
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceσ-Holes and σ-lumps direct the Lewis basic and acidic interactions of noble metal nanoparticles: introducing regium bonds†
Joakim
Halldin Stenlid
 a,
Adam Johannes
Johansson
b and
Tore
Brinck
*a
a,
Adam Johannes
Johansson
b and
Tore
Brinck
*a
aApplied Physical Chemistry, School of Chemical Science and Engineering, KTH Royal Institute of Technology, Teknikringen 30, SE-100 44, Stockholm, Sweden. E-mail: tore@kth.se
bSwedish Nuclear Fuel and Waste Management Company (SKB), Solna, Sweden
First published on 20th December 2017
Abstract
Using local DFT-based probes for electrostatic as well as charge transfer/polarization interactions, we are able to characterize Lewis basic and acidic sites on copper, silver and gold nanoparticles. The predictions obtained using the DFT-probes are compared to the interaction energies of the electron donating (CO, H2O, NH3 and H2S) and the electron accepting (BH3, BF3, HCl [H-down] and Na+) compounds. The probes include the local electron attachment energy [E(r)], the average local ionization energy [Ī(r)], and the electrostatic potential [V(r)] and are evaluated on isodensity surfaces located at distances corresponding to typical interaction distances. These probes have previously been successful in characterizing molecular interactions. Good correlations are found between Lewis acidity and maxima in V(r), appearing as a consequence of σ-holes, as well as minima in E(r), of the noble metal nanoparticles. Similarly are Lewis basic sites successfully described by surface minima in V(r) and Ī(r); the former are indicative of σ-lumps, i.e. regions of enhanced σ-density. The investigated probes are anticipated to function as reliable tools in nanoparticle reactivity and interaction characterization, and may act as suitable descriptors in large-scale screenings for materials of specific properties, e.g. in heterogeneous catalysis. Because of the similarity between the noble metal nanoparticle's interactions with Lewis bases and the concepts of halogen and hydrogen bonding, a new class of bonds is introduced – regium bonds – taking place between a σ-hole of a Cu, Ag or Au compound and an electron donor.
1. Introduction
During the last few decades nano-sized transition metal particles and clusters have emerged as versatile groups of materials for a wide range of applications such as heterogeneous catalysis, medical therapy and solar energy harvesting.1–6 This is largely a reflection of unique properties of the nanoparticles (NPs) in comparison with the corresponding bulk metal, which can be rationalized by perturbations in the electronic structure of the NPs due to atom under-coordination and quantum size effects.7 The objective of the current study is to understand the connection between electronic structures of group 11 NPs and their interactions with Lewis bases and Lewis acids. For this purpose, we have employed common DFT-based concepts from molecular theory, and examined their performance in describing the interactions of various Cu, Ag and Au NPs.We shall first consider the molecular electrostatic potential [V(r)], which has been used extensively in the analysis of chemical reactivity and intermolecular interactions.8 The electrostatic potential has mainly been used for main group compounds, but recently the applicability domain has been extended to Cu, Au and other transition metal NPs.9–13V(r) is rigorously defined by:
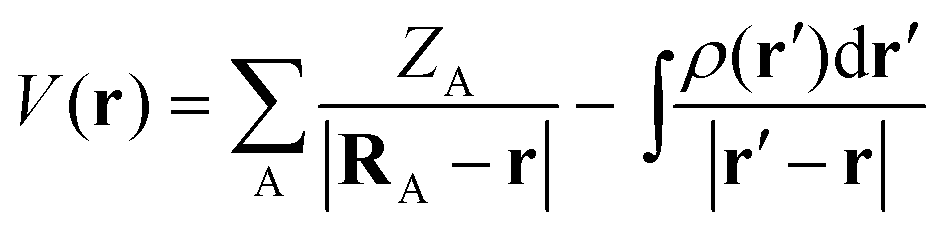 | (1) |
We have recently demonstrated that the size and shape dependent catalytic properties of gold nanoparticles can be explained using the surface electrostatic potentials of the nanoparticles.13 The variations in VS(r) over the nanoparticles were shown to be a consequence of the gold's electron configuration. Here we will use a similar reasoning to explain the interactions of nanoparticles of Cu, Ag, Au with Lewis acids and with Lewis bases.13 As a background to the analysis, we will elaborate on the derivation from ref. 13, which starts from an atomic perspective. The electrostatic potential of an atom is spherically symmetric, everywhere positive, and decreases asymptotically toward zero at increasing distances from the nucleus. When atoms are combined to form molecules or particles, the electron density is redistributed toward the more electronegative atoms and regions of negative potential are formed. As an example, VS(r) of ammonia is negative over the electronegative nitrogen, and the minimum, the VS,min, is located at the lone pair region (Fig. 1a). The magnitude of VS,min gives a direct indication of the strength of interactions with Lewis acids or hydrogen bond donors. For the same reason, the hydrogen atoms of ammonia are positive in VS(r), and the maximum in the surface potential (VS,max) provides a measure of the hydrogen bond donating strength.
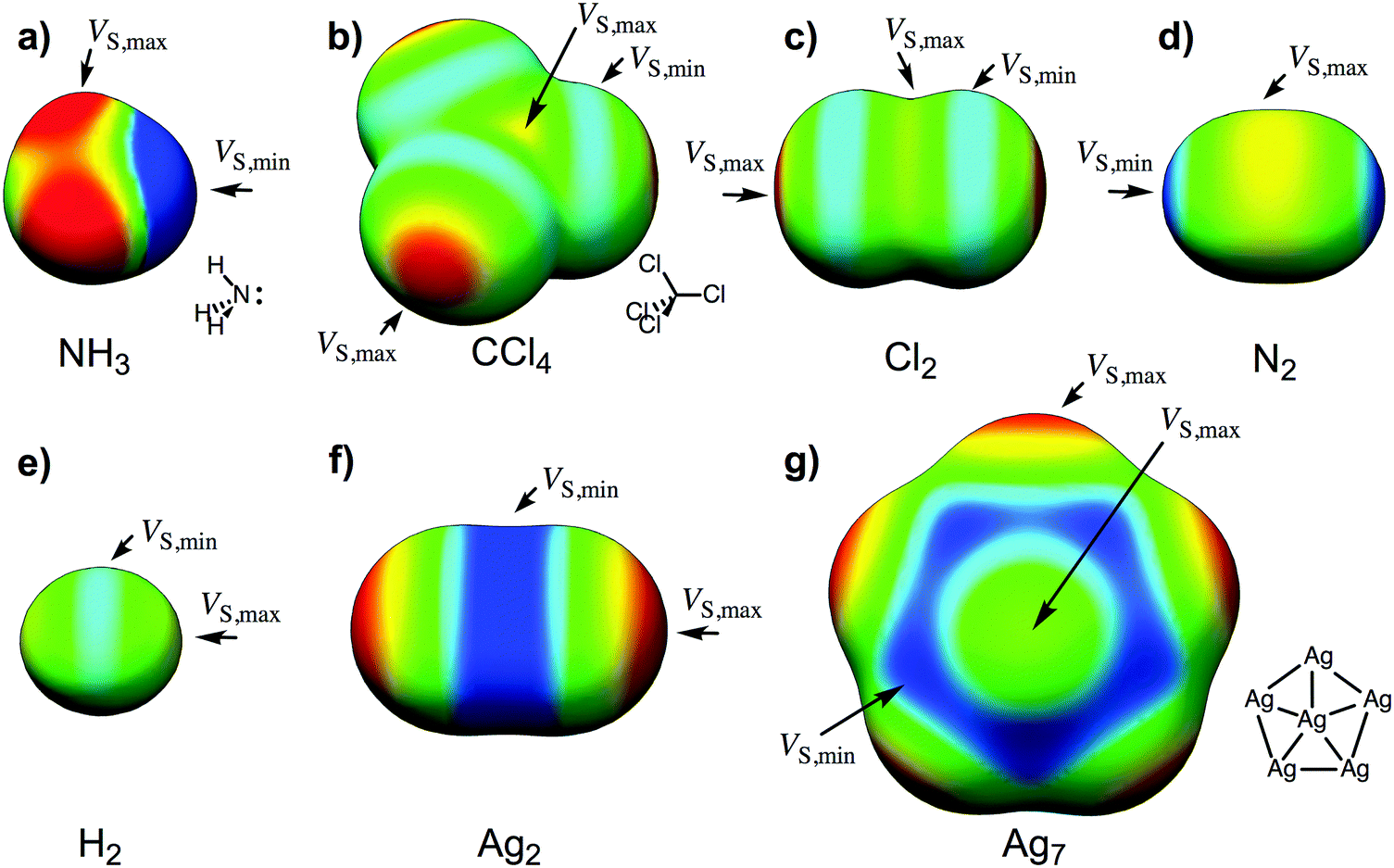 | ||
| Fig. 1 The electrostatic surface potential [VS(r)] computed at the 0.001 a.u. isodensity surface for the (a) NH3, (b) CCl4, (c) Cl2, (d) N2, (e) H2, (f) Ag2, and (g) Ag7 compounds. Coloring from high to low potential, central values of the colors in eV in parentheses: red (0.8) > yellow (0.4) > green (0.0) > cyan (−0.2) > blue (0.4). Selected maxima and minima in the surface potential are marked as VS,max (σ-hole) and VS,min (σ-lump) respectively. | ||
An unexpected feature of VS(r) was first demonstrated by Brinck et al. when analyzing halogenated methanes.15 They found that the charge distribution within a chlorine or heavier halogen can be polarized to such an extent that the potential at the end of the halogen is positive even when the halogen is bonded to a less electronegative atom. (For future reference, we define the end as the outermost region of the halogen atom, X, close to the point that intersects the C–X axis.) Fig. 1b shows this phenomenon on the molecular surface of CCl4, which was one of the molecules of the original study. It was further shown that the positive VS(r) can explain the tendency of halogen compounds to interact with Lewis bases (nucleophiles); this type of interaction is today referred to as halogen bonding.16 Clark et al. later explained the occurrence of the positive potential by a density depletion at the end of the halogen due to polarization of the σ-orbitals; this was denoted a σ-hole.17 A similar type of polarization also exists within the homonuclear diatomics. Fig. 1c shows VS(r) of Cl2, with the characteristic σ-hole(s) (i.e. positive VS,max) at the Cl end(s), and a negative potential region around the middle of each Cl atom. The latter can be traced to the combined contributions from the π and π*-orbitals and the polarized σ-orbitals. The L-shaped form of the Cl2–Cl2 dimer as well as the crystal structure of Cl2 are perfectly consistent with the surface potential of Cl2; the structures are such that VS,min of one molecule aligns with VS,max of another.18–20 Turning toward the N2 molecule, the picture is largely reversed with the most negative potential at the nitrogen tip and the most positive potential at the middle of the bond (Fig. 1d). The latter observation is remarkable considering that the π*-orbitals are unoccupied and the π-orbitals are polarized towards the bonding region. This shows that the surface electrostatic potential is largely governed by the highest σ-orbital. In N2 this orbital is polarized towards the end regions, whereas in Cl2 the orbital is polarized toward the bonding region. Ultimately, this is a reflection of different sp-orbital mixing in the σ-orbitals of the two compounds: in Cl2 the 3s orbitals mix constructively with the phase of the bonding region of the 3pz–3pz σ-orbital, whereas in N2 the 2s orbitals mix deconstructively with the bonding region and constructively with the 2pz lobes at the N2 ends. Figures of the orbitals are provided in the ESI.† Considering the opposite σ-orbital polarization of N2versus Cl2, we find it appropriate to introduce the concept of the σ-lump to rationalize the negative end, the lone pair, of each N in N2. The negative lone pair region of NH3 can also, for obvious reasons, be described as a σ-lump, albeit a much stronger σ-lump than that in N2. In other cases, the σ-lump is not equivalent to a lone pair and the introduction of this new concept is therefore warranted.
