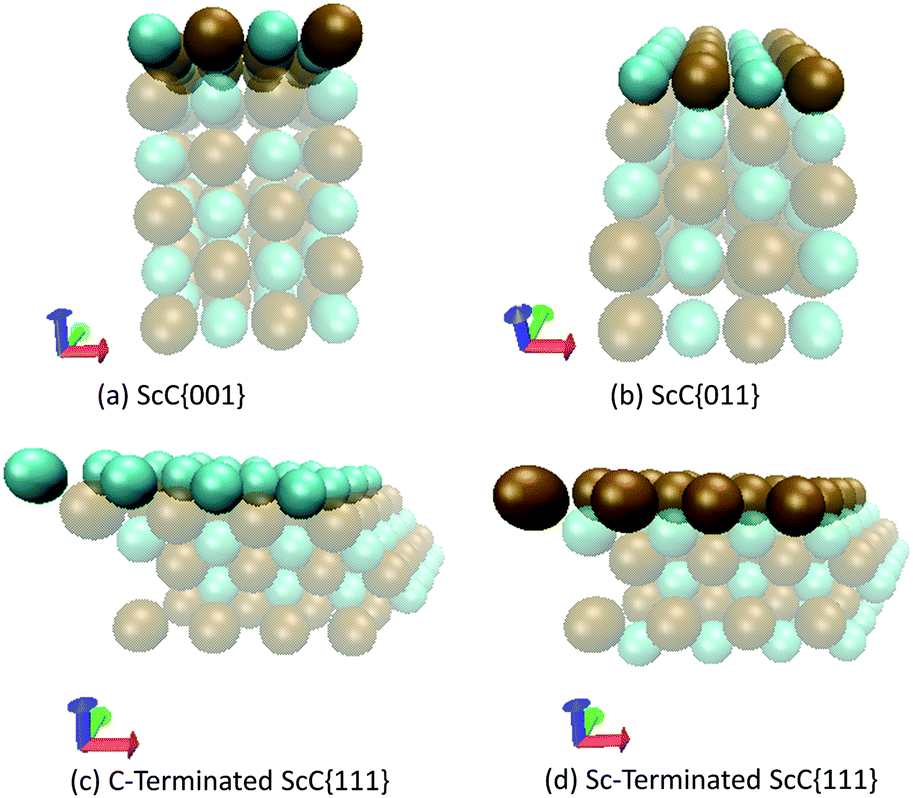
The slab models for all three low-index surfaces were prepared with the aid of the METADISE code.67 Since calculations with all three functionals showed very similar bulk trends, and no obvious systematic errors could be assigned to any functional, it was decided to perform the surface calculation using the PBE functional only. The {001} and {011} faces for each TMC contained six atomic layers terminated by a mixed carbon–metal surfaces.25 The {111} surfaces of each TMC contain alternating layers of metal and carbon atoms and thus they can either be terminated by carbon or by the metal.17,23,25 Each unit cell was multiplied 2 × 2 × 3 to give a total of 96 atoms (with three times the original number of atomic layers in the Z-direction). Subsequently, a vacuum region of 10 Å was added above each surface termination. As an example, the different surfaces of ScC are illustrated in Fig. 2, whilst the energy differences resulting from the relaxation of the layers are included in the ESI.†
After sequentially relaxing each layer, the resulting energies were plotted as a function of the number of relaxed atomic layers which indicated that in all cases a stable plateau was reached between layers three and four. Therefore, it was decided that relaxing just half the slab on each surface would be sufficient to obtain accurate surface properties. A similar protocol has previously been reported by Santos-Carballal et al.68 and it was followed here, because relaxing the whole slab would relax both the carbon- and metal-terminated layers; Thus, any values obtained would represent an average of the two faces. Since it has previously been reported that surface properties may be affected by slab thickness,69–71 we created alternative 2 × 2 × 2 supercells of all {111} surfaces, giving a total of 64 atoms per model. Previous work on TMCs has shown that 4-layer surfaces lead to a sufficiently thick slab for the accurate modelling of surface properties.15,72–74 However, the results obtained using these structures did not correlate very well to those previously determined using 6-layer models; with discrepancies in the values of several work functions of greater than 0.5 eV (see Table S37, ESI†). However, additional test calculation using 8-layer slabs showed excellent correlation with the 6-layer slab models (see Table S39, ESI†). These observations correlate well with previous work by Viñes et al. that also showed a variation in the relaxation of {001} carbide surfaces between their 4- and 8-layer slabs.18 Finally, we decided to benchmark the pristine {111} faces with reconstructed surfaces, whereby migration of surface atoms cancels the perpendicular dipole. Our results are shown in Table S39 (see ESI†). Although the energies of the resulting reconstructed surfaces are slightly lower (=∼1 eV), the relative trends in relaxed surfaces energies remain constant, thereby confirming the validity of our unreconstructed models in this work. In light of the discrepancies between the 4- and 6-layer models only the data from the latter are shown here, with full data sets from the other systems being available in the ESI.†
Once all the different surfaces were fully relaxed, it was possible to assess surface energies (σ), work functions (Φ) and rumpling caused by the uneven displacement of metal and carbon atoms at the surface (r). The surface energy and work function are perhaps the most fundamental characteristics of any material that help to determine the trends in growth rate, surface segregation and catalytic activity.22 However, despite its predictive importance, an experimental determination of the surface energies of various TMCs has proven very difficult due to the very high melting points of this class of materials.22 We therefore begin our analysis of the properties of various surface terminations by relaxing each layer of the surface models sequentially (see ESI†). Surface energies (σ) for the unrelaxed surfaces were calculated using eqn (2). A denotes the area of each surface with Eslab signifying the total energy of the unrelaxed slab and n standing for the number of bulk unit cells used to form the slab volume. The factor of 2 in the denominators is necessary as two surfaces are formed for each system. It is important to note that, in the case of the {111} surfaces, two different surface terminations are considered. Therefore, the energy values are better described as cleavage values ({{111}avg}) that represent an average between the σ of the carbon- and metal-terminated surfaces. These values are calculated in the same way as for σu (see eqn (2)).
| | 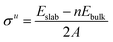 | (2) |
| | 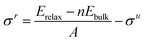 | (3) |
When only half the slab is relaxed, the relaxed surface energy (
σr) must be determined as shown in
eqn (3); here the area of only one surface is included in the denominator and this energy is subtracted from that of the unrelaxed surfaces. The computed
σr for each surface are reported in
Table 3. In the case of the {111} surfaces, the difference in energy between the average cleavage value and the relaxed surface value gives the energy of the purely carbon- or metal-terminated surface relaxation (Δ
σ). The unrelaxed value for the surface energy of a material is roughly equal to half of its cohesive energy (
Ecoh),
75 although, because of variations in dangling bonds, protocols such as the bond-cutting theory
22,76,77 have been used to accurately correlate these two energies. All values are positive since a negative surface energy would imply a negative cohesive energy and any resulting terminations would be unstable and liable to cause the material to disintegrate.
78 Consistent with previous studies,
23 our calculated surface energies confirm the {001} surfaces to be the most stable for all the systems investigated.