The influence of orbital polarization on the surface electrostatic potential is much easier to interpret when only the valence s-orbitals contribute to bonding, such as in the H2 molecule; the bonding σ-orbital is polarized toward the bond, resulting in positive ends and a negative potential in the middle of the bond (Fig. 1e). In other words, there are σ-holes at each end region and a σ-lump in the middle. VS(r) of Li2 (not shown) has a similar pattern, but the σ-holes are stronger and the lump is much more diffuse due to the larger polarizability and the much longer bond length of Li2. The noble metals, Cu, Ag and Au, resemble H and Li in that it is mainly the half-filled s valence orbitals that contribute to chemical bonding. Consequently, the surface electrostatic potential of Ag2 is similar to that of H2, but the magnitudes of the VS,max and VS,min are larger (Fig. 1f). The relevance of the surface potential for the intermolecular interaction tendencies is demonstrated by the Ag2's preference to form T-shaped dimers and to interact end-on with Lewis bases and side-on (middle of the bond) with Lewis acids (see Section S1 of the ESI†).
We can now ask the question whether the σ-orbital polarization, and the positive and negative potential regions resulting from the σ-holes and σ-lumps, respectively, also govern the interactions of larger metal NPs with Lewis bases and Lewis acids. It is anticipated that metal atoms that are not fully coordinated have associated σ-holes that are potential sites for interactions with Lewis bases, and that the magnitude of VS,max at these sites reflects the relative interaction energies at each site. Similarly, we expect that exposed bonds, i.e. σ-lumps, are likely to interact with Lewis acids and that the interaction strengths can be predicted from VS,min (other factors such as charge-transfer and polarization affecting a compound's interaction behavior are discussed below). The surface electrostatic potential of Ag7 is in agreement with the painted picture (Fig. 1g), i.e. the regions of the most positive potential are found at the exposed atoms and are opposite to the chemical bonds, and the negative potential regions are found at the exposed bonds, as well as in the hollow sites in-between atoms. In the following, we will demonstrate that the positions and magnitudes of VS,min and VS,max can be used to identify and rank sites that interact with Lewis acids and Lewis bases, respectively.
It must be emphasized that intermolecular interactions are not solely electrostatic in nature but also have attractive contributions from charge-transfer, polarization and dispersion, as well as repulsive contributions from the Pauli repulsion. Although such a division is artificial, and particularly the differentiation between polarization and charge-transfer is arbitrary, the consideration of different interaction terms can aid the interpretation and prediction of intermolecular interactions. The multifaceted nature of Lewis acid–base interactions was acknowledged already by Pearson who classified Lewis acids and bases as hard or soft.21 Interactions between hard acids and hard bases are considered electrostatically controlled, whereas soft–soft interactions have significant contributions from charge-transfer/polarization. While V(r) for obvious reasons is well suited for the characterization of the electrostatic contribution, it does not capture e.g. charge-transfer/polarization effects. Sjoberg et al.22 recognized this when they introduced the average local ionization energy [Ī(r)]. This property gives a measure of the local cost of ionizing a compound, and, in addition to electrostatics, also reflects the local charge-transfer/polarization capacity of a compound. Surface minima in Ī(r) correspond to sites susceptible to interactions with electron-accepting species (Lewis acids), but cannot be used to characterize interactions with electron donors (Lewis bases). Brinck et al.23 recently introduced the local electron attachment energy, E(r), as a complement to Ī(r). Contrary to Ī(r), surface minima in E(r) correspond to sites where the compound is likely to accept an electron. Hence these sites will, similar to VS,max, correspond to areas susceptible to interactions with Lewis bases. The Ī(r) and E(r) properties have been proven to be useful tools in the study of molecular interactions including non-covalent bonding as well as local reactivity.23–26 The performance of Ī(r) and E(r) in the studied NP interactions will here be compared to V(r) in order to better characterize the origin of the interactions.
Owing to the close resemblance between halogen/hydrogen bonding and the interactions of noble metal NP with Lewis basis, we shall also introduce a new class of bonds – regium bonds, reflecting the royal position of Cu, Ag and Au among the elements in the periodic table. In analogy to halogen or hydrogen bonds, regium bonds are controlled by the polarization of σ-orbitals, and takes place between a charge acceptor (in this case a noble metal atom) displaying a high electrostatic potential, i.e. a σ-hole, and a electron donor with a negative electrostatic potential (for instance the N atom of NH3). Scheme 1 gives a comparison between hydrogen, halogen and regium bonding.
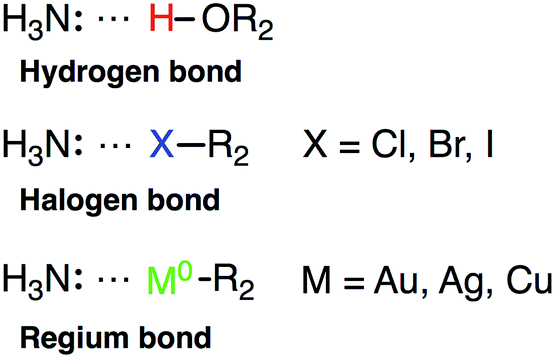 | ||
| Scheme 1 Comparison between hydrogen, halogen and regium bonds. | ||
2. Methods
2.1. Theory
In this section, the theoretical foundation of the Ī(r) and E(r) properties is provided. An account of their practical evaluation and physiochemical significance is also included. Firstly, the use of isodensity surfaces, such as the 0.001 a.u. isosurface, for the evaluation of the descriptor properties used herein deserves an explanation. Chemical interactions and reactions take place at a certain distance from the nuclear framework that constitutes the molecule or particle. In line with this, it has been found that the evaluation of VS(r) and ĪS(r) at a distance from the nuclei corresponding to a constant electron density of 0.001 a.u. [0.004 a.u. for ES(r)], typically marginally more diffuse [dense] than the van der Waals surface, is often a good choice for both qualitative and quantitative studies.8,23,25 In addition by performing the evaluation at a constant density, the analysis is simplified since the Pauli repulsion resulting from the intermolecular electron density overlap will be approximately constant when comparing different adsorption sites of the compound.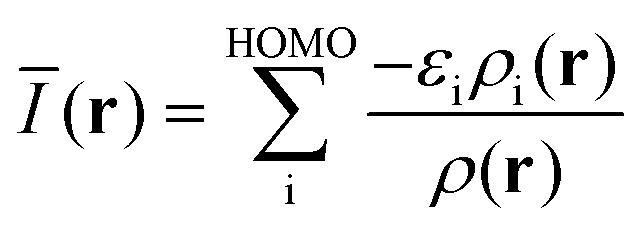 | (2) |
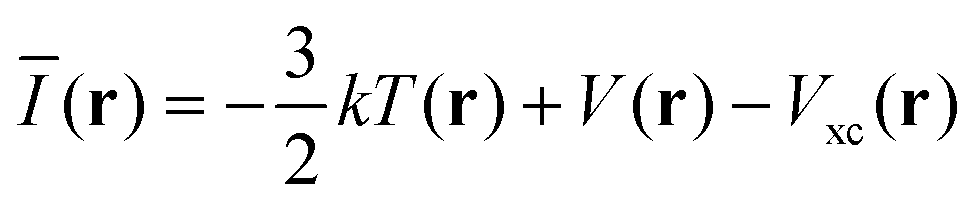 | (3) |
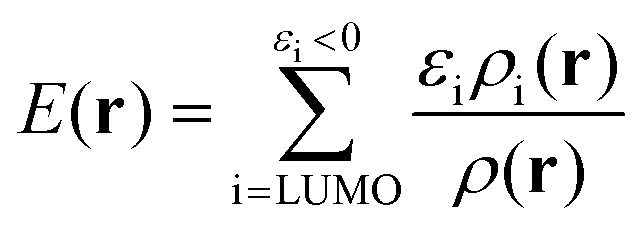 | (4) |
E(r) differs from EAL(r) in the use of a cut-off εi < 0 in the selection of virtual orbitals and in the definition of the denominator, where EAL(r) employs the total virtual orbital density whereas E(r) uses the occupied orbital density.‡ The cut-off is motivated since, within the generalized Kohn–Sham method (GKS-DFT) and given a frozen orbital approximation, only the orbitals with negative eigenvalues will bind a fractional electron, as follows directly from Janak's theorem.23,31 Analogous to Ī(r), E(r) includes contributions from both the electrostatic potential and Vxc, as well as the kinetic energy densities of the virtual orbitals. E(r) can be decomposed as:23
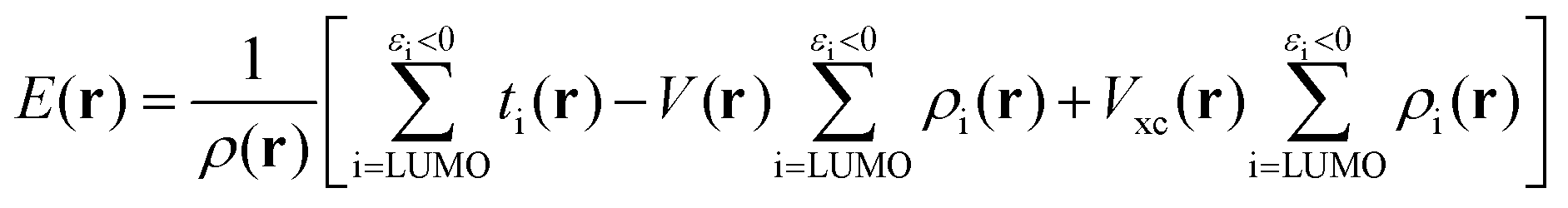 | (5) |
In contrast to VS(r) and ĪS(r), a small benchmark test on organic molecules, comprising aromatic compounds, unsaturated alkenes and halogen bonding molecules, has previously shown that it is more appropriate to evaluate ES(r) on the 0.004 a.u. isodensity surface rather than the 0.001 a.u. surface.23ES(r) is hence obtained at slightly shorter distances from the nucleus than VS(r) and ĪS(r). However, the proper isosurface for interaction analysis on metal NPs remains to be established. In the present study we have thus computed ES(r) on both the 0.001 and 0.004 a.u. isodensity surfaces.
2.2. Computational details
The Cu9, Au9, Ag9, Ag11, Ag17 and Ag18 nanoparticle structures were studied using the Gaussian 09 program suite.32 The structures were chosen on the basis that they have multiple unique adsorption sites and are of a size that is of technical interest while being computationally manageable. In our previous study we studied Au nanoparticles in the size from Au2 to Au561 and found similar variations in VS(r) independent of the particle size.13 All but the Ag18 particles are doublets in their ground states, while Ag18 is a closed-shell singlet. Adsorption energies of the electron donating H2O, NH3 and H2S molecules and electron accepting Na+, HCl (H-down), BH3 and BF3 species were determined at the PBE0-D3(BJ)/def2-TVZPP//PBE0-D3(BJ)/def2-TVZP level of theory33–36 using effective core potentials (ECP)36 for the Ag and Au particles.§ The CO-adsorption energies were obtained from ref. 37. In previous studies it has been found that the use of hybrid functionals, such as PBE0,33 improves the description of transition metal–adsorbate interactions compared to standard GGA functionals.38–43 Likewise, one has found that the inclusion of dispersion (via e.g. Grimme's D3 dispersion corrections34 with Becke–Johnson (BJ) damping35) improves the energetics and structures of e.g. Au nanoparticles34 and other transition metal adsorbate systems.44,45 All structures were characterized as local minima (zero imaginary frequencies) by vibrational analysis employing the harmonic oscillator approximation. Interaction energies, ΔEint, were determined by:| ΔEint = EAds/NP − (EAds + ENP) | (6) |
The modified PBE033 exchange–correlation functional with Hartree–Fock exchange (EHFx) reduced to 10% was used in the evaluation of the V(r), E(r) and Ī(r) properties. The choice of functional was based on an evaluation of a set of functionals ranging from the pure GGA PBE46 to the hybrid PBE0 with different amounts of EHFx (up to 66.67%), see Section S4 of the ESI.† Also included in the evaluation were the range separated HSE06,47,48 the long-range corrected LC-ωPBE49 and the TPSSh50,51 meta hybrid functionals, as well as the hybrid B3LYP52,53 functional and its long-range corrected version CAM-B3LYP.54 The evaluation underlines that a reduced EHFx increases the quality of the E(r) and Ī(r) descriptors, especially at the denser isosurfaces (e.g. 0.004 a.u.). In contrast V(r) is relatively insensitive to the amount of EHFx. The use of a reduced amount of EHFx for DFT calculations of transition-metal compounds is not new, but in-fact a common practice: the so-called B3LYP*55 and PBE0*56 functionals with 10–15% EHFx have for instance been used to describe spin-transitions55,56 and to study enzymatic processes involving redox active open-shell transition metal ions.57,58
We have also found that the combination of the LACV3P*//LANL2-DZ basis sets,59–62 employing the Los Alamos type ECPs for the transition metal atoms, yields results of similar quality for the descriptors as the considerably larger def2 basis set family of Ahlrichs and co-workers36 (i.e. the basis set used for the ΔEint determination). Hence, in order to show that the descriptors can provide accurate predictions at a low computational cost we have employed the LACV3P*//LANL2-DZ basis set combination for the VS(r), ES(r) and ĪS(r) descriptors throughout this study. The in-house program HS95 (T. Brinck) was used to compute the descriptor values; if not otherwise stated, VS(r), ES(r) and ĪS(r) were evaluated on the 0.001 a.u. isodensity surface, since this isosurface was found to generally give the best correlation with computed ΔEint (see the results and discussion section, and the ESI†). NBO,63 Mulliken64 and Bader65 partial charges were determined at the PBE0-D3(BJ)/def2-TVZPP//PBE0-D3(BJ)/def2-TVZP level of sophistication, while the CM5M66 charges were obtained from ref. 37.