Table 3 Calculated unrelaxed (σ) and relaxed (σ) surface energies in J m−2
| Carbide |
{001} |
{011} |
{111}avg |
{111}Cterm |
{111}Mterm |
|
σ (J m−2) |
σ
r
(J m−2) |
σ (J m−2) |
σ
r
(J m−2) |
σ (J m−2) |
Δσ (J m−2) |
Δσ (J m−2) |
|
σ, unrelaxed surface energy; σr, relaxed surface energy; Cave, average cleavage energy; Δσ, relaxation surface energy. |
| ScC |
1.32 |
1.31 |
2.86 |
2.72 |
5.41 |
0.32 |
1.25 |
| TiC |
1.99 |
1.80 |
5.27 |
4.77 |
9.08 |
0.33 |
0.74 |
| VC |
1.95 |
1.57 |
4.97 |
4.27 |
7.94 |
0.70 |
0.99 |
| CrC |
1.64 |
1.20 |
4.25 |
3.11 |
6.50 |
1.06 |
1.44 |
| CoC |
1.09 |
0.57 |
2.79 |
1.73 |
4.82 |
2.56 |
1.99 |
| NiC |
0.67 |
0.12 |
4.01 |
2.85 |
3.44 |
1.46 |
1.49 |
| YC |
1.10 |
1.07 |
2.43 |
2.12 |
4.48 |
0.70 |
1.08 |
| ZrC |
1.88 |
1.73 |
4.75 |
4.27 |
8.06 |
0.52 |
0.59 |
| NbC |
1.90 |
1.56 |
4.62 |
4.09 |
7.28 |
0.98 |
0.45 |
| δ-MoC |
1.00 |
0.49 |
2.79 |
2.16 |
5.43 |
1.31 |
0.99 |
| HfC |
1.83 |
1.74 |
4.91 |
4.58 |
8.22 |
0.25 |
0.43 |
| TaC |
2.22 |
1.79 |
5.07 |
4.46 |
7.68 |
1.35 |
0.43 |
| WC |
1.85 |
1.21 |
4.10 |
3.04 |
5.79 |
1.63 |
0.69 |
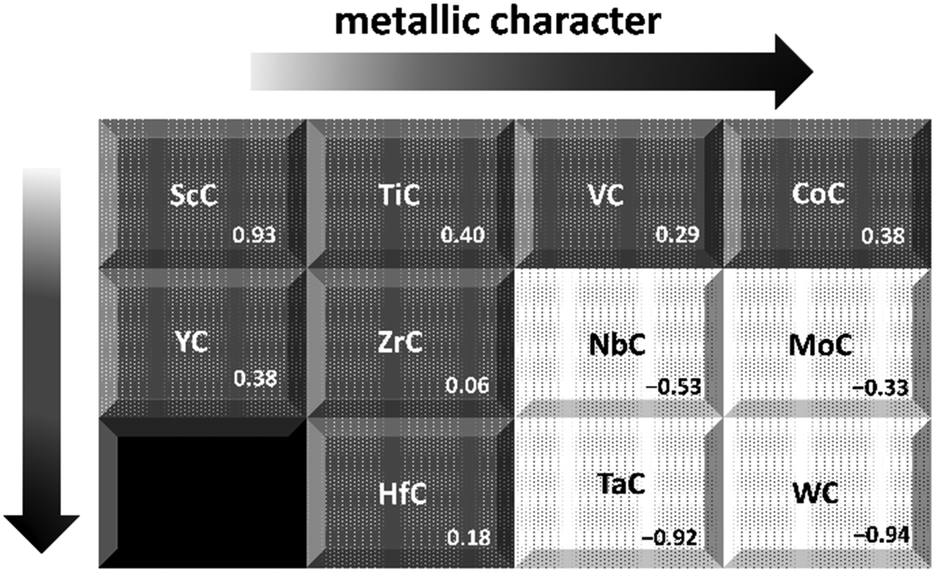
Fig. 3 illustrates the change in preference for the metal- over carbon-terminated {111} surface, across the TMC series. The values were obtained by subtracting the energies of the carbon-terminated surfaces from those of the corresponding metal-terminated surfaces. The results show that the level of preference for a metal-terminated {111} surface decreases across the groups and down the periods. These observations indicate a decreased dominance of the metal-terminated {111} surfaces as the nobleness of the carbide's parent metal increases, to the point where NbC, δ-MoC, TaC and WC demonstrate a preference for carbon-terminated {111} faces. These results help to explain the observation that TiC appears to be exclusively terminated by titanium, when the {111} surface is characterized experimentally.17,79 Also, it has been suggested that, at high temperatures, the preference for termination by the TM could increase due to the higher evaporation rate of the carbon.79 In general, the surface energies are highest for the group 4d carbides, which trend was also reported in 2004 by Hugosson et al.22 They found that one pattern common to all three periodic series was an early maximum for group four TMCs, that may be due to their fully occupied lowest energy d-orbital, which in turn helps to increase the ionic nature of the materials.25 Interestingly, the heats of formation (ΔfE) were found to show a similar (if opposite) trend with an early minimum in ΔfE around group 4d (see Fig. 1).
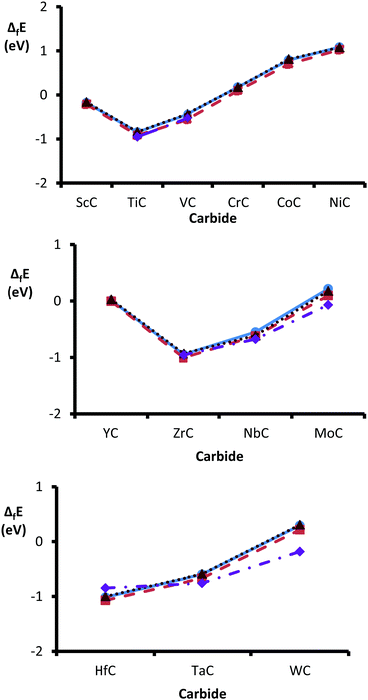 |
| | Fig. 1 Comparison of heats of formation (ΔfE) across the series of group 3d (top), 4d (middle) and 5d (bottom). ΔfE obtained using the PBE method are shown by blue circles (solid lines), values obtained with PBEsol are depicted with orange squares (dashed lines) and by RPBE are shown with brown triangles (dotted lines). Finally, the experimentally determined values are shown by purple diamonds (dash-dotted lines). | |
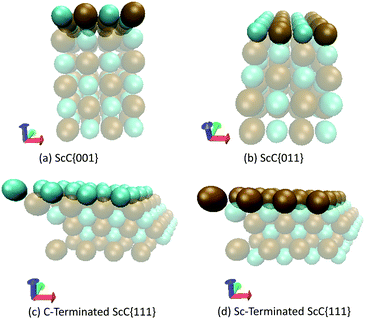 |
| | Fig. 2 Schematic representation of the unreconstructed surface structures of carbon-, metal- and mixed atom-terminated ScC. The green spheres represent carbon atoms and the brown spheres depict scandium. | |
 |
| | Fig. 3 Schematic representation of the degree of preference for metal termination across the carbide series. Values are given in eV and calculated by subtracting the energy of the carbon terminated surfaces from that of the metal. | |
Surface rumpling
As a result of the under-coordination of the surface atoms upon the creation of the surface, a general trend across almost every carbide termination is that atoms making up the surface layer relax into the bulk (see Table 4). Most studies appear to show that often such contraction results in rumpling because of a higher level of inward relaxation of the metal atoms, which leads to a relative displacement of the carbons outwards into the vacuum.17,80–82 These observations are consistent with our calculations, with the two surface terminations that preserve the stoichiometry of the bulk, {001} and {011}, showing a strong tendency for rumpling in a manner that exposes more of the carbons to the vacuum.
Table 4 Surface layer relaxation of the top TMC atomic layer as calculated using PBE level of theory. The initial height of each layer is subtracted from the fully relaxed atomic positions. Values are derived from the bulk (a0) and given in Å
|
|
{001} |
{011} |
{111} |
|
Z
C
|
Z
M
|
Z
C
|
Z
M
|
Z
C
|
Z
M
|
|
Z
C, carbon displacement; ZM, metal displacement. |
| ScC |
−0.01 |
0.00 |
−0.22 |
−0.20 |
−0.44 |
−0.27 |
| TiC |
−0.18 |
−0.30 |
−0.28 |
−0.040 |
−0.41 |
−0.42 |
| VC |
−0.09 |
−0.27 |
−0.11 |
−0.35 |
−0.36 |
−0.36 |
| CrC |
−0.05 |
−0.27 |
−0.08 |
−0.40 |
−0.35 |
−0.35 |
| CoC |
0.11 |
−0.08 |
0.19 |
−0.10 |
0.11 |
−0.34 |
| NiC |
−0.16 |
0.09 |
0.47 |
0.04 |
−0.19 |
−0.16 |
| YC |
−0.19 |
−0.11 |
−0.47 |
−0.31 |
−0.68 |
−0.69 |
| ZrC |
−0.27 |
−0.35 |
−0.38 |
−0.42 |
−0.60 |
−0.59 |
| NbC |
−0.05 |
−0.24 |
−0.11 |
−0.30 |
−0.28 |
−0.28 |
| δ-MoC |
0.09 |
−0.18 |
0.02 |
−0.29 |
−0.36 |
−0.36 |
| HfC |
−0.05 |
−0.14 |
−0.18 |
−0.26 |
−0.30 |
−0.22 |
| TaC |
−0.12 |
−0.32 |
−0.18 |
−0.37 |
−0.34 |
−0.33 |
| WC |
−0.04 |
0.00 |
−0.03 |
−0.38 |
−0.34 |
−0.35 |
Across all TMCs there is consistently a larger contraction of the {011} and metal/carbon-terminated {111} surfaces away from the vacuum, which is to be expected due to the lower coordination of the surface ions – 4 and 3 for the {011} and {111} facets, respectively, compared to 5 for the surface atoms in the {001} surface. The consequence of this effect is that the relative positions of the metals at the TMC {011} faces move further into the bulk than those found on the corresponding {001} surfaces. However, since the carbon atoms also migrate into the bulk, the actual rumpling is similar between the {001} and {011} terminations. The uniform nature of the unreconstructed {111} faces leads to a uniformity in the extent of surface relaxation. However, there is somewhat greater diversity observed in the contraction of the metal and carbon surfaces, than that which is to be seen in the surface energies, due to the varying number of valence d-orbitals and dangling bonds causing different surface compositions at each termination.
Work functions of metal carbides
We now consider the relative redox characteristics of each material. As with the previously discussed bulk moduli, experimental characterization of a TMC's work functions (Φ) is also very susceptible to discrepancies due to variable numbers of carbon vacancies at the surface.83–85 However, it is often a very useful descriptor of the reactivity of a range of different materials. Redox processes are critical to many reaction mechanisms and therefore one may assume that the relative redox potentials of each material will have a strong influence on that material's relative reactivity. To gain insight into the redox behavior of this series of materials we have calculated the work functions of each surface.
For the purposes of this discussion, we will focus on the initial reaction steps required for CO2 reduction by molecular hydrogen.13–16 However, the same principles are relevant to many different redox-driven catalytic reactions. The same carbide surfaces are responsible for reducing electronegative substrates such as CO2 as well as oxidizing electron-donating substrates like H2. A breakdown of the thermochemical contributions to the carbon dioxide adsorption energy are given in eqn (3). Contributions from the ionization energy of the carbide (IPTMC) are subtracted from the sum of the electron affinity of CO2 and the energy associated with the new TMC(CO2) bond formation (ΔGbond). Studies have shown that it is possible to rationalize the respective heights of a very broad range of activation barriers in terms of a valence bond approach.86–89 In theory, the activation energy associated with CO2 adsorption is indicative of an intersection between the endothermic process associated with removing an electron from the bulk carbide and the energy gained in terms of the electron affinity of CO2 and the new bond formed between CO2 and the surface. Since the electron affinity of CO2 is the same for each system and the bond formation energy can only be obtained by a comprehensive study into possible adsorption state, which are not considered here, we will focus on the left-hand side of eqn (3).
| | | ΔGbond = GTMC(CO2) − GTMC − GCO2 | (4) |
| | | BDECO2 = IPTMC − EACO2 − ΔGbond | (5) |
| | | BDECO2 = ΦTMC − EACO2 − ΔGbond | (7) |
When calculating the ionization energies of a solid-state system (IE = Δ
E0 − Δ
E1 + e
−), it is often useful to consider a system's work function (
Φ), which is calculated as shown in
eqn (6). Here
VC refers to the electrostatic potential inside the vacuum and
Ef is the aforementioned Fermi energy (
i.e. the highest energy of electrons in a given bulk volume at zero degrees Kelvin). In essence, this value relates to the minimum amount of energy (in this case in the form of an overpotential) required to excite one electron from the Fermi level out of the finite potential well, that is the carbide bulk, into the vacuum. In non-degenerative semiconductors, the Fermi level is found at least 3
kBT away from the edge of either the valence or conduction bands.
90–92 However, in strongly metallic systems the electrons are degenerative and some density of states is invariably found at the Fermi level, meaning that the work function is in essence a measure of the vertical ionization potentials.
93,94 Therefore, we can substitute work function for ionization potential in
eqn (3), as is seen in
eqn (5). The electron affinity (EA) of a material is defined as the energy between the
VC and the conduction band.
95 In the case of metallic systems, there is no gap between the conduction and valence bands; therefore, a higher work function would indicate a larger electron affinity.