The cross-correlated R2 coefficient of correlation, Q2, which was used in the statistical analysis, was evaluated by the leave-one-out procedure:67
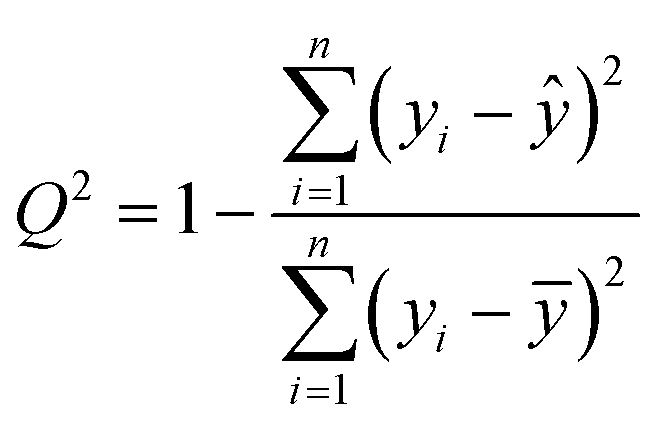 | (7) |
Above, yi is the ith value of y in the data series, ȳ is the average value of y over the data series whilst ŷi/i is an estimated value of y = y(xi) determined from a linear regression analysis of the data series upon excluding the ith data point.
3. Results and discussion
Our results are discussed in the following order: first we will give an overview of the general properties of the studied metal nanoparticles before studying the Lewis acidic and Lewis basic characteristics of the Ag9 nanoparticle in greater detail. This will be compared to the cases of the elementally isomeric Cu9 and Au9 nanoparticles. We will then proceed to assess the performance of the V(r), E(r) and Ī(r) descriptors compared to the results from a previous study of Duanmu et al.37 where site-resolved CO adsorption energies on Ag9, Ag11, Ag17 and Ag18 nanoparticles were correlated with partial atomic charges via the CM5M protocol.663.1. Geometric and electronic structure of the nanoparticles
The Ag nanoparticle structures were obtained from ref. 37. The Ag9, Ag11, Ag17 and Ag18 nanoparticles were re-optimized from the reported geometries while the Au9 and Cu9 nanoparticles were obtained by structural relaxation starting from the geometry of the Ag9 particle. The Ag9 (and hence Cu9 and Au9) and Ag11 structures are based on the D5h Ag7 structure, and formed by extending Ag7 with ad-atoms. Ag11 may, alternatively, be seen as two intertwined Ag7 structures. The Ag17 and Ag18 structures are based on an icosahedral Ag13 substructure. All particles have six or more non-equivalent adsorption sites including sites of three- to six-fold coordination. Over the series of Ag nanoparticles the average coordination number increases with the particle size. As can be seen in Table 1, the average distance between neighboring atoms is quite similar for the Ag particles. There is, however, a slight increase with particle size. For the elemental M9 (M = Cu, Ag, Au) isomers, the Au9 and Ag9 particles are similar in size (similar average bond distances), while the average bond distance in Cu9 is reduced by approximately 12% compared to Ag9 and Au9.
![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) M–M
[Å]
M–M
[Å] |
Sitesb |
![[N with combining macron]](https://www.rsc.org/images/entities/i_char_004e_0304.gif) coord
coord
|
n v-orb | ε LUMO [eV] | ε HOMO(SOMO) [eV] | ΔεHOMO–LUMO [eV] | |
|---|---|---|---|---|---|---|---|
| a Average distance between neighboring atoms. b Number of non-equivalent on top adsorption sites. c Mean coordination number of the particle surface atoms. d Number of virtual spin orbitals below ε = 0. | |||||||
| Cu9 | 2.48 | 7 | 4.67 | 22 | −3.70 | −4.46 | 0.76 |
| Au9 | 2.81 | 7 | 4.67 | 23 | −4.74 | −5.42 | 0.68 |
| Ag9 | 2.82 | 7 | 4.67 | 23 | −3.69 | −4.38 | 0.68 |
| Ag11 | 2.82 | 6 | 4.91 | 29 | −3.72 | −4.31 | 0.59 |
| Ag17 | 2.86 | 8 | 5.88 | 39 | −3.71 | −4.28 | 0.56 |
| Ag18 | 2.84 | 11 | 6.00 | 40 | −3.40 | −4.54 | 1.14 |
The structures of all of the studied nanoparticles are displayed in Fig. 2. VS(r), ES(r) and ĪS(r) determined on the 0.001 a.u. isodensity surfaces are also shown, as well as the valence electronic configuration (valence density of states – DOS). Corresponding figures of the HOMO (or, to be specific, the SOMO, singly occupied molecular orbital) and LUMO orbitals of Ag9 are displayed in Fig. 3. In addition, the HOMO (SOMO) and LUMO orbitals for the other particles are included in the ESI† along with a complete summary of all virtual orbitals used in the evaluation of ES(r) for Ag9, i.e. all unoccupied orbitals of energy lower than the E0 cut-off. The HOMO (SOMO) and LUMO energies can be found in Table 1. An analysis of the frontier orbitals alone is not sufficient to understand the NP interaction behavior (see ESI†). This does not rule out orbital analysis as a tool per se; at the studied level of theory, Ag9 has 23 virtual spin orbitals below the free electron limit (see Table 1 and ESI†). Many of these have energies similar to the LUMO but predict other preferred sites of interactions. Hence, although the use of the LUMO [and HOMOs(SOMOs)] for predicting interactions is limited for these systems, the general use of orbitals for understanding the reactivity is not; by considering the contributions of all relevant orbitals, a better representation is obtained as shown in the following discussion by the use of the E(r) descriptor for local Lewis acidity, and by the Ī(r) descriptor in the case of Lewis basicity.
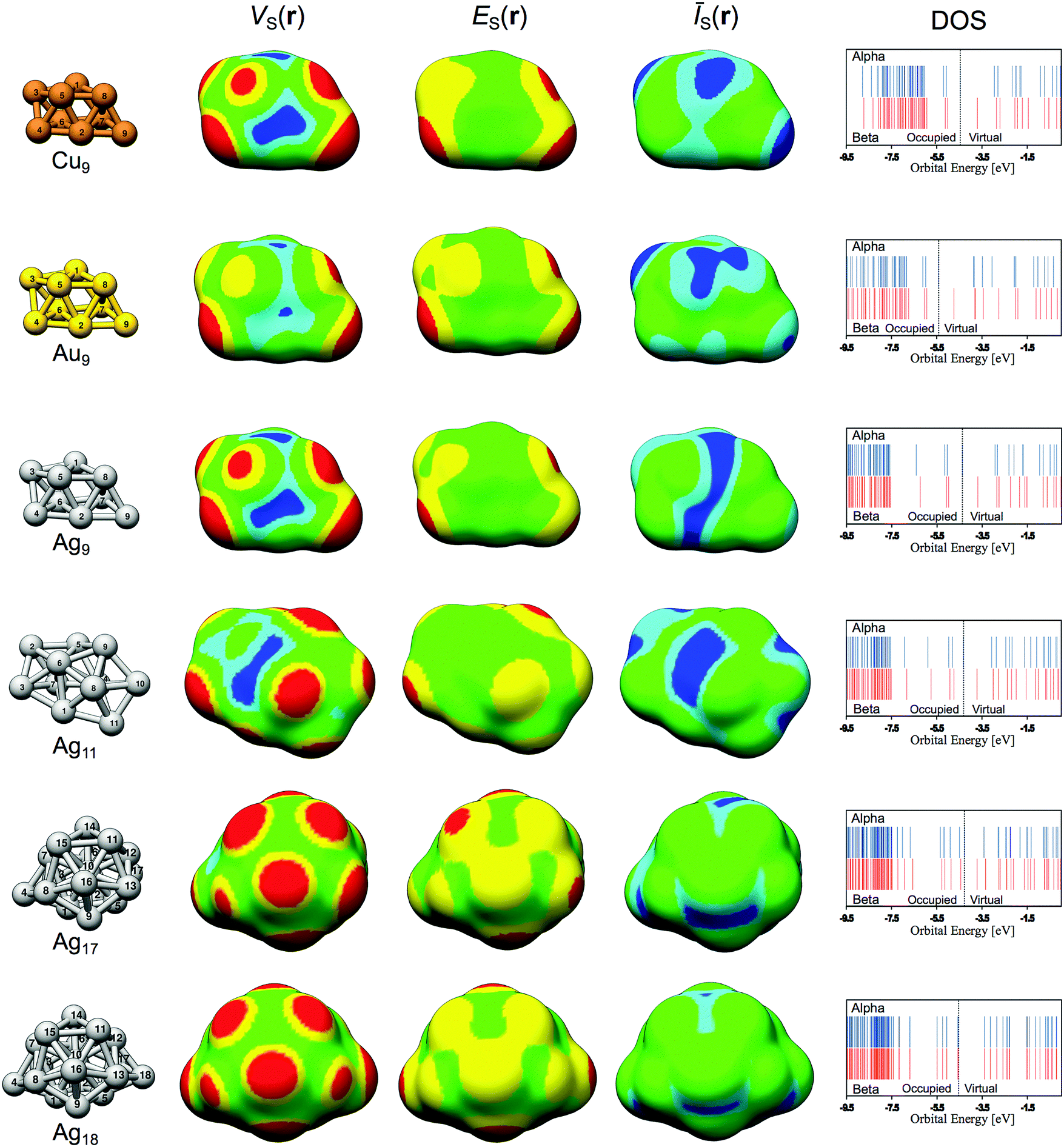 | ||
| Fig. 2 Overview of the Lewis acidic and basic properties of the Cu9, Au9, Ag9, Ag11, Ag17 and Ag18 nanoparticles. The VS(r), ES(r) and ĪS(r) quantities are evaluated on the 0.001 a.u. isodensity surface. Color code for VS(r) in meV: red > 325; 150 < yellow < 325; −150 < green < 150; −325 < cyan < −150; blue < −325. ES(r) in eV: red < −8.0 < yellow < −6.0 < green. ĪS(r) have different color codes for the different metals, in eV; Cu: blue < 5.8 < cyan < 5.9 < green; Ag: blue < 6.1 < cyan < 6.3 < green; Au: blue < 7.2 < cyan < 7.5 < green. The DOS entries show to the electronic valence structure. | ||
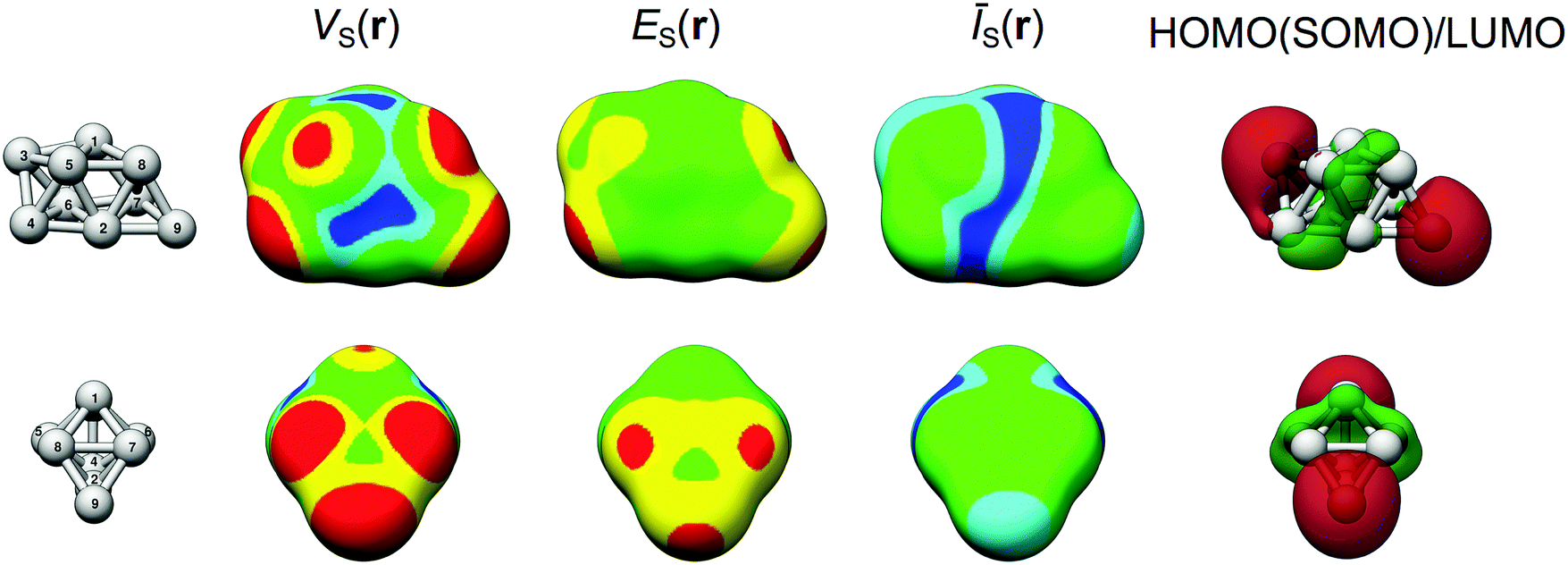 | ||
| Fig. 3 V S(r), ES(r), ĪS(r) at the 0.001 a.u. isodensity surface as well as the LUMO and HOMO (SOMO) orbitals of Ag9. Color code VS(r) in meV: red > 325; 150 < yellow < 325; −150 < green < 150; −325 < cyan < −150; blue < −325. ES(r) in eV: red < −8.0 < yellow < −6.0 < green. ĪS(r) in eV: blue < 6.1 < cyan < 6.3 < green. The HOMO (SOMO) and LUMO orbitals are differentiated by different spin phases (not indicated in figure) with only minor spatial differences. | ||
The VS(r) and ES(r) isodensity maps of Fig. 2 show that Lewis acidic sites (i.e. VS,max sites = σ-holes) can be identified on top of all surface atoms using the VS(r) probe, whereas in a few cases there is no local minimum in ES(r) at the Lewis acidic site. The exceptions include the weak adsorption sites Ag9(2), Ag17(1), Ag17(5), Ag18(1), Ag18(2), Ag18(6), Ag18(7) and Ag18(8). For these sites, the local value of ES(r) at the “on top” site that coincides approximately with the intersection between the particle–adsorbate bond and the isosurface has been used in the interaction analysis. From the VS(r) map we can further identify Lewis basic sites (i.e. VS,min sites = σ-lumps) at bridge and hollow sites formed in-between the atoms of the particle. A more detailed analysis of the Lewis acidic and basic properties of Ag9 follows in Section 3.2 below.