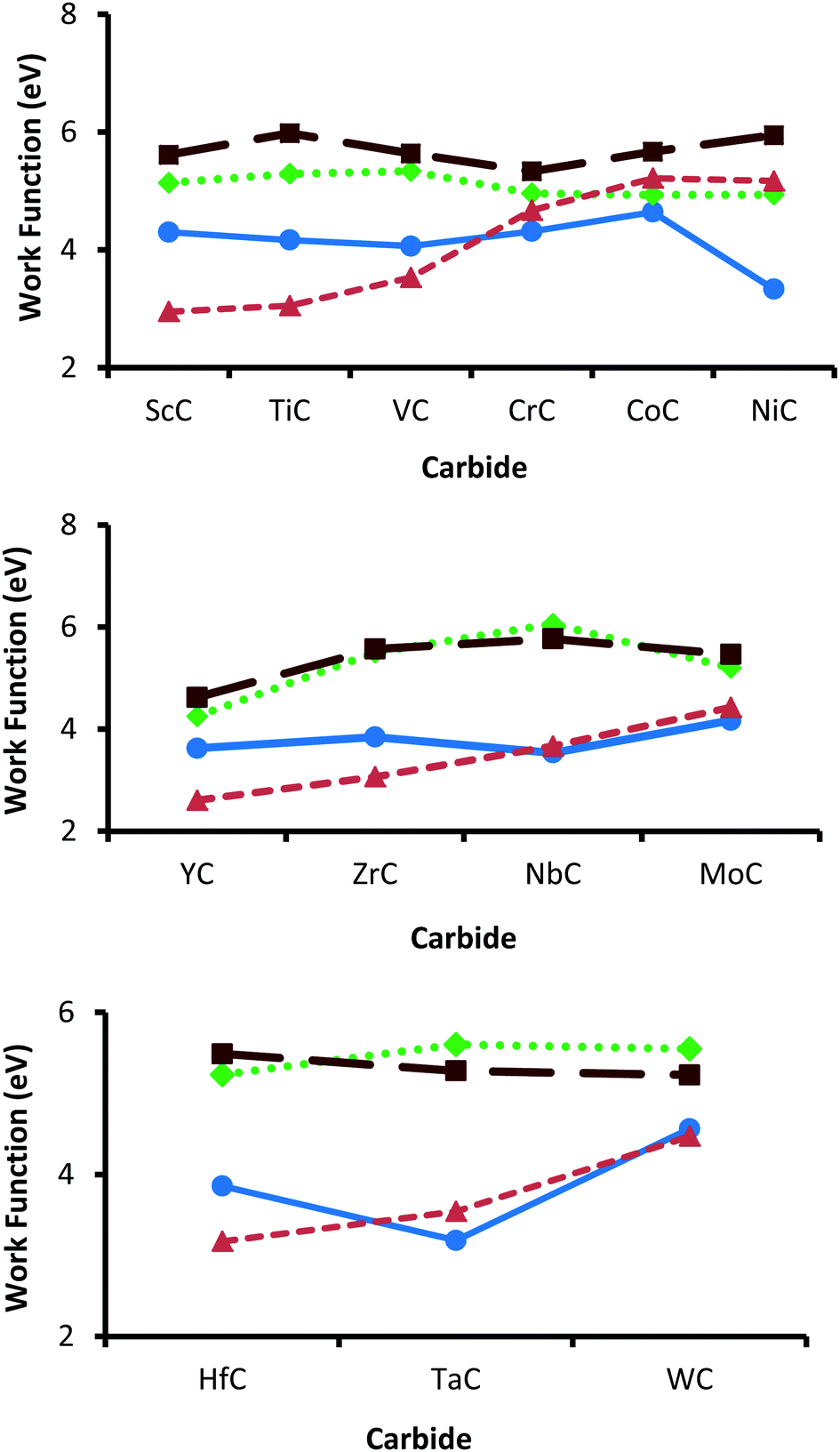
In spite of the inconsistencies in experimentally determined work functions, reported values of 3.8/4.1 eV (TiC),84,85 4.3 eV (VC),85 3.5 eV (ZrC),18 4.1 eV (NbC),85 3.5 eV (δ-MoC),85 4.63 eV (HfC),96 and 4.38 eV (TaC)96 are generally very consistent with the calculated {001} values (see Fig. 4). Experimental studies have determined that the work functions of the parent transition metals steadily increase until a peak is reached towards the end of all three series.97 This trend is replicated reasonably well in the {001} and {011} surfaces of the TMCs. However, this trend is not seen so markedly in any of the {111} terminations, as the work functions of {111}-terminated carbides appear to peak considerably earlier in the series, with maximum values found for VC, NbC and TaC. Since there have not been the same systematic studies into surface properties of the {111} termination,18,22 these results may at first seem surprising. However, it is important to note that even the {001} surfaces show markedly different trends from their parent elemental metals, a point highlighted by Hudosson et al. in 2004.22 Generally, the lowest work functions are found on the {011} terminations followed closely by the {001} surfaces. Importantly, it has been demonstrated that the more open faces such as those found in the {011} terminations are in several cases more reactive than the higher-coordinated ones (such as the {001} faces), regardless of work function.98,99 In terms of redox properties the carbides with the lowest work functions (such as NiC, ScC, YC & HfC) could be expected to be good electron donors. Therefore, these carbides could possibly be suitable reducing agents, for example in the activation of CO2.
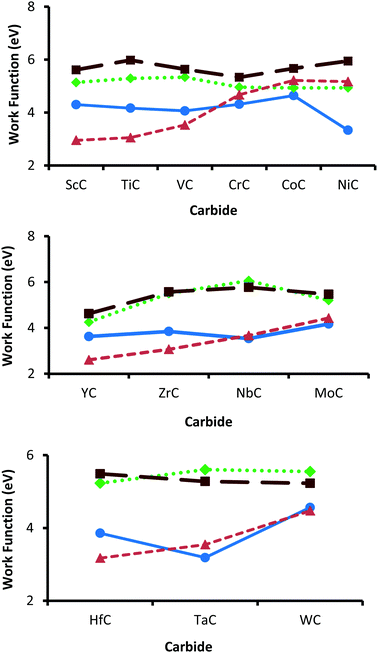 |
| | Fig. 4 Comparison of work functions across the series of group 3d (top), 4d (middle) and 5d (bottom). Values from the {001} surfaces are shown by blue circles (solid line) and work functions obtained from the {011} surfaces are orange triangles (dashed line). The {111} surfaces are either terminated by a carbon layer (dotted line with green diamonds) or by a metal layer (brown squares on a long-dashed line). | |
Density of states
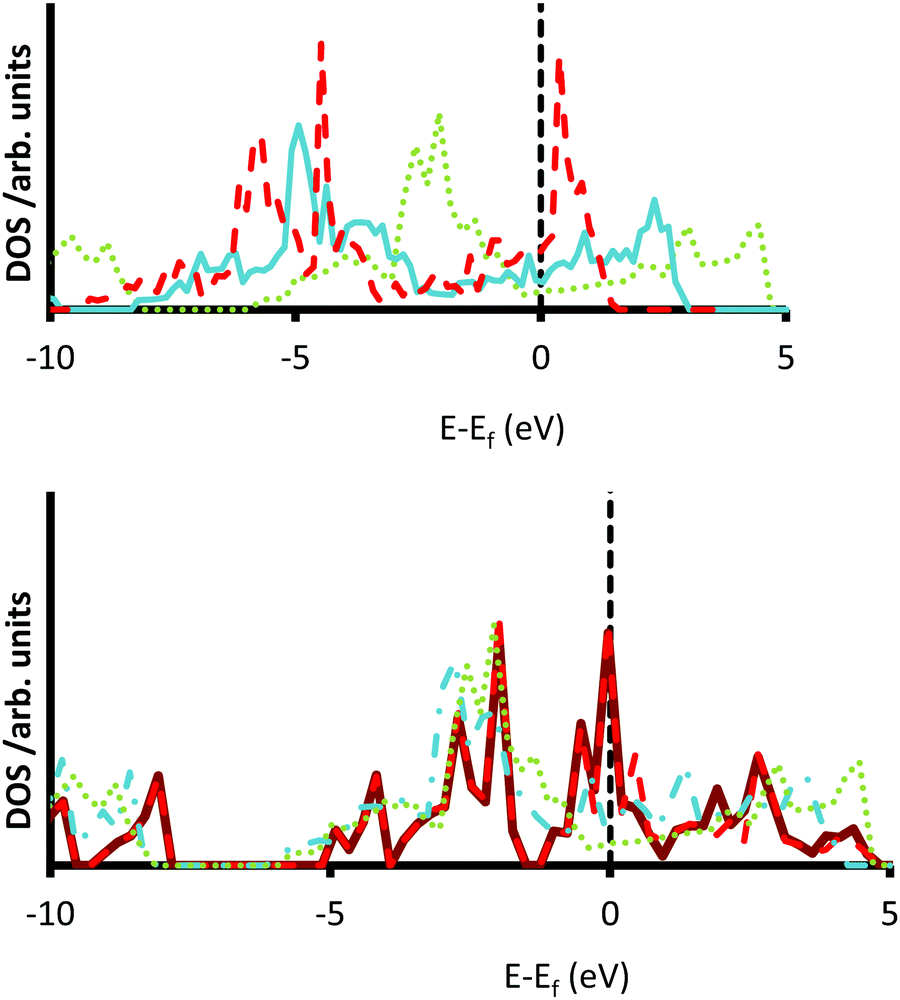
The high work functions seen in the {111} faces of almost all TMCs would be expected to lead to a higher ionization energy and therefore may indicate lower reduction rates of electronegative substrates, as in the case of CO2 redox activation. However, this has not been demonstrated in the experimental studies.17,24 It has been suggested that a reason for the observed increase in reactivity of the {111} surface over the {001} surface is due to the difference in surface states between the surfaces.24 Experimental evidence points to many more surface states close to the Fermi level of the {111} surfaces of a range of carbides.36 We therefore compare the density of states (DOS) of the different materials.