3.2. Lewis acidity and basicity of Ag9
In the following we will elaborate on the details of the local interaction properties of Ag9. This will serve as a prototypical example of the applicability and conceptual transparency of the considered descriptors. Similar analyses are performed in full or in part for the other particles used in the present study (see discussion further below and in the ESI†). Fig. 3 shows how the Lewis basic and acidic character of Ag9 varies over the 0.001 a.u. isosurface from the perspective of the VS(r), ES(r), ĪS(r) properties. In addition, the HOMO (SOMO) and LUMO orbitals are included in the figure for comparison. The reader will now recall that the electrostatic potential VS(r) is able to identify both Lewis acidic (local maxima) and Lewis basic (minima) sites, while minima in ES(r) mark Lewis acidic sites and minima in ĪS(r) mark Lewis basic sites, respectively.| d Mean | d Max | d Min | r wdW | ΔĒintd | Δqnbob | s Δq | V S,min | V S,max | Ī S,min | E S,min | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a van der Waals radius of the interacting atoms, i.e. C, O, S, N, B, B, H, and Na,75 the Bondi van der Waals radii of Ag is 1.72.76 b Average charge-transfer in a.u. upon particle–Lewis base interactions evaluated by NBO charges. c Pooled variance in the atomic partial charge for each interaction over the series. d Obtained at the PBE0-D3(BJ)/def2-TVZPP//PBE0-D3(BJ)/def2-TVZP level of theory. e Note that the NBO analysis suggests that BF3 and HCl donate electrons to Ag9, although they interact via a Lewis basic adsorption mode at the hollow sites. f Could not be determined since no virtual orbital with ε < 0. | |||||||||||
| CO | 2.17 | 2.20 | 2.14 | 1.70 | 8.19 | 0.12 | 0.10 | −13.3 | — | 10.91 | — |
| H2O | 2.51 | 2.63 | 2.46 | 1.52 | 5.56 | 0.05 | 0.03 | −35.1 | — | 9.58 | — |
| H2S | 2.66 | 2.77 | 2.61 | 1.80 | 7.91 | 0.14 | 0.07 | −17.7 | — | 7.99 | — |
| NH3 | 2.36 | 2.41 | 2.33 | 1.55 | 10.55 | 0.09 | 0.05 | −38.9 | — | 7.79 | — |
| BH3 | 2.47 | 2.79 | 2.42 | 1.92 | 24.15 | −0.62 | 0.30 | — | 40.8 | — | −20.18 |
| BF3 | 3.71 | 3.75 | 3.66 | 1.92 | 3.99 | 0.05e | 0.05 | — | 51.2 | — | —f |
| HCl | 2.80 | 2.86 | 2.75 | 1.10 | 5.11 | 0.01e | 0.05 | — | 44.8 | — | —f |
| Na+ | 3.14 | 3.18 | 3.08 | 2.27 | 32.78 | −0.09 | 0.21 | — | 250.1 | — | −87.61 |
We will begin the analysis by discussing H2O and NH3. These are well-behaved probe molecules for evaluation of local Lewis acidity since both H2O and NH3 interact via a single atom (O/N-down) by donation of an electron lone pair without altering the probe molecules or the Ag9 substrate's structure significantly. As described in the Introduction section, this kind of interaction between a noble metal NP σ-hole and an electron donor will henceforth be referred to as a regium bond. The computed regium bond energies of H2O and NH3 for each unique site of Ag9 follow the order predicted by VS(r) with a coefficient of determination (R2) of 0.971 (H2O) and 0.961 (NH3). This is with respect to the local VS,max values determined on the 0.001 a.u. isodensity surface at the sites of interaction. The corresponding R2-coefficents for ES,min are slightly higher, 0.989 and 0.987 for H2O and NH3, respectively (see Fig. 4).
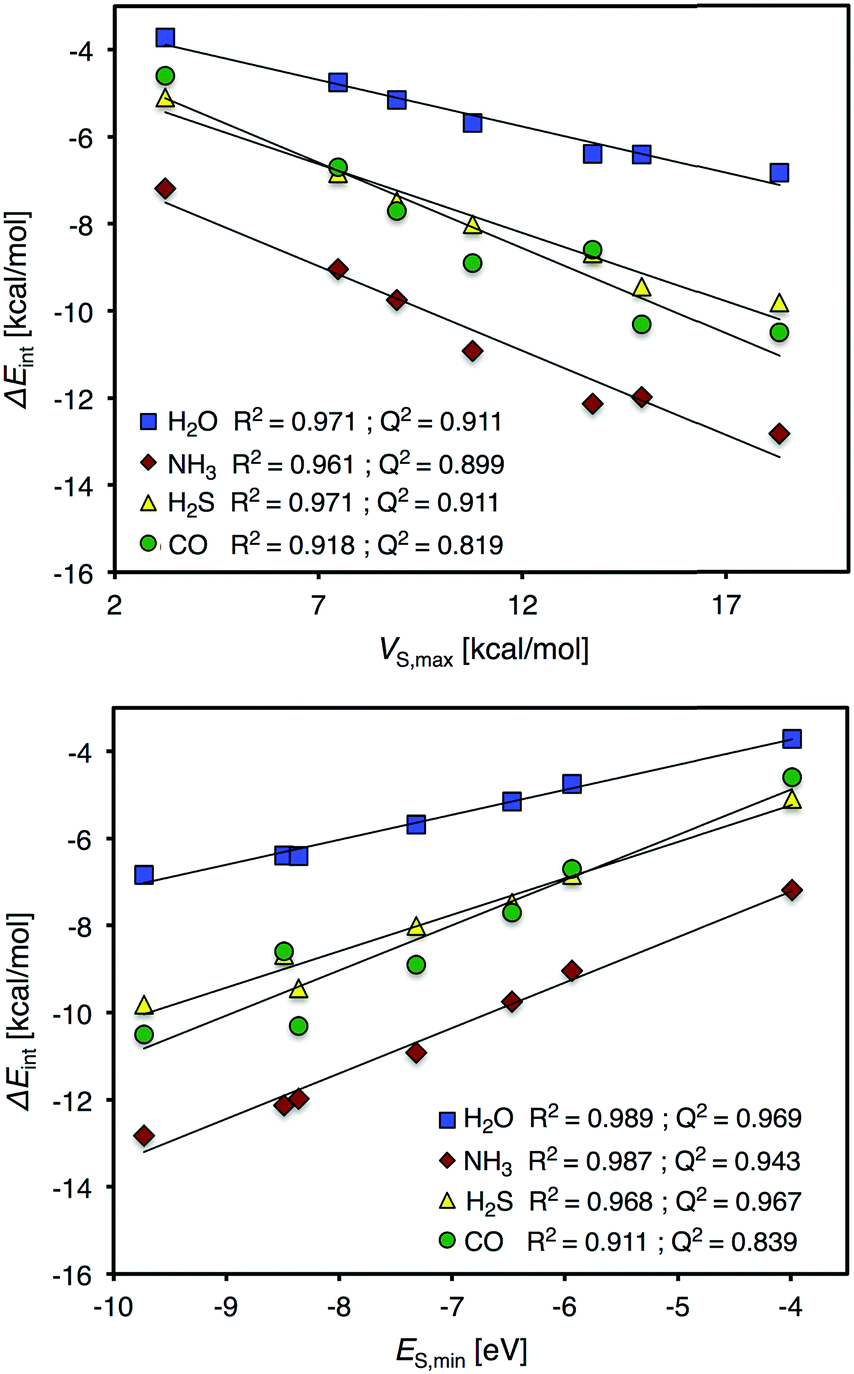 | ||
| Fig. 4 V S,max (top) and ES,min (bottom) values versus local interaction energies of the H2O, NH3, H2S and CO electron donors at the seven unique sites of Ag9. VS,max and ES,min were evaluated at the 0.001 a.u. isodensity surface. | ||
From this isolated comparison one could conclude that ES(r) and VS(r) describe the interactions similarly well and it is difficult to assign the relative charge-transfer/polarization and electrostatic character of the interaction. We can also note that VS,max and ES,min mutually correlate with an R2 value of 0.984. Upon closer analysis it is, however, found that the positions of VS,max better coincide with the adsorption geometries of NH3 and H2O. This can be observed e.g. for position 5 of Ag9 (Fig. 3), or from the fact that there is no local minimum in ES(r) for position 2, which argues for a larger portion of electrostatic control in the interactions. From HSAB theory21 it is known that both H2O and NH3 are considered hard electron donors (Lewis bases); this means that their interactions are expected to be dominated by electrostatics, and not by charge-transfer/polarization. The H2S molecule is located on the other (soft) end of the HSAB scale. We hence expect the regium bonds between the H2S molecule and Ag9 to include a larger portion of charge-transfer/polarization. However, it turns out that VS(r) and ES(r) give similar correlations with the H2S interaction energy as with the H2O and NH3 interaction energies. In addition, VS(r) shows an equally strong correlation with H2S adsorption as ES(r): R2 = 0.971 and 0.968, respectively. Hence, at this point we cannot draw any definitive conclusions regarding electrostatics compared to charge-transfer/polarization effects. It can, nevertheless, be concluded that the interactions are not purely electrostatic since the average interaction energies of the probe molecules (including also CO, vide infra) do not follow the ranking of VS,min of the molecules (Table 2). The comparison between electrostatics and charge-transfer/polarization will be further discussed in connection to the interactions of the Au9 and Cu9 NP of the Section 3.3.
A general conclusion from this study is that the correlation, or the lack of correlation, between descriptor values and interaction energies can partly be explained by the amount of charge-transfer; from NBO analysis we find an inverted correlation between the R2-values for the different series and amounts of charge-transfer Δqnbo (see Table 2 and the ESI†). An even stronger inversed trend is found between R2-vaules for the different series and the variance of the charge-transfer within the series. Taken together, this suggests that the predictive power of the ground-state descriptors VS(r) and ES(r) suffers the more the electronic structure of the ground state is altered by the interactions. The implication is that soft interactions are more difficult to describe than hard interactions since these by nature cause a larger deviation from the ground state. Similarly, and also as a general conclusion, we find that the more the geometries of the adsorbate and NP are distorted upon interactions, the weaker are the correlations between interaction energies and the descriptor values (see the ESI†). In addition to the above, some deviation between descriptor predictions and computed interaction energies is expected since the descriptors do not directly reflect dispersion effects or vibrational energy contributions.
The interactions of CO with the Ag9 have previously been studied by Duanmu et al.37 Although we will here treat CO as a Lewis base, the CO molecule is known to be an ambivalent adsorbate with a mode of interaction that is characterized by both electron donation from CO and back-donation from the substrate as a result of a repulsive σ-interaction and constructive π-orbital overlap,69–72i.e. CO acts as both a Lewis base and, to some degree, a Lewis acid. Upon interactions with metal surfaces it has, moreover, been found that CO undergoes a substantial valence orbital rehybridization.73,74 The density difference plots obtained for H2O, H2S, NH3, and CO adsorption are shown in Fig. 5 illustrating the different interaction modes where CO adsorption leads to a constructive orbital overlap leading to a buildup of electron density; this is indicative of a covalent bond, whereas H2O, H2S and NH3 adsorption instead polarizes the charge densities of the interacting species. The latter suggests an electrostatic interaction enhanced by polarization rather than a charge-transfer complex. On the other hand the interactions with all Lewis bases result in adsorption distances considerably shorter than the sum of the van der Waals radii of the interacting atoms, which suggest interactions stronger than van der Waals interactions. Furthermore, as discussed above, there is no correlation between the average interaction energy and VS,min of the Lewis bases, where, e.g., H2S binds stronger than H2O.
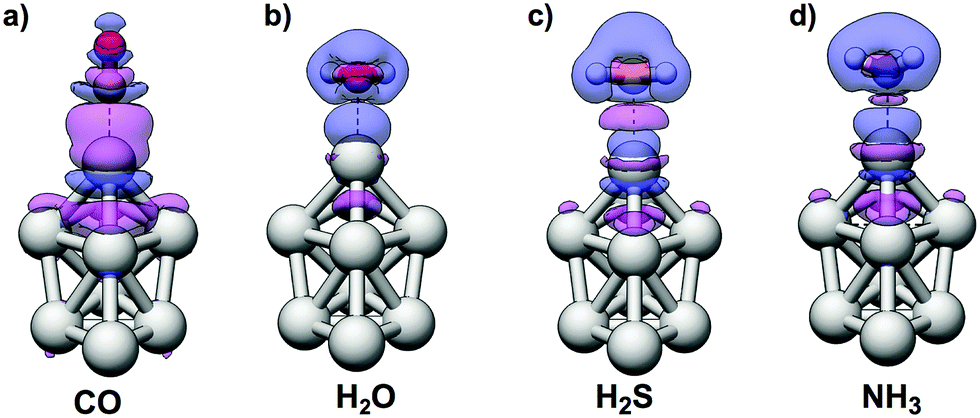 | ||
| Fig. 5 Density difference plots at the 0.00075 a.u. contours for (a) CO, (b) H2O, (c) H2S and (d) NH3 adsorption onto atomic site 4 of Ag9. Color: purple = density accumulation, blue = density depletion. The figures are obtained at the optimized adsorbate structures by subtraction of the individual densities of Ag9 and the adsorbate, with frozen coordinates, from the total density of the Ag9–adsorbate complex. | ||
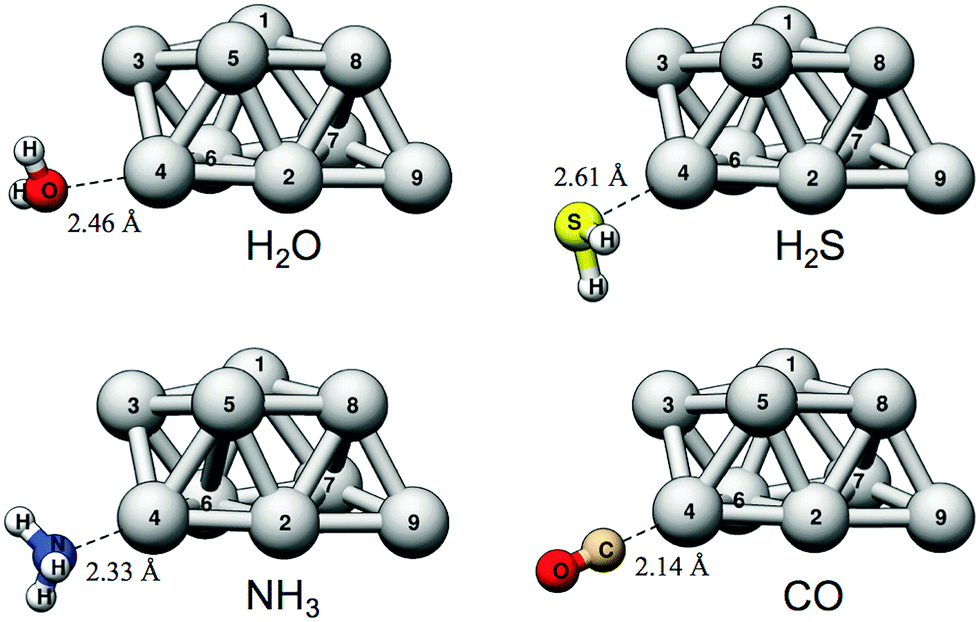 | ||
| Fig. 6 The favored adsorption structures for the H2O, H2S, NH3 and CO Lewis bases onto Ag9. | ||
Given the complex nature of the CO interaction it is expected that neither VS,max nor ES,min will fully capture the interaction by itself. Indeed, among the considered adsorbates the CO interaction energy displays the weakest correlation with respect to the descriptor values [R2 = 0.918 for VS(r) and 0.911 for ES(r)]. Nonetheless, these correlations are significantly better than those obtained by Duanmu et al.37 based on CM5M66 partial atomic charges (R2 = 0.74). The comparison between the VS(r) and ES(r) descriptors and partial charges for CO interactions will be discussed further in Section 3.4.
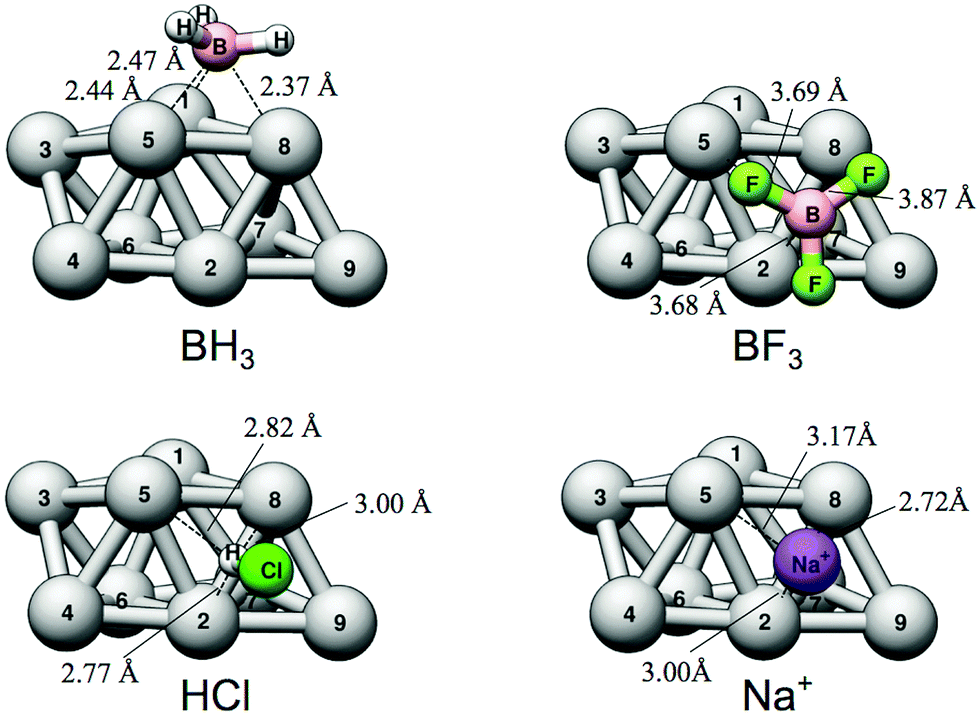 | ||
| Fig. 7 The favored adsorption structures for the BH3, BF3, Na+ and HCl (H-down) electron-acceptors onto Ag9. Note that BH3 favors the h158 site while the remaining compounds prefer the h238 site. The depicted bond distances correspond to those between the central atom of the adsorbent and the adjacent Ag9 atoms. | ||
Similar to the case of Lewis base adsorption, we can employ local descriptor values (the VS,max and ES,min) at the central interacting atom of the probe molecules to better understand the nature of the interaction. We did, for instance, find that ES,min could not be determined on either HCl or BF3 since, at the considered level of theory, they do not have virtual orbitals of negative eigenvalues. Consequently the contribution from charge-transfer to the Lewis acidity is expected to be small, which is corroborated by a NBO analysis (Table 2). Moreover, the relative interaction energies do not follow the ranking of VS,max of the Lewis acids. Again this underlines that the interactions of (some of) the Lewis acids are more complex and not dominated by electrostatics. An interesting observation in connection to this is that in the case of BH3, NBO analysis shows a large charge-transfer of around 0.6e− from Ag9 to BH3. Hence the strong charge-transfer interaction compensates for its comparable small electrostatic driving force giving rise to a comparably strong overall interaction.
3.3. Lewis acidity of Au9, Ag9 and Cu9
As shown above, clearly VS(r), ES(r) and ĪS(r) reflect the interaction behavior of the Ag9 nanoparticle. However, are these descriptors able to capture trends also for other group 11 NPs? This will be investigated by comparing the Lewis acidity of the three elemental isomers Cu9, Ag9 and Au9 probed by H2O and H2S adsorption. Since we are here focusing on Lewis acidity, only the VS(r) and ES(r) descriptors will be considered.Apart from the fact that the Cu particle is considerably smaller in comparison to the other particles, Au9, Ag9 and Cu9 are structurally identical. The similarities are reflected in the VS(r) and ES(r) isodensity maps shown in Fig. 2 that display a close resemblance between the particles. The VS(r) and ES(r) maps indicate that the on top sites are Lewis acidic centers also for the Au and Cu particles. Accordingly, adsorption of H2O and H2S takes place on top of the atomic positions also for the Au and Cu particles. The average binding distances for H2O are 2.17 ± 0.11 Å for Cu9, 2.47 ± 0.15 Å for Ag9, and 2.51 ± 0.12 Å for Au9. On the average, H2O adsorbs strongest onto the Cu particle by −7.4 kcal mol−1, by −6.2 kcal mol−1 and 5.6 kcal mol−1 onto Au9 and Ag9 respectively. H2S adsorbs at the same sites but here Au9 displays the strongest average interactions (−14.8 kcal mol−1) followed by Cu9 and Ag9 (−10.2 and −7.9 kcal mol−1). Upon interaction between Au9 and H2S, the particle geometry is largely distorted. This is a reflection of the well-known fluxionality (i.e. geometric adaptability) of Au NPs, a feature that is often acknowledged as an important factor for the high catalytic activity of Au NPs.77 Due to the large geometric rearrangement upon the H2S–Au9 interactions, the Au9 atoms were constraint at their original positions during optimization of the adsorption structures in order to facilitate the analysis of the correlation between descriptor values and interaction energies.