The increased number of surface states in the {111} terminations of the 5d transition metal carbides are illustrated in Fig. 5, where many peaks in the DOS are found at either side of the Fermi levels. A further trend sees an increase in the intensity of the DOS as the transition metal moves from HfC to WC, as a result of which the most localized peaks are seen for WC (see Fig. 5); a trend that is also replicated for the groups 3–6 in the 3d and 4d carbide series (see Fig. S39 and S40, ESI†). This effect can be explained by the decrease in the strength of the carbon–metal covenant bonds in the group 6 carbides.17 Additionally, as the position of the metal component of the carbide moves from left to right along the series, the resulting negative shift in the DOS causes a shrinking of the conduction band as the covalent character of the material decreases. The decrease of charge transfer in the carbides of the metals later in the series is consistent with experimental studies, where the near-edge X-ray adsorption fine structure (NEXAFS) technique indicated a decrease in ionic character when the position of the parent transition metal moves from the left hand side to the middle of the transition metal block of the periodic table.100
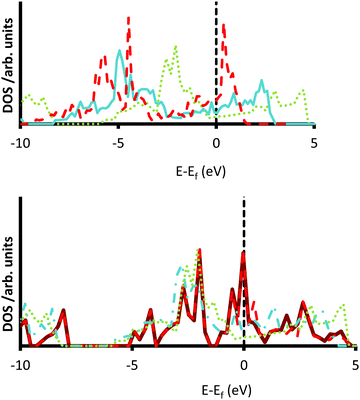 |
| | Fig. 5 Densities of states (DOS) for the 5d transition metal carbides. DOS for HfC gray (dotted) line, TaC blue (solid) line and WC red (dashed) line are shown for the {001} surface (top). DOS are also shown for the {001} grey (dotted) line, {011} blue (dot-dash) line, {111}Cterm red (dashed) line and {111}Mterm green (solid) line termination of HfC (bottom). | |
Kitchin et al.101 and Stottlemyer et al.25 showed a strong correlation between the binding energy of a hydrogen molecule and the d-band center in the density of states calculation. The Fermi level was set to zero and the more negative the center of the d-band density of states the higher the hydrogen binding energy. Importantly, this trend could also be used to explain the increase in reactivity of the TMCs over their parent transition metals,25,101 since the bonds present in the elemental metals are purely metallic. In order to establish the presence of any such trend in the surfaces reported here, we have assessed the d-band centres of each surface, using the established protocol developed by Norskov et al.102,103 The results of this analysis are presented in Table 5 and clearly demonstrate that the centre of the d bands moves away from the Fermi level as more d-electrons are added to the parent metal. More interestingly, it appears that the d-band centres are lowest in the {001} surfaces and highest in the {111} terminations, in a result that mirrors the work function values (see Fig. 4). These results indicate that we could expect the {111} surface to be more active than the {001} faces, in spite of the lower work function in the former, which is related to the potential redox acidity of each surface. These results are in excellent agreement with previous work conducted by Norskov et al., who found that the d-band centre of metallic Platinum changed by ∼1 eV between the {001} and {111} surfaces.103
Table 5 D-Band centres calculated to be of the integrated d-bands for each TMC surface. All values are given in eV, relative to the Fermi level
|
|
{001} |
{011} |
{111}Cterm |
{111}Mterm |
| ScC |
−1.93 |
−1.82 |
−1.09 |
−1.17 |
| TiC |
−4.42 |
−3.92 |
−2.36 |
−2.71 |
| VC |
−7.79 |
−5.79 |
−3.81 |
−4.06 |
| CrC |
−8.39 |
−6.27 |
−4.57 |
−5.73 |
| CoC |
−7.05 |
−5.29 |
−4.54 |
−4.67 |
| NiC |
−6.11 |
−4.54 |
−4.13 |
−2.99 |
| YC |
−1.60 |
−1.56 |
−1.00 |
−0.85 |
| ZrC |
−5.99 |
−5.75 |
−3.65 |
−4.02 |
| NbC |
−9.65 |
−7.87 |
−5.45 |
−5.73 |
| δ-MoC |
−9.73 |
−7.99 |
−5.90 |
−6.29 |
| HfC |
−5.39 |
−5.05 |
−3.24 |
−3.75 |
| TaC |
−7.42 |
−5.80 |
−3.26 |
−3.13 |
| WC |
−9.66 |
−7.75 |
−6.21 |
−6.59 |
In 2005 Viňes et al.18 explored shifts in the core level density of states by comparing the C(2s) bands in diamond and graphite structures to those found in TMCs. Studying TiC, these C(2s) bands were the dominant component in a peak located at −9.5 eV with respect to the Fermi energy.18 Similar peaks at approximately −10 eV from the Fermi level were also found for all surfaces tested in this study. As with the metallic components, these intense peaks indicate more localized electrons, whilst a shift of those peaks towards the Ef indicates a more negative character of the carbons. Djellouli and Aourag demonstrated that TMCs have more ionic character when terminated in the {111} plane,21 which assists in driving this shift in the density of states to higher energy occupations. Therefore, as we increase the ionic character of the carbide, either by moving from right to left across the periodic table100 or by terminating at the {111} surface, the density of surface available states increases.
Another interesting observation is that TMCs containing group 4d metals (TiC, ZrC & HfC) show very little overlap between their valence and conduction bands. In most cases the density of states at the Fermi energy drops to negligible levels. The one group 4 termination that does not follow the same pattern is the {111} face of HfC. Here the Fermi levels are positioned directly in the middle of a very sharp peak in the DOS, which indicates a much more metallic character to these two surfaces. These exceptions notwithstanding, the fact remains that in all other cases, the group four transition metal carbides appear not to have substantial densities of states at their Ef. Stottlemyer et al.25 hypothesized that this may be due to the lack of occupied d-bands, thereby decreasing the amount of charge transfer between the metal and carbon components.
Conclusions
Periodic boundary DFT calculations have been used to undertake an extensive study into the low-index surface properties of a range of 3d, 4d and 5d transition metal carbides with rock-salt structures. In our bulk calculations, almost all the lattice parameters and bulk moduli are in very good agreement with experimentally derived values. Higher levels of surface rumpling are generally observed on surfaces with higher surface energy, i.e. the less stable surfaces. Generally, the lowest energy surfaces were the {001} faces and the lowest work functions appear to be obtained on the {011} terminations. The work functions of the two {111} surface terminations were determined to be higher in energy, whilst shifts in the density of states and d-band centres could indicate interesting activity towards electron-donating substrates. The present study lays the groundwork for more detailed investigations of these systems.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded as part of an EPSRC low carbon fuels grant (EP/N009533/1). Computing facilities for this work were provided by ARCCA at Cardiff University, HPC Wales, and through our membership of the UK's HPC Materials Chemistry Consortium (MCC). The MCC is funded by EPSRC (EP/F067496). All data created as part of this study are openly available at: http://doi.org/10.17035/d.2017.0032992916.
Notes and references
- R. B. Levy and M. Boudart, Platinum-Like Behavior of Tungsten Carbide in Surface Catalysis, Science, 1973, 181, 104–549 Search PubMed.
- P. M. Patterson, T. K. Das and B. H. Davis, Carbon monoxide hydrogenation over molybdenum and tungsten carbides, Appl. Catal., A, 2003, 251, 449–455 CrossRef CAS.
- L. Ping and J. A. Rodriguez, Water-gas-shift reaction on molybdenum carbide surfaces: essential role of the oxycarbide, J. Phys. Chem. B, 2006, 110, 19418–19425 CrossRef PubMed.
- N. M. Schweitzer, J. A. Schaidle, O. K. Ezekoye, X. Pan, S. Linic and L. T. Thompson, High Activity Carbide Supported Catalysts for Water Gas Shift, J. Am. Chem. Soc., 2011, 133, 2378–2381 CrossRef CAS PubMed.
- L. K. Ono, D. Sudfeld and B. Roldan Cuenya, In situ gas-phase catalytic properties of TiC-supported size-selected gold nanoparticles synthesized by diblock copolymer encapsulation, Surf. Sci., 2006, 600, 5041–5050 CrossRef CAS.
- K.-Z. Qi, G.-C. Wang and W.-J. Zheng, A first-principles study of CO hydrogenation into methane on molybdenum carbides catalysts, Surf. Sci., 2013, 614, 53–63 CrossRef CAS.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Carbon capture and storage update, Energy Environ. Sci., 2014, 7, 130–189 CAS.
- M. Aresta and A. Dibenedetto, Utilisation of CO2 as a chemical feedstock: opportunities and challenges, Dalton Trans., 2007, 2975 RSC.
- G. Centi and S. Perathoner, Catalysis: Role and Challenges for a Sustainable Energy, Top. Catal., 2009, 52, 948–961 CrossRef CAS.
- S. Posada-Pérez, F. Viñes, J. A. Rodriguez and F. Illas, Fundamentals of Methanol Synthesis on Metal Carbide Based Catalysts: Activation of CO2 and H2, Top. Catal., 2015, 58, 159–173 CrossRef.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes, Energy Environ. Sci., 2013, 6, 3112 CAS.
- A. Roldan, N. Hollingsworth, A. Roffey, H.-U. Islam, J. B. M. Goodall, C. R. A. Catlow, J. A. Darr, W. Bras, G. Sankar, K. B. Holt, G. Hogarth and N. H. De Leeuw, Bio-inspired CO2 conversion by iron sulfide catalysts under sustainable conditions, Chem. Commun., 2015, 51, 7501–7504 RSC.
- M. D. Porosoff, S. Kattel, W. Li, P. Liu and J. G. Chen, Identifying trends and descriptors for selective CO2 conversion to CO over transition metal carbides, Chem. Commun., 2015, 51, 6988–6991 RSC.
- C. Kunkel, F. Viñes and F. Illas, Transition metal carbides as novel materials for CO2 capture, storage, and activation, Energy Environ. Sci., 2016, 9, 141–144 CAS.
- S. Posada-Pérez, F. Viñes, P. J. Ramirez, A. B. Vidal, J. A. Rodriguez and F. Illas, The bending machine: CO2 activation and hydrogenation on δ-MoC(001) and β-Mo2C(001) surfaces, Phys. Chem. Chem. Phys., 2014, 16, 14912–14921 RSC.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Molybdenum Carbide as Alternative Catalysts to Precious Metals for Highly Selective Reduction of CO2 to CO, Angew. Chem., Int. Ed., 2014, 53, 6705–6709 CrossRef CAS PubMed.
- H. H. Hwu and J. G. Chen, Surface Chemistry of Transition Metal Carbides, Chem. Rev., 2005, 105, 185–212 CrossRef CAS PubMed.
- F. Viñes, C. Sousa, P. Liu, J. A. Rodriguez and F. Illas, A systematic density functional theory study of the electronic structure of bulk and (001) surface of transition-metals carbides, J. Chem. Phys., 2005, 122, 174709 CrossRef PubMed.
- P. M. Stefan, M. L. Shek, I. Lindau, W. E. Spicer, L. I. Johansson, F. Herman, R. V. Kasowski and G. Brogen, Photoemission study of WC(0001), Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 29, 5423–5444 CrossRef CAS.
- L. I. Johansson, A. L. Hagström, B. E. Jacobson and S. B. M. Hagström, ESCA studies of core level shifts and valence band structure in nonstoichiometric single crystals of titanium carbide, J. Electron Spectrosc. Relat. Phenom., 1977, 10, 259–271 CrossRef CAS.
- B. Djellouli and H. Aourag, Theoretical Studies of Stoichiometric TiC, Phys. Status Solidi, 2001, 225, 265–270 CrossRef CAS.
- H. W. Hugosson, O. Eriksson, U. Jansson, A. V. Ruban, P. Souvatzis and I. A. Abrikosov, Surface energies and work functions of the transition metal carbides, Surf. Sci., 2004, 557, 243–254 CrossRef CAS.
- S. Zaima, Y. Shibata, H. Adachi, C. Oshima, S. Otani, M. Aono and Y. Ishizawa, Atomic chemical composition and reactivity of the TiC(111) surface, Surf. Sci., 1985, 157, 380–392 CrossRef CAS.
- T. Noda, M. Yamazaki, K. Ozawa, K. Edamoto and S. Otani, Oxygen adsorption on a ZrC(111) surface: angle-resolved photoemission study, Surf. Sci., 2000, 450, 27–33 CrossRef CAS.
- A. L. Stottlemyer, T. G. Kelly, Q. Meng and J. G. Chen, Reactions of oxygen-containing molecules on transition metal carbides: Surface science insight into potential applications in catalysis and electrocatalysis, Surf. Sci. Rep., 2012, 67, 201–232 CrossRef CAS.
- H. Nowotny and H. Auer-Welsbach, Uber das Scandiumcarbid, Monatshefte fur Chemie, 1961, 92, 789–793 CrossRef CAS.
- K. Nakamura and M. Yashima, Crystal structure of NaCl-type transition metal monocarbides MC (M = V, Ti, Nb, Ta, Hf, Zr), a neutron powder diffraction study, Mater. Sci. Eng., B, 2008, 148, 69–72 CrossRef CAS.
- J. Wang, X. Chen, N. Yang and Z. Fang, Formation of NaCl-type Cr carbide by carbon ion implantation, Appl. Phys. A: Solids Surf., 1993, 56, 307–309 CrossRef.
- M. C. Cadeville and M. F. Lapierre, Etude aux rayons X de la solution solide cobalt–carbone, Scr. Metall., 1972, 6, 399–404 CrossRef CAS.
- J. S. Gibson, J. Uddin, T. R. Cundari, N. K. Bodiford and A. K. Wilson, First-principle study of structure and stability of nickel carbides, J. Phys.: Condens. Matter, 2010, 22, 445503 CrossRef PubMed.