Fig. 8 shows that the trend in H2O interaction energies of the particles closely follows the variations of the relative magnitude of both VS,max and ES,min on the 0.001 a.u. isosurface for all the nanoparticles. For H2O adsorption, ES,min gives similar correlations for all three particles with R2 ≈ 0.985, whereas VS,max gives slightly weaker correlations for Au and Cu (R2 ≈ 0.950) compared to Ag9 (R2 = 0.971). We can, moreover, note that H2O and Cu9 form the strongest interaction amongst the nanoparticles. This is well-reflected by the electrostatic potential since Cu9 has the largest VS,max of the particles. ES,min does, on the other hand, predict the Au9 nanoparticle as the most reactive, which instead is in-line with the H2S adsorption energies. It is interesting to note that the Cu particle, which is considered a harder metal than Au, forms the strongest bonds to the hard H2O adsorbate, while the soft Au9 forms the strongest bonds with the soft H2S adsorbate. As concerning the H2S correlations, the Au9–H2S interaction gives comparable poor R2 values of 0.856 for VS,max and 0.916 for ES,min on the 0.001 a.u. isosurfaces, respectively (see Fig. 8). This can be attributed to a large charge-transfer/polarization effect upon H2S adsorption leading to a redistribution of the electronic structures, which is a reflexing of the large tendency for structural changes of Au9 upon H2S adsorption. For Cu9–H2S interactions we find correlations of R2 = 0.941 for VS,max and R2 = 0.963 for ES,min, comparable to, but slightly weaker than, those of Ag9.
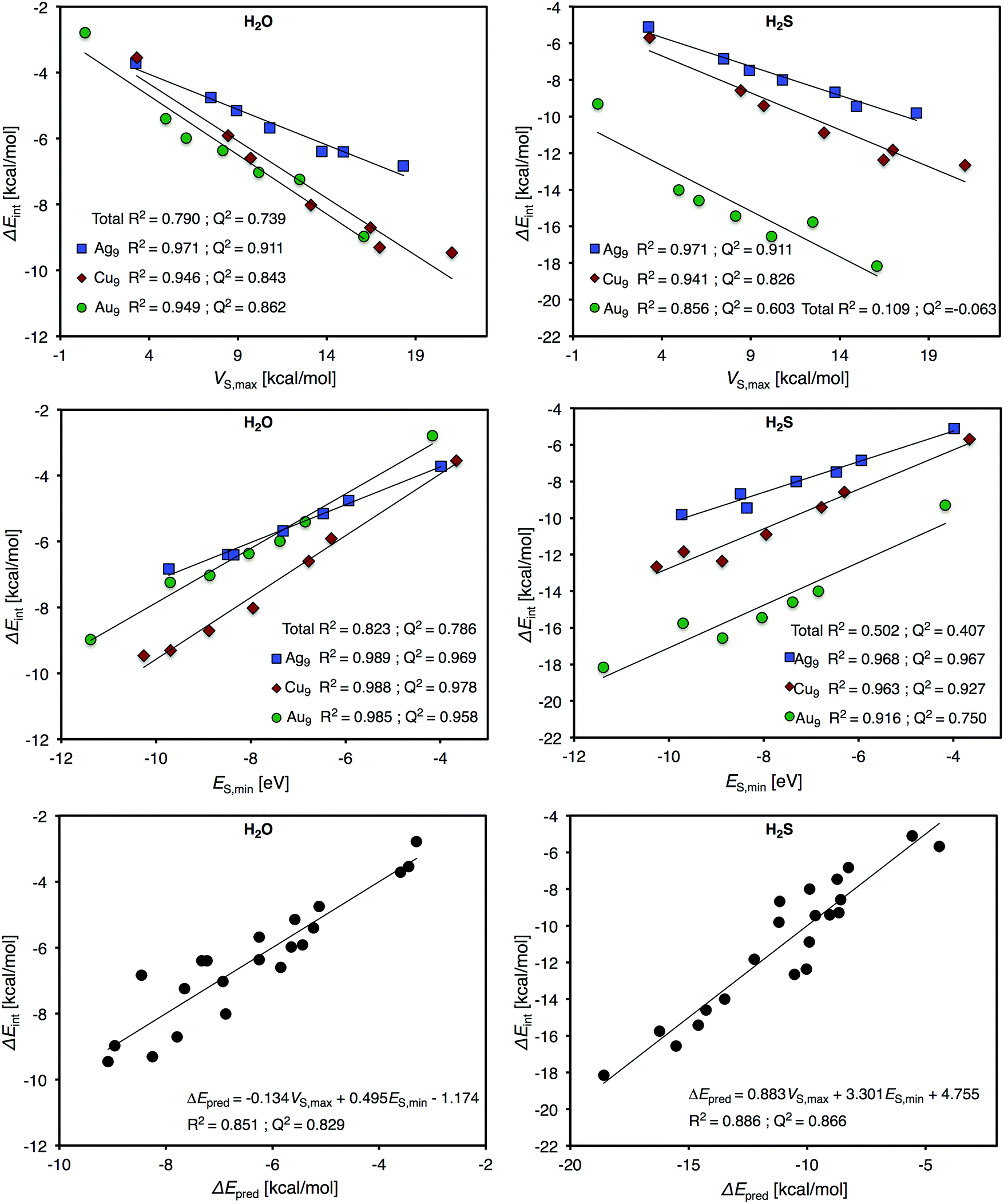 | ||
| Fig. 8 Correlation plots for H2O (left) and H2S (right) interaction energies onto the various sites of the geometrical isomers Au9, Ag9 and Cu9versus the site resolved VS,max (top panel) and ES,min (middle panel) obtained at the 0.001 a.u. isosurface. The bottommost figures show the calculated interaction energy, ΔEint, of H2O and H2S versus predicted interaction energies, ΔEpred, from multi-linear combinations of the VS,max and ES,min. | ||
In order to find an explanation to the differences between the particles, we turn to an electronic structure analysis. The three particles are approximately isoelectronic in the valence shell and display similar orbital configuration profiles, with Au having unoccupied orbitals of slightly lower energy as well as two more unoccupied orbitals below the zero-energy limit compared to the other particles. Apart from that, all orbitals are isolobal for the Au, Ag and Cu particles, with only slight dissimilarities, as can be seen in for instance the LUMO orbitals of Fig. S4 in the ESI.† Hence the variations in the VS(r) and ES(r) amid the particles are due to small differences in their electronic structures, which illustrate the need for a more sophisticated interaction affinity descriptor than e.g. the HOMO and/or LUMO energies. The small deviations between the particles in the correlations of the descriptor values versus adsorption energies can further be explained by geometrical effects upon adsorption, with larger distortions generally giving rise to reduced correlations (see the ESI†).
As mentioned in the introduction, interactions are often rationalized in terms of contributions of different physical characters. While V(r) obviously provides information on the particles’ ability to participate in electrostatic interactions, E(r) also accounts for the ability of the particle to accept electrons in electron-transfer processes taking place upon interactions. The relative influence of electrostatics versus charge-transfer/polarization in the various interactions is attainable by the formation of a multi-variant combination of the VS,max (electrostatics) and ES,min (both electrostatics and charge-transfer/polarization) properties of the different sites of the Au9, Ag9 and Cu9 particles. First of all, one should note that, although the NPs display differences in both the descriptor properties and in the interaction energies, the H2O interaction energies of all the particles put on the same trend line yields a clear correlation with the local values of the individual descriptor, as can be seen in Fig. 8. By this approach, the obtained R2 values for VS,max and ES,min are 0.790 and 0.823 respectively. This can be compared to the corresponding trends for H2S adsorption, which display much weaker correlations with R2 values of 0.109 (VS,max) and 0.502 (ES,min) respectively (see Fig. 8). By the formation of a linear combination between the VS,max and ES,min determined at all the unique adsorption sites of the three NPs, the corresponding H2O interaction energies are predicted with a R2 coefficient of 0.851, i.e. only moderately better than the individual descriptors. For the case of H2S a more pronounced effect is obtained and the correlation is increased to R2 = 0.866 (Fig. 8). From the above one could argue that in order to capture the variations of the H2S interaction over the different particles, one has to account for both their electrostatic and charge-transfer/polarization capabilities. In contrast, the interaction of H2O seems to be of a simpler nature; based on the hard character of H2O, and the generally good performance of VS,max for the estimation of H2O interaction energies, it is reasonable to assume that the interactions are dominated by electrostatics for all the considered NPs. The difference between H2O and H2S can be further demonstrated by assessing the weighting coefficients in their respective multi-linear combinations: for H2O the weighting coefficients of VS,max and ES,min are −0.134 and 0.495, whereas for H2S the coefficients are 0.838 and 3.301 (VS,max in kcal mol−1 and ES,min in eV). Note here that the sign of the VS,max coefficient is changed going from H2O to H2S. Since ES(r) consists of both electrostatic and charge-transfer/polarization contributions, this indicate that, in order to be able to compare the H2S interaction of the different particles, we have to remove a portion of the electrostatic component. This is in contrast with H2O, where the electrostatic component of E(r) instead has to be reinforced.
3.4. CO adsorption onto Ag9–Ag18 particles
In the study of Duanmu et al.,37 CO adsorption energies onto each unique Ag site were determined for the Ag9, Ag11, Ag17 and Ag18 nanoparticles using the N1278 DFT exchange–correlation functional in combination with the 6-311+G(2df,2p) basis set for carbon and oxygen and cc-pVDZ-PP for Ag. As described in Section 3.3, CO interacts C-down and adsorbs at on top sites of the Ag-atoms. The interaction energies range from −3.5 to −16.1 kcal mol−1. By re-optimization of the adsorption structures on the Ag9 particle, we find binding distances of 2.14 ± 0.03 Å.Duanmu et al.37 used partial charges to rationalize the variations of the CO interaction energies at different sites of the Ag nanoparticles. As shown in Table 3, the VS,max descriptor performs better than the CM5M partial atomic charges presented in the work of Duanmu et al., both overall and for each of the particles separately. The same is true when comparing VS,max to atomic charges obtained by other protocols, including Mulliken, Bader and NBO charges. The success of VS,max is not surprising in this context. In contrast to partial charges, the electrostatic potential is a rigorously defined property and provides an unbiased understanding of the interaction. In addition, the local electrostatic potential is a physical observable and its spatial variations are known to be influential in directing chemical interactions.8
| V 0.004 | V 0.001 | E 0.004 | E 0.001 | V + Ed | Ī S,ave | E + Ie | CM5Ma | Mulliken | Bader | NBO | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a From ref. 37. b The Ag9(2), Ag17(1), Ag17(5), Ag18(1), Ag18(6), Ag18(7) and Ag18(8) sites of the 0.004 a.u. isodensity surface have no true minimum in E(r). c The Ag9(2), Ag11(4), Ag17(1), Ag17(5), Ag18(1), Ag18(3), Ag18(6), Ag18(7) and Ag18(8) sites of the 0.001 a.u. isodensity surface have no true minimum in E(r). Instead the E(r) values on the isodensity surface along the Ag-nucleophile bond were used. d Estimated interaction energy obtained by a linear combination of V = V0.001(r) and E = E0.001(r): ΔEint(estimated) = 0.256V − 0.034E − 0.048 [kcal mol−1]. e Estimated interaction energy obtained by a linear combination of E = E0.001(r) and I = ĪS,ave: ΔEint(estimated) = −0.047E + 0.257I − 37.420 [kcal mol−1]. f The corresponding R2 with H2O adsorption energies at the PBE0-D3(BJ)/def2-TVZPP//PBE0-D3(BJ)/def2-TVZP level of theory is V0.004 = 0.950 and V0.001 = 0.938 as well as E0.004 = 0.919 and E0.001 = 0.922. g A similar linear combination of V + I gives a R2 of 0.830. | |||||||||||
| Ag9 | 0.934 | 0.918 | 0.887 | 0.911 | 0.918 | 0.505 | 0.881 | 0.74 | 0.021 | 0.007 | 0.751 |
| Ag11 | 0.907f | 0.868f | 0.743f | 0.761f | 0.832 | 0.516 | 0.794 | 0.79 | 0.135 | 0.074 | 0.614 |
| Ag17 | 0.828 | 0.838 | 0.942 | 0.941 | 0.901 | 0.938 | 0.960 | 0.84 | 0.072 | 0.001 | 0.440 |
| Ag18 | 0.781 | 0.812 | 0.859 | 0.834 | 0.827 | 0.823 | 0.835 | 0.78 | 0.037 | 0.097 | 0.341 |
| Total | 0.817 | 0.819 | 0.835 | 0.819 | 0.839 | 0.731 | 0.850g | 0.73 | 0.047 | 0.030 | 0.427 |
| Q 2 | 0.779 | 0.786 | 0.804 | 0.782 | 0.808 | 0.690 | 0.822 | 0.68 | −0.111 | −0.072 | 0.357 |
| SE | 1.393 | 1.385 | 1.322 | 1.387 | 1.307 | 1.689 | 1.263 | 1.687 | 3.180 | 3.209 | 2.467 |
Regarding the ES(r) descriptor, it gives the best overall correlation amongst all individual descriptors if evaluated at the 0.004 a.u. isosurface, but performs poorly for the Ag11 particle. This can be traced to an underestimation of the interaction strength at the Ag11(7) site, which is also manifested, but to a smaller degree, for the VS(r) descriptor. On the other hand, the ES(r) correlation for the CO adsorption onto the Ag17 particle is very good with a coefficient of determination (R2) of 0.942 on the 0.004 a.u. isodensity surface. Moving on over the series of Ag particles, we find that ES(r) indeed performs well as an indicator of the CO-affinity. The performances of both ES(r) and VS(r) for CO adsorption are, however, slightly inferior to that of e.g. H2O adsorption, and could be linked to the mixed donor–acceptor interaction character of CO, as pointed out previously in Section 3.2, whereas H2O is purely an electron donor. Fig. 9 gives an overview of the performances of VS(r) and ES(r) for the estimations of the local CO-affinity evaluated on the 0.001 a.u. isodensity surfaces.