- P. Soni, G. Pagare and S. P. Sanyal, Structural, high pressure and elastic properties of transition metal monocarbides: a FP-LAPW study, J. Phys. Chem. Solids, 2011, 72, 810–816 CrossRef CAS.
- A. N. Christensen, A neutron-diffraction investigation on single-crystals of titanium-oxide, zirconium carbide and hafnium nitride, Acta Chem. Scand., 1990, 44, 851–852 CrossRef CAS.
- G. Will and R. Platzbecker, Crystal Structure and Electron Density Distribution in Niobium Carbide, Zeitschrift für Anorg. und Allg. Chemie, 2001, 627, 2207 CrossRef CAS.
- O. Matsumoto, Y. Yaguchi, Y. Shiota and Y. Kanzaki, Formation of cubic solid solutions in the Mo–Nb–C and Mo–Ta–C systems by the carbonization of oxides in the plasma arc, High Temp. Sci., 1983, 16, 243–250 CAS.
- J. Yang and F. Gao, First principles calculations of mechanical properties of cubic 5d transition metal monocarbides, Phys. B, 2012, 407, 3527–3534 CrossRef CAS.
- L. Johansson, Electronic and structural properties of transition-metal carbide and nitride surfaces, Surf. Sci. Rep., 1995, 21, 177–250 CrossRef CAS.
- S. T. Oyama, Preparation and catalytic properties of transition metal carbides and nitrides, Catal. Today, 1992, 15, 179–200 CrossRef CAS.
- J. G. Chen, Carbide and Nitride Overlayers on Early Transition Metal Surfaces: Preparation, Characterization, and Reactivities, Chem. Rev., 1996, 96, 1477–1498 CrossRef CAS PubMed.
- E. C. Weigert, A. L. Stottlemyer, M. B. Zellner and J. G. Chen, Tungsten Monocarbide as Potential Replacement of Platinum for Methanol Electrooxidation, J. Phys. Chem. C, 2007, 111, 14617–14620 CAS.
- M. B. Zellner and J. G. Chen, Synthesis, characterization and surface reactivity of tungsten carbide (WC) PVD films, Surf. Sci., 2004, 569, 89–98 CrossRef CAS.
- J. Wang, M. Castonguay, J. Deng and P. H. McBreen, RAIRS and TPD study of CO and NO on β-Mo2C, Surf. Sci., 1997, 374, 197–207 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Two-Dimensional Nanocrystals Produced by Exfoliation of Ti3AlC2, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- C. Xu, L. Wang, Z. Liu, L. Chen, J. Guo, N. Kang, X.-L. Ma, H.-M. Cheng and W. Ren, Large-area high-quality 2D ultrathin Mo2C superconducting crystals, Nat. Mater., 2015, 14, 1135–1141 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Restoring the density-gradient expansion for exchange in solids and surfaces, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Improved adsorption energetics within density-functional theory using revised Perdew–Burke–Ernzerhof functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- M. A. L. Marques, J. Vidal, M. J. T. Oliveira, L. Reining and S. Botti, Density-based mixing parameter for hybrid functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 35119 CrossRef.
- R. Peverati and D. G. Truhlar, Performance of the M11-L density functional for bandgaps and lattice constants of unary and binary semiconductors, J. Chem. Phys., 2012, 136, 134704 CrossRef PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Improved tetrahedron method for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef.
- M. Hellenbrandt, The Inorganic Crystal Structure Database (ICSD) – Present and Future, Crystallogr. Rev., 2014, 10, 17–22 CrossRef.
- P. Janthon, S. M. Kozlov, F. Viñes, J. Limtrakul and F. Illas, Establishing the Accuracy of Broadly Used Density Functionals in Describing Bulk Properties of Transition Metals, J. Chem. Theory Comput., 2013, 9, 1631–1640 CrossRef CAS PubMed.
- P. Janthon, S. (Andy) Luo, S. M. Kozlov, F. Viñes, J. Limtrakul, D. G. Truhlar and F. Illas, Bulk Properties of Transition Metals: A Challenge for the Design of Universal Density Functionals, J. Chem. Theory Comput., 2014, 10, 3832–3839 CrossRef CAS PubMed.
- M. Guemmaz, A. Mosser, R. Ahujab and B. Johansson, Elastic properties of sub-stoichiometric titanium carbides: comparison of FP-LMTO calculations and experimental results, Solid State Commun., 1999, 110, 299–303 CrossRef CAS.
- J. C. Grossman, A. Mizel, M. Côté, M. L. Cohen and S. G. Louie, Transition metals and their carbides and nitrides: trends in electronic and structural properties, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 6343–6347 CrossRef CAS.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Hollow Spheres of Iron Carbide Nanoparticles Encased in Graphitic Layers as Oxygen Reduction Catalysts, Angew. Chemie Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- Q. Wang, K. E. German, A. R. Oganov, H. Dong, O. D. Feya, Y. V. Zubavichus and V. Y. Murzin, Explaining stability of transition metal carbides – and why TcC does not exist, RSC Adv., 2016, 6, 16197–16202 RSC.
- B. Hammer and J. K. Norskov, Why gold is the noblest of all the metals, Nature, 1995, 376, 238–240 CrossRef CAS.
- S. R. Shatynski, The Thermochemistry of Transition Metal Carbides, Oxid. Met., 1979, 13, 105–118 CrossRef CAS.
- J. Yang and F. Gao, First principles calculations of mechanical properties of cubic 5d transition metal monocarbides, Phys. B, 2012, 407, 3527–3534 CrossRef CAS.
- W. P. Leroy, C. Detavernier, R. L. Van, M. Lavoie, R. L. Van Meirhaeghe and C. Lavoie, Thin film solid-state reactions forming carbides as contact materials for carbon-containing semiconductors thin film solid-state reactions forming carbides as contact materials for carbon-containing semiconductors, J. Appl. Phys., 2007, 101, 53714 CrossRef.
- G. W. Watson, E. T. Kelsey, N. H. de Leeuw, D. J. Harris and S. C. Parker, Atomistic simulation of dislocations, surfaces and interfaces in MgO, J. Chem. Soc., Faraday Trans., 1996, 92, 433 RSC.
- D. Santos-Carballal, A. Roldan, R. Grau-Crespo and N. H. de Leeuw, A DFT study of the structures, stabilities and redox behaviour of the major surfaces of magnetite Fe3O4, Phys. Chem. Chem. Phys., 2014, 16, 21082–21097 RSC.
- G. Kresse and J. Hafner, First-principles study of the adsorption of atomic H on Ni(111), (100) and (110), Surf. Sci., 2000, 459, 287–302 CrossRef CAS.
- D. J. Siegel, L. G. Hector and J. B. Adams, Adhesion, stability, and bonding at metal/metal–carbide interfaces: Al/WC, Surf. Sci., 2002, 498, 321–336 CrossRef CAS.
- A. Kiejna, T. Pabisiak and S. W. Gao, The energetics and structure of rutile TiO2(110), J. Phys.: Condens. Matter, 2006, 18, 4207–4217 CrossRef CAS PubMed.
- F. Viñes, C. Sousa, F. Illas, P. Liu and J. A. Rodriguez, A Systematic Density Functional Study of Molecular Oxygen Adsorption and Dissociation on the (001) Surface of Group IV−VI Transition Metal Carbides, J. Phys. Chem. C, 2007, 111, 16982–16989 Search PubMed.