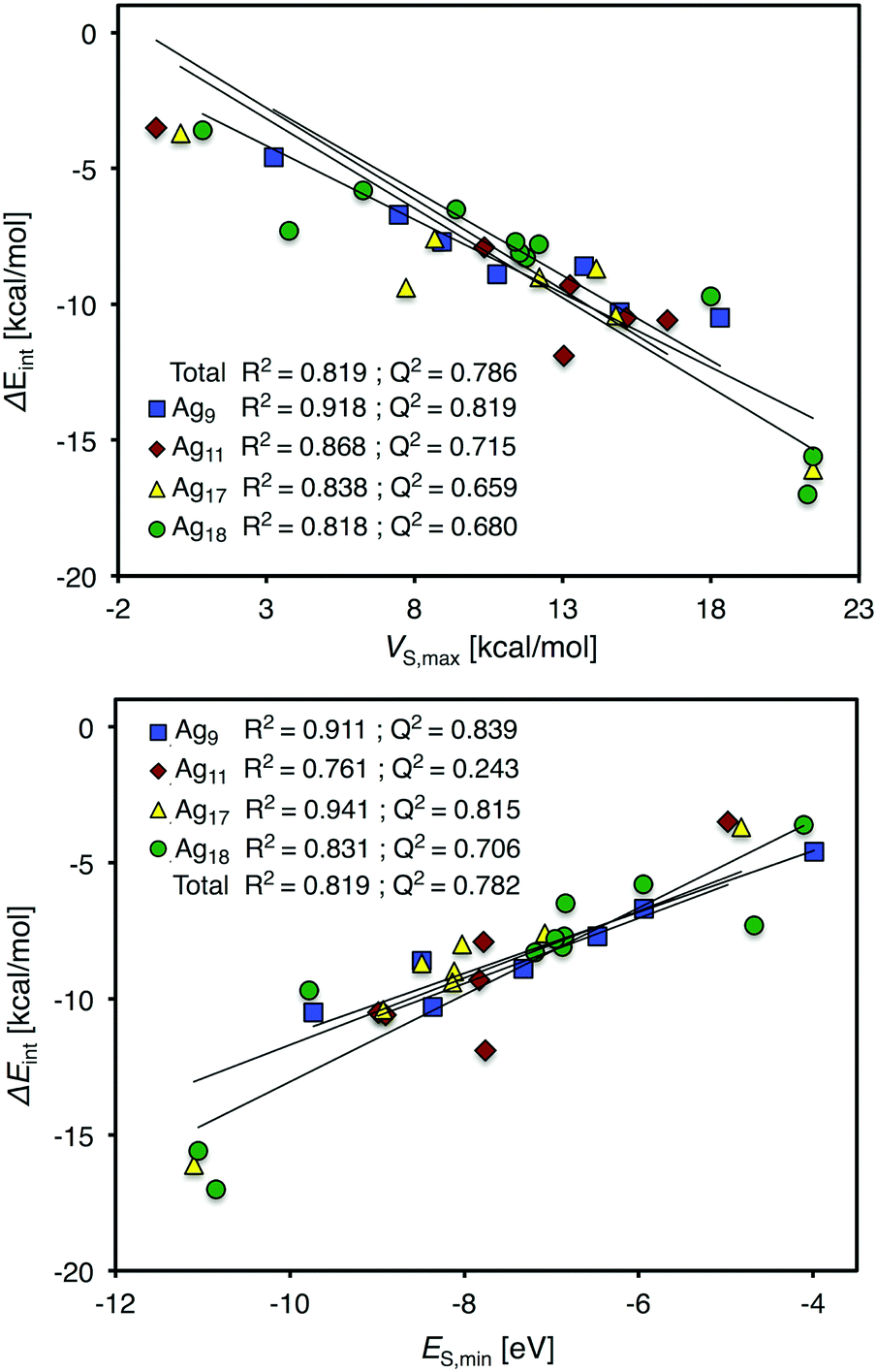 | ||
| Fig. 9 Plots presenting the correlation between site-resolved CO adsorption energies and descriptor values at the adsorption sites for the Ag9, Ag11, Ag17 and Ag18 nanoparticles separately and combined (total). VS,max (top) and ES,min (bottom) were obtained at the 0.001 a.u. isodensity surface. | ||
In addition to the analysis on the 0.004 a.u. isodensity surface, VS,max and ES,min values obtained on the 0.001 a.u. isosurface are also included in Table 3. Although the differences are small, the comparison suggest that the 0.004 a.u. isosurface is slightly better overall for ES(r) while for VS(r) the 0.001 and 0.004 a.u. isosurfaces give similar predictions of the Ag NP CO affinity. However, for the interactions with other adsorbates and including the Au and Cu particles, the general conclusion is that the 0.001 a.u. isosurface gives slightly more accurate results than the 0.004 a.u. isosurface for the group 11 metal NPs.
Since E(r) and V(r) are known to provide complementary information to some degree,23 it is not surprising that a multi-linear combination of E(r) and V(r), as seen in Table 3, increases the correlation with the interaction energies – this is because we are now able to describe a larger portion of the interaction energy. We, furthermore, find that the dual Lewis acid/base character of CO is reflected by the finding that the ĪS(r) Lewis basicity descriptor correlates fairly well with the CO adsorption energies [ĪS(r) is here in the form of the mean average local surface ionization potential at the 0.001 a.u. isosurface, ĪS,ave, of the atom of interaction since minima in ĪS(r) could in general not be identified at the atomic sites]. The mixed electron donor/acceptor character of CO is further supported by the fact that a multi-linear combination of ĪS,ave and ES,min gives the best overall correlation with the CO–Ag-particle interactions (R2 = 0.850).
4. Conclusion
In the current work we have evaluated the performance of a series of isodensity surface based DFT-descriptors for analyzing and predicting local Lewis acidity and basicity. The evaluation was carried out on a selection of Ag, Au and Cu nanoparticles, by correlating descriptor values with site-resolved interaction energies of small probe molecules. The descriptors include the density dependent surface electrostatic potential [VS(r)] as well as two multi-orbital descriptors: the surface average local ionization potential [ĪS(r)] that characterizes the local electron donating capacity of the particles, and the local surface electron attachment energy [ES(r)] that depicts the local electron acceptor capacity of the particle. Qualitatively, both electron donating and accepting properties are well described. From adsorption studies of small probe molecules, we conclude that positions of high susceptibility towards electron-accepting compounds are found at bridge and hollow sites of the noble metal nanoparticles. Upon interactions with electron-donors, the atomic on top sites are instead favored. These findings coincide with areas of high Lewis acidity and basicity defined by the descriptors, e.g., Lewis acidic areas of high VS(r), i.e. σ-holes, correspond to adsorption sites for electron donors, whereas Lewis basic sites of low VS(r), i.e. σ-lumps, adsorb electron acceptors.We find that the interaction energies for the adsorption of CO, H2O, NH3 and H2S Lewis bases can be successfully ranked, both by local maxima in VS(r), σ-holes, as well as minima in ES(r) at the on top sites of the noble metal nanoparticles. The descriptors readily outperform the comparatively simple FMO concept and have a much larger predictive power than partial atomic charges obtained by various protocols. Due to the similarity between halogen and hydrogen bonding and the interaction of the NPs with electron donors (e.g. H2O) via sites of high VS(r), i.e. σ-holes, we have introduced a new class of bonds – regium bonds – where the name reflects the noble character of the Cu, Ag, and Au metals.
As concerning the interactions with the BH3, BF3, HCl, and Na+ electron accepting probe compounds, the same quality of correlation cannot be obtained as for the interactions with electron donors. This is attributed to an increased level of complexity upon interactions, including adsorbate bending, multi-atom interaction and rearrangement of the substrate nanoparticle.
We have further described that the ĪS(r) and ES(r) descriptors provide complementary information to VS(r) with regard to the different contributions to the interaction energy: VS(r) reflects the electrostatic part of the interactions while ĪS(r) and ES(r) also quantify the metal particles’ charge transfer/polarization abilities. Although ES(r) and ĪS(r) display excellent capabilities to identify and rank nanoparticle adsorption sites, we find from our analysis that the adsorption sites are best identified by VS,max and VS,min. Accordingly, electrostatics is expected to be the dominating factor for interactions with hard species. Nevertheless, we also find that in order to reflect trends in the interaction energies of the Cu, Ag and Au particles with e.g. the soft H2S molecule, both electrostatic and charge-transfer/polarization effects have to be included.
As has previously been demonstrated within the field of molecular chemistry, we anticipate that the tested descriptors will be broadly applicable in the nanoparticle and materials sciences. In the near future we will show this for systems ranging from nanoparticles of various sizes and composition to semi-infinite materials and surfaces including metals and metal oxides. The findings of the present study are envisaged to have implications in areas as diverse as heterogeneous catalysis, nucleation/dissolution processes, particle transportation, chromatography, corrosion and nanotoxicology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the Swedish Nuclear Fuel and Waste Management Company (SKB) and the School of Chemical Science and Engineering at KTH (via its Excellence award to J. H. S.). The calculations were performed at resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Centre (NSC) in Linköping University as well as at the PDC Center for High Performance Computing (PDC-HPC).References
- R. Ferrando, J. Jellinek and R. L. Johnston, Nanoalloys:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) From Theory to Applications of Alloy Clusters and Nanoparticles, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
From Theory to Applications of Alloy Clusters and Nanoparticles, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed. - J. Kim, J. E. Dick and A. J. Bard, Advanced Electrochemistry of Individual Metal Clusters Electrodeposited Atom by Atom to Nanometer by Nanometer, Acc. Chem. Res., 2016, 49, 2587–2595 CrossRef CAS PubMed.
- Z. Luo, A. W. Castleman and S. N. Khanna, Reactivity of Metal Clusters, Chem. Rev., 2016, 116, 14456–14492 CrossRef CAS PubMed.
- M. Haruta and M. Daté, Advances in the catalysis of Au nanoparticles, Appl. Catal., A, 2001, 222, 427–437 CrossRef CAS.
- N. Zhou, V. López-Puente, Q. Wang, L. Polavarapu, I. Pastoriza-Santos and Q.-H. Xu, Plasmon-enhanced light harvesting: applications in enhanced photocatalysis, photodynamic therapy and photovoltaics, RSC Adv., 2015, 5, 29076–29097 RSC.
- P. Di Pietro, G. Strano, L. Zuccarello and C. Satriano, Gold and Silver Nanoparticles for Applications in Theranostics, Curr. Top. Med. Chem., 2016, 16, 3069–3102 CrossRef CAS.
- E. Roduner, Size matters: why nanomaterials are different, Chem. Soc. Rev., 2006, 35, 583–592 RSC.
- J. S. Murray and P. Politzer, The electrostatic potential: an overview, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 153–163 CrossRef CAS.
- J. H. Stenlid, A. J. Johansson and T. Brinck, Searching for the thermodynamic limit – a DFT study of the step-wise water oxidation of the bipyramidal Cu7 cluster, Phys. Chem. Chem. Phys., 2014, 16, 2452–2464 RSC.
- D. Farmanzadeh and T. Abdollahi, Investigation on the chemical active sites of copper nanoclusters as nanocatalyst for the adsorption of acetylene: calibration of DFT method and basis set, Theor. Chem. Acc., 2016, 135, 1–14 CrossRef CAS.
- D. Farmanzadeh and T. Abdollahi, a model for the ethylene and acetylene adsorption on the surface of Cun (n = 10–15) nanoclusters: A theoretical study, Appl. Surf. Sci., 2016, 385, 241–248 CrossRef CAS.
- J. H. Stenlid, A. J. Johansson and T. Brinck, σ-Holes on Transition Metal Nanoclusters and Their Influence on the Local Lewis Acidity, Crystals, 2017, 7, 222 CrossRef.
- J. H. Stenlid and T. Brinck, Extending the σ-Hole Concept to Metals: An Electrostatic Interpretation of the Effects of Nanostructure in Gold and Platinum Catalysis, J. Am. Chem. Soc., 2017, 139, 11012–11015 CrossRef CAS PubMed.
- P. Politzer and J. S. Murray, The fundamental nature and role of the electrostatic potential in atoms and molecules, Theor. Chem. Acc., 2002, 108, 134–142 CrossRef CAS.
- T. Brinck, J. S. Murray and P. Politzer, Surface electrostatic potentials of halogenated methanes as indicators of directional intermolecular interactions, Int. J. Quantum Chem., 1992, 44, 57–64 CrossRef.
- P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, An overview of halogen bonding, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- T. Clark, M. Hennemann, J. Murray and P. Politzer, Halogen bonding: the σ-hole, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- T. Brinck, in Theoretical Organic Chemistry, ed. C. Párkányi, Elsevier Science B.V., Amsterdam, 1st edn, 1998, vol. 5 Search PubMed.
- E. D. Stevens, Experimental electron density distribution of molecular chlorine, Mol. Phys., 1979, 37, 27–45 CrossRef CAS.
- S. J. Harris, S. E. Novick, J. S. Winn and W. Klemperer, (Cl2)2: A polar molecule, J. Chem. Phys., 1974, 61, 3866–3867 CrossRef CAS.
- R. G. Pearson, Chemical Hardness, Wiley-VCH Verlag GmbH & Co. KGaA, 1997, pp. 1–27 Search PubMed.
- P. Sjoberg, J. S. Murray, T. Brinck and P. Politzer, Average local ionization energies on the molecular surfaces of aromatic systems as guides to chemical reactivity, Can. J. Chem., 1990, 68, 1440–1443 CrossRef CAS.
- T. Brinck, P. Carlqvist and J. H. Stenlid, Local Electron Attraction Energy and its Use for Predicting Nucleophilic Reactions and Halogen Bonding, J. Phys. Chem. A, 2016, 120, 10023–10032 CrossRef CAS PubMed.
- T. Brinck, J. S. Murray and P. Politzer, Relationships between the aqueous acidities of some carbon, oxygen, and nitrogen acids and the calculated surface local ionization energies of their conjugate bases, J. Org. Chem., 1990, 56, 5012–5015 CrossRef.
- P. Politzer, J. S. Murray and F. A. Bulat, Average local ionization energy: A review, J. Mol. Model., 2010, 16, 1731–1742 CrossRef CAS PubMed.
- J. H. Stenlid and T. Brinck, Nucleophilic Aromatic Substitution Reactions Described by the Local Electron Attachment Energy, J. Org. Chem., 2017, 82, 3072–3083 CrossRef CAS PubMed.
- F. A. Bulat, M. Levy and P. Politzer, Average Local Ionization Energies in the Hartree–Fock and Kohn–Sham Theories, J. Phys. Chem. A, 2009, 113, 1384–1389 CrossRef CAS PubMed.
- T. Brinck, J. S. Murray and P. Politzer, Molecular surface electrostatic potentials and local ionization energies of Group V–VII hydrides and their anions: Relationships for aqueous and gas-phase acidities, Int. J. Quantum Chem., 1993, 48, 73–88 CrossRef CAS.
- B. Ehresmann, B. Martin, A. H. C. Horn and T. Clark, Local molecular properties and their use in predicting reactivity, J. Mol. Model., 2003, 9, 342–347 CrossRef CAS PubMed.
- T. Clark, The local electron affinity for non-minimal basis sets, J. Mol. Model., 2010, 16, 1231–1238 CrossRef CAS PubMed.
- J. F. Janak, Proof that ∂E/∂ni = εi in density-functional theory, Phys. Rev. B: Solid State, 1978, 18, 7165–7168 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: the PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- K. Duanmu and D. G. Truhlar, Partial Ionic Character beyond the Pauling Paradigm: Metal Nanoparticles, J. Phys. Chem. C, 2014, 118, 28069–28074 CAS.
- Y. Zhao and D. G. Truhlar, A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- M. M. Quintal, A. Karton, M. A. Iron, A. D. Boese and J. M. L. Martin, Benchmark Study of DFT Functionals for Late-Transition-Metal Reactions, J. Phys. Chem. A, 2006, 110, 709–716 CrossRef CAS PubMed.
- S. C. Ammal and A. Heyden, Modeling the noble metal/TiO2(110) interface with hybrid DFT functionals: a periodic electrostatic embedded cluster model study, J. Chem. Phys., 2010, 133, 164703 CrossRef PubMed.
- C. Di Valentin, G. Pacchioni and A. Selloni, Electronic Structure of Defect States in Hydroxylated and Reduced Rutile TiO2(110) Surfaces, Phys. Rev. Lett., 2006, 97, 166803 CrossRef PubMed.
- C. M. Lousada, A. J. Johansson, T. Brinck and M. Jonsson, Reactivity of metal oxide clusters with hydrogen peroxide and water – a DFT study evaluating the performance of different exchange–correlation functionals, Phys. Chem. Chem. Phys., 2013, 15, 5539–5552 RSC.
- M. Bühl, C. Reimann, D. A. Pantazis, T. Bredow and F. Neese, Geometries of Third-Row Transition-Metal Complexes from Density-Functional Theory, J. Chem. Theory Comput., 2008, 4, 1449–1459 CrossRef PubMed.
- J. Carrasco, J. Klimeš and A. Michaelides, The role of van der Waals forces in water adsorption on metals, J. Chem. Phys., 2013, 138, 24708 CrossRef PubMed.
- J. Carrasco, B. Santra, J. Klimeš and A. Michaelides, To Wet or Not to Wet? Dispersion Forces Tip the Balance for Water Ice on Metals, Phys. Rev. Lett., 2011, 106, 26101 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid functionals based on a screened Coulomb potential, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Erratum: “Hybrid functionals based on a screened Coulomb potential” [J. Chem. Phys., 118, 8207 (2003)], J. Chem. Phys., 2006, 124, 219906 CrossRef.
- O. A. Vydrov and G. E. Scuseria, Assessment of a long-range corrected hybrid functional, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, Erratum: “Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes” [J. Chem. Phys., 119, 12129 (2003)], J. Chem. Phys., 2004, 121, 11507 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. Reiher, O. Salomon and B. A. Hess, Reparameterization of hybrid functionals based on energy differences of states of different multiplicity, Theor. Chem. Acc., 2001, 107, 48–55 CrossRef CAS.
- A. Slimani, X. Yu, A. Muraoka, K. Boukheddaden and K. Yamashita, Reparametrization Approach of DFT Functionals Based on the Equilibrium Temperature of Spin-Crossover Compounds, J. Phys. Chem. A, 2014, 118, 9005–9012 CrossRef CAS PubMed.
- P. E. M. Siegbahn, The performance of hybrid DFT for mechanisms involving transition metal complexes in enzymes, J. Biol. Inorg. Chem., 2006, 11, 695–701 CrossRef CAS PubMed.
- P. E. M. Siegbahn and F. Himo, Recent developments of the quantum chemical cluster approach for modeling enzyme reactions, J. Biol. Inorg. Chem., 2009, 14, 643–651 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- Jaguar, version 7.9, User Manual, Schrödinger LLC: New York, 2014.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, The NBO Version 3.1, 1990 Search PubMed.
- R. S. Mulliken, Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- R. F. W. W. Bader, Atoms in Molecules – A Quantum Theory, Oxford University Press, 1990 Search PubMed.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases, J. Chem. Theory Comput., 2012, 8, 527–541 CrossRef CAS PubMed.
- A. R. Leach, Molecular Modeling: Principles and Applications, Pearson Education, Ltd, Harlow, England, 2nd edn, 2001 Search PubMed.
- T. Brinck, Modified Interaction Properties Function for the Analysis and Prediction of Lewis Basicities, J. Phys. Chem. A, 1997, 101, 3408–3415 CrossRef CAS.
- G. Blyholder, Molecular Orbital View of Chemisorbed Carbon Monoxide, J. Phys. Chem., 1964, 68, 2772–2777 CrossRef CAS.
- B. Hammer, Y. Morikawa and J. K. Nørskov, CO Chemisorption at Metal Surfaces and Overlayers, Phys. Rev. Lett., 1996, 76, 2141–2144 CrossRef CAS PubMed.
- A. Poater, M. Duran, P. Jaque, A. Toro-Labbé and M. Solà, Molecular Structure and Bonding of Copper Cluster Monocarbonyls CunCO (n = 1–9), J. Phys. Chem. B, 2006, 110, 6526–6536 CrossRef CAS PubMed.
- L. G. M. Pettersson and A. Nilsson, A Molecular Perspective on the d-Band Model: Synergy Between Experiment and Theory, Top. Catal., 2014, 57, 2–13 CrossRef CAS.
- A. Föhlisch, M. Nyberg, J. Hasselström, O. Karis, L. G. M. Pettersson and A. Nilsson, How Carbon Monoxide Adsorbs in Different Sites, Phys. Rev. Lett., 2000, 85, 3309–3312 CrossRef PubMed.
- A. Föhlisch, M. Nyberg, P. Bennich, L. Triguero, J. Hasselström, O. Karis, L. G. M. Pettersson and A. Nilsson, The bonding of CO to metal surfaces, J. Chem. Phys., 2000, 112, 1946–1958 CrossRef.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, Consistent van der Waals Radii for the Whole Main Group, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- A. Bondi, van der Waals Volumes and Radii, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- H. Häkkinen, S. Abbet, A. Sanchez, U. Heiz and U. Landman, Structural, Electronic, and Impurity-Doping Effects in Nanoscale Chemistry: Supported Gold Nanoclusters, Angew. Chem., Int. Ed., 2003, 42, 1297–1300 CrossRef PubMed.
- R. Peverati and D. G. Truhlar, Exchange–Correlation Functional with Good Accuracy for Both Structural and Energetic Properties while Depending Only on the Density and Its Gradient, J. Chem. Theory Comput., 2012, 8, 2310–2319 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp06259a |
| ‡ Note that in KS-DFT most neutral compounds have one or more virtual orbital(s) with negative eigenvalue(s), exceptions are e.g. small and hard molecules such as H2O. The NPs considered in this study have 22–40 virtual orbitals of negative eigenvalue (see Table 1). |
| § Relativistic effects are important, in particular for Au compounds, and affect the geometry and chemical properties significantly. By the use of ECPs scalar relativistic effects are implicitly accounted for. We have previously showed that ECP and relativistic all-electron calculations yield similar results.13 |
| ¶ At the considered level of theory basis set superposition errors (BSSEs) are small. Using a similar computational set up for H2O adsorption onto the Cu7 nanoparticle we have previously found a BSSE of 0.06 eV.9 |
| || It should, nevertheless, be noted that only five points are used in the multi-linear regression, which in-fact is not a sufficient number for a proper analysis. |
| This journal is © the Owner Societies 2018 |