- F. Viñes, C. Sousa, F. Illas, P. Liu and J. A. Rodriguez, Density Functional Study of the Adsorption of Atomic Oxygen on the (001) Surface of Early Transition-Metal Carbides, J. Phys. Chem. C, 2007, 111, 1307–1314 Search PubMed.
- F. Viñes, A. Vojvodic, F. Abild-Pedersen and F. Illas, Brønsted–Evans–Polanyi Relationship for Transition Metal Carbide and Transition Metal Oxide Surfaces, J. Phys. Chem. C, 2013, 111, 4168–4171 Search PubMed.
- I. Vavruch, Stefan's rule as a consequence of cohesive forces, Colloids Surf., 1985, 15, 57–62 CrossRef CAS.
- Q.-L. Tang, X.-X. Duan, B. Liu, A.-Q. Wei, S.-L. Liu, Q. Wang, Y.-P. Liang and X.-H. Ma, A density functional study on properties of a Cu3Zn material and CO adsorption onto its surfaces, Appl. Surf. Sci., 2016, 363, 128–139 CrossRef CAS.
- A. Eichler, J. Hafner, J. Furthmüller and G. Kresse, Structural and electronic properties of rhodium surfaces: an ab initio approach, Surf. Sci., 1996, 346, 300–321 CrossRef CAS.
- Z. Łodziana, N.-Y. Topsøe and J. K. Nørskov, A negative surface energy for alumina, Nat. Mater., 2004, 3, 289–293 CrossRef PubMed.
- K. E. Tan, M. W. Finnis, A. P. Horsfield and A. P. Sutton, Why TiC(111) is observed to be Ti terminated, Surf. Sci., 1996, 348, 49–54 CrossRef CAS.
- J. R. Noonan, H. L. Davis and G. R. Gruzalski, Electronically driven reconstruction of TaC(100), J. Vac. Sci. Technol., A, 1987, 5, 787 CAS.
- M. Tagawa, M. Okuzawa, T. Kawasaki, C. Oshima, S. Otani and A. Nagashima, TiC(100) surface relaxation studied with low-energy electron diffraction intensity analysis, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 73407 CrossRef.
- M. Tagawa, T. Kawasaki, C. Oshima, S. Otani, K. Edamoto and A. Nagashima, TMC(100) surface relaxation studied with low-energy-electron-diffraction intensity analysis, Surf. Sci., 2002, 517, 59–64 CrossRef CAS.
- D. L. Price, B. R. Cooper and J. M. Wills, Effect of carbon vacancies on carbide work functions, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 15311–15315 CrossRef CAS.
- C. Oshima, M. Aono, S. Zaima, Y. Shibata and S. Kawai, The surface properties of TiC(001) and TiC(111) surfaces, J. Less-Common Met., 1981, 82, 69–74 CrossRef CAS.
- P. A. P. Lindberg and L. I. Johansson, Work function and reactivity of some crystal faces of substoichiometric transition-metal carbides, Surf. Sci., 1988, 194, 199–204 CrossRef CAS.
- L. Ji, A. S. Faponle, M. G. Quesne, M. A. Sainna, J. Zhang, A. Franke, D. Kumar, R. Van Eldik, W. Liu and S. P. De Visser, Drug metabolism by cytochrome P450 enzymes: what distinguishes the pathways leading to substrate hydroxylation over desaturation?, Chem. – Eur. J., 2015, 21, 9083–9092 CrossRef CAS PubMed.
- A. S. Faponle, M. G. Quesne, C. V. Sastri, F. Banse and S. P. De Visser, Differences and comparisons of the properties and reactivities of iron(III)–hydroperoxo complexes with saturated coordination sphere, Chem. – Eur. J., 2015, 21, 1221–1236 CrossRef CAS PubMed.
- A. S. Faponle, M. G. Quesne and S. P. de Visser, Origin of the Regioselective Fatty-Acid Hydroxylation versus Decarboxylation by a Cytochrome P450 Peroxygenase: What Drives the Reaction to Biofuel Production?, Chemistry, 2016, 22, 5478–5483 CrossRef CAS PubMed.
- M. G. Quesne, R. Latifi, L. E. Gonzalez-Ovalle, D. Kumar and S. P. De Visser, Quantum mechanics/molecular mechanics study on the oxygen binding and substrate hydroxylation step in AlkB repair enzymes, Chem. – Eur. J., 2014, 20, 435–446 CrossRef CAS PubMed.
- Y. Preezant, Y. Roichman and N. Tessler, Amorphous organic devices – degenerate semiconductors, J. Phys.: Condens. Matter, 2002, 14, 9913–9924 CrossRef CAS.
- M. F. Merriam, R. Smoluchowski and D. A. Wiegand, High-temperature thermal expansion of rocksalt, Phys. Rev., 1962, 125, 65 CrossRef CAS.
- O. A. Aktsipetrov, I. M. Baranova, K. N. Evtyukhov, T. Y. V. Murzina and I. V. Chernyĭ, Reflected second harmonic in degenerate semiconductors: nonlinear electroreflection under surface degeneracy conditions, Sov. J. Quantum Electron., 1992, 22, 807–814 CrossRef.
- G. Mills, M. S. Gordon and H. Metiu, Oxygen adsorption on Au clusters and a rough Au(111) surface: the role of surface flatness, electron confinement, excess electrons, and band gap, J. Chem. Phys., 2003, 118, 4198 CrossRef CAS.
- G. Gui, J. Li and J. Zhong, Band structure engineering of graphene by strain: first-principles calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 75435 CrossRef.
- A. Kahn, Fermi level, work function and vacuum level, Mater. Horiz., 2016, 3, 7–10 RSC.
- G. R. Gruzalski, S.-C. Lui and D. M. Zehner, Work-function changes accompanying changes in composition of (100) surfaces of HfCx and TaCx, Surf. Sci., 1990, 239, L517–L520 CrossRef CAS.
- H. L. Skriver and N. M. Rosengaard, Surface energy and work function of elemental metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 7157–7168 CrossRef CAS.
- W. Dong, V. Ledentu, P. Sautet, A. Eichler and J. Hafner, Hydrogen adsorption on palladium: a comparative theoretical study of different surfaces, Surf. Sci., 1998, 411, 123–136 CrossRef CAS.
- K. Christmann and G. Ertl, Interaction of hydrogen with Pt(111): the role of atomic steps, Surf. Sci., 1976, 60, 365–384 CrossRef CAS.
- J. Chen, J. Eng and S. Kelty, NEXAFS determination of electronic and catalytic properties of transition metal carbides and nitrides: from single crystal surfaces to powder catalysts, Catal. Today, 1998, 43, 147–158 CrossRef CAS.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Trends in the chemical properties of early transition metal carbide surfaces: a density functional study, Catal. Today, 2005, 105, 66–73 CrossRef CAS.
- B. Hammer, O. H. Nielsen and J. K. Norskov, Structure sensitivity in adsorption: CO interaction with stepped and reconstructed Pt surfaces, Catal. Lett., 1997, 46, 31–35 CrossRef CAS.
- T. Bligaard and J. K. Norskov, Ligand effects in heterogeneous catalysis and electrochemistry, Electrochim. Acta, 2007, 52, 5512–5516 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables showing the benchmarking of k-points and cut-offs as well as the relaxation energies of each layer for all surfaces are shown here. Also included are charts and tables for the raw bulk data for each TMC. See DOI: 10.1039/c7cp06336a |
|
| This journal is © the Owner Societies 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Alberto
Roldan
*,
Alberto
Roldan